
Submitted:
20 December 2023
Posted:
20 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Microvascular integrity is a critical factor in myocardial fluid homeostasis. The subtle equilibrium between capillary filtration and lymphatic fluid removal is disturbed during pathological processes leading to inflammation, but also in hypoxia or due to alterations in vascular perfusion and coagulability. The degradation of the glycocalyx as the main component of the endothelial filtration barrier as well as pericyte disintegration result in accumulation of interstitial and intracellular water. Moreover, lymphatic dysfunction evokes an increase in metabolic waste products, cytokines and inflammatory cells in the interstitial space contributing to myocardial oedema formation. This leads to myocardial stiffness and impaired contractility eventually resulting in cardiomyocyte apoptosis, myocardial remodelling and fibrosis.
The following article reviews pathophysiological inflammatory processes leading to myocardial oedema including myocarditis, ischaemia-reperfusion injury and viral infections with a special focus on the pathomechanisms evoked by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. In addition, clinical implications including potential long-term effects due to viral persistence (long-COVID), as well as treatment options are discussed.
Keywords:
myocardial oedema
; glycocalyx
; coronavirus disease 2019
; microcirculation
; platelets
1. Introduction
Myocardial fluid homeostasis is based on a complex interaction between microvascular filtration and absorption, interstitial hydration as well as water uptake by cardiomyocytes, and lymphatic removal [1]. Pathological conditions like ischaemia, ischaemia-reperfusion injury, inflammation, and hypertension disturb the subtle equilibrium and dysregulate myocardial fluid dynamics [2]. As a consequence, tissue oedema, characterized by the accumulation of water in interstitial and intracellular compartments, occurs and results in cardiomyocyte injury, dysfunction, and subsequent cardiac remodelling [3,4,5,6].
Myocardial oedema (MO) has been identified in various cardiac diseases including heart failure (HF) [1], ischaemia-reperfusion injury [7] , and myocarditis [8,9]. MO develops not only due to disturbances to the microvascular barrier leading to increased endothelial permeability through glycocalyx degradation and pericyte detachment, but also due to changes in the composition of the myocardial extracellular matrix (mECM), the cardiac lymphatic system and cardiomyocyte homoeostasis [1]. Myocardial cells play a critical role in fluid homeostasis regulation through the control of ion pumps and membrane-bound proteins [10].
The myocardium is considered to be one of the most vulnerable tissues for oedema formation due to high metabolic demand, low oxygen extraction reserve [11] and autoregulation of blood flow, which relies on close proximity between cardiomyocytes and endothelial cells [12]. Consequently, MO may contribute to myocyte ischaemia by expanding the interstitial space and consecutively increasing distances for oxygen transport [13].
The mECM is integral for the physiologic function of the myocardium [14,15], with its primary components being collagen subtypes I and III, which provide stability and tensile strength [16]. Beyond mechanical support, the mECM is actively involved in signal transduction, regulating cellular differentiation, growth and survival [17].
The disruption of balance between mECM degeneration through enzymatic proteolysis by matrix-metalloproteinases and its inhibition by tissue inhibitors of metalloproteinases (TIMPs) results in increased collagen deposition and manifests as myocardial fibrosis [14]. Degradation and remodelling of the mECM have been observed in acute myocarditis [18] and in chronic heart failure [4,19], with MO playing an active role in promoting these processes [4,20]. MO can facilitate myocardial fibrosis development by increasing mRNA levels of collagen types I and III as well as prolyl 4-hydroxylase [20]. Increased collagen deposition is associated with impaired cardiac contractility, greater left ventricular stiffness and HF [21,22].
Endothelial dysfunction in combination with the breakdown of the endothelial protective layer, i.e. the glycocalyx, can also be induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. This contributes to MO formation through heightened inflammation and generates a pro-thrombotic state via activation of platelets and the coagulation cascade [23,24,25,26,27].
Furthermore, the interaction of pathological processes initiated or exacerbated by MO, including inflammation, endothelial cell dysfunction, release of reactive oxygen species and proinflammatory cytokine signalling, contribute to a vicious circle of immunothrombosis, ultimately leading to tissue necrosis, fibrosis and organ failure [28,29,30].
2. Physiological background of myocardial fluid filtration
2.1. Starling forces and microvascular fluid filtration
A fundamental framework for understanding the forces regulating microvascular fluid exchange, homeostasis and its dysregulation leading to MO can best be described by the Starling´s principle [31] and the revised Starling equation [32]. The rate of fluid filtration (JV) per surface filtration area (S) is dependent on the capillary hydraulic conductivity (LP), the difference in capillary and interstitial hydrostatic pressure (delta P) and the difference in osmotic pressure across the membrane derived from plasma proteins (delta Π) [32]. This rate is strongly influenced by the permeability of the endothelial barrier to various solutes, primarily, which is expressed by the protein reflection coefficient σ [33].
Essentially, the rate of volume filtration per endothelial area is dependent on membrane permeability, differences in intracapillary and interstitial hydrostatic pressure, and variations in intracapillary and interstitial colloid osmotic pressure.

Table 1.
Revised Starling equation and numerical estimates.
| JV =LPS[(PC-PI)-σ(ΠC-ΠG)] |
| LPS: ~ 0.35 ml min-1 mmHg-1 100 g-1 [34,35] |
| S: 500 cm2 g-1 [35,36] |
| PC: end-diastolic 20 - 30 mmHg [37,38] |
| PI: ~120 mmHg during systole, 15 mmHg during diastole [39,40] |
| σ: 0.51 - 0.67 for plasma proteins; 0.41 - 0.59 for albumin [39,41,42] |
| ΠC: 21 - 24 mmHg [39,43,44] |
| ΠG: 13-20 mmHg [45] |
JV = Rate of volume filtration; LP = hydraulic conductivity, i.e. permeability or how easily fluid passes through a certain medium/pores; S = surface filtration area, i.e. endothelial area in case of blood vessels; PC-PI = intracapillary hydrostatic pressure (PC) - interstitial hydrostatic pressure (PI); σ = protein reflection coefficient describes the permeability of a membrane to a given solute; ΠC-ΠG = intracapillary colloid osmotic pressure (ΠC) - subglycocalyx colloid osmotic pressure (ΠG).
PC has a major influence on net fluid flow [2], while PI, which approximates arterial pressure during systole, is its main opposing force. Therefore, myocardial fluid exchange likely takes place predominantly during diastole [2,39]. ΠC is provided by plasma proteins unable to surpass the endothelial barrier and acts as a major opposing force to PC. Alterations of ΠC, for example during hypoalbuminaemia, trigger interstitial fluid extravasation [46]. Furthermore, the glycocalyx as part of the endothelial barrier, not only binds certain proteins such as von Willebrand factor (vWF) or coagulation factor IX, but also regulates passage of molecular substances such as fibrinogen or albumin based on their charge, thereby mediating fluid filtration via σ and ΠG [47,48,49].
A key factor in myocardial fluid homeostasis and preventing interstitial fluid accumulation is the removal of filtered fluid via lymphatic fluid drainage [1,50]. Although myocardial fluid exchange is suspected to occur mainly on the venular side of the capillary bed [51], it is important to recognize that most tissues greatly rely on lymphatic fluid removal rather than venous capillary absorption for interstitial fluid removal and tissue fluid balance [32]. This becomes evident, as impaired lymphatic drainage is commonly accompanied by oedema formation [46]. Therefore, accumulation of interstitial fluid, manifested as oedema, is the result of fluid filtration volume exceeding the lymphatic fluid drainage.
Alteration of starling forces can lead to enhanced fluid filtration and result in MO [32]. Fluid accumulation in the interstitium occurs, when the forces driving the fluid out of the vessel are greater than the opposing forces responsible for intravascular fluid retention. Factors favouring fluid filtration are high LP, high S, high PC, high ΠG, low PI, low ΠC, low σ [32]. Increased capillary pressure can result from conditions such as acute or chronic arterial hypertension, pulmonary hypertension, right ventricular failure, fluid overload in decompensated heart failure or venous obstruction [20,42,46,52,53]. Reduced colloid osmotic pressure typically results from hypoalbuminaemia, which can be attributed to malnutrition, malabsorption, nephrotic syndrome or hepatic failure [46], leading to greater fluid flow and interstitial oedema [6]. This is especially relevant in cardiac surgery and shock management, where crystalloid coronary perfusion and high levels of fluid resuscitation worsen prognosis [2,54,55]. Furthermore, inflammation increases capillary recruitment and leads to vasodilation, which substantially enhances fluid filtration by increasing PC, capillary permeability, blood flow and reducing the colloid osmotic pressure [32]. In addition, inflammation may cause a reduction in PI via changes in the connective tissue and extracellular matrix [56].
2.2. The glycocalyx as key regulator of the endothelial barrier
The endothelial surface layer (ESL) is a network made up of proteoglycans, glycoproteins and glycosaminoglycans, i.e. the glycocalyx, including syndecan-1, glypican, and hyaluronan, in conjunction with associated plasma proteins and soluble glycosaminoglycans [47,57]. The extension of the glycocalyx into the luminal surface of blood vessels varies depending on vessel size, and it projects into gaps between cells and fenestrations [47,57,58,59,60,61]. The glycocalyx exhibits constant change and is in equilibrium between continuous production and shedding due to enzymes or mechanical forces [47,62].
Functionally, the glycocalyx plays a vital role in the proper functioning of small blood vessels and serves critical vasoprotective functions [47]. It physically shields the underlying endothelium and limits immune cell interaction with the vessel walls [47,61,63]. It also regulates endothelial inflammation by capturing cytokines and restricting access to surface receptors [47,61,63]. The glycocalyx is negatively charged and contributes to the endothelial barrier by regulating plasma flow and passage of differently charged molecules [61,64]. In the context of the Frank Starling equation, the glycocalyx and the subglycocalyx space exert opposing forces on volume filtration influencing permeability (LP, σ), and providing an opposing colloid osmotic pressure (ΠG) against the intracapillary colloid osmotic pressure. This function results from anionic sites that repel negatively charged substances and cells, such as red blood cells, while facilitating the passage of positively charged proteins and molecules [48,64,65,66,67]. The removal of glycocalyx-binding proteins and further components increases conductivity adding to microvascular permeability [68,69,70,71,72,73]. Moreover, the glycocalyx regulates microvascular flow via transmission of shear-stress and nitric oxide production (NO) [74,75]. Furthermore, it protects against oxidative stress and endothelial dysfunction by binding antioxidant enzymes like superoxide dismutase to counteract oxygen radicals [76]. The intact glycocalyx also prevents platelet activation and thrombus formation [47,77] by binding and interacting with anticoagulant mediators such as antithrombin III [77] or heparin cofactor II [78].
3. Pathophysiology
3.1. Glycocalyx disintegration
Glycocalyx disruption with concomitant breakdown of the endothelial barrier is described in many critical illnesses and can mostly be attributed to inflammation [79]. The greater exposure of the endothelial surface enables immune cells and harmful agents like proteases and reactive oxygen species (ROS) to interact more readily with the endothelium [79].
The endothelial glycocalyx has a critical role in preventing MO formation [80]. Damage to the coronary glycocalyx results in myocardial fluid accumulation and swelling of the pericapillary interstitial space [81,82,83]. Glycocalyx alteration or neutralization of its negative charge appear to affect its barrier function leading to enhanced permeability [64,84]. Experimental glycocalyx-degradation was found to elevate capillary permeability [85], ultimately leading to myocardial oedema formation [80]. In vivo, alterations of the glycocalyx and microvascular permeability changes have been linked to conditions such as hypertension [52] and hypoproteinaemia [86], sepsis [87], viral infections including SARS-CoV-2 [23,24,25], inflammation and myocarditis [88]. Serious infections and systemic inflammation are known to increase microvascular permeability and induce cardiac dysfunction [89]. Viral infections enhance vascular permeability by upregulating glycocalyx degrading enzymes such as sialidase and heparanase [90,91]. In sepsis, MO formation has been attributed to the shedding of negatively charged glycocalyx molecules [92].
Pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) can disrupt the glycocalyx and increase permeability independent of leukocyte adhesion [93]. In addition, initial degradation of the glycocalyx eases leukocyte and platelet adhesion to endothelial cells [47,63]. Cytokines activate both leukocytes and endothelial cells, leading to upregulation of adhesion molecules and facilitate recruitment of inflammatory cells to sites of inflammation [94]. After adhesion of leukocytes to the endothelium, release of proteases and free radicals can exacerbate endothelial damage that can contribute to oedema formation [95,96,97,98]. Inflammatory stimuli also affect the endothelial barrier by targeting downstream signalling molecules that ultimately control intercellular junctions [99] (see also 3.2).
Hypoxia has been linked to increased permeability via glycocalyx disruption and MO formation [100]. Additionally, in the context of inflammation and atherosclerosis, oxidized low-density lipoprotein (LDL) has been shown to degrade the glycocalyx [101], increase microvascular permeability, [102,103] and facilitate adhesion of leucocytes potentially enhancing atherosclerotic plaque development [104,105,106].
Moreover, damage to the endothelial surface layer can also be observed in the setting of inflammation related to ischaemia-reperfusion injury [107,108,109]. The generation of ROS triggers NLRP3 inflammasome activation leading to caspase-1 mediated pyroptosis [110,111,112].
Ischaemia-related changes and chronic inflammation serve as potent triggers for proangiogenic factors such as vascular endothelial growth factor (VEGF) and hypoxia-inducible factor (HIF)-1 alpha [113,114,115,116]. VEGF can increase vascular permeability via SRC signalling, facilitating protein and cell migration for angiogenesis [99,117]. Beside VEGF, matrix metalloproteinases (MMP) play a key role in angiogenesis, as they degrade the extracellular matrix [118]. Inflammatory conditions and exposure to inflammatory mediators, such as TNF-α, can enhance MMP expression and activity [119]. Importantly, MMP9 is associated with a greater risk of coronary atherosclerosis and cardiovascular events [120].
Cardiac natriuretic peptides have a significant impact on circulatory balance via their strong natriuretic and diuretic effects [121]. Natriuretic peptides like A-type natriuretic peptide (ANP) can also increase capillary permeability by increasing capillary filtration [122,123]. Experimental ANP administration caused degradation of the glycocalyx contributing to enhanced fluid filtration [82]. Besides ANP, vascular barrier function was also influenced by B- and C-type natriuretic peptide (BNP and CNP) leading to shedding of glycocalyx components such as syndecan-1 and heparan sulphate [124]. NP-related shedding of glycocalyx components was accompanied by enhanced permeability and significant fluid extravasation [124,125]. Interestingly, NT-proBNP levels have been shown to be independently linked with in-hospital mortality of COVID-19 patients with pneumonia, but without HF [126]. Moreover, NT-proBNP serves as an efficient biomarker for identifying patients at risk of cardiac events, as demonstrated in patients with type 2 diabetes without preexisting cardiac disease [127].
3.2. Intercellular junctions and key signalling processes
Besides the glycocalyx, the endothelial barrier is upheld by intercellular junctions, i.e. tight and adherens junctions, which cover the intercellular gap between endothelial cells [61,64,99].
Tight junctions (TJ) consist of the three main components claudins, occludins and junction adhesion molecules [128]. They are attached to the cellular actin cytoskeleton via cingulin, which interacts with one of the zonula occludens (ZO) proteins (ZO-1, 2, or 3) [129].
TJ are controlled by Rho GTPases, kinases and phosphatases [99,130,131,132,133,134]. Adherens junctions (AJ) are also connected to the actin cytoskeleton via vascular endothelial-cadherin (VE-cadherin), a key protein in controlling and maintaining the endothelial barrier [135]. VE-cadherin also interacts with several kinases, phosphatases and other signalling molecules, which control its function and ultimately mediate VE-cadherin stability [99,136,137]. Herein, actin plays a decisive role in regulating the endothelial barrier [138]. Actin filaments attached to tight and adherens junction proteins can open intercellular gaps via contraction after a rise in cytosolic Ca2+ and activation of the myosin-light-chain kinase with concomitant inhibition of the myosin-light-chain-phosphatase through RhoA signalling [139]. This reorganization of actin filaments can be triggered by various inflammatory mediators [140,141,142]. The Rho-GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) is a signalling molecule that coordinates actin-binding proteins to stabilize intercellular junctions [143]. Under physiological conditions Rac1 increases endothelial barrier function, via signalling pathways involving sphingosine-1-phosphate (S1P) [144,145,146] or angiopoietin-1 (Ang-1) [99,147,148]. S1P stabilizes the glycocalyx by decreasing the activity of MMPs, hereby reducing the shedding of syndecan-1, chondroitinsulphate and heparan sulphate [149]. Signalling pathways involving guanine nucleotide exchange factors (GEFs) such as T-lymphoma invasion and metastasis inducing protein 1 (Tiam1), guanine nucleotide exchange factor 2 (Vav2), and Trio Rho guanine nucleotide exchange factor (Trio) activate Rac1 [99,150,151]. These GEFs are regulated by cyclic adenosine monophosphate (cAMP) signalling, predominantly through guanine nucleotide exchange factor 3 (Epac1) dependent pathways as opposed to those initiated by protein kinase A (PKA) [152,153]. Elevated levels of cAMP enhance the endothelial barrier via activation of Rac1 and can protect against external barrier compromising factors [150,154,155].
Inflammation disrupts the endothelial barrier by opening of TJ and AJ resulting in increased permeability, which facilitates the transport of fluids, soluble substances and cells [99,150,156,157,158,159,160]. There are two main factors contributing to this process: the phosphorylation of AJ components [137,161] and an imbalance in the signalling of Rac1 and RhoA in favour of RhoA, which enables actin-contraction on VE-cadherin and opening of junctions [99,139,162,163]. Both factors result in the endocytosis of VE-cadherin and disassembly of junctions. In systemic inflammation several inflammatory mediators are associated with AJ and TJ internalization and increased permeability [99]. For example, TNF-α, lipopolysaccharide (LPS) or thrombin can lead to a reduction in cAMP levels and deactivate Rac1 resulting in barrier breakdown and increased permeability [150,156,157,158,159]. TNF-α and LPS signalling involves activation of the metalloprotease a disintegrin and metalloproteinase 10 (ADAM10) [164]. Phosphorylation of VE-cadherin is facilitated by the activation of Src and other kinases [165].
3.3. Inflammation and myocardial oedema
MO can occur as the consequence of acute or sub-acute events and herein as a response to inflammatory, ischemic or prothrombotic stimuli [10].
The impact of inflammation on the endothelial barrier and fluid filtration is profound, with the potential to multiply net fluid filtration several times (up to 17 fold) [32]. Moreover, pro-inflammatory cytokines can cause a systemic capillary leak syndrome, rarely resulting in myocardial involvement [166]. This is mostly due to interstitial oedema, which may progress to acute hypotension with cardiac shock and acute ventricular dysfunction [166]. Systemic capillary leak syndrome can be triggered by viral infections such as parvovirus B19, dengue virus or SARS-CoV2 [166,167,168]. The latter has been reported to be driven by the SARS-CoV-2 spike protein via involvement of glycosaminoglycans, integrins, as well as the TGF-β signalling axis, altogether mediating endothelial dysfunction and extracellular matrix reorganization [168].
Importantly, the inflammation and oedema formation, amplify the activity of enzymes that degrade the mECM [169]. Usually beta1-integrin bound collagen fibrils to fibroblasts exert a compressive force countering interstitial expansion. However, during inflammation these collagen fibrils can detach from fibroblasts, which greatly affects PI [32,56]. Inflammation and MO also result in impaired myocardial contraction and relaxation [55,170,171]. Moreover, MO disturbs ventricular filling through increased diastolic chamber stiffness [169]. The extent of MO has prognostic value in various myocardial disease entities including acute heart failure [53,172], myocardial infarction [173], aortic stenosis [174], pulmonary arterial hypertension [175], and myocarditis [176,177]. MO seen in HF can result from impaired myocardial contractility reducing lymph flow rate [178]. Additionally, it can be attributed to increased venous congestion affecting fluid dynamics [179] as well as inflammation and oxidative stress, which increase microvascular permeability [180,181].
In addition, pro-inflammatory cytokines disrupt the endothelial cells' ability to protect against platelet activation through mechanisms like NO, prostacyclin production, and expression of CD39 (ecto-ADPase) [182,183,184,185]. Various cytokines such as TNF-α and IL-6 have been found to reduce the expression of CD39 in blood vessels, contributing to ADP-triggered platelet activation and an enhanced interaction between platelets and white blood cells [186,187]. Moreover, IL-6 can induce angiogenesis via VEGF [188,189], leading to greater vascular permeability [163,190]. Further, TNF and interleukin-1 (IL-1) can facilitate activation of endothelial cells, which can increase local blood flow via NO-mediated relaxation of vascular smooth muscle tone and increase leukocyte recruitment [191,192,193]. Inflammation effectively induces a collapse of the endothelial barrier and leads to increased movement of fluids, molecules and proteins into the interstitium [194], caused by increased capillary permeability, increased blood flow and reduced colloid osmotic pressure [32].
Cardiac pericytes, which interact with microvascular endothelium, are also implicated in myocardial remodelling associated with MO and concomitant inflammation [195]. Pericytes, located around endothelial cells in microvessels, are vital for capillary maintenance and functionality [196,197]. Pericytes regulate blood flow through an alteration of the capillary diameter in response to a series of vasoactive molecules [198,199]. Myocardial capillary pericytes also express ACE-2 [200], making them susceptible to SARS-CoV-2. Upon activation and inflammatory injury, pericytes may transdifferentiate into myofibroblast, which secrete mECM promoting myocardial fibrosis [201]. Additionally, pericytes can regulate the entry of immune cells [197] by either relaxing or widening gaps and allowing diapedesis of leukocytes [202,203]. Under certain circumstances pericytes can become migratory, for example after increased angiopoietin-2 signalling, hereby contributing to vascular dysfunction and increased permeability [204].
3.4. Detection of myocardial oedema and myocarditis
For the non-invasive evaluation of myocarditis, the updated Lake Louise Criteria are applied [207]. Therein, the combination of at least one finding indicative of MO and one finding of non-ischaemic myocardial injury are utilised to make a diagnosis of acute myocarditis with sufficient sensitivity and specificity [207]. Various cardiac magnetic resonance imaging (CMR) sequences are employed to evaluate MO, hyperaemia, capillary congestion or leakage, necrosis, and fibrosis [207].
MO is represented by prolonged T1 and T2 relaxation times due to increased extracellular fluid content. However, prolonged T1 times are also seen in fibrotic areas [208] and in the setting of acute decompensated heart failure with concurrent volume overload due to venous congestion [53]. However, unlike myocarditis, MO linked to heart failure-related congestion is relieved upon successful recompensation [53]. Similar to other tissues, hyperaemia and capillary leak are hallmarks of inflammation in the myocardium and are represented by early T1 mapping in CMR [209]. Contrast agents based on gadolinium, which mark the extracellular space expansion, show distinct myocardial uptake patterns in inflamed and non-inflamed areas of the myocardium [209]. Late gadolinium enhancement (LGE) in a non-subendocardial pattern is considered indicative of severe inflammation or fibrosis [207]. While less specific, ventricular dysfunction and pericardial involvement are supportive criteria for myocarditis [207]. Caution is advised in the context of myocardial infarction, as myocardial injury, including MO due to ischaemia and reperfusion injury may mimic some features also seen in myocarditis. However, these features typically exhibit different distribution patterns and may change during recovery [210,211].
While less specific, the updated Lake Louise Criteria can also be utilised to identify chronic viral myocarditis [212]. However, it's worth noting that these criteria are not fully applicable in COVID-19 [213]. Also, CMR in acute viral myocarditis is yet unable to differentiate between myocarditis due to direct infection of the myocardium and secondary immune responses with cardiac involvement [212].
When routine cardiac workup and CMR are not sufficient to diagnose myocarditis, an endomyocardial biopsy (EMB) is recommended [214]. This has the advantage that subsequent analysis of EMB samples can not only help define the aetiology, but also facilitates the decision on specific treatment options [215].
3.5. SARS-CoV-2 Infection
Recently, SARS-CoV-2 infection has become a major global burden with complications primarily impacting the respiratory and cardiovascular system [216,217].
SARS-CoV-2 can enter host cells utilizing the ACE-2 receptors [218] (Figure 1). Viral entry gets facilitated by CD209L, NRP1, TMPRSS2, heparan sulphate and cathepsin B/L [219,220,221,222,223].
Additionally, viral S protein is recognized as PAMP (pathogen-associated molecular pattern) by toll-like receptor (TLR) 2 on monocytes, macrophages, and lung epithelial cells [224].
Virus-cell interactions are suspected to decrease ACE-2 activity and elevate angiotensin II (AngII), a potent vasoconstrictor [225,226,227] that promotes thrombogenicity, oxidative stress and inflammation [218]. The ACE-2 receptor and TMPRSS2 are abundantly expressed throughout the body, found in the respiratory tract and small intestine epithelium, smooth muscle and endothelial cells in arteries, and in cells like pericytes and myocytes [218,228,229]. The wide distribution of ACE-2 and TMPRSS2 expression could explain the variety of symptoms, changes and complications during and after SARS-CoV-2 infection. For instance, in patients suffering respiratory symptoms, SARS-CoV-2 molecules could be found within capillary endothelial and alveolar epithelial cells, both cell types that express ACE-2 receptors [230,231].
SARS-CoV-2 particles have even been discovered in myocardial interstitial cells, either through transient viremia or the migration of infected macrophages, in cases of COVID-19-related cardiogenic shock [232]. Additionally, SARS-CoV-2 antigens were also detected in cardiomyocytes [233].
Cardiovascular diseases (CVD) themselves have been linked to negative effects on the microvasculature, leading to impaired capillary perfusion and glycocalyx integrity as reported in previous studies [234,235,236,237]. When considering preexisting CVD, it is noteworthy that SARS-COV-2 infection is associated with even more adverse outcomes and greater disease severity [238], as COVID-19 has the potential to exacerbate CVD and evoke microvascular complications [239].
Severe COVID-19 infection can be linked to myocarditis, heart failure, cardiogenic shock, and renal failure [232,240,241,242,243]. These conditions can result in pronounced tissue inflammation, hypoxia, and imbalances in electrolytes, which can contribute to complications such as arrhythmias [244].
SARS-CoV-2 is suggested to interact directly with cardiac cells that have ACE-2 receptors, like pericytes or endothelial cells [229] and impair vascular integrity, leading to damage of the microcirculation which can lead to more inflammation, cardiac fibrosis, and prothrombotic processes [200]. In addition, SARS-CoV-2 infection could enhance microvascular permeability by disrupting signalling processes between pericytes and endothelial cells [245,246]. As a response, pericytes are supposed to release various factors including Angiopoietin-2 (Ang-2), transforming growth factor-β1, microRNA-132, and hepatocyte growth factor, which are linked to atrial fibrillation (AF) and promote local tissue inflammation as well as changes in atrial structure and electrophysiology [246,247,248,249,250,251,252,253]. Elevated plasma level of VEGF, Ang-2, and vWF in patients with AF indicate the contribution to a prothrombotic state [254]. Oedema formation leads to an increase in hydrostatic pressure in heart tissue, which can alter electrical properties and functions [255,256]. Elevated hydrostatic pressure also promotes the risk of AF by enhanced activation of the renin-angiotensin system (RAAS) [257]. This effect has been observed in both human tissues and the atria of hypertensive rats, where the onset of AF coincided with higher levels of angiotensin II, lower levels of angiotensin 1-7 and less ACE-2 receptor activation [257]. In addition, elevated levels of angiotensin themselves can lead to MO [258]. In cardiac tissue, angiotensin II can induce the release of reactive oxygen species (ROS) and activate redox-sensitive transcription factors such as NFkB, AP-1, and HIF-1, which stimulates the secretion of VEGF and prostaglandin, ultimately leading to greater vascular permeability [258].
SARS-CoV-2 enters endothelial cells via the interaction of the spike protein and ACE-2 receptor [218]. Entry gets facilitated by CD209L, NRP1, TMPRSS2, heparan sulphate, cathepsin B/L and triggers an endothelial inflammatory response [26,182,219,220,221,222,223,230,259,260]. Viral infection upregulates cytokines production (“cytokine storm”) [261], ROS production [262] and enhances the activity of glycocalyx degrading enzymes such as MMPs, heparanases and hyaluronidases [90,91,119,263,264,265]. Hereby the infection leads to endothelial cell injury, dysfunction and contributes to glycocalyx breakdown [23,24,25,26,182,230,259,260]. Moreover, exposure of the endothelial surface enables immune cells and harmful agents such as ROS to interact more readily with the endothelium [79]. As a result of pro-inflammatory intracellular signalling processes, intercellular junctions are dismantled leading to increased capillary fluid extravasation resulting in oedema formation [99,150,156,157,158,159,160]. Additionally, SARS-CoV-2 might also influence pericytes, which also express ACE-2 [200]. Infection could disrupt vital signalling processes between pericytes and endothelial cells [245,246]. This can stimulate pericyte transdifferentiation into myofibroblast promoting fibrosis [201], induce pericyte migration [204] and result in wider gaps allowing for greater diapedesis of leukocytes [202,203]. SARS-CoV-2 induced endothelial injury and dysfunction also creates a pro-thrombotic state with greater expression of TF, vWF, increased production of thromboxane and endothelin-1, ET formation as well as decreased prostacyclin and NO [182,183,184,185,254,266,267,268,269].
Abbreviations: ACE-2 receptor = angiotensin-converting enzyme 2 receptor; CD209L = cluster of differentiation 209; ET = extracellular trap; MMPs = matrix metalloproteinases; NO = nitric oxide; NRP1 = neuropilin 1; ROS = reactive oxygen species; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; TF = tissue factor; TMPRSS2 = transmembrane protease serine 2; vWF = von Willebrand factor.
Myocarditis in SARS-CoV-2 infection
In the context of the COVID-19 pandemic, the incidence, pathophysiology and prevalence of SARS-CoV-2-induced viral myocarditis has been controversially discussed. The exact mechanisms leading to myocardial injury and potentially myocarditis are yet only poorly understood, but two direct mechanisms (injury by infection of cardiomyocytes and infection of endothelial cells with subsequent endotheliitis) and three indirect mechanisms (hypercoagulability, injury from cytokine storm and systemic inflammation as well as auto-immune mediated damage) are currently under scrutiny [217]. Recent research has suggested that the extremely rare cases of (peri-) myocarditis associated with COVID-19 messenger ribonucleic acid based vaccine are likely caused by cytotoxic T-cells, pro-fibrotic monocytes, and up-regulation of pro-inflammatory cytokines [270]. However, whether these findings also apply to SARS-CoV-2 induced myocarditis are currently unclear.
The United States Centre for Disease Control calculated the risk to develop myocarditis in the course of COVID-19 to be 0.146% in both in- as well as out-patients, close to 16 times the risk for myocarditis compared to patients without COVID-19 [271]. In one retrospective study involving 6439 patients hospitalized for COVID-19, 37 (0.6%) developed de novo heart failure [272]. Patients recovering from COVID-19 have also reduced cardiac ejection fractions and increased left ventricular volumes compared to healthy controls [273]. Clinical presentations of patients with COVID-19 myocarditis vary from mild to fulminant with concomitant haemodynamic compromise, but most patients make a full clinical recovery [217]. However, CMR revealed that between 30% and 78% of recovered COVID-19 patients show signs of persistent myocardial injury including LGE, prolonged T1 and T2 times indicative of interstitial fibrosis and MO [274,275], as well as signs of ongoing inflammation [273,276].
The long-term consequences of myocardial injury in the course of COVID-19 have yet to be fully established and ought to be monitored regularly with clinical follow-ups, evaluation of cardiac arrhythmia and optimally repeated CMR [217].
Arrhythmias in SARS-CoV-2 infection
Arrhythmias, conduction disturbances and changes in the ST-segment are reported in more than half of COVID-19 patients three- and six-months after discharge from hospital [277]. Especially noteworthy is the potential association between SARS-CoV-2 infection and AF, both during the acute infection phase and potentially during recovery [278,279]. COVID-19 is linked to an increased incidence of AF and a detrimental impact on outcomes of hospitalized patients [280]. The reported frequency of arrhythmias and AF in the context of SARS-COV-2 infection and hospitalization varies, with studies showing prevalence rates ranging from 16.7% in Wuhan, China [281], compared to 27.5% of COVID-19 patients admitted to the ICUs in the USA [282].
As aetiology of electrophysiological changes different pathomechanisms are discussed. Electrical instability and remodelling mediated by SARS-COV-2 might be a result of mitochondrial dysfunction resulting in a cellular energy deficit as well as oxidative stress [283,284,285]. SARS-COV-2 can directly affect cellular functions by binding to the ACE-2 receptors on cardiomyocytes, cardiofibroblasts, pericytes and endothelial cells [200,286]. Immunhostochemical detection of SARS-CoV-2 spike and nucleocapsid protein in cardiac atrioventricular node has been reported in a patient with lethal COVID-19 infection, suggesting direct interaction with the cardiac conduction system [287]. In addition, the existence of the angiotensin-(1-7)/Mas receptor axis has been reported in rat sinoatrial cells [288]. This pathway is responsible for anti-inflammatory, anti-fibrotic and anti-hypertrophic signalling, a reduction of ROS formation and protection against arrhythmias during reperfusion injury [286,288]. Dysregulation can be observed during acute and possibly also chronic COVID-19 [289,290,291].
Moreover, myocardial oedema formation due to endothelial barrier disruption with activated pericytes is discussed as a major pathomechanism during SARS-CoV-2 infection [292]. The rise in hydrostatic pressures reduces the peak current density of the L-type calcium current, and upregulates the transient outward K+ current as well as the ultra-rapid delayed rectifier K+ current, resulting in a shortened action potential duration [256,257].
Though not yet proven specifically for COVID-19, the occurrence of arrhythmias has been associated with LGE in CMR, as seen in a relevant proportion of recovered COVID-19 patients [293].
Glycocalyx changes and inflammation in SARS-CoV-2 infection
Recent studies established the role of SARS-CoV-2 induced injury to the vasculature [26,182,230,259,260]. The invasion of SARS-CoV-2 into cells triggers endotheliitis, inducing endothelial damage and dysfunction [26,182,260]. A key part of the COVID-19 disease process driven by SARS-CoV-2 is glycocalyx disintegration, resulting in endothelial dysfunction [23,24,25,26]. This, combined with an unregulated host response that involves both proinflammatory and prothrombotic pathways, significantly contributes to the development of severe COVID-19 [26]. As a consequence, microvascular dysfunction leading to (micro-) thrombosis occurs, which may even result in potentially lethal ischemic multiorgan failure [294,295,296].
Infection with COVID-19 can induce a hyperinflammatory state and unleash a so called “cytokine storm”, leading to excessive production of various inflammatory mediators including numerous cytokines and chemokines [261]. Overproduction of these substances, including IL- 1α, IL- 1β, IL- 2, IL- 7, IL- 6, IL- 8, IL- 10, TNF, interferon (IFN)-γ, granulocyte colony- stimulating factor (G-CSF), IFN-inducible protein-10, monocyte chemotactic protein - 1, macrophage inflammatory protein 1 alpha and CXC- chemokine ligand 10 has been described [182,297,298,299,300,301,302,303].
Overexpression can have a multitude of effects as molecular signalling pathways of these factors can be quite complex and involve several cell types and proteins. Additionally, during inflammation endothelial derived angiopoietin-2 enhances vascular leakage in response to cytokines and inflammatory mediators such as histamine, bradykinin and VEGF [304]. Overexpression of pro-inflammatory factors, especially TNF-α and IL-6, have been observed in patients with dilated CMP and are greatly associated with activation of fibroblasts, collagen deposition and cardiac fibrosis [305,306,307]. Increased levels of IL-6, IL-8, and TNF-α have also been linked to worse outcomes in hospitalized COVID-19 patients [303].
These enhanced inflammatory responses promote the disintegration of the endothelial surface layer. Normally, the glycocalyx is in a balance of constant shedding due to flow and degrading enzymes, and renewal via production of new components [308]. However, during inflammation ROS [262] and the enzymes capable of degrading the glycocalyx such as hyaluronidases [263], heparinases [264] and MMPs [265] exhibit enhanced activity. In addition, there is increased leucocyte adhesion and diapedesis due to endothelial activation and upregulation of cellular adhesion molecules [83]. The impact of inflammation on the glycocalyx in COVID-19 is evident in the extent of damage it incurs, and may be associated with disease severity [309]. Patients with more severe COVID-19 infection, requiring ventilation showed a thinner glycocalyx layer with higher levels of circulating glycocalyx components than non-ventilated patients or healthy controls [310,311,312]. This can be explained by the fact, that patients with more severe COVID-19 had higher activity levels of MMPs, hyaluronidase or heparanase [312,313].
Immunothrombosis in SARS-CoV-2 infection
SARS-CoV-2 triggers a potent immunothrombotic response by the above described mechanisms. Endothelial injury and dysfunction results in simultaneous activation of platelets and the coagulation cascade as well as proinflammatory pathwyays [23,24,25,26,27]. Consequently, COVID-19 manifests as an extremely pro-thrombotic condition. The highly pro-thrombotic nature of COVID-19 is evident from the finding of microthrombi in the myocardium and widespread thrombosis in pulmonary vessels in COVID-19 patients [230,314]. Furthermore, thrombus formation has been demonstrated in both large and small blood vessels of arteries and veins [182,315].
Endothelial dysfunction results in decreased prostacyclin and NO production, while increasing the production of thromboxane and endotheliln-1, which greatly promotes vasoconstriction and thrombosis [267]. In addition, endothelial injury induces the expression of tissue factor (TF), hereby activating the coagulation cascade[184]. Inflammation also promotes the release von Willebrand factor (vWF) from endothelial cells, which can remain anchored to the endothelial surface eventually forming vWF- platelet aggregates[266,316,317].
NET formation can also contribute to immunothrombosis during COVID-19 [318]. Inflammation and infection activates neutrophil granulocytes and induces the formation of NETs, which is a defense mechanism against extracellular organisms [319,320]. Activated neutrophils release their granular and nuclear content to eliminate pathogens [319]. NET formation can be induced by the inflammatory response to the virus [182], and is thought to play a key role in the disease course and severity, as patients exhibit higher markers of NETs in comparison to healthy controls and have worse outcome [268]. Additionally, NET markers like citrullinated histone H3, cell-free DNA and neutrophil elastase are also associated with greater levels of leukocytes, inflammatory cytokines, and in vivo markers of coagulation, fibrinolysis, and endothelial damage [268]. In the context of SARS-CoV-2 infection, NET formation is increased and impairs endothelial function and vascular integrity hereby worsening patient outcomes [182,268,269].
Possible long-term effects of SARS-CoV-2 infection
Following recovery from acute SARS-CoV-2 infection, many patients continue to suffer a wide range of symptoms, commonly referred to as “long COVID” [296,321]. Recent research suggests that up to 30 % of patients may still experience symptoms nine months after COVID-19 infection [322]. In patients without previous heart disease 73% reported cardiac symptoms after approximately 100 days of mild SARS-CoV-2 infection related to signs of inflammation and MO in the heart in CMR [323]. Symptoms persisted in 57% of participants after approximately 1 year and were more likely in females and those with initial widespread heart inflammation [323]. Furthermore, persistent cardiovascular effects due to COVID-19 infection, such as myocardial fibrosis and oedema, as well as changes in the microvasculature like capillary damage, capillary blood flow alterations, tissue hypoxia, and inflammation have been described [217,324]. The potential mechanisms involved explaining these post-recovery phenomena might include viral persistence, interaction with other viruses, the triggering of autoimmune reactions, vascular damage culminating in chronic inflammation and microthrombosis [325].
Other emerging studies suggest that SARS-CoV-2 induces chronic inflammation, increased ROS production and a dysbalanced metabolic state due to mitochondrial dysfunction, especially in patients with previous suboptimal mitochondrial function and low mitochondrial reserve due to factors like comorbidities or age [326]. In this context, the release of oxidized DNA fragments from damaged mitochondria activating the NLRP3 inflammasome, is thought to be a driving force of chronic inflammation [327].
3.6. Cardiotrophic viruses and myocarditis
While myocarditis can arise from various pathogens, viruses are suspected to be the most common cause [328,329,330]. In addition, viral and postviral myocarditis are prominent factors contributing to both acute and chronic dilated cardiomyopathy [331]. Viral myocarditis is characterized by cardiac inflammation, necrosis and fibrosis with MO as a frequent complication, which can be detected via CMR [332], and is part of the Lake Louise Criteria [207]. In the literature several other viruses besides SARS-CoV-2, the so called cardiotrophic viruses are also associated with myocarditis and cardiomyopathies [333]. Highly prevalent are cardiotropic viruses such as parvovirus B19 [333,334] and human herpesvirus-6 [335]. Human cytomegalovirus [336,337,338], Epstein–Barr virus [339] and enteroviruses, particularly coxsackie viruses B [340], are described as moderately frequent [333]. Other rare cardiotropic viruses include adenovirus [340,341] and hepatitis C virus [342,343,344].
These cardiotropic viruses invade the myocardium using distinct host-cellular pathways resulting in endocytosis [333]. Prior to binding to the virus-specific receptor that initiates entry, the majority of viruses interact with glycocalyx components (Table 2) [345].
Herein, the negatively charged heparan sulphate proteoglycans are used by a large variety of viruses for attachment [346,347]. However, the intact glycocalyx hinders virus-cell receptor interaction, meaning, that glycocalyx disruption and removal can uncover the needed receptors and facilitate for viral entry [347]. For example, the interaction of ACE-2 receptors with the SARS-CoV-2 spike protein is dependent on the condition of the glycocalyx [348].
Viral entry can trigger a wide range of host-cell responses and activate the innate immune system, as viral proteins can be recognized by TLR1, TLR2, TLR4, TLR6, and TLR10 [349]. For instance, the spike protein of SARS-CoV-2 showed significant binding to TLR1, TLR6 and especially TLR4 [349,350]. Activation of TLR4 can cause cardiac injury and has been linked to cardiac hypertrophy, fibrosis and apoptosis, however the exact mechanism remains elusive [351]. Active viral replication and its transcription products have been shown to directly cause cardiomyocyte damage or apoptosis in coxsackievirus B3 and adenovirus infection [352,353]. In addition, parvovirus B19 and human herpesvirus 6 can cause vascular endothelial dysfunction, resulting in poor cardiomyocyte function [335,354,355]. Furthermore, myocarditis may occur, when infected immune cells introduce viral genetic material into the myocardial tissue, with viruses such as human herpesvirus 6 and Epstein–Barr virus playing a role [356]. Viral entry follows an inflammatory phase driven by immunological activation, primarily involving natural killer cells and macrophages, leading to cytokine-triggered inflammation, including IL-1, IL-2, TNF, and INF-gamma [357,358]. Initial cardiac damage facilitates the release of inflammatory cytokines and damage-associated molecular patterns (DAMPs) [333,359] initiating the infiltration of mononuclear cells such as lymphocytes and monocytes [360]. The most common inflammatory infiltrate in proven myocarditis is lymphocytic, followed by borderline, granulomatous, giant cell, and eosinophilic [361]. Ultimately, CD4+ and clonal B cell activation sustain local inflammation, leading to further myocyte necrosis, myocardial dysfunction, and additional negative inotropy, as TNF-mediated activation of endothelial cells amplifies cytokine production and inflammation, including inducible nitric oxide synthase activation [362,363]. The increase in proinflammatory chemokines and cytokines activates cardiac myofibroblasts and increases fibrous tissue formation [305]. If the virus persists, viral myocarditis can play a role in the gradual decline of cardiac function and the development of dilated cardiomyopathy [364]. Viral persistence is also associated with a progressive worsening of left ventricular ejection fraction, whereas viral elimination is linked to improved patient outcome as shown for enterovirus-associated cardiomyopathy [365,366]. Active viral replication could play a crucial role in this process [367,368,369]. Viral infections can also induce autoimmune processes leading to chronic inflammation, tissue remodelling, and the development of dilated cardiomyopathy [370,371,372,373,374]. Herein, autoimmune reactions can activate T cells that target the myocardium, potentially based on cross-reactivity/molecular mimicry phenomenon of viral epitopes and cardiac structures [375,376] . In addition, the production of cytokines like TNF, IL-1a, IL-1b, IL-2 and IFN- γ increases. Cytokines propagate myocardial damage in combination with antibodies against viral and cardiac proteins and, by affecting the contractile apparatus and matrix proteins, worsen systolic heart function [375,377,378,379,380].
Table 2.
Viruses and interaction with the glycocalyx.
| Virus | Glycocalyx components | Literature reference |
|---|---|---|
| Parvovirus B19 | heparan sulphate, sialic acid | [381] |
| Human herpesvirus 1, 6 | heparan sulphate, syndecan-1 | [382,383] |
| Epstein–Barr virus | glycoproteins, hyaluronan synthesis | [384,385] |
| Human cytomegalovirus | heparan sulphate | [386,387] |
| Enteroviruses | heparan sulphate, P-selectin glycoprotein ligand-1, sialylated glycan | [388,389,390] |
| Adenovirus | heparan sulphate, sialic acid | [391,392,393] |
| Hepatitis C virus | heparan sulphate, syndecan-1 | [394,395] |
| SARS-CoV-2 | heparan sulphate proteoglycans | [223,396] |
| Influenza virus | sialic acids, hyaluronan synthesis | [391,397] |
4. Clinical implications and treatment options
In addition to established evidence-driven therapeutic guidelines, novel translational approaches to cardiac inflammatory processes are needed to improve outcome and ameliorate associated complications in these patients.
4.1. Myocardial oedema and myocarditis
Patients with myocardial inflammation, myocarditis and MO have an increased risk of suffering potentially lethal arrhythmia and sudden cardiac death independent of myocardial dysfunction [398,399,400,401]. Both in ischaemic as well as in non-ischaemic cardiomyopathies, positive LGE is associated with a stark increase in incidence of arrhythmia [293]. This association is especially pronounced in patients with reduced left ventricular ejection fraction ≤ 30% (HFrEF) [293]. The cumulative evidence suggests a close correlation of LGE and T1-mapping in CMR with myocardial fibrosis, inflammation and oedema [207,208,399,402,403], which has also been validated by endocardial voltage mapping and histopathologic analysis [208,404]. As inflammation, oedema and the subsequent disturbance of cardiac conduction constitutes a substrate for serious arrhythmia [293,401,405,406,407], anti-inflammatory treatment strategies may help to alleviate the burden of arrhythmia in addition to guideline directed medical (GDM) therapy (e.g. anticoagulation, antiarrhythmics) . Upon persistence of AF or bradycardia/VT electrical CV or transient/permanent device-therapy may be required [408,409].
In addition to (critical) arrhythmia, myocarditis may also promote heart failure and cardiogenic shock [214,410,411]. In these patients, GDM HF treatment is recommended [214]. In the acute phase characterized by prominent congestion and fluid overload, diuretic treatment with loop diuretics is essential to remove excess water [214]. In severe cases of cardiogenic shock, inotropes and vasopressors, as well as mechanical circulatory support (MCS) such as veno-arterial extra-corporal membrane oxygenation (VA-ECMO) has to be considered [412,413].
Another frequent factor aggravating the inflammatory processes is anemia, which accounts for worse clinical outcome in hospitalized COVID-19 patients [414,415]. Anemia leads to enhanced platelet reactivity [416,417] and by contribution to prothrombotic processes and ischaemia its impact on myocardial oedema formation may be assumed. Therefore, prevention of anaemia should be a cornerstone in primary prevention.
In order to improve cardiac function and alleviate excessive inflammation in acute myocarditis, immuno-modulatory anti-cytokine therapy utilizing targeted biologics may prove viable depending on aetiology [418,419,420,421]. However, it should be noted that - though there were no safety concerns- IL-1ß inhibition by canakinumab did not show clinical improvement in the Canakinumab in COVID-19 Cardiac Injury trial at day 14 [422]. This was a randomized controlled trial comparing canakinumab (at a dose of 600mg or 300mg) to placebo in 45 patients with myocardial injury [422]. Future studies regarding dosing regimens and longer follow up periods are required for canakinumab and other immunotherapies [422,423,424].
In the context of auto-immune mediated myocarditis, (e.g., giant cell myocarditis or systemic autoimmune diseases including sarcoidosis, systemic lupus erythematosus (SLE), thyrotoxicosis, or granulomatosis with polyangiitis) corticosteroids, azathioprine, and cyclosporine are utilized [418,421]. However, evidence for these treatment strategies are often weak and recommendations are solely based on expert consensus and pathophysiologic considerations [418,425].
In a retrospective cohort of biopsy-proven virus-negative chronic inflammatory cardiomyopathy, immunosuppression on top of heart failure therapy resulted in improved survival compared to standard heart failure treatment alone [426]. In patients with ventricular arrhythmia, there are some signals that immunosuppression may alleviate arrythmia burden, but evidence remains tenuous [401,409,427].
Experimental and clinical evidence suggests a decisive role for inflammatory cytokines in regulation of cardiac remodelling, as biomarkers of inflammation are elevated in heart failure patients, even more so during acute cardiac decompensation [428,429,430]. In heart failure associated with disease other than myocarditis, immunomodulation has failed to demonstrate significant positive effects on mortality. [431].
The targeted biologic immunomodulators etanercept and infliximab, both inhibiting TNF-α, have been tested in HF with reduced ejection fraction (HFrEF), but provided no clinical benefit [432,433]. Left ventricular ejection fraction and left ventricular end-diastolic diameter were improved in a meta-analysis of 19 randomized controlled trials of 1341 patients, therefore indicating a potential role for immunomodulation in HFrEF [431]. In heart failure with preserved ejection fraction (HFpEF), a relevant role for inflammation in the pathogenesis is also assumed, but effective translational therapeutic approaches remain to be explored [428,429,434].
As previously mentioned, CMR is the gold-standard for the assessment of myocardial inflammation, oedema and fibrosis [207]. Hence, CMR may also be utilized to guide immunomodulatory therapy in chronic myocarditis [418]. Especially in the context of systemic inflammatory disease, a combination of CMR with functional imaging including fluorodeoxyglucose positron emission tomography could improve diagnosis, management and tapering regimes of immunosuppressive agents [418,435,436,437,438].
Decongestion with diuretics and standard HF treatment also relieves MO [53]. Recent guidelines recommend sodium-glucose cotransporter 2 inhibitors (SGLT-2i) in heart failure patients to reduce the risk for hospitalisation and cardiovascular death [214,439,440]. Though SGLT-2i are suggested to have anti-inflammatory properties and counteract myocardial fibrosis, an improvement of patient survival during hospitalised COVID-19 infection could hitherto not be shown in a metaanalysis including the DARE-19, RECOVERY and ACTIV-4A trial [29,441,442,443,444,445]. Another drug class recommended for heart failure treatment are mineralocorticoid receptor antagonists [214,440]. Herein, finerenone is suggested to preserve endothelial glycocalyx and to protect against COVID-19 associated adverse events in patients with type 2 diabetes and chronic kidney disease [446].
Additional future treatment options could target stimulation of lymphatic water removal. Experimental increase of lymphangiogenesis in a rat model of heart failure post myocardial infarction has been shown to improve restoration of myocardial fluid balance, and reduce cardiac inflammation, fibrosis, and dysfunction [447]. Also targeting intravascular pressure, colloid osmotic pressure and intravascular permeability could prove useful.
In myocarditis due to viral infection, direct antiviral therapy, interferon, and intravenous immunoglobulins may be considered depending on the viral pathogen [399]. In chronic viral myocarditis with entero-, or adenovirus, interferon-β treatment increased viral clearance of entero-, adeno, and parvovirus B19 and reduced endothelial damage in parvovirus B19 infection [340,399,448,449].
Rather than direct viral infection of cardiomyocytes, which has only been demonstrated for enterovirus, (e.g., coxsackievirus), molecular mimicry and subsequent auto-immune reaction is thought to contribute to cardiomyocyte injury [399,450,451,452]. However, the therapeutic implications of these findings remain a matter of debate as evidence is currently scarce and randomized controlled trials would be required [399,425].
Currently, several specific antiviral agents are available [399]. Pocapavir and pleconaril as well as intravenous immunoglobulin therapy have been explored for neonatal enteroviral myocarditis [453,454,455]. Anti-herpesvirus drugs such as ganciclovir can be used against persistent Epstein–Barr virus, cytomegalovirus or human herpesvirus 6 to reduce viral load [456]. Antiviral therapy against hepatitis C virus-associated myocarditis consists of established antiviral drugs such as ombitasvir, paritaprevir, ritonavir and dasabuvir [457]. Influenza positive myocarditis can be treated with the neuraminidase inhibitors peramivir and zanamivir [458,459]. Intravenous immunoglobulin therapy is often used in parvovirus B19 infection, with new treatment strategies such as synthetic nucleotide analogues cidofovir and brincidofovir (broad-range antivirals), synthetic coumarin derivates, flavonoid molecules, and hydroxyurea currently being explored [460]. Another approach consists of targeting autoantibodies via immunoadsorption or aptamers (synthetic oligonucleotides that can bind specific molecules like antibodies) [461].
Since SARS-CoV-2 induced COVID-19 is associated with cardiovascular injury, besides direct strategies targeting the virus itself, protection of the endothelium and the glycocalyx, as well as prevention of complications from endothelial injury and dysfunction may prove advantageous [182,216,217,238,239]. Several approaches to combat COVID-19 infection are currently being explored [462]. These include targeting the viral entry mechanisms, immune regulation pathways, or the lifecycle of the virus [462,463].
At the beginning of the COVID-19 pandemic, drug repurposing was explored, but with limited success [463]. Chloroquine and hydroxychloroquine prevent viral entry into the cell via the inhibition of glycosylation of host receptor proteins and manipulation of endosomal proteolytic processing [463]. Furthermore, both agents are also demonstrating ani-inflammatory effects by inhibiting cytokine production by reducing T cell activation [463,464]. Camostat mesylate and arbidol also inhibit host cell entry by inhibiting a host serine protease and interacting with the angiotensin converting enzyme 2 receptor and the S protein, respectively [463]. Lopinavir, darunavir, and remdesivir, agents that interfere with RNA synthesis, were also considered for trial in severe COVID-19 [463].
As of mid-2023, several direct antiviral therapeutics are available [462]. These include inhibitors of RNA-dependent RNA polymerase (remdesivir, molnupiravir, JT001), inhibitors of SARS-CoV-2 main protease (nirmatrelvir–ritonavir, ensitrelvir), and agents, which interfere with the interaction of the S protein and the angiotensin converting enzyme 2 receptor (bebtelovimab, regdanvimab, sotrovimab and others), the latter group being discontinued due to resistance of more recent virus strains [462]. In addition to antiviral agents, in some situations, immunomodulators are also recommended, including glucocorticoids, janus kinase inhibitors, and targeted cytokine antagonists against IL-6 and IL-1β [462].
4.2. Endothelial damage and glycocalyx disintegration
In COVID-19, endothelial cell infection leads to dysfunction of the endothelial surface layer and subsequent disturbances of haemostasis, thrombocyte aggregation and MO formation [25,26]. While this may also be observed in early disease stages, endothelial damage is thought to be a major contributor to multi-organ failure in severe COVID-19 [23,24,25,26].
Detailed pathophysiologic insight into the exact processes and deleterious stimuli, which the endothelium is exposed to, both in the context of ischaemia and reperfusion injury as well as viral infection, may inspire several techniques aiming to ameliorate glycocalyx disintegration [465,466,467]. While there are currently no established agents for this indication, various compounds have undergone testing [60].
The administration of nitric oxide during postischemic reperfusion was demonstrated to reduce vascular leakage and vascular resistance, as well as preserve glycocalyx integrity in guinea pig hearts in vitro [468]. Hawthorn extract WSS 1442 has the ability to increase coronary flow by boosting nitric oxide release from vascular endothelium [469], which aligns safeguarding or augmentation of the glycocalyx [470]. Moreover, WSS 1442 thickens the glycocalyx, which is linked to significantly reduced sodium permeability in vitro [471].
Hyperbaric oxygen, as a preconditioning stimulus, was shown to provide benefits and protection against ischaemia and reperfusion injury [466]. The presumed mechanism involves improving endothelial function and oxygenation while reducing local inflammation, vascular permeability, and tissue oedema [466]. The effect of preconditioning may be appreciated in nuclear magnetic resonance imaging and spectroscopy, where muscle metabolism is positively influenced by preconditioning during reperfusion, with increased production of phosphocreatine and greater oxygen consumption [472].
Furthermore, various agents resembling glycocalyx components are being explored [473,474,475,476,477]. Sulodexide is a natural glycosaminoglycan which regenerates the glycocalyx by boosting glycosaminoglycan synthesis and reducing degradation [473]. In the setting of type 2 diabetes and chronic venous disease it has been shown to have beneficial effects by regenerating the glycocalyx and combating endothelial dysfunction with anti-inflammatory effects [478,479]. Pentosan polysulphate is an oral heparin-like substance without notable anticoagulant properties and currently approved by the US Food and Drug Administration for the treatment of interstitial cystitis [480]. Research indicates it boosts glycosaminoglycan levels in diabetic mice and reduces glycocalyx breakdown via decreased MMP activity [60,474]. Wheat germ agglutinin lectin attaches to heparan sulphate and hyaluronic acid and has been shown to decrease albumin filtration and albuminuria in a rat model of chronic kidney dysfunction [475]. Rhamnan sulphate is a heparin-like compound [476] that resulted in an improved glycocalyx and decreased permeability in vitro [481]. Cationic copolymers were designed to specifically boost endothelial barrier function [477]. They have been demonstrated to diminish the increase in hydraulic conductivity caused by shear stress and pressure, as well as to decrease capillary filtration in an isolated perfused mouse lung model, suggesting potential utility in the treatment of pulmonary oedema [477]. As parts of the glycocalyx are often involved in viral entry, this could provide a potential new approach in targeting these components/developing new antibodies targeting these components. For example, interfering with the binding of a virus such as SARS-CoV-2 with heparan sulphate via neutralizing antibodies recovered from COVID-19 patients could be used to combat further infection [223].
Furthermore, substances that interfere with molecular signalling involved in leukocyte diapedesis and migration are also being explored [482]. The inhibition of intercellular adhesion molecule (ICAM)-1 to limit neutrophil infiltration prior to reperfusion exhibited protective properties as it demonstrated to reduce neutrophil recruitment and smaller infarct size following ischaemia [482].
In a murine model, the extracellular matrix protein Secreted Protein Acidic and Rich in Cysteine (SPARC) was shown to play a crucial role in protecting against cardiac inflammation and mortality in cases of viral myocarditis by preserving the integrity and barrier function of the endothelial glycocalyx [88]. The absence of SPARC lead to increased inflammation, reduced cardiac function, and mortality, but administering recombinant SPARC could reverse these effects, highlighting its potential therapeutic significance in viral myocarditis [88].
Plasma proteins like albumin can be used to treat conditions such as subarachnoid haemorrhage, shock, and trauma [483,484]. In a rodent model of haemorrhagic shock, the degradation of the glycocalyx and subsequent restoration by infusion of plasma was demonstrated in comparison to Ringer lactate [483]. Plasma-treated rodents showed increased syndecan-1 mRNA expression and reduced lung injury. This restoration aligns with S1P effects, where albumin carries sphingosine-1 phosphate (S1P) that protects against matrix metalloproteinase mediated glycocalyx degradation [149,485]. Therefore, plasma proteins like S1P could be crucial for safeguarding the glycocalyx. However, the glycocalyx and its implications in various diseases from atherosclerosis to shock and infection remain to be fully understood. Further research in this field is crucial and may provide new insights and novel treatment options.
4.3. Thrombosis
Thrombosis in COVID-19 is a critical aspect of disease progression and closely linked to endothelial dysfunction [182]. As a consequence of glycocalyx degradation and excessive inflammation, platelets are more likely to adhere to the endothelium, provoking both micro- as well as macrovascular thrombosis [27,486,487]. In addition to degradation of the glycocalyx, TLR-4 dependent mechanisms are also thought to contribute to immunothrombosis in COVID-19 [28,488]. While anticoagulation as therapeutic measure was accepted consensus early on in the pandemic, the exact timing, dosage, and duration remain still a matter of debate [489,490,491]. Recent data indicate that the establishment of a therapeutic anticoagulation could potentially improve clinical outcome in COVID-19 hospitalized patients in non-critical condition, particularly those at higher risk [492]. However, further research is needed to establish a consensus on antithrombotic therapy in the context of COVID-19 [489,490,491].
5. Discussion
Viral infections lead to systemic inflammatory processes affecting (micro-) vascular homeostasis [28,29,30,296]. During the acute phase, rapid cellular entry is facilitated through the destabilization of the protective endothelial shield, the glycocalyx [296]. The breakdown of the endothelial barrier leads to enhanced permeability shifting inflammatory processes to adjacent tissues [296]. The chronic phase comprises viral persistence and sub-clinical inflammation triggering immunothrombosis, oedema formation, tissue necrosis and, more specifically, myocardial remodelling resulting in heart failure [1,4,28,29,183,269,365,380].
Many viruses contribute to endoplasmic reticular stress by using the cells machinery for the production of large amounts of viral proteins as well as double-stranded RNA intermediates as part of their replication cycle [493]. Consequently, dsRNA is recognized by cytosolic pattern recognition receptors (PRRs) triggering pro-inflammatory responses, which include protein kinase R (PKR) and oligoadenylate-ribonuclease L (OAS-RNase L) activation as well as interferon production [494]. PKR activation is also central in the signalling cascade mediated by TLR-4 after sensing of viral glycoproteins [28]. This pathway seems to have a regulatory function, while promoting the activation of the transcription factor nuclear factor erythroid-2-related factor 2 (NRF2), which enhances the expression of anti-oxidant enzymes, such as superoxide dismutase 1 (SOD-1) or heme oxygenase 1 (HO-1) [495,496]. HO-1 induction has been shown to ameliorate reperfusion patterns, reduce I/R injury and oedema formation [497,498,499].
Immunothrombosis is a crucial pathway during acute SARS-COV-2 infection and initiated by TLR-signalling [28]. Herein, extracellular trap (ET) formation results in the release of nuclear as well as mitochondrial DNA and histones. The latter are recognized as DAMPs and promote thrombin generation through TLR-2 and TLR-4 on platelets [500,501]. Thrombin can activate platelets even at subnanomolar concentrations and despite P2Y12 inhibition it still promotes the formation of a considerable and stable amount of platelet aggregates [502,503,504,505,506]. Herein, it could be shown that ticagrelor inhibits thrombin-mediated platelet activation more strongly than prasugrel in patients with acute coronary syndrome and dual antiplatelet therapy [507].
(Micro-) thrombotic complications during SARS-CoV-2 infection are furthermore driven by the TLR-vWF-NETosis axis, destabilizing microvascular integrity [28,296].
Interestingly, in patients with acute lung injury/ acute respiratory distress syndrome higher angiopoetin-2 as well as vWF levels were associated with pulmonary permeability oedema [508]. Moreover, oedema formation during SARS-CoV-2 was also described in the brain of COVID-19 patients with neurological symptoms. Herein, widespread white matter volume shifts corresponding to vasogenic oedema were detected by multi-compartment diffusion microstructure imaging sequences of the MRI [509]. These changes together with fibrinogen leakage suggest the disruption of the blood-brain barrier [509,510]. The latter is characterized by a breakdown of pericyte homeostasis and perivascular inflammation, as documented in brain tissue samples from autopsies of COVID-19 patients [510]. SARS-CoV-2 infection of pericytes triggers in consequence viral entry into the central nervous system [510]. Moreover, viral persistence in the central and peripheral nervous system accounts for different post COVID sequelae including symptoms of cognitive concerns and dysautonomia such as postural orthostatic tachycardia syndrome (POTS) and small fibre neuropathy [511,512,513]. In this context it is hypothesized that microvascular dysfunction with interruption of pericyte integrity is one of the underlying major causes of post COVID sequelae [514].
Also in the heart, SARS- COV-2 affects the microvascular barrier including pericytes, and, further, cardiomyocytes and fibroblasts [515,516,517]. In a model of COVID-19 in hamsters the occurrence of fibrin-rich microthrombi and loss of pericytes was associated with oedematous cardiomyocyte swelling [518]. In addition, the disruption of human cardiac pericytes is caused by the SARS-CoV-2 S- protein in an extracellular signal-regulated kinase 1/2 (ERK1/2) dependent manner through the CD147 receptor [519]. Though the phosphorylation of HIF-1 by ERK 1/2 promotes its nuclear accumulation and control of HIF-1 target gene expression. Surprisingly, however, SARS-CoV-2 infection inhibits HIF1α translocation in cultured cardiomyocytes, as well as in an epithelial cell line [518]. Therefore, it can be assumed that local hypoxia, which is conferred by prothrombotic and inflammatory stimuli together with impaired hypoxia resolvability is responsible for cardiomyocyte oedema formation [518].
In summary, microvascular homeostasis including glycocalyx and pericyte integrity plays a key role in maintenance of the endothelial barrier and interstitial fluid balance [80]. The impairment of the endothelial surface layer during SARS-CoV-2 infection due to inflammation and ischaemia significantly enhances vascular permeability resulting in oedema formation [64,80,84,85,518].
Further research on the pathophysiology and treatment concepts is warranted to obtain a more detailed insight into unique signalling pathways and possible therapeutic opportunities during acute and chronic SARS-CoV-2 infection.
Author Contributions
Conceptualization: P.P.W.; Writing—Original Draft Preparation: N.G.P., M.P. and P.P.W.; Writing—Review and Editing: N.G.P., M.P., M.A., M.H., A.A.K., C.W.K., A. P.- P., A.W., N.P., P.P.W; Supervision: C.W.K. and P.P.W.; All authors have contributed substantially to the work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
All authors declare no conflict of interest directly related to this work.
References
- Vasques-Nóvoa F, Angélico-Gonçalves A, Alvarenga JMG, et al. Myocardial oedema: pathophysiological basis and implications for the failing heart. ESC Hear Fail. 2022;9(2):958-976. [CrossRef]
- Mehlhorn U, Geissler HJ, Laine GA, Allen SJ. Myocardial fluid balance. Eur J Cardio-Thoracic Surg. 2001;20(6):1220-1230. [CrossRef]
- Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res. 1991;68(6):1713-1721. [CrossRef]
- Desai K, V. , Laine GA, Stewart RH, et al. Mechanics of the left ventricular myocardial interstitium: effects of acute and chronic myocardial edema. Am J Physiol Heart Circ Physiol. 2008;294(6):H2428-34. [CrossRef]
- Rubboli A, Sobotka PA, Euler DE. Effect of acute edema on left ventricular function and coronary vascular resistance in the isolated rat heart. Am J Physiol - Hear Circ Physiol. 1994;267(3 36-3). [CrossRef]
- Miyamoto M, McClure DE, Schertel ER, et al. Effects of hypoproteinemia-induced myocardial edema on left ventricular function. Am J Physiol. 1998;274(3):H937-44. [CrossRef]
- Karolle BL, Carlson RE, Aisen AM, Buda AJ. Transmural distribution of myocardial edema by NMR relaxometry following myocardial ischemia and reperfusion. Am Heart J. 1991;122(3 Pt 1):655-664. [CrossRef]
- Röttgen R, Christiani R, Freyhardt P, et al. Magnetic resonance imaging findings in acute myocarditis and correlation with immunohistological parameters. Eur Radiol. 2011;21(6):1259-1266. [CrossRef]
- Friedrich MG, Strohm O, Schulz-Menger J, Marciniak H, Luft FC, Dietz R. Contrast media-enhanced magnetic resonance imaging visualizes myocardial changes in the course of viral myocarditis. Circulation. 1998;97(18):1802-1809. [CrossRef]
- Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I. Myocardial edema: A translational view. J Mol Cell Cardiol. 2012;52(5):931-939. [CrossRef]
- Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88(3):1009-1086. [CrossRef]
- Bassenge E, Heusch G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol. 1990;116:77-165. [CrossRef]
- Ziegler WH, Goresky CA. Transcapillary exchange in the working left ventricle of the dog. Circ Res. 1971;29(2):181-207. [CrossRef]
- Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;68-69:490-506. [CrossRef]
- Frangogiannis, NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res. 2019;125(1):117-146. [CrossRef]
- Weber, KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13(7):1637-1652. [CrossRef]
- Valiente-Alandi I, Schafer AE, Blaxall BC. Extracellular matrix-mediated cellular communication in the heart. J Mol Cell Cardiol. 2016;91:228. [CrossRef]
- Al-Kofahi M, Omura S, Tsunoda I, et al. IL-1β reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed Pharmacother. 2018;107:1591-1600. [CrossRef]
- Abassi Z, Khoury EE, Karram T, Aronson D. Edema formation in congestive heart failure and the underlying mechanisms. Front Cardiovasc Med. 2022;9:2720.
- Davis KL, Laine GA, Geissler HJ, Mehlhorn U, Brennan M, Allen SJ. Effects of myocardial edema on the development of myocardial interstitial fibrosis. Microcirculation. 2000;7(4):269-280. [CrossRef]
- Van Heerebeek L, Hamdani N, Handoko ML, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117(1):43-51. [CrossRef]
- Van Heerebeek L, Borbély A, Niessen HWM, et al. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation. 2006;113(16):1966-1973. [CrossRef]
- Fraser DD, Patterson EK, Slessarev M, et al. Endothelial Injury and Glycocalyx Degradation in Critically Ill Coronavirus Disease 2019 Patients: Implications for Microvascular Platelet Aggregation. Crit care Explor. 2020;2(9):e0194. [CrossRef]
- Rapkiewicz A, V. , Mai X, Carsons SE, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine. 2020;24:100434. [CrossRef]
- Ortega-Paz L, Capodanno D, Montalescot G, Angiolillo DJ. Coronavirus Disease 2019-Associated Thrombosis and Coagulopathy: Review of the Pathophysiological Characteristics and Implications for Antithrombotic Management. J Am Heart Assoc. 2021;10(3):1-24. [CrossRef]
- Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet (London, England). 2020;395(10234):1417-1418. [CrossRef]
- Zhang S, Liu Y, Wang X, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13(1). [CrossRef]
- Panzer B, Kopp CW, Neumayer C, et al. Toll-like Receptors as Pro-Thrombotic Drivers in Viral Infections: A Narrative Review. Cells. 2023;12(14):1865. [CrossRef]
- Poledniczek M, Neumayer C, Kopp CW, et al. Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches. Biomedicines. 2023;11(8). [CrossRef]
- Poledniczek M, Neumayer C, Kopp CW, et al. Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease – Pathophysiology and Translational Therapeutic Approaches. Published online July 17, 2023. [CrossRef]
- Starling, EH. On the Absorption of Fluids from the Connective Tissue Spaces. J Physiol. 1896;19(4):312-326. [CrossRef]
- Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res. 2010;87(2):198-210. [CrossRef]
- Staverman, AJ. The theory of measurement of osmotic pressure. Recl des Trav Chim des Pays-Bas. 1951;70(4):344-352. [CrossRef]
- MICHEL, CC. Fluid movements through capillary walls. In: Handbook of Physiology. The Cardiovascular System Microcirculation, Section 2. Vol 0. American Physiology Society; 1984:375-409.
- Chilian WM, Eastham CL, Layne SM, Marcus ML. Small vessel phenomena in the coronary microcirculation: phasic intramyocardial perfusion and coronary microvascular dynamics. Prog Cardiovasc Dis. 1988;31(1):17-38. [CrossRef]
- Bassingthwaighte, JB. A concurrent flow model for extraction during transcapillary passage. Circ Res. 1974;35(3):483-503. [CrossRef]
- Chilian, WM. Microvascular pressures and resistances in the left ventricular subepicardium and subendocardium. Circ Res. 1991;69(3):561-570. [CrossRef]
- Koyama T, Kikuchi Y, Kakiuchi Y, Nagashima C. An analysis of water movement between myocardial tissue and capillary blood during reactive hyperemia. Jpn J Physiol. 1979;29(1):1-13. [CrossRef]
- Laine GA, Granger HJ. Microvascular, interstitial, and lymphatic interactions in normal heart. Am J Physiol. 1985;249(4 Pt 2). [CrossRef]
- Stein PD, Marzilli M, Sabbah HN, Lee T. Systolic and diastolic pressure gradients within the left ventricular wall. Am J Physiol. 1980;238(5). [CrossRef]
- Pilati, CF. Macromolecular transport in canine coronary microvasculature. Am J Physiol. 1990;258(3 Pt 2). [CrossRef]
- Mehlhorn U, Davis KL, Laine GA, Geissler HJ, Allen SJ. Myocardial fluid balance in acute hypertension. Microcirculation. 1996;3(4):371-378. [CrossRef]
- Mehlhorn U, Allen SJ, Davis KL, Geissler HJ, Warters RD, Rainer de Vivie E. Increasing the colloid osmotic pressure of cardiopulmonary bypass prime and normothermic blood cardioplegia minimizes myocardial oedema and prevents cardiac dysfunction. Cardiovasc Surg. 1998;6(3):274-281. [CrossRef]
- Navar PD, Navar LG. Relationship between colloid osmotic pressure and plasma protein concentration in the dog. Am J Physiol. 1977;233(2). [CrossRef]
- Curry FE, Michel CC. The Colloid Osmotic Pressure Across the Glycocalyx: Role of Interstitial Fluid Sub-Compartments in Trans-Vascular Fluid Exchange in Skeletal Muscle. Front Cell Dev Biol. 2021;9. [CrossRef]
- Mortimer PS, Levick JR. Chronic peripheral oedema: the critical role of the lymphatic system. Clin Med. 2004;4(5):448-453. [CrossRef]
- Reitsma S, Slaaf DW, Vink H, et al. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch Eur J Physiol. 2007;454(3):345-359. [CrossRef]
- Vink H, Duling BR. Capillary endothelial surface layer selectively reduces plasma solute distribution volume. Am J Physiol Heart Circ Physiol. 2000;278(1). [CrossRef]
- Ori A, Wilkinson MC, Fernig DG. A Systems Biology Approach for the Investigation of the Heparin/Heparan Sulfate Interactome. J Biol Chem. 2011;286(22):19892. [CrossRef]
- Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev. 2012;92(3):1005-1060. [CrossRef]
- Wearn, JT. THE EXTENT OF THE CAPILLARY BED OF THE HEART. J Exp Med. 1928;47(2):273-290. [CrossRef]
- Laine, GA. Microvascular changes in the heart during chronic arterial hypertension. Circ Res. 1988;62(5):953-960. [CrossRef]
- Verbrugge FH, Bertrand PB, Willems E, et al. Global myocardial oedema in advanced decompensated heart failure. Eur Hear journal Cardiovasc Imaging. 2017;18(7):787-794. [CrossRef]
- Takegawa R, Kabata D, Shimizu K, et al. Serum albumin as a risk factor for death in patients with prolonged sepsis: An observational study. J Crit Care. 2019;51:139-144. [CrossRef]
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med. 2011;39(2):259-265. [CrossRef]
- Wiig H, Rubin K, Reed RK. New and active role of the interstitium in control of interstitial fluid pressure: potential therapeutic consequences. Acta Anaesthesiol Scand. 2003;47(2):111-121. [CrossRef]
- Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121-167. [CrossRef]
- Squire JM, Chew M, Nneji G, Neal C, Barry J, Michel C. Quasi-periodic substructure in the microvessel endothelial glycocalyx: a possible explanation for molecular filtering? J Struct Biol. 2001;136(3):239-255. [CrossRef]
- Rostgaard J, Qvortrup K. Electron microscopic demonstrations of filamentous molecular sieve plugs in capillary fenestrae. Microvasc Res. 1997;53(1):1-13. [CrossRef]
- Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med. 2016;280(1):97-113. [CrossRef]
- Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch. 2000;440(5):653-666. [CrossRef]
- Lipowsky, HH. Microvascular rheology and hemodynamics. Microcirculation. 2005;12(1):5-15. [CrossRef]
- Vink H, Constantinescu AA, Spaan JAE. Oxidized lipoproteins degrade the endothelial surface layer : implications for platelet-endothelial cell adhesion. Circulation. 2000;101(13):1500-1502. [CrossRef]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279-367. [CrossRef]
- Busch B, Ljungman C, Heldin CM, Waskson E, Öbrink B. Surface properties of cultured endothelial cells. Haemostasis. 1979;8(3-5):142-148. [CrossRef]
- Damiano, ER. The effect of the endothelial-cell glycocalyx on the motion of red blood cells through capillaries. Microvasc Res. 1998;55(1):77-91. [CrossRef]
- Adamson RH, Huxley VH, Curry FE. Single capillary permeability to proteins having similar size but different charge. Am J Physiol. 1988;254(2 Pt 2). [CrossRef]
- Henry CBS, Duling BR. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol. 1999;277(2). [CrossRef]
- Adamson, RH. Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J Physiol. 1990;428(1):1-13. [CrossRef]
- Powers MR, Blumenstock FA, Cooper JA, Malik AB. Role of albumin arginyl sites in albumin-induced reduction of endothelial hydraulic conductivity. J Cell Physiol. 1989;141(3):558-564. [CrossRef]
- Schneeberger EE, Hamelin M. Interaction of serum proteins with lung endothelial glycocalyx: its effect on endothelial permeability. Am J Physiol. 1984;247(2 Pt 2). [CrossRef]
- Turner MR, Clough G, Michel CC. The effects of cationised ferritin and native ferritin upon the filtration coefficient of single frog capillaries. Evidence that proteins in the endothelial cell coat influence permeability. Microvasc Res. 1983;25(2):205-222. [CrossRef]
- Levick JR, Michel CC. The effect of bovine albumin on the permeability of frog mesenteric capillaries. Q J Exp Physiol Cogn Med Sci. 1973;58(1):87-97. [CrossRef]
- Lopez-Quintero S, V. , Amaya R, Pahakis M, Tarbell JM. The endothelial glycocalyx mediates shear-induced changes in hydraulic conductivity. Am J Physiol Circ Physiol. 2009;296(5):H1451-H1456. [CrossRef]
- Jacob M, Rehm M, Loetsch M, et al. The endothelial glycocalyx prefers albumin for evoking shear stress-induced, nitric oxide-mediated coronary dilatation. J Vasc Res. 2007;44(6):435-443. [CrossRef]
- Li Q, Bolli R, Qiu Y, Tang XL, Murphree SS, French BA. Gene therapy with extracellular superoxide dismutase attenuates myocardial stunning in conscious rabbits. Circulation. 1998;98(14):1438-1448. [CrossRef]
- Shimada K, Kobayashi M, Kimura S, et al. Anticoagulant heparin-like glycosaminoglycans on endothelial cell surface. Jpn Circ J. 1991;55(10):1016-1021. [CrossRef]
- Tovar AMF, De Mattos DA, Stelling MP, Sarcinelli-Luz BSL, Nazareth RA, Mourão PAS. Dermatan sulfate is the predominant antithrombotic glycosaminoglycan in vessel walls: implications for a possible physiological function of heparin cofactor II. Biochim Biophys Acta. 2005;1740(1):45-53. [CrossRef]
- Patterson EK, Cepinskas G, Fraser DD. Endothelial Glycocalyx Degradation in Critical Illness and Injury. Front Med. 2022;9:898592. [CrossRef]
- Van den Berg BM, Vink H, Spaan JAE. The Endothelial Glycocalyx Protects Against Myocardial Edema. Circ Res. 2003;92(6):592-594. [CrossRef]
- Jacob M, Bruegger D, Rehm M, Welsch U, Conzen P, Becker BF. Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology. 2006;104(6):1223-1231. [CrossRef]
- Bruegger D, Jacob M, Rehm M, et al. Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am J Physiol Circ Physiol. 2005;289(5):H1993-H1999. [CrossRef]
- van den Berg BM, Vink H, Spaan JAE. The Endothelial Glycocalyx Protects Against Myocardial Edema. Circ Res. 2003;92(6):592-594. [CrossRef]
- Malik AB, Siflinger-Birnboim A. Vascular Endothelial Barrier Function and Its Regulation. Published online 1993:231-267. [CrossRef]
- Adamson, RH. Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J Physiol. 1990;428(1):1-13. [CrossRef]
- Mann, GE. Alterations of myocardial capillary permeability by albumin in the isolated, perfused rabbit heart. J Physiol. 1981;319(1):311-323. [CrossRef]
- Wiesinger A, Peters W, Chappell D, et al. Nanomechanics of the Endothelial Glycocalyx in Experimental Sepsis. Sokolov I, ed. PLoS One. 2013;8(11):e80905. [CrossRef]
- Rienks M, Carai P, van Teeffelen J, et al. SPARC preserves endothelial glycocalyx integrity, and protects against adverse cardiac inflammation and injury during viral myocarditis. Matrix Biol. 2018;74:21-34. [CrossRef]
- Epstein FH, Parrillo JE. Pathogenetic Mechanisms of Septic Shock. 1993;328(20):1471-1477. [CrossRef]
- Tang THC, Alonso S, Ng LFP, et al. Increased Serum Hyaluronic Acid and Heparan Sulfate in Dengue Fever: Association with Plasma Leakage and Disease Severity. Sci Rep. 2017;7. [CrossRef]
- Puerta-Guardo H, Glasner DR, Harris E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016;12(7):e1005738. [CrossRef]
- Bansch P, Nelson A, Ohlsson T, Bentzer P. Effect of charge on microvascular permeability in early experimental sepsis in the rat. Microvasc Res. 2011;82(3):339-345. [CrossRef]
- Henry CBS, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000;279(6):H2815-23. [CrossRef]
- Ley K, Tedder TF. Leukocyte interactions with vascular endothelium. New insights into selectin-mediated attachment and rolling. J Immunol. 1995;155(2):525-528. [CrossRef]
- Peterson MW, Stone P, Shasby DM. Cationic neutrophil proteins increase transendothelial albumin movement. J Appl Physiol. 1987;62(4):1521-1530. [CrossRef]
- Hoover RL, Briggs RT, Karnovsky MJ. The adhesive interaction between polymorphonuclear leukocytes and endothelial cells in vitro. Cell. 1978;14(2):423-428. [CrossRef]
- Morita Y, Clemens MG, Miller LS, et al. Reactive oxidants mediate TNF-alpha-induced leukocyte adhesion to rat mesenteric venular endothelium. Am J Physiol. 1995;269(6 Pt 2). [CrossRef]
- Carden DL, Smith JK, Korthuis RJ. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ Res. 1990;66(5):1436-1444. [CrossRef]
- Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol. 2018;222(1):e12860. [CrossRef]
- Ward BJ, Donnelly JL. Hypoxia induced disruption of the cardiac endothelial glycocalyx: implications for capillary permeability. Cardiovasc Res. 1993;27(3):384-389. [CrossRef]
- Constantinescu AA, Vink H, Spaan JAE. Elevated capillary tube hematocrit reflects degradation of endothelial cell glycocalyx by oxidized LDL. Am J Physiol Heart Circ Physiol. 2001;280(3):H1051-7. [CrossRef]
- Gardner G, Banka CL, Roberts KA, Mullick AE, Rutledge JC. Modified LDL–Mediated Increases in Endothelial Layer Permeability Are Attenuated With 17β-Estradiol. Arterioscler Thromb Vasc Biol. 1999;19(4):854-861. [CrossRef]
- Rangaswamy S, Penn MS, Saidel GM, Chisolm GM. Exogenous Oxidized Low-Density Lipoprotein Injures and Alters the Barrier Function of Endothelium in Rats In Vivo. Circ Res. 1997;80(1):37-44. [CrossRef]
- Lehr HA, Frei B, Olofsson AM, Carew TE, Arfors KE. Protection From Oxidized LDL–Induced Leukocyte Adhesion to Microvascular and Macrovascular Endothelium In Vivo by Vitamin C but Not by Vitamin E. Circulation. 1995;91(5):1525-1532. [CrossRef]
- Liao L, Harris NR, Granger DN. Oxidized low-density lipoproteins and microvascular responses to ischemia-reperfusion. 1996;271(6 40-6). [CrossRef]
- Lehr HA, Becker M, Marklund SL, et al. Superoxide-dependent stimulation of leukocyte adhesion by oxidatively modified LDL in vivo. Arterioscler Thromb A J Vasc Biol. 1992;12(7):824-829. [CrossRef]
- Beręsewicz A, Czarnowska E, Mączewski M. Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Myocard Ischemia Reperfus. Published online 1998:87-97. [CrossRef]
- Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286(5):H1672-80. [CrossRef]
- Kurzelewski M, Czarnowska E, Beresewicz A. Superoxide- and nitric oxide-derived species mediate endothelial dysfunction, endothelial glycocalyx disruption, and enhanced neutrophil adhesion in the post-ischemic guinea-pig heart. J Physiol Pharmacol. 2005;56(2):163-178. http://www.ncbi.nlm.nih. 1598.
- Yu H, Kalogeris T, Korthuis RJ. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radic Biol Med. 2019;135:182-197. [CrossRef]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/Reperfusion. Compr Physiol. 2016;7(1):113-170. [CrossRef]
- Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol 2021 189. 2021;18(9):2114-2127. [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653-660. [CrossRef]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407(6801):242-248. [CrossRef]
- Polverini, PJ. The pathophysiology of angiogenesis. Crit Rev Oral Biol Med. 1995;6(3):230-247. [CrossRef]
- Zimna A, Kurpisz M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res Int. 2015;2015. [CrossRef]
- Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4(6):915-924. [CrossRef]
- Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18(5):1135-1149. [CrossRef]
- Ben-Yosef Y, Miller A, Shapiro S, Lahat N. Hypoxia of endothelial cells leads to MMP-2-dependent survival and death. Am J Physiol Cell Physiol. 2005;289(5). [CrossRef]
- Sun J, Singh P, Shami A, et al. Spatial Transcriptional Mapping Reveals Site-Specific Pathways Underlying Human Atherosclerotic Plaque Rupture. J Am Coll Cardiol. 2023;81(23):2213-2227. [CrossRef]
- Goetze JP, Bruneau BG, Ramos HR, Ogawa T, de Bold MK, de Bold AJ. Cardiac natriuretic peptides. Nat Rev Cardiol 2020 1711. 2020;17(11):698-717. [CrossRef]
- Ando SI, Imaizumi T, Harada S, Hirooka Y, Takeshita A. Atrial natriuretic peptide increases human capillary filtration and venous distensibility. J Hypertens. 1992;10(5):451-457. [CrossRef]
- Huxley VH, Tucker VL, Verburg KM, Freeman RH. Increased capillary hydraulic conductivity induced by atrial natriuretic peptide. Circ Res. 1987;60(2):304-307. [CrossRef]
- Jacob M, Saller T, Chappell D, Rehm M, Welsch U, Becker BF. Physiological levels of A-, B- and C-type natriuretic peptide shed the endothelial glycocalyx and enhance vascular permeability. Basic Res Cardiol. 2013;108(3). [CrossRef]
- Oberleithner H, Peters W, Kusche-Vihrog K, et al. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. 2011;462(4):519-528. [CrossRef]
- Selcuk M, Keskin M, Cinar T, et al. Prognostic significance of N-Terminal Pro-BNP in patients with COVID-19 pneumonia without previous history of heart failure. Eur Heart J. 2021;42(Supplement_1). [CrossRef]
- Huelsmann M, Neuhold S, Resl M, et al. PONTIAC (NT-proBNP Selected PreventiOn of cardiac eveNts in a populaTion of dIabetic patients without A history of Cardiac disease): A Prospective Randomized Controlled Trial. J Am Coll Cardiol. 2013;62(15):1365-1372. [CrossRef]
- Hartsock A, Nelson WJ. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta - Biomembr. 2008;1778(3):660-669. [CrossRef]
- Yano T, Torisawa T, Oiwa K, Tsukita S. AMPK-dependent phosphorylation of cingulin reversibly regulates its binding to actin filaments and microtubules. Sci Reports 2018 81. 2018;8(1):1-10. [CrossRef]
- González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778(3):729-756. [CrossRef]
- Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16(2):209-221. [CrossRef]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2(1). [CrossRef]
- Guillemot L, Paschoud S, Pulimeno P, Foglia A, Citi S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim Biophys Acta. 2008;1778(3):601-613. [CrossRef]
- Schossleitner K, Rauscher S, Gröger M, et al. Evidence That Cingulin Regulates Endothelial Barrier Function In Vitro and In Vivo. Arterioscler Thromb Vasc Biol. 2016;36(4):647-654. [CrossRef]
- Dejana E, Vestweber D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci. 2013;116:119-144. [CrossRef]
- Bravi L, Dejana E, Lampugnani MG. VE-cadherin at a glance. Cell Tissue Res. 2014;355(3):515-522. [CrossRef]
- Gavard, J. Endothelial permeability and VE-cadherin: a wacky comradeship. Cell Adh Migr. 2014;8(2):158-164. [CrossRef]
- Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol. 2005;288(3). [CrossRef]
- Stevens T, Garcia JGN, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol. 2000;279(3). [CrossRef]
- Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res. 2009;77(1):53-63. [CrossRef]
- Dudek SM, Garcia JGN. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487-1500. [CrossRef]
- Shasby DM, Shasby SS, Sullivan JM, Peach MJ. Role of endothelial cell cytoskeleton in control of endothelial permeability. Circ Res. 1982;51(5):657-661. [CrossRef]
- García-Ponce A, Citalán-Madrid AF, Velázquez-Avila M, Vargas-Robles H, Schnoor M. The role of actin-binding proteins in the control of endothelial barrier integrity. Thromb Haemost. 2015;113(01):20-36. [CrossRef]
- Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77(1):39-45. [CrossRef]
- Xu M, Waters CL, Hu C, Wysolmerski RB, Vincent PA, Minnear FL. Sphingosine 1-phosphate rapidly increases endothelial barrier function independently of VE-cadherin but requires cell spreading and Rho kinase. Am J Physiol Cell Physiol. 2007;293(4). [CrossRef]
- Wilkerson BA, Argraves KM. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim Biophys Acta. 2014;1841(10):1403-1412. [CrossRef]
- David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 requires IQ domain GTPase-activating protein 1 to activate Rac1 and promote endothelial barrier defense. Arterioscler Thromb Vasc Biol. 2011;31(11):2643-2652. [CrossRef]
- Frye M, Dierkes M, Küppers V, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. 2015;212(13):2267-2287. [CrossRef]
- Zeng Y, Adamson RH, Curry FRE, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. 2014;306(3). [CrossRef]
- Schlegel N, Waschke J. cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 2014;355(3):587-596. [CrossRef]
- Lampugnani MG, Zanetti A, Breviario F, et al. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell. 2002;13(4):1175-1189. [CrossRef]
- Kopperud RK, Rygh CB, Karlsen T V., et al. Increased microvascular permeability in mice lacking Epac1 (Rapgef3). Acta Physiol (Oxf). 2017;219(2):441-452. [CrossRef]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105(5):1950-1955. [CrossRef]
- Michel CC, Curry FE. Microvascular permeability. Physiol Rev. 1999;79(3):703-761. [CrossRef]
- Curry FRE, Adamson RH. Tonic regulation of vascular permeability. Acta Physiol (Oxf). 2013;207(4):628-649. [CrossRef]
- Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit Care Med. 2009;37(5):1735-1743. [CrossRef]
- Schlegel N, Waschke J. Impaired cAMP and Rac 1 signaling contribute to TNF-alpha-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation. 2009;16(6):521-533. [CrossRef]
- Baumer Y, Spindler V, Werthmann RC, Bünemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol. 2009;220(3):716-726. [CrossRef]
- Aslam M, Tanislav C, Troidl C, Schulz R, Hamm C, Gündüz D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol Rep. 2014;2(10). [CrossRef]
- Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. 2013;126(Pt 12):2545-2549. [CrossRef]
- Küppers V, Vockel M, Nottebaum AF, Vestweber D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014;355(3):577-586. [CrossRef]
- Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87(2):243-253. [CrossRef]
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39(4-5):187-199. [CrossRef]
- Flemming S, Burkard N, Renschler M, et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res. 2015;107(1):32-44. [CrossRef]
- Orsenigo F, Giampietro C, Ferrari A, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3. [CrossRef]
- Garatti L, Wu MA, Ammirati E, Sacco A. Systemic leak capillary syndrome with myocardial involvement and cardiogenic shock: a case report. Eur Hear J - Case Reports. 2022;6(7). [CrossRef]
- Yacoub S, Wertheim H, Simmons CP, Screaton G, Wills B. Cardiovascular manifestations of the emerging dengue pandemic. Nat Rev Cardiol 2014 116. 2014;11(6):335-345. [CrossRef]
- Biering SB, Gomes de Sousa FT, Tjang L V., et al. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-β signaling. Nat Commun 2022 131. 2022;13(1):1-19. [CrossRef]
- Dongaonkar RM, Stewart RH, Quick CM, Uray KL, Cox CS, Laine GA. Award article: Microcirculatory Society Award for Excellence in Lymphatic Research: time course of myocardial interstitial edema resolution and associated left ventricular dysfunction. Microcirculation. 2012;19(8):714-722. [CrossRef]
- Pratt JW, Schertel ER, Schaefer SL, et al. Acute transient coronary sinus hypertension impairs left ventricular function and induces myocardial edema. Am J Physiol. 1996;271(3 Pt 2):H834-41. [CrossRef]
- Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res. 2010;87(2):331-339. [CrossRef]
- Taherzadeh M, Das AK, Warren JB. Nifedipine increases microvascular permeability via a direct local effect on postcapillary venules. Am J Physiol. 1998;275(4). [CrossRef]
- Monmeneu J V, Bodí V, Sanchis J, et al. Cardiac magnetic resonance evaluation of edema after ST-elevation acute myocardial infarction. Rev Esp Cardiol. 2009;62(8):858-866. [CrossRef]
- Fehrmann A, Treutlein M, Rudolph T, et al. Myocardial T1 and T2 mapping in severe aortic stenosis: Potential novel insights into the pathophysiology of myocardial remodelling. Eur J Radiol. 2018;107:76-83. [CrossRef]
- Alabed S, Saunders L, Garg P, et al. Myocardial T1-mapping and extracellular volume in pulmonary arterial hypertension: A systematic review and meta-analysis. Magn Reson Imaging. 2021;79:66-75. [CrossRef]
- Spieker M, Haberkorn S, Gastl M, et al. Abnormal T2 mapping cardiovascular magnetic resonance correlates with adverse clinical outcome in patients with suspected acute myocarditis. J Cardiovasc Magn Reson. 2017;19(1). [CrossRef]
- Gräni C, Eichhorn C, Bière L, et al. Prognostic Value of Cardiac Magnetic Resonance Tissue Characterization in Risk Stratifying Patients With Suspected Myocarditis. J Am Coll Cardiol. 2017;70(16):1964. [CrossRef]
- Stevenson LW, Tillisch JH. Maintenance of cardiac output with normal filling pressures in patients with dilated heart failure. Circulation. 1986;74(6):1303-1308. [CrossRef]
- Laine GA, Allen SJ, Katz J, Gabel JC, Drake RE. Effect of systemic venous pressure elevation on lymph flow and lung edema formation. J Appl Physiol. 1986;61(5):1634-1638. [CrossRef]
- Robinson AD, Ramanathan KB, McGee JE, Newman KP, Weber KT. Oxidative stress and cardiomyocyte necrosis with elevated serum troponins: pathophysiologic mechanisms. Am J Med Sci. 2011;342(2):129-134. [CrossRef]
- Colombo PC, Onat D, Harxhi A, et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur Heart J. 2014;35(7):448-454. [CrossRef]
- Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038-3044. [CrossRef]
- Libby, P. The Heart in COVID-19: Primary Target or Secondary Bystander? JACC Basic to Transl Sci. 2020;5(5):537-542. [CrossRef]
- Folco EJ, Mawson TL, Vromman A, et al. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38(8):1901-1912. [CrossRef]
- Marcus AJ, Broekman MJ, Drosopoulos JHF, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99(6):1351-1360. [CrossRef]
- Roy C, Tabiasco J, Caillon A, et al. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase-1/CD39 in hypertension. Purinergic Signal. 2018;14(1):73-82. [CrossRef]
- Robson SC, Kaczmarek E, Siegel JB, et al. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J Exp Med. 1997;185(1):153-163. [CrossRef]
- Wei LH, Kuo ML, Chen CA, et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22(10):1517-1527. [CrossRef]
- Ishii K, Sasaki T, Iguchi K, et al. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate. 2018;78(11):849-856. [CrossRef]
- Soga N, Namba N, McAllister S, et al. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res. 2001;269(1):73-87. [CrossRef]
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803-815. [CrossRef]
- Busse R, Fleming I. Vascular endothelium and blood flow. Handb Exp Pharmacol. 2006;176(PART2):43-78.
- Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006;176(PART1):213-254.
- McDONALD DM, THURSTON G, BALUK P. Endothelial Gaps as Sites for Plasma Leakage in Inflammation. Microcirculation. 1999;6(1):7-22. [CrossRef]
- Siao CJ, Lorentz CU, Kermani P, et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J Exp Med. 2012;209(12):2291-2305. [CrossRef]
- Armulik A, Genové G, Betsholtz C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev Cell. 2011;21(2):193-215. [CrossRef]
- Wang S, Cao C, Chen Z, et al. Pericytes Regulate Vascular Basement Membrane Remodeling and Govern Neutrophil Extravasation during Inflammation. PLoS One. 2012;7(9):e45499. [CrossRef]
- Díaz-Flores L, Gutiérrez R, Madrid JF, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24(7):909-969. [CrossRef]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nat 2006 4437112. 2006;443(7112):700-704. [CrossRef]
- Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116(6):1097-1100. [CrossRef]
- Johnsson C, Hällgren R, Elvin A, Gerdin B, Tufveson G. Hyaluronidase ameliorates rejection-induced edema. Transpl Int. 1999;12(4):235-243. [CrossRef]
- Jackson, DG. Leucocyte Trafficking via the Lymphatic Vasculature- Mechanisms and Consequences. Front Immunol. 2019;10(MAR). [CrossRef]
- Schwager S, Detmar M. Inflammation and Lymphatic Function. Front Immunol. 2019;10. [CrossRef]
- Pfister F, Feng Y, Hagen F Vom, et al. Pericyte MigrationA Novel Mechanism of Pericyte Loss in Experimental Diabetic Retinopathy. Diabetes. 2008;57(9):2495-2502. [CrossRef]
- Curry FRE. Atrial natriuretic peptide: an essential physiological regulator of transvascular fluid, protein transport, and plasma volume. J Clin Invest. 2005;115(6):1458-1461. [CrossRef]
- Chappell D, Bruegger D, Potzel J, et al. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. 2014;18(5):538. [CrossRef]
- Ferreira VM, Schulz-Menger J, Holmvang G, et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J Am Coll Cardiol. 2018;72(24):3158-3176. [CrossRef]
- Iles LM, Ellims AH, Llewellyn H, et al. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur Hear journal Cardiovasc Imaging. 2015;16(1):14-22. [CrossRef]
- Palmisano A, Benedetti G, Faletti R, et al. Early T1 Myocardial MRI Mapping: Value in Detecting Myocardial Hyperemia in Acute Myocarditis. Radiology. 2020;295(2):316-325. [CrossRef]
- Beijnink CWH, van der Hoeven NW, Konijnenberg LSF, et al. Cardiac MRI to Visualize Myocardial Damage after ST-Segment Elevation Myocardial Infarction: A Review of Its Histologic Validation. Radiology. 2021;301(1):4-18. [CrossRef]
- Masci PG, Pavon AG, Muller O, et al. Relationship between CMR-derived parameters of ischemia/reperfusion injury and the timing of CMR after reperfused ST-segment elevation myocardial infarction. J Cardiovasc Magn Reson. 2018;20(1). [CrossRef]
- Gutberlet M, Spors B, Thoma T, et al. Suspected chronic myocarditis at cardiac MR: diagnostic accuracy and association with immunohistologically detected inflammation and viral persistence. Radiology. 2008;246(2):401-409. [CrossRef]
- Luo Y, Liu BT, Yuan WF, Zhao CX. Frontiers of COVID-19-related myocarditis as assessed by cardiovascular magnetic resonance. World J Clin Cases. 2022;10(20):6784. [CrossRef]
- McDonagh TA, Metra M, Adamo M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42(36):3599-3726. [CrossRef]
- Caforio ALP, Pankuweit S, Arbustini E, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(33):2636-2648. [CrossRef]
- Wadowski PP, Piechota-Polańczyk A, Andreas M, Kopp CW. Cardiovascular Disease Management in the Context of Global Crisis. Int J Environ Res Public Health. 2022;20(1):689. [CrossRef]
- Siripanthong B, Asatryan B, Hanff TC, et al. The Pathogenesis and Long-Term Consequences of COVID-19 Cardiac Injury. JACC Basic to Transl Sci. 2022;7(3):294-308. [CrossRef]
- Liu PP, Blet A, Smyth D, Li H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation. 2020;142(1):68-78. [CrossRef]
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280.e8. [CrossRef]
- Prasad K, AlOmar SY, Almuqri EA, Rudayni HA, Kumar V. Genomics-guided identification of potential modulators of SARS-CoV-2 entry proteases, TMPRSS2 and Cathepsins B/L. PLoS One. 2021;16(8). [CrossRef]
- Amraei R, Yin W, Napoleon MA, et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent Sci. 2021;7(7):1156-1165. [CrossRef]
- Cantuti-Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518). [CrossRef]
- Bermejo-Jambrina M, Eder J, Kaptein TM, et al. Infection and transmission of SARS-CoV-2 depend on heparan sulfate proteoglycans. EMBO J. 2021;40(20). [CrossRef]
- Dai J, Wang Y, Wang H, et al. Toll-Like Receptor Signaling in Severe Acute Respiratory Syndrome Coronavirus 2-Induced Innate Immune Responses and the Potential Application Value of Toll-Like Receptor Immunomodulators in Patients With Coronavirus Disease 2019. Front Microbiol. 2022;13:948770.
- Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol - Hear Circ Physiol. 2011;300(1):397-407.
- Kawamura H, Kobayashi M, Li Q, et al. Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J Physiol. 2004;561(Pt 3):671-683. [CrossRef]
- Tilton RG, Kilo C, Williamson JR, Murch DW. Differences in pericyte contractile function in rat cardiac and skeletal muscle microvasculatures. Microvasc Res. 1979;18(3):336-352. [CrossRef]
- Bertram S, Heurich A, Lavender H, et al. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS One. 2012;7(4):e35876. [CrossRef]
- Sakamoto A, Kawakami R, Kawai K, et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arterioscler Thromb Vasc Biol. 2021;41(1):542-544. [CrossRef]
- Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. [CrossRef]
- Carsana L, Sonzogni A, Nasr A, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020;20(10):1135-1140. [CrossRef]
- Tavazzi G, Pellegrini C, Maurelli M, et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. 2020;22(5):911-915. [CrossRef]
- Nakamura Y, Katano H, Nakajima N, et al. SARS-CoV-2 is localized in cardiomyocytes: a postmortem biopsy case. Int J Infect Dis. 2021;111:43. [CrossRef]
- Kim YH, Nijst P, Kiefer K, Tang WHW. Endothelial Glycocalyx as Biomarker for Cardiovascular Diseases: Mechanistic and Clinical Implications. Curr Heart Fail Rep. 2017;14(2):117-126. [CrossRef]
- Wadowski PP, Hülsmann M, Schörgenhofer C, et al. Sublingual functional capillary rarefaction in chronic heart failure. Eur J Clin Invest. 2018;48(2):e12869. [CrossRef]
- Wadowski PP, Steinlechner B, Zimpfer D, et al. Functional capillary impairment in patients with ventricular assist devices. Sci Reports 2019 91. 2019;9(1):1-8. [CrossRef]
- Wadowski PP, Schörgenhofer C, Rieder T, et al. Microvascular rarefaction in patients with cerebrovascular events. Microvasc Res. 2022;140. [CrossRef]
- Xu J, Xiao W, Liang X, et al. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health. 2021;21(1):1533. [CrossRef]
- Gerotziafas GT, Catalano M, Colgan MP, et al. Guidance for the Management of Patients with Vascular Disease or Cardiovascular Risk Factors and COVID-19: Position Paper from VAS-European Independent Foundation in Angiology/Vascular Medicine. Thromb Haemost. 2020;120(12):1597-1628. [CrossRef]
- Ronco C, Reis T, Husain-Syed F. Management of acute kidney injury in patients with COVID-19. Lancet Respir Med. 2020;8(7):738-742. [CrossRef]
- Ullah W, Saeed R, Sarwar U, Patel R, Fischman DL. COVID-19 Complicated by Acute Pulmonary Embolism and Right-Sided Heart Failure. JACC Case reports. 2020;2(9):1379-1382. [CrossRef]
- Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet (London, England). 2020;395(10235):1517-1520. [CrossRef]
- Siripanthong B, Nazarian S, Muser D, et al. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Hear Rhythm. 2020;17(9):1463-1471. [CrossRef]
- Antzelevitch C, Burashnikov A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card Electrophysiol Clin. 2011;3(1):23-45. [CrossRef]
- Avolio E, Madeddu P. Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascul Pharmacol. 2016;86:53-63. [CrossRef]
- Sweeney M, Foldes G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front Cardiovasc Med. 2018;5:413767.
- Bontekoe J, Lee J, Bansal V, et al. Biomarker Profiling in Stage 5 Chronic Kidney Disease Identifies the Relationship between Angiopoietin-2 and Atrial Fibrillation. Clin Appl Thromb. 2018;24(9_suppl):269S-276S. [CrossRef]
- Li J, Yang Y, Ng CY, Zhang Z, Liu T, Li G. Association of Plasma Transforming Growth Factor-β1 Levels and the Risk of Atrial Fibrillation: A Meta-Analysis. PLoS One. 2016;11(5):e0155275. [CrossRef]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017;3(5):425-435. [CrossRef]
- Chang S-H, Yeh Y-H, Lee J-L, Hsu Y-J, Kuo C-T, Chen W-J. Transforming growth factor-β-mediated CD44/STAT3 signaling contributes to the development of atrial fibrosis and fibrillation. Basic Res Cardiol. 2017;112(5):58. [CrossRef]
- Qiao G, Xia D, Cheng Z, Zhang G. miR-132 in atrial fibrillation directly targets connective tissue growth factor. Mol Med Rep. 2017;16(4):4143-4150. [CrossRef]
- Lee LL, Chintalgattu V. Pericytes in the Heart. Adv Exp Med Biol. 2019;1122:187-210. [CrossRef]
- Li M, Yi X, Ma L, Zhou Y. Hepatocyte growth factor and basic fibroblast growth factor regulate atrial fibrosis in patients with atrial fibrillation and rheumatic heart disease via the mitogen-activated protein kinase signaling pathway. Exp Ther Med. 2013;6(5):1121-1126. [CrossRef]
- Freestone B, Chong AY, Lim HS, Blann A, Lip GYH. Angiogenic factors in atrial fibrillation: a possible role in thrombogenesis? Ann Med. 2005;37(5):365-372. [CrossRef]
- Doubt TJ, Hogan PM. Effects of hydrostatic pressure on conduction and excitability in rabbit atria. J Appl Physiol. 1978;45(1):24-32. [CrossRef]
- Li X, Xue YM, Guo HM, et al. High hydrostatic pressure induces atrial electrical remodeling through upregulation of inflammatory cytokines. Life Sci. 2020;242. [CrossRef]
- Li X, Deng CY, Xue YM, et al. High hydrostatic pressure induces atrial electrical remodeling through angiotensin upregulation mediating FAK/Src pathway activation. J Mol Cell Cardiol. 2020;140:10-21. [CrossRef]
- Granger DN, Rodrigues SF, Yildirim A, Senchenkova EY. Microvascular Responses to Cardiovascular Risk Factors. Microcirculation. 2010;17(3):192-205. [CrossRef]
- Wadowski PP, Jilma B, Kopp CW, Ertl S, Gremmel T, Koppensteiner R. Glycocalyx as Possible Limiting Factor in COVID-19. Front Immunol. 2021;12:607306. [CrossRef]
- Huertas A, Montani D, Savale L, et al. Endothelial cell dysfunction: a major player in SARS-CoV-2 infection (COVID-19)? Eur Respir J. 2020;56(1). [CrossRef]
- Huang KJ, Su IJ, Theron M, et al. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185-194. [CrossRef]
- Šoltés L, Mendichi R, Kogan G, Schiller J, Stankovská M, Arnhold J. Degradative action of reactive oxygen species on hyaluronan. Biomacromolecules. 2006;7(3):659-668. [CrossRef]
- Maciej-Hulme, ML. New Insights Into Human Hyaluronidase 4/Chondroitin Sulphate Hydrolase. Front cell Dev Biol. 2021;9. [CrossRef]
- Matzner Y, Bar-Ner M, Yahalom J, Ishai-Michaeli R, Fuks Z, Vlodavsky I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J Clin Invest. 1985;76(4):1306. [CrossRef]
- Endo K, Takino T, Miyamori H, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278(42):40764-40770. [CrossRef]
- Wagner, DD. The Weibel-Palade body: The storage granule for von Willebrand factor and P-selectin. Thromb Haemost. 1993;70(1):105-110. [CrossRef]
- Gupta RM, Libby P, Barton M. Linking regulation of nitric oxide to endothelin-1: The Yin and Yang of vascular tone in the atherosclerotic plaque. Atherosclerosis. 2020;292:201-203. [CrossRef]
- Ng H, Havervall S, Rosell A, et al. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arterioscler Thromb Vasc Biol. 2021;41(2):988-994. [CrossRef]
- Hidalgo, A. A NET-thrombosis axis in COVID-19. Blood. 2020;136(10):1118-1119. [CrossRef]
- Barmada A, Klein J, Ramaswamy A, et al. Cytokinopathy with aberrant cytotoxic lymphocytes and profibrotic myeloid response in SARS-CoV-2 mRNA vaccine–associated myocarditis. Sci Immunol. 2023;8(83). [CrossRef]
- Boehmer TK, Kompaniyets L, Lavery AM, et al. Association Between COVID-19 and Myocarditis Using Hospital-Based Administrative Data - United States, March 2020-January 2021. MMWR Morb Mortal Wkly Rep. 2021;70(35):1228-1232. [CrossRef]
- Alvarez-Garcia J, Jaladanki S, Rivas-Lasarte M, et al. New Heart Failure Diagnoses Among Patients Hospitalized for COVID-19. J Am Coll Cardiol. 2021;77(17):2260-2262. [CrossRef]
- Puntmann VO, Carerj ML, Wieters I, et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5(11):1265. [CrossRef]
- Li H, Zhu H, Yang Z, Tang D, Huang L, Xia L. Tissue Characterization by Mapping and Strain Cardiac MRI to Evaluate Myocardial Inflammation in Fulminant Myocarditis. J Magn Reson Imaging. 2020;52(3):930-938. [CrossRef]
- Hinojar R, Varma N, Child N, et al. T1 Mapping in Discrimination of Hypertrophic Phenotypes: Hypertensive Heart Disease and Hypertrophic Cardiomyopathy: Findings From the International T1 Multicenter Cardiovascular Magnetic Resonance Study. Circ Cardiovasc Imaging. 2015;8(12). [CrossRef]
- Wang H, Li R, Zhou Z, et al. Cardiac involvement in COVID-19 patients: mid-term follow up by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2021;23(1):14. [CrossRef]
- Liu T, Wu D, Yan W, et al. Twelve-Month Systemic Consequences of Coronavirus Disease 2019 (COVID-19) in Patients Discharged From Hospital: A Prospective Cohort Study in Wuhan, China. Clin Infect Dis An Off Publ Infect Dis Soc Am. 2022;74(11):1953. [CrossRef]
- Seecheran R, Narayansingh R, Giddings S, et al. Atrial Arrhythmias in a Patient Presenting With Coronavirus Disease-2019 (COVID-19) Infection. J Investig Med high impact case reports. 2020;8. [CrossRef]
- Guzik TJ, Mohiddin SA, Dimarco A, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116(10):1666-1687. [CrossRef]
- Inciardi RM, Adamo M, Lupi L, et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur Heart J. 2020;41(19):1821-1829. [CrossRef]
- Wang D, Hu B, Hu C, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA. 2020;323(11):1061-1069. [CrossRef]
- Colon CM, Barrios JG, Chiles JW, et al. Atrial Arrhythmias in COVID-19 Patients. JACC Clin Electrophysiol. 2020;6(9):1189-1190. [CrossRef]
- Kim YM, Guzik TJ, Zhang YH, et al. A Myocardial Nox2 Containing NAD(P)H Oxidase Contributes to Oxidative Stress in Human Atrial Fibrillation. Circ Res. 2005;97(7):629-636. [CrossRef]
- Shang C, Liu Z, Zhu Y, et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol. 2022;12. [CrossRef]
- Muszyński P, Bonda TA. Mitochondrial Dysfunction in Atrial Fibrillation—Mechanisms and Pharmacological Interventions. J Clin Med 2021, Vol 10, Page 2385. 2021;10(11):2385. [CrossRef]
- Oudit GY, Wang K, Viveiros A, Kellner MJ, Penninger JM. Angiotensin-converting enzyme 2—at the heart of the COVID-19 pandemic. Cell. 2023;186(5):906-922. [CrossRef]
- Jakovac H, Ferenčić A, Stemberger C, Mohar Vitezić B, Cuculić D. Detection of SARS-CoV-2 Antigens in the AV-Node of a Cardiac Conduction System-A Case Report. Trop Med Infect Dis. 2022;7(3). [CrossRef]
- Ferreira AJ, Moraes PL, Foureaux G, Andrade AB, Santos RAS, Almeida AP. The angiotensin-(1-7)/Mas receptor axis is expressed in sinoatrial node cells of rats. J Histochem Cytochem. 2011;59(8):761-768. [CrossRef]
- Nádasy GL, Balla A, Cojocaru E, Cojocaru C, Vlad C-E, Eva L. Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries. Biomed 2023, Vol 11, Page 2004. 2023;11(7):2004. [CrossRef]
- Khazaal S, Harb J, Rima M, et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules. 2022;27(9). [CrossRef]
- Carpenter RM, Young MK, Petri WAO, et al. Repressed Ang 1-7 in COVID-19 Is Inversely Associated with Inflammation and Coagulation. mSphere. 2022;7(4). [CrossRef]
- Stone E, Kiat H, McLachlan CS. Atrial fibrillation in COVID-19: A review of possible mechanisms. FASEB J. 2020;34(9):11347-11354. [CrossRef]
- Disertori M, Rigoni M, Pace N, et al. Myocardial Fibrosis Assessment by LGE Is a Powerful Predictor of Ventricular Tachyarrhythmias in Ischemic and Nonischemic LV Dysfunction: A Meta-Analysis. JACC Cardiovasc Imaging. 2016;9(9):1046-1055. [CrossRef]
- McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020;2(7):e437-e445. [CrossRef]
- Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438-e440. [CrossRef]
- Wadowski PP, Panzer B, Józkowicz A, et al. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. Int J Mol Sci. 2023;24(3):2492. [CrossRef]
- Mehta P, Porter JC, Manson JJ, et al. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: challenges and opportunities. Lancet Respir Med. 2020;8(8):822-830. [CrossRef]
- Hojyo S, Uchida M, Tanaka K, et al. How COVID-19 induces cytokine storm with high mortality. Inflamm Regen. 2020;40(1). [CrossRef]
- Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020;80(6):607-613. [CrossRef]
- Zizzo G, Cohen PL. Imperfect storm: is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol. 2020;2(12):e779-e790. [CrossRef]
- Chen Y, Wang J, Liu C, et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol Med. 2020;26(1). [CrossRef]
- Xu ZS, Shu T, Kang L, et al. Temporal profiling of plasma cytokines, chemokines and growth factors from mild, severe and fatal COVID-19 patients. Signal Transduct Target Ther. 2020;5(1). [CrossRef]
- Del Valle DM, Kim-Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26(10):1636-1643. [CrossRef]
- Benest A, V. , Kruse K, Savant S, et al. Angiopoietin-2 is critical for cytokine-induced vascular leakage. PLoS One. 2013;8(8). [CrossRef]
- Frangogiannis, NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450-1488. [CrossRef]
- Sivakumar P, Gupta S, Sarkar S, Sen S. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol Cell Biochem. 2008;307(1-2):159-167. [CrossRef]
- Vanhoutte D, Van Almen GC, Van Aelst LNL, et al. Matricellular proteins and matrix metalloproteinases mark the inflammatory and fibrotic response in human cardiac allograft rejection. Eur Heart J. 2013;34(25):1930-1941. [CrossRef]
- Vanhoutte D, Schellings MWM, Götte M, et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115(4):475-482. [CrossRef]
- Zhang D, Li L, Chen Y, et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol Med. 2021;27(1). [CrossRef]
- Rovas A, Osiaevi I, Buscher K, et al. Microvascular dysfunction in COVID-19: the MYSTIC study. Angiogenesis. 2021;24(1):145. [CrossRef]
- Goonewardena SN, Grushko OG, Wells J, et al. Immune-Mediated Glycocalyx Remodeling in Hospitalized COVID-19 Patients. Cardiovasc Drugs Ther. 2023;37(2):307. [CrossRef]
- Queisser KA, Mellema RA, Middleton EA, et al. COVID-19 generates hyaluronan fragments that directly induce endothelial barrier dysfunction. JCI Insight. 2021;6(17). [CrossRef]
- Buijsers B, Yanginlar C, de Nooijer A, et al. Increased Plasma Heparanase Activity in COVID-19 Patients. Front Immunol. 2020;11. [CrossRef]
- Roshdy A, Zaher S, Fayed H, Coghlan JG. COVID-19 and the Heart: A Systematic Review of Cardiac Autopsies. Front Cardiovasc Med. 2021;7. [CrossRef]
- Godoy LC, Goligher EC, Lawler PR, Slutsky AS, Zarychanski R. Anticipating and managing coagulopathy and thrombotic manifestations of severe COVID-19. CMAJ. 2020;192(40):E1156-E1161. [CrossRef]
- Denorme F, Vanhoorelbeke K, De Meyer SF. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Front Immunol. 2019;10:495067.
- Kalagara T, Moutsis T, Yang Y, et al. The endothelial glycocalyx anchors von Willebrand factor fibers to the vascular endothelium. Blood Adv. 2018;2(18):2347-2357. [CrossRef]
- Middleton EA, He X-Y, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169-1179. [CrossRef]
- Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535. [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134-147. [CrossRef]
- Aiyegbusi OL, Hughes SE, Turner G, et al. Symptoms, complications and management of long COVID: a review. J R Soc Med. 2021;114(9):428-442. [CrossRef]
- Logue JK, Franko NM, McCulloch DJ, et al. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw open. 2021;4(2):e210830. [CrossRef]
- Puntmann VO, Martin S, Shchendrygina A, et al. Long-term cardiac pathology in individuals with mild initial COVID-19 illness. Nat Med. 2022;28(10):2117-2123. [CrossRef]
- Østergaard, L. SARS CoV-2 related microvascular damage and symptoms during and after COVID-19: Consequences of capillary transit-time changes, tissue hypoxia and inflammation. Physiol Rep. 2021;9(3):e14726. [CrossRef]
- Mantovani A, Morrone MC, Patrono C, et al. Long Covid: where we stand and challenges ahead. Cell Death Differ. 2022;29(10):1891-1900. [CrossRef]
- Nunn AVW, Guy GW, Brysch W, Bell JD. Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems. Biomedicines. 2022;10(12). [CrossRef]
- Xian H, Watari K, Sanchez-Lopez E, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. 2022;55(8):1370-1385.e8. [CrossRef]
- Leone O, Pieroni M, Rapezzi C, Olivotto I. The spectrum of myocarditis: from pathology to the clinics. Virchows Arch 2019 4753. 2019;475(3):279-301. [CrossRef]
- Andréoletti L, Lévêque N, Boulagnon C, Brasselet C, Fornes P. Viral causes of human myocarditis. Arch Cardiovasc Dis. 2009;102(6-7):559-568. [CrossRef]
- Kühl U, Pauschinger M, Seeberg B, et al. Viral Persistence in the Myocardium Is Associated With Progressive Cardiac Dysfunction. Circulation. 2005;112(13):1965-1970. [CrossRef]
- Cooper, LT. Myocarditis. N Engl J Med. 2009;360(15):1526-1538. [CrossRef]
- Jeserich M, Brunner E, Kandolf R, et al. Diagnosis of viral myocarditis by cardiac magnetic resonance and viral genome detection in peripheral blood. Int J Cardiovasc Imaging. 2013;29(1):121-129. [CrossRef]
- Schultheiss H-P, Baumeier C, Aleshcheva G, Bock C-T, Escher F. Viral Myocarditis—From Pathophysiology to Treatment. J Clin Med. 2021;10(22):5240. [CrossRef]
- Verdonschot J, Hazebroek M, Merken J, et al. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: review of the literature. Eur J Heart Fail. 2016;18(12):1430-1441. [CrossRef]
- Escher F, Kühl U, Gross U, et al. Aggravation of left ventricular dysfunction in patients with biopsy-proven cardiac human herpesvirus A and B infection. J Clin Virol. 2015;63:1-5. [CrossRef]
- Bonavita CM, Cardin RD. Don’t Go Breaking My Heart: MCMV as a Model for HCMV-Associated Cardiovascular Diseases. Pathog (Basel, Switzerland). 2021;10(5). [CrossRef]
- Magno Palmeira M, Umemura Ribeiro HY, Garcia Lira Y, et al. Heart failure due to cytomegalovirus myocarditis in immunocompetent young adults: a case report. BMC Res Notes. 2016;9(1). [CrossRef]
- Roubille C, Brunel AS, Gahide G, Kovacsik HV, Le Quellec A. Cytomegalovirus (CMV) and acute myocarditis in an immunocompetent patient. Intern Med. 2010;49(2):131-133. [CrossRef]
- Watanabe M, Panetta GL, Piccirillo F, et al. Acute Epstein-Barr related myocarditis: An unusual but life-threatening disease in an immunocompetent patient. J Cardiol cases. 2019;21(4):137-140. [CrossRef]
- Schultheiss HP, Piper C, Sowade O, et al. Betaferon in chronic viral cardiomyopathy (BICC) trial: Effects of interferon-β treatment in patients with chronic viral cardiomyopathy. Clin Res Cardiol. 2016;105(9):763-773. [CrossRef]
- Pauschinger M, Bowles NE, Fuentes-Garcia FJ, et al. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation. 1999;99(10):1348-1354. [CrossRef]
- Ilyas SZ, Tabassum R, Hamed H, Rehman SU, Qadri I. Hepatitis C Virus-Associated Extrahepatic Manifestations in Lung and Heart and Antiviral Therapy-Related Cardiopulmonary Toxicity. Viral Immunol. 2017;30(9):633-641. [CrossRef]
- Matsumori A, Matoba Y, Sasayama S. Dilated cardiomyopathy associated with hepatitis C virus infection. Circulation. 1995;92(9):2519-2525. [CrossRef]
- Matsumori A, Shimada T, Chapman NM, Tracy SM, Mason JW. Myocarditis and heart failure associated with hepatitis C virus infection. J Card Fail. 2006;12(4):293-298. [CrossRef]
- Mathez G, Cagno V. Viruses Like Sugars: How to Assess Glycan Involvement in Viral Attachment. Microorganisms. 2021;9(6). [CrossRef]
- Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses. 2019;11(7). [CrossRef]
- Pickles RJ, Fahrner JA, Petrella JM, Boucher RC, Bergelson JM. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J Virol. 2000;74(13):6050-6057. [CrossRef]
- Targosz-Korecka M, Kubisiak A, Kloska D, Kopacz A, Grochot-Przeczek A, Szymonski M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci Rep. 2021;11(1). [CrossRef]
- Zhou R, Liu L, Wang Y. Viral proteins recognized by different TLRs. J Med Virol. 2021;93(11):6116-6123. [CrossRef]
- Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105-2113. [CrossRef]
- Katare PB, Nizami HL, Paramesha B, Dinda AK, Banerjee SK. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci Reports 2020 101. 2020;10(1):1-15. [CrossRef]
- Garmaroudi FS, Marchant D, Hendry R, et al. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015;10(4):629-653. [CrossRef]
- Hsu TC, Wu WJ, Chen MC, Tsay GJ. Human parvovirus B19 non-structural protein (NS1) induces apoptosis through mitochondria cell death pathway in COS-7 cells. Scand J Infect Dis. 2004;36(8):570-577. [CrossRef]
- Bachelier K, Biehl S, Schwarz V, et al. Parvovirus B19-induced vascular damage in the heart is associated with elevated circulating endothelial microparticles. PLoS One. 2017;12(5):e0176311. [CrossRef]
- Vallbracht KB, Schwimmbeck PL, Kühl U, Seeberg B, Schultheiss HP. Endothelium-dependent flow-mediated vasodilation of systemic arteries is impaired in patients with myocardial virus persistence. Circulation. 2004;110(18):2938-2945. [CrossRef]
- Belkin MN, Uriel N. Heart health in the age of highly active antiretroviral therapy: A review of HIV cardiomyopathy. Curr Opin Cardiol. 2018;33(3):317-324. [CrossRef]
- Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Heart. 1994;72(6):561-566. [CrossRef]
- Kawai, C. From Myocarditis to Cardiomyopathy: Mechanisms of Inflammation and Cell Death. Circulation. 1999;99(8):1091-1100. [CrossRef]
- Neu C, Thiele Y, Horr F, et al. DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR. Int J Mol Sci. 2022;23(24). [CrossRef]
- Lavine KJ, Pinto AR, Epelman S, et al. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series. J Am Coll Cardiol. 2018;72(18):2213. [CrossRef]
- Magnani JW, Suk Danik HJ, Dec GW, DiSalvo TG. Survival in biopsy-proven myocarditis: A long-term retrospective analysis of the histopathologic, clinical, and hemodynamic predictors. Am Heart J. 2006;151(2):463-470. [CrossRef]
- Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343(19):1388-1398. [CrossRef]
- Zaragoza C, Ocampo C, Saura M, et al. The role of inducible nitric oxide synthase in the host response to Coxsackievirus myocarditis. Proc Natl Acad Sci. 1998;95(5):2469-2474. [CrossRef]
- Kühl U, Lassner D, Pauschinger M, et al. Prevalence of erythrovirus genotypes in the myocardium of patients with dilated cardiomyopathy. J Med Virol. 2008;80(7):1243-1251. [CrossRef]
- Kühl U, Pauschinger M, Noutsias M, et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation. 2005;111(7):887-893. [CrossRef]
- Kühl U, Lassner D, Von Schlippenbach J, Poller W, Schultheiss HP. Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J Am Coll Cardiol. 2012;60(14):1295-1296. [CrossRef]
- Pauschinger M, Doerner A, Kuehl U, et al. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation. 1999;99(7):889-895. [CrossRef]
- Pietsch H, Escher F, Aleshcheva G, Lassner D, Bock CT, Schultheiss HP. Detection of parvovirus mRNAs as markers for viral activity in endomyocardial biopsy-based diagnosis of patients with unexplained heart failure. Sci Rep. 2020;10(1). [CrossRef]
- Kuhl U, Lassner D, Dorner A, et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res Cardiol. 2013;108(5). [CrossRef]
- Fairweather DL, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15(1):17-27. [CrossRef]
- Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127-155. [CrossRef]
- Huber, S. Viral Myocarditis and Dilated Cardiomyopathy: Etiology and Pathogenesis. Curr Pharm Des. 2016;22(4):408-426. [CrossRef]
- Rose, NR. Viral myocarditis. Curr Opin Rheumatol. 2016;28(4):383-389. [CrossRef]
- Bracamonte-Baran W, Čiháková D. Cardiac Autoimmunity: Myocarditis. Adv Exp Med Biol. 2017;1003:187-221. [CrossRef]
- Kawai, C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99(8):1091-1100. [CrossRef]
- Cunningham, MW. T cell mimicry in inflammatory heart disease. Mol Immunol. 2004;40(14-15):1121-1127. [CrossRef]
- Elamm C, Fairweather DL, Cooper LT. Republished: pathogenesis and diagnosis of myocarditis. Postgrad Med J. 2012;88(1043):539-544. [CrossRef]
- Monda E, Palmiero G, Rubino M, et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int J Mol Sci. 2020;21(18):1-14. [CrossRef]
- Heymans S, Eriksson U, Lehtonen J, Cooper LT. The Quest for New Approaches in Myocarditis and Inflammatory Cardiomyopathy. J Am Coll Cardiol. 2016;68(21):2348-2364. [CrossRef]
- Westermann D, Lindner D, Kasner M, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4(1):44-52. [CrossRef]
- Huang LY, Halder S, Agbandje-Mckenna M. Parvovirus glycan interactions. Curr Opin Virol. 2014;7(1):108-118. [CrossRef]
- Conti C, Cirone M, Sgro R, Altieri F, Zompetta C, Faggioni A. Early interactions of human herpesvirus 6 with lymphoid cells: role of membrane protein components and glycosaminoglycans in virus binding. J Med Virol. 2000;62(4):487-497. [CrossRef]
- Hadigal S, Koganti R, Yadavalli T, Agelidis A, Suryawanshi R, Shukla D. Heparanase-Regulated Syndecan-1 Shedding Facilitates Herpes Simplex Virus 1 Egress. J Virol. 2020;94(6). [CrossRef]
- Connolly SA, Jardetzky TS, Longnecker R. The structural basis of herpesvirus entry. Nat Rev Microbiol. 2021;19(2):110. [CrossRef]
- Stuhlmeier, KM. Hyaluronan production in synoviocytes as a consequence of viral infections: HAS1 activation by Epstein-Barr virus and synthetic double- and single-stranded viral RNA analogs. J Biol Chem. 2008;283(24):16781-16789. [CrossRef]
- Mitra D, Hasan MH, Bates JT, et al. The degree of polymerization and sulfation patterns in heparan sulfate are critical determinants of cytomegalovirus entry into host cells. PLoS Pathog. 2021;17(8). [CrossRef]
- Compton T, Nowlin DM, Cooper NR. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology. 1993;193(2):834-841. [CrossRef]
- McLeish NJ, Williams ÇH, Kaloudas D, Roivainen MM, Stanway G. Symmetry-related clustering of positive charges is a common mechanism for heparan sulfate binding in enteroviruses. J Virol. 2012;86(20):11163-11170. [CrossRef]
- Tan CW, Poh CL, Sam I-C, Chan YF. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol. 2013;87(1):611-620. [CrossRef]
- Kobayashi K, Koike S. Cellular receptors for enterovirus A71. J Biomed Sci. 2020;27(1). [CrossRef]
- Ramos-Martínez IE, Ramos-Martínez E, Segura-Velázquez RÁ, et al. Heparan Sulfate and Sialic Acid in Viral Attachment: Two Sides of the Same Coin? Int J Mol Sci. 2022;23(17). [CrossRef]
- Schröer K, Alshawabkeh M, Schellhorn S, Bronder K, Zhang W, Ehrhardt A. Influence of Heparan Sulfate Proteoglycans and Factor X on species D Human Adenovirus Uptake and Transduction. Viruses. 2022;15(1). [CrossRef]
- Fender P, Schoehn G, Perron-Sierra F, Tucker GC, Lortat-Jacob H. Adenovirus dodecahedron cell attachment and entry are mediated by heparan sulfate and integrins and vary along the cell cycle. Virology. 2008;371(1):155-164. [CrossRef]
- Grigorov B, Reungoat E, Gentil dit Maurin A, et al. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell Microbiol. 2017;19(5). [CrossRef]
- Zhang F, Sodroski C, Cha H, Li Q, Liang TJ. Infection of Hepatocytes With HCV Increases Cell Surface Levels of Heparan Sulfate Proteoglycans, Uptake of Cholesterol and Lipoprotein, and Virus Entry by Up-regulating SMAD6 and SMAD7. Gastroenterology. 2017;152(1):257-270.e7. [CrossRef]
- Cerezo-Magaña M, Bång-Rudenstam A, Belting M. Proteoglycans: a common portal for SARS-CoV-2 and extracellular vesicle uptake. Am J Physiol Cell Physiol. 2023;324(1):C76-C84. [CrossRef]
- Bell TJ, Brand OJ, Morgan DJ, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. 2019;80:14-28. [CrossRef]
- Corrado D, Basso C, Thiene G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res. 2001;50(2):399-408. [CrossRef]
- Tschöpe C, Ammirati E, Bozkurt B, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021;18(3):169-193. [CrossRef]
- Baksi AJ, Kanaganayagam GS, Prasad SK. Arrhythmias in viral myocarditis and pericarditis. Card Electrophysiol Clin. 2015;7(2):269-281. [CrossRef]
- Peretto G, Sala S, Rizzo S, et al. Ventricular Arrhythmias in Myocarditis: Characterization and Relationships With Myocardial Inflammation. J Am Coll Cardiol. 2020;75(9):1046-1057. [CrossRef]
- Ferreira VM, Piechnik SK, Dall’Armellina E, et al. Native T1-mapping detects the location, extent and patterns of acute myocarditis without the need for gadolinium contrast agents. J Cardiovasc Magn Reson. 2014;16(1). [CrossRef]
- Ammirati E, Moroni F, Sormani P, et al. Quantitative changes in late gadolinium enhancement at cardiac magnetic resonance in the early phase of acute myocarditis. Int J Cardiol. 2017;231:216-221. [CrossRef]
- Harrison JL, Jensen HK, Peel SA, et al. Cardiac magnetic resonance and electroanatomical mapping of acute and chronic atrial ablation injury: a histological validation study. Eur Heart J. 2014;35(22):1486-1495. [CrossRef]
- Dickfeld T, Tian J, Ahmad G, et al. MRI-Guided ventricular tachycardia ablation: integration of late gadolinium-enhanced 3D scar in patients with implantable cardioverter-defibrillators. Circ Arrhythm Electrophysiol. 2011;4(2):172-184. [CrossRef]
- Fernández-Armenta J, Berruezo A, Andreu D, et al. Three-dimensional architecture of scar and conducting channels based on high resolution ce-CMR: insights for ventricular tachycardia ablation. Circ Arrhythm Electrophysiol. 2013;6(3):528-537. [CrossRef]
- Perez-David E, Arenal Á, Rubio-Guivernau JL, et al. Noninvasive identification of ventricular tachycardia-related conducting channels using contrast-enhanced magnetic resonance imaging in patients with chronic myocardial infarction: comparison of signal intensity scar mapping and endocardial voltage mappin. J Am Coll Cardiol. 2011;57(2):184-194. [CrossRef]
- Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death Developed by the task force for the management of patients with death of the European Society of Cardiology ( ESC ) Endorsed by the. Eur Heart J. Published online 2022:1-130. [CrossRef]
- Nagai T, Inomata T, Kohno T, et al. JCS 2023 Guideline on the Diagnosis and Treatment of Myocarditis. Circ J. 2023;87(5):674-754. [CrossRef]
- Roth GA, Mensah GA, Johnson CO, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J Am Coll Cardiol. 2020;76(25):2982-3021. [CrossRef]
- Stolfo D, Collini V, Sinagra G. Advanced Heart Failure in Special Population: Cardiomyopathies and Myocarditis. Heart Fail Clin. 2021;17(4):661-672. [CrossRef]
- Pozzi M, Banfi C, Grinberg D, et al. Veno-arterial extracorporeal membrane oxygenation for cardiogenic shock due to myocarditis in adult patients. J Thorac Dis. 2016;8(7):E495. [CrossRef]
- Combes A, Price S, Slutsky AS, Brodie D. Temporary circulatory support for cardiogenic shock. Lancet. 2020;396(10245):199-212. [CrossRef]
- Means, RT. The anaemia of infection. Baillieres Best Pract Res Clin Haematol. 2000;13(2):151-162. [CrossRef]
- Bellmann-Weiler R, Lanser L, Barket R, et al. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J Clin Med. 2020;9(8):1-11. [CrossRef]
- Giustino G, Kirtane AJ, Baber U, et al. Impact of Anemia on Platelet Reactivity and Ischemic and Bleeding Risk: From the Assessment of Dual Antiplatelet Therapy With Drug-Eluting Stents Study. Am J Cardiol. 2016;117(12):1877-1883. [CrossRef]
- Wadowski PP, Kopp CW, Koppensteiner R, et al. Decreased platelet inhibition by P2Y12 receptor blockers in anaemia. Eur J Clin Invest. 2018;48(1):e12861. [CrossRef]
- Ammirati E, Bizzi E, Veronese G, et al. Immunomodulating Therapies in Acute Myocarditis and Recurrent/Acute Pericarditis. Front Med. 2022;9:838564.
- Schultheiss HP, Khl U, Cooper LT. The management of myocarditis. Eur Heart J. 2011;32(21):2616-2625. [CrossRef]
- Lampejo T, Durkin SM, Bhatt N, Guttmann O. Acute myocarditis: aetiology, diagnosis and management. Clin Med (Northfield Il). 2021;21(5):e505. [CrossRef]
- Bang V, Ganatra S, Shah SP, et al. Management of Patients With Giant Cell Myocarditis: JACC Review Topic of the Week. J Am Coll Cardiol. 2021;77(8):1122-1134. [CrossRef]
- Cremer PC, Sheng CC, Sahoo D, et al. Double-blind randomized proof-of-concept trial of canakinumab in patients with COVID-19 associated cardiac injury and heightened inflammation. Eur Hear J Open. 2021;1(1). [CrossRef]
- Xu J, Zhou Z, Zheng Y, Yang S, Huang K, Li H. Roles of inflammasomes in viral myocarditis. Front Cell Infect Microbiol. 2023;13:1149911.
- van de Veerdonk FL, Giamarellos-Bourboulis E, Pickkers P, et al. A guide to immunotherapy for COVID-19. Nat Med 2022 281. 2022;28(1):39-50. [CrossRef]
- Ammirati E, Frigerio M, Adler ED, et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ Heart Fail. 2020;13(11):e007405. [CrossRef]
- Merken J, Hazebroek M, Van Paassen P, et al. Immunosuppressive Therapy Improves Both Short- and Long-Term Prognosis in Patients With Virus-Negative Nonfulminant Inflammatory Cardiomyopathy. Circ Heart Fail. 2018;11(2):e004228. [CrossRef]
- Peretto G, Sala S, Luca G De, et al. Immunosuppressive Therapy and Risk Stratification of Patients With Myocarditis Presenting With Ventricular Arrhythmias. JACC Clin Electrophysiol. 2020;6(10):1221-1234. [CrossRef]
- Abernethy A, Raza S, Sun JL, et al. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure With Preserved Ejection Fraction. J Am Hear Assoc Cardiovasc Cerebrovasc Dis. 2018;7(8). [CrossRef]
- Wijk SS-V, Empel V Van, Davarzani N, et al. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail. 2015;17(10):1006-1014. [CrossRef]
- Hanna A, Frangogiannis NG. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc Drugs Ther. 2020;34(6):849. [CrossRef]
- Bajaj NS, Gupta K, Gharpure N, et al. Effect of immunomodulation on cardiac remodelling and outcomes in heart failure: a quantitative synthesis of the literature. ESC Hear Fail. 2020;7(3):1319-1330. [CrossRef]
- Mann DL, McMurray JJV, Packer M, et al. Targeted Anticytokine Therapy in Patients With Chronic Heart Failure. Circulation. 2004;109(13):1594-1602. [CrossRef]
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (AT. Circulation. 2003;107(25):3133-3140. [CrossRef]
- Paulus WJ, Zile MR. FROM SYSTEMIC INFLAMMATION TO MYOCARDIAL FIBROSIS: THE HFPEF PARADIGM REVISITED. Circ Res. 2021;128(10):1451. [CrossRef]
- Ning N, Guo HH, Iagaru A, Mittra E, Fowler M, Witteles R. Serial Cardiac FDG-PET for the Diagnosis and Therapeutic Guidance of Patients With Cardiac Sarcoidosis. J Card Fail. 2019;25(4):307-311. [CrossRef]
- Shelke AB, Aurangabadkar HU, Bradfield JS, Ali Z, Kumar KS, Narasimhan C. Serial FDG-PET scans help to identify steroid resistance in cardiac sarcoidosis. Int J Cardiol. 2017;228:717-722. [CrossRef]
- Terasaki F, Azuma A, Anzai T, et al. JCS 2016 Guideline on Diagnosis and Treatment of Cardiac Sarcoidosis - Digest Version. Circ J. 2019;83(11):2329-2388. [CrossRef]
- Birnie DH, Sauer WH, Bogun F, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Hear Rhythm. 2014;11(7):1304-1323. [CrossRef]
- Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):E876-E894. [CrossRef]
- McDonagh TA, Metra M, Adamo M, et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2023;44(37). [CrossRef]
- Zeng S, Delic D, Chu C, et al. Antifibrotic effects of low dose SGLT2 Inhibition with empagliflozin in comparison to Ang II receptor blockade with telmisartan in 5/6 nephrectomised rats on high salt diet. Biomed Pharmacother. 2022;146:112606. [CrossRef]
- Li C, Zhang J, Xue M, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18(1):1-13.
- Kosiborod MN, Esterline R, Furtado RHM, et al. Dapagliflozin in patients with cardiometabolic risk factors hospitalised with COVID-19 (DARE-19): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9(9):586-594. [CrossRef]
- SGLT2 inhibitors not linked with improved survival in hospitalised COVID-19 patients. Accessed December 3, 2023. https://www.escardio.org/The-ESC/Press-Office/Press-releases/SGLT2-inhibitors-notlinked-with-improved-survival-in-hospitalised-COVID-19-patients.
- Abani O, Abbas A, Abbas F, et al. Empagliflozin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet Diabetes Endocrinol. 2023;11(12):905-914. [CrossRef]
- Pitt B, Agarwal R, Anker SD, et al. Association of Finerenone Use With Reduction in Treatment-Emergent Pneumonia and COVID-19 Adverse Events Among Patients With Type 2 Diabetes and Chronic Kidney Disease: A FIDELITY Pooled Secondary Analysis. JAMA Netw Open. 2022;5(10):e2236123-e2236123. [CrossRef]
- Henri O, Pouehe C, Houssari M, et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation. 2016;133(15):1484-1497. [CrossRef]
- Schultheiss HP, Bock CT, Aleshcheva G, Baumeier C, Poller W, Escher F. Interferon-β Suppresses Transcriptionally Active Parvovirus B19 Infection in Viral Cardiomyopathy: A Subgroup Analysis of the BICC-Trial. Viruses. 2022;14(2). [CrossRef]
- Schmidt-Lucke C, Spillmann F, Bock T, et al. Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus b19 infection. J Infect Dis. 2010;201(6):936-945. [CrossRef]
- Anzai A, Mindur JE, Halle L, et al. Self-reactive CD4+ IL-3+ T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J Exp Med. 2019;216(2):369. [CrossRef]
- Ammirati E, Varrenti M, Veronese G, et al. Prevalence and outcome of patients with acute myocarditis and positive viral search on nasopharyngeal swab. Eur J Heart Fail. 2021;23(7):1242-1245. [CrossRef]
- Gil-Cruz C, Perez-Shibayama C, de Martin A, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science. 2019;366(6467):881-886. [CrossRef]
- Amdani SM, Kim HS, Orvedahl A, John AO, Said A, Simpson K. Successful treatment of fulminant neonatal enteroviral myocarditis in monochorionic diamniotic twins with cardiopulmonary support, intravenous immunoglobulin and pocapavir. BMJ Case Rep. 2018;2018. [CrossRef]
- Abzug MJ, Michaels MG, Wald E, et al. A Randomized, Double-Blind, Placebo-Controlled Trial of Pleconaril for the Treatment of Neonates With Enterovirus Sepsis. J Pediatric Infect Dis Soc. 2016;5(1):53-62. [CrossRef]
- Yen MH, Huang YC, Chen MC, et al. Effect of intravenous immunoglobulin for neonates with severe enteroviral infections with emphasis on the timing of administration. J Clin Virol. 2015;64:92-96. [CrossRef]
- Kühl U, Lassner D, Wallaschek N, et al. Chromosomally integrated human herpesvirus 6 in heart failure: prevalence and treatment. Eur J Heart Fail. 2015;17(1):9-19. [CrossRef]
- Sanchez MJ, Bergasa N V. Hepatitis C associated cardiomyopathy: potential pathogenic mechanisms and clinical implications. Med Sci Monit. 2008;14(5):RA55-63. https://pubmed.ncbi.nlm.nih. 1844.
- Baik SH, Jeong HS, Kim SJ, Yoon YK, Sohn JW, Kim MJ. A Case of Influenza Associated Fulminant Myocarditis Successfully Treated with Intravenous Peramivir. Infect Chemother. 2015;47(4):272-277. [CrossRef]
- Ito N, Sato M, Momoi N, et al. Influenza A H1N1 pdm09-associated myocarditis during zanamivir therapy. Pediatr Int. 2015;57(6):1172-1174. [CrossRef]
- Manaresi E, Gallinella G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses. 2019;11(7). [CrossRef]
- Düngen HD, Dordevic A, Felix SB, et al. β1-Adrenoreceptor Autoantibodies in Heart Failure: Physiology and Therapeutic Implications. Circ Heart Fail. 2020;13(1). [CrossRef]
- Li G, Hilgenfeld R, Whitley R, Clercq E De. Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov. 2023;22(6):449. [CrossRef]
- Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;323(18):1824-1836. [CrossRef]
- Zhou D, Dai SM, Tong Q. COVID-19: a recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J Antimicrob Chemother. 2020;75(7):1667-1670. [CrossRef]
- Rout A, Tantry US, Novakovic M, Sukhi A, Gurbel PA. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin Pharmacother. 2020;21(15):1851-1865. [CrossRef]
- Hentia C, Rizzato A, Camporesi E, et al. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018;8(5):e00959. [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17(12):773-789. [CrossRef]
- Bruegger D, Rehm M, Jacob M, et al. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit Care. 2008;12(3). [CrossRef]
- Koch E, Malek FA. Standardized Extracts from Hawthorn Leaves and Flowers in the Treatment of Cardiovascular Disorders – Preclinical and Clinical Studies. Planta Med. 2011;77(11):1123-1128. [CrossRef]
- Ebong EE, Lopez-Quintero S V., Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol. 2014;6(3):338-347. [CrossRef]
- Peters W, Drueppel V, Kusche-Vihrog K, Schubert C, Oberleithner H. Nanomechanics and Sodium Permeability of Endothelial Surface Layer Modulated by Hawthorn Extract WS 1442. PLoS One. 2012;7(1):e29972. [CrossRef]
- Andreas M, Schmid AI, Keilani M, et al. Effect of ischemic preconditioning in skeletal muscle measured by functional magnetic resonance imaging and spectroscopy: a randomized crossover trial. J Cardiovasc Magn Reson. 2011;13(1):32. [CrossRef]
- Broekhuizen LN, Lemkes BA, Mooij HL, et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. 2010;53(12):2646-2655. [CrossRef]
- Illien-Junger S, Grosjean F, Laudier DM, Vlassara H, Striker GE, Iatridis JC. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLoS One. 2013;8(5):e64302. [CrossRef]
- Salmon AHJ, Ferguson JK, Burford JL, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23(8):1339-1350. [CrossRef]
- Harada N, Maeda M. Chemical structure of antithrombin-active rhamnan sulfate from monostrom nitidum. Biosci Biotechnol Biochem. 1998;62(9):1647-1652. [CrossRef]
- Giantsos-Adams K, Lopez-Quintero V, Kopeckova P, Kopecek J, Tarbell JM, Dull R. Study of the therapeutic benefit of cationic copolymer administration to vascular endothelium under mechanical stress. Biomaterials. 2011;32(1):288-294. [CrossRef]
- Masola V, Zaza G, Onisto M, Lupo A, Gambaro G. Glycosaminoglycans, proteoglycans and sulodexide and the endothelium: biological roles and pharmacological effects. Int Angiol. 2014;33(3):243-254. Accessed August 30, 2023. https://pubmed.ncbi.nlm.nih.gov/24936533/.
- Frati Munari, AC. [Medical significance of endothelial glycocalyx. Part 2: Its role in vascular diseases and in diabetic complications]. Arch Cardiol Mex. 2014;84(2):110-116. [CrossRef]
- Taneja, R. Current status of oral pentosan polysulphate in bladder pain syndrome/interstitial cystitis. Int Urogynecol J. 2021;32(5):1107-1115. [CrossRef]
- Cancel LM, Tarbell JM. Rhamnan sulfate enhances the endothelial glycocalyx and decreases the LDL permeability of human coronary artery endothelial cells in vitro. FASEB J. 2013;27(S1):896.3-896.3. [CrossRef]
- Merchant SH, Gurule DM, Larson RS. Amelioration of ischemia-reperfusion injury with cyclic peptide blockade of ICAM-1. Am J Physiol Heart Circ Physiol. 2003;284(4):H1260-8. [CrossRef]
- Kozar RA, Peng Z, Zhang R, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112(6):1289-1295. [CrossRef]
- Suarez JI, Martin RH, Calvillo E, Bershad EM, Venkatasubba Rao CP. Effect of human albumin on TCD vasospasm, DCI, and cerebral infarction in subarachnoid hemorrhage: the ALISAH study. Acta Neurochir Suppl. 2015;120:287-290. [CrossRef]
- Curry F-RE, Adamson RH. Sphingosine-1-phosphate and the “albumin effect” on rat venular microvessels. FASEB J. 2013;27(S1):896.2-896.2. [CrossRef]
- Bonaventura A, Vecchié A, Dagna L, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21(5):319-329. [CrossRef]
- Conway EM, Mackman N, Warren RQ, et al. Understanding COVID-19-associated coagulopathy. Nat Rev Immunol. 2022;22(10):639-649. [CrossRef]
- Carnevale R, Cammisotto V, Bartimoccia S, et al. Toll-Like Receptor 4-Dependent Platelet-Related Thrombosis in SARS-CoV-2 Infection. Circ Res. 2023;132(3):290-305. [CrossRef]
- Zhang H, Lao Q, Zhang J, Zhu J. Coagulopathy in COVID-19 and anticoagulation clinical trials. Best Pract Res Clin Haematol. 2022;35(3):101377. [CrossRef]
- Chandra A, Chakraborty U, Ghosh S, Dasgupta S. Anticoagulation in COVID-19: current concepts and controversies. Postgrad Med J. 2022;98(1159):395-402. [CrossRef]
- Yadav P, Kumar D, Meena DS, et al. Post-Discharge Prophylactic Anticoagulation in COVID-19 Patients: A Clinical Dilemma. Cardiovasc Hematol Disord Drug Targets. 2021;21(3):206-209. [CrossRef]
- Stone GW, Farkouh ME, Lala A, et al. Randomized Trial of Anticoagulation Strategies for Noncritically Ill Patients Hospitalized With COVID-19. J Am Coll Cardiol. 2023;81(18):1747. [CrossRef]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13(3):393-403. [CrossRef]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu Rev Immunol. 2019;37:349-375. [CrossRef]
- Liddell, JR. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, Vol 6, Page 65. 2017;6(3):65. [CrossRef]
- Vivarini áislan de C, Calegari-Silva TC, Saliba AM, et al. Systems approach reveals nuclear factor erythroid 2-related factor 2/protein kinase r crosstalk in human cutaneous leishmaniasis. Front Immunol. 2017;8(SEP):283936.
- Andreas M, Schmid AI, Doberer D, et al. Heme arginate improves reperfusion patterns after ischemia: a randomized, placebo-controlled trial in healthy male subjects. J Cardiovasc Magn Reson. 2012;14(1). [CrossRef]
- Hangaishi M, Ishizaka N, Aizawa T, et al. Induction of heme oxygenase-1 can act protectively against cardiac ischemia/reperfusion in vivo. Biochem Biophys Res Commun. 2000;279(2):582-588. [CrossRef]
- Li W, Zhang B, Wei X, Cui X, Kobayashi T. Effects of heme oxygenase 1 on brain edema and neurologic outcome after cardiopulmonary resuscitation in rats. Anesthesiology. 2008;109(2):260-268. [CrossRef]
- Ligi D, Lo Sasso B, Giglio RV, et al. Circulating histones contribute to monocyte and MDW alterations as common mediators in classical and COVID-19 sepsis. Crit Care. 2022;26(1). [CrossRef]
- Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952-1961. [CrossRef]
- Wadowski PP, Eichelberger B, Kopp CW, et al. Disaggregation Following Agonist-Induced Platelet Activation in Patients on Dual Antiplatelet Therapy. J Cardiovasc Transl Res. 2017;10(4):359-367. [CrossRef]
- Panzer B, Wadowski PP, Huber K, Panzer S, Gremmel T. Protease-activated receptor-mediated platelet aggregation in patients with type 2 diabetes on potent P2Y12 inhibitors. Diabet Med. 2022;39(8). [CrossRef]
- Wadowski PP, Pultar J, Weikert C, et al. Protease-activated receptor-mediated platelet aggregation in acute coronary syndrome patients on potent P2Y12 inhibitors. Res Pract Thromb Haemost. 2019;3(3):383-390. [CrossRef]
- Ofosu FA, Dewar L, Craven SJ, et al. Coordinate activation of human platelet protease-activated receptor-1 and -4 in response to subnanomolar alpha-thrombin. J Biol Chem. 2008;283(40):26886-26893. [CrossRef]
- Gremmel T, Michelson AD, Wadowski PP, et al. Sex-specific platelet activation through protease-activated receptor-1 in patients undergoing cardiac catheterization. Atherosclerosis. 2021;339:12-19. [CrossRef]
- Wadowski PP, Weikert C, Pultar J, et al. Ticagrelor Inhibits Toll-Like and Protease-Activated Receptor Mediated Platelet Activation in Acute Coronary Syndromes. Cardiovasc drugs Ther. 2020;34(1):53-63. [CrossRef]
- Van Der Heijden M, Van Nieuw Amerongen GP, Koolwijk P, Van Hinsbergh VWM, Groeneveld ABJ. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax. 2008;63(10):903-909. [CrossRef]
- Rau A, Schroeter N, Blazhenets G, et al. Widespread white matter oedema in subacute COVID-19 patients with neurological symptoms. Brain. 2022;145(9):3203-3213. [CrossRef]
- Bocci M, Oudenaarden C, Sàenz-sardà X, et al. Infection of brain pericytes underlying neuropathology of covid-19 patients. Int J Mol Sci. 2021;22(21):11622.
- Stein SR, Ramelli SC, Grazioli A, et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature. 2022;612(7941):758-763. [CrossRef]
- Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023 213. 2023;21(3):133-146. [CrossRef]
- Panagiotides NG, Zimprich F, Machold K, et al. A Case of Autoimmune Small Fiber Neuropathy as Possible Post COVID Sequelae. Int J Environ Res Public Health. 2023;20(6). [CrossRef]
- Jones OY, Yeralan S. Is Long COVID a State of Systemic Pericyte Disarray? J Clin Med. 2022;11(3). [CrossRef]
- Abdi A, AlOtaiby S, Badarin F Al, Khraibi A, Hamdan H, Nader M. Interaction of SARS-CoV-2 with cardiomyocytes: Insight into the underlying molecular mechanisms of cardiac injury and pharmacotherapy. Biomed Pharmacother. 2022;146:112518. [CrossRef]
- Delorey TM, Ziegler CGK, Heimberg G, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellulartargets. Nature. 2021;595(7865):107. [CrossRef]
- Katwa LC, Mendoza C, Clements M. CVD and COVID-19: Emerging Roles of Cardiac Fibroblasts and Myofibroblasts. Cells. 2022;11(8). [CrossRef]
- Daems M, Liesenborghs L, Boudewijns R, et al. SARS-CoV-2 infection causes prolonged cardiomyocyte swelling and inhibition of HIF1α translocation in an animal model COVID-19. Front Cardiovasc Med. 2022;9:964512.
- Avolio E, Carrabba M, Milligan R, et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: a potential non-infective mechanism of COVID-19 microvascular disease. Clin Sci (Lond). 2021;135(24):2667-2689. [CrossRef]
Figure 1.
Inflammation during SARS-CoV-2 infection.
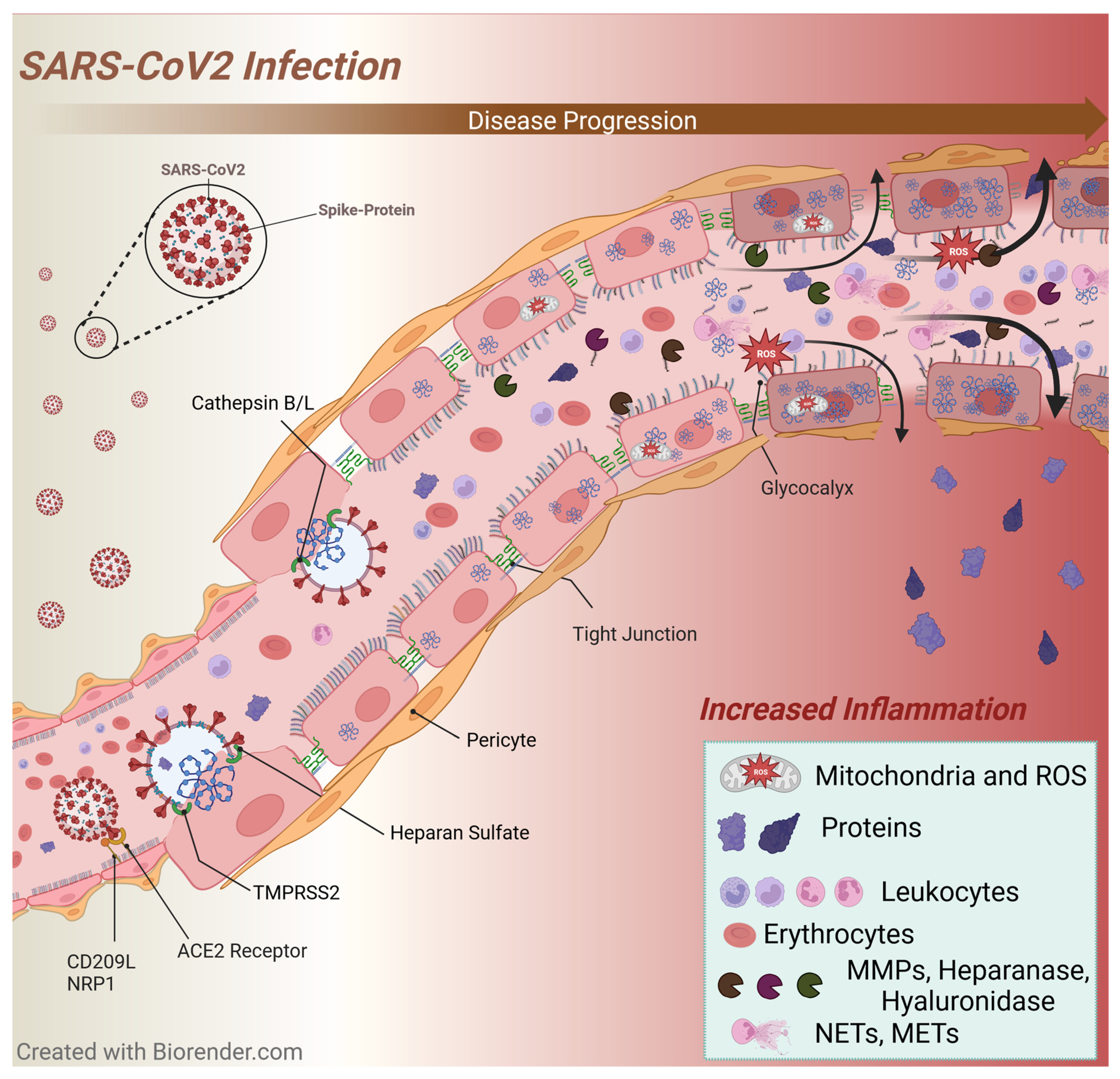
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



