
Submitted:
20 December 2023
Posted:
20 December 2023
You are already at the latest version
Abstract
Radical reactions in water or aqueous media are important for organic synthesis, realizing high yielding processes under non-toxic and environmentally friendly conditions. This overview includes (i) a general introduction to organic chemistry in water and aqueous media, (ii) synthetic approaches in-, on- and with-water as well as in heterogeneous phase, (iii) reaction of carbon-centered radicals with water (or deuterium oxide) activated through coordination with various Lewis acids, (iv) photocatalysis in water and aqueous media, and (v) synthetic applications bioinspired from naturally occurring processes. A wide range of chemical processes and synthetic strategies under different experimental conditions have been reviewed that lead to important functional group translocation and transformation reactions, leading to the preparation of complex molecules. These results reveal how water as a solvent/medium/reagent in radical chemistry has matured over the last two decades and how further discoveries are foreseen to come in the near future.
Keywords:
organic synthesis
; radical reactions
; water and aqueous media
; on water reactions
; water coordination with Lewis acids
; photocatalysis
; bioinspired reactions
1. Organic Reactions in Aqueous Media
The use of water as solvent for organic reactions, initially proposed by Breslow [1], became very popular in the last three decades for various reasons, not only being inexpensive and nontoxic, but mainly being an environmental friendly substitute for organic solvents [2,3,4,5,6,7]. In addition, reactions “in water” may bring about many benefits, among them, easy product separation based on lower solubility in water, lack of toxicity and inflammability, high polarity, heat capacity, solubility of inorganic compounds and, often, increased reaction rate and selectivity. A further boost for the use of this solvent came from the concept of the reactivity “on water”, introduced by Sharpless and co-workers to describe reactions of water-insoluble organic compounds that take place in aqueous suspensions [8]. The development of organic microenvironments in aqueous phase allows the reaction to proceed considerably faster in the presence of water than in organic solvents, and tremendous research progress on chemical reactions under aqueous conditions has been reported. Recently, the differences in reactivity between homogeneous and heterogeneous systems, that is, “in-water” and “on-water” systems, respectively, have attracted considerable attention as testified by numerous reviews [8,9,10,11,12,13,14,15].
Despite these findings, the percentage of synthetic organic radical reactions carried out in the presence of water remained very small [16,17,18], until Macmillan and coworkers introduced the concept of photoredox catalysis [19]. In this review we will present a variety of radical reactions where water exerts significant influence on the outcome.
2. Free Radicals in Water
2.1. Neutral Carbon-Centered Radicals Do Not React with Water
An enormous amount of data in the literature indicate that neutral carbon-centered radicals do not react with water. The reaction products in aqueous solutions often are a consequence of oxidation followed by reaction with water. For example, the C1′ radical formation by exposure to enediyne antibiotics (e.g., NCS-Chrom) in the presence of O2 leads to an abasic site, with the formation of the 2-deoxyribonolactone residue (Figure 1) as a consequence of C1′ carbon oxidation [20]. The mechanistic details for the formation of 2-deoxyribonolactone, elucidated at the nucleoside level by oxygen-18 labeling and a laser flash photolysis experiment, indicated the involvement of an heterolytic cleavage with release of O2•– (k = 1.5 x 104 s–1) followed by hydrolysis [21,22].
On the other hand, radical cations are prone to react with nucleophiles [23]. An example is the well know guanine moiety G in nucleosides, ds-oligonucleotides or DNA that can be oxidized to G•+ by a variety of oxidants such as SO4•−, Br2•−, Cl2•−, and CO3•−, including various metal complexes. Figure 2 shows the case of 2′-deoxyguanosine (dG) and the resulting radical cation, which reacts with water to give the adduct 8-HO-dG• in competition with a deprotonation step [24,25]. 8-HO-dG• is precursor of 8-oxo-guanine moiety, the most relevant modification in genetic material [26].
Occasionally it is reported that alkyl radicals may react with water directly by hydrogen abstraction [27]. Considering that the bond dissociation energy (BDE) of water is ca. 117 kcal mol–1, these reactions are highly endothermic and do not occur. However, it is well known that coordination with various Lewis Acids can provide a substantial decrease of BDE and favor the reactions with alkyl radical. We have summarized this area of research in Section 5 below.
2.2. Polar Effects and the Increase of Reaction Rates
The hydrogen abstraction from thiols by carbon-centered radicals (R•) is one of the most-important reactions in free-radical chemistry. The intracellular concentration of glutathione (GSH) is in the range of 1-10 mM (depending on the cell type) and GSH serves as an H-atom donor in the repair of R• produced in biological systems [28].
Kinetic data on the reactions of thiols with R• are numerous in organic, aqueous, and mixed solutions [29,30]. In organic solvents for example, primary-, secondary-, or tertiary-alkyl radicals abstract hydrogen from Me(CH2)7SH with rate constants of ca. 107 M–1 s–1 and an α-hydroxyl substituent has no net kinetic effect on the thiol trapping [31]. All the rate constants were found to be essentially the same for a specific thiol, despite the change of the reaction thermochemistry. On the other hand, a peculiarity on the radical substituents is found in aqueous solutions. Table 1 shows the rate constants for the reaction of the α-hydroxyalkyl radicals with 2-mercaptoethanol (Reaction 1). The rate of reaction increases by replacing H-atom with Me-group, despite the fact that the exothermicity of the reaction decreases in the same direction [30,32]. Other water-soluble alkanethiols behave similarly [33]. The observed order of reactivity, which is contrary to the BDE of the radical’s parents, has been discussed in terms of increasing polarization in the transition states [31,33].
HOCH2CH2SH + R• → HOCH2CH2S• + RH
Free radical-based chemistry in water has been thoroughly studied in radiation chemistry for several decades. Kinetic data for many radical reactions in water as the medium are obtained by pulse radiolysis [30].
3. Synthetic Approaches Using Radical Intermediates in Water and Aqueous Media
Radical reactions have become popular among organic chemists with the introduction of reducing agents, like Bu3SnH or (TMS)3SiH, and led to a rapid growth in the number of organic transformations in organic solvents [34,35,36]. Figure 3a shows a generic radical chain processes for the reduction of a functional group X by a reducing agent MH, where M• radicals are generated by some initiation processes. The reduction chain propagated from the removal of the X atom in the organic substrate (RX) by the M• radical. The radical R• then reacts with MH giving the reduced product, RH, and a “fresh” M• radical to propagate the chain. The chain is terminated by radical−radical combination or disproportionation reactions. The radical R• may undergo further transformation, like intra- or intermolecularly addition to an unsaturated moiety, giving a new radical, which is finally reduced by MH.
In earlier work, ad hoc synthesized water-soluble group 14 hydrides have been used for the reduction of halides [37,38], whereas Barton and coworkers introduced hypophosphorous acid (H3PO2) and its salts, or dialkyl phosphites, as radical-based reducing agent for organic halides and for the deoxygenation of alcohols through thionocarbonates or xanthates [39,40]. Often hydrophobic substrates limit the scope of these reactions and therefore, reactions employ a mixture of water and organic solvents or water and a phase-transfer agent [41]. The details of this research area go beyond this article. Several reviews appeared in the last two decades and the reader is addressed to them [16,18,42]. Two reactions are reported in Figure 4 as examples connected to the mechanism mentioned above. The debromination of nucleoside 1 using H3PO2 in aqueous acetonitrile afford the reduction product in 97% yield; since HBr is produced, triethylamine (Et3N) was used as a base to prevent the decomposition of the acid-labile substrate and product [43]. Using diethylphosphine oxide as reducing reagent, the iodobenzene derivative 2, in the presence of the water-soluble azo compound V-501 as radical initiator, afforded the indole derivative in 98% yield, although a large amount of Et2P(O)H and V-501, and long reaction time are needed [44].
A protocol for the Minisci-type C-H alkylation of heteroarenes using the widely available and structurally diverse pool of inactivated alkyl bromides mediated by photoredox catalysis has been reported [45]. The example of quinazolinone and 4-bromopiperidine is shown in Figure 5a. It is proposed that the method utilizes the inexpensive oxidant K2S2O8 as photocatalyst for the photoredox cycle and rearomatizes the N-heterocycle after the Minisci addition (inset of Figure 5a), whereas the R• is generated by the reaction of (TMS)3Si• with the alkyl bromide. The presence of water as co-solvent improved considerably the yields by facilitating the dissolution of persulfate and purification of final products.
A synthetic strategy for the fluorination of alkyl bromides using N-fluorobenzenesulfonimide (NFSI) has been disclosed [46]. Figure 5b shows the optimized conditions and few examples obtained in good yields by a radical chain propagation. The initiation step is the photoexcitation of benzophenone (BP) in its triplet state (3BP), which reacts with (TMS)3SiOH to generate the resulting silyl radical, via oxidation and deprotonation [47]. Subsequent bromine atom abstraction from RBr would then yield the corresponding alkyl radical. In its turn, fluorine-atom transfer from NFSI leads to the corresponding nitrogen-centered radical which is polarity matched to abstract a hydrogen atom from the hydridic silane (TMS)3SiOH, thereby propagating this chain mechanism via the silyl radical (TMS)2Si(•)OTMS generated by 1,2-SiMe3 shift from silicon to oxygen [46].
Halogen atom-transfer cyclization and addition have also been extensively studied in radical-based organic synthesis [48,49]. Triethylborane (Et3B) in the presence of a trace of O2 is an efficient radical initiator [50] and has been widely used for synthetic radical reactions. Et3B is also stable in water and aqueous media. Et3B-induced halogen atom-transfer is used in a variety of radical reactions. Two examples are shown in Figure 6a where the radical cyclization of the iodo derivative 3 and the intermolecular bromine atom-transfer addition reaction of bromo ester 4 to an alkene proceed smoothly in good yields [16,51,52]. In the case of the iodo derivative 3, by replacing H2O with MeOH/H2O (v/v, 3:1) as the solvent, a yield of 75% (88/12) was obtained. The reaction mechanism is reported in Figure 3b. Using similar approaches, imine [53] and alkenylsilane [54] derivatives are alkyl radical acceptors for the construction of new carbon–carbon bonds. In particular, a tandem intermolecular radical addition–oxidation sequence can convert vinylsilanes into ketones, via silyl hydroperoxides, in good yields (Figure 6b) [54].
The incorporation of CF3-groups into organic molecules has a profound effect on their lipophilicity, permeability, and metabolic stability, which are key elements in pharmaceuticals and agrochemicals [55]. Useful methods for the trifluoromethylation of a variety or organic compounds using (bpy)CuIII(CF3)3 (bpy = 2,2′-bipyridine) [56] in aqueous acetone have been recently reported and the yields varied from moderate to very good [57]. In Figure 7 are reported three classes of reactions, i.e., the trifluoromethylation of alkyl bromides [58], of alcohol via the O-alkyl thiocarbonates [59] and of benzylic C−H bonds [60], with the yields of some selected examples. Three different mechanistic schemes are proposed, although the key step involving trifluoromethyl group transfer from CuII(CF3)2 intermediates to alkyl radicals is in common. In the case of Figure 7a it is suggested that the combination of Et3SiH/K2S2O8/light generate alkyl radicals through the reaction of Et3Si• with RBr [58]. In the case of Figure 7b the combination of (TMS)3SiH/Na2S2O8/light generate alkyl radical through the reaction of (TMS)3Si• with thiocarbonate and in parallel the photolysis of (bpy)CuIII(CF3)3 produce CF3• and CuII(CF3)2 species; while CF3• is generating (TMS)3Si• by a second path, the CuII(CF3)2 species react with the carbon-centered radical derived from the thiocarbonate derivative to give the desired product [61]. In the case of Figure 7c, trifluoroacetic acid (TFA) was used as an additive to the combination of i-Pr3SiH/(NH4)2S2O8/light; it was suggested that UV light serves to generate SO4•–, that abstracts H-atom from the benzylic C−H bond, and for the homolysis of (bpy)CuIII(CF3)3 to form CF3• and the active CuII(CF3)2 species, whereas i-Pr3SiH serves for quenching of CF3• and perhaps the resulting silyl radical could form the benzylic radical [60]. In the above-described reactions, the medium acetone/H2O varied from 2/1 to 8/1 to 1/1, respectively, and the beneficial effect of water has been attributed to the improved solubility of K2S2O8 [58] or to the CuII–complex stabilization [60]. C(sp2)–H trifluoromethylation of aldehydes through acyl radicals under conditions similar to the ones for alkyl radicals (cf. Figure 7a) was also reported [61].
An analogous protocol has been reported for the decarboxylative trifluoromethylation of aliphatic carboxylic acids with (bpy)CuIII(CF3)3 using AgNO3 as the catalyst for the formation of alkyl radicals, and the combination ZnMe2/K2S2O8 in aqueous acetonitrile at 40 °C for 10 h [62].
4. Radical Reactions Occurring “on Water” and in Heterogeneous Phase
Reactions “on water” involve water-insoluble organic compounds and take place in aqueous suspensions [9,10,11,12,13]. They have received considerable attention because of their high efficiency and application to straightforward synthetic protocols. In this section, we will give some examples of free radical chemistry useful to organic synthesis that proceeds in a heterogeneous phase. In particular, we will consider: (i) “on water” reactions with lack of solubility of the reactants, and (ii) “in and on water” reactions with some reactants soluble in water and some others suspended, where an amphiphilic co-reactant is employed in order to transfer the radical reactivity.
Trifluoromethylation based on radical intermediates in water or aqueous medium has been developed by different groups, functions broadly on a variety of substrates and demonstrates high functional group tolerance [63,64,65,66]. These reports are based on the discovery of a general procedure that combines CF3SO2Na (known as Langlois reagent), tert-butyl hydroperoxide (TBHP) and catalytic amounts of metal catalyst generating CF3• radical, as shown Figure 8a [53]. Optimization of the method shows that catalysts like FeSO4 or CuSO4 in CH2Cl2/H2O (2.5:1) work well, but also the trace metals found in the reagents were enough. Similar results are observed for the system with water alone as solvent. Subsequent addition of the in situ-formed CF3• to heterocycles or electron-rich arenes affords products of trifluoromethylation. Two examples of trifluoromethylation of uracil and xanthine are shown in Figure 8a, obtained in CH2Cl2/H2O (2.5:1) and water respectively, where the yields refer to 1 g scale [53].
Trifluoromethylation of a variety of aryl and heteroaryl boronic acids 5 is shown in Figure 8b, using CuCl as catalyst and CF3SO2Na and TBHP in aqueous medium at room temperature [64]. These reagents are suggested to react in situ to generate CF3• as the active trifluoromethylating reagent. In the 18 examples the corresponding products are obtained in moderate to high yields.
A CuSO4•5H2O-catalyzed decarboxylative trifluoromethylation of various α,β-unsaturated carboxylic acids 6 by using CF3SO2Na and TBHP was developed in aqueous medium at 50 °C (Figure 8c) [65]. In particular, the decarboxylative coupling reactions of various cinnamic acids start with the addition of CF3• radical at the α-position of the double bond, then proceeds via an elimination of carbon dioxide and Cu(I) to generate the product. In 24 examples the corresponding products are obtained in moderate to high yields and an E/Z ratio up to 99:1. The same authors also show a similar radical process for iron-catalyzed difluoromethylation of arylsubstituted acrylic acids by using (CF2HSO2)2Zn [65].
A Cu-catalyzed sequential difunctionalization/trifluoromethylation of N-arylacrylamides 7 that leads to oxindole derivatives has been reported “on water” at room temperature (Figure 8d) [66]. Optimized conditions were found for Cu(NO3)2•2.5H2O and TMEDA used in a 1:1 ratio (TMEDA= Tetramethylethylenediammine) as catalyst. Yields of 18 examples vary from moderate to high, depending on the substituents R1, R2 and R3, the quantities of CF3SO2Na and TBHP, and the reaction time [66]. This protocol for the introduction of the CF3-group exhibits several noteworthy features, such as inexpensive and readily available catalyst and trifluoromethylating reagent, and in some cases, reaction conditions that rely on water and ambient temperatures, in air.
Hydrophobic aliphatic and aromatic aldehydes undergo facile oxidation upon simply stirring their aqueous emulsions in air to give the corresponding carboxylic acids in high yields. Two examples are given in Figure 9, where the insoluble in water products can be extracted with CH2Cl2 and isolated after solvent evaporation [67]. The mechanism involves the formation of acyl peroxide via a radical chain (addition of acyl radical to molecular oxygen followed by hydrogen abstraction from aldehyde) and subsequent reaction with aldehyde to afford the carboxylic acid. The authors further explored the possibility of using an aldehyde as the source of both carbonyl and ester functions in the Passerini reaction. Figure 9 shows an example of hydrophobic cyclohexyl aldehyde and pentyl isocyanide in a 3:1 ratio, for 4 h, at 40 °C [67].
A mild, metal-free photochemical method for the hydroacylation of olefins was also developed [27,68]. This protocol was employed for a variety of aliphatic and aromatic aldehydes and olefins. Figure 10 shows the optimized conditions and few examples obtained in moderate to good yields, by a radical chain propagation. The initiation step is the photoexcitation of phenylglyoxylic acid that generates PhC(O)• and exchanges aldehydic H with RC(O)H [27]. The propagation step is straightforward involving addition of acyl radical to alkene and the adduct alkyl radical abstracts the aldehydic hydrogen to complete the chain reaction [69].
Tris(trimethylsilyl)silane, (TMS)3SiH, is a well-known reducing agent in organic chemistry based on free radical reactivity in organic solvents [36,70,71,72]. It was noticed that (TMS)3SiH is insoluble and does not decomposed in water, even refluxing for hours, at 100 °C [73,74]. Two methods are reported for the use of (TMS)3SiH as reducing agent in an aqueous environment depending on the hydrophilic or hydrophobic character of substrates (Figure 11a). For water-soluble substrates the amphiphilic β-mercaptoethanol was necessary as co-reactant. As the initiator, the water-insoluble 1,1′-azobis(cyclohexane-carbonitrile) (ACCN; half-life of 2.33 h at 100 °C) was found to give the best performance with both hydrophobic and hydrophilic substrates [73,74].
The reduction of organohalides and different thiocarbonyl alcohol derivatives (Barton-McCombie reaction) was obtained successfully. It has been suggested that all water-insoluble materials (substrate, reagents and initiator) suspended in the aqueous medium can interact due to the vigorous stirring, that creates an efficient vortex and dispersion. The reduction chain proceeds in the usual way: (i) removal of the X atom or group from the organic substrate (RX) by the (TMS)3Si• radical, and (ii) the radical R• reacts with the silane giving the reduced product, RH, and a “fresh” (TMS)3Si• radicals to propagate the chain. In the hydrosilylation of water-insoluble alkenes, alkynes, and aldehydes suspended together with (TMS)3SiH and the radical initiator ACCN, in aqueous medium at 100 °C under vigorous stirring, these substrates can be transformed into (TMS)3Si-containing compounds in good yields [75].
Figure 12 shows few examples of high yield reductions under the conditions of (TMS)3SiH/HOCH2CH2SH couple at reflux, with catalytic ACCN initiation. In particular, the reduction of water-soluble iodo-containing compounds 8, the role of the amphiphilic thiol is to act as the hydrogen donor and then to be regenerated by reaction the resulting thiyl radical with silane [74]. Water soluble alkyl and aryl azides can be reduced to the corresponding primary amines by the couple (TMS)3SiH/HOCH2CH2SH in aqueous media in the presence of a lipophilic initiator. The nucleoside derivative 9 is an example. The mechanistic steps for this transformation involve the addition of silyl radical to the azide function, liberation of nitrogen, and formation of a silyl-substituted aminyl radical, which abstracts hydrogen from the thiol. The hydrolysis of the silylamine rapidly occurred in water and afforded the amine as final product [74].
The azido reactivity is used for the synthesis of bicyclo [1.1.1]-pentan-1-amine (11), a unique and important fragment in medicinal chemistry. By the above mentioned procedures this scaffold was synthesized in one-pot, starting from the easily available intermediate, 1-azido-3-iodobicyclo[1.1.1]pentane (10) that contains both the iodo and azide functions [76]. (TMS)3SiH (2 equiv) is needed for fully reducing both moieties. With optimized conditions in hands, the authors then explored the practicality of the protocol on multigram scales, and consistent outcomes were obtained in all cases (78-82%) [76].
(TMS)3SiH in water was also successfully employed for the construction of C-C bonds in the synthesis of perfluoroalkylated aliphatic compounds [77]. In particular, the coupling of polyfluoroalkyl radicals to olefins was found to be mediated by (TMS)3Si• radical in water (Figure 13a). Initiation of the process was carried out using oxygen or ACCN as the initiator, forming the silyl radical ready to abstract the iodine atom from RfI precursors (12), thus generating the corresponding perfluoroalkyl radicals. Fast addition of the latter to 1-hexene, followed by fast reduction of the resulting C-centered radical with (TMS)3SiH led to the desired perfluoroalkylated products 13 in good yield. The fast addition of electrophilic radical Rf• to olefin, as compared to its reduction by TTMSS, is key to the success of this process.
Figure 13b shows the formation of a nitrogen-containing heterocycle in good yield through a radical cascade reaction [78]. The reaction proceeds through the Fe(II)-catalyzed decomposition of t-BuOOH and formation of a t-BuO• radical, which then abstracts a hydrogen from (TMS)3SiH. The vinyl radical is generated through addition of (TMS)3Si• onto an alkynone 14, followed by a 5-exo-trig cyclization at the ipso- position, leading to a cyclohexadienyl radical, finally trapped by the Fe(III)−OH species, affording the final product with the regeneration of the Fe(II) catalyst.
The (TMS)3Si• radical can be generated by direct oxidation of the corresponding silane ([Eox (TMS)3SiH/(TMS)3SiH•+ = +0.73 V vs SCE in MeCN) followed by deprotonation [79]. A visible light-promoted hydrosilylation of alkynes by (TMS)3SiH has been achieved by using catalytic amounts of Eosin Y (1 mol %) as a photocatalyst, the thiol i-Pr3SiSH as a radical quencher, and potassium carbonate as a base additive in dioxane/H2O (100/1 v/v) [79]. The corresponding alkenylsilanes were provided with high regio- and stereo- selectivites in the reactions of various terminal and internal alkynes. The role of H2O is to facilitate the regeneration of thiol by protonation of thiolate derived from the reaction of thiyl radical with reduced Eosin Y. The iodo derivative 15 has been successfully functionalized via a (TMS)3SiH mediated carbon-carbon bond formation [80]. An example of the optimization procedure is reported in Figure 14a, using the iridium catalyst Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 as radical initiator. The reaction is thought to proceed via a common radical chain reaction. Interestingly, using MeOH as the solvent, the yield is quite low, but the presence of 10% H2O afforded the highest yield.
Figure 14b shows the hydrophobic substrate 16 in water with combination of water-soluble radical initiator 2,2’-azobis[2-(2-imidazolin-2-yl)propane] (VA-061), and surfactant cetyltrimethylammonium bromide (CTAB). For comparison, three protocols are reported using the hydrophobic (TMS)3SiH, the water-soluble 1-ethylpiperidine hypophosphite (EPHP) and water-soluble H3PO2/NaHCO3, as chain carrier. With (TMS)3SiH the target product was obtained in good yield within a short period of time [41].
The radical addition/reduction reaction of alkyl iodides (R’I) to the C=N bond of hydrazones has been developed “on-water” (Figure 14c) [81]. The reaction was optimized for R = Ph and R′ = i-Pr and was faster “on water” than in organic solvents, reaching 92% yield. Good yields were obtained for a variety of substituents on both reagents. The developed protocol can be applied to the synthesis of 3-substituted isoindolinone derivatives. Extension of this protocol, to radical addition to the C=N bond in hydrazone derivatives of α-ketoesters has also been described [82].
It is also worth mentioning the achievement of a radical polymerization reaction involving Lewis pair catalysts “on-water”, at room temperature [83]. Various (meth)-acrylates, diethyl acrylamide, and 4-vinylpyridine, initiated by Lewis pair catalysts, specifically PPh3/Cu(OTf)2 or PPh3/Sn(OTf) 2, are prepared with significantly higher efficiency than the same reactions in organic solvents or in bulk.
5. Reaction of Carbon-Centered Radicals with Water-Activated by Lewis Acids
The most accurate value for the bond dissociation energy (BDE) of the H—OH bond homolytic cleavage has been estimated as 117.59 kcal/mol [84]. It has been estimated that even the highly reactive phenyl radical reaction with water is endothermic [85]. These facts preclude the reaction of water with carbon free radicals under standard free radical reaction conditions and has allowed for all the chemistry in and on-water that was described in the previous sections. Starting around 2005, there have been increasing reports of water (or deuterium oxide) activation through coordination with various Lewis Acids that decrease the H–OH BDE of water and make the participation of water in radical chain reactions possible.
The subject of coordination-induced bond weakening of X-H bonds (where X is mainly O, N or C) is one of increasing importance in recent years [86]. Below we describe each system with emphasis in these systems that have been well studied in terms of mechanism and applications in synthesis. We are focusing on the application of H–OH homolytic bond weakening methods and their application in synthesis.
Although proton-coupled electron transfer (PCET) reactions are only formal hydrogen atom transfer (HAT) reactions and do not involve a homolytic H–OH bond cleavage, reactions of this type are also included, as they constitute formal HAT processes. In a recent review of PCET reagent thermochemistry by R.G. Agarwal et al. [87], there is an interesting and enlightening discussion of the concept of coordination-induced bond weakening and how it correlates with the reducing ability of the coordinating metal, referring mostly to titanium (III) and the work of Cuerva [88] and samarium (II) and the works of J. M. Mayer [89], and R. A. Flowers [90], that will be described below. But first, the chemistry of boron and the works from the groups of J. L. Wood [91] and P. Renaud [92] will be described, that opened the studies in this field and eventually in the field of thiol assisted radical reductions [93] and this section will be completed with the most recent reports on germanium [94], molybdenum [95] and bismuth [96], which have not been applied yet in synthesis, as well as a very recent report on phosphorus [97].
5.1. Boron derivatives
The first system which claimed H-OH bond weakening through coordination, was initially described in 2005 by Wood and coworkers [91]. It involved a variant of the Barton-McCombie deoxygenation of xanthates in the presence of trialkylboranes, utilizing water or deuterium oxide as the hydrogen or deuterium atom source, respectively. Τreatment of a thioxanthate 17 with trimethylborane and an excess of D2O, under free radical producing conditions, generated the deoxygenated product in high yield (90%) and high deuterium incorporation (94%) (Figure 15). The authors [91] proposed that a free radical mechanism was operating and that D (or H) atom abstraction from D2O (or H2O) was assisted by complexation with trimethylborane, a proposal corroborated by ab initio calculations that estimated an 86 kcal/mol BDE for the H–OH bond when oxygen was bound to boron. The reaction was also shown to operate in the presence of other alkyl boranes and other thioxanthate substrates [91] and later was expanded to the reduction of alkyl iodides with H2O (but not D2O) [98].
Around the same time, P. Renaud and coworkers [99] made a similar observation in the radical reduction of β-alkylcatecholboranes to alkanes proposing a MeO–H bond weakening when coordinated to a catecholborate intermediate. The mechanistic scheme proposed (Figure 16) was corroborated by deuteration experiments. Also, J. Boivin and coworkers reported, in a series of papers [100,101,102], that reduction of S-alkylthionocarbonates, including 2-oxoalkyl derivatives, could be effected under radical conditions in the presence of triethylborane and air. It was combined with mechanistic studies that suggested that the origin of the hydrogen atom could be an OH group complexed with boron as suggested by Wood, the solvent or triethylborane, since the reduction gave good yields even under anhydrous conditions. J. Jin and M. Newcomb [103] came to a similar conclusion and in addition they measured the apparent rate of the reaction with either water or methanol, by using a radical clock and determined that the reaction is entropically disfavored and its rate is enhanced at low temperatures. The low entropy of the reaction was later corroborated and measured (ΔS° ~ -24 kcal/mol) by P. Renaud and coworkers [104] who also corrected the rate constants when they determined that (a) the complex Et3B·MeOH is formed in very small quantities (1-2%) and (b) increasing complexation with excess methanol (or water) to species such as Et3B·MeO-H…O(H)Me leads to a diminished reduced hydrogen donor character that explains all the radical chemistry that has been observed in or on water in the presence of Et3B.
Dennis P. Curran and T. R. McFadden, in 2016 [50], helped to explain initiation with Et3B and O2, by defining two regimes, one of low and one of high-oxygen concentration. They explained that the radical reduction of alkyl iodides with Et3B·OH2 [98] and the chemistry described above by P. Renaud and J. Boivin worked through a common-features transition state (Figure 17a) primed for ejection of an alkyl radical in a rather inefficient radical chain, operating under the high-oxygen regime. They also predicted that reductions with Et3B·RO-H, (R = H or Me), where a Lewis acid/Lewis base interaction between oxygen and boron is important, would not be able to be optimized due to three other destructive radical chains interlocked with the reduction deiodination chain (Figure 17b), unless the radicals Bu• and R• exhibited different selectivities.
In 2018, thirteen years after the initial report by J. L. Wood [91], P. Renaud and coworkers [92], revisited the reaction of xanthates and alkyl iodides with Et3B·OH2, or Et3B·OD2. They corroborated Curran’s proposal that hydrogen (or deuterium) atom abstraction from H2O (or D2O) was only a minor and inefficient radical chain path [50]. They discovered that the main reduction involved, in the case of xanthates, in situ adventitious generation of thiol from partial hydrolysis of xanthates. The thiol acted as a catalyst for the majority of the observed reaction products, according to Figure 18. In this mechanistic scheme the role of D2O was to simply generate a deuterated thiol through a proton-deuterium exchange that would provide the deuterium atom required in an otherwise typical radical reduction mechanism. Alkyl iodides could also be efficiently deuterated in the presence of thiols and D2O [92]. In the light of P. Renaud’s work, every mechanistic proposal of radical deuteration with D2O in an environment that there is a possibility of a generation of a thiyl radical or related intermediate, should be seen with skepticism, when the proposed mechanism does not involve H-D exchange between D2O and a thiol.
Although the assistance of thiols in radical reactions was well known, the in situ deuteration of thiols with D2O provides an easy and effective way to incorporate deuterium in stable C-D bonds, a process that finds applications in the deuterium enrichment for the metabolic stabilization of drugs [105]. There are many applications that have been reviewed recently [106], but we can mention some notable recent examples that of course go beyond the use of Et3B as initiator. In recent work from the group of A. Studer [93], they applied the RSH/D2O protocol to an elegant remote site-selective radical C(sp3)-H monodeuteration of amides. In Figure 19 the proposed mechanism and scope of the reaction are described.
Q. Liu, Y. Li and coworkers [107] developed a reversible, xanthate (EtOCS2K) mediated, H-D exchange reaction of arene-conjugated (E)-alkenes using inexpensive D2O. The proposed mechanism involves thermal generation of trisulfur radical anion (S3•–), from xanthate decomposition, which after reversible addition-deuteration-elimination provides the deuterated products. Also in this case, a xanthate S-D bond generated by reaction with D2O is responsible for the radical deuteration step. Complex substrates could be deuterated, and some polyphenols were also ortho-deuterated under the same conditions, although not consistently, since in the examples presented some phenols do not get ortho-deuterated (Figure 20).
5.2. Titanium Complexes
Around the same time that the boron induced activation of water was first suggested, a similar observation was reported by J. M. Cuerva and coworkers [88] in the case of Ti(III). Specifically, it was proposed that aqua complexes of Cp2Ti(III)Cl (Nugent’s reagent) mediate the reductive opening of epoxides with a mechanism involving a HAT or DAT (deuterium atom transfer) from normal or heavy water to an intermediate free radical. A rate constant was later measured for the process [110], which was consistent with the role of water as the hydrogen atom donor and a kinetic isotope effect kH/kD of 3.35 was measured [88]. It was shown with UV-VIS spectrophotometry that aliphatic and aromatic alcohols and amines exhibit a similar X-H bond weakening upon complexation with titanocene dichloride [111]. This particular reactivity of Ti(III)-water complex was further studied with ESR and theoretical calculations, from which the active reductant forms of the titanocene-aqua complex were determined [112]. Theoretical calculations using DFT estimated a rather low BDE of 60 kcal/mol and a radical mechanism was proposed (Figure 21a-c). The proposed mechanism [88], involved a competition between a slower anionic process and a faster radical process as shown in Figure 22.
Synthetically, the Cp2Ti(III)Cl•D2O system has been utilized as a deuterating reductant in a series of transformations. In an interesting application [113], the system was applied to the reduction of propargyl halides which through a proposed radical mechanism were transformed to exocyclic allenes. Similarly, A. Rosales et al. [114], in a mechanistic study of the titanocene(III)/Mn-promoted reduction of aromatic ketones in aqueous media, revised the previously proposed mechanism and proposed the intermediacy of a titanaoxirane (Figure 23) which in the presence of water undergoes hydrolysis towards the observed alcohol product. The authors cautioned that the free-radical character conventionally assumed for these chemical processes should be reconsidered.
5.3. Samarium(II) Iodide (SmI2)
Low-valent samarium is an important single electron reductant in organic synthesis and samarium(II) iodide (SmI2) is the most common reagent used in reactions. A prominent category of additives used in reactions of samarium diiodide is small protic molecules. Water is of particular interest and has been used as a proton source in a wide range of reactions including functional group reductions as well as reductive coupling reactions. The rates of Sm-mediated-reductions were examined in the presence of increasing water concentrations. The O−H BDFE of water bound to Sm(II) could be estimated by comparison with the BDFE of the C−H bond formed upon an initial PCET reduction of an arene by SmI2- water. The reduction of trans-stilbene by SmI2-water allowed for the determination of O−H bond weakening of at least 73.9 kcal/mol upon coordination of water to SmI2 [116].
Although these reactions have been proposed to proceed through sequential electron transfer with proton transfer from SmI2-water, the results obtained for the reduction of arenes suggests that PCET may be operative in other challenging reductions by SmI2-water. To study this supposition, Flowers and co-workers found that the rates of reduction of a model aldehyde, ketone, and lactone were consistent with an initial PCET from the samarium(II)-water reagent proceeding through a highly ordered transition state shown in Figure 24 to form a ketyl radical intermediate [117].
Although SmI2 is often employed as a reductant for oxygen-containing functional groups such as carbonyls, samarium has recently been found to be significantly azaphilic. This was utilized by Mayer and co-workers to reduce a range of tertiary enamines, shown in Figure 25a [89]. Both sequential ET PT and PT ET mechanisms were considered; however, both pathways were ruled out through competition reaction experiments. This reduction was thus proposed to proceed through either a concerted PCET or formal HAT, both illustrated in Figure 25b [89].
The O-H homolytic bond weakening of alcohols and water were estimated experimentally from the reduction of anthracene and benzyl chloride [117] but a poorly defined speciation of SmI2 in THF/alcohol mixtures limits reliable thermodynamic analyses of these systems. Recently, E.A. Boyd and J. C. Peters [115] utilized a bulky and strongly chelating ligand to facilitate a clean electrochemical behavior. They revealed an exceptionally weak N-H and O-H BDFEs of 27.2 and <24.1 kcal/mol, respectively, cementing the view that Sm(II) coordination induces the most significant bond weakening reported to date.
5.4. Molybdenum Complexes
In 2016, the group of P. J. Chirik [95] reported on a number of molybdenum complexes with ammonia, hydrazine or water that promoted hydrogen gas evolution due to a coordination-induced X-H (where X is nitrogen or oxygen) bond weakening. Calculations performed for the O−H bond of (PhTpy)(PPh2Me)2Mo(OH2)+ (Figure 26) determined a bond dissociation free energy (BDFE) value of 33.7 kcal/mol. The [1-OH2]+ compound evolved hydrogen gas too rapidly to allow for characterization, leading to more stable and isolable [1-OH]+ compound (Figure 26) [95].
5.5. Germanium Corrole Complex
In 2015, X. Fu, S. Ye and coworkers [94] reported a germanium (III) corrole complex, [(TPFC)Ge-(TEMPO)] (TPFC = tris(pentafluorophenyl)corrole, TEMPO = 2,2,6,6-tetramethylpiperidine-1-oxyl), that was found to react with water and produce (TPFC)Ge-OH and TEMPOH. Similar results were observed with methanol and ammonia, as well as primary aliphatic and aromatic and secondary aliphatic amines. Although the reaction proceeded slowly under thermal conditions (5 h) it was accelerated by visible light photolysis (1.4 h). The reaction was studied theoretically, and a mechanism was proposed, corroborated by DFT calculations (B3LYP and BP86), that involved water insertion between germanium and TEMPO, followed by a PCET rather than a HAT process, with an ET from the non-innocent corrole ligand to the TEMPO N-O bond, coupled with a proton transfer to TEMPO, as shown in Figure 27 below [94]:
5.6. Organobismuth(II) Species
In a similar to the above study published in 2022, the group of J. Cornella reported on the radical activation of N–H and O–H bonds at bismuth [96]. They reported the synthesis and characterization of a radical equilibrium complex based on bismuth featuring an extremely weak Bi–O bond, which permitted the in situ generation of an organobismuth(II) species (Figure 28). As a result, radical activation of N–H and O–H bonds, including water, occurs in seconds at room temperature leading to a Bi(III) hydroxy complex. Theoretical calculations estimated a BDFEO-H(H2O) = 52.1 kcal/mol [96].
The recent work of A. Studer and coworkers [97] on an interesting photocatalytic system based on a phosphine-mediated water activation for radical hydrogenation, that will be described in the following section of photoredox processes, also falls within the area of coordination-induced bond weakening of O–H bonds, described in this section.
6. Photocatalysis in Water and Aqueous Media
The beginning of the era of photocatalytic reactions dates back to the 1900s, when Giacomo Ciamician clarified the influence of light in chemical reactions [118]. However, for nearly a century, photocatalysis received limited attention from researchers probably due to the perception that controlling radical reactions was challenging. It was only in 2008 that Macmillan [19], Yoon [119], and Stephenson [120] and their coworkers introduced the concept of photoredox catalysis. In this groundbreaking approach, transition-metal complexes were employed as photocatalysts, operating under mild reaction conditions, and demonstrating unprecedented utility across a range of chemical transformations. Since that pivotal moment, the field of photocatalysis has undergone substantial growth and diversification [121,122,123,124,125,126,127,128].
Recent applications of photoredox reactions using visible light have become increasingly popular not only because they operate under mild conditions but also because they eliminate the need for radical chemical initiators or the use of stoichiometric quantities of chemical reductants or oxidants in redox-neutral processes. The utilization of visible light as an irradiation source helps minimize waste and reduces overall reaction costs significantly. Photoredox catalysis also exhibits tolerance to various functional groups present in organic structures [124]. In a parallel vein, the combination of water or water-based mixtures with organic solvents aligns with the principles of green chemistry, receiving support from both organic chemists and the industry. In this context, harnessing the synergy between light and water presents exciting opportunities and expands the toolkit available to chemists for functionalization reactions driven by visible light photoredox catalysis. The appeal lies in the safety, affordability, and environmental friendliness of both water and visible light [10].
Most photocatalytic processes conducted in water are focused on eliminating contaminants, whether they are organic or organometallic in nature [129]. This is often achieved by harnessing the well-known redox properties of photocatalysts when they are in their excited states. These properties convert photocatalysts into highly oxidizing or reducing substances, creating reactive intermediates that facilitate the breakdown of contaminants. Importantly, this degradation occurs without the necessity of potent chemical oxidizers or reductants [130].
Nevertheless, the development of preparative organic photoredox catalytic methodologies in water or aqueous environments faces challenges primarily due to the limited solubility of many organic substrates and catalysts in water or aqueous solvents. Additionally, conducting photoreactions in heterogeneous settings also presents difficulties. Recent review articles have focused on visible-light-mediated organic transformations in water or aqueous environments, placing particular emphasis on the choice of photocatalyst [131], the nature of the reaction medium [132], and the types of synthetic transformations achieved.[18,19]
In this section, we will explore representative organic synthetic transformations towards the formation of C-C bonds, achieved through visible light photoredox catalysis in water or aqueous conditions while also outlining the contributions by some of us in the field. Representative examples of radical additions to carbon-carbon multiple bonds and homolytic aromatic substitution reactions, including arylations and cyclization processes, will be discussed. Instances where water exerts significant influence on the outcomes of reactions will be presented. Water can also act as a proton or oxygen donor and may be necessary in stoichiometric quantities as a co-reactant in some cases. In other cases, water is utilized to dissolve specific reactants or photocatalysts. It is also adopted due to protocol improvements or more eco-friendly methods when contrasted with performing the same reaction in organic solvents. The overall objective of these various roles of water, whether as a reaction medium or merely as a component in photocatalytic processes, is to reduce the environmental consequences of both synthetic and photocatalytic procedures.
6.1. Photocatalyzed Radical Additions to Carbon-Carbon Multiple Bonds in Aqueous Media
Radical additions to carbon-carbon multiple bonds are a very useful synthetic transformations specially for producing medium-molecular-weight scaffolds from smaller molecules [135]. In this subsection representative examples of radical alkylation [136], arylation [136], acylation [138] and fluoroalkylation [139], including atom transfer radical additions (ATRA) reactions [140] on carbon-carbon multiple bonds, will be presented.
1,2-Difunctionalization reactions of alkenes involving radicals are quite versatile, and various photocatalytic techniques have been utilized for this purpose [141]. Chen, Guo, and collaborator developed a novel methodology involving photocatalysis and the use of water as co-solvent to achieve hydroxytrifluoroethylation of styrenes [142]. In a standard procedure, the authors employed fac-Ir(ppy)3 as photoredox catalyst, N,N-diisopropylethylamine as a reductive sacrificial donor in acetonitrile : water (8:1) as reaction media and under oxygen atmosphere. The presence of water played a pivotal role in the outcome of the reaction, as the desired product failed to form without it. The authors explored various styrene derivatives, including those with electron-donating or halogen substituents, and found that the reaction proceeded smoothly in all cases, as shown in Figure 29a. The authors based on different mechanistic studies, proposed a reaction mechanism (Figure 29b), where initially the photocatalyst fac-IrIII(ppy)3 is promoted to its excited state and further reduced by N,N-diisopropylethylamine generating fac-IrIII(ppy)2(ppy)•-. A CF3CH2I molecule is reduced by fac-IrIII(ppy)2(ppy)•- regenerating the photocatalyst and affording I– and a trifluoroethyl radical (CF3CH2•). The CF3CH2• radical is subsequently captured by the styrene derivative, generating benzyl-type radical. In the presence of oxygen and water, this radical reacts with molecular oxygen leading to the production of the hydroxytrifluoroethylation reaction product (Figure 29b). Water plays a crucial role in the reaction, as its absence leads to the formation of 2,2,2-trifluoroethanol as the product, with no observation of the difunctionalized product. Furthermore, experiment using 18O2 or H218O suggesting that the oxygen in the product comes from molecular oxygen present in the reaction media and not from water [142].
Ever since the Meerwein reaction was initially introduced, aryl radicals have demonstrated their versatility as intermediates in organic synthesis, especially in processes involving functional group interconversions and the formation of C–C bonds [134,143]. In 2014, König and colleagues reported a photocatalytic arylation of styrene derivatives using tetrafluoroborate aryldiazonium salts as substrates [144]. They employed [Ru(bpy)3]Cl2 as the photocatalyst and irradiated the reaction with a 440 nm light source in a reaction medium consisting of CH3CN and water. The versatility of this transformation with various aryl diazonium salts is outlined in Figure 30a. The proposed a mechanism based on experimental evidence and prior research findings, as depicted in Figure 30b. Initially, an aryl radical is generated through electron transfer from the excited state of the photocatalyst [Ru(bpy)3]2+* to the diazonium salt and generating [Ru(bpy)3]3+. The aryl radical adds to the olefin, forming radical intermediate I, which is subsequently oxidized by Ru(bpy)3]3+ to produce carbocation II and regenerating the photocatalyst. Finally, intermediate II reacts with the solvent (acetonitrile), leading to the formation of intermediate III, followed by hydrolysis to yield the amino-arylated product. The involvement of water in the reaction is essential as it provides the hydrogen and oxygen atoms necessary for the attack by water in the transformation of intermediate III to the final reaction product (Figure 320) [144].
Ketones are versatile building blocks in organic synthesis due to their inherent ability to participate in a wide array of bond-forming reactions as electrophilic compounds. As a result, the synthesis of ketone groups has been a point of extensive research for many years. Traditional approaches for generating ketones can be broadly categorized into the following four groups, depending on the active intermediate derived from the starting materials: (1) the oxidation of alcohols, (2) the acylation of carbon-centered nucleophiles, (3) the addition of acyl radicals to unsaturated moieties, and (4) the acylation of carbon-centered radicals [138]. Indeed, the past decades have been a flourishing period in the field of organic radical chemistry that delivered ground-breaking results, especially in organic synthesis. Consequently, a diverse array of radical acylation reagents and catalytic systems have been meticulously crafted and advanced, following the publication of a seminal review on radical acylation authored by S. Kim and colleagues in 2004 [145].
Zhang, Xie, and Zhu have presented a visible light-mediated photocatalytic process that involves the deoxygenation of aryl carboxylic acids facilitated by PPh3 for the generation of acyl radicals [146]. This transformation results in the deoxygenative coupling of aryl carboxylic acids with olefins, occurring in an aqueous environment to yield aromatic ketones. The authors determined the optimal reaction conditions using 0.2 mmol of aryl carboxylic acid, 1.5 equivalents of alkene, 1 mol% of the photocatalyst Ir[dF(CF3)ppy]2(dtbby)PF6, 20 mol% of K2HPO4 as base, and 1.2 equivalents of PPh3. They also explored the reaction′s applicability with various aryl carboxylic acids and alkenes, and the outcomes of this exploration are summarized in Figure 31a. After conducting mechanistic investigations, the authors propose a plausible reaction mechanism, as depicted in Figure 31b. Upon visible light excitation, the photocatalyst enters a triplet charge transfer state which oxidizes Ph3P to its radical cation while simultaneously reducing the photocatalyst. The resulting Ph3P·+ species combines with the aryl carboxylate ion, forming a P-centered radical. This process triggers the cleavage of the O-C=O bond, yielding an acyl radical and Ph3P=O. The acyl radical subsequently attacks the double bond of the olefin, resulting in the formation of a radical adduct. This adduct is further reduced by the lower oxidation state of the photocatalyst, generating a carbanion. Upon protonation from water, the carbanion ultimately produces the final reaction product (Figure 31b). Deuterium labeling experiments conducted in D2O revealed that water served as the proton source. When the aromatic acid was labeled with 18O, Ph3P=18O was formed, indicating that the oxygen atom in Ph3P=18O originated from the carboxylic acid and not from water [146].
Atom transfer radical addition (ATRA) reactions are pivotal processes in synthetic chemistry, allowing for the efficient dual functionalization of alkenes while maximizing the atom economy [140]. Stephenson and coworkers presented a seminal study on photocatalyzed ATRA reactions conducted in an aqueous environment [147]. This publication marks the first documented instance of intermolecular ATRA reactions involving haloalkanes and α-halocarbonyls with olefins, all facilitated by photoredox catalysis activated by visible light in aqueous media. The authors identified the optimal reaction conditions, utilizing Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst, 1 mmol of the olefin, 2 equivalents of the atom transfer agent, LiBr as an additive in DMF : H2O (1:4) as the reaction medium under blue LED irradiation in an argon atmosphere (Figure 32a). The versatility of this reaction was demonstrated with various olefins featuring diverse functional groups, including alcohols, benzyl esters, alkyl bromides, silyloxy esters, esters, enones, carbamates, and aromatic rings. Regarding the atom transfer agent, the reaction exhibited good compatibility with a range of haloalkanes and α-halocarbonyls, including CF3I (Figure 32a). Taking into consideration multiple mechanistic studies, the authors proposed the reaction mechanism depicted in Figure 32b. The reaction commences with photoexcitation of the Ir(III) photocatalyst, resulting in the formation of an excited species of Ir(III)*, which undergoes a single electron transfer process with the haloalkane (or α-halocarbonyl), leading to the generation of Ir(IV) (with a halide as a counterion) and the electrophilic alkyl radical. The alkyl radical subsequently adds to the olefin to form the radical adduct. The formation of the ATRA reaction product can occur through oxidation of the radical adduct by Ir(IV) associated with the halide counterion, resulting in the formation of a carbocation. This carbocation then rapidly reacts with the halide anion to yield the desired reaction product. It is important to note that external nucleophiles, including water (which serves as a major co-solvent), do not participate in reactions with the proposed carbocation intermediate, as evidenced by mechanistic experiments conducted by the authors [147].
The iodoperfluorohexylation of olefins and alkynes in water has been recently reported [148]. These represent the first examples of photocatalyzed ATRA reactions conducted entirely in water as the solvent. Optimized reaction conditions were achieved when employing alkene or alkynes (0.2 mmol) as substrates, perfluorohexyl iodide (3 equiv) as fluoroalkyl radical source, Rose Bengal (RB, 5 mol%) as photocatalyst, N,N,N′,N′-tetramethylethylenediamine (TMEDA, 3 equiv) as sacrificial electron donor, under green LEDs irradiation and Ar atmosphere. Both alkenes and alkynes rendered products derived from the ATRA pathway, and in the case of alkynes, exclusively as E-stereoisomers (Figure 33). This synthetic approach was also applied to the late-stage functionalization of the pharmacologically active alkyne drug (D)-(−)-Norgestrel acetate, in 50% using the aqueous media MeOH:H2O (1:2), a hormonal medication and contraceptive. Mechanistic investigations provided evidence for the involvement of a radical pathway. However, the authors were unable to conduct optical experiments to assess the operation of an oxidative or reductive photocatalytic quenching cycle [148]. This limitation stems from the poor solubility of C6F13-I in the reaction medium, preventing the measurement of reliable optical spectroscopy values for Stern–Volmer kinetic analysis or triplet quenching experiments.
Qing and collaborators have recently developed the first methodology for the hydrofluoromethylation of unactivated alkenes [149]. This innovative method utilizes fluoroiodomethane and hydrosilanes, merging photoredox catalysis with silane-mediated deiodination processes. Key aspects of the procedure involve the utilization of water as the solvent, ICH2F for CH2F radical generation, PhSiH3 as the H-donor, and (TMS)3SiH as an additive to achieve higher yields. The mild reaction conditions enable the tolerance of various functional groups such as phenol, ether, aldehyde, carboxylic acid, ester, sulfone, trifluoromethyl, and trifluoromethoxy among others (Figure 34). Initial mechanistic investigations suggest that employing water as the solvent facilitates the addition of CH2F radicals to unactivated alkenes, and the incorporation of (TMS)3SiH significantly enhances chemoselectivity.
Liu and colleagues[ reported on a direct and site-specific C(sp3)–F bond alkylation in polyfluorinated iminosulfides using alkenes and water through photoredox catalysis, yielding a diverse range of 3-fluoro-3-perfluoroalkyl-γ-lactams, accompanied by the simultaneous formation of a C(sp3)–C(sp3) bond, a C(sp3)–N bond, and a C=O bond (Figure 35) [150]. The study revealed that various substituted 2-vinylpyridines effectively participated in the reaction, producing the corresponding products in moderate to high yields and high to excellent diastereoselectivities. The conditions proved compatible with diverse functional groups such as methyl, bromo, chloro, ketone, ester, aldehyde, methoxyl, cyano, among others (Figure 35a). Notably, a range of perfluoroalkyl units (Rf), including C2F5, C3F7, C4F9, and C5F11, underwent site-specific defluorofunctionalization. This approach demonstrated precise chemoselectivity control and exhibited outstanding tolerance toward various functional groups. Considering several mechanistic studies, the authors proposed a plausible reaction mechanism outlined in Figure 35b [150]. Initially, the Ir(III) photocatalyst, upon irradiation, is promoted to its excited state, Ir(III)*. Subsequently, Ir(III)* reduces A through single electron transfer (SET), producing the Ir(IV) species and radical anion A•–. Following this, a spin-center shift process takes place, leading to the formation of the corresponding radical I with the cleavage of a C–F bond. I is captured by 2-vinylpyridine derivative to form the alkyl radical intermediate II. This intermediate is then intercepted by the C=N double bond of the imine group via a 5-endo-trig cyclization, resulting in the formation of the C-centered radical intermediate III. The photoredox cycle is subsequently closed by SET between III and Ir(IV), giving rise to the carbocationic intermediate IV which is then trapped by the hydroxyl anion, yielding intermediate V. Finally, V undergoes elimination, prompted by the base, resulting in the formation of a polyfluorinated γ-lactam B and thiophenol. It must be pointed out that by 18O-labeling experiments the authors confirmed that the oxygen atom in the amide group originates from water.
4.2. Photocatalyzed Homolytic Aromatic Substitutions in Aqueous Media
Homolytic aromatic substitution (HAS) is a practical synthetic approach used to exchange aromatic hydrogen atoms with appropriate substituents, facilitating the formation of new C-C or C-heteroatom bonds. Currently, due to the advances in the field or synthetic radical organic chemistry, HAS have become the preferred synthetic approach, competing with organometallic, transition-metal, and electrophilic aromatic substitution methodologies. In the last decade, visible-light photoredox catalysis has emerged as a pivotal approach for aromatic substitution. The main photoredox catalysts employed include polypyridine complexes of Ru(II) and Ir(III), as well as organic photoredox catalysts, providing a metal-free option for HAS [151].
Xue, Xiao, and collaborators have introduced a photocatalytic arylation methodology for electron-deficient heteroarenes in water as solvent [152]. Optimized reaction conditions were achieved using 4-methoxybenzenediazonium tetrafluoroborate (0.3 mmol) as aryl radical source, Ru(bpy)3Cl2 as photocatalyst (2.5 mol%), pyridine hydrochlorides (1.5 mmol) as radical acceptors, and water as the solvent. The reaction was carried out under irradiation with a 40 W fluorescent lamp under Ar atmosphere. As depicted in Figure 36a, pyridines featuring electron-withdrawing groups (such as CF3, CN, COOEt, Br) or electron-donating groups (such as CH3) resulted in reasonably good yields of arylated products. The authors subsequently examined the substrate scope by evaluating different aryl diazonium salts, functionalized with groups such as cyano, carboxyethyl, chlorine, bromine, and fluorine, as aryl radical precursor. In these studies, the authors utilized 4-trifluoromethylpyridine hydrochloride as the radical acceptor, successfully yielding the corresponding arylation products in satisfactory yields. Furthermore, the authors explored a one-pot approach by synthesizing the aryldiazonium salt and conducting the in situ arylation of the heteroaromatic compound in water. This approach also yielded reasonable yields of the coupling products [152]. Based on mechanistic experiments performed and established literature precedents, the authors proposed a mechanism as illustrated in Figure 36b. The process initiates with the excitation of the Ru(bpy)32+ photocatalyst (Scheme X11) upon exposure to visible light, resulting in the formation of [Ru(bpy)32+]*. A reductive electron transfer process between [Ru(bpy)32+]* and the aryldiazonium salt generates an aryl radical Ar•. This aryl radical engages a homolytic aromatic substitution process with a pyridinium chloride, leading to the formation of radical intermediate I. Subsequent oxidation by an additional aryldiazonium salt species generates the cationic intermediate II and aryl radicals, which then participate in a chain reaction cycle. Deprotonation of intermediate II ultimately yields the arylated pyridine product. Although this reaction appears straightforward and has been conducted in various organic solvents under different radical conditions, the authors′ enhanced methodology, employing water as the reaction medium and utilizing photocatalysis, offers distinct advantages. Notably, the use of water has improved the reactivity of pyridine nuclei as pyridinium salts in arylation reactions, resulting in advantages such as enhanced regioselectivity (substitution of pyridine rings at the 2-position), utilization of aryl precursors from soluble benzenediazonium salts, and improved arylation product yields [152].
Natarajan and colleagues successfully developed a protocol for synthesizing phenanthridine-6-carboxylates from N-biarylglycine esters in water as solvent [153]. This reaction proceeds via an intermolecular HAS ensuing a cyclization process. The authors determined the optimal reaction conditions using N-biarylglycine esters (0.3 mmol) as the substrate, Rose Bengal as the photocatalyst (5 mol%), and performing the reaction in water as solvent under blue LEDs irradiation in the presence of air. To explore the substrate scope for this transformation, the authors examined various N-biarylglycine methyl esters with different substituents on the aromatic rings (Figure 37a). As shown in figure, both electron-withdrawing and electron-donating substituents on the biaryl moiety resulted in high yields of substituted phenanthridines. The authors proposed a reaction mechanism for the synthesis of phenanthridine-6-carboxylates from N-biarylglycine esters, as illustrated in Figure 37b. Initially, the excited photocatalyst Rose Bengal (RB*) oxidizes the N-biarylglycine ester substrate to form radical cation I, concomitantly generating the radical anion of RB·-, which then reduces oxygen present in the reaction medium to superoxide anion regenerating the photocatalyst. The superoxide anion subsequently deprotonates intermediate radical cation I, yielding radical II and a hydroperoxyl radical. Through a HAS process, radical II produces cyclohexadienyl radical intermediate III. The hydroperoxyl radical previously formed abstracts a hydrogen atom from intermediate III, yielding dihydrophenanthridine IV. In a second catalytic cycle, the oxidation of IV occurs, ultimately yielding the phenanthridine reaction product. Independent experiments conducted with dihydrophenanthridine IV under irradiation with Rose Bengal as the photocatalyst, using the optimized reaction conditions, resulted in the quantitative yield of the phenanthridine product. This demonstrates the effective oxidation of dihydrophenanthridine IV within the photocatalytic cycle to yield the phenanthridine final reaction product (Figure 37) [153].
Figure 37.
(a) Photocatalyzed synthesis of phenanthridine-6-carboxylates from N-biarylglycine esters in water and representative examples; (b) Proposed reaction mechanism.
Figure 37.
(a) Photocatalyzed synthesis of phenanthridine-6-carboxylates from N-biarylglycine esters in water and representative examples; (b) Proposed reaction mechanism.
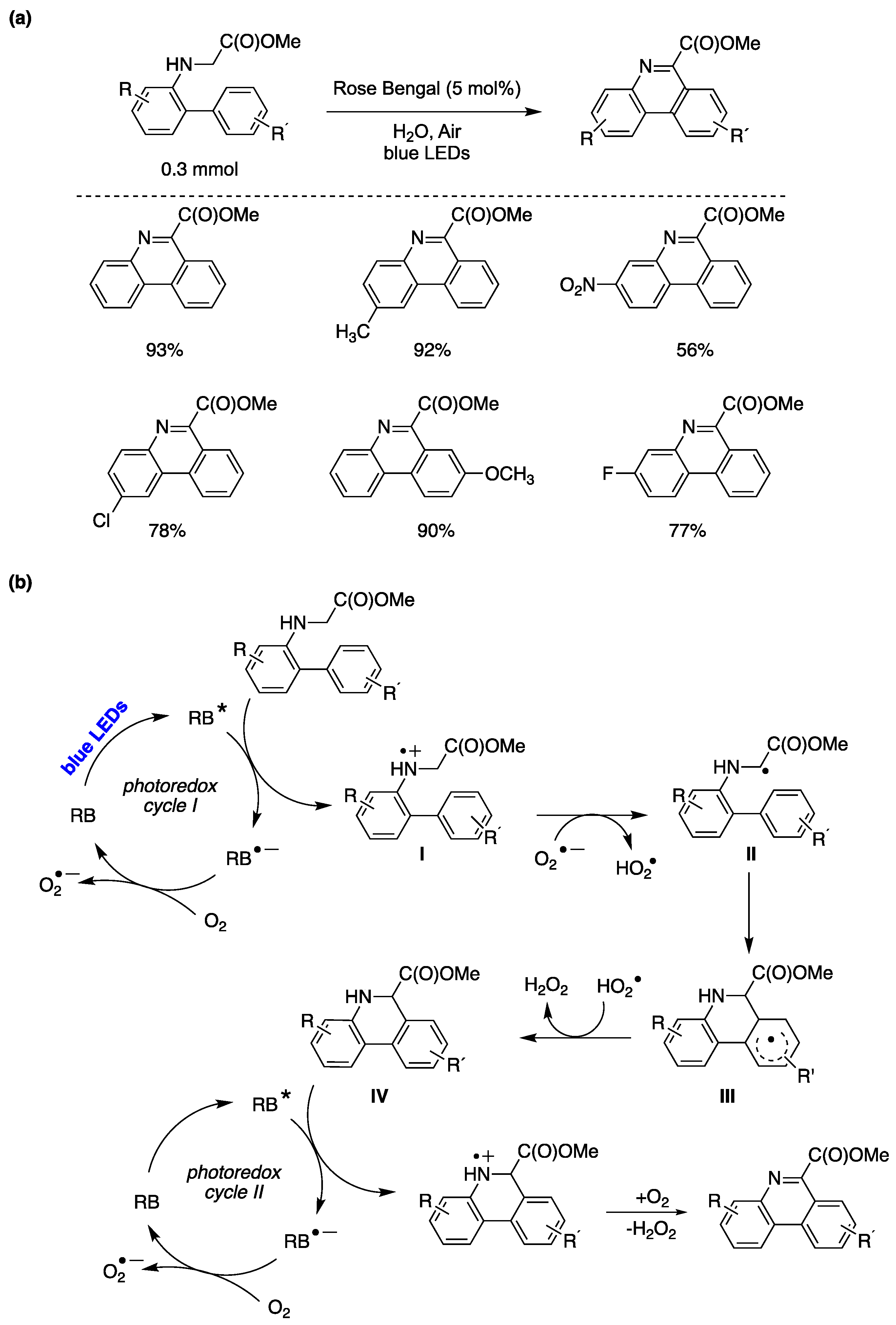
Figure 38.
(a) for the Photocatalyzed C-H fluoroalkylation of arenes in water promoted by Vitamin B12 and Rose Bengal, and representative examples; (a) water : acetonitrile (1:1); (b) Proposed reaction mechanism.
Figure 38.
(a) for the Photocatalyzed C-H fluoroalkylation of arenes in water promoted by Vitamin B12 and Rose Bengal, and representative examples; (a) water : acetonitrile (1:1); (b) Proposed reaction mechanism.
Innovative synthetic techniques for producing fluoroalkylated aromatic compounds are in great demand because of their distinctive characteristics, which enable their use in various fields such as medicinal chemistry, agrochemistry, and materials science. Within this context, radical fluoroalkylation reactions mediated by catalytic cycles driven by light have gained significant attention over the past decade [154]. The first perfluoroalkylation reaction of activated arenes in water has been recently reported [155]. Optimized reaction conditions were achieved when employing activated arenes (0.2 mmol) as substrates, perfluorohexyl bromide (3 equiv) as perfluoroalkyl radical source, TMEDA (3 equiv) as sacrificial electron donor, Rose Bengal (5 mol%) as catalyst, vitamin B12 (cyanocobalamin, 5 mol%) as co-catalyst, in water as solvent under green LEDs irradiation in an Ar atmosphere. The reaction scope was extended to different amino-substituted arenes and alkoxyarenes bearing electron donating or withdrawing groups, yielding the corresponding perfluoroalkyl substituted products in good to excellent yields (Figure 38a) [155]. The authors, based on the mechanistic investigations performed and information available in the literature, proposed a plausible reaction mechanism outlined in Figure 38b [155]. The sequence begins with vitamin B12 undergoing a one-electron reduction process facilitated by a Rose Bengal (RB) oxidative photocatalytic cycle. This reduction leads to the formation of cob(II)alamin I upon cyanide loss. The reaction proceeds with the further reduction of I via an additional RB oxidative photocatalytic cycle, resulting in the generation of cob(I)alamin II, which rapidly reacts with n-C6F13Br, producing the Co(III)-C6F13 complex III and a bromide anion. Upon exposure to light, the complex III releases an n-C6F13• radical and regenerates I, thereby completing the cobalt-mediated co-catalytic cycle (Figure 40b). The n-C6F13• radical formed reacts with the arene via a HAS mechanism affording the perfluorohexylated reaction product.
Modified crown ethers are essential building blocks in supramolecular chemistry, finding uses in phase transfer catalysis, metal extraction, intelligent materials, and molecular machines [156]. A successful protocol for the late-stage incorporation of fluoroalkyl moieties into (di)benzo crown ethers has been reported [157]. The photocatalyzed reactions were carried out in mixed aqueous-organic solvents, as the inclusion of water was found to be essential for maximizing the yield. Optimized reaction conditions were achieved employing aromatic crown ethers as substrates (0.2 mmol), fluoroalkyl iodides (3 equiv) as fluoroalkyl radical source, Eosin Y (EY, 5 mol%) as photocatalyst, TMEDA (3 equiv) as electron donor, alkali chloride (1.2 equiv) as additive, in MeCN:water (1:1) as solvent and under green LEDs irradiation (Figure 39a). The capacity of crown ethers to form complexes with metal ions played a crucial role in enhancing the solubility of the substrates within the reaction mixture. This, in turn, facilitated perfluoroalkyl group substitution with high yields, along with remarkable chemo- and regioselectivity. Different fluoroalkyl-substituted crown ethers were prepared and isolated in yields ranging from 60 to 99%, proving the wide scope of the protocol. The authors also showed that a (di)benzo crown ether within a complex rotaxane structure can efficiently undergo direct perfluoroalkylation with high chemo- and regioselectivity using the described method. This underscores the ability of the methodology for incorporating stereogenic and/or functional elements into mechanically interlocked structures with remarkable ease and efficiency [157]. Based on mechanistic investigations and information available literature, the authorshave put forth a plausible reaction mechanism illustrated in Figure 39b. Under the studied reaction conditions, EY, which contains two relatively acidic protons (pKa 2.0, 3.8, in water), readily undergoes deprotonation by TMEDA, leading to the quantitative formation of EY2–. In contrast to EY, EY2– exhibits strong absorption in the green region of the UV–vis spectra. In the context of photoredox catalysis, and upon green light irradiation, the triplet state is typically considered the most relevant excited state for EY2– due to its very brief singlet lifetime. Furthermore, the 3[EY2–]* state can serve as both a moderate oxidant and reductant. It was suggested that, under the investigated reaction conditions, EY2– functions as an oxidant by accepting an electron from TMEDA. This is supported by a favorable spontaneous electron transfer, indicated by a ΔGPET value of -0.36 eV. Subsequently, perfluoroalkyl radicals (RF•) could be generated by the reaction of EY•3– with the corresponding perfluoroalkyl iodides (RFI). The RF• radical formed then reacts with the (di)benzo crown ether metal cation complex through a homolytic aromatic substitution mechanism, resulting in the formation of the perfluorohexylated reaction product (Figure 39b) [157].
4.3. Miscellaneous
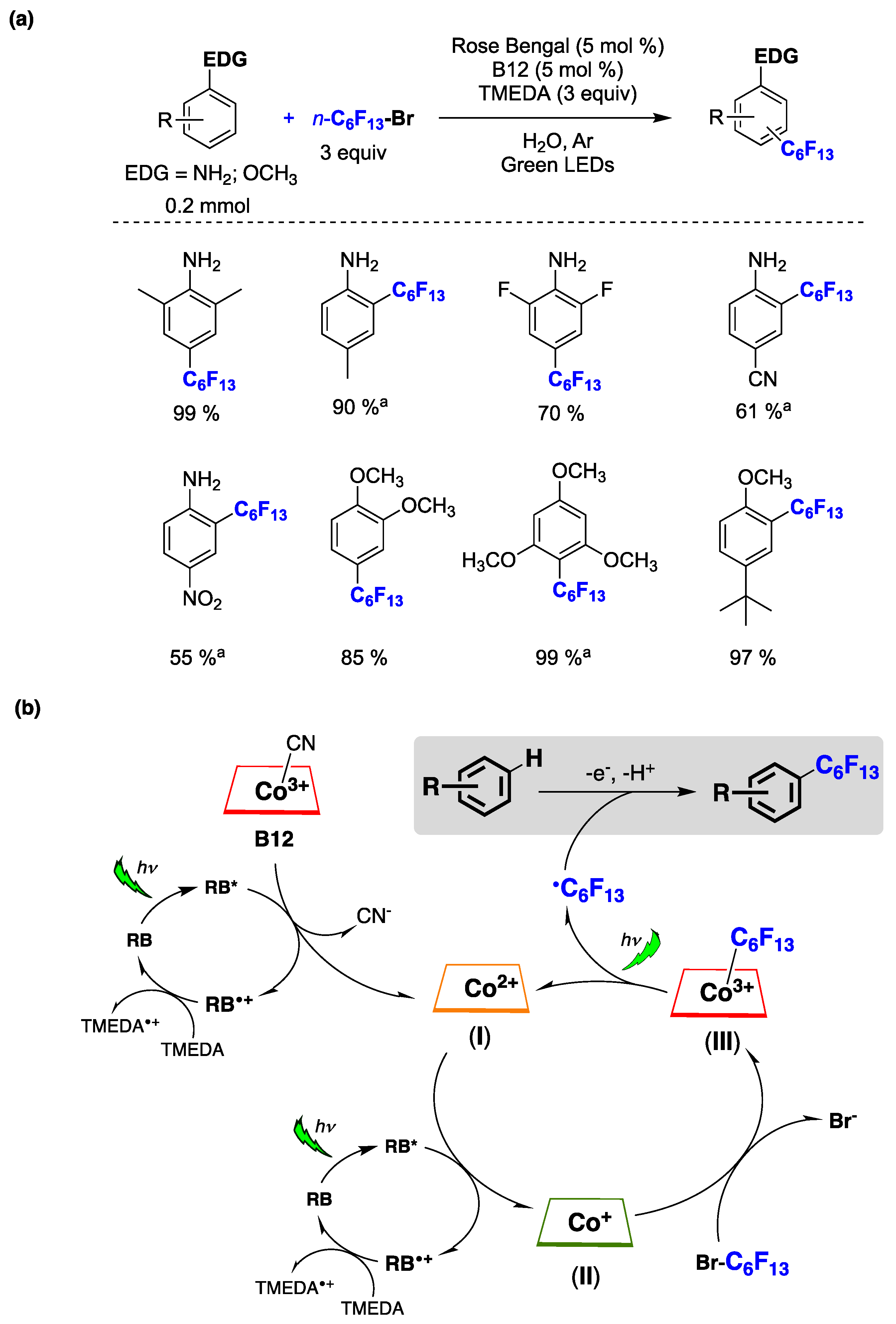
Guo, Liu, and collaborators [158] recently reported a method for the diastereoselective synthesis of trifluoromethylated cyclobutane derivatives under visible light irradiation. This approach involves a one-pot process that combines [2+2]-photocycloaddition with water-assisted hydrodebromination (Figure 40a). Various compounds, including quinolinones, isoquinolinones, and coumarins, can successfully participate in this one-pot process with 1-bromo-1-trifluoromethylethene. Furthermore, stereo-defined trisubstituted trifluoromethylated cyclobutane alcohols, carboxylic acids, and amines can be straightforwardly obtained by the ring opening of lactone or lactam, maintaining the original high diastereoselectivity facilitated by water−tristrimethylsilylsilane coordination. Thioxanthone (TX) functioned as both a photosensitizer and a hydrogen atom transfer (HAT) agen). The diastereoselectivity in the hydrogen atom abstraction process was regulated by (TMS)3SiH and assisted by water. This approach[44] served as a complementary method to achieve a novel mode of hydrodebromination under mild conditions. TX played a pivotal role in facilitating this one-pot transformation, acting as a suitable substitute for transition metal and radical initiators [158].
Figure 40.
(a) Diastereoselective synthesis of trifluoromethylated cyclobutane derivatives by [2+2]-Photocycloaddition followed by water-assisted hydrodebromination. (b) Photoredox cross-electrophile coupling of alkyl bromides with DNA- tagged aryl iodides in aqueous solution.
Figure 40.
(a) Diastereoselective synthesis of trifluoromethylated cyclobutane derivatives by [2+2]-Photocycloaddition followed by water-assisted hydrodebromination. (b) Photoredox cross-electrophile coupling of alkyl bromides with DNA- tagged aryl iodides in aqueous solution.
Kölmel, Ratnayake and Flanagan [159] developed a catalytic process that allows the photoredox cross-electrophile coupling of alkyl bromides with DNA-tagged aryl iodides in an aqueous environment (Figure 40b). The success of this metallaphotoredox transformation relies on the utilization of novel pyridyl bis(carboxamidine) ligand crucial to the nickel catalytic cycle (see right part of Figure 40b for chemical structures). The described C(sp2)-C(sp3) coupling exhibits broad tolerance for both DNA-tagged aryl iodides and alkyl bromides. Significantly, the reaction has been optimized for parallel synthesis, a key requirement for the efficient preparation of combinatorial libraries, utilizing a 96-well plate-compatible blue LED array as the light source. Consequently, this mild and DNA-compatible transformation is well-suited for the construction of DNA-encoded libraries.
Photoredox catalysis has also been applied for the in situ deuteration of thiols with D2O that provides an easy and effective way to incorporate deuterium in stable C-D bonds, a process that, as already mentioned in the previous section, allows the deuterium enrichment for the metabolic stabilization of drugs [105]. This subject has been recently reviewed [106], and here we mention a couple of recent examples. The scope of the radical H/D exchange of unactivated C(sp3)-H bonds as well as the multi-deuteration of C(sp3)-H bonds with D2O as cheap deuderium source was studied by utilizing a synergistic photoredox catalysis and organocatalysis system in detail, including a scale-up experiment [107]. Figure 41a shows the optimized example of a remote site-selective radical C(sp3)-H deuteration of PMP amides by utilizing a photoredox catalytic system made on iridium(III) complex with Bu4N+ (BuO)2P(O)– together with a RSSR/D2O system [108]. A visible-light mediated catalytic asymmetric radical deuteration at non-benzylic positions was also developed through a series of radical addition reactions of mainly N-heterocyclic carbene-borane complexes, and of silicon and phosphine radicals, to exocyclic double bonds coupled with an asymmetric deuteration of the resulting radical with a chiral thiol/D2O system [109]. Two examples are shown in Figure 41b, the first is the optimized conditions for the addition of N-heterocyclic carbene–borane complex onto exocyclic olefin and the second being a deuterosilylation with (TMS)3SiH. In both cases, cysteine-derived β-turn-containing peptidic (R*SH) as the deuterium atom transfer catalyst and readily available 4DPAIPN as the organophotocatalyst in a binary solvent mixture of toluene and D2O (3:1) were used (see right side of Figure 41b for the structures). The mechanism of the reaction was studied theoretically and a favored transition state is proposed for the origin of enantioselectivity, while the study of the scope of the reaction indicated greater than 95:5 enantiomeric ratios in many cases [109].
It is also worth mentioning the most recent report on phosphorous chemistry by the group of A. Studer [97], who described an interesting photocatalytic system based on a phosphine-mediated water activation for radical hydrogenation. Figure 42a shows the optimized conditions for this transformation and some selected examples of alkene hydrogenation. It is based on an iridium(II) photocatalyst that generates the radical Ar3P(•)–OH as intermediate, which can be considered a formal ‘free’ H atoms donor. Arylthiols are used as catalytic co-reductants for the radical hydrogenation of π systems. This co-catalysis approach ensures that both H atoms of water can be used as H atom donors in the reduction of π systems, as shown in Figure 42b. Indeed, control experiments revealed that H2O is the exclusive hydrogen source in the hydrogenation and the solvent (acetonitrile) is not involved. The preparation of few deuterated compounds using cheap D2O, was also reported (see Figure 42a for an example) [97].
7. Bioinspiration from Biological Reactivity
There are many examples in the literature of bioinspired reactions in organic synthesis and some of them are connected with free radical reactivity [34,160]. Knowledge of free radical reactivity taking place in biological processes can be very important for organic chemistry in two main directions: (i) the design of biomimetic models to simulate free radical damage and provide molecular libraries for mechanistic and biomarker discovery, and (ii) the inspiration for new synthetic methods based on the same mechanisms that nature uses to prepare biomolecules. In this section, we report two examples of such approaches of interest in our laboratory: the synthesis of purine 5’,8-cyclo-2’-deoxynucleosides via radical cyclization and the reduction of carbonyl moieties by disulfide radical anion.
3.1. Purine 5’,8-Cyclo-2’-Deoxynucleosides
Purine 5’,8-cyclo-2’-deoxynucleosides or 5’,8-cyclopurines (cPu) are important modified nucleosides which represent tandem type lesions observed among the DNA modifications and have been identified in mammalian cellular DNA in vivo [161,162,163,164]. Figure 43a shows the four cPu structures, i.e., 5’,8-cyclo-2’-deoxyadenosine (cdA) and 5’,8-cyclo-2’-deoxyguanosine (cdG) in their 5’R and 5’S diastereomeric forms, whereas Figure 43b shows the formation of the C5’ radical by H-atom abstraction from HO• radical and the subsequent steps of the mechanism of cPu formation.
Biomimetic experiments for the cPu formation and related mechanism were performed using γ-radiolysis as HO• radical-generating sources. γ-Radiolysis of neutral water leads to the following primary reactive species: solvated electrons (e–aq), hydroxyl radicals (HO•) and hydrogen atoms (H•). In N2O-saturated solution (~0.02 M of N2O), e–aq are efficiently transformed into HO• radicals and therefore HO• and H• account for 90% and 10%, respectively, of the reactive species. Under these conditions, it was found that both dG and dA afford ~10% yields of cdG or cdA in R/S product ratios of 8.3:1 and 6:1, respectively [165,166,167]. The reactivities of HO• with dG or dA are unselective and the obtained yields correspond to the H-atom abstraction from C5’–H which is ~10% [26]. Hydrated 5’-aldehydes were formed instead of the cPu in the presence of oxygen, indicating the trapping of C5’ radicals by oxygen prior to cyclization [168]. The rate constants of radical cyclization were determined by time-resolved spectroscopy to be 1.6 × 105 and 6.9 × 105 s–1 at room temperature for dA and dG, respectively (Figure 43b) [167,169].
Synthetic procedures for cPu nucleosides were established starting from the corresponding 8-bromopurine derivatives in aqueous solutions, with a radical cascade mechanism that mimics DNA damage (Figure 43c). Indeed, UV photolysis of 8-Br-2’-dG afforded cdG in 26% yield and a R/S ratio of 8:1 [165], whereas the gamma-radiolysis of 8-Br-2’-dA in the presence of K4Fe(CN)6 afforded of cdA in 67% yield (based on the starting material conversion) and a R/S ratio of 6:1 [167]. It is worth mentioning that the pro-5’R conformer is stabilized in water when the 3’OH and 5’OH groups are free, whereas the pro-5’S conformer prevails in aprotic solvents when the 3’OH and 5’OH groups are protected with a TBDMS group [170,171].
Analogous reactions have been employed: (i) for the isotopically synthesis of 15N5-labeled cPu derivatives necessary as reference material for quantification of oxidative DNA lesions in biological samples by liquid chromatography-tandem mass spectrometry [172,173,174]. Such quantification has been a subject of interest in our laboratory for the last 5 years; we provided evidence for cPu formation in cells [175,176,177,178], animals [179,180], and humans [181] as valuable biomarkers for free radical activity. (ii) for the synthesis of the phosphoramidite synthones of the cPu [182] for further incorporation to model DNA for biological studies [183,184,185,186].
3.2. Disulfide Radical Anion as a Reductant
2′-Deoxy-ribonucleotides diphosphates are the building blocks for the de novo synthesis of DNA. Ribonucleotide diphosphates undergo a selective but complex radical mechanism by the ribonucleotide reductase class I, active in eukaryotes and microorganisms. (Figure 44a) [187]. Figure 44b summarizes this multistep mechanism [187], which initially involves a reversible H-atom abstraction from C3’–H by the thiyl radical generated from one of the cysteine residues at the active site (18). The subsequent steps involve: elimination of H2O with translocation of the radical center to C-2′, quenching of the newly formed C-2′ radical by another cysteine residue, and formation of a disulfide radical anion (19). The reduction of ketone follows by disulfide radical anion (19 → 20) and the restoration of the initial cysteine residue (20 → 21). The restoration of the other two cysteine residues occurs by the intervention of thioredoxin reductase (TRR) and NADPH [188].
In chemical synthesis, the selective deoxygenation of a 1,2-diol to an alcohol is a difficult conversion that can be accomplished by dehydration of a 1,2-diol to an aldehyde or ketone followed by a reduction. For example, the Barton−McCombie approach is an example of multistep reactivity that is applicable to sugar substrates and involves protection and derivatization to an intermediate that undergoes a radical-mediated deoxygenation reaction [36,189]. The enzymatic transformation shown in Figure 44 has been taken as inspiration for using sulfur radical chemistry in the cis-1,2-cyclopentanediol (18) transformation to cyclopentanol (20) as well as the reduction of carbonyl compounds by disulfide radial anion [190] (Figure 45b).
The method employs H2S/HS– generated in situ by dissolving Na2S in water at suitable pH and the corresponding transient species HS•/S•− and HSSH•−/HSS•2− (Figure 45a) [191]. The initial HS•/S•− radical abstracts an H-atom from cis-1,2-cyclopentanediol (22) to give the radical 25, which undergoes a β-C—O scission and H2O elimination to give radical 26 that completes the catalytic cycle by reacting with H2S/HS−, regenerating HS•/S•− and forming cyclopentanone (23) (left side of Figure 45b). The ketone 23 is reduced by the dimeric radical anion HSSH•−/HSS•2− to the radical anion 6, followed by protonation and H-atom abstraction of 28 from H2S/HS− affords the product cyclopentanol (24) completing the radical chain (right side of Figure 45b). pH Dependence experiments demonstrated that the catalytic cycle is more efficient under acidic conditions (pH < 7) whereas the radical chain is more effective under basic conditions (pH > 7) [190]. Indeed, a variety of carbonyl compounds are reduced to the corresponding alcohols in excellent yields (90-99%) using UV light for 1h at pH 9 and 42 °C [190]. It is worth also mentioning that the reduction of 2-hydroxycyclohexanone (8) under the same conditions proceeds via a dual radical chain mechanism, affording first the ketone 9 and then the alcohol 10 (Figure 45c). The reduction potential E(HSS–/HSS•2−) is estimated to be –1.13 V [192], indicating the efficiency of the species HSS•2− for one-electron transfer to a carbonyl group. The reactive species derived from H2S and the reported mechanistic steps in Figure 45 can also be discussed in terms prebiotic photochemical transformations, with H2S and UV light being both present in the primitive earth environment [193,194].
The bioinspired conversion of ketones to alcohols was also achieved by the intermediacy of disulfide radical anion of cysteine (CysSSCys)•− in water [195]. A variety of ketones are reduced to the corresponding alcohols in excellent yields (91-99%) using UV light (low-pressure Hg lamp, 5.5 W), for 1h, at pH 10.6 and 42 °C, in N2-saturated aqueous solutions. However, high concentration of cysteine and pH 10.6 are necessary for for shifting the equilibrium to (CysSSCys)•− and providing high-yielding reactions. The reaction mechanism for the reduction of a ketone to the corresponding alcohol is also shown in Figure 46a. The ketone is reduced to ketyl radical anion via single-electron transfer, followed by protonation from the aqueous medium and H-atom abstraction from CysSH affording the alcohol, thus completing the radical chain mechanism. Figure 46b shows the reduction of 2-cyclopenten-1-one (32) under identical experimental conditions. The reported time profile of the reduction of compound 32 at pH 10.6 shows clearly that the reaction 32 → 36 occurs stepwise, with formation of the cyclopentanone (35) as the intermediate stable product, i.e., proceeds via a dual radical chain mechanism affording first the ketone 35 and then the alcohol 36.
The standard reduction potential E0(CysSSCys/CysSSCys•−) = –1.38 V [196,197] indicates the efficiency of CysSSCys•− for single electron transfer to a carbonyl group. Recently, the standard reduction potential of a variety of alkyl and aromatic disulfides have been reported [198]. The absorption spectra of (CysSSCys)•− have a λmax at 420 nm [199]; pulse radiolysis studies of the decay of (CysSSCys)•− at various concentrations of ketones, determined the rate constants of three cyclic ketones to be in the range of 104 –105 M−1 s−1 at ~22 ◦ C [195].
8. Conclusions
An overview of radical reactions in water or aqueous media achieved in the last two decades have been presented. The number of applications of radical reactions “in-water” and “on-water” has been continuously increasing, either using known radical chemistry or bioinspired processes from nature, towards biomarker development. Many methodologies of coordination-induced water H–OH bond weakening and photocatalysis using water as solvent or reagent have been developed finding new radical chain processes. Some of them have already been applied in the development of novel synthetic methodologies. In addition, the emergence of molybdenum, germanium, bismuth and phosphorous inducing lowering of H–OH or H–OR BDEs enlarges this promising field, and is expected to provide other useful and novel synthetic applications in the near future.
Author Contributions
All authors have contributed to conceptualization, data curation, writing—original draft preparation and revision, and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Breslow, R. Hydrophobic Effects on Simple Organic Reactions in Water. Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- Li, C.-J. Organic Reactions in Aqueous Media with a Focus on Carbon-Carbon Bond Formations. Chem. Rev. 1993, 93, 2023–2035. [Google Scholar] [CrossRef]
- Lindström, U.M. Stereoselective Organic Reactions in Water. Chem. Rev. 2002, 102, 2751–2772. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. Determining the Geometries of Transition States by Use of Antihydrophobic Additives in Water. Acc. Chem. Res. 2004, 37, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-J. Organic Reactions in Aqueous Media with a Focus on Carbon-Carbon Bond Formations: A Decade Update. Chem. Rev. 2005, 105, 3095–3165. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. (Ed.) Water in Organic Synthesis; Georg Thieme Verlag KG: Stuttgart, 2012. [Google Scholar]
- Levin, E.; Ivry, E.; Diesendruck, C.E.; Lemcoff, N.G. Water in N-Heterocyclic Carbene-Assisted Catalysis. Chem. Rev. 2015, 115, 4607–4692. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Muldoon, J.; Finn, M. G.; Fokin, V. V.; Kolb, H. C.; Sharpless, K. B. Angew. Chem., Int. Ed. 2005, 44, 3275-3279. [CrossRef] [PubMed]
- Shapiro, N.; Vigalok, A. Highly Efficient Organic Reactions “on Water”, “in Water”, and Both. Angew. Chem., Int. Ed. 2008, 47, 2849–2852. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef]
- Butler, R.N.; G. Coyne, A.G. Water: Nature’s Reaction Enforcer–Comparative Effects for Organic Synthesis “In-Water” and “On-Water”. Chem. Rev. 2010, 110, 6302–6337. [Google Scholar] [CrossRef]
- Simon, M.O.; Li, C.J. Green Chemistry Oriented Organic Synthesis in Water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef]
- Butler, R.N.; Coyne, A.G. Organic Synthesis Reactions on- Water at the Organic−Liquid Water Interface. Org. Biomol. Chem. 2016, 14, 9945–9960. [Google Scholar] [CrossRef] [PubMed]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Romney, D.K.; Arnold, F.H.; Lipshutz, B.H.; Li, C.-J. Chemistry Takes a Bath: Reactions in Aqueous Media. J. Org. Chem. 2018, 83, 7319–7322. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Shinokubo, H.; Oshima, K. Synthetic Radical Reactions in Aqueous Media. Synlett 2002, 674–686. [Google Scholar] [CrossRef]
- Postigo, A. Synthetically useful carbon–carbon and carbon–sulphur bond construction mediated by carbon- and sulphur-centred radicals in water and aqueous media. RSC Adv. 2011, 1, 14–32. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Oshima, K. Free-Radical Reactions. In Water in Organic Synthesis; Kobayashi, S., Ed.; Georg Thieme Verlag KG: Stuttgart, 2012; pp. 645–677. [Google Scholar]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science. 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Dedon, P.C. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem. Res. Toxicol. 2008, 24, 206–219. [Google Scholar] [CrossRef]
- Emanuel, C.J.; Newcomb, M.; Ferreri, C.; Chatgilialoglu, C. Kinetics of 2′-deoxyuridin-1′-yl radical reactions. J. Am. Chem. Soc. 1999, 121, 2927–2928. [Google Scholar] [CrossRef]
- Gimisis, T.; Chatgilialoglu, C. Fate of the C-1′ peroxyl radical in the 2′-deoxyuridine system. Chem. Commun. 1998, 1249–1250. [Google Scholar]
- Schmittel, M.; Burghart, A. Understanding Reactivity Patterns of Radical Cations. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550–2589. [Google Scholar] [CrossRef]
- Chatgilialoglu, C. The Two Faces of the Guanyl Radical: Molecular Context and Behavior. Molecules 2021, 26, 3511. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Capobianco, A.; Bobrowski, K.; Peluso, A.; Chatgilialoglu, C. Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules 2023, 13, 1493. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Lloyd, R.S. (Eds.) DNA Damage, DNA Repair and Disease; Royal Society of Chemistry: Croydon, UK, 2021. [Google Scholar]
- Voutyritsa, E.; Kokotos, C.G. Green Metal-Free Photochemical Hydroacylation of Unactivated Olefins. Angew. Chem. Int. Ed. 2020, 59, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Thiyl Radicals in Biology: Their Role as a "Molecular Switch" Central to Cellular Oxidative Stress. In S-Centered Radicals; Alfassi, Z.B., Ed.; J. Wiley & Sons: Chichester, 1999; p. 289. [Google Scholar]
- Chatgilialoglu, C. RSH/(TMS)3SiH Reducing System: A Tool for Measuring Rate Constants for Reactions of Radicals with Thiols. Helv. Chim. Acta 2006, 89, 2387–2398. [Google Scholar] [CrossRef]
- Ross, A.B.; Mallard, W.G.; Helman, W.P.; Buxton, G.V.; Huie, R.E.; Neta, P. 5NDRL-NIST Solution Kinetic Database – Vers. 36; Notre Dame Radiation Laboratory, Notre Dame, IN and NIST Standard Reference Data, Gaithersburg, MD, 1998.
- Tronche, C.; Martinez, F.N.; Horner, J.H.; Newcomb, M.; Senn, M.; Giese, B. Polar Substituent and Solvent Effects on the Kinetics of Radical Reactions with Thiols. Tetrahedron Lett. 1996, 37, 5845–5848. [Google Scholar] [CrossRef]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair, A Chemical Perspective; Springer: Berlin, Germany, 2006; p. 144. [Google Scholar]
- Reid, D.L.; Shustov, G.V.; Armstrong, D.A.; Rauk, A.; Schuchmann, M.N.; Akhlaq, M.S.; von Sonntag, C. H-atom abstraction from thiols by C-centered radicals. A theoretical and experimental study of reaction rates. Phys. Chem. Chem. Phys. 2002, 4, 2965–2974. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Studer, A. (Eds.) Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley: Chichester, UK, 2012. [Google Scholar]
- Baguley, P.A.; Walton, J.C. Flight from the Tyranny of Tin: The Quest for Practical Radical Sources Free from Metal Encumbrances. Angew. Chem., Int. Ed. 1998, 37, 3072–3082. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Landais, Y.; Timokhin, V.I. Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev. 2018, 118, 6516–6572. [Google Scholar] [CrossRef]
- Light, J.; Breslow, R. A water soluble tin hydride reagent. Tetrahedron Lett. 1990, 31, 2957–2958. [Google Scholar] [CrossRef]
- Rai, R.; Collum, D.B. Reductions and radical cyclizations of aryl and alkyl bromides mediated by NaBH4 in aqueous base. Tetrahedron Lett. 1994, 35, 6221–6224. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Hypophosphorous acid and its salts: New reagents for radical chain deoxygenation, dehalogenation and deamination. Tetrahedron Lett. 1992, 33, 5709–5712. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.C. The invention of radical reactions. Part 32. Radical deoxygenations, dehalogenations, and deaminations with dialkyl phosphites and hypophosphorous acid as hydrogen sources. J. Org. Chem. 1993, 58, 6838–6842. [Google Scholar] [CrossRef]
- Kita, Y.; Nambu, H.; Ramesh, N.G.; Anilkumar, G.; Matsugi, M. A Novel and Efficient Methodology for the C−C Bond Forming Radical Cyclization of Hydrophobic Substrates in Water. Org. Lett. 2001, 3, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Guyon, C.; Metay, E.; Popowycz, F.; Lemaire, M. Synthetic applications of hypophosphite derivatives in reduction. Org. Biomol. Chem. 2015, 13, 7879–7906. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Katayama, S.; Hirose, N.; Naito, M.; Izawa, K. Radical deoxygenation and dehalogenation of nucleoside derivatives with hypophosphorous acid and dialkyl phosphites. Tetrahedron Lett. 2001, 42, 7605–7608. [Google Scholar] [CrossRef]
- Khan, T.A.; Tripoli, R.; Crawford, J.J.; Martin, C.G.; Murphy, J.A. Diethylphosphine Oxide (DEPO): High-Yielding and Facile Preparation of Indolones in Water. Org. Lett. 2003, 5, 2971–2974. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.J.; Schubert, J.W.; Streckfuss, E.C.; Balsells, J.; ElMarrouni, A. Photoredox catalysis for silyl-mediated C-H alkylation of heterocycles with non-activated alkyl bromides. Eur. J. Org. Chem. 2020, 2020, 1515–1522. [Google Scholar] [CrossRef]
- Lovett, G.H.; Chen, S.; Xue, X.S.; Houk, K.N.; MacMillan, D.W.C. Open-shell fluorination of alkyl bromides: unexpected selectivity in a silyl radical-mediated chain process. J. Am. Chem. Soc. 2019, 141, 20031–20036. [Google Scholar] [CrossRef]
- Le, C.; Chen, T.Q.; Liang, T.; Zhang, P.; MacMillan, D.W.C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. [Google Scholar] [CrossRef]
- Jasperse, C.P.; Curran, D.P.; Fevig, T.L. Radical reactions in natural product synthesis. Chem. Rev. 1991, 91, 1237–1286. [Google Scholar] [CrossRef]
- Juliá, F.; Constantin, T.; Leonori, D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122, 2292–2352. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; McFadden, T.R. Understanding Initiation with Triethylboron and Oxygen: The Differences between Low-Oxygen and High-Oxygen Regimes. J. Am. Chem. Soc. 2016, 138, 7741–7752. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Nakamura, T.; Shinokubo, H.; Oshima, K.; Omoto, K.; Fujimoto, H. Powerful Solvent Effect of Water in Radical Reaction: Triethylborane-Induced Atom-Transfer Radical Cyclization in Water. J. Am. Chem. Soc. 2000, 122, 11041–11047. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K. Triethylborane-Induced Bromine Atom-Transfer Radical Addition in Aqueous Media: Study of the Solvent Effect on Radical Addition Reactions. J. Org. Chem. 2001, 66, 7776–7785. [Google Scholar] [CrossRef] [PubMed]
- McNabb, S.B.; Ueda, M.; Naito, T. Addition of Electrophilic and Heterocyclic Carbon-Centered Radicals to Glyoxylic Oxime Ethers. Org. Lett. 2004, 6, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; H. Shinokubo, H.; Oshima, K. From Alkenylsilanes to Ketones with Air as the Oxidant. Angew. Chem., Int.Ed. 2003, 42, 849–851. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Romine, A.M.; Nebra, N.; Konovalov, A.I.; Martin, E.; Benet-Buchholz, J.; Grushin, V.V. Easy Access to the Copper(III) Anion [Cu(CF3)4]−. Angew. Chem., Int. Ed. 2015, 54, 2745–2749. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, Z.; Fang, Y.; Zhu, L.; Li, C. Radical trifluoromethylation. Chem. Soc. Rev. 2021, 50, 6308–6319. [Google Scholar] [CrossRef]
- Shen, H.; Liu, Z.; Zhang, P.; Tan, X.; Zhang, Z.; Li, C. Trifluoromethylation of Alkyl Radicals in Aqueous Solution. J. Am. Chem. Soc. 2017, 139, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Cook, S.P. Interrupting the Barton–McCombie Reaction: Aqueous Deoxygenative Trifluoromethylation of O-Alkyl Thiocarbonates. Org. Lett. 2021, 23, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; AbuSalim, D.I.; Cook, S.P. Aqueous Benzylic C−H Trifluoromethylation for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 12378–12382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shen, H.; Zhu, L.; Cao, W.; Li, C. Radical C(sp2)–H Trifluoromethylation of Aldehydes in Aqueous Solution. Org. Lett. 2018, 20, 7062–7065. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, Z.; Shen, H.; Zhang, P.; Zhang, Z.; Li, C. Silver-Catalyzed Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2017, 139, 12430–12433. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.N.; Brueckl, T.; Baxter, R.D.; Fujiwara, Y.; Seiple, I.B.; Su, S.; Blackmond, D.G.; Baran, P.S. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411–14415. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.D.; Kuenzi, S.A.; Sanford, M.S. Practical Method for the Cu-Mediated Trifluoromethylation of Arylboronic Acids with CF3 Radicals Derived from NaSO2CF3 and tert-Butyl Hydroperoxide (TBHP). Org. Lett. 2012, 14, 4979–4981. [Google Scholar] [CrossRef]
- Li, Z.J.; Cui, Z.L.; Liu, Z.Q. Copper- and Iron-Catalyzed Decarboxylative Tri- and Difluoromethylation of α,β-Unsaturated Carboxylic Acids with CF3SO2Na and (CF2HSO2)2Zn via a Radical Process. Org. Lett. 2013, 15, 406–409. [Google Scholar] [CrossRef]
- Yang, F.; Klumphu, P.; Liang, Y.-M.; Lipshutz, B.H. Copper-catalyzed trifluoromethylation of N-arylacrylamides “on water” at room temperature. Chem. Commun. 2014, 50, 936–938. [Google Scholar] [CrossRef]
- Shapiro, N.; Vigalok, A. Highly Efficient Organic Reactions “on Water”, “in Water”, and Both. Angew. Chem. Int. Ed. 2008, 47, 2849–2852. [Google Scholar] [CrossRef]
- Papadopoulos, G.N.; Voutyritsa, E.; Kaplaneris, N.; Kokotos, C.G. Green Photo-Organocatalytic C—H Activation of Aldehydes: Selective Hydroacylation of Electron-Deficient Alkenes. Chem. Eur. J. 2018, 24, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chemistry of acyl radicals. Chem. Rev. 1999, 99, 1991–2070. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C. Organosilanes in Radical Chemistry; Wiley: Chichester, UK, 2004. [Google Scholar]
- Chatgilialoglu, C. (Me3Si)3SiH: Twenty years after its discovery as a radical-based reducing agent. Chem.—Eur. J. 2008, 14, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Lalevée, J. Recent Applications of the (TMS)3SiH Radical-Based Reagent. Molecules 2012, 17, 527–555. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.; Ferreri, C.; Navacchia, M.L.; Chatgilialoglu, C. The radical-based reduction with (TMS)3SiH “on water”. Synlett 2005, 2854–2856. [Google Scholar]
- Postigo, A.; Kopsov, S.; Ferreri, C.; Chatgilialoglu, C. Radical reactions in aqueous medium using (Me3Si)3SiH. Org. Lett. 2007, 9, 5159–5162. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.; Kopsov, S.; Zlotsky, S.S.; Ferreri, C.; Chatgilialoglu, C. Hydrosilylation of C-C Multiple Bonds Using (Me3Si)3SiH in Water. Comparative Study of the Radical Initiation Step. Organometallics 2009, 28, 3282–3287. [Google Scholar] [CrossRef]
- Goh, Y.L.; Tam, E.K.W.; Bernardo, P.H.; Cheong, C.B.; Johannes, C.W.; William, A.D.; Adsool, V.A. A New Route to Bicyclo[1.1.1]pentan-1-amine from 1-Azido-3-iodobicyclo[1.1.1]pentane. Org. Lett. 2014, 16, 1884–1887. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Postigo, A. (Me3Si)3SiH-Mediated Intermolecular Radical Perfluoroalkylation. Reactions of Olefins in Water. J. Org. Chem. 2010, 75, 6141–6148. [Google Scholar] [CrossRef]
- Wu, L.-J.; Tan, F.-L.; Li, M.; Song, R.-J.; Li, J.-H. Fe-Catalyzed oxidative spirocyclization of N-arylpropiolamides with silanes and TBHP involving the formation of C−Si bonds. Org. Chem. Front. 2017, 4, 350–353. [Google Scholar] [CrossRef]
- Zhu, J.; Cui, W.-C.; Wang, S.; Yao, Z.-J. Radical Hydrosilylation of Alkynes Catalyzed by Eosin Y and Thiol under Visible Light Irradiation. Org. Lett. 2018, 20, 3174–3178. [Google Scholar] [CrossRef] [PubMed]
- Pickford, H.D.; Nugent, J.; Owen, B.; Mousseau, J.J.; Smith, R.C.; Anderson, E.A. Twofold Radical-Based Synthesis of N,C-Difunctionalized Bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2021, 143, 9729–9736. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.K.; Jang, D.O. Radical “On Water” Addition to the C═N Bond of Hydrazones: A Synthesis of Isoindolinone Derivatives. J. Org. Chem. 2018, 83, 7373–7379. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.K.; Jang, D.O. A synthetic route toward α,α-dialkyl α-amino ester derivatives via radical addition to hydrazone derivatives of α-ketoesters under ‘‘on-water” conditions. Tetrahedron Lett. 2020, 61, 151411. [Google Scholar] [CrossRef]
- Mori, K.; Shimizu, A.; Horibe, M.; Takei, M.; Awano, N.; Matsuoka, S.; Suzuki, M. Lewis Pair Radical Polymerization “On-Water”. Macromolecules 2021, 54, 3–10. [Google Scholar] [CrossRef]
- Ruscic, B.; Wagner, A.F.; Harding, L.B.; Asher, R.L.; Feller, D.; Dixon, D.A.; Peterson, K.A.; Song, Y.; Qian, X.; Ng, C.-Y.; Liu, J.; Chen, W.; Schwenke, D.W. On the Enthalpy of Formation of Hydroxyl Radical and Gas-Phase Bond Dissociation Energies of Water and Hydroxyl. J. Phys. Chem. A 2002, 106, 2727–2747. [Google Scholar] [CrossRef]
- Mardyukov, A.; Sanchez-Garcia, E.; Crespo-Otero, R.; Sander, W. Interaction and Reaction of the Phenyl Radical with Water: A Source of OH Radicals. Angew. Chem. Int. Ed. 2009, 48, 4804–4807. [Google Scholar] [CrossRef]
- Boekell, N.G.; Flowers, R.A. Coordination-Induced Bond Weakening. Chem. Rev. 2022, 122, 13447–13477. [Google Scholar] [CrossRef]
- Agarwal, R.G.; Coste, S.C.; Groff, B.D.; Heuer, A.M.; Noh, H.; Parada, G.A.; Wise, C.F.; Nichols, E.M.; Warren, J.J.; Mayer, J.M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Campaña, A.G.; Justicia, J.; Rosales, A.; Oller-López, J.L.; Robles, R.; Cárdenas, D.J.; Buñuel, E.; Oltra, J.E. Water: The Ideal Hydrogen-Atom Source in Free-Radical Chemistry Mediated by TiIII and Other Single-Electron-Transfer Metals? Angew. Chem. Int. Ed. 2006, 45, 5522–5526. [Google Scholar] [CrossRef]
- Kolmar, S.S.; Mayer, J.M. SmI2 (H2O) n Reduction of Electron Rich Enamines by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2017, 139, 10687–10692. [Google Scholar] [CrossRef] [PubMed]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. Proton-Coupled Electron Transfer in the Reduction of Carbonyls by Samarium Diiodide-Water Complexes. J. Am. Chem. Soc. 2016, 138, 8738–8741. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, D.A.; Wiberg, K.B.; Schacherer, L.N.; Medeiros, M.R.; Wood, J.L. Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source. J. Am. Chem. Soc. 2005, 127, 12513–12515. [Google Scholar] [CrossRef] [PubMed]
- Soulard, V.; Villa, G.; Vollmar, D.P.; Renaud, P. Radical Deuteration with D2O: Catalysis and Mechanistic Insights. J. Am. Chem. Soc. 2018, 140, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, Y.; Derdau, V.; Studer, A. Remote Site-Selective Radical C(sp3 )−H Monodeuteration of Amides Using D2O. Angew. Chem. Int. Ed. 2021, 60, 18645–18650. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Jing, H.; Ge, H.; Brothers, P.J.; Fu, X.; Ye, S. The Mechanism of E–H (E = N, O) Bond Activation by a Germanium Corrole Complex: A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2015, 137, 7122–7127. [Google Scholar] [CrossRef] [PubMed]
- Bezdek, M.J.; Guo, S.; Chirik, P.J. Coordination-Induced Weakening of Ammonia, Water, and Hydrazine X–H Bonds in a Molybdenum Complex. Science 2016, 354, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Reijerse, E.J.; Bhattacharyya, K.; Leutzsch, M.; Kochius, M.; Nöthling, N.; Busch, J.; Schnegg, A.; Auer, A.A.; Cornella, J. Radical Activation of N–H and O–H Bonds at Bismuth(II). J. Am. Chem. Soc. 2022, 144, 16535–16544. [Google Scholar] [CrossRef]
- Medeiros, M.R.; Schacherer, L.N.; Spiegel, D.A.; Wood, J.L. Expanding the Scope of Trialkylborane/Water-Mediated Radical Reactions. Org. Lett. 2007, 9, 4427–4429. [Google Scholar] [CrossRef]
- Pozzi, D.; Scanlan, E.M.; Renaud, P. A Mild Radical Procedure for the Reduction of B -Alkylcatecholboranes to Alkanes. J. Am. Chem. Soc. 2005, 127, 14204–14205. [Google Scholar] [CrossRef]
- Boivin, J.; Nguyen, V.T. Part 1. Reduction of S -Alkyl-Thionocarbonates and Related Compounds in the Presence of Trialkylboranes/Air. Beilstein J. Org. Chem. 2007, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Allais, F.; Boivin, J.; Nguyen, V.T. Part 2. Mechanistic Aspects of the Reduction of S -Alkyl-Thionocarbonates in the Presence of Triethylborane and Air. Beilstein J. Org. Chem. 2007, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boivin, J.; Nguyen, V.T. Part 3. Triethylborane-Air: A Suitable Initiator for Intermolecular Radical Additions of S -2-Oxoalkyl-Thionocarbonates ( S -Xanthates) to Olefins. Beilstein J. Org. Chem. 2007, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Newcomb, M. Rate Constants for Reactions of Alkyl Radicals with Water and Methanol Complexes of Triethylborane. J. Org. Chem. 2007, 72, 5098–5103. [Google Scholar] [CrossRef]
- Povie, G.; Marzorati, M.; Bigler, P.; Renaud, P. Role of Equilibrium Associations on the Hydrogen Atom Transfer from the Triethylborane-Methanol Complex. J. Org. Chem. 2013, 78, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Deuterated Drugs Draw Heavier Backing. Nat. Rev. Drug Discov. 2016, 15, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Y.; Wu, X.; Zhu, C.; Xie, J. Radical Deuteration. Chem. Soc. Rev. 2022, 51, 6291–6306. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Ji, X.; He, R.; Liu, Y.; Chen, Z.; Huang, Y.; Liu, Q.; Li, Y. Synthesis of Deuterated ( E )-Alkene through Xanthate-Mediated Hydrogen–Deuterium Exchange Reactions. Org. Lett. 2021, 23, 7412–7417. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, J.; Qin, M.; Li, J.; Han, J.; Zhu, C.; Li, W.; Xie, J. Highly Selective Single and Multiple Deuteration of Unactivated C(Sp3)-H Bonds. Nat. Commun. 2022, 13, 4224. [Google Scholar] [CrossRef]
- Shi, Q.; Xu, M.; Chang, R.; Ramanathan, D.; Peñin, B.; Funes-Ardoiz, I.; Ye, J. Visible-Light Mediated Catalytic Asymmetric Radical Deuteration at Non-Benzylic Positions. Nat. Commun. 2022, 13, 4453. [Google Scholar] [CrossRef]
- Zhang, J.; Mück-Lichtenfeld, C.; Studer, A. Photocatalytic Phosphine-Mediated Water Activation for Radical Hydrogenation. Nature 2023, 619, 506–513. [Google Scholar] [CrossRef]
- Jin, J.; Newcomb, M. Rate Constants for Hydrogen Atom Transfer Reactions from Bis(Cyclopentadienyl)Titanium(III) Chloride-Complexed Water and Methanol to an Alkyl Radical. J. Org. Chem. 2008, 73, 7901–7905. [Google Scholar] [CrossRef]
- Paradas, M.; Campaña, A.G.; Jiménez, T.; Robles, R.; Oltra, J.E.; Buñuel, E.; Justicia, J.; Cárdenas, D.J.; Cuerva, J.M. Understanding the Exceptional Hydrogen-Atom Donor Characteristics of Water in Ti III -Mediated Free-Radical Chemistry. J. Am. Chem. Soc. 2010, 132, 12748–12756. [Google Scholar] [CrossRef]
- Gansäuer, A.; Behlendorf, M.; Cangönül, A.; Kube, C.; Cuerva, J.M.; Friedrich, J.; van Gastel, M. H2O Activation for Hydrogen-Atom Transfer: Correct Structures and Revised Mechanisms. Angew. Chem. Int. Ed. 2012, 51, 3266–3270. [Google Scholar] [CrossRef]
- Muñoz-Bascón, J.; Hernández-Cervantes, C.; Padial, N.M.; Álvarez-Corral, M.; Rosales, A.; Rodríguez-García, I.; Oltra, J.E. Ti-Catalyzed Straightforward Synthesis of Exocyclic Allenes. Chem. – Eur. J. 2014, 20, 801–810. [Google Scholar] [CrossRef]
- Rosales, A.; Muñoz-Bascón, J.; Roldan-Molina, E.; Castañeda, M.A.; Padial, N.M.; Gansäuer, A.; Rodríguez-García, I.; Oltra, J.E. Selective Reduction of Aromatic Ketones in Aqueous Medium Mediated by Ti(III)/Mn: A Revised Mechanism. J. Org. Chem. 2014, 79, 7672–7676. [Google Scholar] [CrossRef]
- Boyd, E.A.; Peters, J.C. Sm(II)-Mediated Proton-Coupled Electron Transfer: Quantifying Very Weak N–H and O–H Homolytic Bond Strengths and Factors Controlling Them. J. Am. Chem. Soc. 2022, 144, 21337–21346. [Google Scholar] [CrossRef]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. Reversibility of Ketone Reduction by SmI 2 –Water and Formation of Organosamarium Intermediates. Organometallics 2017, 36, 4579–4583. [Google Scholar] [CrossRef]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. High-Affinity Proton Donors Promote Proton-Coupled Electron Transfer by Samarium Diiodide. Angew. Chem. Int. Ed. 2016, 55, 6033–6036. [Google Scholar] [CrossRef]
- Ciamician, G. The Photochemistry of the Future. Science (80-. ). 1912, 36, 385–394. [Google Scholar] [CrossRef]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient visible light photocatalysis of [2+2] enone cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef]
- Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R.J. Electron-transfer photoredox catalysis: Development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685. [Google Scholar] [CrossRef]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef]
- Rueda-Marquez, J.J.; Levchuk, I.; Fernández Ibañez, P.; Sillanpää, M. A critical review on application of photocatalysis for toxicity reduction of real wastewaters. J. Clean. Prod. 2020, 258, 120694. [Google Scholar] [CrossRef]
- Arias-Rotondo, D.M.; McCusker, J.K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef]
- Sun, K.; Lv, Q.-Y.; Chen, X.-L.; Qu, L.-B.; Yu, B. Recent advances in visible-light-mediated organic transformations in water. Green Chem. 2021, 23, 232–248. [Google Scholar] [CrossRef]
- Russo, C.; Brunelli, F.; Tron, G.C.; Giustiniano, M. Visible-Light Photoredox Catalysis in Water. J. Org. Chem. 2023, 88, 6284–6293. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Yerien, D.E.; Postigo, A. Advances in Photocatalytic Organic Synthetic Transformations in Water and Aqueous Media. ACS Sustain. Chem. Eng. 2021, 9, 10016–10047. [Google Scholar] [CrossRef]
- Dou, Q.; Zeng, H. Recent advances in photo-induced organic synthesis in water. Curr. Opin. Green Sustain. Chem. 2023, 40, 100766. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Yu, J.; Lu, C.; Wu, Z.; Wu, X.; Xue, X.; Zhu, C. Polarity Umpolung Strategy for the Radical Alkylation of Alkenes. Angew. Chemie Int. Ed. 2020, 59, 8195–8202. [Google Scholar] [CrossRef]
- Crespi, S.; Fagnoni, M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833. [Google Scholar] [CrossRef]
- Kvasovs, N.; Gevorgyan, V. Contemporary methods for generation of aryl radicals. Chem. Soc. Rev. 2021, 50, 2244–2259. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Lin, J.; Li, Z.; Huang, H. Radical acylation: concepts, synthetic applications and directions. Org. Chem. Front. 2023, 10, 1056–1085. [Google Scholar] [CrossRef]
- Yerien, D.E.; Barata-Vallejo, S.; Postigo, A. Radical Perfluoroalkylation of Aliphatic Substrates. ACS Catal. 2023, 7756–7794. [Google Scholar] [CrossRef]
- Wallentin, C.J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef]
- Yao, H.; Hu, W.; Zhang, W. Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions. Molecules 2020, 26, 105. [Google Scholar] [CrossRef]
- Li, L.; Huang, M.; Liu, C.; Xiao, J.C.; Chen, Q.Y.; Guo, Y.; Zhao, Z.G. 2,2,2-Trifluoroethylation of Styrenes with Concomitant Introduction of a Hydroxyl Group from Molecular Oxygen by Photoredox Catalysis Activated by Visible Light. Org. Lett. 2015, 17, 4714–4717. [Google Scholar] [CrossRef]
- Hari, D.P.; König, B. The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light. Angew. Chemie Int. Ed. 2013, 52, 4734–4743. [Google Scholar] [CrossRef]
- Prasad Hari, D.; Hering, T.; König, B. The photoredox-catalyzed meerwein addition reaction: Intermolecular amino-arylation of alkenes. Angew. Chemie - Int. Ed. 2014, 53, 725–728. [Google Scholar] [CrossRef]
- Kim, S. Free Radical-Mediated Acylation and Carboxylation Reactions. Adv. Synth. Catal. 2004, 346, 19–32. [Google Scholar] [CrossRef]
- Zhang, M.; Xie, J.; Zhu, C. A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Nguyen, J.D.; Tucker, J.W.; Konieczynska, M.D.; Stephenson, C.R.J. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef]
- Yerien, D.E.; Barata-Vallejo, S.; Mansilla, D.; Postigo, A. Rose Bengal-photocatalyzed perfluorohexylation reactions of organic substrates in water. Applications to late-stage syntheses. Photochem. Photobiol. Sci. 2022, 21, 803–812. [Google Scholar] [CrossRef]
- Hu, C.C.; Tong, C.L.; Zhang, Y.Y.; Xu, X.H.; Qing, F.L. Photoredox-Catalyzed and Silane-Mediated Hydrofluoromethylation of Unactivated Alkenes with Fluoroiodomethane in Water. Org. Lett. 2023, 25, 1035–1039. [Google Scholar] [CrossRef]
- Wang, T.; Zong, Y.Y.; Huang, T.; Jin, X.L.; Wu, L.Z.; Liu, Q. Photocatalytic redox-neutral selective single C(sp3)-F bond activation of perfluoroalkyl iminosulfides with alkenes and water. Chem. Sci. 2023, 14, 11566–11572. [Google Scholar] [CrossRef]
- Gurry, M.; Aldabbagh, F. A new era for homolytic aromatic substitution: replacing Bu 3 SnH with efficient light-induced chain reactions. Org. Biomol. Chem. 2016, 14, 3849–3862. [Google Scholar] [CrossRef]
- Xue, D.; Jia, Z.H.; Zhao, C.J.; Zhang, Y.Y.; Wang, C.; Xiao, J. Direct arylation of n-heteroarenes with aryldiazonium salts by photoredox catalysis in water. Chem. - A Eur. J. 2014, 20, 2960–2965. [Google Scholar] [CrossRef]
- Natarajan, P.; Chuskit, D. Priya Metal-free, visible-light-promoted oxidative radical cyclization of: N-biarylglycine esters: One-pot construction of phenanthridine-6-carboxylates in water. Green Chem. 2019, 21, 4406–4411. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Cooke, M.V.; Postigo, A. Radical Fluoroalkylation Reactions. ACS Catal. 2018, 8, 7287–7307. [Google Scholar] [CrossRef]
- Yerien, D.E.; Postigo, A.; Baroncini, M.; Barata-Vallejo, S. Bioinspired photocatalysed C-H fluoroalkylation of arenes in water promoted by native vitamin B12and Rose Bengal. Green Chem. 2021, 23, 8147–8153. [Google Scholar] [CrossRef]
- Nicoli, F.; Baroncini, M.; Silvi, S.; Groppi, J.; Credi, A. Direct synthetic routes to functionalised crown ethers. Org. Chem. Front. 2021, 8, 5531–5549. [Google Scholar] [CrossRef]
- Yerien, D.E.; Groppi, J.; Postigo, A.; Credi, A.; Conde, R.S.; Baroncini, M.; Barata-Vallejo, S. Late-Stage Photocatalytic Fluoroalkylation of Aromatic Crown Ethers in Aqueous Media. European J. Org. Chem. 2023, 26, e202300478. [Google Scholar] [CrossRef]
- Hu, X.; Xu, W.; Liu, Y.; Guo, H. Visible Light-Induced Diastereoselective Construction of Trifluoromethylated Cyclobutane Scaffolds through [2+2]-Photocycloaddition and Water-Assisted Hydrodebromination. J. Org. Chem. 2023, 88, 2521–2534. [Google Scholar] [CrossRef]
- Kölmel, D.K.; Ratnayake, A.S.; Flanagan, M.E. Photoredox cross-electrophile coupling in DNA-encoded chemistry. Biochem. Biophys. Res. Commun. 2020, 533, 201–208. [Google Scholar] [CrossRef]
- McLean, J.T.; Benny, A.; Nolan, M.D.; Swinand, G.; Scanlan, W.M. Cysteinyl radicals in chemical synthesis and in nature. Chem. Soc. Rev. 2021, 50, 10857–10894. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radical Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5′,8-cyclonucleoside lesions: chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Geacintov, N.E.; Krokidis, M.G.; Liu, Y.; Masi, A.; Shafirovich, N.; Terzidis, M.A.; Tsegay, P.S. 5′,8-Cyclopurine lesions in DNA damage: Chemical, analytical, biological and diagnostic significance. Cells 2019, 8, 513. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Krokidis, M.G.; Masi, A.; Terzidis, M.A. On the relevance of hydroxyl radical to purine DNA damage. Free Radical Res. 2021, 55, 384–404. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Bazzanini, R.; Jimenez, L.B.; Miranda, M.A. (5′S)- and (5′R)-5′,8-Cyclo-2′-deoxyguanosine: Mechanistic insights on the 2′-deoxyguanosin-5′-yl radical cyclization. Chem. Res. Toxicol. 2007, 20, 1820–1824. [Google Scholar] [CrossRef]
- Flyunt, R.; Bazzanini, R.; Chatgilialoglu, C.; Mulazzani, Q.G. Fate of the 2′-deoxyadenosin-5′-yl radical under anaerobic conditions. J. Am. Chem. Soc. 2000, 122, 4225–4226. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Guerra, M.; Mulazzani, Q.G. Model studies of DNA C5′ radicals. Selective generation and reactivity of 2′-deoxyadenosin-5′-yl radical. J. Am. Chem. Soc. 2003, 125, 3839–3848. [Google Scholar]
- Boussicault, F.; Kaloudis, P.; Caminal, C.; Mulazzani, Q.G.; Chatgilialoglu, C. The fate of C5′ radicals of purine nucleosides under oxidative conditions. J. Am. Chem. Soc. 2008, 130, 8377–8385. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; D’Angelantonio, M.; Kciuk, G.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem. Res. Toxicol. 2011, 24, 2200–2206. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Chatgilialoglu, C.; Montevecchi, P.C. C5′-adenosinyl radical cyclization. A stereochemical investigation. J. Org. Chem. 2006, 71, 4445–4452. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Chatgilialoglu, C. Radical cascade protocol for the synthesis of (5′S)- and (5′R)-5′,8-cyclo-2′-deoxyguanosine derivatives. Aust. J. Chem. 2013, 66, 330–335. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Chatgilialoglu, C. An ameliorative protocol for the quantification of purine 5′,8-cyclo-2′-deoxynucleosides in oxidized DNA. Front. Chem. 2015, 3, 47. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Terzidis, M.A.; Szreder, T.; Bobrowski, K. New Insights into the Reaction Paths of Hydroxyl Radicals with Purine Moieties in DNA and Double-Stranded Oligodeoxynucleotides. Molecules 2019, 24, 3860. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Eriksson, L.A.; Krokidis, M.G.; Masi, A.; Wang, S.; Zhang, R. Oxygen Dependent Purine Lesions in Double-Stranded Oligodeoxynucleotides: Kinetic and Computational Studies Highlight the Mechanism for 5′,8-Cyclopurine Formation. J. Am. Chem. Soc. 2020, 142, 5825–5833. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Terzidis, M.A.; Efthimiadou, E.; Zervou, S.; Kordas, G.; Papadopoulos, K.; Hiskia, A.; Kletsas, D.; Chatgilialoglu, C. Purine 5′,8-cyclo-2′-deoxynucleoside lesions: formation by radical stress and repair in human breast epithelial cancer cells. Free Radic. Res. 2017, 51, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Parlanti, E.; D’Errico, M.; Pascucci, B.; Pino, A.; Alimonti, A.; Pietraforte, D.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Purine DNA Lesions at Different Oxygen Concentration in DNA Repair-Impaired Human Cells (EUE-siXPA). Cells 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; D’Errico, M.; Pascucci, B.; Parlanti, E.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Oxygen-Dependent Accumulation of Purine DNA Lesions in Cockayne Syndrome Cells. Cells 2020, 9, 1671. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Pascucci, B.; D’Errico, M. Assessing the Formation of Purine Lesions in Mitochondrial DNA of Cockayne Syndrome Cells. Biomolecules 2022, 12, 1630. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Louka, M.; Efthimiadou, E.K.; Zervou, S.K.; Papadopoulos, K.; Hiskia, A.; Ferreri, C.; Chatgilialoglu, C. Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study. Cancers 2019, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Prasinou, P.; Efthimiadou, E.K.; Boari, A.; Ferreri, C.; Chatgilialoglu, C. Effects of Aging and Disease Conditions in Brain of Tumor-Bearing Mice: Evaluation of Purine DNA Damages and Fatty Acid Pool Changes. Biomolecules 2022, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Fortini, P.; Krokidis, M.G.; Romeo, E.F.; Bascietto, C.; De Angelis, P.; Guglielmi, V.; Chatgilialoglu, C. Increased Levels of 5′,8-Cyclopurine DNA Lesions in Inflammatory Bowel Diseases. Redox Biol. 2020, 34, 101562. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Masi, A.; Sansone, A.; Terzidis, M.A.; Tsakos, M. A problem solving approach for the diastereoselective synthesis of (5′S)- and (5′R)-5′,8-cyclopurine lesions. Org. Chem. Front. 2014, 1, 698–702. [Google Scholar] [CrossRef]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; Shafirovich, V. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucl. Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef]
- Xu, M.; Lai, Y.; Jiang, Z.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. A 5’, 8-cyclo-2’-deoxypurine lesion induces trinucleotide repeat deletion via a unique lesion bypass by DNA polymerase b. Nucl. Acids Res. 2014, 42, 13749–13763. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Xu, M.; Lai, Y.; Laverde, E.E.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. Bypass of a 5’,8-cyclopurine-2’-deoxynucleoside by DNA polymerase b during DNA replication and base excision repair leads to nucleotide misinsertions and DNA strand breaks. DNA Repair 2015, 33, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Shafirovich, V.; Kolbanovskiy, M.; Kropachev, K.; Liu, Z.; Cai, Y.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Amin, S.; Dadali, A.; Broyde, S.; Geacintov, N.E. Nucleotide Excision Repair and Impact of Site-Specific 5’,8-Cyclopurine and Bulky DNA Lesions on the Physical Properties of Nucleosomes. Biochemistry 2019, 58, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Minnihan, E.C.; Nocera, D.G.; Stubbe, J. Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res. 2013, 46, 2524–2535. [Google Scholar] [CrossRef]
- Greene, B.L.; Taguchi, A.T.; Stubbe, J.; Nocera, D.G. Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc. 2017, 139, 16657–16665. [Google Scholar] [CrossRef]
- McCombie, S.W.; Motherwell, W.B.; Tozer, M.J. The Barton-McCombie Reaction. Org. React. 2012, 77, 161–438. [Google Scholar]
- Barata-Vallejo, S.; Ferreri, C.; Golding, B.T.; Chatgilialoglu, C. Hydrogen Sulfide: A Reagent for pH-Driven Bioinspired 1,2-Diol Mono-deoxygenation and Carbonyl Reduction in Water. Org. Lett. 2018, 20, 4290–4294. [Google Scholar] [CrossRef] [PubMed]
- Das, T.N.; Huie, R.E.; Neta, P.; Padmaja, S. Reduction Potential of the Sulfhydryl Radical: Pulse Radiolysis and Laser Flash Photolysis Studies of the Formation and Reactions of ‚SH and HSSH·- in Aqueous Solutions. J. Phys. Chem. A 1999, 103, 5221–5226. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L. Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys. 2017, 617, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Straub, K.D. The Role of Hydrogen Sulfide in Evolution and the Evolution of Hydrogen Sulfide in Metabolism and Signaling. Physiology 2016, 31, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Green, N.J.; Xu, J.; Sutherland, J.D. Illuminating Life’s Origins: UV Photochemistry in Abiotic Synthesis of Biomolecules. J. Am. Chem. Soc. 2021, 143, 7219–7236. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Skotnicki, K.; Ferreri, C.; Marciniak, B.; Bobrowski, K.; Chatgilialoglu, C. Biomimetic Ketone Reduction by Disulfide Radical Anion. Molecules 2021, 26, 5429. [Google Scholar] [CrossRef]
- Mezyk, S.P.; Armstrong, D.A. Disulfide anion radical equilibria: Effects of NH +, -CO −, -NHC(O)- and -CH groups. J. Chem. Soc. Perkin Trans. 1999, 2, 1411–1419. [Google Scholar] [CrossRef]
- Zhu, Q.; Nocera, D.G. Catalytic C(β)−O Bond Cleavage of Lignin in a One-Step Reaction Enabled by a Spin-Center Shift. ACS Catal. 2021, 11, 14181–14187. [Google Scholar] [CrossRef]
- Zhu, Q.; Costentin, C.; Stubbe, J.; Nocera, D.G. Disulfide radical anion as a super-reductant in biology and photoredox chemistry. Chem Sci. 2023, 14, 6876–6881. [Google Scholar] [CrossRef]
- Hoffman, M.Z.; Hayon, E. One-Electron Reduction of the Disulfide Linkage in Aqueous Solution. Formation, Protonation, and Decay Kinetics of the RSSR- Radical. J. Am. Chem. Soc. 1972, 94, 795–7957. [Google Scholar] [CrossRef]
Figure 1.
Mechanistic details for the formation of 2-deoxyribonolactone from 2′-deoxynucleosides: The C1′ radical is oxidized by addition to molecular oxygen, heterolytic cleavage with release of O2•–, followed by hydrolysis.
Figure 1.
Mechanistic details for the formation of 2-deoxyribonolactone from 2′-deoxynucleosides: The C1′ radical is oxidized by addition to molecular oxygen, heterolytic cleavage with release of O2•–, followed by hydrolysis.
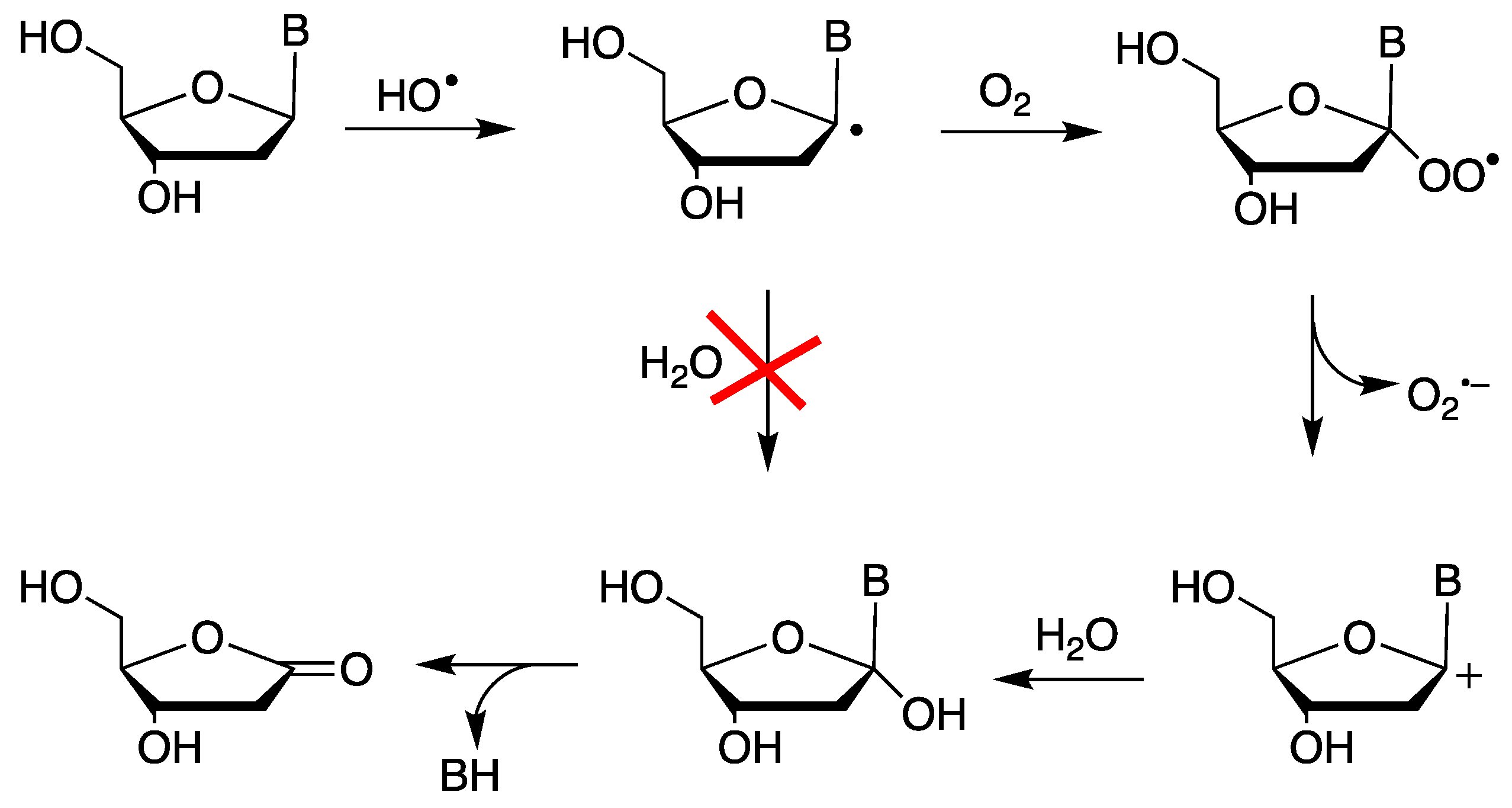
Figure 2.
Oxidation of guanine moiety and the reaction with water, ultimately affording 8-oxo-dG.
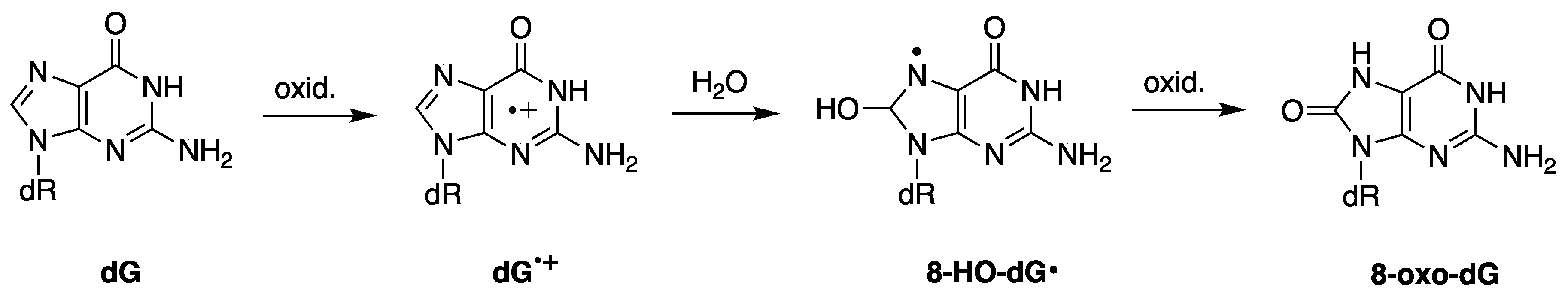
Figure 3.
Radical chain mechanisms of (a) functional group reduction (X = atom or group) by a reducing agent MH; (b) halogen atom-transfer in the new C–C bond formation.
Figure 3.
Radical chain mechanisms of (a) functional group reduction (X = atom or group) by a reducing agent MH; (b) halogen atom-transfer in the new C–C bond formation.
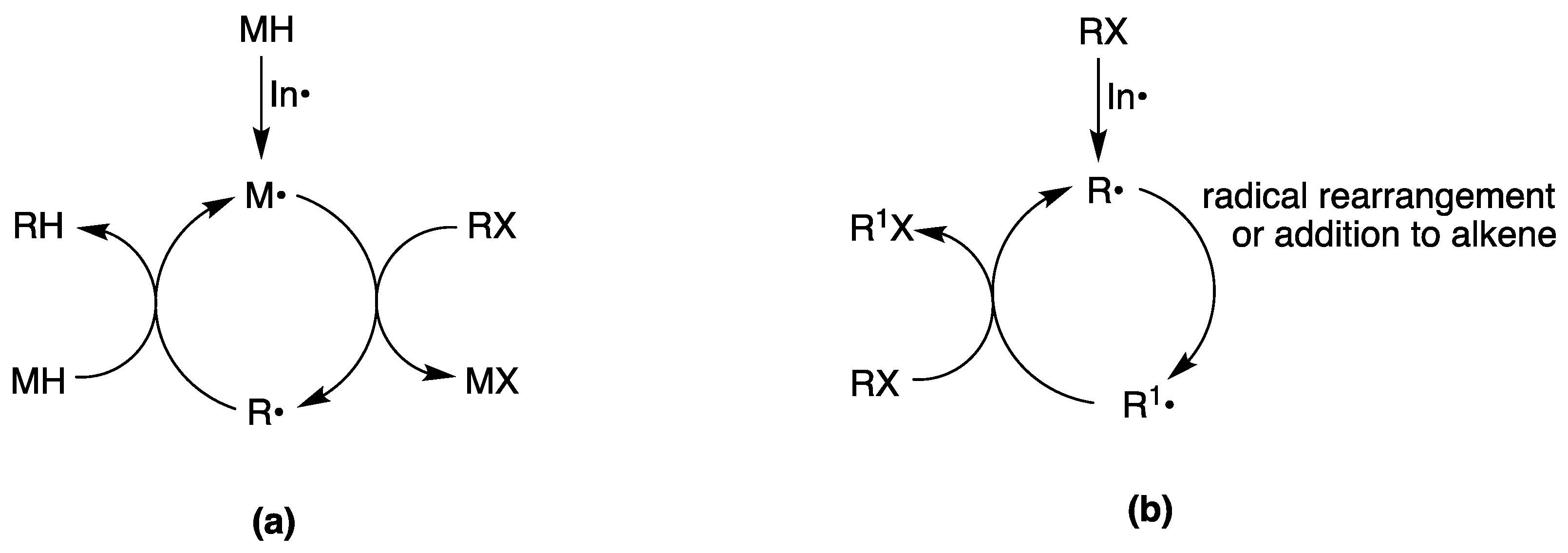
Figure 4.
Radical-based reactions using H3PO2 or Et2P(O)H as reducing reagents in aqueous environment.
Figure 4.
Radical-based reactions using H3PO2 or Et2P(O)H as reducing reagents in aqueous environment.
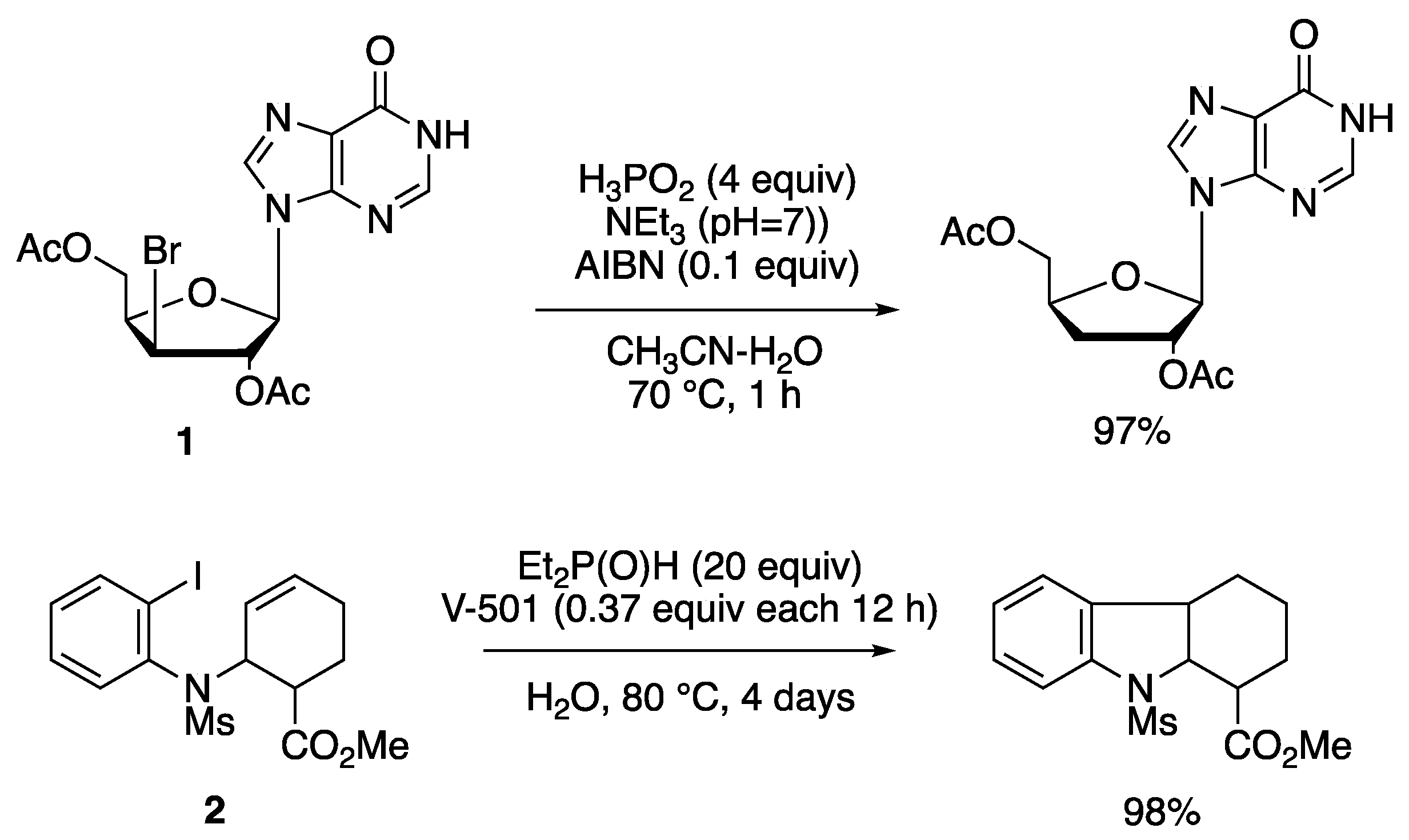
Figure 5.
(a) Silyl radical-mediated C–H alkylation of heterocycles with non-activated alkyl bromides. (b) Silyl radical-mediated protocol for the fluorination of secondary alkyl bromides.
Figure 5.
(a) Silyl radical-mediated C–H alkylation of heterocycles with non-activated alkyl bromides. (b) Silyl radical-mediated protocol for the fluorination of secondary alkyl bromides.
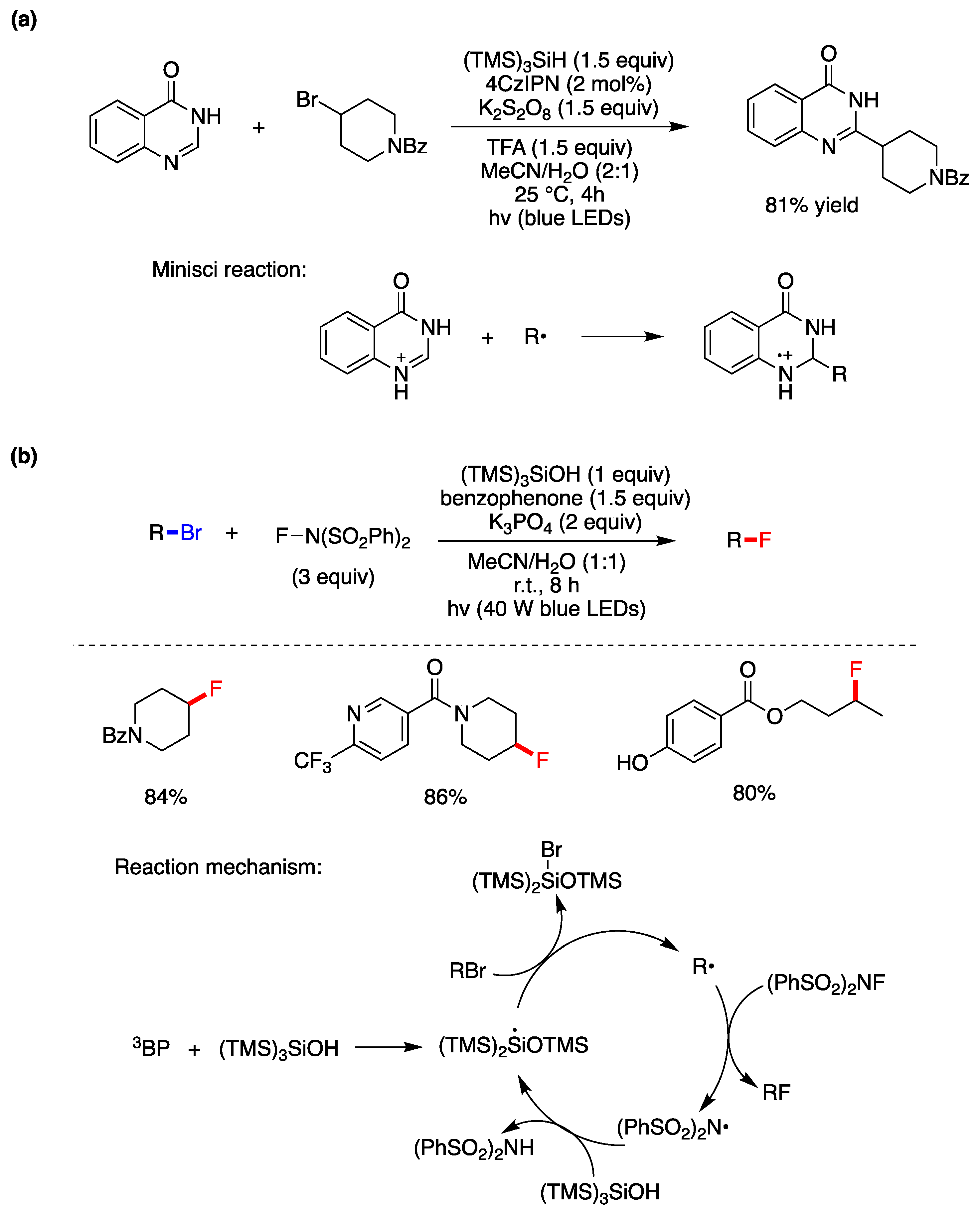
Figure 6.
(a) Two examples of halogen atom-transfer in the intramolecular and intermolecular C–C bond formation, respectively. (b) Synthesis of ketones from alkenylsilanes under radical conditions with air as the oxidant.
Figure 6.
(a) Two examples of halogen atom-transfer in the intramolecular and intermolecular C–C bond formation, respectively. (b) Synthesis of ketones from alkenylsilanes under radical conditions with air as the oxidant.
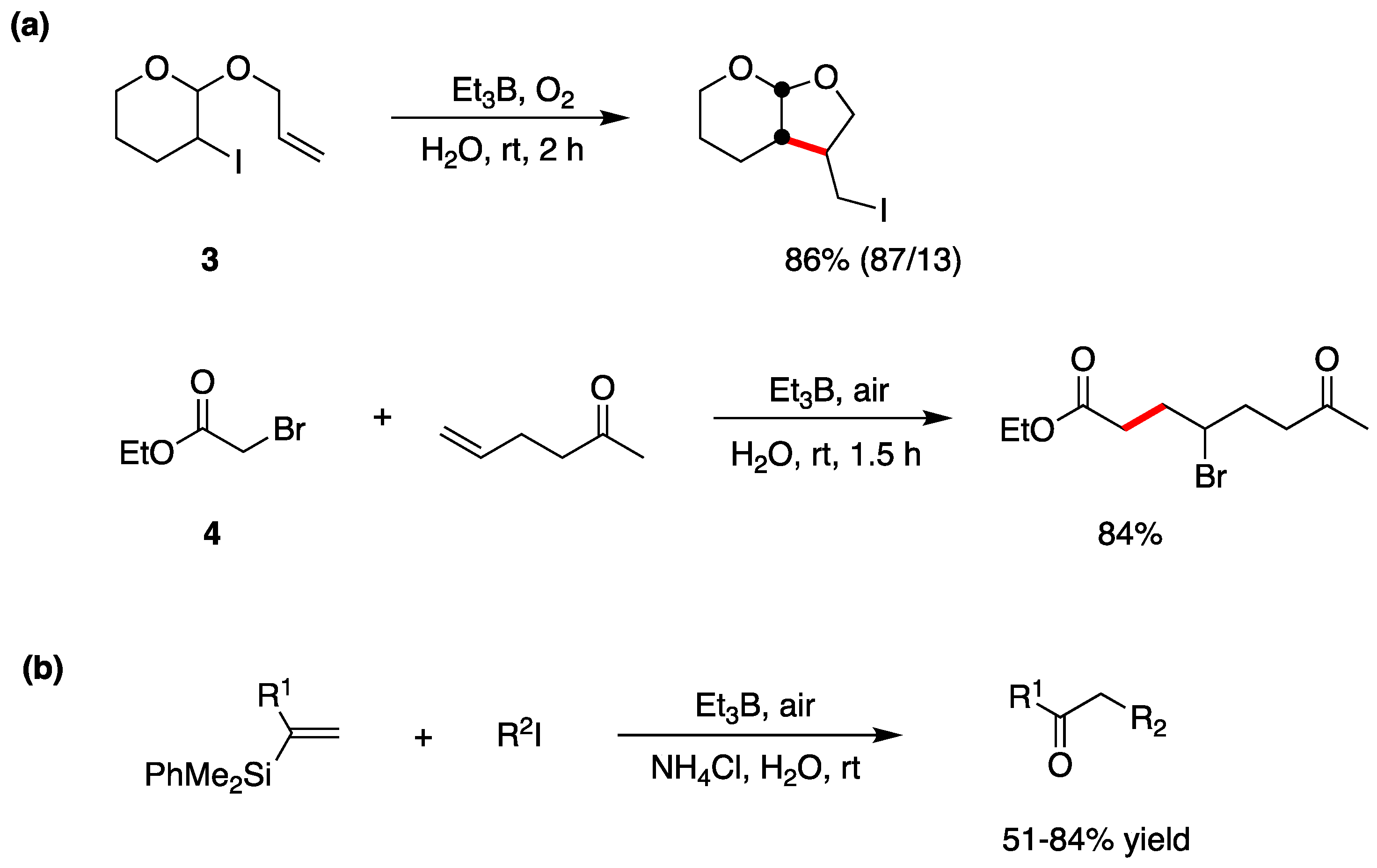
Figure 7.
A light-driven, copper-mediated site-selective trifluoromethylation for C(sp3)−CF3 formation in aqueous solution: (a) Starting from alkyl bromides and Et3SiH; (b) starting from O-alkyl thiocarbonates and (TMS)3SiH; (c) Starting from benzylic C–H and i-Pr3SiH; bpp =2,2’-bipyridine.
Figure 7.
A light-driven, copper-mediated site-selective trifluoromethylation for C(sp3)−CF3 formation in aqueous solution: (a) Starting from alkyl bromides and Et3SiH; (b) starting from O-alkyl thiocarbonates and (TMS)3SiH; (c) Starting from benzylic C–H and i-Pr3SiH; bpp =2,2’-bipyridine.
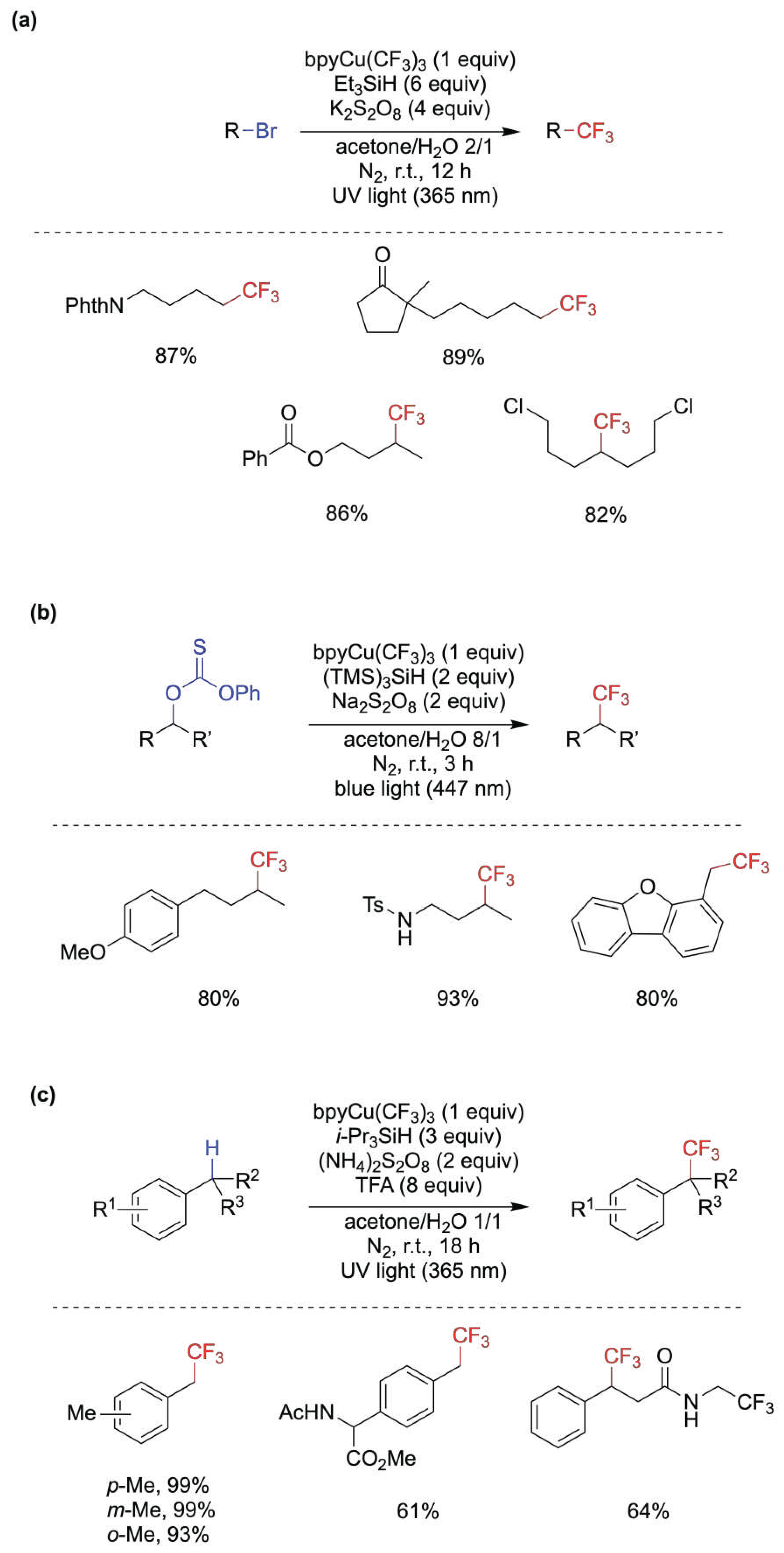
Figure 8.
Copper-catalyzed trifluoromethylation using CF3SO2Na: (a) the mechanism of CF3• generation from CF3SO2Na and tert-butyl hydroperoxide (TBHP), and the addition products of two heterocycles where the yields refer to 1 g scale; (b) trifluoromethylation of aryl and heteroaryl boronic acids; (c) decarboxylative trifluoromethylation of cinnamic acids; (d) synthesis of a variety of CF3-containing oxindoles from N-arylacrylamides ‘‘on water’’ at room temperature.
Figure 8.
Copper-catalyzed trifluoromethylation using CF3SO2Na: (a) the mechanism of CF3• generation from CF3SO2Na and tert-butyl hydroperoxide (TBHP), and the addition products of two heterocycles where the yields refer to 1 g scale; (b) trifluoromethylation of aryl and heteroaryl boronic acids; (c) decarboxylative trifluoromethylation of cinnamic acids; (d) synthesis of a variety of CF3-containing oxindoles from N-arylacrylamides ‘‘on water’’ at room temperature.
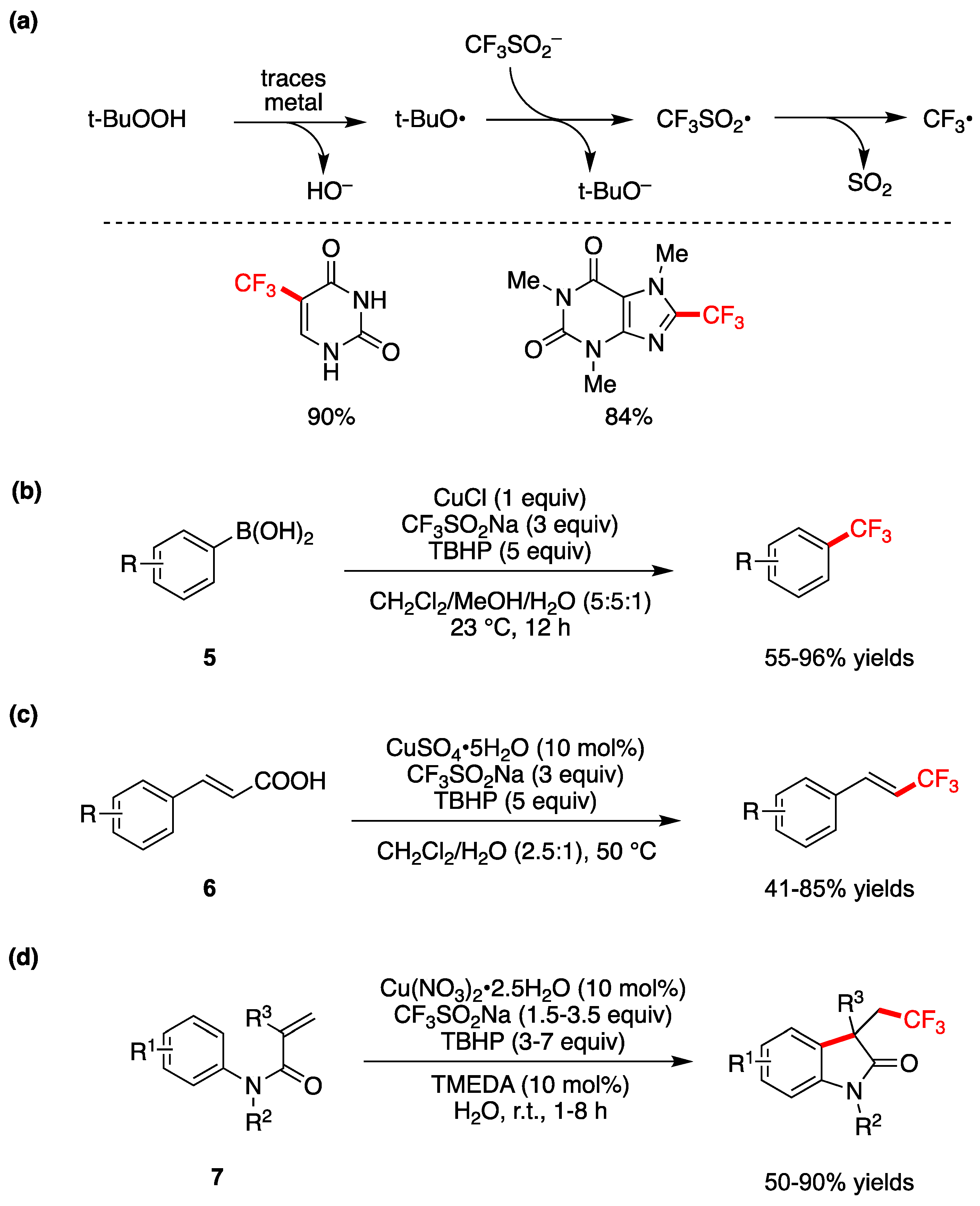
Figure 9.
Aldehyde oxidation and tandem aldehyde oxidation/Passerini reaction “on water”.
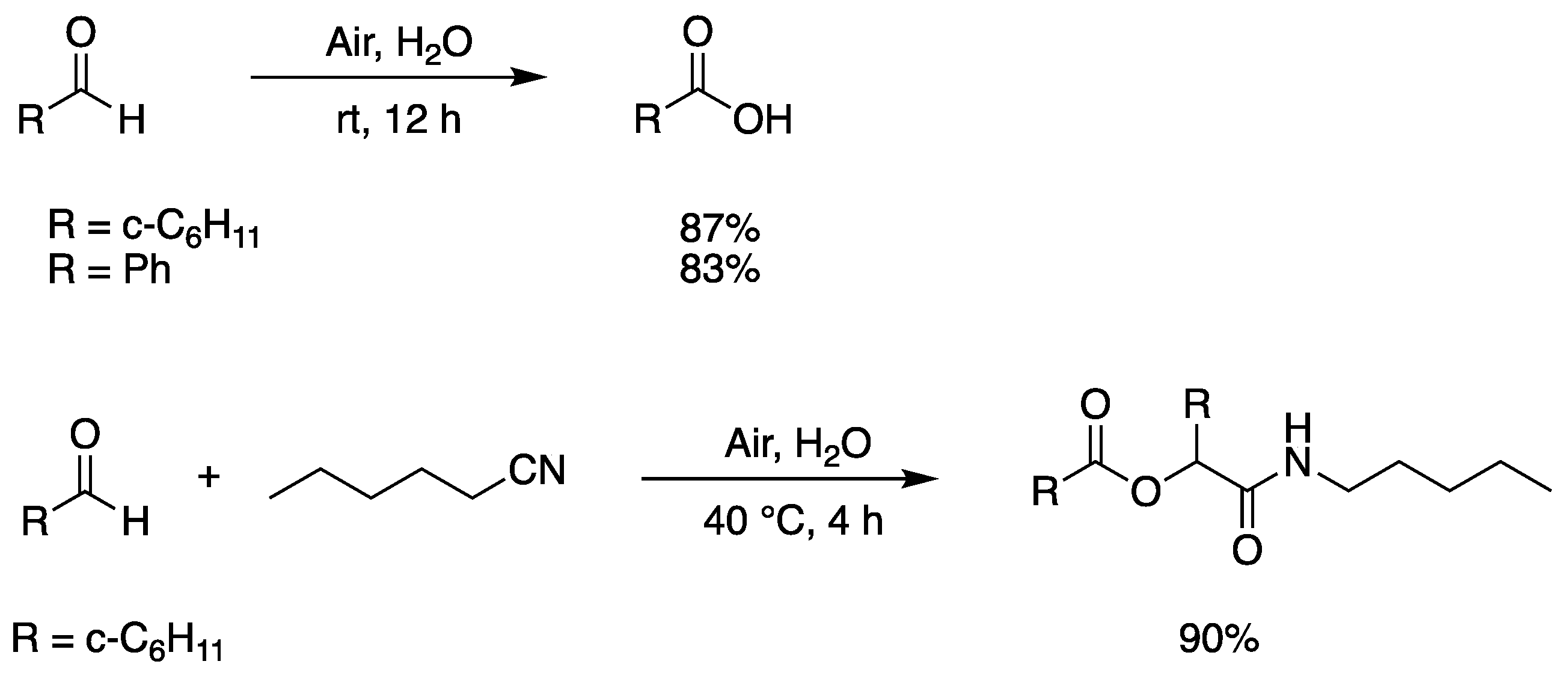
Figure 10.
Photochemical hydroacylation of unactivated olefins "on water".
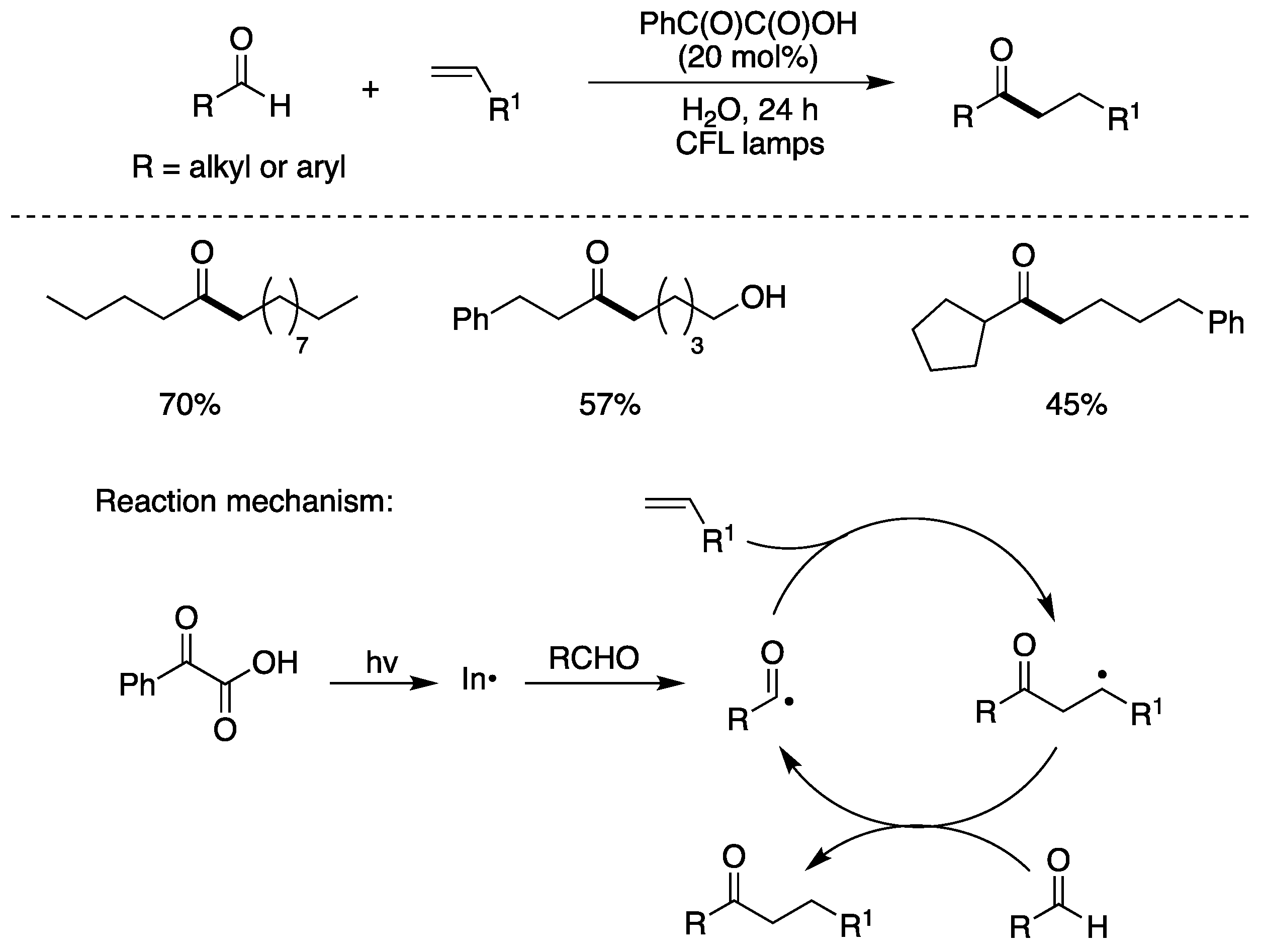
Figure 11.
The use of (TMS)3SiH as reducing agent in aqueous environment: (a) two different experimental approaches depending on the substrate being water-insoluble or water-soluble where an amphiphilic thiol is needed; (b) Mechanism of reduction of a functional Group (X = atom or group) by the (TMS)3SiH/HOCH2CH2SH couple, where the hydrogen atom donor to R• is the thiol.
Figure 11.
The use of (TMS)3SiH as reducing agent in aqueous environment: (a) two different experimental approaches depending on the substrate being water-insoluble or water-soluble where an amphiphilic thiol is needed; (b) Mechanism of reduction of a functional Group (X = atom or group) by the (TMS)3SiH/HOCH2CH2SH couple, where the hydrogen atom donor to R• is the thiol.
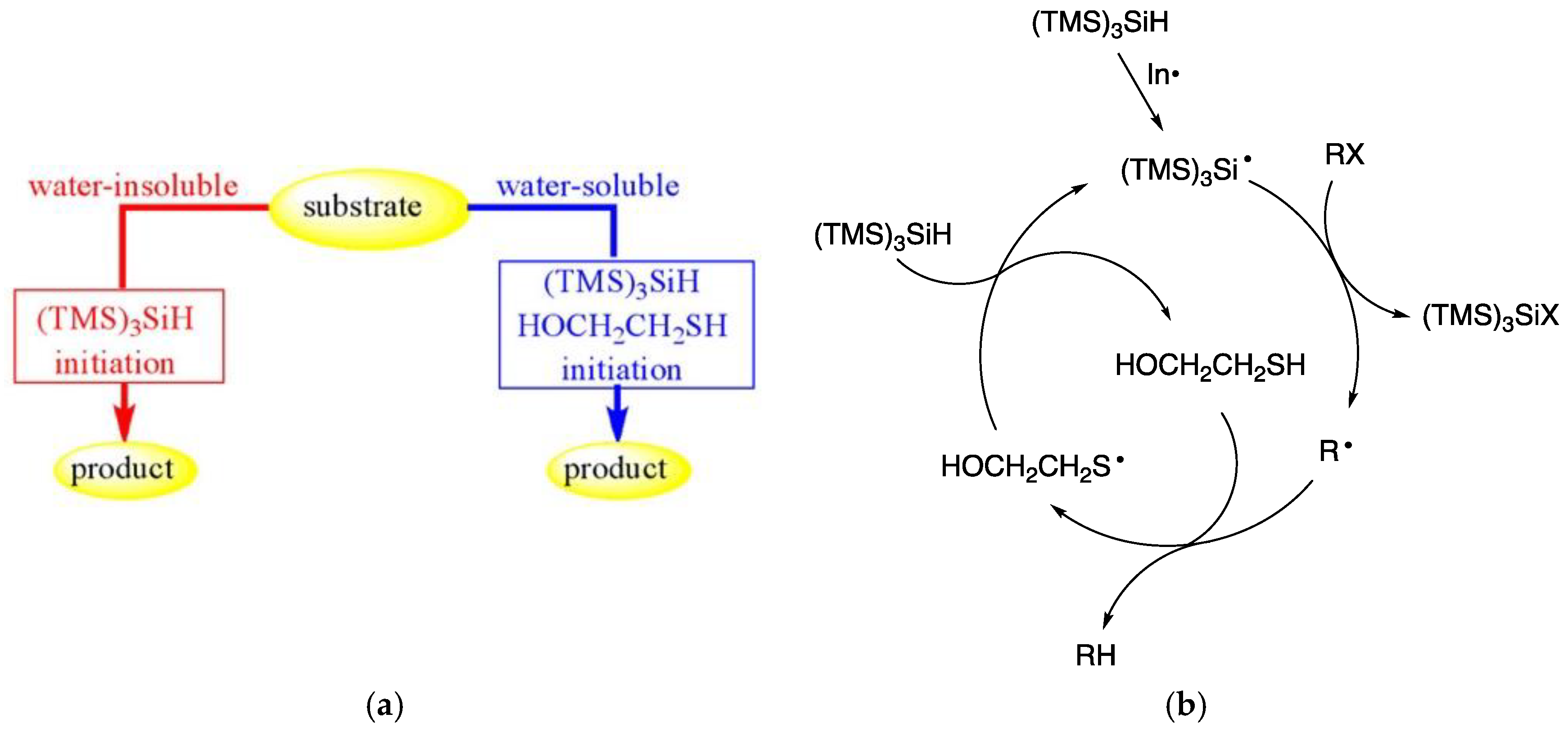
Figure 12.
Radical reduction of a variety of compounds by the couple (TMS)3SiH/ HOCH2CH2SH.
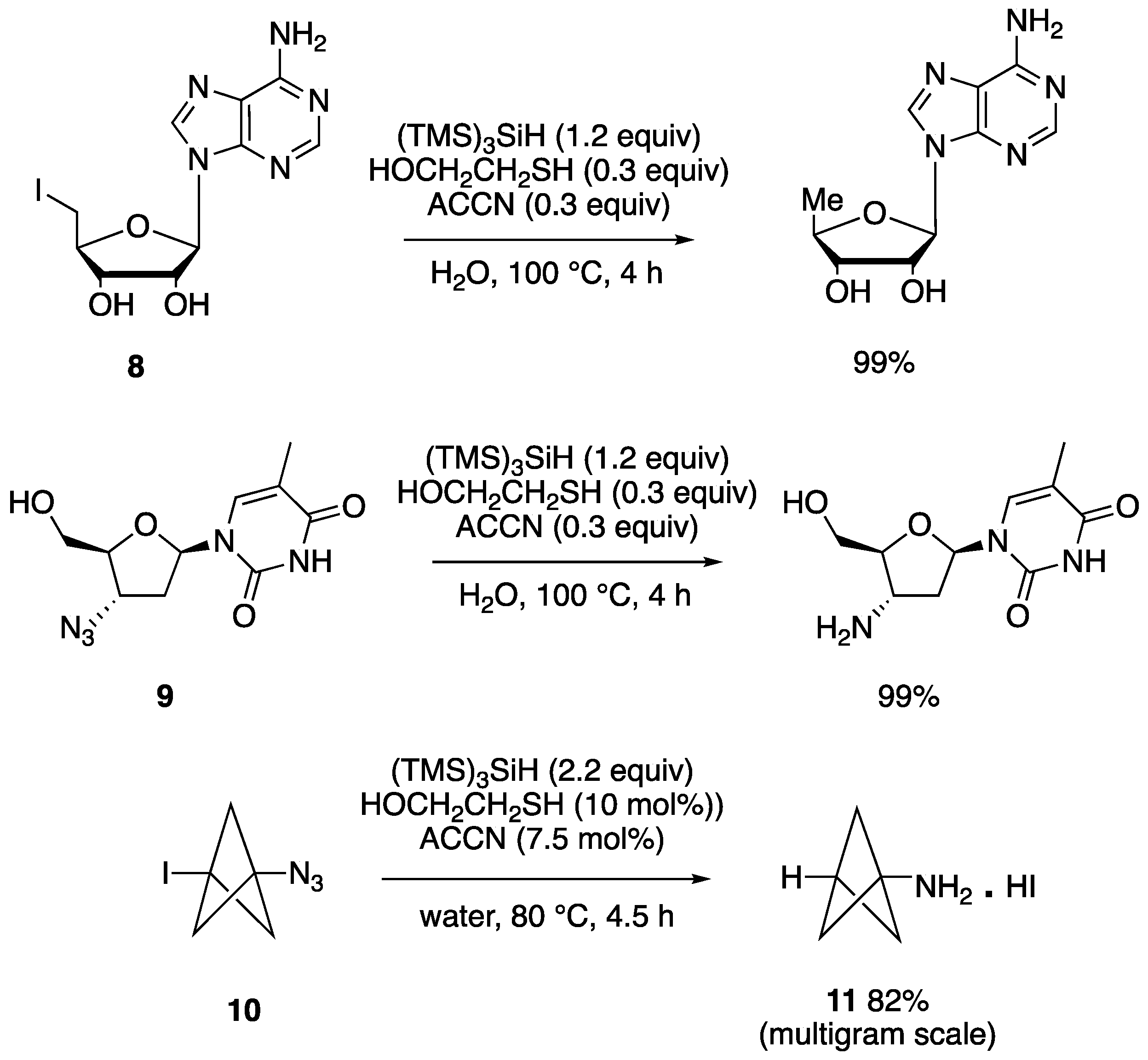
Figure 13.
Carbon-carbon bond formation mediated by (TMS)3SiH: (a) perfluoroalkylation of olefins; (b) Synthesis of a nitrogen-containing heterocycle.
Figure 13.
Carbon-carbon bond formation mediated by (TMS)3SiH: (a) perfluoroalkylation of olefins; (b) Synthesis of a nitrogen-containing heterocycle.
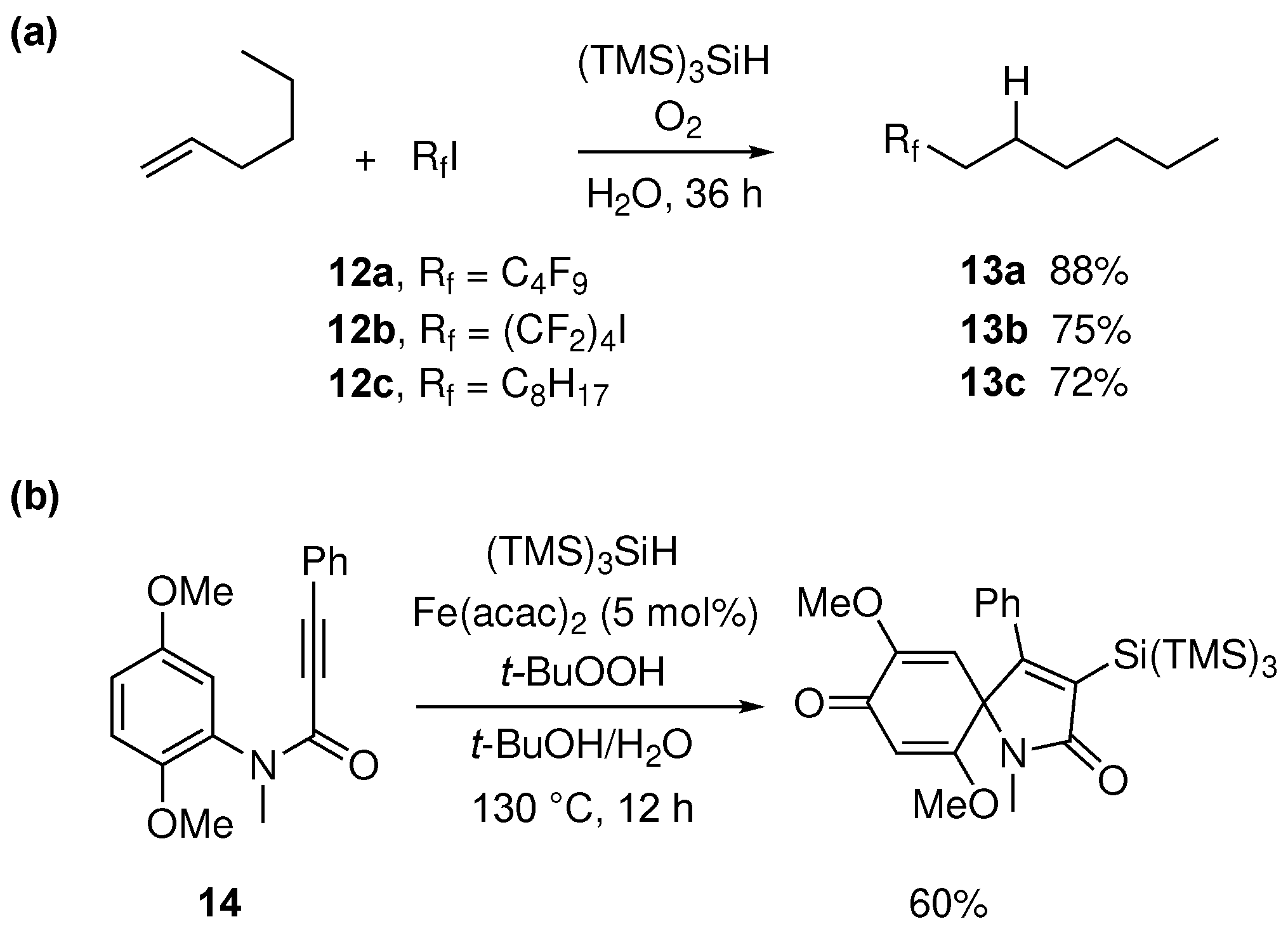
Figure 14.
(a) An example of synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. (b) comparison of three protocols using (TMS)3SiH or the water-soluble 1-ethylpiperidine hypophosphite (EPHP) or H3PO2/NaHCO3, respectively. (c) Radical addition to the C=N bond in hydrazone derivatives.
Figure 14.
(a) An example of synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. (b) comparison of three protocols using (TMS)3SiH or the water-soluble 1-ethylpiperidine hypophosphite (EPHP) or H3PO2/NaHCO3, respectively. (c) Radical addition to the C=N bond in hydrazone derivatives.
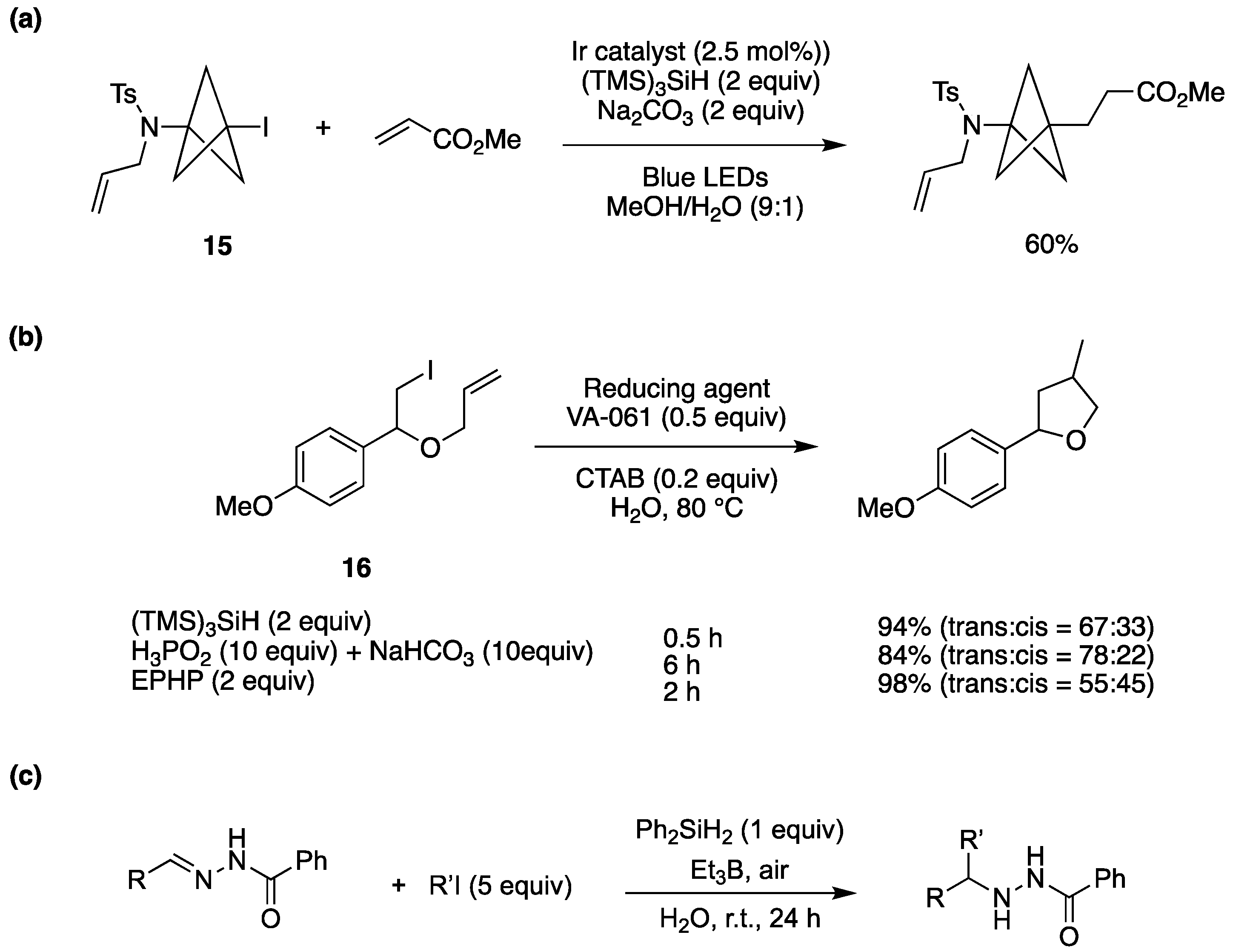
Figure 15.
Thioxanthate radical deoxygenation by J. L. Wood and coworkers and proposed water “activation” [91].
Figure 15.
Thioxanthate radical deoxygenation by J. L. Wood and coworkers and proposed water “activation” [91].
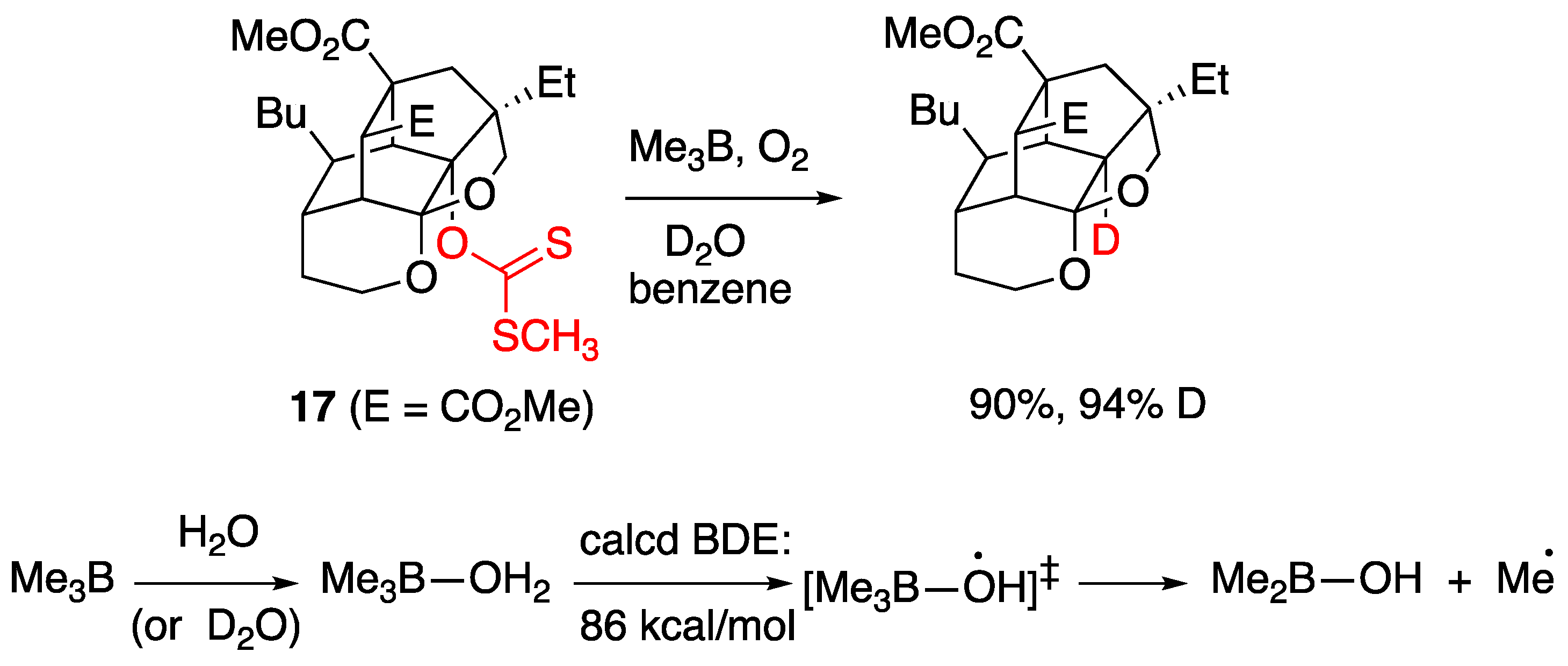
Figure 16.
Mechanistic proposal for the MeO–H bond weakening [99].
Figure 16.
Mechanistic proposal for the MeO–H bond weakening [99].
Figure 17.
(a) Valence bond representation of common-features TSs accessed from three different classes of reactions. Though these are radical reactions, the TS features a Lewis acid/base interaction between O and B. (b) Three chains interlock with the reduction deiodination chain and with each other [50].
Figure 17.
(a) Valence bond representation of common-features TSs accessed from three different classes of reactions. Though these are radical reactions, the TS features a Lewis acid/base interaction between O and B. (b) Three chains interlock with the reduction deiodination chain and with each other [50].
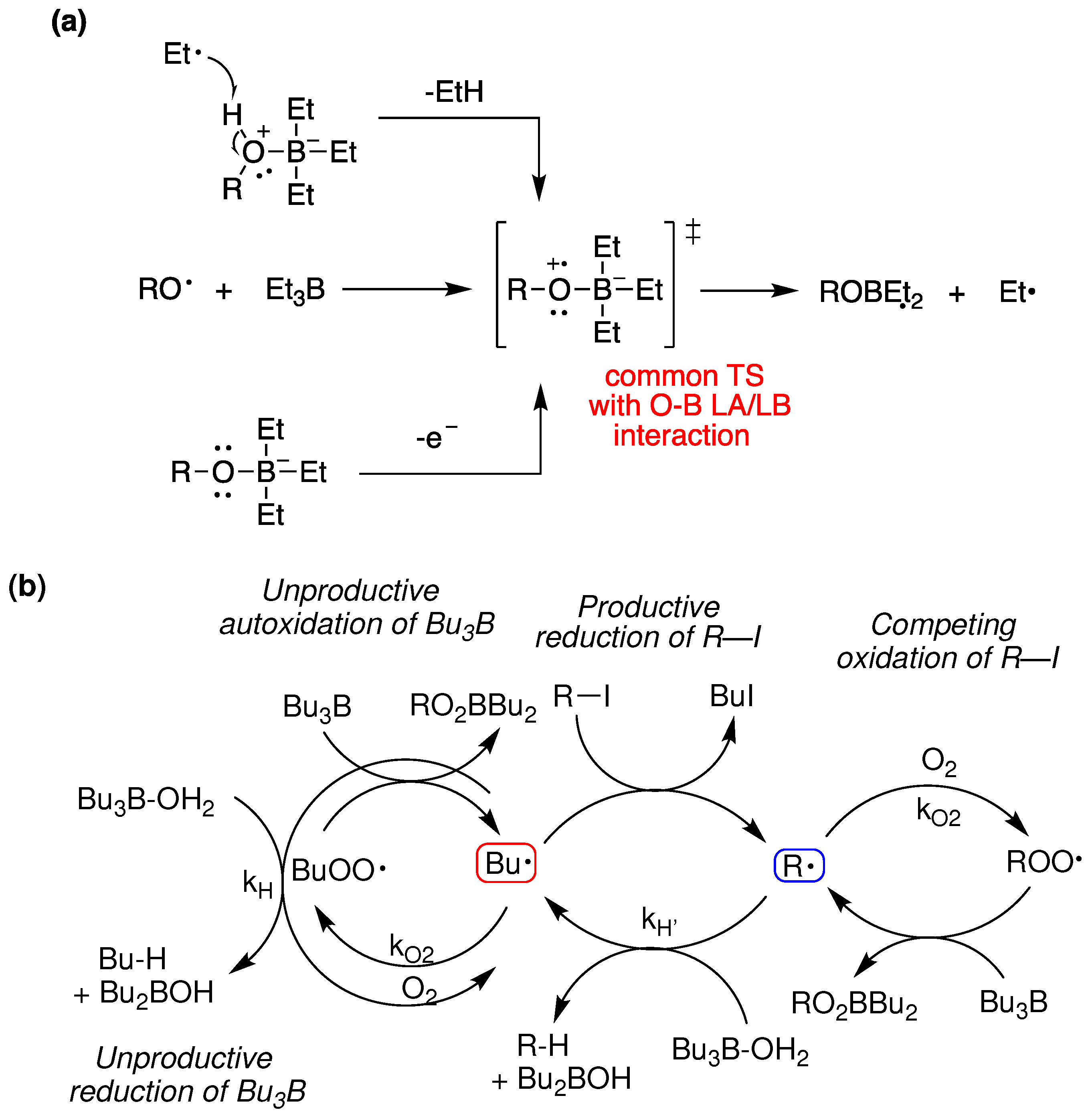
Figure 18.
Revised mechanism for the deoxygenation of xanthate employing D2O as the deuterium atom source [92].
Figure 18.
Revised mechanism for the deoxygenation of xanthate employing D2O as the deuterium atom source [92].
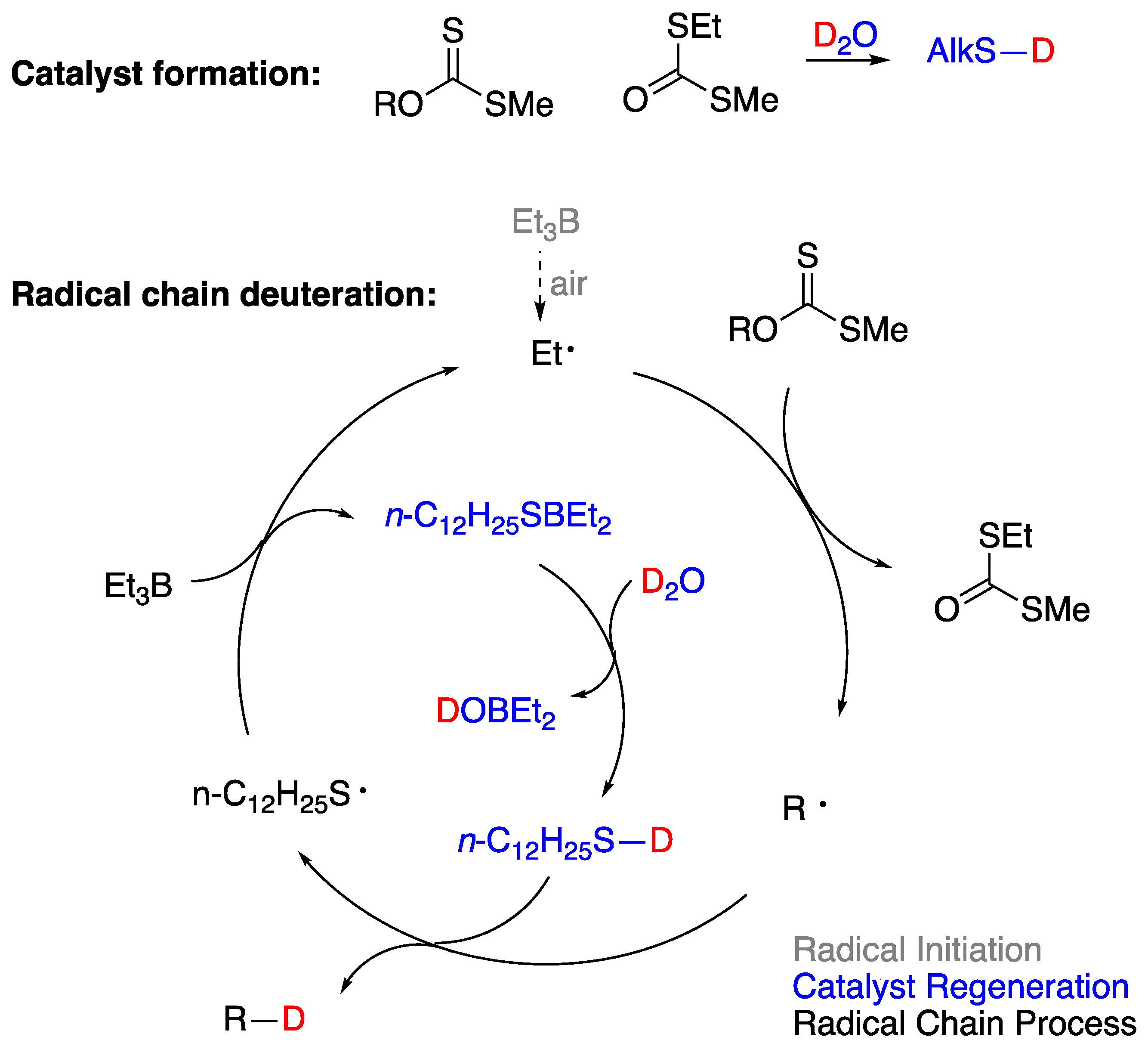
Figure 19.
(a) Remote site-selective radical C(sp3 )−H monodeuteration of amides and selected examples. (b) Proposed mechanism [93].
Figure 19.
(a) Remote site-selective radical C(sp3 )−H monodeuteration of amides and selected examples. (b) Proposed mechanism [93].
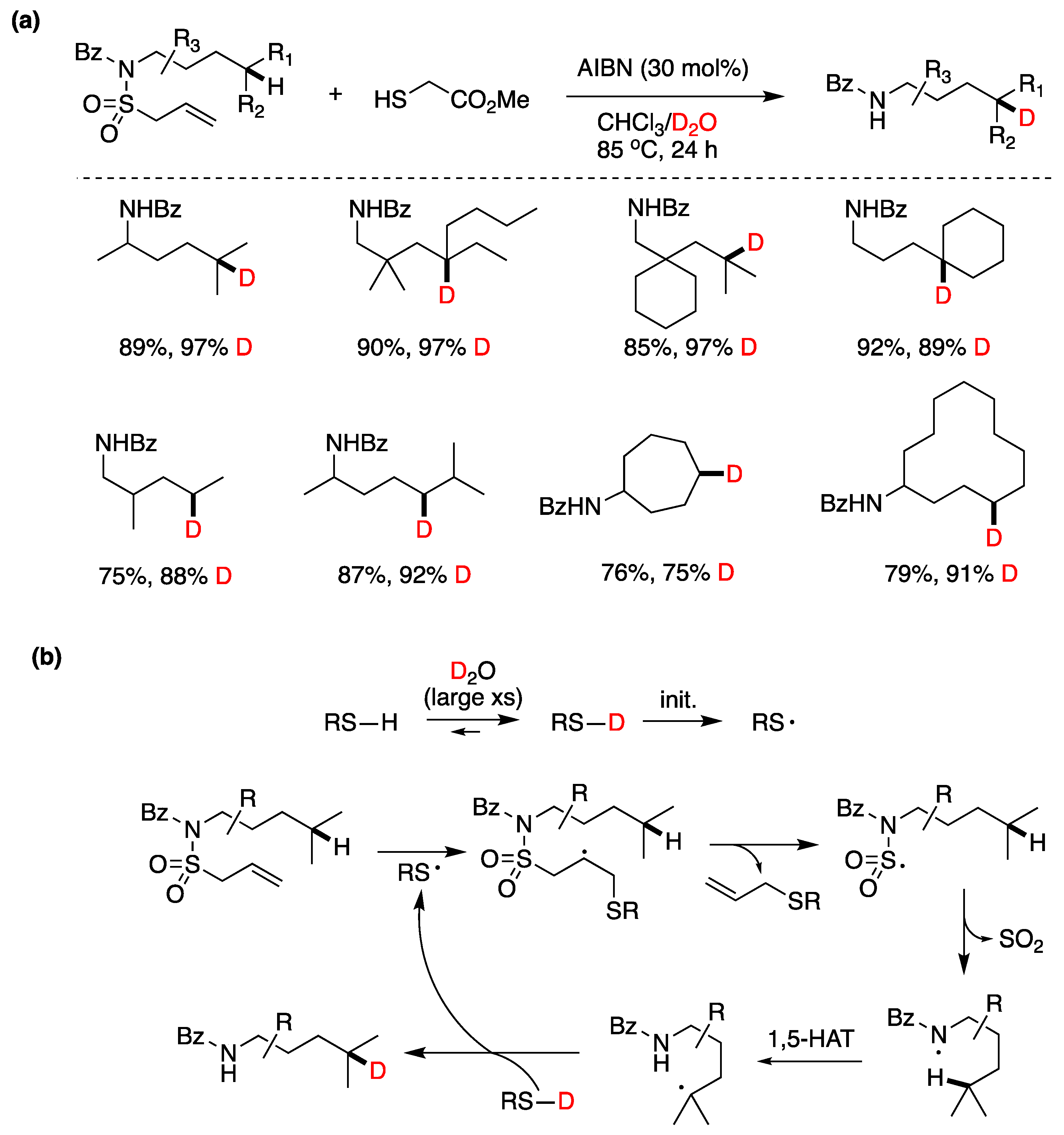
Figure 20.
Synthesis of deuterated (E)-alkenes [107].
Figure 20.
Synthesis of deuterated (E)-alkenes [107].
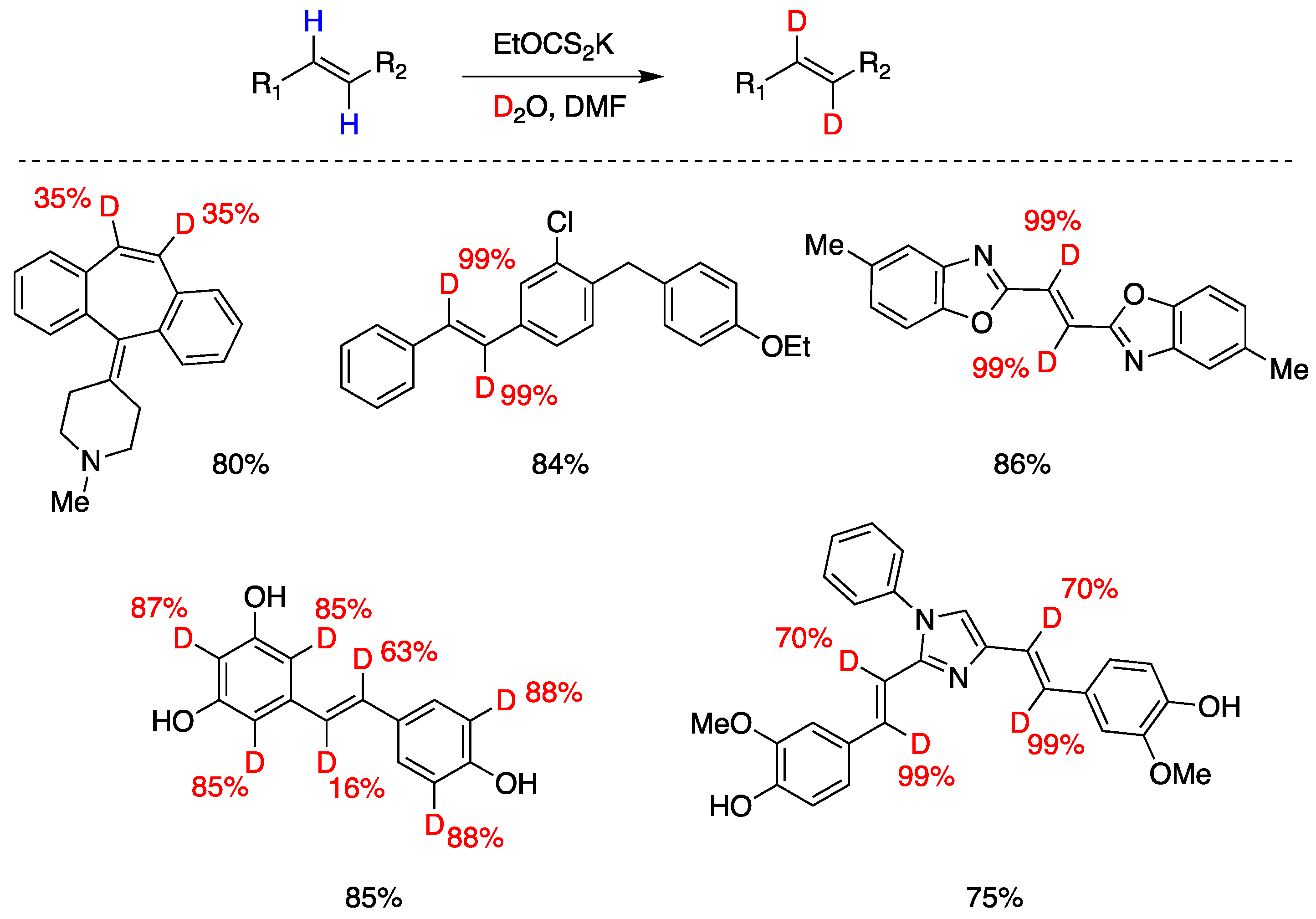
Figure 21.
(a) Calculated BDE for the O-H bond of water complexed with Cp2Ti(III)Cl [88]; (b) Active reductant forms according to ESR and theoretical calculations [112]; (c) proposed mechanism and intermediates [88].
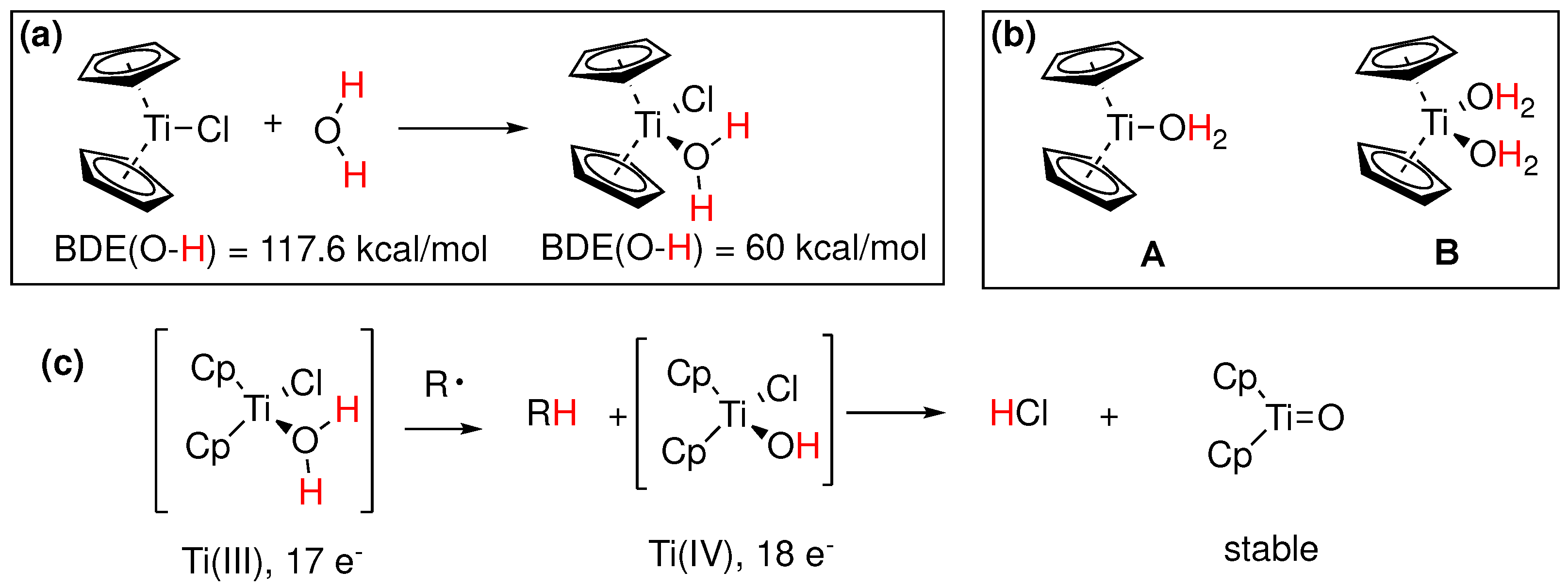
Figure 22.
Proposed mechanism in a tricyclic system [114].
Figure 22.
Proposed mechanism in a tricyclic system [114].
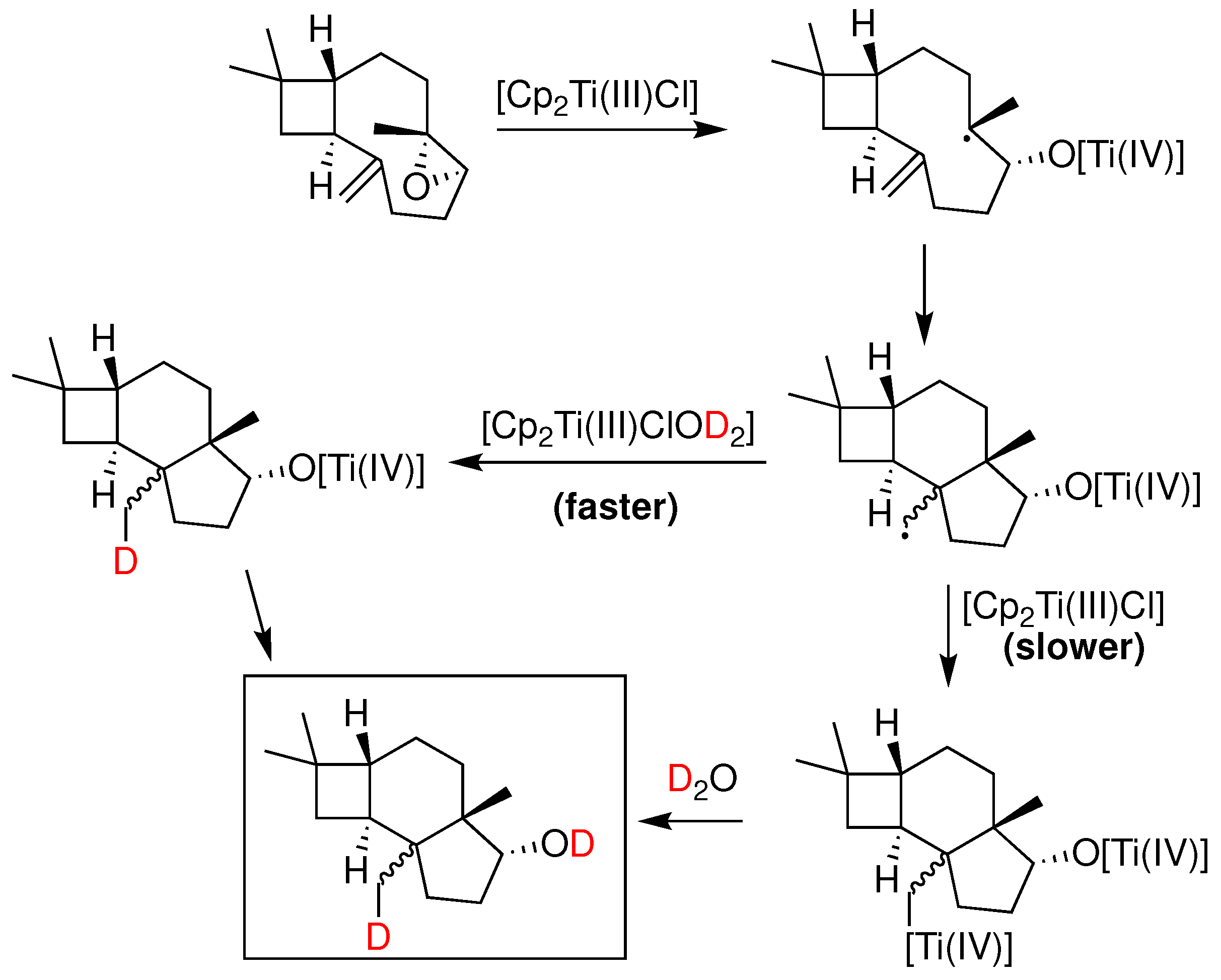
Figure 23.
Revised mechanism for the Titanocene(III)/Mn-promoted reduction of ketones in aqueous media [114].
Figure 23.
Revised mechanism for the Titanocene(III)/Mn-promoted reduction of ketones in aqueous media [114].
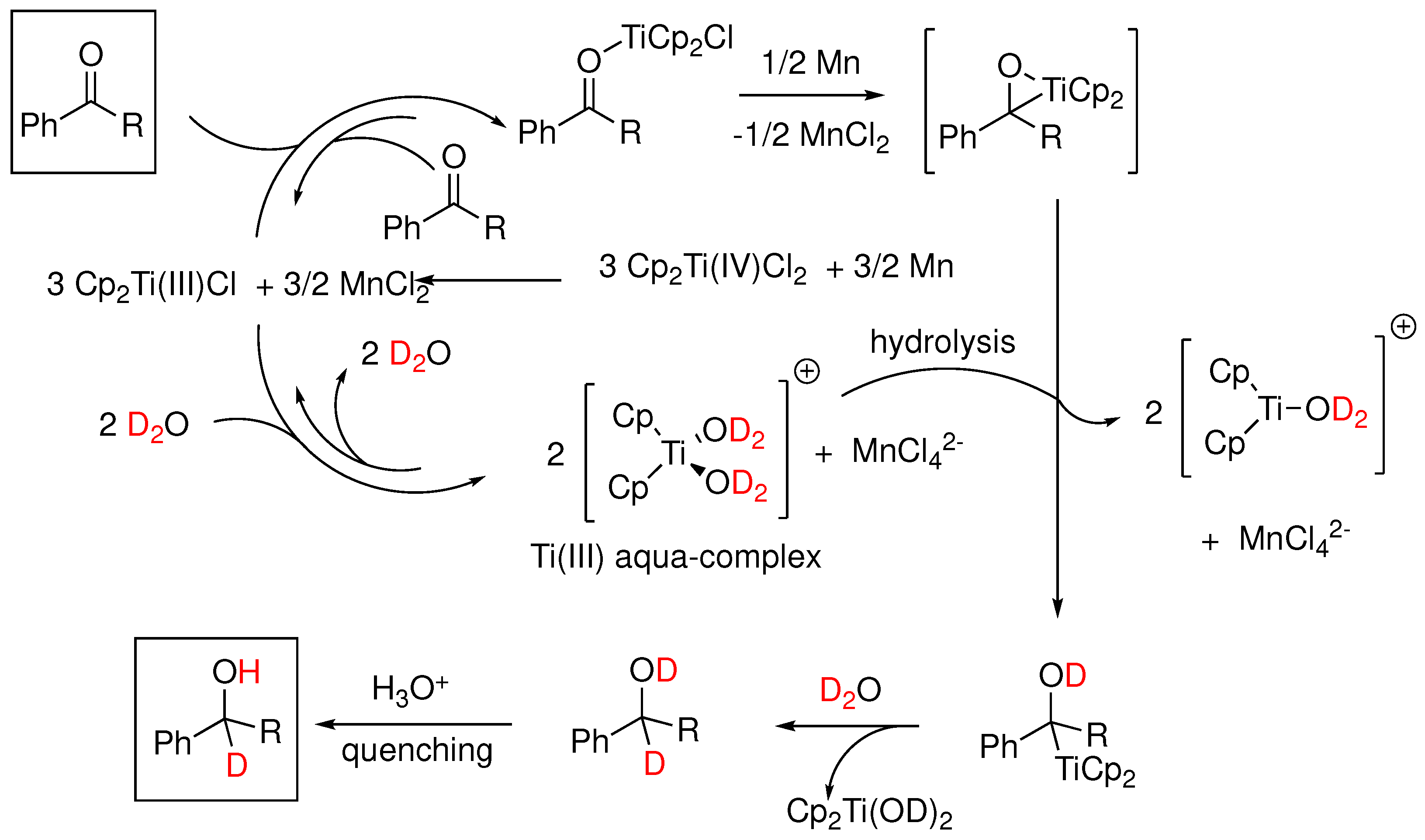
Figure 24.
Proposed reduction of a ketone by Sm(II)I2-water proceeding through a highly ordered transition state.
Figure 24.
Proposed reduction of a ketone by Sm(II)I2-water proceeding through a highly ordered transition state.
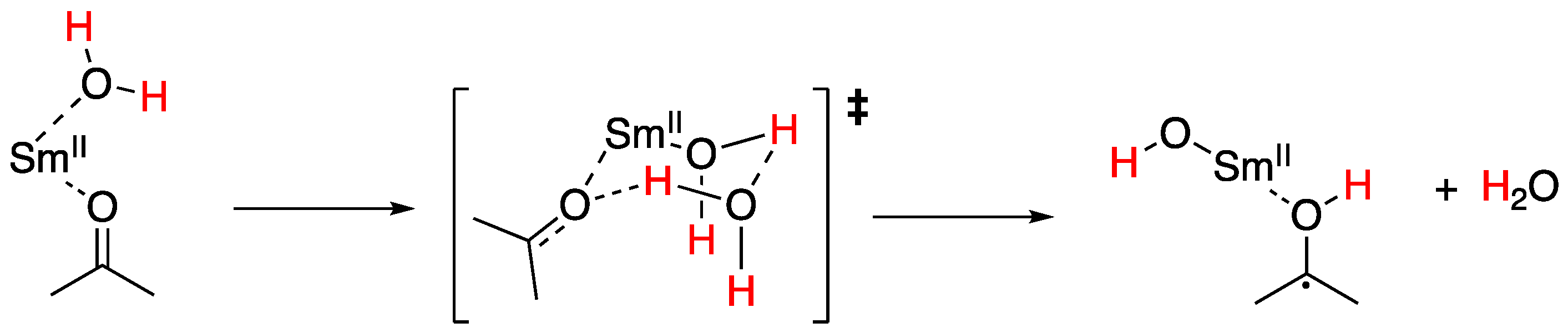
Figure 25.
(a) Reaction Scheme and Substrate Scope for the SmI2-Water-Mediated Reduction of Enamines. (b) Proposed PCET and HAT Mechanisms for the Reduction of Enamines by SmI2-H2O [89].
Figure 25.
(a) Reaction Scheme and Substrate Scope for the SmI2-Water-Mediated Reduction of Enamines. (b) Proposed PCET and HAT Mechanisms for the Reduction of Enamines by SmI2-H2O [89].
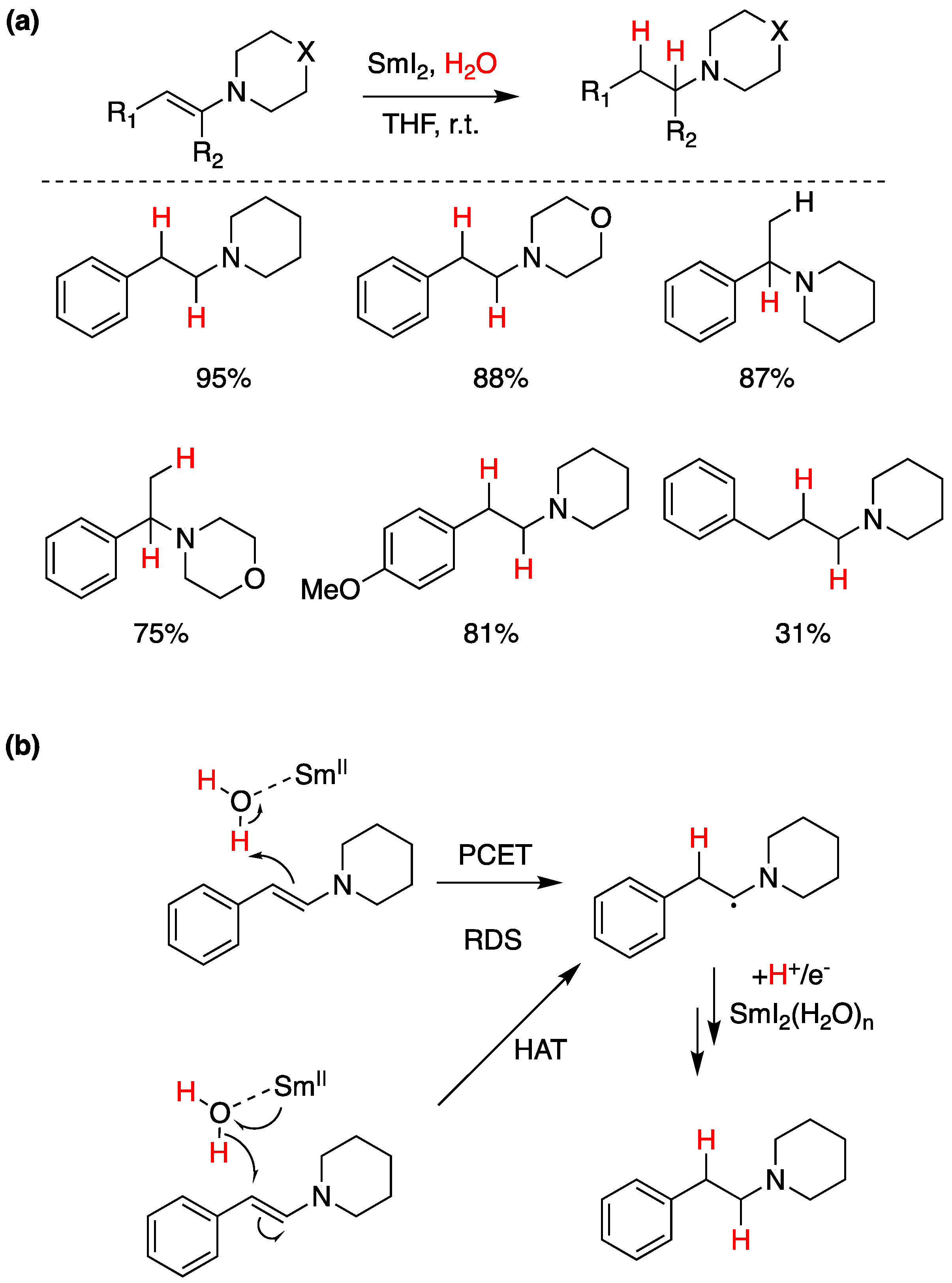
Figure 26.
Spontaneous hydrogen evolution from a molybdenum Mo-aquo complex [95].
Figure 26.
Spontaneous hydrogen evolution from a molybdenum Mo-aquo complex [95].
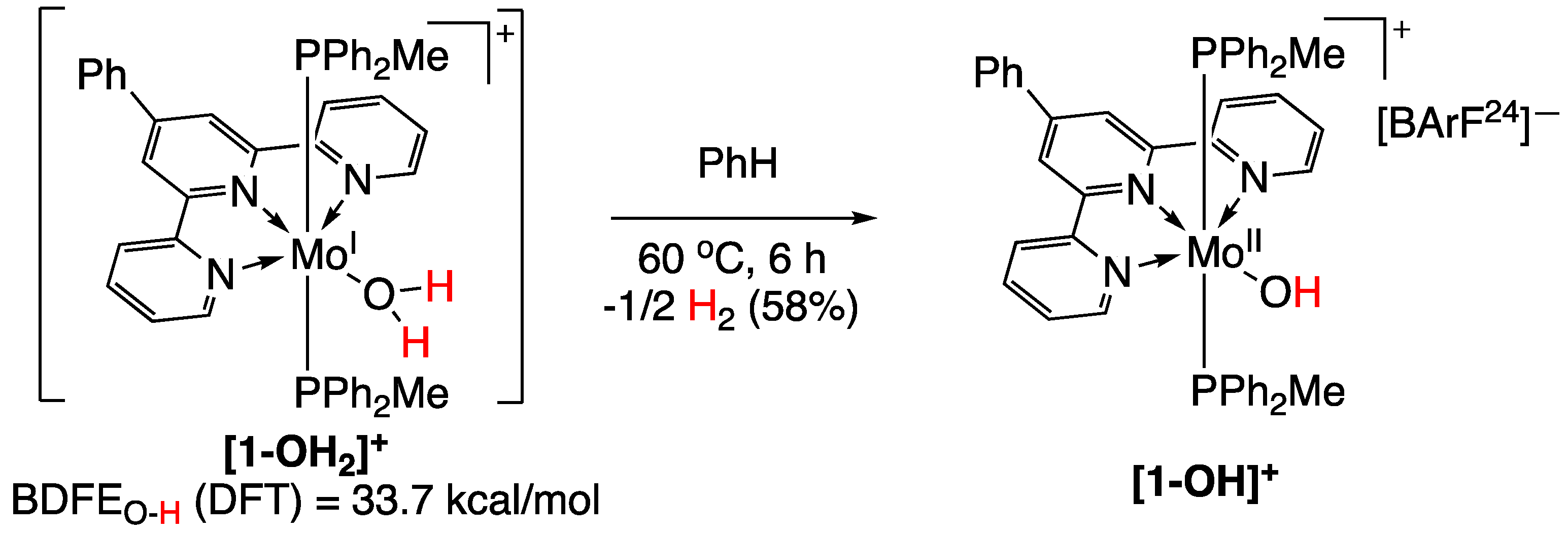
Figure 27.
Reaction of a germanium corrole complex with water [94].
Figure 27.
Reaction of a germanium corrole complex with water [94].
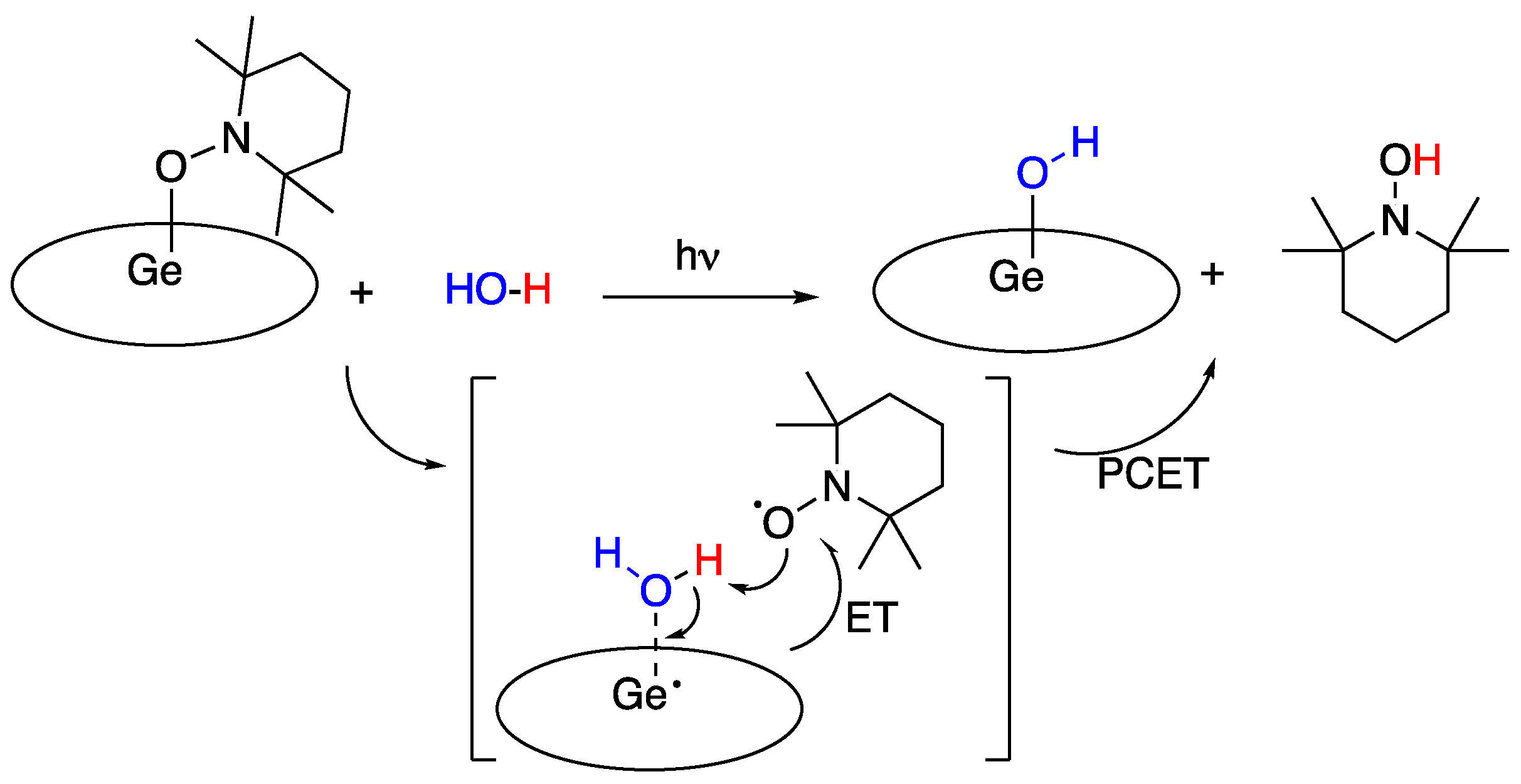
Figure 28.
Reaction of H2O with a Bi(II) complex generates H2 and a Bi(III) hydroxy complex [96].
Figure 28.
Reaction of H2O with a Bi(II) complex generates H2 and a Bi(III) hydroxy complex [96].
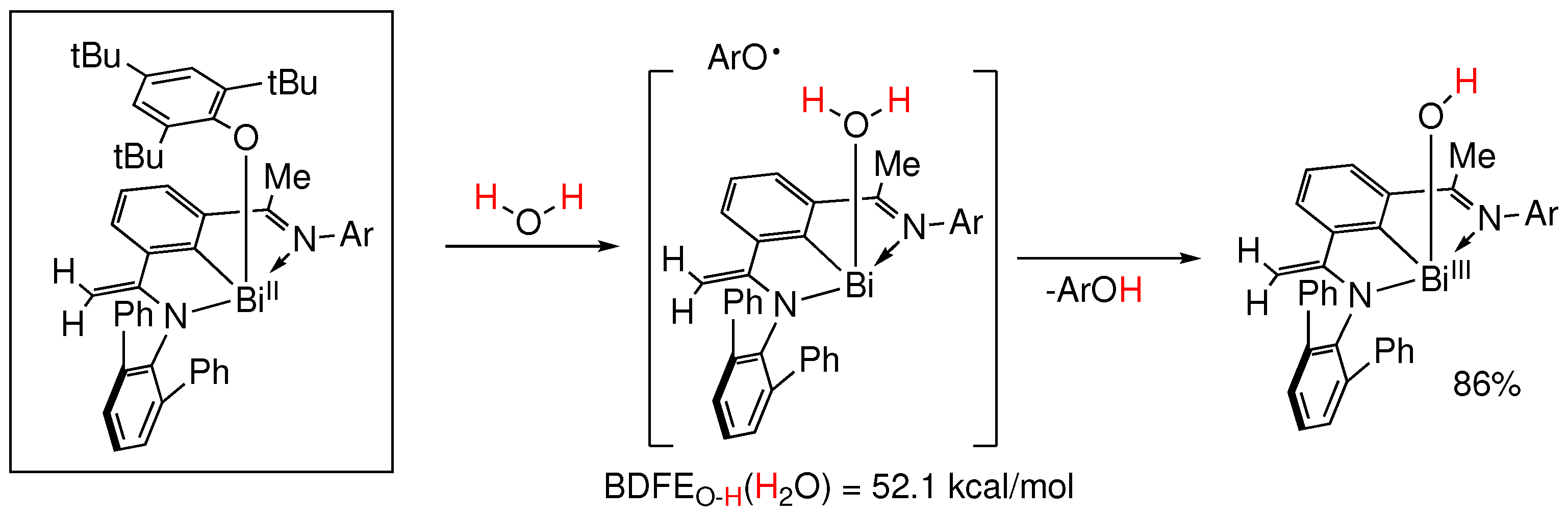
Figure 29.
(a) Hydroxytrifluoroethylation of styrenes by photoredox catalysis in aqueous media and representative examples; (b) Proposed reaction mechanism.
Figure 29.
(a) Hydroxytrifluoroethylation of styrenes by photoredox catalysis in aqueous media and representative examples; (b) Proposed reaction mechanism.
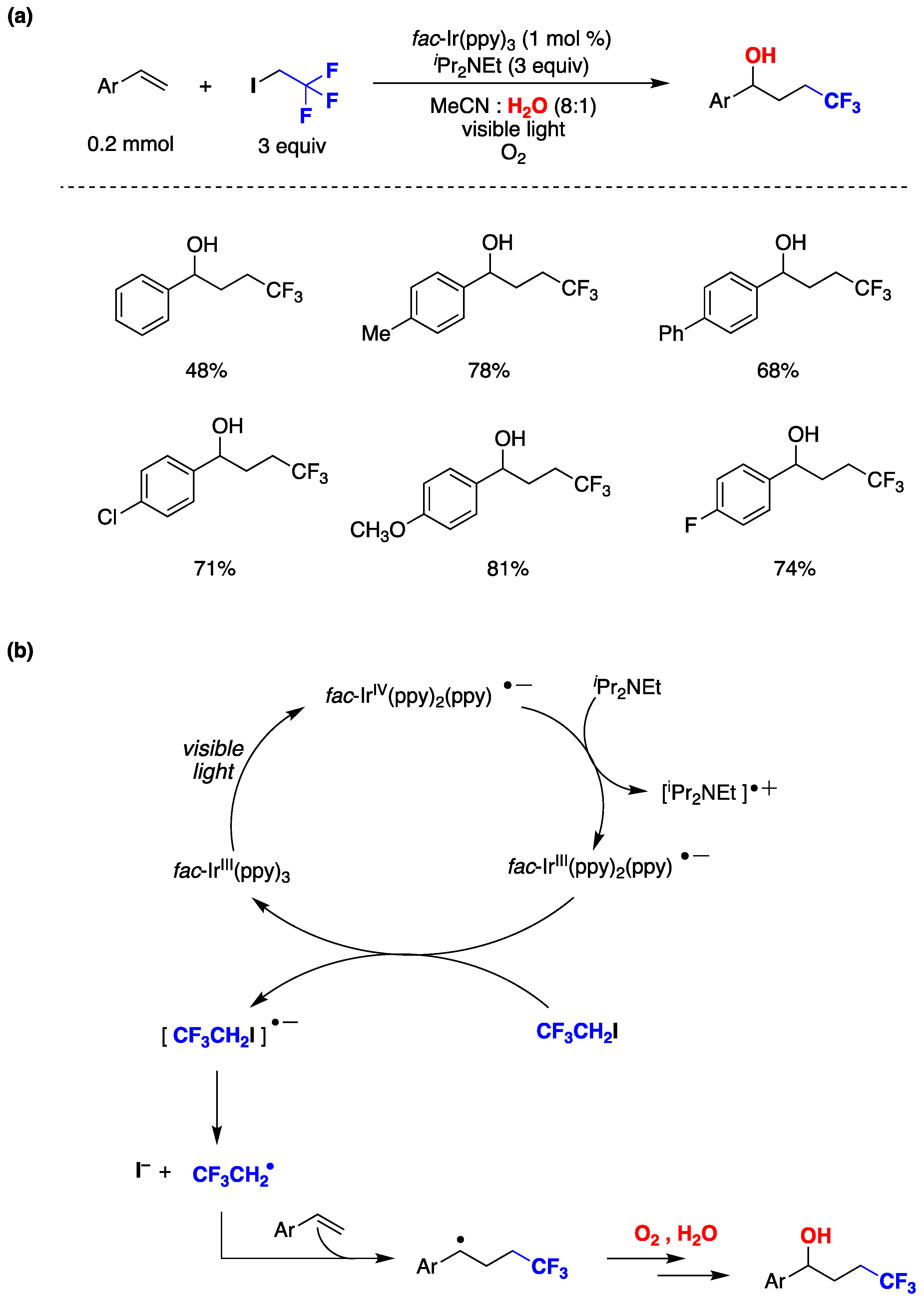
Figure 30.
(a) Arylation of styrene by photoredox catalysis in MeCN:H2O as reaction media and representative examples; (b) Proposed reaction mechanism.
Figure 30.
(a) Arylation of styrene by photoredox catalysis in MeCN:H2O as reaction media and representative examples; (b) Proposed reaction mechanism.
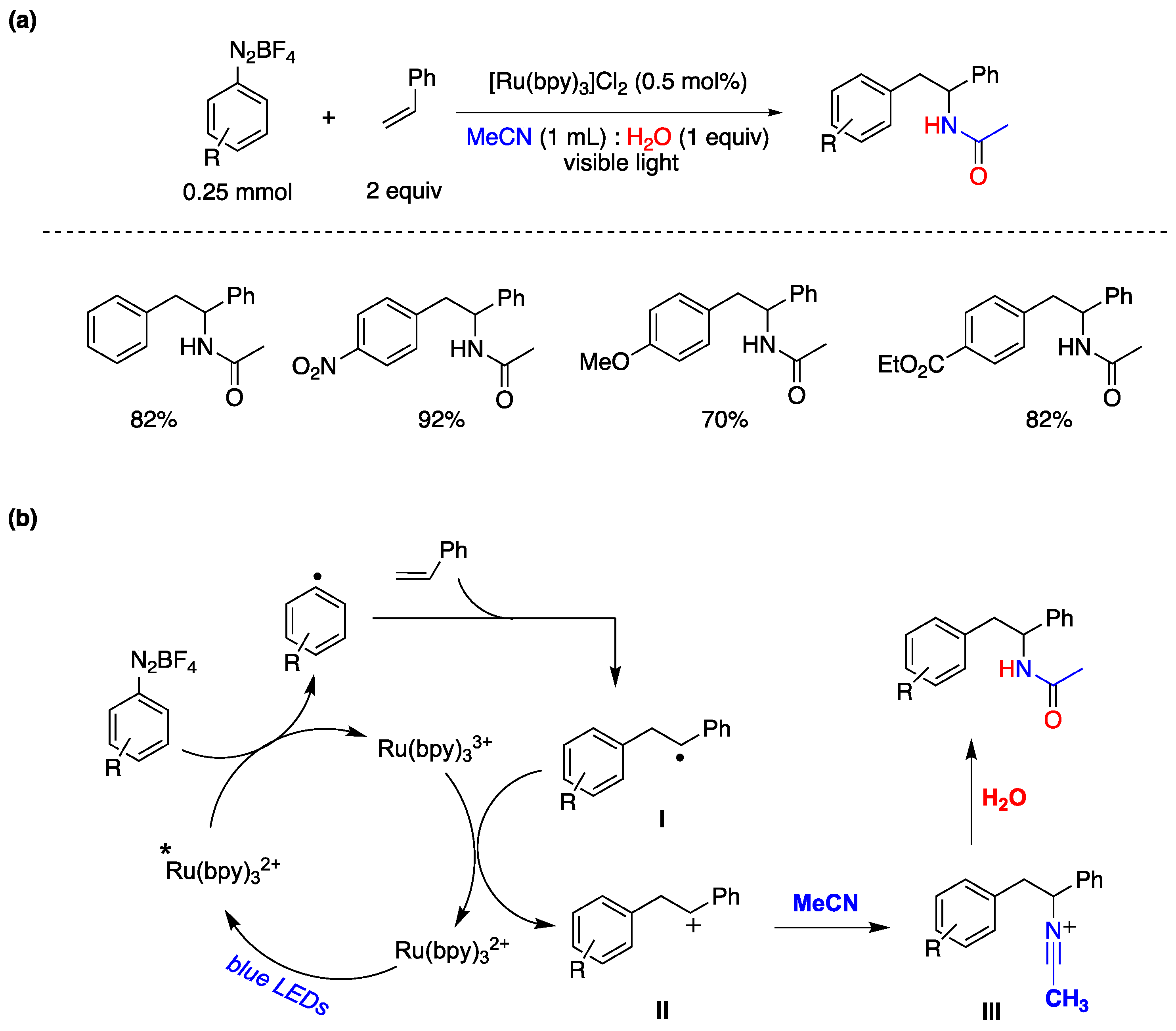
Figure 31.
(a) Synthesis of aromatic ketones by deoxygenative carbon-carbon coupling of aryl-carboxylic acids with olefins in aqueous phase and representative examples; (b) Proposed reaction mechanism.
Figure 31.
(a) Synthesis of aromatic ketones by deoxygenative carbon-carbon coupling of aryl-carboxylic acids with olefins in aqueous phase and representative examples; (b) Proposed reaction mechanism.
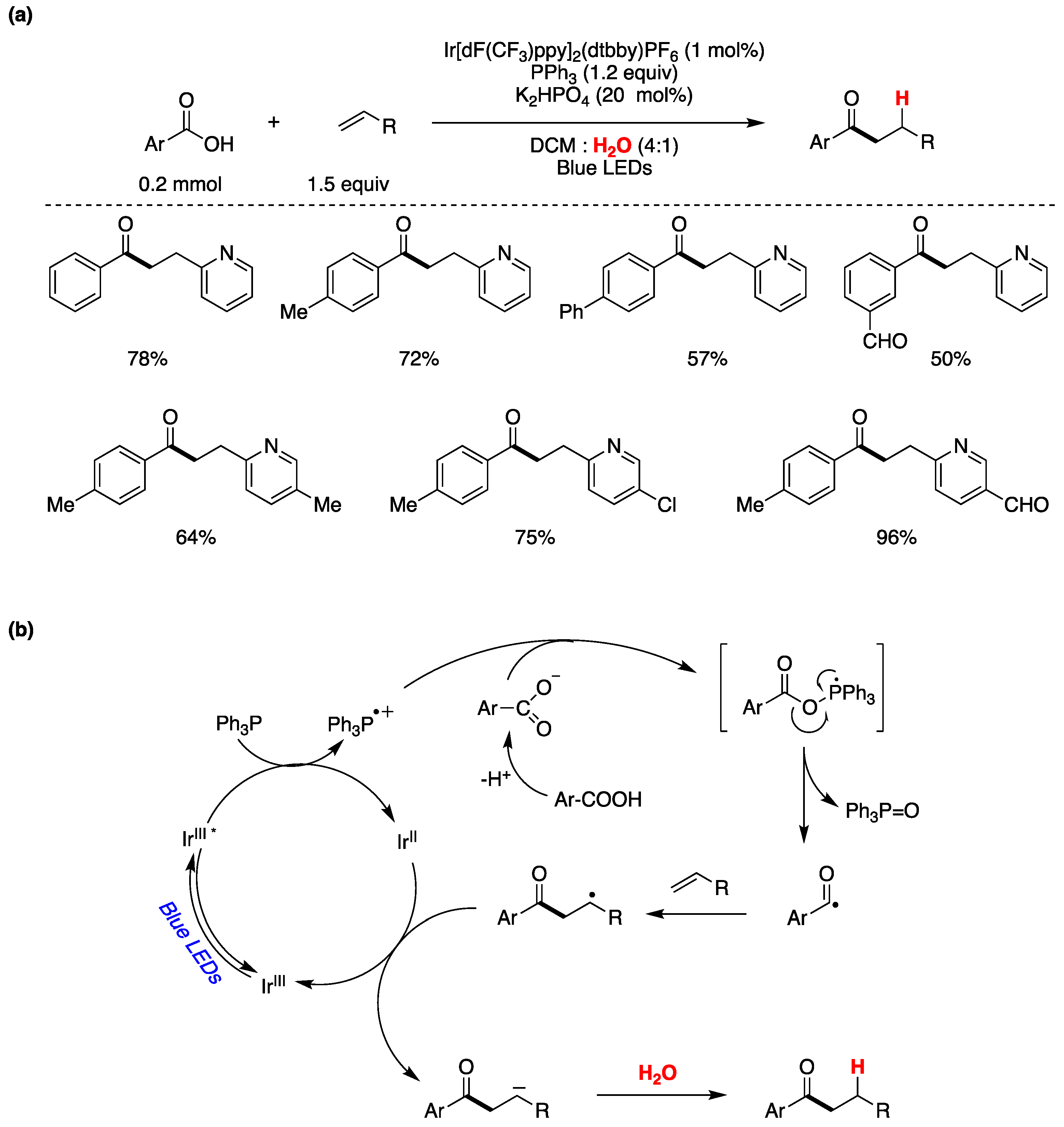
Figure 32.
(a) Intermolecular atom transfer radical addition (ATRA) to olefins by photoredox catalysis in aqueous media and representative examples, (a) without LiBr; (b) Proposed reaction mechanism.
Figure 32.
(a) Intermolecular atom transfer radical addition (ATRA) to olefins by photoredox catalysis in aqueous media and representative examples, (a) without LiBr; (b) Proposed reaction mechanism.
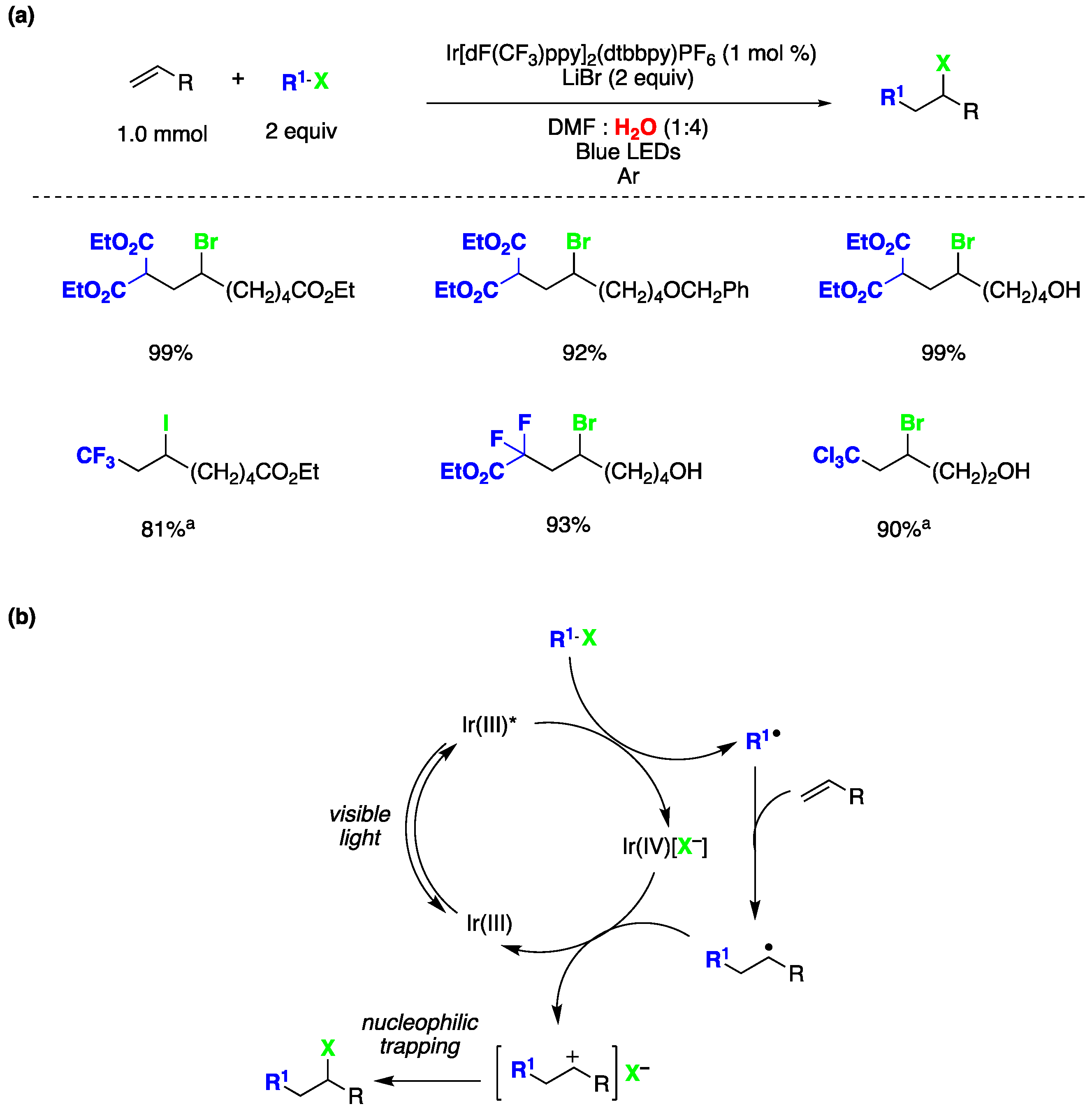
Figure 33.
Representative examples for the photocatalyzed iodoperfluorohexylation of olefins and alkynes in water; (a) in MeOH:H2O (1:2).
Figure 33.
Representative examples for the photocatalyzed iodoperfluorohexylation of olefins and alkynes in water; (a) in MeOH:H2O (1:2).
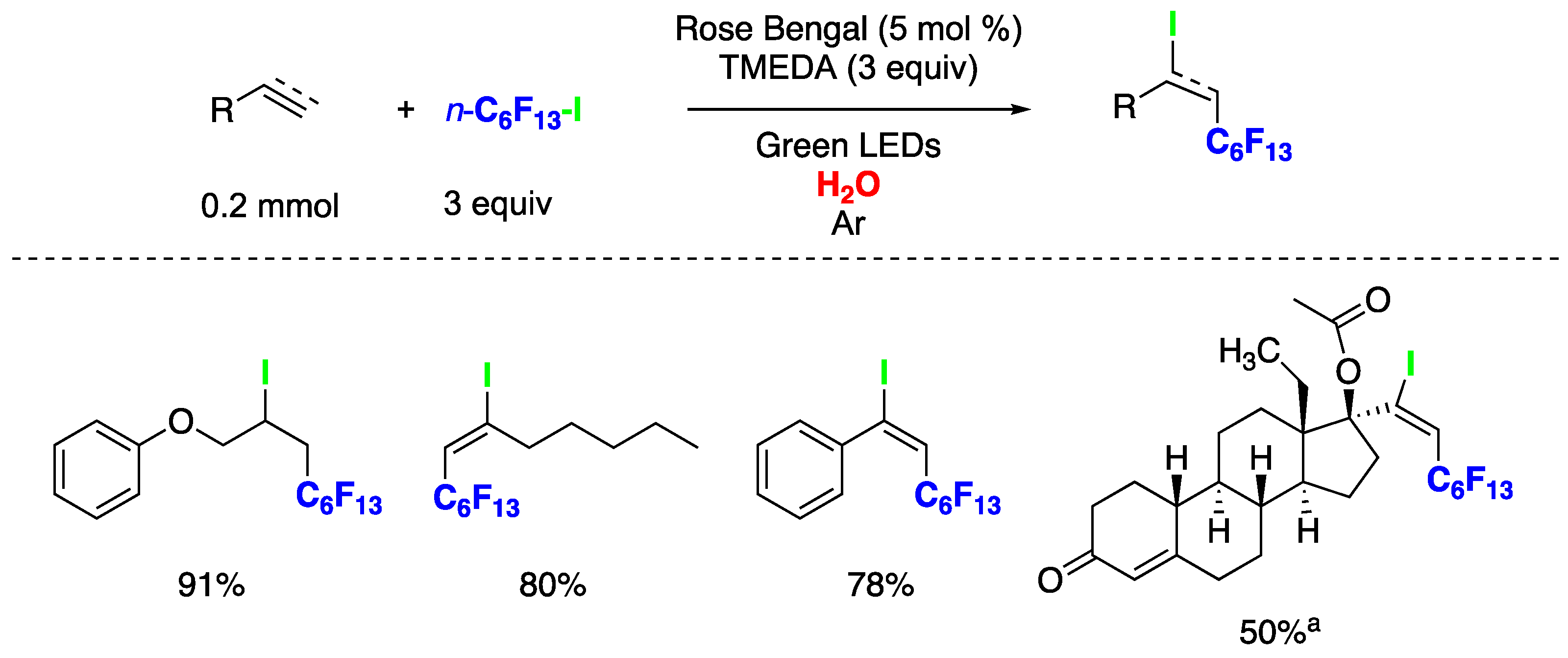
Figure 34.
Photoredox-catalyzed and silane-mediated hydrofluoromethylation of unactivated alkenes in water, and representative examples.
Figure 34.
Photoredox-catalyzed and silane-mediated hydrofluoromethylation of unactivated alkenes in water, and representative examples.
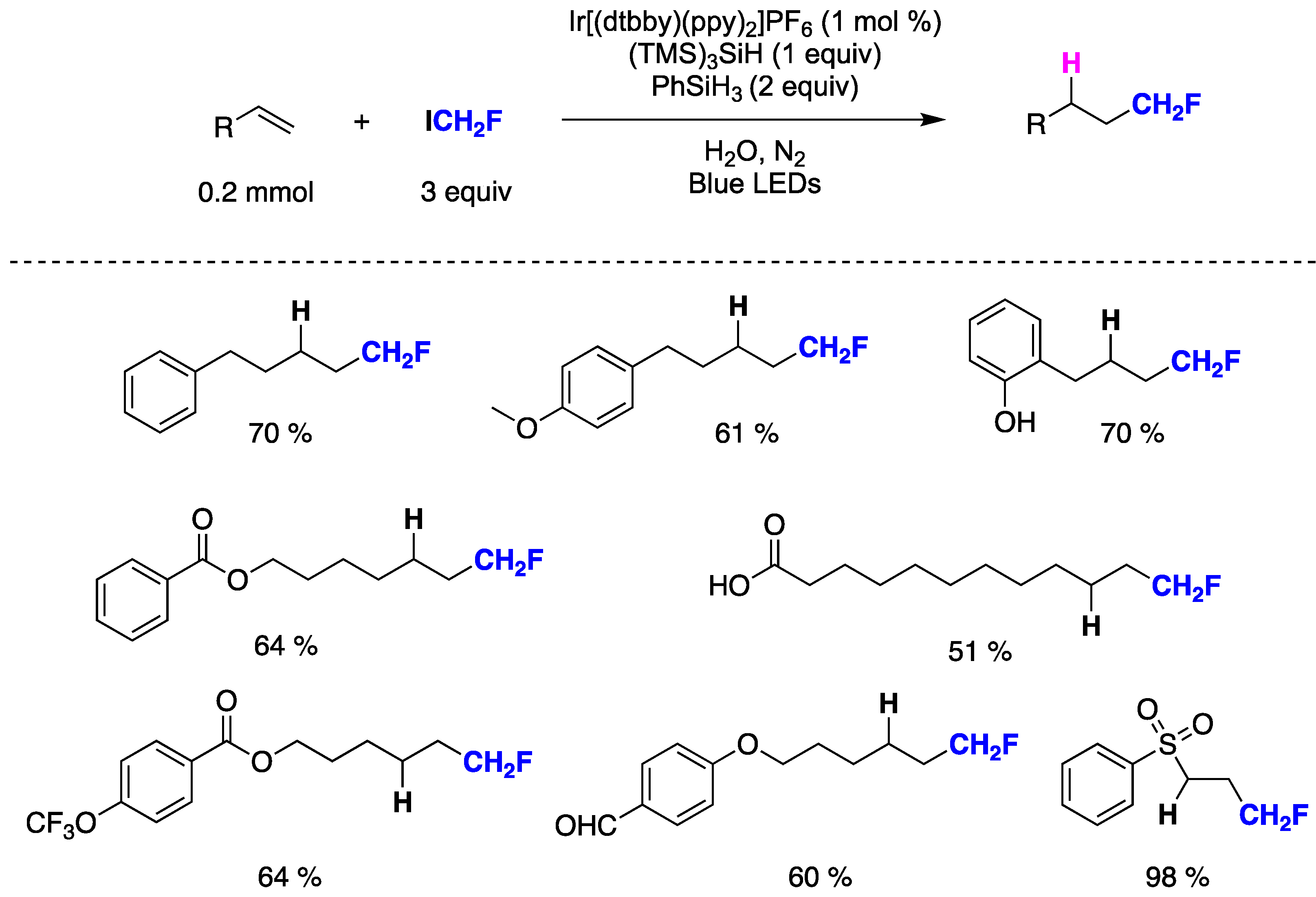
Figure 35.
(a) Photocatalystic single C(sp3)-F bond activation of perfluoroalkyl iminosulfides with alkenes in water and selected examples; (b) Proposed reaction mechanism.
Figure 35.
(a) Photocatalystic single C(sp3)-F bond activation of perfluoroalkyl iminosulfides with alkenes in water and selected examples; (b) Proposed reaction mechanism.
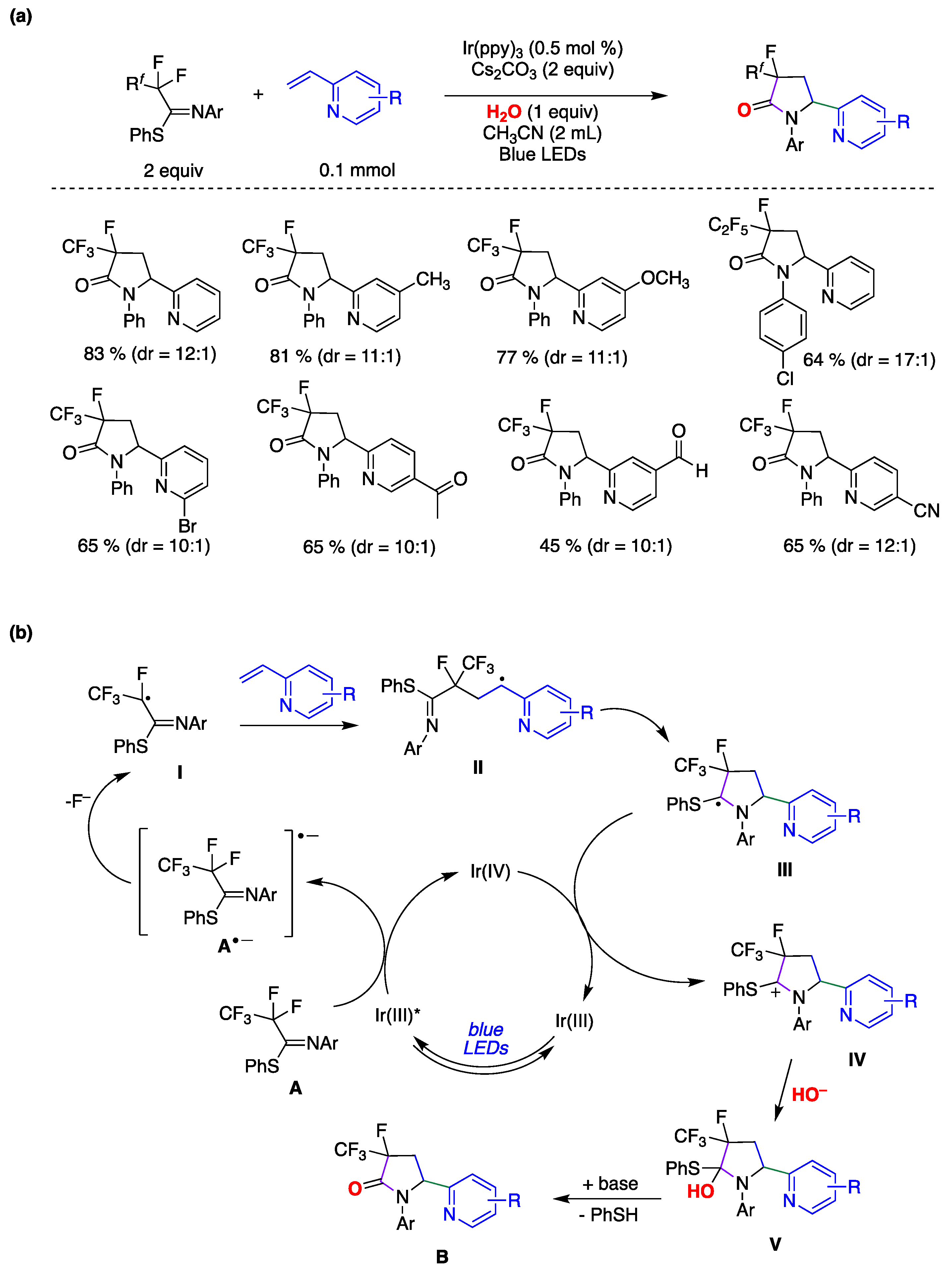
Figure 36.
(a) Photocatalyzed arylation of pyridine hydrochlorides derivatives in water and representative examples; (b) Proposed reaction mechanism.
Figure 36.
(a) Photocatalyzed arylation of pyridine hydrochlorides derivatives in water and representative examples; (b) Proposed reaction mechanism.
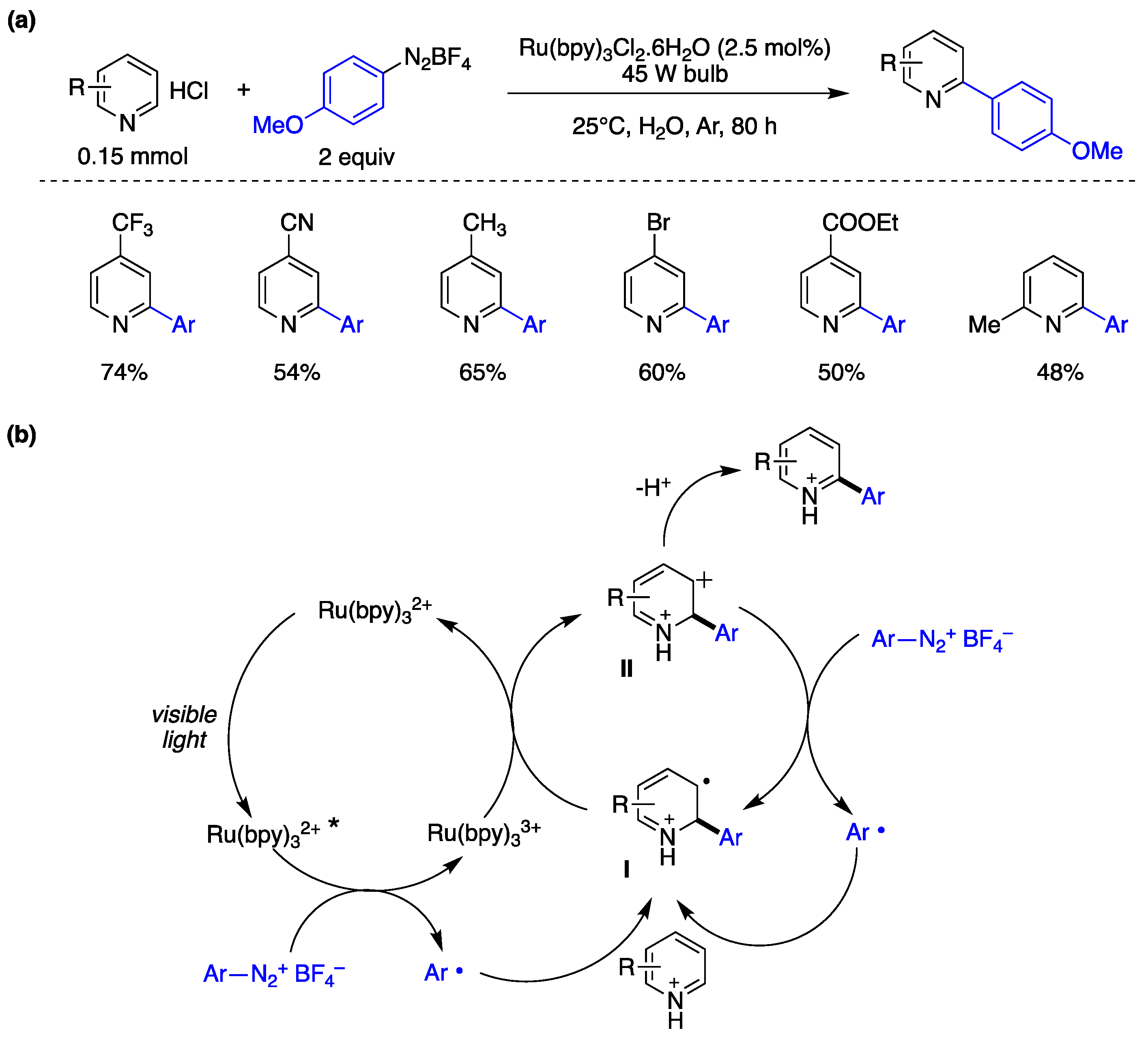
Figure 39.
(a) Late-stage photocatalytic fluoroalkylation of aromatic crown ethers in aqueous media and representative examples; (b) Proposed reaction mechanism.
Figure 39.
(a) Late-stage photocatalytic fluoroalkylation of aromatic crown ethers in aqueous media and representative examples; (b) Proposed reaction mechanism.
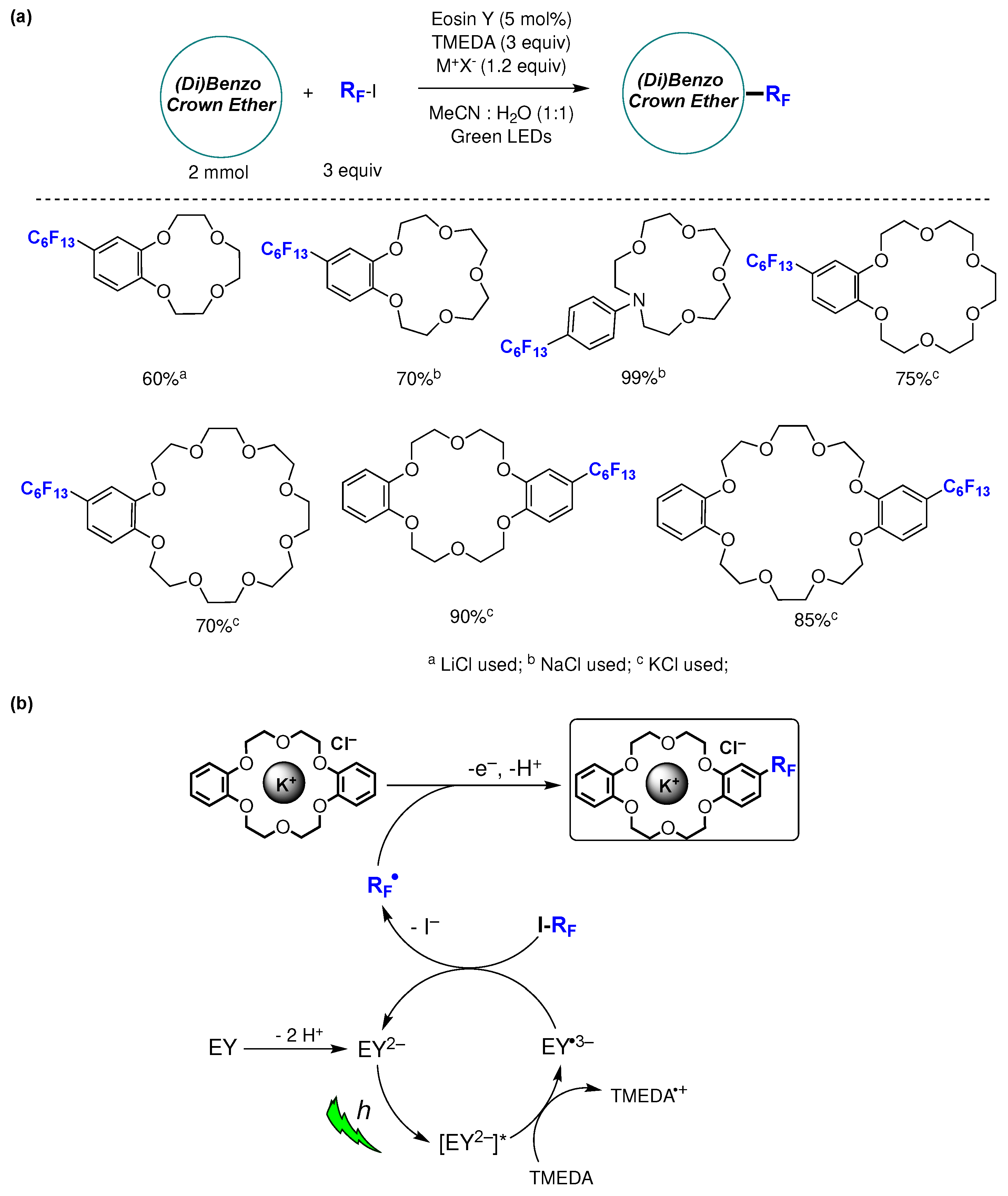
Figure 41.
Photoredox catalysis for radical deuteration: (a) An example of H/D exchange of unactivated C(sp3)-H bond; (a) Two examples of site- and enantioselective incorporation of deuterium into organic compounds.
Figure 41.
Photoredox catalysis for radical deuteration: (a) An example of H/D exchange of unactivated C(sp3)-H bond; (a) Two examples of site- and enantioselective incorporation of deuterium into organic compounds.
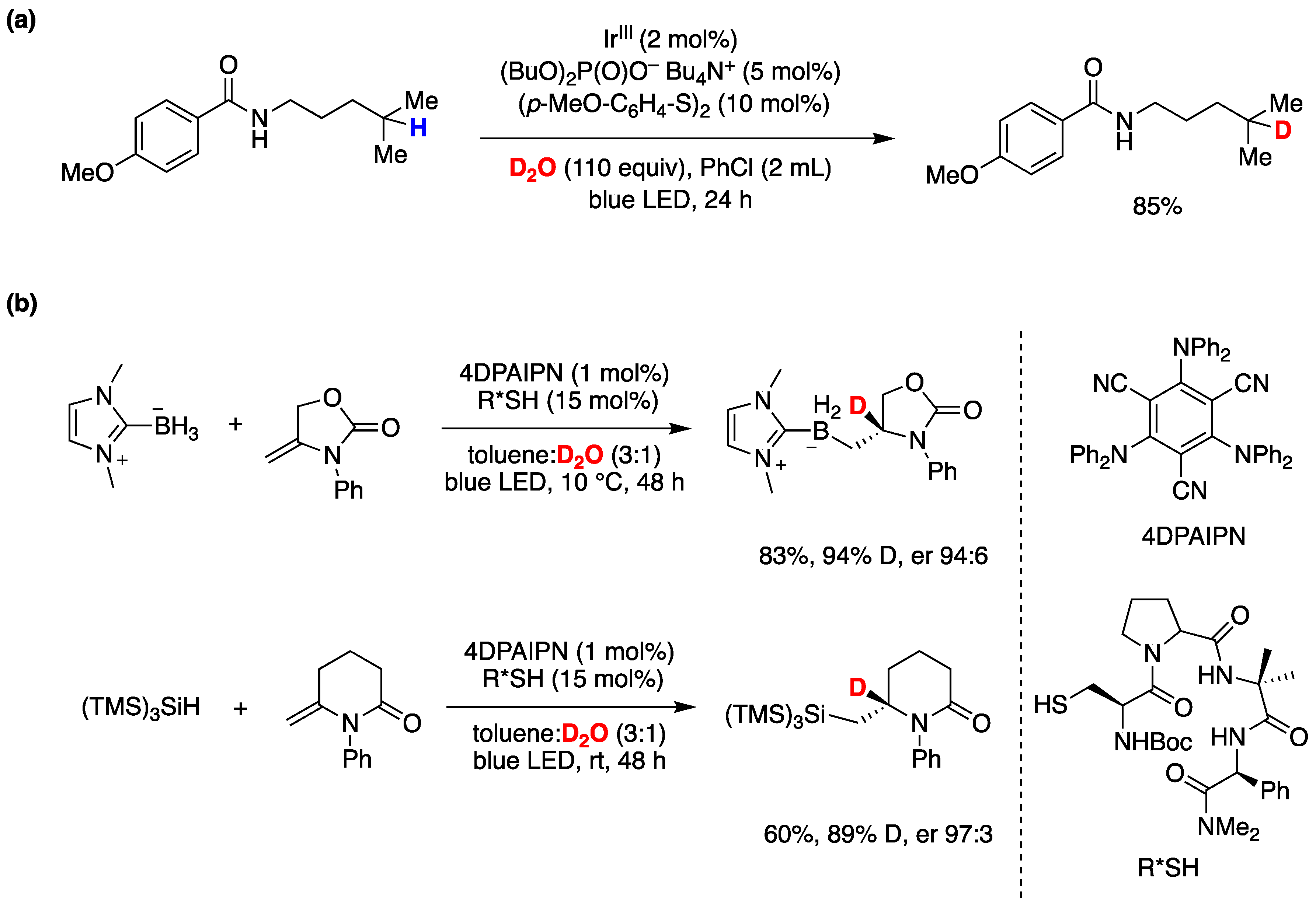
Figure 42.
(a) Optimized conditions for the photocatalytic phosphine-mediated water activation for radical hydrogenation and selected examples; (b) Proposed mechanistic scheme.
Figure 42.
(a) Optimized conditions for the photocatalytic phosphine-mediated water activation for radical hydrogenation and selected examples; (b) Proposed mechanistic scheme.
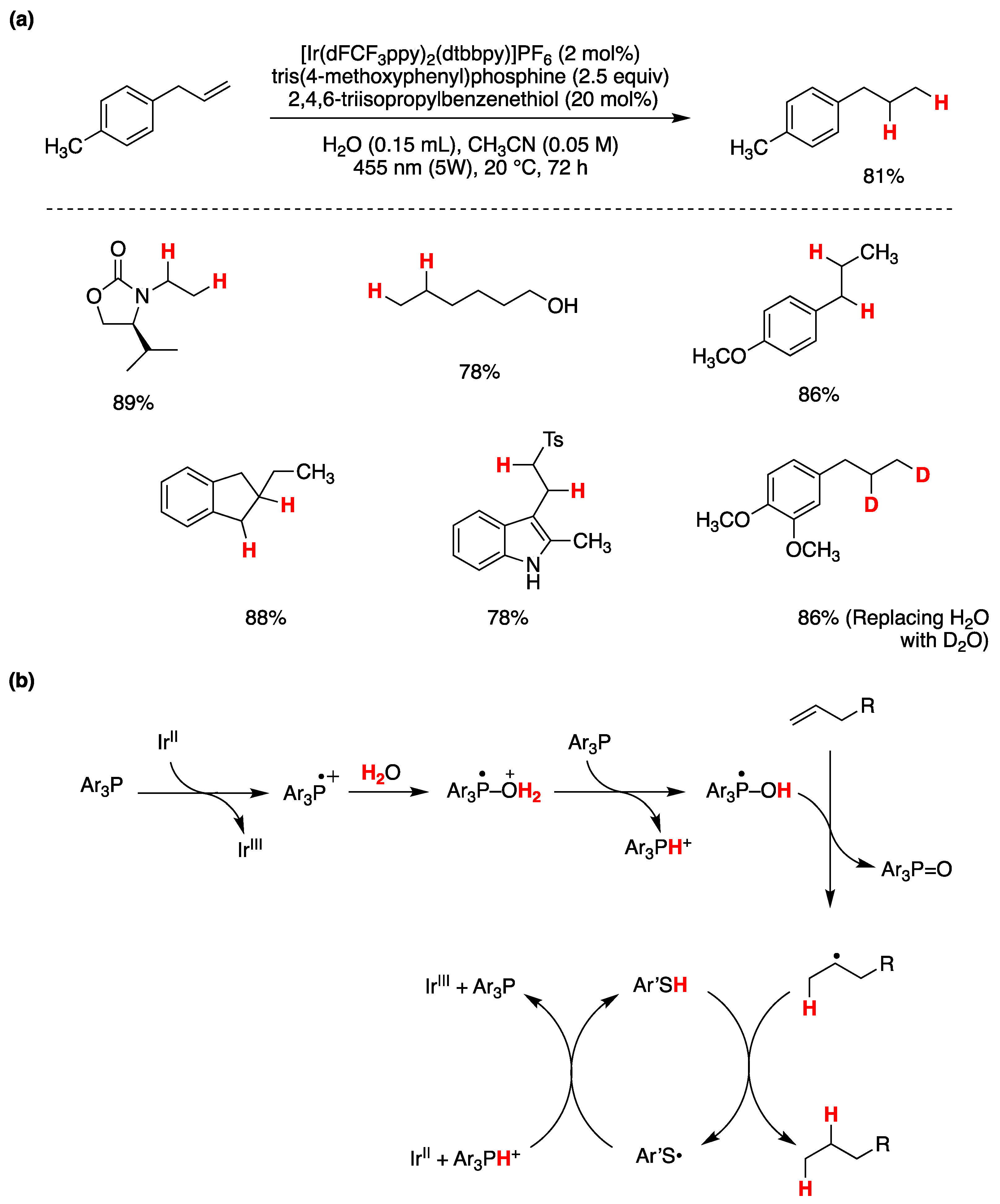
Figure 43.
(a) Structures of 5′,8-cyclo-2′-deoxyadenosine (cdA) and 5′,8-cyclo-2′-deoxyguanosine (cdG) in their 5′R and 5′S diastereomeric forms. (b) Bioinspired radical transformations for the synthesis of 5′,8-cyclopurines (cPu). (c) Radical cascade reaction that mimics the DNA damage for the synthesis of 5′,8-cyclopurines (cPu).
Figure 43.
(a) Structures of 5′,8-cyclo-2′-deoxyadenosine (cdA) and 5′,8-cyclo-2′-deoxyguanosine (cdG) in their 5′R and 5′S diastereomeric forms. (b) Bioinspired radical transformations for the synthesis of 5′,8-cyclopurines (cPu). (c) Radical cascade reaction that mimics the DNA damage for the synthesis of 5′,8-cyclopurines (cPu).
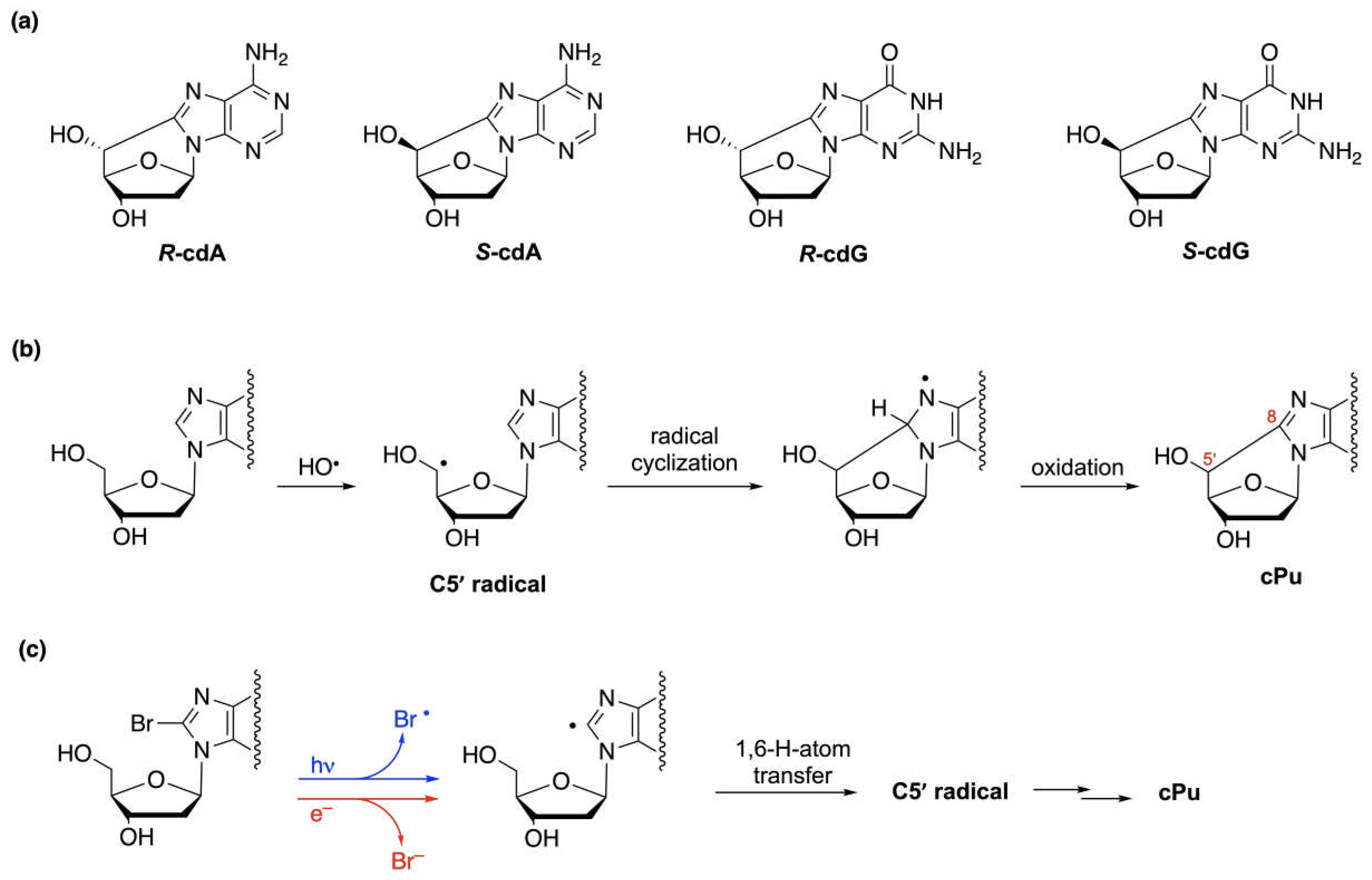
Figure 44.
(a) Transformation of ribonucleotides to 2′-deoxy-ribonucleotides by ribonucleotide reductases class Ia, active in eukaryotes and microorganisms (Taken from ref [190]). (b) Some details of the complex radical mechanism at the active site of enzyme; in particular, the reaction 19 → 20 is the reduction of ketone by disulfide radical anion (Taken from ref [195]).
Figure 44.
(a) Transformation of ribonucleotides to 2′-deoxy-ribonucleotides by ribonucleotide reductases class Ia, active in eukaryotes and microorganisms (Taken from ref [190]). (b) Some details of the complex radical mechanism at the active site of enzyme; in particular, the reaction 19 → 20 is the reduction of ketone by disulfide radical anion (Taken from ref [195]).
Figure 45.
(a) Hydrogen sulfide (H2S/HS−) affords the sulfhydryl radical (HS•/S•−) by a variety of methods and adds reversibly to the parent compound to form the dimeric radical anion. (b) Dual catalytic/radical chain mechanism for the deoxygenation of cis-1,2-cyclopentanediol (22) to cyclopentanol 24) via cyclopentanone (23) in aqueous solutions (Taken from ref [190]). (c) Dual radical chain mechanism for the deoxygenation of 2-hydroxycyclohexanone (29).
Figure 45.
(a) Hydrogen sulfide (H2S/HS−) affords the sulfhydryl radical (HS•/S•−) by a variety of methods and adds reversibly to the parent compound to form the dimeric radical anion. (b) Dual catalytic/radical chain mechanism for the deoxygenation of cis-1,2-cyclopentanediol (22) to cyclopentanol 24) via cyclopentanone (23) in aqueous solutions (Taken from ref [190]). (c) Dual radical chain mechanism for the deoxygenation of 2-hydroxycyclohexanone (29).
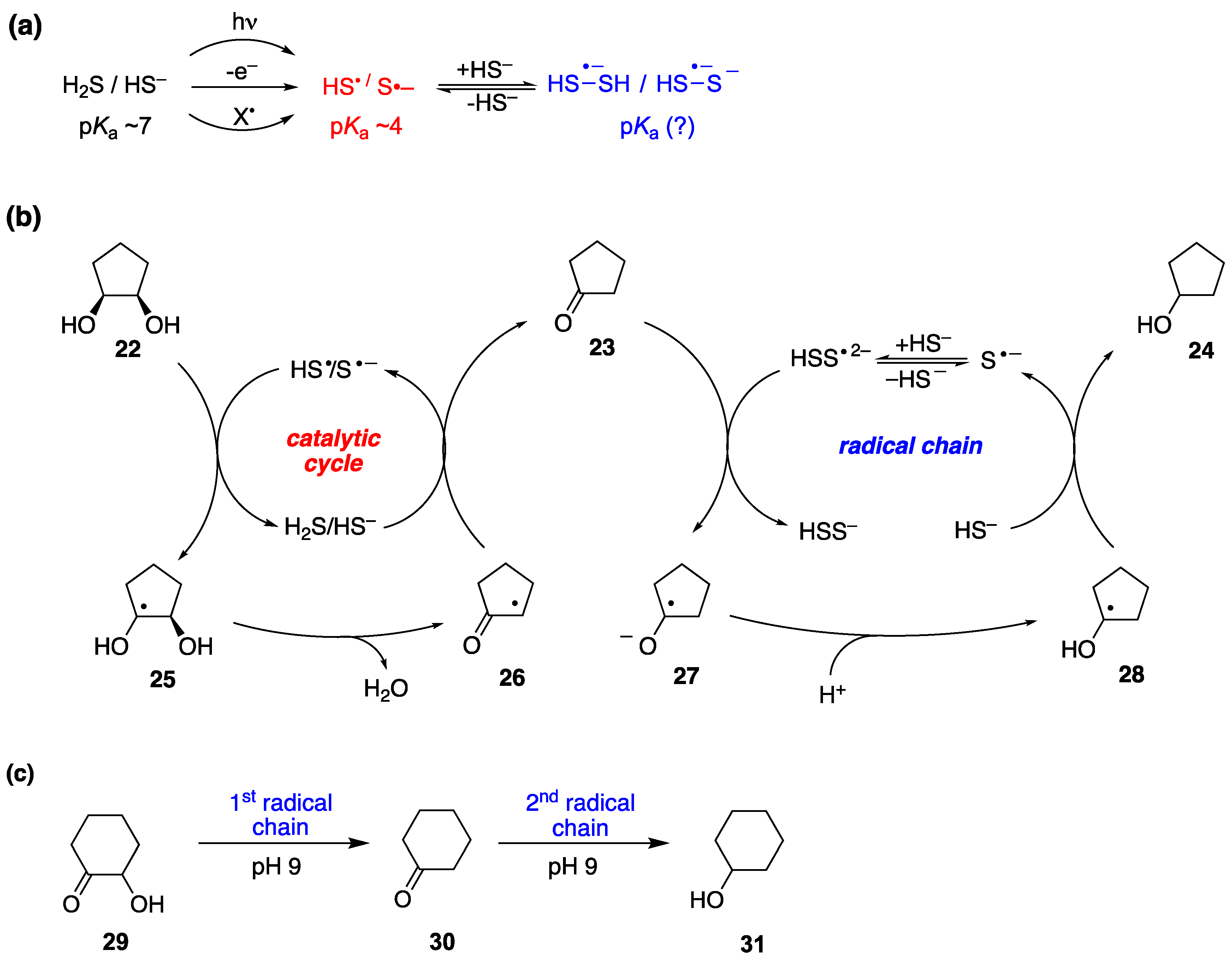
Figure 46.
(a) The radical chain reaction for the reduction of ketones to the corresponding alcohols; (b) The mechanism for the reduction of 2-cyclopenten-1-one (19) involves dual radical chain reactions A and B (Taken from ref [195]).
Figure 46.
(a) The radical chain reaction for the reduction of ketones to the corresponding alcohols; (b) The mechanism for the reduction of 2-cyclopenten-1-one (19) involves dual radical chain reactions A and B (Taken from ref [195]).
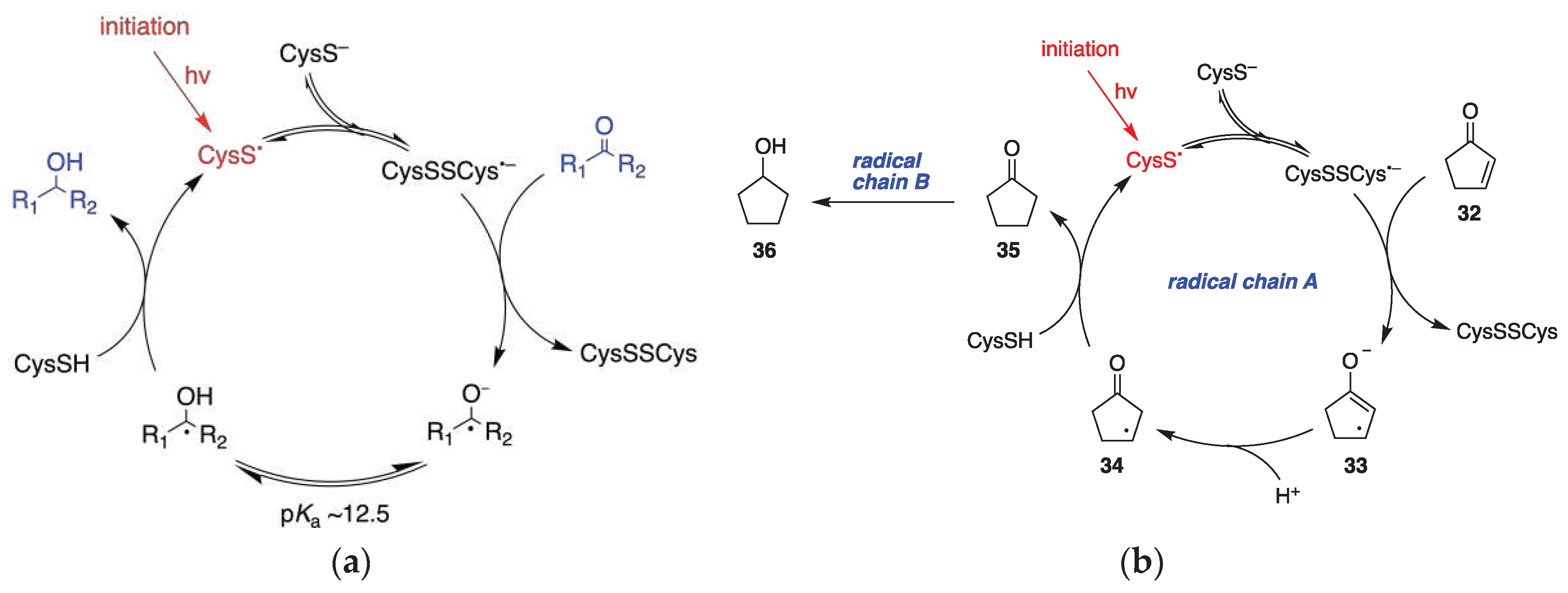

Table 1.
Rate constants for the reaction of α-hydroxyalkyl radicals with 2-mercaptoethanol in water as the solvent.1.
Table 1.
Rate constants for the reaction of α-hydroxyalkyl radicals with 2-mercaptoethanol in water as the solvent.1.
| Alkyl Radical | kH, M–1 s–1 |
| HOCH2• | 1.3 x 108 |
| HOC(•)Me | 2.3 x 108 |
| HOC(•)Me2 | 5.1 x 108 |
1 At room temperature.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



