
Submitted:
21 December 2023
Posted:
22 December 2023
You are already at the latest version
Abstract
Intermediate CAG expansions in the gene ataxin-2 (ATXN2) are a known risk factor for ALS, but little is known about their role in FTD risk. Moreover, their contribution to the risk and phenotype of patients might vary in populations with different genetic background. The aim of this study was to assess the relationship of intermediate CAG expansions in ATXN2 with the risk and phenotype of ALS and FTD in the Spanish population. Repeat-Primed PCR was performed in 620 ALS and 137 FTD patients in three referral centers of Spain to determine the exact number of CAG repeats. In our cohort, ≥27 CAG repeats in ATXN2 was associated with a higher risk of developing ALS (odds ratio [OR] = 2.666 [1.471-4.882]; p = 0.0013), but not FTD (odds ratio [OR] = 1.446 [0.558-3.574]; p = 0.44). Moreover, ALS patients with ≥27 CAG repeats in ATXN2 showed shorter survival rate compared to those with <27 repeats (hazard ratio [HR] 1.74 [1.18, 2.56], p = 0.005), more frequent limb onset (odds ratio [OR] = 2.34 [1.093-4.936]; p = 0.028), as well as family history of ALS (odds ratio [OR] = 2.538 [1.375-4.634]; p = 0.002). Intermediate CAG expansions of ≥27 repeats in ATXN2 are associated with ALS risk, but not with FTD, in the Spanish population. ALS patients carrying an intermediate expansion in ATXN2 show more frequent limb onset, but worse prognosis than those without expansions. In patients carrying C9orf72 expansions, the intermediate ATXN2 expansion might increase the penetrance and modify the phenotype.
Keywords:
amyotrophic lateral sclerosis
; ATXN2
; risk factor
; frontotemporal dementia
; mutation
; poli-Q expansion
1. Introduction
Amyotrophic Lateral Sclerosis (ALS) is a progressive adult-onset neurodegenerative disease affecting both upper (UMN) and lower motor neurons (LMN) [1,2]. Disease onset is usually focal, but it spreads to other body regions, causing weakness, muscle atrophy, cramps, brisk reflexes, dyspnoea and dysphagia, and finally leading to death due to respiratory failure [3,4]. Sixty five percent of ALS patients have limb onset, thirty percent bulbar onset and five percent other phenotypes [5].
Over 10% of ALS cases report family history of ALS or frontotemporal dementia (FTD), considered as familial ALS (FALS) [6], while those without family history are called sporadic ALS (SALS). Remarkably, both FALS and SALS patients largely share genetic causes, and mendelian mutations can be found in up to 10% of SALS. Mendelian mutations causing ALS show considerable founders effects, resulting in variable genetic background even among populations of European ancestry [7]. Moreover, aggregation studies have identified an overlap between ALS and other neuropsychiatric diseases [8].
FTD is an insidious neurodegenerative syndrome characterised pathologically by degeneration of frontal and temporal lobes and clinically by deficits in behaviour, executive function, and/or language [9,10].
In the last decades, an increasing evidence of the tight relationship between ALS and frontotemporal dementia (FTD) based on clinical, genetic and histopathological aspects has been established [10,11]. Histopathologically, over 95% of ALS patients and about 50% of FTD patients are characterised by the presence of intracellular aggregates of misfolded TDP-43 [12]. Thus, ALS and FTD-TDP43 are considered the two phenotypic extremes of a single clinicopathologic spectrum [10,13].
Genetically, some genes have been described to cause both diseases following a mendelian (usually autosomal dominant) inheritance, such as an hexanucleotide expansion in C9orf72 or pathogenic sequence variants in the TBK1, VCP and TARDBP genes. However, other genes are particularly associated with only one disease, such as SOD1 for ALS or GRN and MAPT for FTD.
Besides mendelian genes, several genetic risk factors have been described to contribute to the development of these diseases. Genetic risk factor is any variant that interacting with the environment or other genetic variants confers a greater susceptibility for a disease [14].
Expansions of CAG repeats in the first exon of ATXN2 gene were described as an ALS genetic risk factor over a decade ago [15]. This abnormal CAG expansion had been previously described as the causative molecular mechanism of spinocerebellar ataxia type 2 (SCA2) [16,17], a neurodegenerative disease characterized by uncoordinated movements, postural tremor, decreased muscle tone, weakness and cognitive impairment [18,19]. ATXN2 is localized on chromosome 12 (12q23-24) and comprises 25 exons. The protein encoded by this gene is named ataxin-2. This protein takes part in several cellular processes like RNA metabolism, endocytosis, cytoskeleton reorganization and calcium-mediated signalling, among others [18,19].
Alleles below or equal to 31 repeats are present in normal individuals [20]. In contrast, 33 and 34 CAG repeats are related to reduced-penetrance SCA2, whereas alleles with a repeat number >34 are considered full-penetrant for SCA2, presenting an autosomal dominant pattern of inheritance [16,17,18,19].
CAG-triplet expansion encodes for a polyglutamine region (polyQ) in Ataxin-2. This classifies SCA2 within the group of polyQ diseases such as Huntington disease [21]. This polyglutamine region is thought to result in a toxic gain of function affecting specific types of neurons as Purkinje cells, basal ganglia neurons and motor neurons of the brain and spinal cord [19]. In fact, in SCA2, patients motor neurons are known to degenerate [22]. This data suggested ATXN2 could be a suitable candidate as genetic risk factor for ALS.
Since the initial description, the association between an intermediate CAG repeat expansion in ATXN2 and an increased risk for ALS has been replicated in several studies, but the threshold for increased risk ranges from 27 to 33 repeats [15,22,23,24,25,26,27]. It is unknown if variations on the reported expansion lengths could be due to the genetic background of the population [28,29,30].
Moreover, only few studies [27,31,32] have assessed the role of CAG expansions in ATXN2 as a factor of bad prognosis in ALS patients, with contradictory results [33].
In FTD, the role of ATXN2 intermediate CAG expansions in predisposing to FTD has not been established, but one study suggested that ATXN2 may act as a phenotype modifier [28,34].
This study aims to assess ATXN2 CAG expansions as genetic risk factor and phenotypic modifier for ALS and FTD in the Spanish population.
2. Materials and Methods
2.1. Patient Population
ALS and FTD patients were recruited from three referral centres in Spain (12 de Octubre Hospital Research Institute (Madrid), La Fe Hospital (Valencia) and Bellvitge University Hospital (Barcelona). Controls, which included healthy individuals as well as patients with other neuromuscular diseases (congenital myopathies, Steinert’s myotonic dystrophy, familial periodic paralysis non-dystrophic myotonia and spinal muscular atrophy), were recruited in HU12O and HUB.
2.2. Genetic Analysis
The CAG repeat expansion in ATXN2 and the C9orf72 GGGGCC repeat expansion were analysed in all patients and controls in the Genetic Molecular Laboratories of all three hospitals.
2.2.1. Determination of CAG Repeat Expansion Length in ATXN2 and C9orf72
DNA from whole peripheral blood was extracted with Illustra Nucleon Genomic kit (GE Healthcare Life Science). CAG repeats in ATXN2 and GGGGCC repeats in C9orf72 were determined by polymerase chain reaction (PCR) followed by capillary electrophoresis.
- Standard PCR for ATXN2
Primers used in ATXN2 gene amplification are shown in Table A1 and volumes are shown in Table A2. PCR procedure for ATXN2 involved an initial stage of 95 ºC for 5 minutes, followed by 42 cycles (97 ºC for 15 seconds, 53 ºC for 20 seconds and 72 ºC for 1 minute), and a final stage at 72 ºC for 10 minutes.
PCR products are diluted to 1/10 and mixed with formamide and GeneScanTM 400HD RoxTM dye Size Standard. We used 3130 Genetic Analyzer to 3130xl Genetic Analyzer Upgrade (Applied BiosystemTM). Results were analysed by Peak Scanner 2.0 version (ThermoFisher Scientific). We used two internal controls of 27 and 37 CAG repeats, provided by the Immunology Service of 12 de Octubre Hospital.
When PCR product analysis showed two peaks, we assumed those lengths for both alleles. Conversely, if we obtained a single peak it could be due to two main reasons: this patient could carry both alleles with the same length or one of the alleles is large enough not to be amplified by a conventional PCR. Therefore, single peak samples were amplified by repeat-primed-PCR procedure (See primers in Table A1).
- Repeat-Primed PCR for ATXN2
PCR procedure involved an initial stage of 95 ºC for 5 minutes, followed by 30 cycles (94 ºC for 30 seconds, 60 ºC for 30 seconds and 72 ºC for 30 seconds), and a final stage at 72 ºC for 10 minutes. Afterwards, capillary electrophoresis was performed as detailed in A.
RP-PCR pattern is presented as a succession of peaks with 3 base pairs of difference corresponding to each of the CAG triplet repeats. The first peak corresponds to 7 repeats (which are included in the forward primer SCA-P4), and each subsequent peak adds one more repeat. In this way, normal alleles will show a few large peaks, while expanded alleles will show a multitude of peaks, each of them decreasing in size (PCR conditions are available in Table A3).
2.3. Data Analysis
Data are summarized using mean and standard deviation for the continuous variables, and relative and absolute frequencies for the categorical variables.
One-tailed Fisher test evaluated the association between different intermediate CAG repeat expansions (RE) size in ATXN2 and both ALS and FTD diseases. Subsequently, the relationships between demographic (sex), clinical (type of onset and presence of familial history) and genetic variables were explored with the chi-square test.
A means comparison of age of onset between carriers and non-carriers of intermediate RE in ATXN2 was performed using the U de Mann-Whitney test. To do this, a Kolmogorov-Smirnov test was first performed to check the normality. Kaplan–Meier curve, log-rank test, and a Cox regression model, including the CAG intermediate expansion length in ATXN2 as a random effect, were used to assess the effect on survival. Statistical analyses were performed through SPSS Statistics 24 (IBM, Spain) and graphics were made with Graph Pad Prism Software support.
2.4. Ethics Approval
Participants expressed their consent for data collection and genetic studies in their respective centers. Information for all patients was de-identified, and data collection and processing were approved by the Ethics Committee for Biomedical Research of the 12 de Octubre Hospital Research Institute (Madrid), La Fe Hospital (Valencia) and Bellvitge University Hospital (Barcelona). The study has been conducted according to the principles expressed in the Declaration of Helsinki. This genetic study was approved by the local CEIm at Hospital 12 de Octubre Research Institute. All patients received a patient information sheet, and an informed consent was obtained from all subjects involved in the study.
3. Results
3.1. RE in ATXN2
A total of 757 patients (620 ALS and 137 FTD patients) and 362 controls were included in the study. Their demographic, clinical and genetic characteristics are summarized in Table 1. In ALS patients, CAG repeat expansion ranged from 22 to 36 repeats, in FTD from 22 to 30 repeats and in controls from 22 to 33 repeats. Regarding allele frequencies, in ALS patients the most common allele was (CAG)22 (52.1%) followed by (CAG)23 (32.1%). The most frequent alleles in FTD patients were (CAG)22 (48.2%) and (CAG)23 (27.0%). Finally, the predominant allele among the control group was also (CAG)22 (51.1%) followed by (CAG)23 (31.8%) (Figure 1).
3.2. RE in ATXN2 as a Risk Factor for ALS
3.3. Clinical Characteristics of ALS patients with RE in ATXN2
Fourteen percent of ALS patients with an intermediate CAG repeat expansion (≥27 CAG) developed ALS-FTD compared to 12.5% of those not carrying the expansion. In addition, 78.85% of them showed limb onset ALS vs 66.31% of those with less than 27 repetitions, although these differences were not statistically significant. A statistically significant association was found between limb onset and being a carrier of ≥28 repetitions (p = 0.0139). Finally, an association was found between being a carrier of ≥28 repetitions and having a positive familial history of ALS (p = 0.0009). No differences in age, male/female ratio nor co-mutations were observed (Table 3).
Mean survival of ALS patients with intermediate CAG expansion (29.23 ± 12.77) was shorter than that of ALS patients with normal CAG repeats (40.69 ± 28.13) and this difference was statistically significant (p = 0.0458). The Kaplan-Meier curves (Figure 2) and log-rank tests showed shorter median survival in patients with ≥27 CAG repeats in ALS patients than those with shorter lengths (34.5 vs 29.1 months, p = 0.004). In the Cox regression model, be a carrier of ≥27 CAG was an independent factor for poor survival (hazard ratio [HR] 1.74 [1.18, 2.56], p = 0.005), together with bulbar onset and age of onset (Table 4 and Figure 3).
The longest survival is observed in the group of patients with limb onset who are non-carriers of an intermediate ATXN2 expansion (red), followed by the group with bulbar onset who are non-carriers of an intermediate expansion (blue). Among the intermediate expansion-carrying groups (≥27 CAG repeats), patients with bulbar onset (green) also exhibit shorter survival.
3.4. RE in ATXN2 as a Risk Factor for FTD
We also analysed the presence of an intermediate CAG repeat expansion alleles in FTD patients and controls. Intermediate repeat expansion alleles (27-33) were numerically more frequent in FTD patients than controls (Figure 1), but this difference was not statistically significant (p >0.05) (Table 5).
Finally, no statistically significant association was found between age of onset or family history of FTD and being a carrier of intermediate repeats (p > 0.05).
3.5. Concomitant RE in ATXN2 and Expansions in C9ORF72
Six (10%) of the 60 ALS patients, and one of the 7 FTD (14.28%) patients, carrying a full-length C9orf72 RE were also carriers of the intermediate expansion in ATXN2 (Table 1). All ALS patients carrying co-expansions had family history of ALS and/or FTD vs 57% of ALS patients carrying the C9orf72 RE only. The mean age of onset of patients with co-expansios was 55 years, with 16% of patients showing bulbar onset (16.67%) and a median survival of 36.4 months (data available for only 2 patients). In comparison, patients with C9orf72 RE only, had a mean age of onset of 58 years, with 31% of them having a bulbar onset and a median survival of 39 months. The single FTD patient exhibiting co-expansion also had a positive family history.
4. Discussion
4.1. RE in ATXN2 Gene in ALS patients
Since Elden and colleagues [15] reported the association between an intermediate CAG repeat expansion (27-33) in ATXN2 and an increased risk for ALS, several studies have widely reaffirmed such association (Table 6). Even though most studies have focused on populations of European ancestry, there is no consensus length established.
The results of this study carried out in Spanish population are consistent with previous studies that have proposed an intermediate length CAG repeat expansion in ATXN2 as genetic risk factor for ALS. Specifically, this study suggests ≥27 repeat expansion alleles as risk factor for ALS, although stronger association was found in ≥28 and ≥29 alleles. Thus, our findings are similar to previous research done in populations of European ancestry. However, further investigation to understand why the threshold of CAG repeats is higher in other populations also of European ancestry is needed (Table 6). Environmental factors are good candidates to be explored in future research studies.
Our study also confirms the association of the intermediate RE and the presence of family history. This had been found previously in some studies [37,41,42], but not in others [39].
Regarding survival results, ≥27 repeat ALS patients showed a strikingly reduced survival (29.23 ± 12.77) compared to ALS patients with non-expanded alleles (40.70 ± 28.13). Our results are consistent with previous reports in Italian population [27,31], but opposed to studies done in UK, Netherlands and USA [32,33]. This differences may be due to the diverse genetic background (Mediterranean vs north European) or to selection bias in the studied populations (i.e. our cohort was enriched for familial cases). Interestingly, we confirm here that the effect of the RE in survival is independent of other variables. Moreover, our data suggest that patients with spinal onset carrying an intermediate RE in ATXN2 have even worse prognosis than bulbar onset patients not carrying the RE. Thus, this information might be useful when stratifying patients in clinical trials.
Elden and colleagues [15] also reported an increased toxicity of TDP-43 due to intermediate CAG repeat expansion in ATXN2 gene. Intermediate polyglutamine region enhances Ataxin-2 interaction with TDP-43 and makes TDP-43 more prone to mislocalize from the nucleus to cytoplasm under stress situations. This mechanism could explain the pathogenesis involved in intermediate CAG repeat expansion in ATXN2 gene for ALS. Besides, a more recent study carried out by Becker and colleagues [43] reported an increased survival and motor neuron function in transgenic murine models with lower Ataxin-2 levels. This suggests Ataxin-2 has therapeutic potential for ALS and in some FTD, as both show TDP-43 positive inclusions in neurons.
4.2. Association of Intermediate CAG Repeat Expansion in ATXN2 Gene with FTD
As mentioned above, ALS and FTD are closely associated not just based on histopathology (TDP-43 inclusions), but also in clinical and genetic aspects [10,11]. Despite these similitudes, we were not able to corroborate any effect of ATXN2 intermediate RE in FTD risk. Specifically, although more FTD patients than controls were carrying intermediate RE, this difference was not statistically significant. However, it must be highlighted that the sample size of FTD was considerably lower than that of ALS patients. Other studies, probably underpowered, also failed to find this association [29,41]. However, larger studies have confirmed that intermediate RE in ATXN2 are most frequent among FTD patients than controls [28,32], although with lower ORs than those found for ALS. This reduced association is not surprising, given that about 50% of FTD patients do not show TDP-43 inclusions. We didn’t find differences in age of onset or family history in FTD patients carrying the intermediate RE. Conversely, other study has suggested intermediate alleles in ATXN2 as phenotypic modifiers in FTD [29], reporting a positive association between an intermediate number of CAG repeats and an earlier onset of the disease, parkinsonism and psychotic symptoms at disease onset.
4.3. Concomitant RE in ATXN2 and Expansions in C9orf72
In our cohort, 10% of ALS patients and 14.3% of FTD patients carrying C9orf72 expansions harboured also an intermediate RE in ATXN2. Thus, the frequency of intermediate RE in ALS patients carrying C9orf72 expansions, was similar to that found in sporadic patients. This is in contrast with two studies that found lower (0.6-2%) frequency of co-expansions [44,45]. Despite the small sample size, our data suggest that co-expansions are not infrequent in Spanish population and may act as a modifier factor of the penetrance and phenotype of C9orf72. Firstly, 100% ALS patients with both expansions vs only 57% of those with C9orf72 RE only reported family history of ALS or FTD. Recently, the penetrance of C9orf72 has been found to vary between families [46].Thus, the presence of ATXN2 intermediate RE might be one of the factors that determines this variable penetrance. Secondly, bulbar onset was twice more frequent in patients with both expansions than in those with only the C9orf72 expansion. Thirdly, patients carrying both expansions showed slightly earlier disease onset and shorter disease duration than those carrying only the C9orf72 expansion. These data must be taken with caution, given that only 7 patients carried both expansions, but again suggest an additive toxic effects of both expansions.
Summarizing, this study confirms that intermediate CAG RE in ATXN2 are a genetic risk factor for ALS onset and poor survival also in the Spanish population.
5. Conclusions
- Intermediate CAG repeat expansions in ATXN2 gene acts as genetic risk factor for ALS in the Spaniard population, with statistical significant risk in those carrying 27 repeats or above.
- Expanded alleles in ATXN2 are associated with shorter survival in ALS patients of our cohort.
- Intermediate CAG repeat expansion in ATXN2 gene has not relation with FTD patients in the Spaniard population.
- The presence of ≥28 repetitions is associated with limb onset and positive family history.
- Co-expansion of intermediate RE in ATXN2 and C9orf72 expanded hexanucleotide patients might influence the penetrance of the C9orf72 expansion.
Author Contributions
Conceptualization, Daniel Borrego-Hernández, Juan Francisco Vazquez-Costa and Raúl Domínguez; Data curation, Daniel Borrego-Hernández, Ariadna Padró-Miquel, Pilar García-Casanova and María José Corominas; Formal analysis, Daniel Borrego-Hernández, Laura Expósito-Blázquez and Cristina Martin Arriscado; Funding acquisition, Juan Francisco Vazquez-Costa, Mònica Povedano and Alberto García-Redondo; Investigation, Daniel Borrego-Hernández and Juan Francisco Vazquez-Costa; Methodology, Daniel Borrego-Hernández, Laura Expósito-Blázquez, Elena Aller, Ariadna Padró-Miquel, Pilar García-Casanova, María José Corominas and Rosario Osta; Project administration, Juan Francisco Vazquez-Costa, Mònica Povedano and Alberto García-Redondo; Resources, Elena Aller, Pilar Cordero-Vázquez and Jesús Esteban-Pérez; Software, Daniel Borrego-Hernández, Juan Francisco Vazquez-Costa and Cristina Martin Arriscado; Supervision, Alberto García-Redondo; Validation, Raúl Domínguez, Ariadna Padró-Miquel, Pilar García-Casanova, María José Corominas, Mònica Povedano and Alberto García-Redondo; Visualization, Daniel Borrego-Hernández; Writing – original draft, Daniel Borrego-Hernández; Writing – review & editing, Daniel Borrego-Hernández, Juan Francisco Vazquez-Costa and Raúl Domínguez.
Funding
ALS research Laboratory at Hospital 12 de Octubre was funded by grants of the Instituto de Salud Carlos III (ISCIII), through the projects “17/00491” and “21/00286” co-funded by the European Union. Dr. Borrego-Hernández is funded by “Líneas de financiación de: estrategias frente a enfermedades neurodegenerativas (incluido ELA)” from Spanish Health Ministry, through a grant of Comunidad de Madrid “Estudio genético de la población con ELA de la Comunidad de Madrid” (agreement: BOCM142–17/09/2019 pg.105). L.E-B is funded by Asociación Española de ELA (ADELA). Dr Vazquez-Costa is funded by grants of the Instituto de Salud Carlos III (ISCIII) grant number “JR19/00030” and “FIS a2021/00737”, co- funded by European Regional Development Fund (‘A way to make Europe’), and STOPELA (2017/0653). The part of the study done in Hospital Universitari de Bellvitge, was funded by the Motorneuron functional unit and the genetics laboratory own funds.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the ethics committees of Hospital 12 de Octubre (protocol 18/067 and date of approval 27/02/2018) and Hospital la Fe (protocol code 2017/0653 and date of approval 17/01/2018). Hospital Universitari de Bellvitge ethics comitte give the approval for the study named "ESTUDIO SOBRE RIESGO ASOCIADO A LA EXPANSIÓN INTERMEDIA DEL TRIPLETE "CAG" EN EL GEN ATXN2 EN EL DESARROLLO DE LA ESCLEROSIS LATERAL AMIOTRÓFICA EN NUESTRA POBLACIÓN" with code BB21-029 and project reference PR145/21, in July 12th 2021.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.
Acknowledgments
We want to particularly acknowledge patients, the Biobank La Fe (PT13/0010/0026) and Biobank HUB-ICO-IDIBELL (PT20/00171) integrated in the ISCIII Biobanks and Biomodels Platform and Xarxa Banc de Tumors de Catalunya (XBTC) for their collaboration.
Conflicts of Interest
Dr Vázquez-Costa has served on advisory boards for Biogen and Hoffmann La-Roche, and has received travel and speaker honoraria from Biogen and Hoffmann La-Roche, outside the submitted work. The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Appendix A

Table A1.
PCR amplification primers for ATXN2.
| Primer | PCR | Sequence |
|---|---|---|
| SCA2-F | Standard | FAM-CGTGCGAGCCGGTGTATGGG |
| SCA2-R | Standard & RP-PCR | GGCCGACGCTAGAAGGCCGCT |
| AF-P3 | RP-PCR | TACGCATCCCAGTTTGAGACG |
| SCA-P4 | RP-PCR | FAM-TACGCATCCCAGTTTGAGCAGCAGCAGCAGCAGCAGCAGCAG |
Table A2.
Standard PCR.
| Reactive | Volume |
|---|---|
| Buffer 10X | 2.5 |
| dNTPs 1.25 mM | 4 |
| GC Rich | 5 |
| MgCl2 | 1.5 |
| DMSO | 2.5 |
| H2O | 4 |
| *Primer F* | 1.5 |
| Primer R | 1.5 |
| FastStart Taq | 0.5 |
| ADN | 2 |
| MIX | 23 |
* Volumes expressed in microliters (µL).
Table A3.
Repeat-primed PCR.
| Reactive | Volume |
|---|---|
| Buffer 10X | 2.5 |
| dNTPs 1.25 mM | 4 |
| GC Rich | 5 |
| MgCl2 | 1,5 |
| H2O | 1,6 |
| Primer R | 2 |
| AF-P3 | 2 |
| *SCA-P4* | 2 |
| Taq FastStart | 0,4 |
| ADN | 4 |
* Volumes expressed in microliters (µL).
References
- Chiò, A.; et al. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Marangi, G.; Traynor, B.J. Genitic cause of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015, 1607, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mora, G.; Restagno, G.; Brunetti, M.; Ossola, I.; Fuda, G. UNC13A influences survival in Italian ALS patients: a population-based study. Bone 2013, 34. [Google Scholar]
- Gowland, A.; et al. Predicting the future of ALS: the impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom, 2020–2116. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 264–274. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; et al. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef]
- Byrne, S.; et al. Aggregation of neurologic and neuropsychiatric disease in ALS kindreds: A population based case controlled cohort study of Familial and Sporadic ALS. . Ann. Neurol. 2013, 1–33. [Google Scholar]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 248–264. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clinical Epidemiology 2015, 7. [Google Scholar]
- Neumann, M. Frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Molecular similarities and differences. Rev. Neurol. (Paris) 2013, 169, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: from genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Imbert, G.; et al. Cloning of the gene for spinooerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef]
- K. Sanpei et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Rüb, U.; Auburger, G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008, 7, 115–124. [Google Scholar] [CrossRef]
- Magaña, J.J.; Velázquez-Pérez, L.; Cisneros, B. Spinocerebellar ataxia type 2: Clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 2013, 47, 90–104. [Google Scholar] [CrossRef]
- Sequeiros, J.; Seneca, S.; Martindale, J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur. J. Hum. Genet. 2010, 18, 1188–1195. [Google Scholar] [CrossRef]
- Fan, H.C.; et al. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef]
- Lee, T.; et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 2011, 20, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Van Langenhove, T.; et al. Ataxin-2 polyQ expansions in FTLD-ALS spectrum disorders in Flanders-Belgian cohorts. Neurobiol. Aging 2012, 33, 1004.e17–1004e20. [Google Scholar] [CrossRef]
- Neuenschwander, A.G.; Thai, K.K.; Figueroa, K.P.; Pulst, S.M. Amyotrophic Lateral Sclerosis Risk for Spinocerebellar Ataxia Type 2 ATXN2 CAG Repeat Alleles: A Meta-analysis. JAMA Neurol. 2014, 71, 1529–1534. [Google Scholar] [CrossRef]
- Lahut, S.; et al. ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS - A systematic review and meta-analysis of observational studies. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Borghero, G.; et al. ATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestry. Neurobiol. Aging 2015, 36, 2906–e1. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.; et al. Role for ATXN1, ATXN2, and HTT intermediate repeats in frontotemporal dementia and Alzheimer’s disease. Neurobiol. Aging 2020, 87, 139–e1. [Google Scholar] [CrossRef]
- Rubino, E.; et al. ATXN2 intermediate repeat expansions influence the clinical phenotype in frontotemporal dementia. Neurobiol. Aging 2019, 73, 231–e7. [Google Scholar] [CrossRef]
- Fournier, C.; et al. Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol. Commun. 2018, 6, 41. [Google Scholar] [CrossRef]
- Chio, A.; et al. Exploring the phenotype of Italian patients with ALS with intermediate ATXN2 polyQ repeats. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.D.; et al. ATXN2 intermediate expansions in amyotrophic lateral sclerosis. Brain 2022, 145, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, W.; et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 2017, 51, 178–e1. [Google Scholar] [CrossRef]
- Henden, L.; et al. Short tandem repeat expansions in sporadic amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Adv. 2023, 9. [Google Scholar] [CrossRef]
- Dejesus-Hernandez, M.; et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Cleary, E.M.; et al. Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol. Cell. Probes 2016, 30, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Daoud, H.; Belzil, V.; Martins, S.; Sabbagh, M. Association of Long ATXN2 CAG Repeat Sizes With Increased Risk of Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 739–742. [Google Scholar] [CrossRef]
- Gispert, S.; et al. The modulation of Amyotrophic Lateral Sclerosis risk by Ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol. Dis. 2012, 45, 356–361. [Google Scholar] [CrossRef]
- Conforti, F.L.; et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012, 79, 2315–2320. [Google Scholar] [CrossRef] [PubMed]
- Tavares de Andrade, H.M.; et al. Intermediate-length CAG repeat in ATXN2 is associated with increased risk for amyotrophic lateral sclerosis in Brazilian patients. Neurobiol. Aging 2018, 69, 292–e15. [Google Scholar] [CrossRef]
- Lattante, S.; et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 2014, 83, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 2011, 20, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.A.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; et al. ATNX2 is not a regulatory gene in Italian amyotrophic lateral sclerosis patients with C9ORF72 GGGGCC expansion. Neurobiol. Aging 2016, 39, 218–e5. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014, 35, 2421–e13. [Google Scholar] [CrossRef]
- Van Wijk, I.F. Assessment of risk of ALS conferred by the GGGGCC hexanucleotide repeat expansion in C9orf72 among first-degree relatives of patients with ALS carrying the repeat expansion. Amyotroph. Lateral Scler. Front. Degener. 2023, 9. [Google Scholar] [CrossRef]
Figure 1.
(a) Graphical representation of the percentage of patients carrying each CAG expansion allele in ATXN2 gene. (b) Frequency of CAG expansion alleles in ATXN2 in ALS, FTD and control groups. Close-up view of 27 to 36 region.
Figure 1.
(a) Graphical representation of the percentage of patients carrying each CAG expansion allele in ATXN2 gene. (b) Frequency of CAG expansion alleles in ATXN2 in ALS, FTD and control groups. Close-up view of 27 to 36 region.
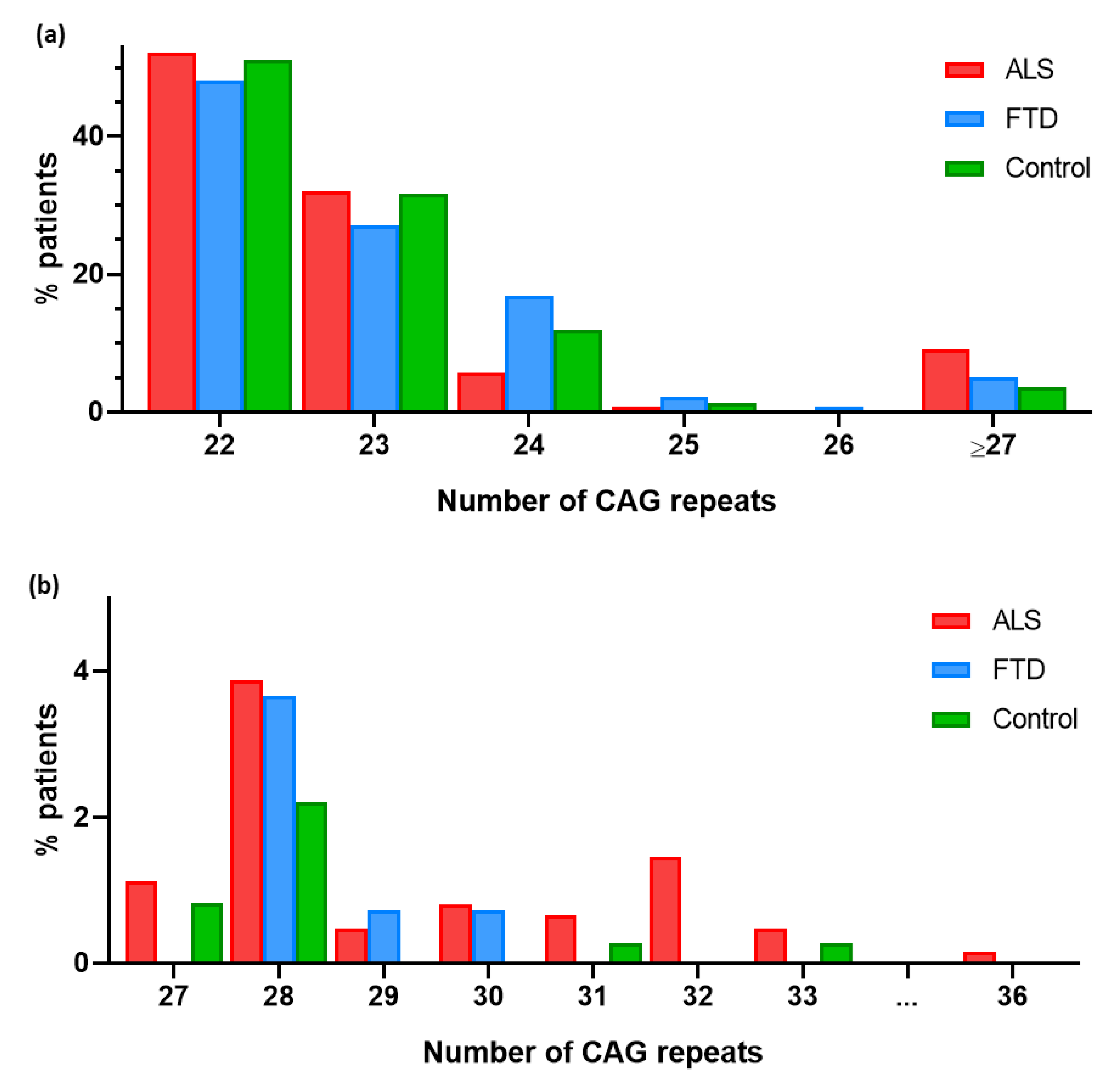
Figure 2.
Kaplan–Meier curves representing the survival of amyotrophic lateral sclerosis patients according to number of ATXN2 repeats (≥ or <27). Survival data was available from 29 out of 56 ALS patients with ≥ 27 RE and from 295 out of 564 <27 RE.
Figure 2.
Kaplan–Meier curves representing the survival of amyotrophic lateral sclerosis patients according to number of ATXN2 repeats (≥ or <27). Survival data was available from 29 out of 56 ALS patients with ≥ 27 RE and from 295 out of 564 <27 RE.
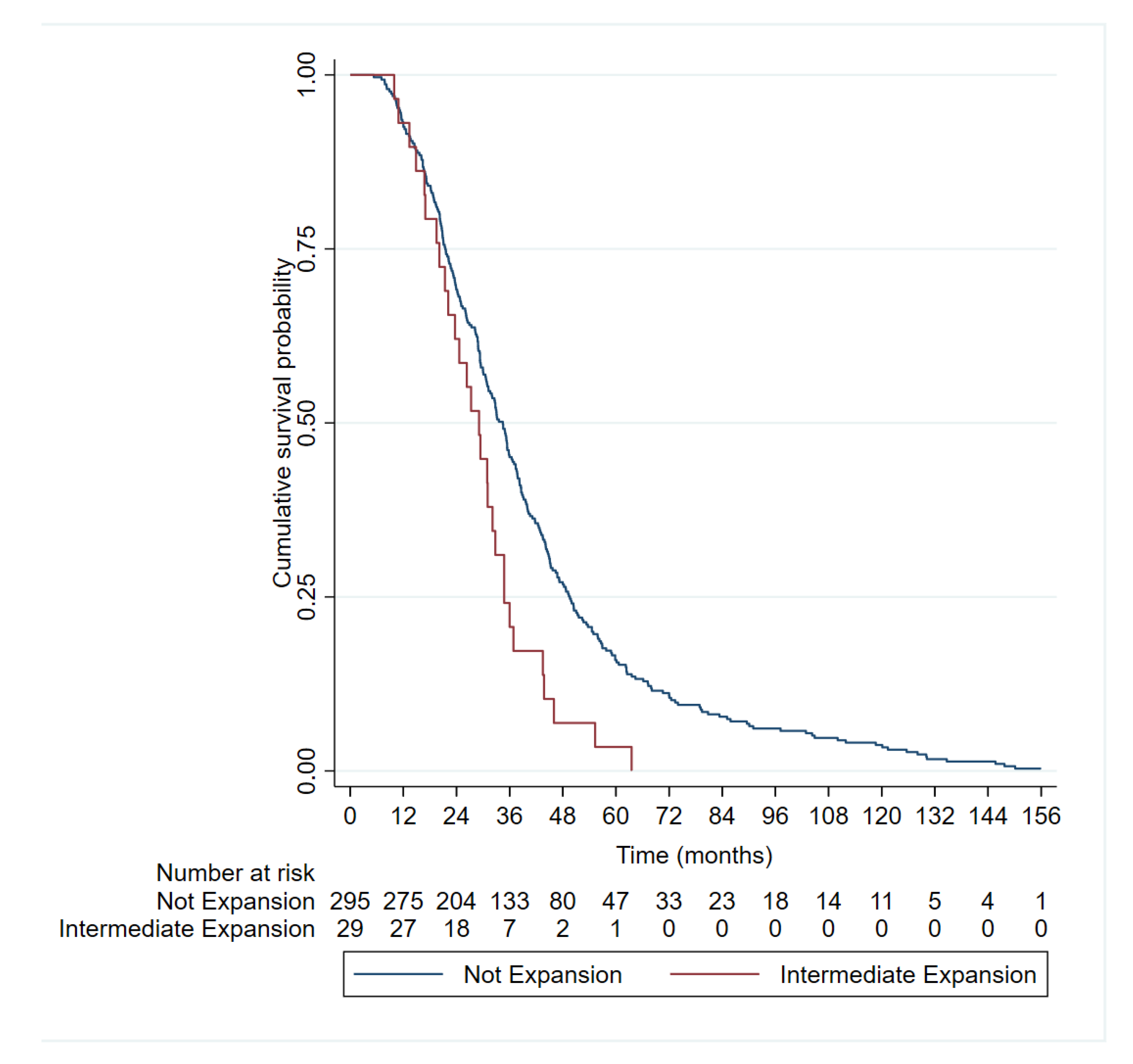
Figure 3.
Kaplan-Meier curves representing the categorical variables included in the multivariable Cox regression survival model.
Figure 3.
Kaplan-Meier curves representing the categorical variables included in the multivariable Cox regression survival model.
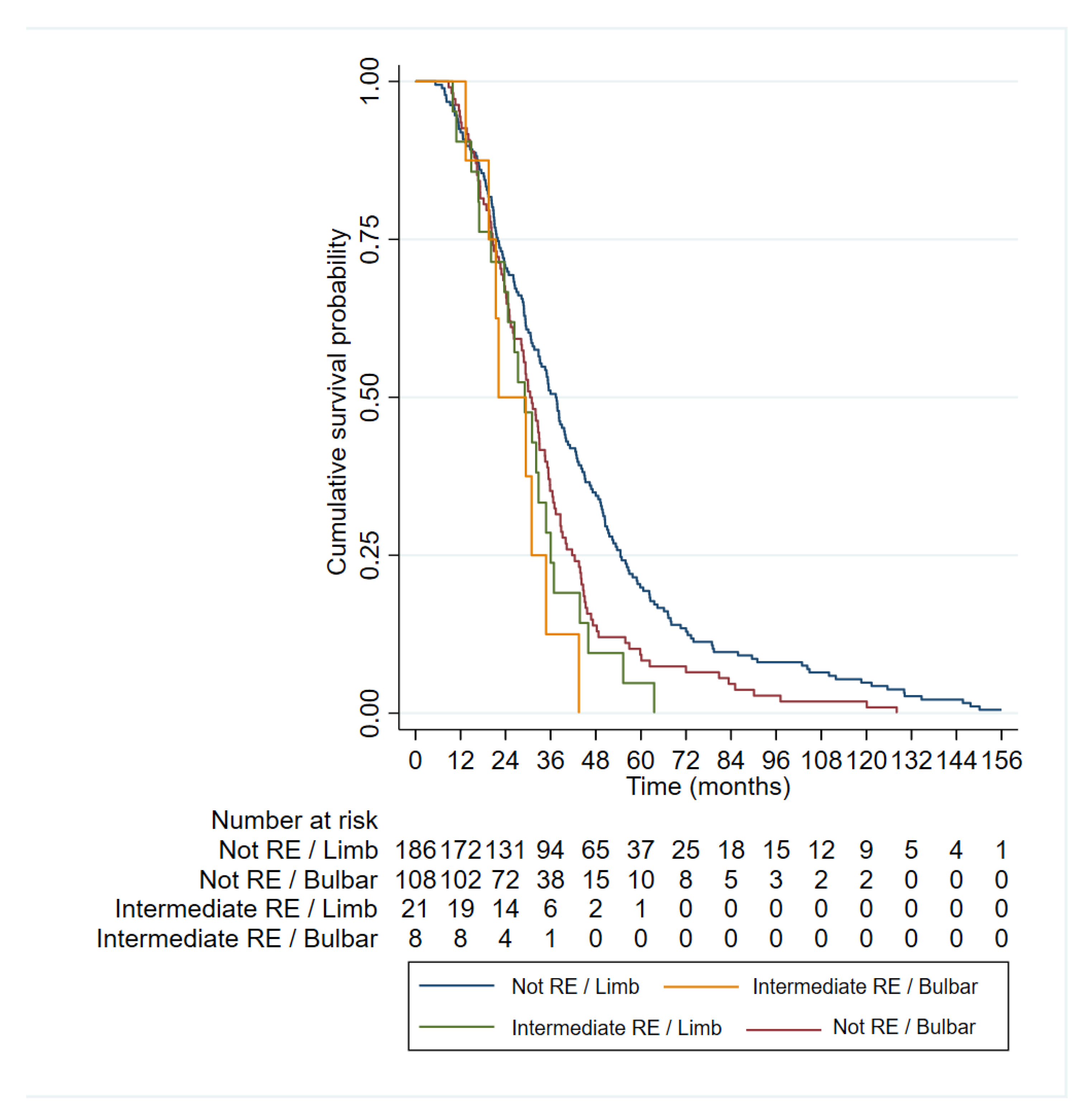
Table 1.
Demographic and clinical characteristics of ALS patients, FTD patients and controls.
| Clinical Characteristics | ALS | FTD | Controls |
|---|---|---|---|
| Comorbidity, n (%) | 87 (14.03%) | 18 (13.14%) | - |
| Male sex, n (%) | 337 (54.35%) | 81 (59.12%) | 175 (48.34%) |
| Limb onset, n (%) | 415 (66.94%) | - | - |
| Familial, n (%) | 142 (22.90%) | 38 (27.74%) | - |
| ATXN2 ≥27 CAG repeats, n (%) | 56 (9.03%) | 7 (5.11%) | 13 (3.59%) |
| C9orfF72 expansion, n (%) | 60 (9.68%) | 7 (5.11%) | 0 (0.00%) |
| ATXN2 ≥27 CAG repeats and C9orfF72 expansion, n (%) | 6 (10.0%) | 1 (14.3%) | 0 (0.00%) |
| Age of onset (years, mean ± SD (range) | 59.87 ± 13.45 (16.4 – 95.6) | 63.16 ± 9.85 (39.3 – 81.8) | 56.32 ± 15.64 (18.5 – 85.8)* |
| Survival (months, mean ± SD) (range) | 39.67 ± 27.30 (5.3 – 156.1) |
99.32 ± 51.90 (21.8 – 216.0) | - |
| Total ALS samples | 620 | 137 | 362 |
*In control group, age at sample collection.
Table 2.
CAG repeat expansion alleles in ATXN2 in control group and ALS patients, p-value and odds ratio.
Table 2.
CAG repeat expansion alleles in ATXN2 in control group and ALS patients, p-value and odds ratio.
| CAG repeats | ALS | Control | p-valuea | Odds ratiob |
|---|---|---|---|---|
| ≥27 CAG repeats | 56 (9.03%) | 13 (3.59%) | 0.0006 | 2.666 [1.471-4.882] |
| ≥28 CAG repeats | 49 (7.90%) | 10 (2.76%) | 0.0005 | 3.021 [1.547-6.309] |
| ≥29 CAG repeats | 25 (4.03%) | 2 (0.55%) | 0.0005 | 7.563 [2.098-32.550] |
a Fisher test (statistical significance p <0.01); b 95% confidence interval.
Table 3.
Clinical and genetic characteristics of ALS patients with the different positive cut-offs for RE ATXN2 gene.
Table 3.
Clinical and genetic characteristics of ALS patients with the different positive cut-offs for RE ATXN2 gene.
| ALS | ||||||
|---|---|---|---|---|---|---|
| <27 | ≥27 | <28 | ≥28 | <29 | ≥29 | |
| Mean age of onset (years) | 59.95 | 59.16 | 59.96 | 58.66 | 59.81 | 61.35 |
| Mean Survival (months) | 40.69 | 29.23 | 40.47 | 29.68 | 39.90 | 33.66 |
| C9orf72+ (n) | 54 (9.57%) | 6 (10.71%) | 54 (9.46%) | 6 (12.24%) | 58 (9.75%) | 2 (8%) |
| Other genes + (n) | 94 (16.66%) | 10 (18.52%) | 94 (16.46%) | 10 (20.41%) | 100 (16.81%) | 4 (16%) |
| Male/Female (n/n; Ratio) | 309/254 (1.217) | 28/28 (1) | 310/260 (1.192) | 27/22 (1.227) | 324/270 (1.2) | 13/12 (1.083) |
| Limb/Bulbar (n/n; Ratio) | 374/176 (2.125) | 41/11 (3.727) | 378/179 (2.111) | 37/8 (4.625) | 396/184 (1.467) | 29/3 (6.333) |
| Family history (+/-; %) | 122/442 (21.63%) | 20/36 (35.71%) | 122/449 (21.37%) | 20/29 (40.28%) | 131/464 (22.02%) | 11/14 (44.00%) |
| Cognitive impairment (yes/no; %) | 79/485 (14.01%) | 7/49 (12.5%) | 79/492 (13.84%) | 7/42 (14.29%) | 83/512 (13.95%) | 3/22 (12.00%) |
+/-: Refers to the number of patients with and without family history, respectively. n/n: Refers to the number of males and females; limb and bulbar onset, respectively, within those categories.
Table 4.
Cox model assessing those co-variables influencing the survival in ALS patients.
| Univariable | Multivariable | |||||
|---|---|---|---|---|---|---|
| Variable | HR | CI (95%) | p-value | HR | CI (95%) | p-value |
| ≥27 repeats | 1.74 | 1.181 - 2.563 | 0.005 | 1.782 | 1.209 - 2.628 | 0.004 |
| Bulbar onset | 1.415 | 0.538 - 0.856 | 0.001 | 1.400 | 1.105 – 1.775 | 0.004 |
| Age of onset | 1.016 | 1.007 - 1.026 | 0.001 | 1.015 | 1.005 - 1.824 | 0.003 |
| FTD | 0.992 | 0.7748 - 1.270 | 0.949 | - | - | - |
| Female Sex | 1.253 | 1.003 - 1.565 | 0.046 | - | - | - |
HR: Hazard Ratio. CI: Confidence interval.
Table 5.
CAG repeat expansion in ATXN2 in control group and FTD patients, p-value and odds ratio.
| CAG repeats | FTD | Control | p-valuea | Odds ratiob |
|---|---|---|---|---|
| ≥27 CAG repeats | 7 (5.11%) | 13 (3.59%) | 0.220 | 1.446 [0.558 -3.574] |
| ≥28 CAG repeats | 7 (5.11%) | 10 (2.76%) | 0.099 | 1.895 [0.7295 -5.241] |
| ≥29 CAG repeats | 2 (1.46%) | 2 (0.55%) | 0.155 | 2.667 [0.4134 -17.12] |
a Fisher test, b 95% confidence interval.
Table 6.
Intermediate CAG repeat expansion in ATXN2 gene associated with ALS extracted from 12 studies with statistical determinations.
Table 6.
Intermediate CAG repeat expansion in ATXN2 gene associated with ALS extracted from 12 studies with statistical determinations.
| Study | CAG repeat number | p-valuea | Odds ratiob |
|---|---|---|---|
| North America [15] | 27-33 | 0.000036 | 2.8 [1.54-5.12] |
| Europe [22] | >30 | 0.0062 | - |
| France and Canada [37] | ≥29 | 0.00024 | 5.5 [1.9-15.9] |
| Europe [38] | >30 | 0.004 | 5.74 [4.26-7.22] |
| Flanders [23] | 27-33 | 0.012 | - |
| South Italy [39] | ≥28 | 0.001 | 5.832 [1.71–9.78] |
| Turkey [25] | >30 | 0.01721 | - |
| Meta-analysis [24] | 29-33 | - | >1 |
| Europe and North America [26] | 30-33 | 0.0001 | 4.44 [2.91-6.76] |
| Sardinia [27] | ≥31 | 0.0001 | - |
| Brasil [40] | ≥26 | 0.005 | 2.56 [1.29-5.08] |
| Europe [32] | ≥31 | 9.50 x 10-7 | 6.93 [3.19-15.02] |
| Spain (present study) | ≥27 | 0.0013 | 2.666 [1.471-4.882] |
Statistical determinations include p-value and Odds ratio (if present). a Fisher test, b 95% confidence interval.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



