
Submitted:
25 December 2023
Posted:
26 December 2023
You are already at the latest version
Abstract
Malaxidinae is one of the confusing groups with classification in Orchidaceae. Previous phylogenetic analysis reveals that the relationships among the taxa in Malaxidinae have not yet been reliably established using a few plastome regions or nuclear ribosomal internal transcribed spacer (nrITS). In the present study, the complete plastomes of Oberonia integerrima and Crepidium purpureum were assembled using high-throughput sequencing. Combined with publicly available complete plastome data, this resulted in a data set of 19 plastomes, including 17 species of Malaxidinae. The plastome feature and phylogenetic relationship were compared and analyzed. The results showed that: (1) The plastomes of Malaxidinae species possessed the quadripartite structure of typical angiosperms, with sizes ranging from 142,996 bp to 158,787 bp and encoding from 125 to 133 genes. The ndh genes were lost or pseudogenized with varying degrees in six species. An unusal inversion was detected in the Large Single-Copy region (LSC) of Oberonioides microtatantha. (2) A total of 8 regions, including ycf1, matK, rps16, rpl32, ccsA-ndhD, clpP-psbB, trn-FGAA-ndhJ, and trnSGCU-trnGUCC, were identified as mutational hotspots. (3) Based on complete plastomes, 68 protein-coding genes and 51 intergenic regions, respectively, the phylogenetic analyses resolved genera-level relationships in this subtribe with strong support. The Liparis was supported as polyphyletic.
Keywords:
Malaxidinae
; plastid genome
; comparative genomics
; phylogeny
1. Introduction
The subtribe Malaxidinae (Orchidaceae, Malaxideae), consisting of approximately 1,250 species in 14 genera, is sister to Dendrobiinae [1,2,3]. The species of Malaxidinae are terrestrial, occasionally epiphytic, rarely holomycotrophic, and characterized by fleshy stems, terminal and racemose inflorescences, and small flowers [4]. This subtribe is primarily distributed in the tropics and subtropics, with only a few genera extending into temperate regions (Liparis, Malaxis, and Oberonia) [3]. Malaxidinae possesses remarkable ornamental value due to its distinctive flowers. Additionally, this subtribe holds importance in traditional Chinese medicine, plant chemistry, and pharmacology [5,6].
Malaxidinae species show limited morphological variations, leading to unclear taxonomic boundaries at species and genera level, making them a challenging group for taxonomic research [4,7,8,9,10]. Previous studies based on traditional molecular markers have consistently shown that the phylogenetic relationships of Malaxidinae had moderate to low support values and highly unstable topological structures. Cameron et al. [4] proposed the division of Malaxidinae into at least seven genera using the nuclear ITS and plastid matK regions. The phylogenetic analysis also revealed the Oberonia was monophyletic, while Liparis s.l. (including terrestrial and epiphytic) plus Malaxis s.l. were polyphyletic. Based on four markers, ITS, matK, trnS-trnG and trnL-trnF, the result of Tsutsumi et al. [11] showed Liparis was polyphyletic, as Malaxis species were nested within Liparis. Other phylogenetic analyses of Malaxideae mainly employed ITS and matK, with extended sampling include Crepidium, Crossoglossa, Crossoliparis, Dienia, Hippeophyllum, Stichorkis, and Tamayorkis [2,12,13,14,15,16,17]. Despite the expansion of genera sampling in these studies, the issue of non-monophyly in Liparis, Malaxis, and Crepidium, as well as low support values and unstable topological structures, remains unresolved. The phylogenetic relationships of Malaxidinae were still difficult to determine, necessitating alternative approaches to increase resolution.
The plastome, characterized by a high copy number per cell and a comparatively smaller size, which makes complete sequencing much more tractable. Due to the lack of recombination, low rates of nucleotide substitutions, and usually uniparental inheritance [18,19], the plastome has found widespread application in addressing issues such as node collapse and low support values based on traditional molecular markers [20,21,22,23]. A limited number of Malaxidinae plastome sequences, including Liparis, Oberonia, and Oberonioides, have been published in GenBank database (https://www.ncbi.nlm.nih.gov/) [24,25,26,27,28]. However, detailed analyses of plastome composition and structure, and comparisons with each other have not yet been conducted. In this study, we present two newly sequenced plastomes of Oberonia integerrima and Crepidium purpureum and compared with a total of 17 previously published Malaxidinae plastome sequences (from 15 species). Following this, we analyzed the differences in genome size, content, and structure, the inverted repeats (IR) contraction and expansion, and codon-usage bias, identifying the sequence divergence, along with variant hotspot regions. In addition, we reconstructed the phylogenetic trees of Malaxidinae based on plastome datasets. The present results provide a useful genetic resource for molecular identification and evolutionary studies of Malaxidinae.
2. Results
2.1. Plastome Feature and Genome Rearrangement
The lengths of newly sequenced plastomes of Oberonia integerrima and Crepidium purpureum were 146,620 bp and 158,780 bp, the GC content was 36.8% and 37.4%, respectively (Figure 1). Among the typical quadripartite structure of these two species, the length and percentage of the large single copy (LSC) region were 83,791 bp (57.15%) and 86,383 bp (54.40%), the inverted repeat (IR) region were 25,821 bp (17.61%) and 27,015 bp (17.01%), the small single copy (SSC) region were 11,187 bp (7.63%) and 18,367 bp (11.57%), respectively.
In O. integerrima, a total of 126 annotated genes and 75 protein-coding genes were detected, ndhC, ndhF, ndhG, ndhI, ndhA, and ndhH were lost, while ndhJ, ndhK, ndhB, and ndhD were pseudogenized. However, the ndh gene was neither lost or pseudogenized in C. purpureum, a total of 132 annotated genes and 86 protein-coding genes were detected. A total of 38 transfer RNA (tRNA) and eight ribosomal RNA (rRNA) were annotated in these two species (Supplementary Table S1).
In total, 19 plastomes of Malaxidinae, including Liparis, Crepidium, Oberonioides, and Oberonia, were analyzed in this study. The plastomes in length ranged from 142,996 bp (O. japonica) to 158,787 bp (L. nervosa), with a GC content of 36.8% to 37.4%. All plastomes displayed a typical quadripartite structure, the length and percentage of the LSC region ranged from 81,669 bp (57.11%) to 86,752 bp (54.82%), the IR region ranged from 24,003 bp (16.56%) to 27,268 bp (17.17%), and the SSC region ranged from 10,224 bp (7.15%) to 18,367 bp (11.57%).
The annotated genomes encoded 125–132 genes, of which O. japonica and O. seidenfadenii were the least genes, containing 74–86 protein-coding genes, 38 tRNA genes, and eight rRNA genes (Supplementary Table S1). The ndh gene was lost or pseudogenized to varying degrees in six species, including Oberonia integerrima, O. japonica, O. seidenfadenii, Oberonioides microtatantha, L. japonica, and L. yongnoana. Furthermore, the rearrangements of plastome were analyzed by Mauve. An inversion, approximately 18.5 kb of rpl33-rps3 region, was identified in the LSC region of Oberonioides microtatantha (Figure 2).
2.2 IR Region Border Analysis
The IR/single copy (SC) region border in Malaxidinae plastomes showed a relatively conserved structure (Figure 3). Except for rpl22 of L. japonica was located in the LSC region, rpl22 of other species spanned the IRb/LSC (JLB) boundary. The IRb/SSC (JSB) boundary was located in ndhF, except for four species of Oberonia and Oberonioides—the JSB boundary was located between the trnNGUU and rps15. The ycf1 gene was located in the SSC region in Oberonioides microtatantha and O. integerrima, but in other species were spanned the IRa/SSC (JSA) junction. The IRa/LSC (JLA) border of all species was located between rps19 and psbA.
2.3 Codon Usage Analysis
After extracting the protein-coding genes and removing the repeated genes, the matrices of 68 unique coding genes were obtained. The 68 protein-coding genes encoded 19,247–19,464 codons among the 19 plastomes (Supplementary Table S2). The most frequent amino acid was leucine (Leu), while the lowest was cysteine (Cys). Additionally, the synonymous codon usage (RSCU) values were calculated, ranging from 0.360 to 1.899, and the heatmap revealed that codon frequencies were largely similar within Malaxidinae. The GCU codon had the highest RSCU value (1.844–1.899), while GAC exhibited the lowest RSCU value (0.360–0.381) (Figure 4, Supplementary Table S2). Among three termination codons, UAA, UAG and UGA, the RSCU value of UAA codon (1.368–1.544) was highest (Supplementary Table S2).
2.4 Nucleotide Mutation Hotspots
The sequence identity was compared and plotted using the program mVISTA. The results showed that the protein-coding genes were highly conserved than the intergenic regions (Figure 5). The IR regions were more conserved than the LSC and SSC regions. The total length of 68 unique protein-coding genes was 69,810 bp, variable sites (V) were 7,153 (accounting for 10.24% of the total length) and parsimony informative sites (Pi) were 3,798 (5.44%). Among the individual protein-coding gene, the variable sites values ranged from 0.81% (rps12) to 23.86% (ycf1) and the Pi values ranged from 0% (psbF) to 14.53% (ycf1) (Figure. 6, Supplementary Table S3), and the ycf1, matK, rps16, and rpl32 genes exhibited higher Pi values (14.53%, 11.84%, 11.53%, and 10.92%). Regarding 51 unique intergenic regions, the combined dataset had a length of 30,003 bp, of which the variable sites was 7,694 (25.64%) and the Pi was 4,137 (13.79%) (Figure. 6, Supplementary Table S3). The Pi values ranged from 2.54% (rps7-ndhB) to 24.00% (ccsA-ndhD), notably, the ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC regions had higher Pi values (24.00%, 22.31%, 22.05% and 21.62%).
2.5 Phylogenetic Analysis
Three matrices, comprising 68 protein-coding genes (Figure 7A), 51 intergenic regions (Figure 7B) and complete plastomes (Figure 8), were employed to reconstruct the phylogenetic relationship of Malaxidinae. Three phylogenetic trees exhibited consistent topologies, except for the relationship among three species of Oberonia (Figure 7, Figure 8). The bootstrap values for most nodes were ≥ 85 in maximum likelihood (ML) and maximum parsimony (MP) analyses and were ≥ 0.99 in Bayesian inference (BI) analysis. The phylogenetic relationship of complete plastome (Figure 8) indicated that newly sequenced O. integerrima formed monophyletic group with other two species (O. japonica and O. seidenfadenii). This monophyletic group was sister to the clade containing L. viridiflora, L. bootanensis, and L. pingtaoi. Additionally, the other newly sequenced species, C. purpureum, was strongly supported as sister to L. vivipara and L. nervosa. Furthermore, the Oberonioides was sister to the clade of Liparis containing L. auriculata, L. loeselii, L. pauliana, L. kumokiri, L. japonica, L. yongnoana, and L. makinoana. All these results supported that Liparis was polyphyletic.
Phylogenetic trees were constructed using the top four hotspots of protein-coding genes (ycf1, matK, rps16, and rpl32) and the top four hotspots of intergenic regions (ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC), respectively, as selected by Pi value. Apart from the phylogenetic relationship within Oberonia, the topological structures of the two ML trees exhibited three distinctions compared to the reconstruction based on the complete plastomes. In the ML tree based on the top-four hotspots of protein-coding genes, L. bootanensi was sister to L. pingtaoi with weak support and short branch length, and this pair was sister to L. viridiflora (Figure 9A). In the ML tree using the top-four hotspots of intergenic regions, the clade, including C. purpureum, L. vivipara and L. nervosa, had strong support as sister to the Oberonia, L. pingtaoi, L. bootanensis, and L. viridiflora (Figure 9B). And Oberonioides microtatantha was sister to L. auriculata, moreover, this pair was sister to L. loeselii and its allies (Figure 9B). A total of 23 branch nodes, the bootstrap values for 20 nodes were ≥ 85 in ML tree based on the combination of four protein-coding genes, while, there were 21 nodes in ML tree based on the combination of four intergenic regions.
3. Discussion
3.1 Plastome Structure Conservation and Divergence
In this study, the lengths of sequenced plastome of O. integerrima and C. purpureum were 146,620 bp and 158,780 bp, respectively, falling well within the range of autotrophic Orchidaceae plastome size, from 142,859 bp (Schoenorchis seidenfadenii) to 178,131 bp (Cypripedium formosanum) [20,29]. In terms of the plastome structure feature, Malaxidinae species exhibited a typical quadripartite structure observed in angiosperm plastomes, containing LSC, SSC and two copies of IR. The size of the plastome ranged from 142,996 bp (O. japonica) to 158,787 bp (L. nervosa); the range of genes number was 125 to 132; all the species possessed 38 tRNA and 8 rRNA genes (Supplementary Table S1). All of these characteristics were consistent with previously reported autotrophic species of Orchidaceae [21,30,31]. The plastome size of Oberonia and Oberonioides (<14.7 kb) was smaller than that of Liparis and Crepidium (>15.0 kb) (Supplementary Table S1). This discrepancy could be attributed to the loss or pseudogenization of the ndh genes, which were related to photosynthesis.
Previous studies have indicated that the loss or pseudogenization of ndh genes in the plastid genome was a common phenomenon in autotrophic Orchidaceae species [29,32,33]. Such patterns were generally associated with lifestyle and they have occurred more frequently in plastid of epiphytic or lithophytic species, such as Aeridinae, Bulbophyllum, Cymbidium and Dendrobium [20,30,34,35]. In this study, Oberonia and Oberonioides were epiphytic or lithophytic, suggesting a significant association between the ndh gene loss or pseudogenization and epiphytic life forms. While it was noteworthy that L. japonica and L. yongnoana, two terrestrial species, also displayed the loss of ndh genes, which was worth to further investigation to better understand this phenomenon.
It was generally assumed that the changes in IR regions serve as the main factors for the difference in the plastome size. Moreover, the expansion and contraction at the IR/SC boundary were the common evolutionary phenomenon [36,37]. Among Malaxidinae plastomes, the JLA and JLB boundaries—locating in psbA and rpl22 (except L. japonica), respectively—were highly conserved (Figure 3). In comparison to the Liparis and Crepidium, the loss of ndhF of Oberonia and Oberonioides resulted in the location of the JSB boundary shifting to trnNGUU-rps15 and leading to a contraction of the SSC region. Consequently, the plastomes of Oberonia and Oberonioides exhibited smaller genome sizes (Figure 3, Supplementary Table S1).
The codons played a crucial role in genetic information transmission, mRNA stability, and the accurate expression of protein functions [38]. Different species exhibited varying degrees of codon bias, influenced by selective pressures and translation efficiency during evolution [39,40]. In this study, the codon usage bias of Malaxidinae was similar overall. The Leu precented the highest frequency of amino acids and showed it was closely associated with photosynthesis-related metabolism [41]. Conversely, the Cys showed the lowest, as excessive accumulation of Cys could lead to the occurrence of cellular oxidative damage and toxic responses [42]. For termination codons, UAA codon was the more preferred codon than UAG and UGA (Supplementary Table S2), which was similar to other Orchidaceae species [40,43].
Previous studies provided evidence that the combined dataset of mutational hotspots regions was effective for identification analyses in some groups [30,44]. In our study, the protein-coding genes displayed more conserved than the intergenic regions. Specifically, the ycf1, matK, rps16, and rpl32 genes among the 68 protein-coding genes and the ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC regions among the 51 intergenic regions were identified as mutation hotspots, respectively (Figure 6, Supplementary Table S3). According to ML analyses with the two combinations, the most branch nodes had strong support. About the four protein-coding genes combinations, the phylogenetic relationship for the main clades of Malaxidinae showed well correspond with the complete plastome-based phylogeny. While, the topology of the phylogenetic tree based on the combination of intergenic regions indicated more significant inconsistencies. Therefore, the combination, containing ycf1, matK, rps16, and rpl32, was more ideal molecular markers or phylogenetic and species identification analyses within the genus of Malaxidinae.
3.2 Phylogenetic Relationships
The molecular-based phylogenetic relationships of Malaxidinae were inconsistent with the traditional morphological classification. Cameron et al. [4] constructed the phylogenetic relationships and combined with the lifestyle to support the division of Malaxidinae into two major clades: one of terrestrial clade and the other of epiphytic clade. Likewise, Margońska et al. [10], Salazar et al. [13], Li et al. [16], and Kumar et al. [17] expanded the sampling to include additional genera. The molecular phylogenetic reconstructions exhibited that Liparis, Malaxis, Oberonia, and Crepidium were embedded within each other, and were multiple genera confirmed as polyphyletic. However, the phylogenetic relationships commonly showed unstable topologies with moderate to weak support. Some authors have proposed to exclude the non-monophyly taxa of Liparis and Malaxis into several small genera to ensure the monophyly of this group. While the unreliable topologies might reflect the unreliable phylogenetic relationships, resulting in wrong taxonomic revision.
Our phylogenetic study utilized high-throughput sequencing to obtain complete platome. The phylogenetic relationships were performed using ML, MP, and BI methods with complete plastomes, protein-coding genes and intergenic regions. The results showed that the topology structures were almost the same within ML, MP and BI analysis, further most branch nodes were strongly supported (BS ≥ 85, PP ≥ 0.99) in three phylogenetic trees (Figure 7, Figure 8). This finding provides robust evidence supporting the utility of plastid genomes as molecular markers and a practical approach for phylogenetics in this subtribe.
Considering the lifestyle of the species (Supplementary Table S4), our phylogenetic trees demonstrated that Malaxidinae was clustered into two major clades: epiphytic clade and terrestrial clade (Figure 8), which strongly congruence with previously results [4,10,17]. In three phylogenetic analyses, the relationships among O. integerrima, O. japonica, and O. seidenfadenii varied slightly with moderate support values (Figure 7, Figure 8). The analysis confirmed that Crepidium, Oberonia, and Oberonioides were nested within Liparis, indicating that Liparis was polyphyletic (Figure 8). These results suggested the need for redefining the relationships among these genera, which required broader sampling to extend the phylogenomic datasets. Overall, our phylogenomic analyses significantly improved the stability of topology structure and support values of the plastid phylogenomic than previous studies.
4. Materials and Methods
4.1 Sample Preparation, Sequencing and Data Acquisition
In this study, Oberonia integerrima and Crepidium purpureum were sampled and newly sequenced. Voucher specimen was deposited in the herbarium of the Forestry College of Fujian Agriculture and Forestry University (FJFC) under the specimen code MHLi or119 and MHLi or110, respectively. Additionally, 17 published sequences (15 species) of Malaxidinae and six sequences as outgroup were downloaded from GenBank database (Supplementary Table S4).
The total DNA was extracted from fresh leaves by Plant Mini Kit (Qiagen. CA. USA) based on the manufacturer’s protocol. The DNA integrity was evaluated by electrophoresis on 1% agarose gels, and DNA sample preparations with over 1 μg DNA per sample were selected. The total DNA samples were randomly sheared, and the libraries for paired-end 150 bp sequencing were conducted using an Illumina HiSeq 4000 platform to generate approximately 20 Gb of raw data for per sample.
4.2 Plastome DNA Assembly, Annotation and Comparison
The raw data was filtered for low-quality sequencing reads to obtain plastid-like reads. The whole plastome sequences were assembled by GetOrganelle pipeline v1.7.1 [45] with a reference of published plastome, L. bootanensis (MN627759). The “fastg” and “cvs” files were imported to Bandage [46], then the low-quality fragments were filtered and edited to obtain the circular plastomes. With the reference of L. bootanensis, the assembled plastomes were annotated by Plastid Genome Annotator (PGA) [47], and the results were further corrected and adjusted using the Dual Organellar GenoMe Annotator (DOGMA) [48]. For high-quality annotation plastomes, we used Geneious 11.1.5 (https://www.geneious.com) to align with L. bootanensis as the reference, including manually adjusting the position of the initiation and termination codons and examining the loss or pseudogenization about ndh genes. The published sequences of GenBank were re-annotated by the same steps. The 19 visible circle annotation results were drawn with the online tool OGDRAW (http://ogdraw.mpimp-golm.mpg.de/) [49].
4.3 Plastome Features Analysis
The annotated plastome genome information about 19 species of Malaxidinae and six species of outgroup was collected, including the gene length, GC content, size of inverted repeat (IR), large single copy (LSC) and small single copy (SSC), as well as the number of genes and ndh gene loss/pseudogenization. With the reference, L. bootanensis, the complete plastomes of Malaxidinae were aligned using mVISTA in Shuffle-LAGAN mode [50] to assess the variability of sequences. The boundary regions of LSC/IRb/SSC/IRa were analyzed with IRscope (https://irscope.shinyapps. io/irapp) [51]. The rearrangement and inversion of plastome were detected and graphed with Mauve [52].
4.4 Codon Usage and Nucleotide Mutation Hotspots Analysis
All protein-coding genes and intergenic regions of 25 samples (23 species) were extracted by PhyloSuit v1.2.1 [53]. Then we deleted one copy gene of the IR and ndh genes, and retained the matrix with more than 20 class groups, resulting in a dataset of 68 protein-coding genes and 51 intergenic regions. These genes and regions were aligned individually by MAFFT v7 [54] with default parameters, and concatenated using Phylosuite v1.2.1. The variable site (V) and parsimony information site (Pi) were counted using MAFFT v7 to determine highly variable regions of Malaxidinae. The relative synonymous codon usage (RSCU) of 19 plastomes of Malaxidinae and codon frequencies were calculated using DAMBE [55]. And a heatmap was generated using Tbtools [56].
4.5 Phylogenetic Analysis
The phylogenetic relationships were analyzed with three alignment matrices: complete plastomes, protein-coding genes and intergenic regions using maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI). Under the GTRGAMMA model of evolution [57], ML analyses were conducted on the CIPRES Science Gateway (RAxML-HPC2 on XSEDE 8.2.12) [58]. Bootstrap iterations were performed with 1000 bootstrap replicates using heuristic searches [59], with the other parameters to default values. For MP analysis, 1000 random addition sequence replicates were used with a heuristic search about 1000 tree-bisection-reconnection (TBR) branch-swapping, treating all characters as unordered and of equal weight [60]. BI analysis was performed using MrBayes v.3.2.6 with the GTR + I + Γ substitution model [61]. For the reliability of results, the Markov chain Monte Carlo (MCMC) algorithm was run for 10,000,000 generations, with one tree sampled every 100 generations. The first 25% of the resulting trees were discarded as burn-in to construct majority-rule consensus trees and estimate posterior probabilities (PP).
Based on nucleotide mutation hotspots analysis, the top four mutational hotspots from both protein-coding genes and intergenic regions were identified separately by Pi value. Two sets of these hotspots were concatenated using Phylosuite v1.2.1 [53]. And the phylogenetic relationships were constructed with these two alignment matrices using ML, the detailed steps were referred to in the preceding text. Then the ideal molecular marks were identified by the comparison with the complete plastome-based phylogeny.
5. Conclusions
In this study, two plastomes of O. integerrima and C. purpureum were newly sequenced. Except for an inversion with rpl33-rps3 region in Oberonioides microtatantha, the plastome structures in Malxidinae were highly conserved. The plastome size of Oberonia and Oberonioides was smaller than Liparis and Crepidium due to the loss or pseudogenization of ndh genes and the contraction of the SSC region. A total of four protein-coding genes (ycf1, matK, rps16, rpl32) and four intergenic spacers (ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC) were identified as mutation hotspots. The combination for ycf1, matK, rps16, and rpl32, could be used in the phylogeny and identification with the geus of Malxidinae. The phylogenomic analysis strongly supported the determination that Malaxidinae was clustered into two clades, i.e., the epiphytic clade and the terrestrial clade. The genus Liparis was shown to be polyphyletic.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Z.-J.L. and S.L.: Conceptualization. M.-Y.Z.: Methodology, Software; M.-Y.Z., M.-H.L. and J.S.: Data curation, Writing—Original draft preparation, Writing—Reviewing and editing. J.S., J.Z. and Y.Z.: Validation; Resources. All authors read and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Key Research and Development Program of China (2023YFD1600504).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are provided within this manuscript and supplementary materials.
Acknowledgments
We acknowledge the technical support by lab staff during the conduction of lab experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pridgeon, A.M.; Cribb, P.J.; Chase, M.W.; Rasmussen, F.N. Genera Orchidacearum Volume 6: Epidendroideae (Part 1). Oxford University Press: Oxford, UK, 2005.
- Chase, M.W.; Cameron, K.M.; Freudestein, J.V.; Pridgeon, A.M.; Salazar, G.; Van den Berg, C.; Schuiteman, A. An update classification of Orchidaceae. Bot. J. Linn. Soc. 2015, 177, 151–174. [Google Scholar] [CrossRef]
- Govaerts, R.J.; Bernet, P.; Kratochvil, K.; Gerlach, G.; Carr, G.; Alrich, P.; Pridgeon, A.M.; Pfahl, J.; Campacci, M.A.; Holland Baptista, D.; et al. 2021. World checklist of Orchidaceae. The Board of Trustees of the Royal Botanic Gardens, Kew. Available at http://www.kew.org/wcsp/monocots/ [accessed November 2021].
- Cameron, K.M. Leave it to the leaves: a molecular phylogenetic study of Malaxideae (Epidendroideae, Orchidaceae). Am. J. Bot. 2005, 92, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Guo, X.; Nagle, D.G.; Zhang, W.D.; Tian, X.H. Genus Liparis: A review of its traditional uses in China, phytochemistry and pharmacology. J. Ethnopharmacol. 2019, 234, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xie, Y.G.; Guo, Y.G.; Yan, S.K.; Jin, H.Z. Chemical constituents of Liparis viridiflora. Chem. Nat. Compd. 2019, 55, 552–554. [Google Scholar] [CrossRef]
- Szlachetko, D.L. Systema orchidalium. Fragm. Florist. Geobot. Pol. 1995, 3, 1–152. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evol. Int. J. Org. Evol. 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Margońska, H.B. Crossoliparis—a new genus of Malaxidinae (Orchidaceae, Malaxideae), from neotropic. Acta Soc. Bot. Pol. 2009, 78, 297–299. [Google Scholar] [CrossRef]
- Margońska, H.B.; Kowalkowska, A.K.; Górniak, M.; Rutkowski, P. Taxonomic redefinition of the subtribe Malaxidinae (Orchidales, Malaxideae). Koeltz Scientific Books: Koenigstein, Germany, 2012; pp. 1–606.
- Tsutsumi, C.; Yukawa, T.; Lee, N.S.; Lee, C.S.; Kato, M. Phylogeny and comparative seed morphology of epiphytic and terrestrial species of Liparis (Orchidacea) in Japan. J. Plant Res. 2007, 120, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yan, H. A remarkable new species of Liparis (Orchidaceae) from China and its phylogenetic implications. PLoS ONE 2013, 8, e78112. [Google Scholar] [CrossRef]
- Salazar, J.R.; Salazar, G.A.; Cabrera, L.I.; Nez-Machorro, R.; Batista, J.A. A new paludicolous species of Malaxis (Orchidaceae) from Argentina and Uruguay. Phytotaxa 2014, 175, 121–132. [Google Scholar] [CrossRef]
- Tang, G.D.; Zhang, G.Q.; Hong, W.J.; Liu, Z.J.; Zhuang, X.Y. Phylogenetic analysis of Malaxideae (Orchidaceae: Epidendroideae): Two new species based on the combined nrDNA ITS and chloroplast matK sequences. Guihaia 2015, 35, 447–463. [Google Scholar]
- Li, M.H.; Zhang, G.Q.; Lan, S.R.; Liu, Z.J. A molecular phylogeny of Chinese orchids. J. Syst. Evol. 2016, 54, 349–362. [Google Scholar] [CrossRef]
- Li, L.; Chung, S.W.; Li, B.; Zeng, S.J.; Yan, H.F.; Li, S.J. New insight into the molecular phylogeny of the genus Liparis s.l. (Orchidaceae: Malaxideae) with a new generic segregate: Blepharoglossum. Plant Syst. Evol. 2020, 306, 1–10. [Google Scholar] [CrossRef]
- Kumar, P.; Li, J.; Gale, S.W. Integrative analyses of Crepidium (Orchidaceae, Epidendroideae, Malaxideae) shed more light on its relationships with Dienia, Liparis and Malaxis and justify reinstatement of narrow endemic C. allanii. Bot. J. Linn. Soc. 2022, 198, 285–305. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Kai, F.M.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Liu, D.K.; Tu, X.D.; Zhao, Z.; Zeng, M.Y.; Zhang, S.; Ma, L.; Zhang, G.Q.; Wang, M.M.; Liu, Z.J.; Lan, S.R; et al. Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma–Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 2020, 145, 106729. [Google Scholar] [CrossRef]
- Tu, X.D.; Liu, D.K.; Xu, S.W.; Zhou, C.Y.; Gao, X.Y.; Zeng, M.Y.; Zhang, S.; Chen, J.L.; Ma, L.; Zhou, Z.; et al. Plastid phylogenomics improves resolution of phylogenetic relationship in the Cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, Orchidaceae). Mol. Phylogenet. Evol. 2021, 164, 107269. [Google Scholar] [CrossRef]
- Chen, J.L.; Wang, F.; Zhou, C.Y.; Ahmad, S.; Zhou, Y.Z.; Li, M.H.; Liu, Z.J.; Peng, D.H. Comparative phylogenetic analysis for Aerides (Aeridinae, Orchidaceae) based on six complete plastid genomes. Int J Mol Sci. 2023, 24, 12473. [Google Scholar] [CrossRef]
- Yan, R.; Gu, L.; Qu, L.; Wang, X.; Hu, G. New Insights into phylogenetic relationship of Hydrocotyle (Araliaceae) based on plastid genomes. Int. J. Mol. Sci. 2023, 24, 16629. [Google Scholar] [CrossRef]
- Huang, H.X.; Chen, L.J; Liu, Z.J.; Li, M.H. Liparis vivipara (Orchidaceae: Malaxideae), a new species from China: evidence from morphological and molecular analyses. Phytotaxa 2018, 351, 289–295. [Google Scholar] [CrossRef]
- Ha, Y.H.; Gil, H.Y.; Lee, J.; Kim, D.K.; Choi, K.; Chang, K.S.; Oh, S.H. The complete chloroplast genome sequence of Liparis yongnoana, an endemic orchid of Korea. Mitochondrial DNA B Resour. 2019, 4, 2666–2667. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, J.F.; Chen, M.H. The complete chloroplast genome sequence of Oberonia seidenfadenii (Orchidaceae), a rare plant species endemic to China. Mitochondrial DNA B Resour. 2019, 4, 3362–3363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.H.; Wang, X.T.; Zheng, S.Z.; Zhu, W.Y.; Song, Z. The complete chloroplast genome of Liparis nervosa (Orchidaceae). Mitochondrial DNA B Resour. 2020, 5, 123–124. [Google Scholar] [CrossRef]
- Liu, C.Q.; Kang, N.; Liu, X.; Chen, Y.; Tao, Y.; Zhang, Y.; Zhang, Y.Y.; Li, Y.L.; Tang, G.D.; Li, Y.L. Complete plastid genome sequence of Oberonioides microtatantha (Schltr.) Szlach.(Orchidaceae), an endemic herb in China. Mitochondrial DNA B Resour. 2021, 6, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.W.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Zhu, S.; Pan, J.; Li, L.; Sun, J.; Ding, X. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 2017, 7, 2073. [Google Scholar]
- Zhao, Z.; Zeng, M.Y.; Wu, Y.W.; Li, J.W.; Zhou, Z.; Liu, Z.J.; Li, M.H. Characterization and comparative analysis of the complete plastomes of five Epidendrum (Epidendreae, Orchidaceae) species. Int. J. Mol. Sci. 2023, 24, 14437. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.W.; Chiu, C.C.; Hsiao, H.C.W.; Yang, C.J.; Jin, X.H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.T.; Yang, L.H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017, 90, 994–1006. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Zeng, M.Y.; Gao, X.; Zhao, Z.; Li, R.; Wu, Y.; Liu, Z.J.; Zhang, D.; Li, M.H. Characteristics and comparative analysis of seven complete plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae). Int. J. Mol. Sci. 2023, 24, 14544. [Google Scholar] [CrossRef]
- Zavala-Páez, M.; Vieira, L.D.N.; Baura, V.A.D.; Balsanelli, E.; Souza, E.M.D.; Cevallos, M.C.; Smidt, E.D.C. Comparative plastid genomics of neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front. Plant Sci. 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.P.; Dou, S.J.; Ji, Z.J.; Xue, Q.Z. Synonymous codon usage and gene function are strongly related in Oryza sativa. Biosystems 2005, 80, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.F.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon bias as a means to fine-tune gene expression. Molec. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, H.; Zhang, N.; Ma, J.; Wang, J.; Hu, G.; Li, M.; Zhao, P. Comparative analyses of chloroplast genomes provide comprehensive insights into the adaptive evolution of Paphiopedilum (Orchidaceae). Horticulturae 2022, 8, 391. [Google Scholar] [CrossRef]
- Knill, T.; Reichelt, M.; Paetz, C.; Gershenzon, J.; Binder, S. Arabidopsis thaliana encodes a bacterial-type heterodimeric isopropylmalate isomerase involved in both Leu biosynthesis and the Met chain elongation pathway of glucosinolate formation. Plant Mol. Biol. 2009, 71, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.; Nunes-Nesi, A.; Araujo, W.; Braun, H.-P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef]
- Xiao, T.; He, L.; Yue, L.; Zhang, Y.; Lee, S.Y. Comparative phylogenetic analysis of complete plastid genomes of Renanthera (Orchidaceae). Front. Genet. 2022, 13, 998575. [Google Scholar] [CrossRef]
- Smidt, E.C.; Páez, M.Z.; Vieira, L.D.N.; Viruel, J.; De Baura, V.A.; Balsanelli, E.; Maltempi de Souza, E.; Chase, M.W. Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS ONE 2020, 15, e227991. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Li, W.X.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z. DAMBE: software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools-an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 289660. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Freudenstein, J.V.; Rasmussen, F.N. What does morphology tell us about orchid relationships?—A cladistic analysis. Am. J. Bot. 1999, 86, 225–248. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP: phylogenetic analysis using parsimony and other methods. Version 4. Sinauer Associates, Sunderland, MA, USA, 2003.
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
Figure 1.
Annotation map of the plastomes of Oberonia integerrima (A) and Crepidium purpureum (B).
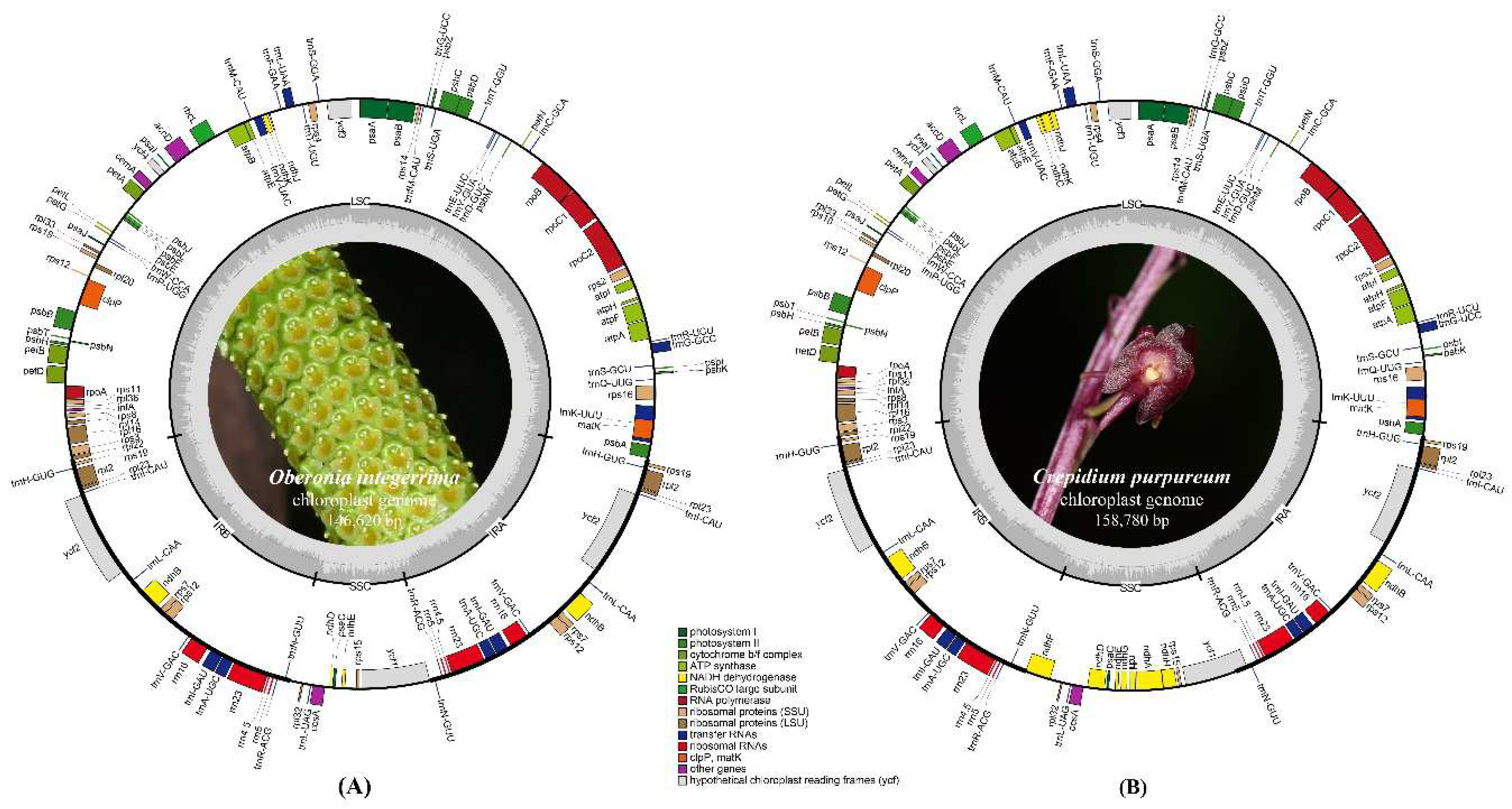
Figure 2.
Alignment of 19 Malaxidinae plastomes using MAUVE. Comparative gene maps showed an inversion in rpl33-rps3 region of Oberonioides microtatantha.
Figure 2.
Alignment of 19 Malaxidinae plastomes using MAUVE. Comparative gene maps showed an inversion in rpl33-rps3 region of Oberonioides microtatantha.
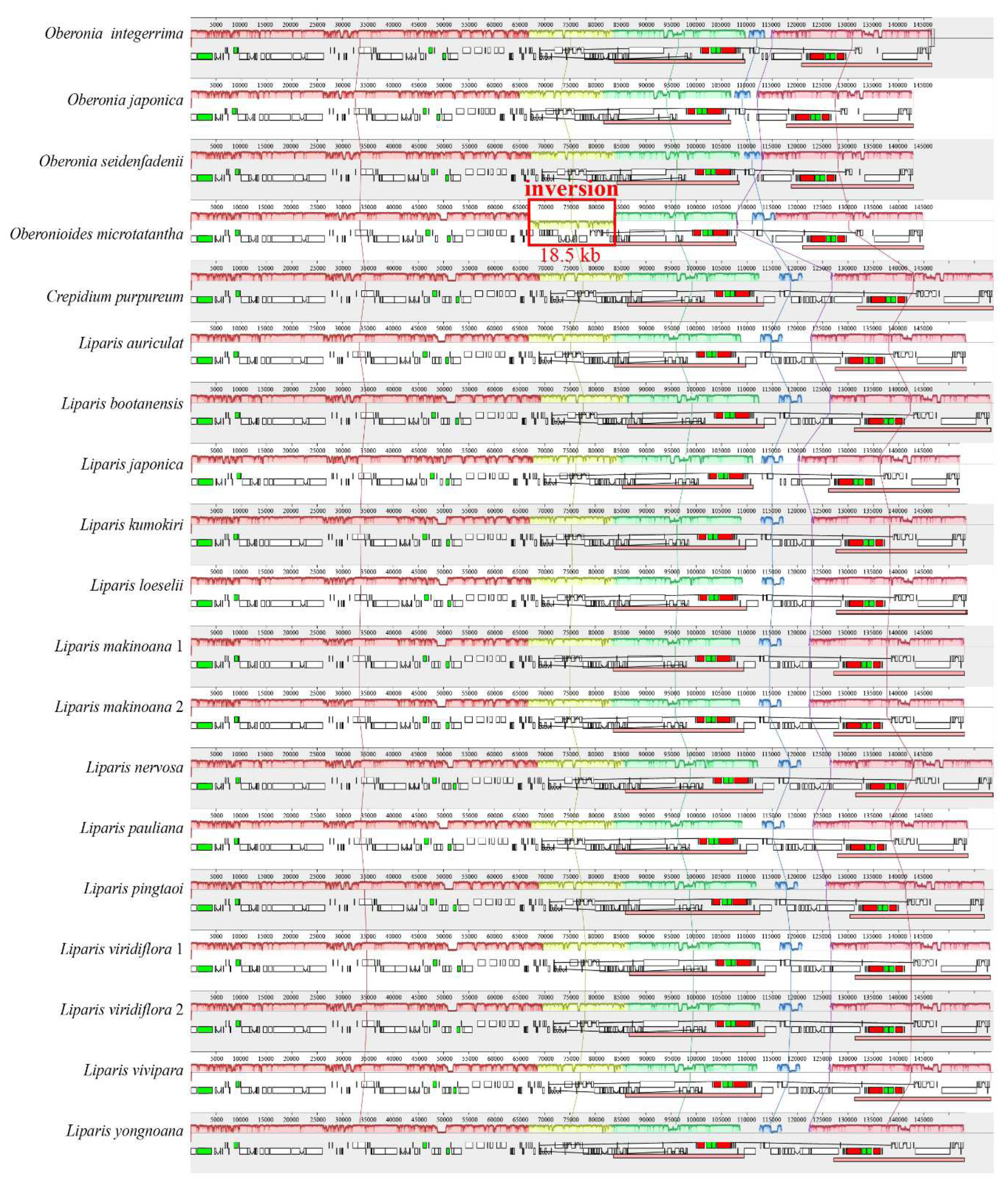
Figure 3.
Comparison of boundries between the LSC, SSC, and IR regions among 19 Malaxidinae plastomes.
Figure 3.
Comparison of boundries between the LSC, SSC, and IR regions among 19 Malaxidinae plastomes.

Figure 4.
The RSCU values of concatenated 68 protein-coding genes for Malaxidinae plastomes. Color key: the red values mean higher RSCU values and the blue values mean lower RSCU values.
Figure 4.
The RSCU values of concatenated 68 protein-coding genes for Malaxidinae plastomes. Color key: the red values mean higher RSCU values and the blue values mean lower RSCU values.

Figure 5.
mVISTA map of Malaxidinae plastomes with L. bootanensis as reference. The y-axis shows the coordinates between the plastomes.
Figure 5.
mVISTA map of Malaxidinae plastomes with L. bootanensis as reference. The y-axis shows the coordinates between the plastomes.
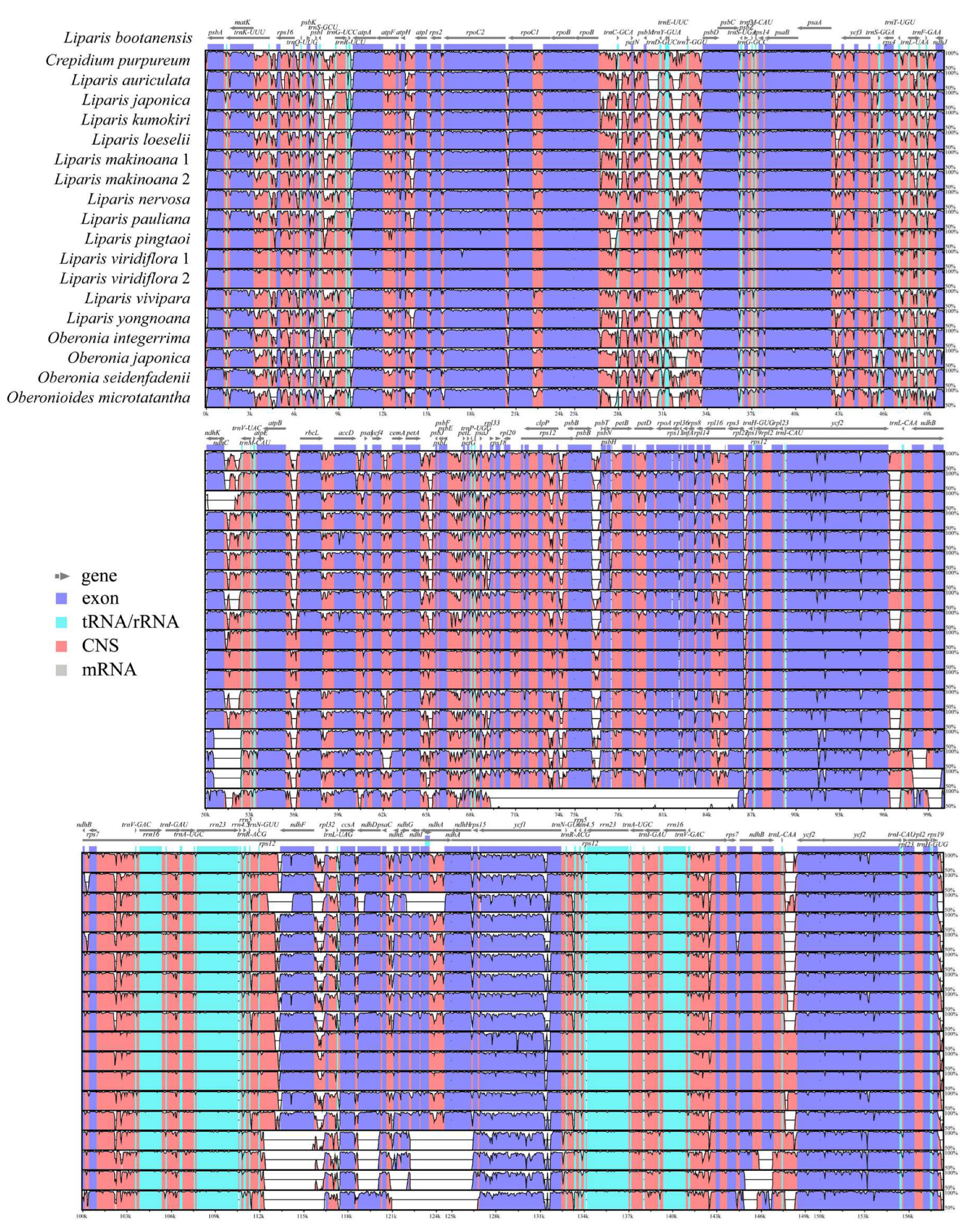
Figure 6.
The length of protein-coding region (A) and intergenic region (B), and the proportion of Variable-site (orange) and Parsimony-information site (green).
Figure 6.
The length of protein-coding region (A) and intergenic region (B), and the proportion of Variable-site (orange) and Parsimony-information site (green).
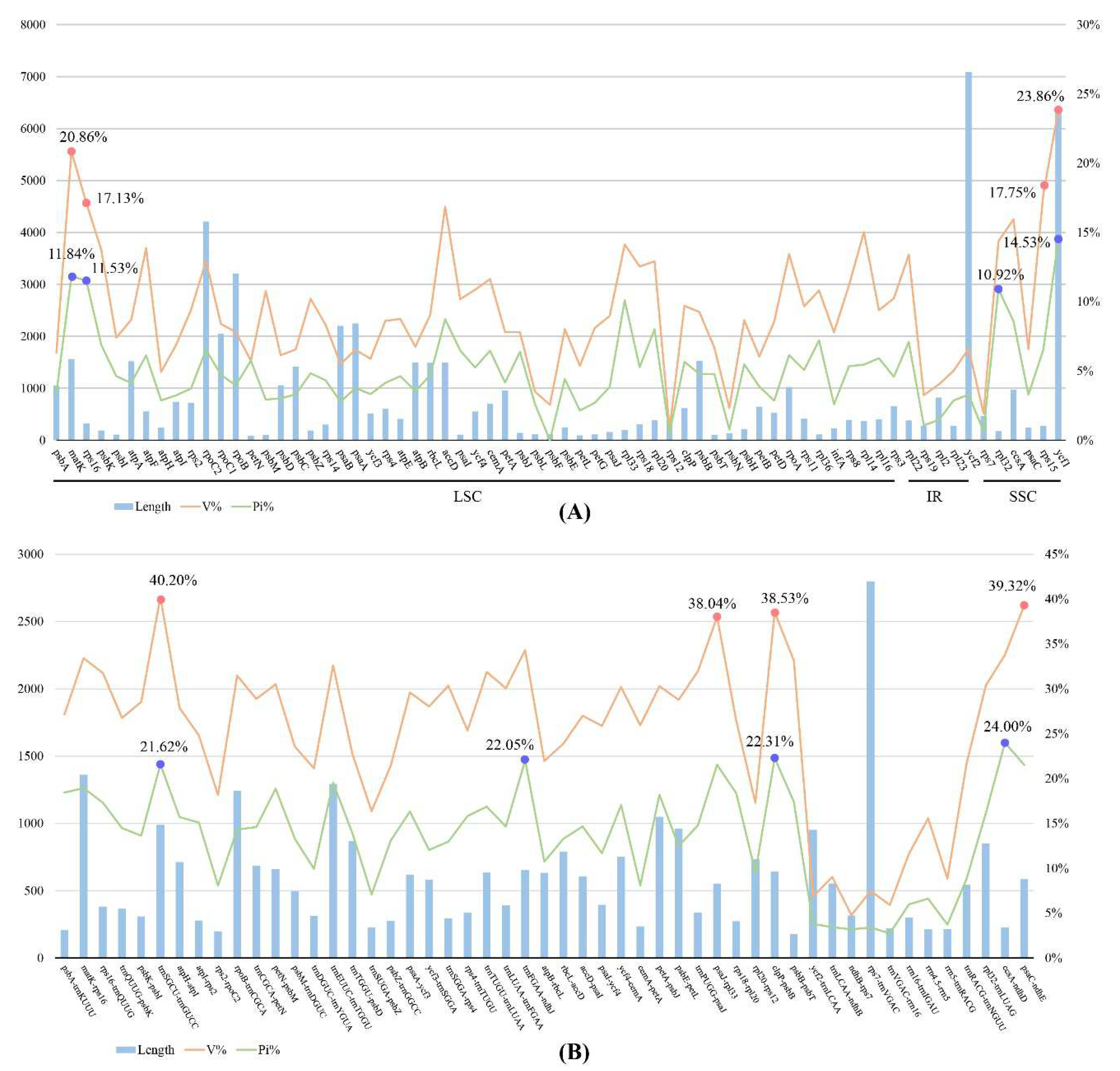
Figure 7.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood analysis based on 68 protein-coding region (A) and 51 intergenic regions (B). Numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BSML left, BSMP middle, and PP right). An asterisk (*) indicates the node has 100% bootstrap or 1.00 posterior probability.
Figure 7.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood analysis based on 68 protein-coding region (A) and 51 intergenic regions (B). Numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BSML left, BSMP middle, and PP right). An asterisk (*) indicates the node has 100% bootstrap or 1.00 posterior probability.
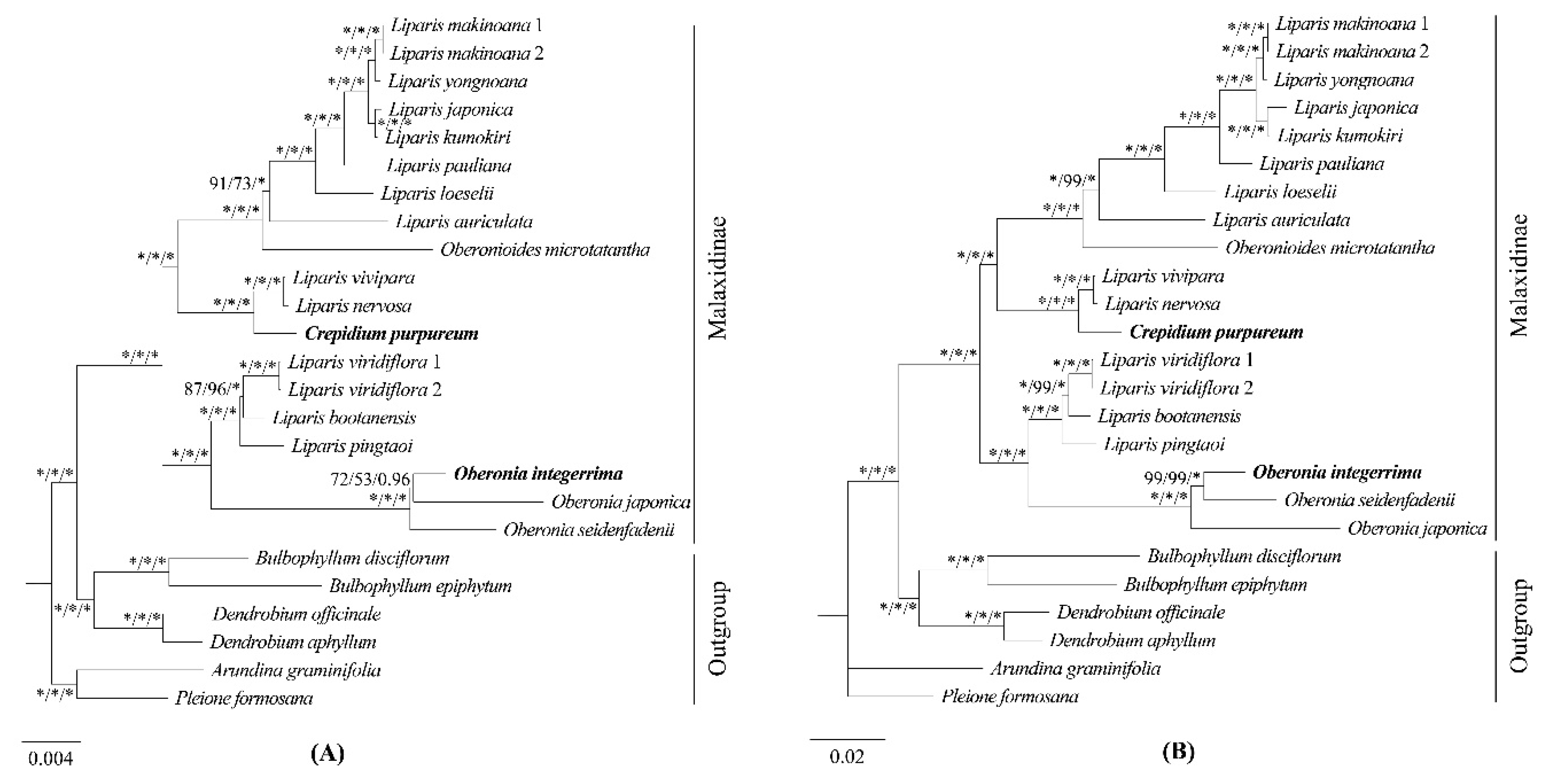
Figure 8.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood analysis based on whole plastome dataset. Numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BSML left, BSMP middle, and PP right). An asterisk (*) indicates the node has 100% bootstrap or 1.00 posterior probability. Green represents terrestrial clade, while orange represents epiphytic clade. The species names in bold indicate sequenced in this study.
Figure 8.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood analysis based on whole plastome dataset. Numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BSML left, BSMP middle, and PP right). An asterisk (*) indicates the node has 100% bootstrap or 1.00 posterior probability. Green represents terrestrial clade, while orange represents epiphytic clade. The species names in bold indicate sequenced in this study.
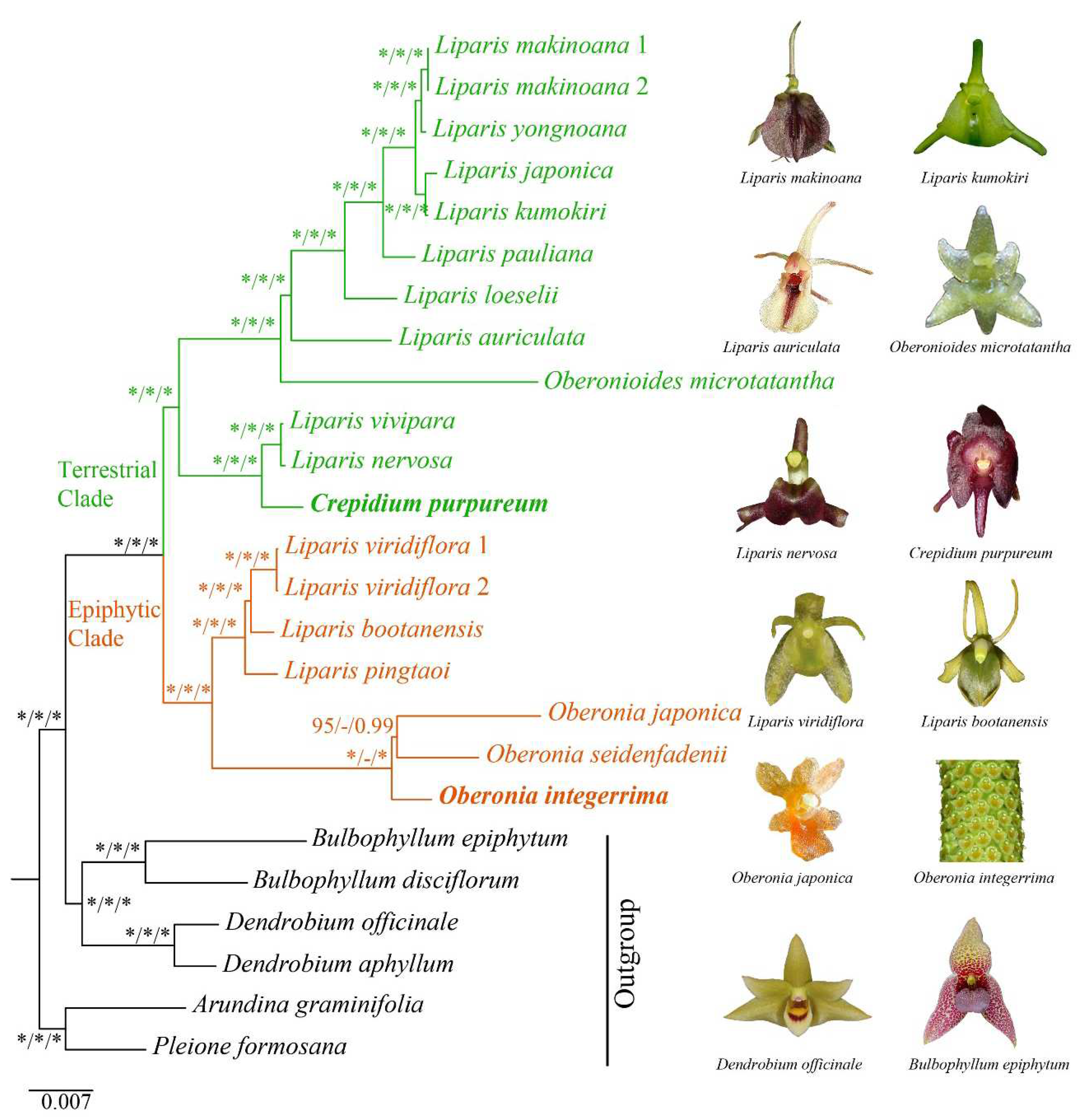
Figure 9.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood (ML) analysis based on the top-four hotspots of protein-coding genes (ycf1, matK, rps16, and rpl32) (A) and the top-four hotspots of intergenic regions (ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC) (B). Numbers near the nodes are bootstrap percentages of ML analysis. An asterisk (*) indicates the node has 100% bootstrap.
Figure 9.
Phylogenetic tree of Malaxidinae obtained by Maximum Likelihood (ML) analysis based on the top-four hotspots of protein-coding genes (ycf1, matK, rps16, and rpl32) (A) and the top-four hotspots of intergenic regions (ccsA-ndhD, clpP-psbB, trnFGAA-ndhJ, and trnSGCU-trnGUCC) (B). Numbers near the nodes are bootstrap percentages of ML analysis. An asterisk (*) indicates the node has 100% bootstrap.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



