
Submitted:
26 December 2023
Posted:
26 December 2023
You are already at the latest version
Abstract
Currently, the chemistry of conjugated nitrodienes is becoming more and more popular. These molecules are successfully applied in cycloaddition to synthesize six-membered rings in Diels-Alder reactions. Nitrodienes can be also applied to obtain bis-compounds in [3+2] cycloaddition. Moreover, the presence of a nitro group in the structure provides a possibility of further modification of the products. The simplest symmetrical representative of conjugated nitrodienes is (1E,3E)-1,4-dinitro-1,3-butadiene. Although the first mentions of the compound date back to the early 1950s, the compound has not yet been examined thoroughly enough. Therefore, in this article a comprehensive study of (1E,3E)-1,4-dinitro-1,3-butadiene has been described. For this purpose, an experimental study including synthesis process as well as spectral characteristic has been conducted. So as to better understand the properties of this compound a computational study of reactivity indices based on MEDT and also assessment of pharmacokinetics and biological activity according to ADME and PASS methodologies have been made. On this basis, some future application trends of (1E,3E)-1,4-dinitro-1,3-butadiene have been proposed.
Keywords:
nitrodiene
; synthesis
; spectral characteristics
; DFT reactivity indices
; ADME
; PASS
1. Introduction
Chemistry of nitro group containing compounds has been of great interest to scientists for many years [1]. These kinds of structures are extremely attractive building blocks in modern organic synthesis. It is due to the presence of a nitro group in the molecule, which creates many possibilities for its transformation. Thanks to this, it is possible to synthesize many nitrogen-containing connections like amines, hydroxylamines, oximes, nitriles, diazocompounds and others as well as to obtain carbonyl compounds [2]. Among all nitro compounds conjugated nitroalkenes (CNAs) deserve a special attention [3,4]. These compounds exhibit various biological activities such as antibacterial [5,6] or antifungal [7]. This makes CNAs often used in many reactions such as Michael addition [8,9] as well as cycloaddition (CA) reactions, to synthesize heterocycles, especially for the preparation of five-membered rings [10] like pyrazolines [11,12], isoxazolines [13,14,15,16] or pyrrolidine [17,18], and six-membered rings in the Diels-Alder reaction [19,20,21].
Recently, the chemistry of conjugated nitro dienes (CNDs) has gained greater interest [22,23]. Due to their structure, these compounds offer much more possibilities of transformation compared to CNAs [24].
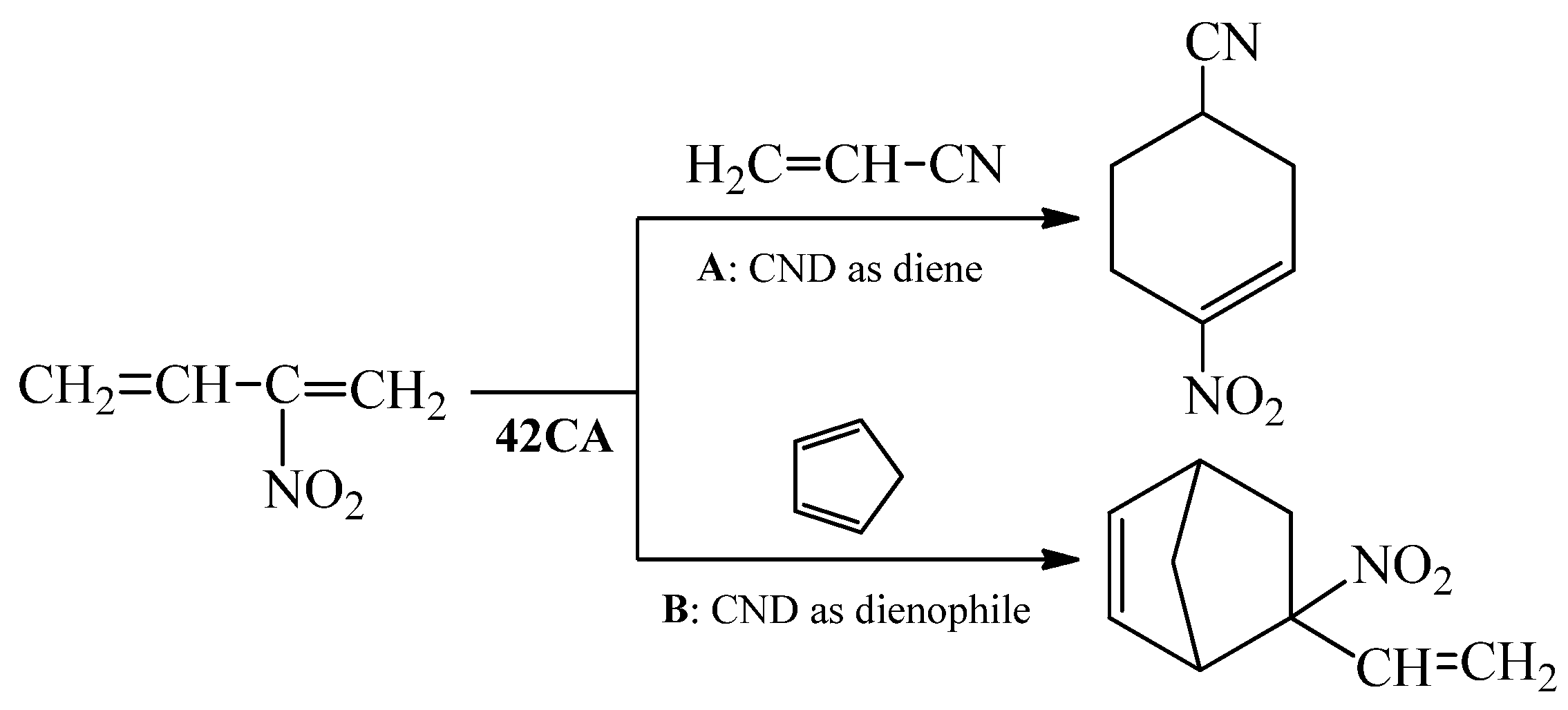
Undoubtedly, CNDs are primary component in the Diels-Alder ([4+2] cycloaddition, 42CA) reactions [25]. Most often, these compounds play a role of standard diene (Scheme 1, pathway A). However, there are known cases of reactions where CNDs are applied in Diels-Alder reaction as dienophiles (Scheme 1, pathway B).
Scheme 1.
Application of conjugated nitrodiene (CND) in Diels-Alder reactions in a double role.
What is more, CNDs can be also applied in reactions of [3+2] cycloaddition (32CA) with various three atoms components (TACs) [26]. In a consequence, it is possible to obtain a symmetric biheterocycles (Scheme 2) which may successfully use in medicine [27] as well as in optoelectronics [28] and also determine a useful ingredient for dielectric industry [29,30,31].
Scheme 2.
Application of conjugated nitrodiene (CND) in [3+2] cycloaddition reaction to obtain symmetric biheterocyclic system.
Scheme 2.
Application of conjugated nitrodiene (CND) in [3+2] cycloaddition reaction to obtain symmetric biheterocyclic system.
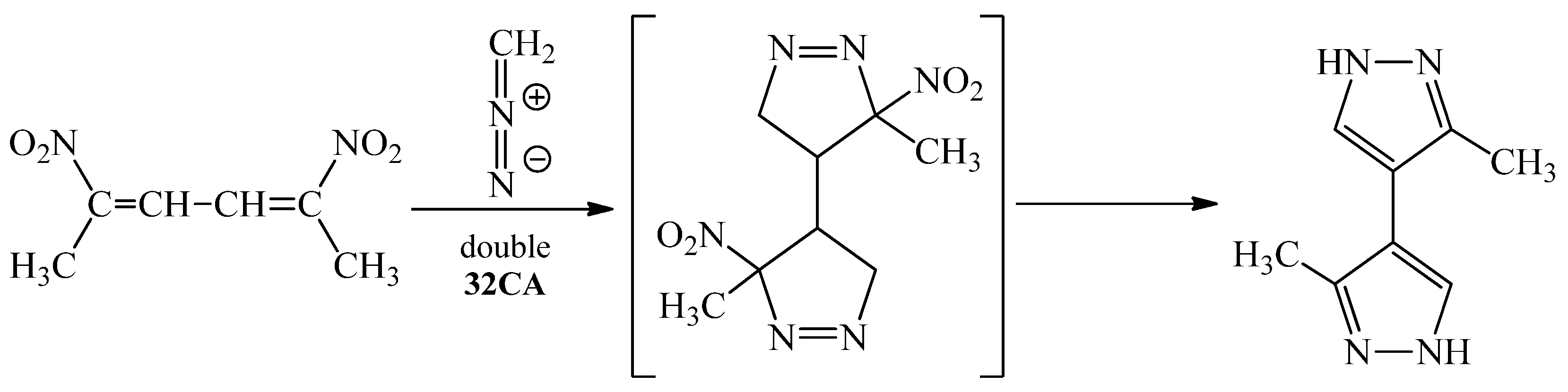
Examples of heterocycles obtained via processes of 6π-electrocyclization (6π-eˉ) of CNDs are also found in the literature [22]. This protocol of ring closure can be useful to synthesize cyclic nitronates, which may react with ethylene systems creating final products (Scheme 3) [32].
Scheme 3.
Application of conjugated nitrodiene CND 6π-electrocyclization processes to obtain cyclic nitronate.
Scheme 3.
Application of conjugated nitrodiene CND 6π-electrocyclization processes to obtain cyclic nitronate.
However, the number of examples of the use of CNDs in CA reactions as well as in 6π-eˉ processes is strongly limited. Reactions described in the literature that tackles the application of these compounds in organic synthesis are predominantly limited to nucleophilic additions and substitutions with thiols and amines, and less numerous examples of electrophilic additions of halogens [22,23,24,25]. Therefore, the use of CNDs as building blocks to obtain heterocyclic systems is an attractive course of research and determines one of the most promising trend in current organic synthesis.
Despite the fact that the chemistry of CNDs has been known since the middle of the twentieth century, there are still many gaps in the synthesis and spectral characteristics of these compounds [23]. What is more, numerous protocols and reports presented in the scientific literature are incomplete.
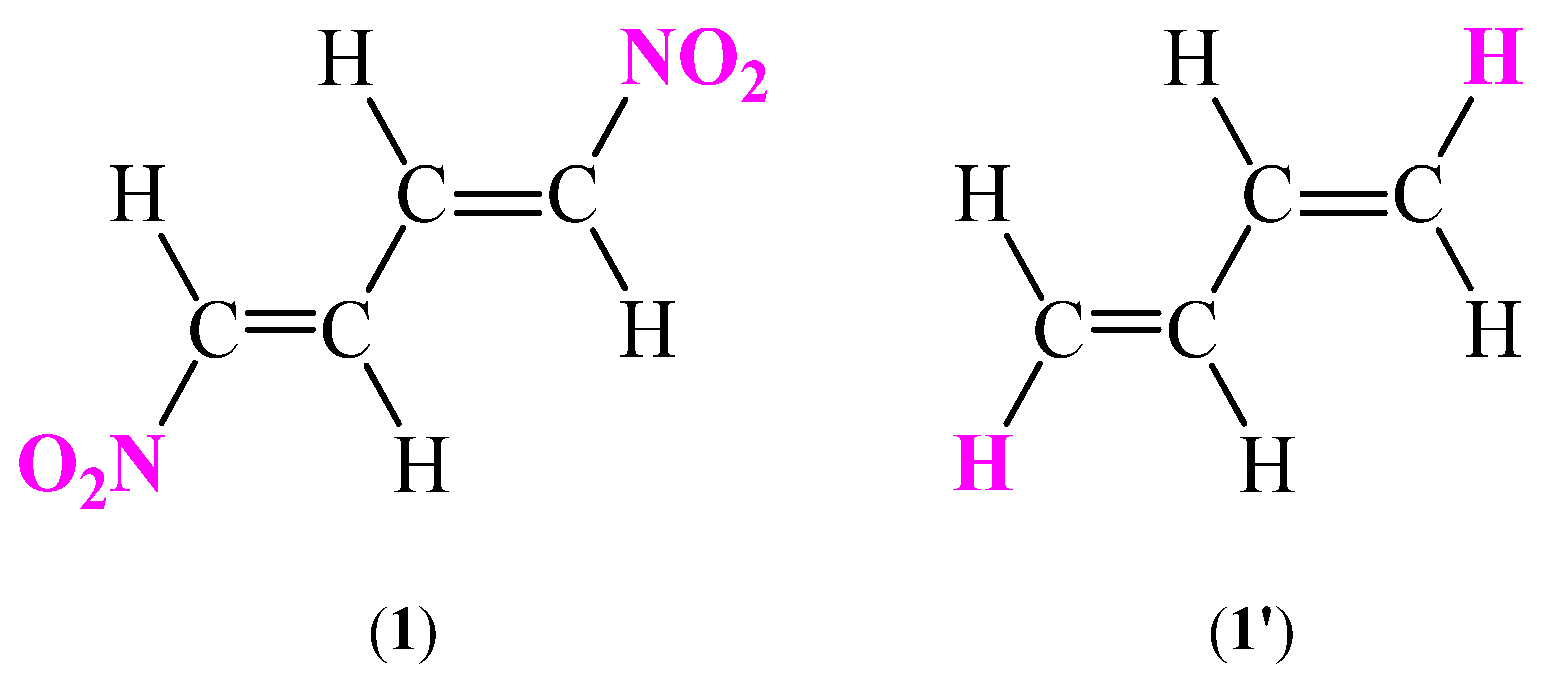
Seeing the wide applicational potential of CNDs in a modern heterocyclic chemistry, we decided to carry out comprehensive study of one compound from this class. (1E,3E)-1,4-dinitro-1,3-butadiene (1) was selected as the research object (Figure 1). Compound 1 was chosen for several reasons. Firstly, nitrodiene 1 is a representative of the simplest symmetrical CND system. The compound 1 was successfully tested in several reactions, mostly in nucleophilic addition and nucleophilic substitution [23,25]. However, also two examples of application of (1E,3E)-1,4-dinitro-1,3-butadiene (1) in Diels-Alder reactions are available in the literature [26]. What is more, there are reports that compound 1 is one of the few nitrodienes that has biological properties. In 1970 Durden et al. [33] described that (1E,3E)-1,4-dinitro-1,3-butadiene (1) can to be active against Staphylococcus Aureus and Aspergillus Niger. In the same research, the Authors reported that nitrodiene 1 was tested against Uromyces Phaseoli. According to in-vivo tests the compound 1 is a promising candidate to be applied in agrochemical industry as a mean prevented the bean rust [33]. In view of the above, systematizing information and filling information gaps regarding the (1E,3E)-1,4-dinitro-1,3-butadiene (1) is justified.
Figure 1.
Figure 1. The structures of tested (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) as the simplest analogues of nitrodiene 1.
Figure 1.
Figure 1. The structures of tested (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) as the simplest analogues of nitrodiene 1.
As a part of the research, a comprehensive experimental-computational study was carried out. For this purpose, the (1E,3E)-1,4-dinitro-1,3-butadiene (1) was synthesized. During the obtaining process the necessary modifications of synthesis protocol were introduced to increase the efficiency of obtaining nitrodiene 1. Then, the spectral analyses were performed to confirm the structure of the obtained system. Research of the geometric isomerism aspects was also carried out. In turn, in the computational part, the reactivity of title compound 1 was thoroughly studied. For this purpose, selected aspects of Molecular Electron Density Theory (MEDT) [34] were used. The influence of the presence of the nitro group on the reactivity properties was also examined by comparing nitrodiene 1 with its analogue, the simplest representative of dienes, which is S-trans-1,3-butadiene (1’) (Figure 1). Finally, in order to address the biological potential of (1E,3E)-1,4-dinitro-1,3-butadiene (1), analyses of physicochemical descriptors, pharmacokinetic properties, druglike nature and medicinal chemistry friendliness, based on selected aspects of ADME (Absorption, Distribution, Metabolism, Excretion) and PASS (Prediction of Activity Spectra for Substances) were performed.
2. Results and Discussion
2.1. Synthetic and Computational Aspects of (1E,3E)-1,4-dinitro-1,3-butadiene Preparation
2.1.1. Synthesis Protocol and its Necessary Modification
Generally, in the literature two synthesis methods of (1E,3E)-1,4-dinitro-1,3-butadiene (1) are available [23,25]. One of them includes an electrochemical process of direct oxidation of the of 1,4-dinitrobut-2-ene (2) (Scheme 4) by bromine or iodine in the presence of methanolic solution of KOH [35].
Scheme 4.
Synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) via electrochemical processes.
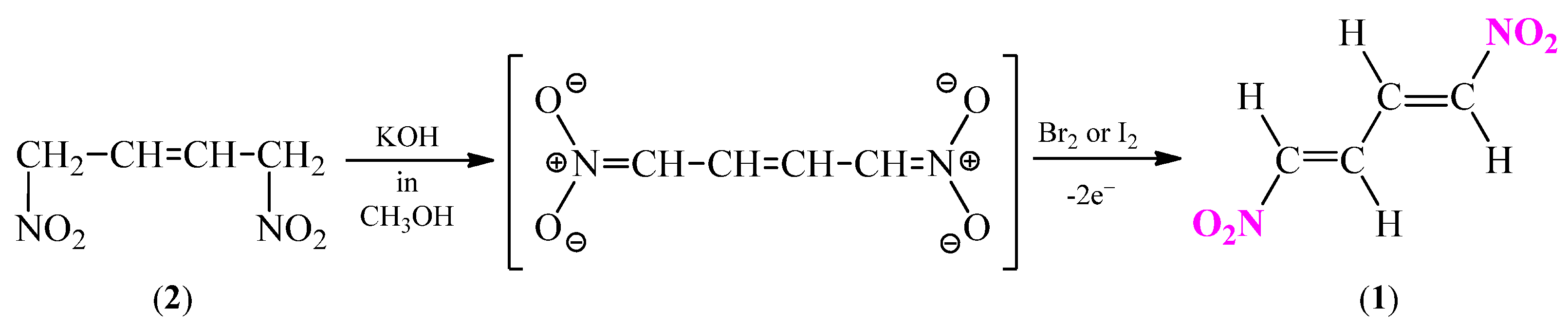
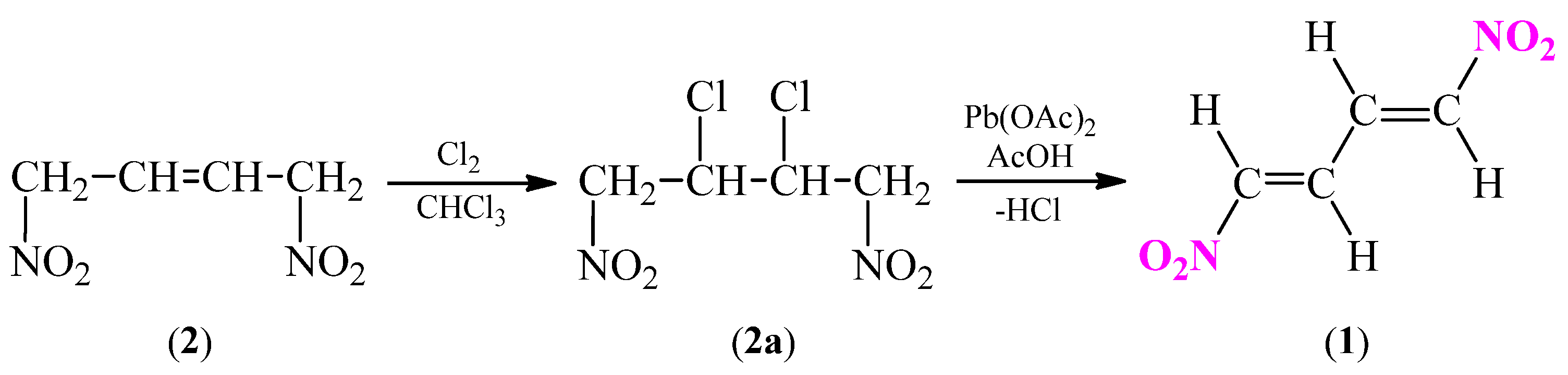
Definitely, the most popular alternative methods are the thermal eliminations of different fragments like hydrogen halides or acetic acid from 1,4-dinitrobut-2-ene analogues [23,25]. The reaction can be carried out by two different paths. First alternative is started from 1,4-dinitrobut-2-ene (2), similarly to previous example (Scheme 5). In this case, dinitroalkene 2 is chlorinated with gaseous chlorine, in the presence of iodine to obtain 2,3-dichloro-1,4-dinitrobutane 2a. In the next stage, the process of dehydrochlorinated of compound 2a in the presence of lead(II) acetate in glacial acetic acid is carried out [36].
Scheme 5.
Synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) via dehydrochlorination reaction of 2,3-dichloro-1,4-dinitrobutane (2a).
Scheme 5.
Synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) via dehydrochlorination reaction of 2,3-dichloro-1,4-dinitrobutane (2a).
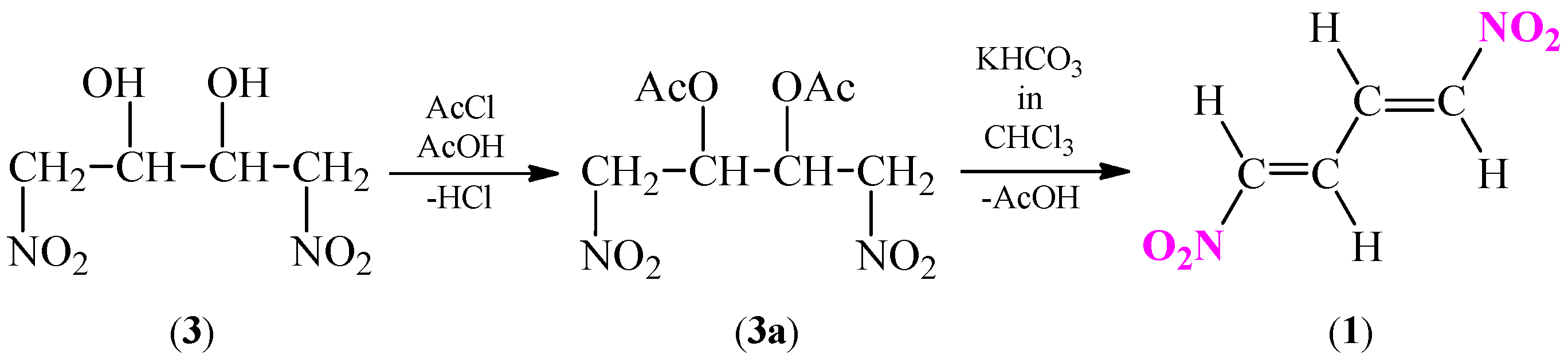
Another alternative based on elimination reaction and creation of conjugated system based on 1,4-dinitrobutane-2,3-diol (3) (Scheme 6). First step includes an acylation process of diol 3 to 2,3-diacetoxy-1,4-dinitrobutane (3a). In the next stage, an elimination of two acetic acid molecules from ester 3a in the presence of potassium bicarbonate in chloroform is carried out [33,37,38,39].
Scheme 6.
Synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) via dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a).
Scheme 6.
Synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) via dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a).
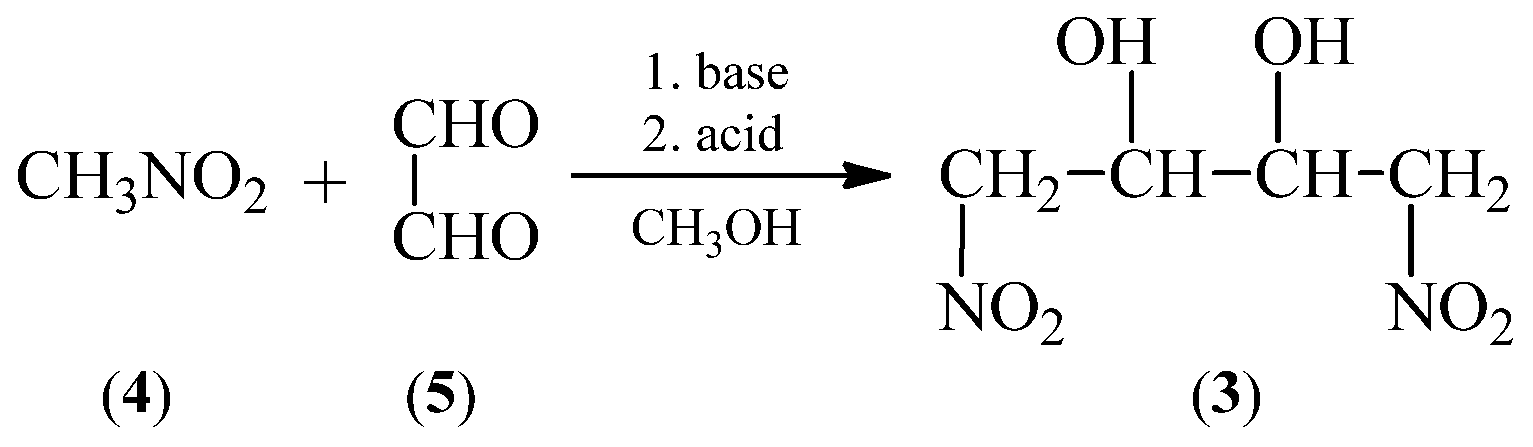
Due to the determined popularity of the last presented method, we decided to check the protocol of synthesis (1E,3E)-1,4-dinitro-1,3-butadiene (1) based on Scheme 6. In order to obtain the 1,4-dinitrobutane-2,3-diol (3) a procedure of condensation reaction between nitromethane (4) and glyoxal (5) (Scheme 7) was applied [33,37,38,39].
Scheme 7.
Synthesis of 1,4-dinitrobutane-2,3-diol (3) via condensation reaction between nitromethane (4) and glyoxal (5).
Scheme 7.
Synthesis of 1,4-dinitrobutane-2,3-diol (3) via condensation reaction between nitromethane (4) and glyoxal (5).
In the first stage of the synthesis of nitrodiene 1, the condensation reaction between nitromethane (4) and glyoxal (5) (Scheme 7) was carried out. In the literature this procedure was described four times [33,37,38,39]. The differences were in the acidic and basic agents used (Table 1). Taking into account both acidic and basic factors as well as the yield, it was decided to synthesize nitrodiol 3 using the modified method presented by Novikov et al. [37].The only change that was made was to use glacial acetic acid instead of SO2 as the acidic agent. Invariably, NaOH was used as the basic agent. It was this stage of synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) which proved to be the most problematic.

Table 1.
Acidic and basic agents together with yields for condensation reaction between nitromethane (4) and glyoxal (5).
Table 1.
Acidic and basic agents together with yields for condensation reaction between nitromethane (4) and glyoxal (5).
| Acidic agent | Basic agent | Yield | Reference |
|---|---|---|---|
| SO2 | NaOH | 80.5 | Novikov et al. [37] |
| SO2 | NaOH | 61.0 | Durden et al. [33] |
| SO2 | KOH | 36.4 | Carroll [38] |
| H2SO4 or H3PO4 or AcOH or (COOH)2 | KOH | 24.0 | Plaut [39] |
An aqueous solution of NaOH and glyoxal (5) solution were simultaneously added dropwise to an equivolume mixture of nitromethane (4) and methanol the temperature was maintained at 0-2 °C. The mixture was maintained at 0 °C for one more hour, acidified with 50.0 % solution of acetic acid in water, until pH of 5 was reached. The organic phase was separated and the organic one was extracted with nitromethane as described by Novikov et al. [37].
The solvent from extracts was evaporated, the crystals washed with minimal amount of water, yielding 15.5 %. Novikov et al. [37] claim that the process yields 80.5 % of nitrodiol 3 stereoisomers combined. The yield obtained by modification of the procedure was improbable and unsatisfying. Another series of four extractions with nitromethane followed, the evaporation of the extracts from the second round yielded further 16.8 % of compound 3. The total yield after primal and additional extraction equated to 32.3 % which is still lower than 80.5 % claimed by Novikov et al. [37].
Therefore, an attempt to obtain diol 3 by the method described by Plaut [39] was undertaken. During the synthesis it was found that the leaving of the reaction mixture unacidified overnight and acidifying it with acetic acid the next day led to unexpected self-heating of the mixture after an hour since addition of the acidic agent. The mixture heated itself to about 40 °C what lead to complete disappearance of diol 3 (as controlled by TLC eluent cyclohexane–ethyl acetate Cy:EtOAc 80:20 v/v, developed with iodine).
The condensation of nitromethane (4) and glyoxal (5) was once again conducted similarly as described by Novikov et al. [37] with the substitution of SO2 to acetic acid until pH reached 5. KOH was also used instead of NaOH, as proposed by Carroll and collaborators [38,40]. The time of reaction after the addition of all substrates was also greatly reduced, and the mixture was diluted and acidified right after the substrates were added. This treatment prevented the darkening of the reaction mixture. The acidified mixture was divided, and a few approaches for extraction of 1,4-dinitrobutane-2,3-diol (3) were tested.
Next, an effort to find an alternative method of extraction was undertaken. After the acidification of acetic acid until pH 5 the mixture split into two phases, and they were separated. The aqueous phase was extracted with nitromethane, toluene, and diethyl ether separately. The process was monitored by TLC technique (eluent cyclohexane–ethyl acetate Cy:EtOAc 80:20 v/v, developed with iodine). During the research it was found that neither of the tested solvents was appropriate. It was rationalized that due to the glycolic nature of the diol 3 extraction from aqueous phase might be a problematic stage. Therefore, the aqueous phase was evaporated under vacuum at 40 °C. The evaporation yielded a viscous amber-coloured liquid that formed crystals when cooled to -18 °C overnight. A priori, the evaporated mixture could contain diol 3, potassium acetate, and colored substances. Samples of the dried mixture were washed with various solvents and the solubility of mixture’s ingredients was assessed (Table 2). Based on information in Table 2 it can be concluded that among all tested solvents the diethyl ether is most suitable because it dissolved the diol, while the coloured fraction as well as potassium acetate were practically insoluble.
Table 2.
Results for choice of solvent for extraction of 1,4-dinitrobutane-2,3-diol (3) from reaction mixture (✓✓✓-soluble, ✓✓-slightly soluble, ✓-barely soluble, x-insoluble). The solubility of diol 3 was controlled by TLC, the solubility of “coloured substances” was assessed by the colour of the washings (it ranged from brown to pale-yellow), while the solubility of potassium acetate was tested on a pure substance.
Table 2.
Results for choice of solvent for extraction of 1,4-dinitrobutane-2,3-diol (3) from reaction mixture (✓✓✓-soluble, ✓✓-slightly soluble, ✓-barely soluble, x-insoluble). The solubility of diol 3 was controlled by TLC, the solubility of “coloured substances” was assessed by the colour of the washings (it ranged from brown to pale-yellow), while the solubility of potassium acetate was tested on a pure substance.
| Fraction | Solvent | |||||
|---|---|---|---|---|---|---|
| DMFA | Nitromethane | Methanol | Acetone | Chloroform | Diethyl Ether | |
| (ε=37.781) | (ε=36.562) | (ε=32.613) | (ε=20.493) | (ε=4.7113) | (ε=4.240) | |
| 1,4-dinitrobutane-2,3-diol (3) | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | x | ✓✓ |
| Coloured substances | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓ |
| Potassium acetate | ✓✓✓ | x | ✓✓✓ | ✓✓✓ | x | x |
Based on the conducted research a protocol for extracting diol 3 was proposed and tested. For this purpose, the viscous amber concentrate was mixed with anhydrous sodium sulphate, so that the mixture had a consistency of wet sand. The mixture was covered and set aside for 30 minutes. After that time, the solid was transferred into a Soxhlet extractor and extracted with diethyl ether under an inert gas. After the extraction the extract was cooled, solid that had fallen out was separated and washed sparingly with diethyl ether. The filtrate was evaporated dry on a rotatory evaporator in a bath of 40 °C. The dry crystals were washed with diethyl ether. In total, the isomer mixture of the 1,4-dinitro-2,3-butanediol (3) was obtained in the form of white lumpy and needle-like crystals.
Definitely, the hardest challenge for presented experimental part of study was the step of obtaining 1,4-dinitro-2,3-butanediol (3). Both of the 2,3-diacetoxy-1,4-dinitrobutane (3a) as well as (1E,3E)-1,4-dinitro-1,3-butadiene (1) synthesis were carried out based on the procedures described by Novikov et al. [37]. The introduced changes in the synthesis protocols are not drastic and were intended only to improve the processes of obtaining ester 3a and nitrodiene 1. All modified procedures are collected and presented in section 3.1 "Materials and Methods".
2.1.2. Spectral characteristic
In order to confirm the structure of the obtained compound as well as to supplement the missing in the literature information about (1E,3E)-1,4-dinitro-1,3-butadiene (1), the spectral analyses such as UV-Vis, IR, 1H NMR, 13C NMR and 2D 1H-13C HMQC NMR were performed.
In particular, the UV-Vis, showed in Figure S1, confirmed [33,40] the presence of only one maximum of absorption in a range of 500-190 nm, which is located at λ = 281 nm (CH3OH). In turn, in IR spectrum, presented in Figure S2, allowed for finding the signals coming from ~C–H fragments, nitro groups as well as fragments characteristic for conjugated alkene connections. signals coming from trans alkene fragments also turned out to be Significant for analysis.
The analysis of 13C NMR spectrum, shown in Figure S4, indicates that the carbon C1 and C2 atoms, directly connected with nitro groups, are strongly shifted towards the weaker area to δ = 146.60 ppm. In turn, the signal δ = 129.50 ppm can be assigned to the central C2 and C3 carbon atoms. On the other hand, thanks to 1H NMR analysis (Figure S3 together with 2D 1H-13C HMQC NMR analysis (Figure S5), it was possible to assign signals to all hydrogen atoms in the (1E,3E)-1,4-dinitro-1,3-butadiene (1).
2.1.3. Understanding the geometric isomerism based on DFT calculation
In 1970 Durden et al. [33] reported that the dinitrodiene 1 occurs in (1E,3E) conformation. In order to confirm the presented hypothesis, a comprehensive analysis of dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a) to 1,4-dinitro-1,3-butadiene (1) (Scheme 8) using quantum chemical tools was performed.
Scheme 8.
Possible substrates and products of dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a) to 1,4-dinitro-1,3-butadiene (1).
Scheme 8.
Possible substrates and products of dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a) to 1,4-dinitro-1,3-butadiene (1).
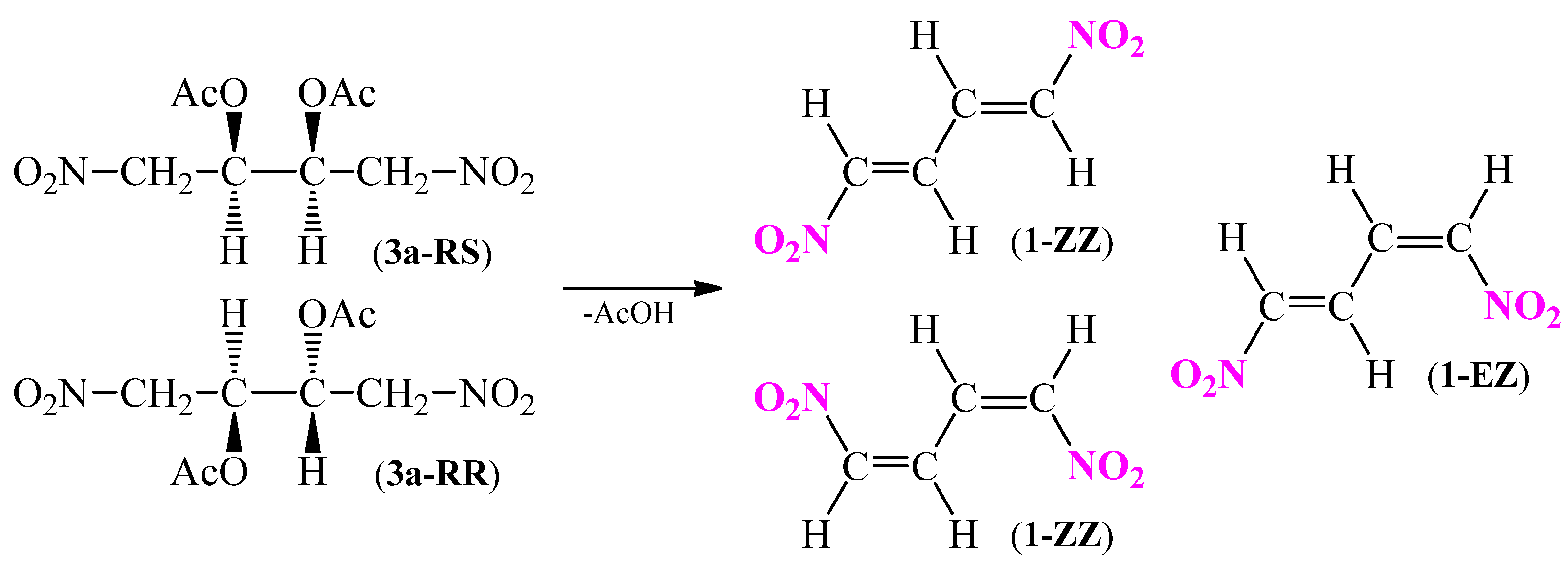
For this purpose, the computational calculation using B3LYP/6-31G(d) model in a gas phase was used. Firstly, the optimization process of two forms (RR) and (RS) of 2,3-diacetoxy-1,4-dinitrobutane (3a) as well as three forms (EE), (EZ=ZE), (ZZ) of 1,4-dinitro-1,3-butadiene (1), and acetic acid molecules was conducted. Then, the thermodynamic properties of all possible combination of decomposition reactions (Scheme 8) were calculated (Table 3).
Table 3.
Thermodynamic parameters of dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a) to 1,4-dinitro-1,3-butadiene (1), calculated in gas phase according to B3LYP/6-31G(d) (ΔH and ΔG are given in kcal·mol-1, ΔS is given in cal·mol-1·K-1).
Table 3.
Thermodynamic parameters of dehydro-acetylation reaction of 2,3-diacetoxy-1,4-dinitrobutane (3a) to 1,4-dinitro-1,3-butadiene (1), calculated in gas phase according to B3LYP/6-31G(d) (ΔH and ΔG are given in kcal·mol-1, ΔS is given in cal·mol-1·K-1).
| Transformation | ΔH | ΔG | ΔS |
|---|---|---|---|
| 3a-RR → 1-ZZ + 2AcOH | 33.64 | 11.98 | 72.61 |
| 3a-RR → 1-EZ + 2AcOH | 36.09 | 14.27 | 73.17 |
| 3a-RR → 1-ZZ + 2AcOH | 37.31 | 15.11 | 74.45 |
| 3a-RS → 1-ZZ + 2AcOH | 30.31 | 7.65 | 75.99 |
| 3a-RS → 1-EZ + 2AcOH | 32.76 | 9.94 | 76.54 |
| 3a-RS → 1-ZZ + 2AcOH | 33.98 | 10.78 | 77.83 |
According to the results presented in Table 3 it can be observed that regardless of the isomer of 2,3-diacetoxy-1,4-dinitrobutane (3a), the more favored product for dehydro-acetylation reaction of ester 3a from the thermodynamic point of view is (1E,3E)-1,4-dinitro-1,3-butadiene (1). The conclusion stands in agreement with observations made by Durden et al. [33] and with the results of IR analysis.
2.2. Study of electron density distribution, bioactivity and pharmacokinetics indices based on MEDT, ADME and PASS
The computational study has been conducted for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and its simpler analogue S-trans-1,3-butadiene (1’) to show changes in the structure of the molecule introduced by the presence of the nitro group. The obtained results have been divided into four parts. Firstly, a topological analysis of Electron Localization Function (ELF) together with Natural Population Analysis (NPA), and Molecular Electrostatic Potential (MEP) for molecules 1 and 1’ at the ground states (GS) has been done in order to characterize their electronic structures. Secondly, an analysis of reactivity indices based on Conceptual Density Functional Theory (CDFT) for compounds 1 and 1’ at the ground state has been carried out. In the next part, the Non-Covalent Interactions (NCI) analysis is performed in order to obtain a deeper insight of the structure of molecules 1 and 1’. Finally, the comprehensive evaluation of the chemical structure of (1E,3E)-1,4-dinitro-1,3-butadiene (1) has been studied to determine the likeness of nitrodiene 1 activity as pharmaceutics. To assess the potential biological activity an in-silico protocol of Absorption, Distribution, Metabolism, Excretion (ADME) has been used. In order to better research the biological significance of nitrodiene 1 a simple protocol of Prediction of Activity Spectra for Substances (PASS) was carried out.
2.2.1. Analysis of the Electronic Structure of the Molecules 1 and 1’ based on ELF, NPA and MEP
The ELF is a technique which allows prediction of the likelihood of finding an electron in the neighborhood space of a reference electron located at a given point and with the same spin. The technique is based on electron density distribution in molecules and allows both to characterize the electronic structure of compounds, and to predict their reactivity in chemical processes [41]. The ELF attractor positions of the core and valence basins together with ELF localization domains for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) are shown in Figure 2, while the most relevant valence basin populations are given in Table 4.
Figure 2.
B3LYP/6-31G(d) ELF attractor positions of the core and valence basins for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) together with ELF localization domains of represented at an isosurface value of ELF = 0.75. For ELF localization domains, protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green, and core basins in magenta. In the first row the ELF attractors are shown as pink spheres.
Figure 2.
B3LYP/6-31G(d) ELF attractor positions of the core and valence basins for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) together with ELF localization domains of represented at an isosurface value of ELF = 0.75. For ELF localization domains, protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green, and core basins in magenta. In the first row the ELF attractors are shown as pink spheres.
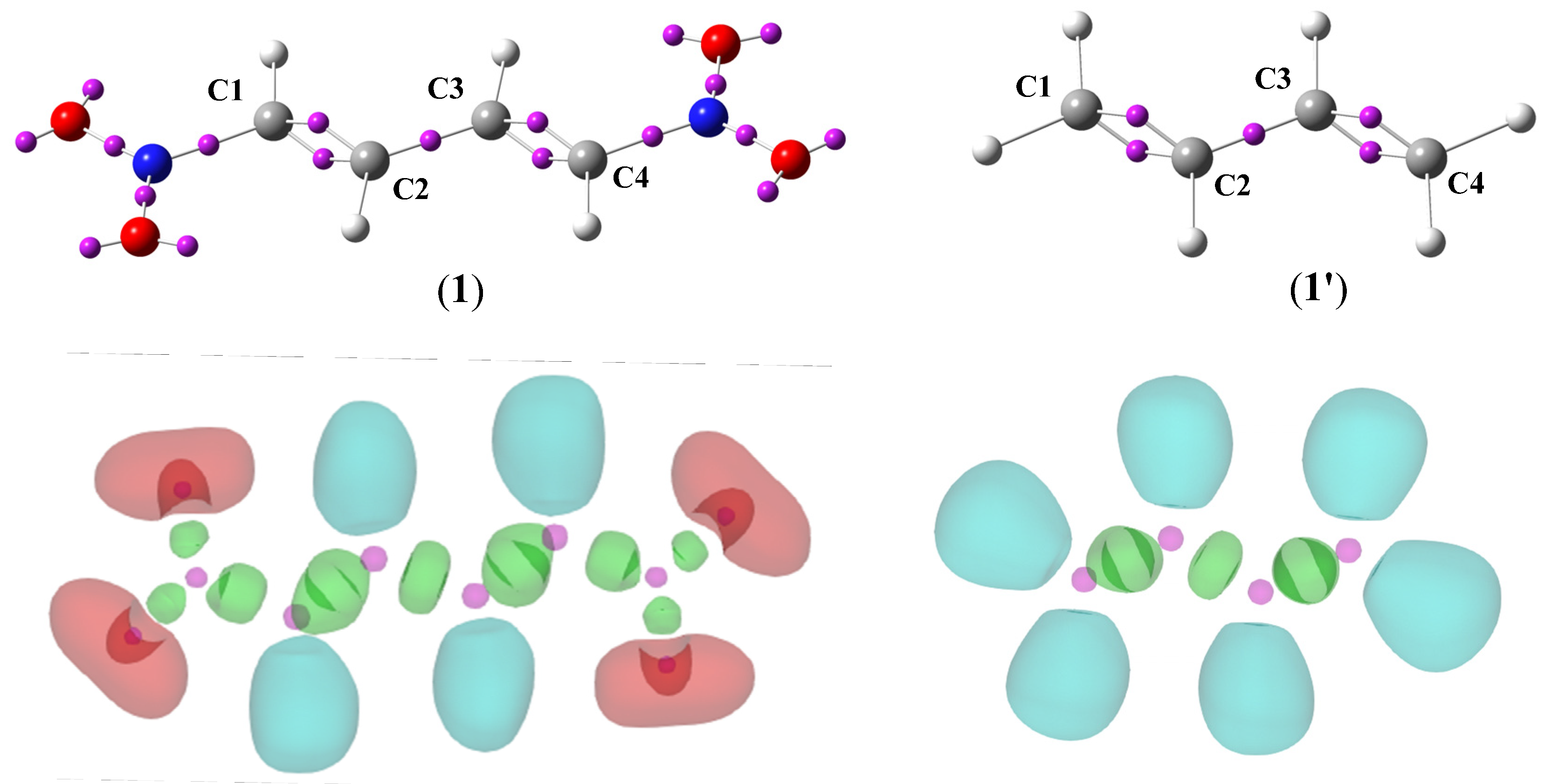
The ELF topology of S-trans-1,3-butadiene (1’) presents two pairs of disynaptic basins V(C1,C2) and V’(C1,C2) (Figure 2), integrating a total electron population of 3.44 e (Table 4) as well as V(C3,C4) and V’(C3,C4) (Figure 2), integrating the same value of total electron population of 3.44 e (Table 4). The presence of these pairs is associated with somewhat depopulated C1-C2 and C3-C4 double bonds. In turn, the presence of disynaptic basin V(C2,C3) (Figure 2), integrating a population of 2.30 e (Table 4), is associated with C2-C3 single bond.
Table 4.
B3LYP/6-31G(d) the most significant ELF valence basin populations N giving in average number of electrons [e].
Table 4.
B3LYP/6-31G(d) the most significant ELF valence basin populations N giving in average number of electrons [e].
| 1 | 1’ | |
|---|---|---|
| ELF basins | N [e] | N [e] |
| V(C1,C2) | 1.73 | 1.72 |
| V’(C1,C2) | 1.73 | 1.72 |
| V(C2,C3) | 2.23 | 2.20 |
| V(C1,C2) | 1.73 | 1.72 |
| V’(C1,C2) | 1.73 | 1.72 |
In turn, the ELF topology for (1E,3E)-1,4-dinitro-1,3-butadiene (1) is practically identical. when compared with the S-trans-1,3-butadiene (1’), the total electron population associate with the presents of two double bonds C1-C2 as well as C3-C4 is slightly higher and the total electron population associate with the presents of single bond C2-C3 is slightly smaller (Table 4). Therefore, the introduction of two nitro groups located symmetrically to the structure does not affect the electron localization. Based on ELF analysis the proposed Lewis-like structures together with the natural atomic charges for compounds 1 and 1’ are given in Figure 3.
Figure 3.
B3LYP/6-31G(d) proposed ELF-based Lewis-like structures with the natural atomic charges for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) together with the molecular electrostatic potential maps. Negative charges are coloured in red, while negligible charges are coloured in green. Natural atomic charges are given in average number of electrons [e].
Figure 3.
B3LYP/6-31G(d) proposed ELF-based Lewis-like structures with the natural atomic charges for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) together with the molecular electrostatic potential maps. Negative charges are coloured in red, while negligible charges are coloured in green. Natural atomic charges are given in average number of electrons [e].
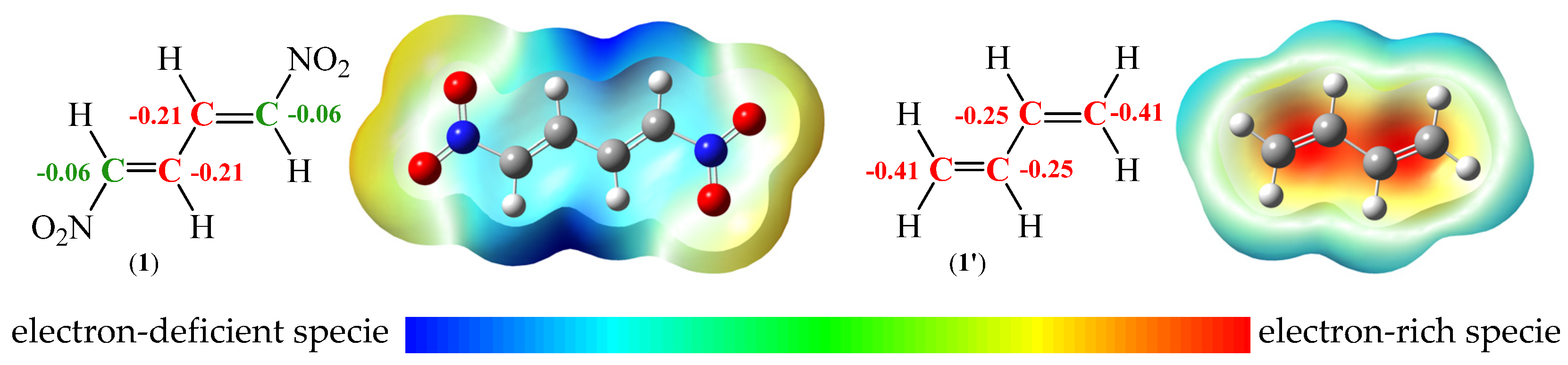
Charge distributions for molecules 1 and 1’ were examined through the NPA [42,43]. The analysis indicates a similar negative distribution located at the central carbon atoms C2 and C3 which form a single bond. For S-trans-1,3-butadiene (1’) both natural atomic charges are -0.25 e (Figure 3). In a case of (1E,3E)-1,4-dinitro-1,3-butadiene (1) for analogous carbon atoms C2 and C3, a slight decrease of electron-rich specie to -0.21 e is noticeable (Figure 3). A different situation is observed in the case of terminal carbon atoms. Both carbon atoms C1 and C4 in diene 1’ are represented by highly electronegative region, -0.41 e (Figure 3). On the other hand, the introduced nitro groups at nitrodiene 1 strongly interact with C1 and C4 carbon atoms. As a result, these centres are depleted of electrons to a value practically typical of neutral regions, -0.06 e (Figure 3). The presented results correlate well with MEP [44]. The analysis of a surface localized a high negative electrostatic potential around all carbon atoms in diene 1' (in red) and poorly electropositive region within the vicinity of the hydrogen atoms (in green-blue). In turn, the positive electrostatic potential is both found around the carbon atoms (in light-blue) in nitrodiene 1 as well as stronger electropositive region within the vicinity of the hydrogen (Figure 3).
2.2.2. Analysis of the CDFT Reactivity Indices for the compounds 1 and 1’
The CDFT is a very important tool in understanding the reactivity of molecules in polar processes. The theory is to connect well established chemical concepts, like ionization potential l, electron affinity A, electronic chemical potential μ, Milliken electronegativity X, chemical hardness η and chemical softness S with electronic structure of a molecule. Based on those, the indication of global electronic properties of substrates, such as global electrophilicity ω, and global nucleophilicity N for molecules, can be established. As an effect, it is possible to assign addends either the role of electrophile or nucleophile in studied reactions [45,46,47]. Furthermore, with application of either Parr functions, not only global, but also local electronic properties of a molecule can be estimated, thus allowing to predict reactivity of molecules in the studied reactions, based only on substrate structures [48,49]. The global reactivity indices for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) are given in Table 5, while the local electronic properties are shown in Figure 4.
Table 5.
B3LYP/6-31G(d) HOMO and LUMO energy as well as the global reactivity indices giving in electronvolts [eV].
Table 5.
B3LYP/6-31G(d) HOMO and LUMO energy as well as the global reactivity indices giving in electronvolts [eV].
| [eV] | 1 | 1’ | |
|---|---|---|---|
| HOMO energy | -8.31 | -6.23 | |
| LUMO energy | -3.91 | -0.61 | |
| Energy gap, ΔE | 4.40 | 5.62 | |
| Ionization potential, l | 8.31 | 6.23 | |
| Electron affinity, A | 3.91 | 0.61 | |
| Electronic chemical potential, μ | -6.11 | -3.42 | |
| Milliken electronegativity, X | -4.40 | -5.62 | |
| Chemical hardness, η | 4.40 | 5.62 | |
| Chemical softness, S | 0.23 | 0.18 | |
| Global electrophilicity, ω | 4.24 | 1.04 | |
| Global nucleophilicity, N | 0.81 | 2.89 |
The calculated HOMO energy [45] of S-trans-1,3-butadiene (1’) is -6.23 eV and the calculated LUMO energy [45] for diene 1’ is 0.61 eV (Table 5). In turn, the presence in the structure two strongly electron withdrawing nitro groups to molecule in (1E,3E)-1,4-dinitro-1,3-butadiene (1) causes a reduction of both HOMO energy, to -8.31 eV, and LUMO energy, to 3.91 eV (Table 5). Consequently, the HOMO-LUMO energy gap shrinks. For diene 1' it equates to 5.62 eV and for nitrodiene 1 it is 4.40 eV (Table 5). It means that the (1E,3E)-1,4-dinitro-1,3-butadiene (1) is a less stable compound compared to S-trans-1,3-butadiene (1’).
The calculated global electrophilicity [50] ω index of S-trans-1,3-butadiene (1’) is 1.04 eV and the calculated global nucleophilicity [51] N index for this diene 1’ is 2.89 eV (Table 5). These values give the conclusion that diene 1’ can be classified as moderate electrophile as well as moderate nucleophile in polar reactions, within the electrophilicity and nucleophilicity scale [45]. In turn, the introduction of two strongly electron withdrawing nitro groups to the molecule, in (1E,3E)-1,4-dinitro-1,3-butadiene (1) completely changes the preference of reactivity indices. In particular, the calculated global electrophilicity [50] ω index is significantly increased to 4.24 eV and also the calculated global nucleophilicity [51] N index is significantly reduced to 0.81 eV (Table 5). These values give the conclusion that nitrodiene 1 can be classified as super strong electrophile and marginal nucleophile in a polar reaction, within the electrophilicity and nucleophilicity scale [45]. The presented information is an important conclusion, as in the reactions, including the participation of non-symmetric reagents, the regioselectivity can be defined through interaction between the most electrophilic centre of the electrophile and the most nucleophilic centre of the nucleophile [48]. To characterise the most nucleophilic and the most electrophilic centres for (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) the electrophilic Pk+ and nucleophilic Pk⁻ Parr functions together with local electrophilicity ωk and local nucleophilicity Nk of diene 1’ and nitrodiene 1 were analysed (Figure 4) [49].
Figure 4.
B3LYP/6-31G(d) local electronic properties of diene 1’ and nitrodiene 1 presented as three-dimensional representations (3D) of Mulliken atomic spin densities for radical anions 1˙ˉ and 1’˙ˉ and also radical cations 1˙+ and 1’˙+ together with the electrophilic Pk+ and the nucleophilic Pkˉ Parr functions values (given in yellow) as well as the indexes of the local electrophilicity ωk of 1˙ˉ and 1’˙ˉ (given in red, in eV) and the local nucleophilicity Nk of 1˙+ and 1’˙+ (given in blue, in eV).
Figure 4.
B3LYP/6-31G(d) local electronic properties of diene 1’ and nitrodiene 1 presented as three-dimensional representations (3D) of Mulliken atomic spin densities for radical anions 1˙ˉ and 1’˙ˉ and also radical cations 1˙+ and 1’˙+ together with the electrophilic Pk+ and the nucleophilic Pkˉ Parr functions values (given in yellow) as well as the indexes of the local electrophilicity ωk of 1˙ˉ and 1’˙ˉ (given in red, in eV) and the local nucleophilicity Nk of 1˙+ and 1’˙+ (given in blue, in eV).
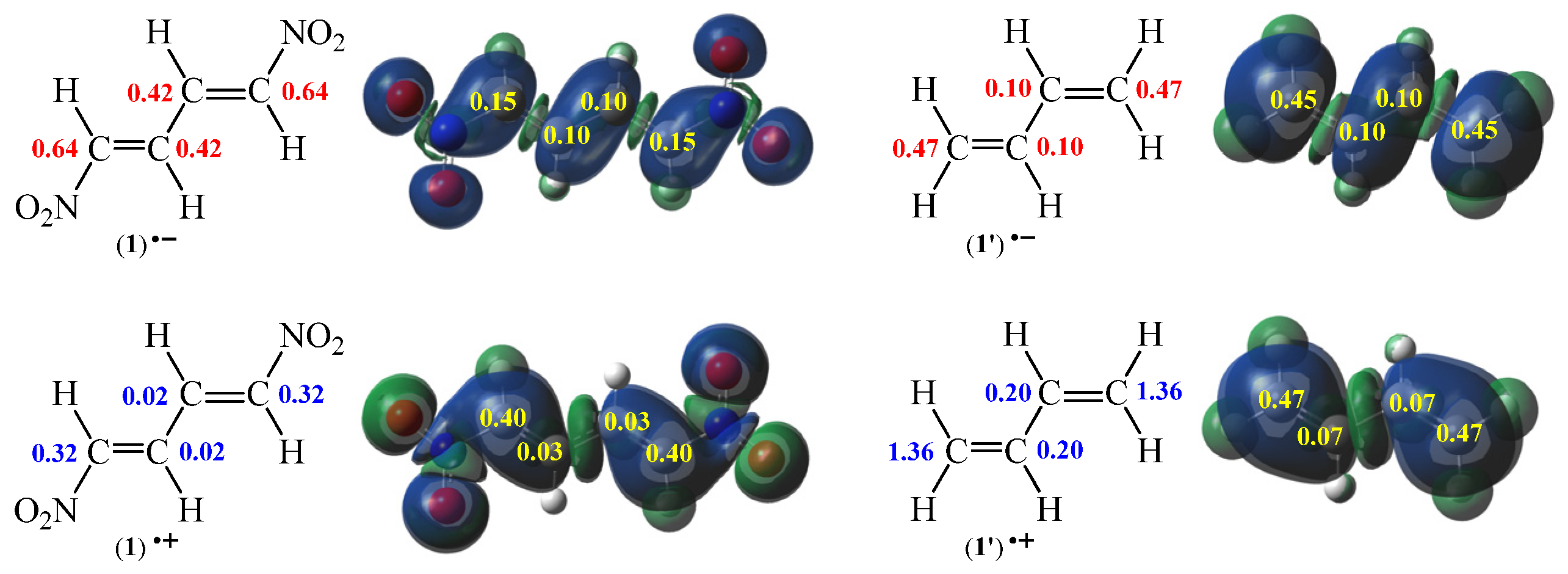
Analysis of the electrophilic Pk+ Parr function [49] of S-trans-1,3-butadiene (1’) indicates that the terminal carbon atoms are the most electrophilic centres of this species, both presenting the value PC+ = 0.45. At these atoms, the value of the local electrophilicity ωk index is ωC = 0.47 eV (Figure 4). In turn, electrophilic Pk+ Parr functions for the centric atoms of diene 1’ are visibly reduced to PC+ = 0.10 (ωC = 0.10 eV) (Figure 4). Analysis of the electrophilic Pk+ Parr function of (1E,3E)-1,4-dinitro-1,3-butadiene (1) show a similar tendency. In particular, the most electrophilic centres in this molecule 1 are situated on the terminal carbon atoms, presenting the value PC+ = 0.15. At these atoms, the value of the local electrophilicity ωk index is ωC = 0.64 eV (Figure 4). In turn, the values of electrophilic Pk+ Parr function for the centric atoms of nitrodiene 1 are slightly reduced to PCˉ = 0.10 (ωC = 0.42 eV) (Figure 4). Based on presented analysis it can be concluded that the direct neighbourhood of strongly electron withdrawing nitro groups causes a significant reduction of local electrophilic properties in dinitrodiene 1 when compared to diene 1’.
On the other hand, analysis of the nucleophilic Pkˉ Parr functions [49] of S-trans-1,3-butadiene (1’) indicates that the terminal carbon atoms are also the most nucleophilic centres of this species, presenting the values PCˉ = 0.47. At these atoms, the values of the local nucleophilicity Nk index is NC = 1.36 eV (Figure 4). In turn, values of nucleophilic Pkˉ Parr functions for the centric atoms of diene 1’ are extremely reduced to PCˉ = 0.07 (NC = 0.20 eV) (Figure 4). The differences between nucleophilic properties for nonsymetric atoms in molecule 1’ are a slightly greater than for electrophilic properties. Another situation is observed in a case of (1E,3E)-1,4-dinitro-1,3-butadiene (1). Analysis of the nucleophilic Pkˉ Parr function of nitrodiene 1 show that the most nucleophilic centres in molecule 1 are also situated on the terminal carbon atoms, presenting the value PCˉ = 0.40. At these atoms, the values of the local nucleophilicity Nk index is NC = 0.32 eV (Figure 4). In turn, values of nucleophilic Pkˉ Parr function for the centric atoms of nitrodiene 1 are reduced practically to zero, PCˉ = 0.03 (NC = 0.02 eV) (Figure 4). Based on presented analysis it can be concluded that the direct neighbourhood of strongly electron withdrawing nitro group does not cause a significant reduction of local nucleophilic properties of nitrodiene 1 in comparison to diene 1’.
2.2.3. Analysis of the Non-covalent Interaction of the Molecules 1 and 1’ based on NCI
The NCI is a very useful and popular method for revealing regions and types of weak interactions in chemical systems. The analysis is related with the electron density (ρ) and the reduced density gradient (s). Due to the size of molecules, the most common approach is assigning of van der Waals interactions (vdW), steric effects (SEs), and hydrogen bonds (HBs) based on pairwise distances between atoms according to their vdW radii. In an effect, the direct representation and characterization of non-covalent interactions in three-dimensional space is possible. Therefore, NCI is an useful tool in understanding many chemical, biological, and technological problems [52,53]. The non-covalent interaction scatter diagram together with the reduced density gradient (RDG) analysis for diene 1’ and nitrodiene 1 are presented in Figure 5. A negative sign, indicates attractive forces connected with hydrogen and halogen bonding, is marked in blue colour; van der Waals forces are coloured in green, whereas red fragment, to the right from zero, expresses the repulsive forces, which mostly occur in steric interactions as well as in presence of rings.
Figure 5.
B3LYP/6-31G(d) non-covalent interaction scatter diagram together with the reduced density gradient (RDG) analysis for diene 1’ and nitrodiene 1.The strong attractive interactions like H-bond or halogen-bond are colored in blue, the van der Waals interactions are colored in green while the repulsive or steric interactions are colored in red.
Figure 5.
B3LYP/6-31G(d) non-covalent interaction scatter diagram together with the reduced density gradient (RDG) analysis for diene 1’ and nitrodiene 1.The strong attractive interactions like H-bond or halogen-bond are colored in blue, the van der Waals interactions are colored in green while the repulsive or steric interactions are colored in red.
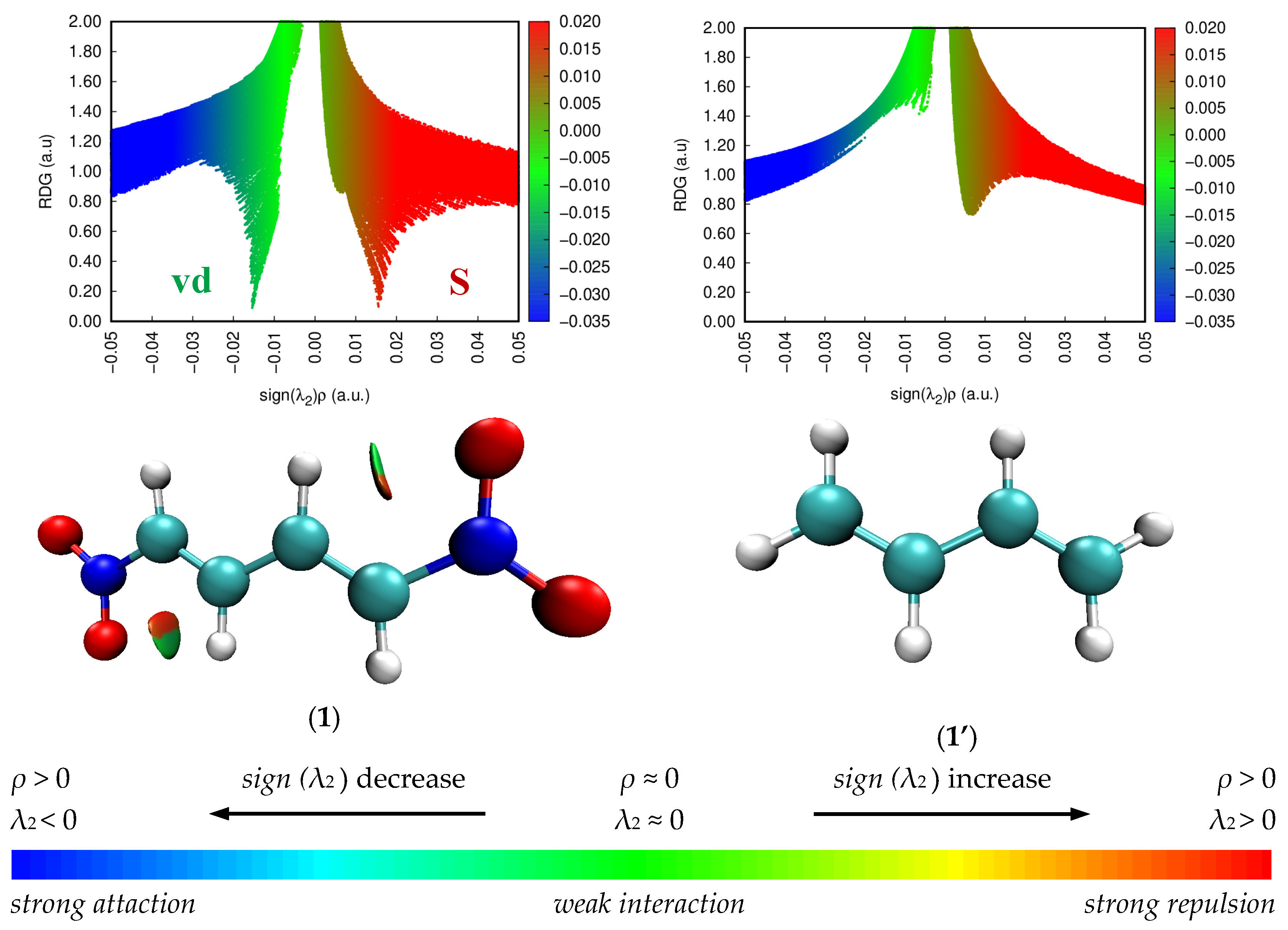
The NCI analysis of S-trans-1,3-butadiene (1’) indicates that molecule 1’ does not have any characteristic non-covalent interactions (Figure 5). A completely different situation occurs in the case of (1E,3E)-1,4-dinitro-1,3-butadiene (1). The NCI analysis shows that the introduction of two strongly electron withdrawing nitro groups significantly effects intramolecular interactions inside the molecule 1. Particularly in the area between nitrogen atom, the terminal carbon atom and the centric carbon atom (O2N−CH=CH− fragment) an intramolecular interaction, located symmetrically on both sides, is observed. The occurrence of these interactions is related to two types of non-covalent effects. The first of them is due to steric interactions (part of red colour, sign (λ2 )ρ between -0.02 and -0.01 a.u.) (Figure 5). The second component relates to van der Waals interactions (part of green colour, sign (λ2 )ρ between 0.02 and 0.01 a.u.). It can be identified with the interaction of the oxygen atom of the nitro group and the hydrogen atom in the diene part (Figure 5).
2.2.4. Analysis of Drug-Likeness and ADME Studies of the Molecule 1
ADME studies are critical in modern drug discovery [54]. The process of predicting a good drug by analysing its pharmacokinetic properties in the laboratory is expensive, and labour intensive. So, to accelerate the research and more rationally dispense the financing ADME parameters can be inspected using in silico tools. The undoubted advantage of the ADME approach is the fact that the analysis is based on structure of compound, including the atoms presence in the molecule and their connections. Therefore, ADME is one of the simplest and most useful methodology for the initial examination of drug-likeness [55,56]. Within the ADME approach for (1E,3E)-1,4-dinitro-1,3-butadiene (1) some physicochemical properties like lipophilicity, water solubility, pharmacokinetics and also medicinal chemistry assessments were performed by online server SwissADME [54] and summarized in Table 6.
Table 6.
Drug-likeness parameters for (1E,3E)-1,4-dinitro-1,3-butadiene (1).
| Physicochemical Properties | ||||||
| Formula | C4H4N2O4 | |||||
| Molecular weight MW | 144.09 g/mol | |||||
| #heavy atoms | 10 | |||||
| #aromatic heavy atoms | 0 | |||||
| #rotatable bonds | 3 | |||||
| #H-bond acceptors | 4 | |||||
| #H-bond donors | 0 | |||||
| Molar refractivity MR | 36.6 | |||||
| Topological polar surface area TPSA | 91.64 Ų | |||||
| Lipophilicity Log Po/w | ||||||
| iLOGP | XLOGP | WLOGP | MLOGP | SILICOS-IT | Consensus | |
| -1.9 | 0.81 | 1.61 | -0.78 | -2.34 | -0.52 | |
| Water Solubility Log S | ||||||
| Log S (ESOL) | Solubility | Class | Log S (Ali) | Solubility | Class | |
| -1.05 | 1.300 mg/ml | very soluble | -2.32 | 0.696 mg/ml | soluble | |
| Pharmacokinetics | ||||||
| CYP1A2 INH | CYP2C19 INH | CYP2C9 INH | CYP2D6 INH | CYP3A4 INH | Skin permeation | |
| No | No | No | No | No | -6.60 cm/s | |
| Medicinal Chemistry Friendliness | ||||||
| PAINS | Brenk | Synthetic accessibility | ||||
| 0 alert | 2 alerts | 29.5% | ||||
According to the obtained parameters the assessment using models developed by Lipinski et al. [57], Ghose et al. [58], Veber et al. [59], Egan et al. [60] and Muegge et al. [61] was performed, to evaluate the drug-likeness. The main features and conditions of these rules and filters are collected in Table 7.
Table 7.
Main features of the five druglikeness rules evaluated throughout this work.
| Lipinski et al. [57] (Pfizer) |
Ghose et al. [58] (Amgen) |
Veber et al. [59] (GSK) |
Egan et al. [60] (Pharmacia) |
Muegge et al. [61] (Bayer) |
| MW ≤ 500 Da MLOGP ≤ 4.15 #H-bond donors ≤ 5 #H-bond acceptors ≤ 10 |
160 Da ≤ MW ≤ 480 Da -0.4 ≤ WLOGP ≤ 5.6 40 ≤ MR ≤ 130 20 ≤ #atoms ≤ 70 |
#rotatable bonds ≤ 10 TPSA ≤ 140 Å2 |
WLOGP ≤ 5.88 TPSA ≤ 131.6 Å2 |
200 Da ≤ MW ≤ 600 Da -0.4 ≤ XLOGP ≤ 5.6 TPSA ≤ 150 Å2 #rings ≤ 7 #carbons > 4 #heteroatoms > 1 #rotatable bonds ≤ 15 #H-bond donors ≤ 5 #H-bond acceptors ≤ 10 |
Based on physicochemical descriptors, predicted ADME parameters shown in Table 6, and the main features of the five druglikeness rules shown in Table 7 it can be concluded that (1E,3E)-1,4-dinitro-1,3-butadiene (1) is a good drug candidate according to the filters by Lipinski et al. [57], Veber et al. [59], and Egan et al. [60]. The compound 1 has appropriate: molecular weight, number of rotatable bonds, H-bond donor – acceptor ratio, and satisfactory value of topological polar surface area (TPSA), which is commonly used parameter for the optimization of a drug's ability to permeate cells [62]. What is more, dinitrodiene 1 has excellent lipophilicity properties.
However, according to more demanding filters such as Ghose et al. [58] and Muegge et al. [61] the (1E,3E)-1,4-dinitro-1,3-butadiene (1) has low potential as a drug candidate. Both of filters are define a minimum molecular weight. The nitrodiene 1 does not meet their criteria due to too low weight and molar refractivity [63]. Nitrodiene 1 has only 14 atoms in total, only 4 of which are carbon atoms.
The analysis of another drug-likeness parameters (Table 6), not included in the tested filters, shows that (1E,3E)-1,4-dinitro-1,3-butadiene (1) not only has good lipophilic properties but also is water-soluble. These physicochemical properties are crucial to drugs’ uptake and their further metabolism [64,65]. What is more, nitrodiene 1 not is Pan-Assay Interference Compounds (PAINS) but due to its structure causes two alerts, namely nitro group and absence of organic ring. The (1E,3E)-1,4-dinitro-1,3-butadiene (1) is also not a good option as a drug candidate based on pharmacokinetic aspects. In particular, nitrodiene 1 is not an inhibitor candidate in the case of all studied isoforms CYP.
Finally, the Bioavailability Radar which allows us to assess the similarity of the drug-likeness of a nitrodiene 1 is presented in Figure 6. Based on analysis it can be concluded that the main problem presented in Bioavailability Radar is totall insaturation in the molecule of nitrodiene 1. It is caused by the sp2 hybrydization of all the carbon atoms. It should be underlined that the optimal ratio of carbon atoms in the sp3 hybridization to all carbon atoms lays in the range of 0.25 – 1 [55,56].
Figure 6.
The Bioavailability Radar of the drug-likeness for (1E,3E)-1,4-dinitro-1,3-butadiene (1). On the pink area represents the optimal range for each property like lipophilicity, size, polarity, insolubility, insaturation and flexibility.
Figure 6.
The Bioavailability Radar of the drug-likeness for (1E,3E)-1,4-dinitro-1,3-butadiene (1). On the pink area represents the optimal range for each property like lipophilicity, size, polarity, insolubility, insaturation and flexibility.

2.2.5. Assessment of Antimicrobial Activities based on PASS for the Molecule 1
At the end, the antimicrobial activity of (1E,3E)-1,4-dinitro-1,3-butadiene (1) was predicted applying PASS which is component of Way2Drug portal [66]. The PASS is a simple and very useful tool that calculates the potential biological activity of a molecule and allows to preliminary assess molecule’s drug-likeness, possible mechanisms of its action, pharmacological effects, toxicity, adverse effects, and other parameters. The assessment is made based on structural formula. The results of PASS analysis are expressed as probabilities to be active (Pa) or inactive (Pi). The values of Pa and Pi are in a range from 0.000 to 1.000, where the value of 0.000 means complete lack of probability, while a value of 1.000 means certainty. The compound can be assigned as probably active when Pa > Pi. The obtained data is useful to decide (I) if the synthesis and characterization of new compounds is justified prior to preparing them; and (II) if the screened compounds are good candidates for further biological research [67]. In order to evaluate primary biological properties of nitrodiene 1 the assessment of antimicrobial activities was performed by online server PASS [66] and summarized in Table 8.
Table 8.
Prediction of the main antimicrobial activity of the (1E,3E)-1,4-dinitro-1,3-butadiene (1) using PASS. The results are expressed as probability to be active (Pa) or inactive (Pi).
Table 8.
Prediction of the main antimicrobial activity of the (1E,3E)-1,4-dinitro-1,3-butadiene (1) using PASS. The results are expressed as probability to be active (Pa) or inactive (Pi).
| Antimicrobial activity | Pa | Pi | |
| Antiviral (Picornavirus) | 0,581 | 0,024 | |
| Antifungal | 0,416 | 0,047 | |
| Antibacterial | 0,396 | 0,031 | |
| Antiparasitic | 0,274 | 0,065 |
According to the PASS analysis, presented in Table 8, (1E,3E)-1,4-dinitro-1,3-butadiene (1) has a very low antimicrobial activity. All Pa parameters against microorganisms, namely antiviral, antifungal, antibacterial as well as antiparasitic are lower than 0.6. So as to deem a compound potentially active the Pa parameter should be higher than 0.7 [68]. However, nitrodiene 1 shows partial biological activity. For example, compound 1 can be tested in a role of an inhibitor against enzyme of Saccharopepsin (Pa = 0.912), Acrocylindropepsin (Pa = 0.912), Polyporopepsin (Pa = 0.864) or applied in pancreatic disorders treatment (Pa = 0.826) as well as many others. The full information about biological activity and potential application direction of nitrodiene 1 (Pa > 0.7) is collected in Table S3 in Supplementary Materials. It should be underlined that in all cases Pa > Pi. Therefore, the mentioned activities are possible.
3. Materials and Methods
3.1. Materials
Commercially available (Sigma–Aldrich, Szelągowska 30, 61-626 Poznań, Poland) reagents and solvents were used. All solvents were tested with high pressure liquid chromatography before use.
3.2. General Procedure for 1,4-dinitro-2,3-butanediol (3) Isomers Synthesis
In a flask 35 cm3 of nitromethane (4) and 35 cm3 of methanol were added and stirred. Then, everything was cooled to temperature 0 °C. To the cold mixture in an alternating way, drop by drop, 5.8 cm3 of glyoxal (5) as well as a solution of 6.4 g KOH in 6.0 cm3 distilled water was added, all time maintaining the temperature below 5 °C. Immediately after the glyoxal and KOH solution, to the flask 25 cm3 of distilled water was added, the mixture was neutralized by 12.0 cm3 of glacial acetic acid, to pH = 5. At the end, the mixture was delivered to the ambient temperature.
After the reaction, the post-reaction mixture was concentrated by vacuum evaporator. The obtained concentrate in the form of thick syrup, slowly darkening at the room temperature. When the concentrate must be stored it should be stored frozen at around -18°C. Anhydrous Na2SO4 (5.0 g of Na2SO4 for 1.0 g of concentrate) was mixed with the concentrate and left under cover for 30 minutes so as the inorganic salt could absorb the residual water. The final mixture should have the consistency of slightly damp sand.
Extract the solid with diethyl ether in a Soxhlet extractor, under an inert gas, for around 90 minutes. After extraction, the solvent was cooled. If solid have precipitated from the diethyl ether, it should be separated by filtering and washing with a small amount of diethyl ether, the filtrate should be evaporated dry in rotatory evaporator, and obtained solids washed sparingly with diethyl ether. If not, the extract should be evaporated in a vacuum evaporator, and the solids washed sparingly with diethyl ether. In total, 4.0 g (44.5 %) of the mixed isomers of the 1,4-dinitro-2,3-butanediol (3) was obtained in the form of white lumpy and needle-like crystals. The melting points of the products are respectively 87.5–88.0 °C and 92.5 –93.0 °C.
3.3. General Procedure for 2,3-diacetoxy-1,4-dinitrobutane (3a) Isomers Synthesis
In a flask a 7.7 g of isomers mixture of 1,4-dinitro-2,3-butanediol (3), 19.3 cm3 of acetyl chloride and 95 cm3 of glacial acetic acid were added and stirred. Then, all was heated under reflux until the end of release of hydrogen chloride gas. This moment was monitored through clouding of a drop of 3.0 % solution of AgNO3 under the influence of HCl fumes. Next, the heating was stopped, and mixture was delivered to ambient temperature. After cooling, the mixture was added to 400 g of finely crushed ice. When the ice completely melted, the obtained solid was ground under the liquid, then the mixture was filtered and the solid washed sparingly with water. The wet ester was dried in a desiccator over phosphorus pentoxide under vacuum until the powder was completely dry (when tested with cobalt chloride test strip). In total, 9.2 g (81.0 %) of the isomers mixture of the 2,3-diacetoxy-1,4-dinitrobutane (3a) was obtained in the form of a white dust. According to the melting point measurement the product was a mixture of two fractions. The melting points of those are respectively 71.0–73.0 °C and 75.0 –77.0 °C (after recrystallization from ethanol).
3.4. General Procedure for (1E,3E)-1,4-dinitro-1,3-butadiene (1) Synthesis
In a 500 cm3 flask, equipped with a magnetic stirrer, a reflux condenser, and a heating mantle 8.19 g of a mixture of 2,3-diacetoxy-1,4-dinitrobutane (3a) isomers, 0.3 g of KHCO3 and 230 cm3 of chloroform were added. The reagents were heated together under reflux for 4 hours. After that, the post-reaction mixture was cooled and filtered under vacuum. The obtained sediment was washed with chloroform. Then, the filtrate was evaporated on a vacuum evaporator to obtain about 4.0 g of slightly moist, yellow crystals, smelling strongly of vinegar. The crystals should be washed with a small amount of distilled water to remove the rest of acetic acid. Immediately after rinsing, the crystals must be dried in a desiccator over phosphorus oxide, under vacuum and without access to light. In total, 3.0 g (65.0 %) of (1E,3E)-1,4-dinitro-1,3-butadiene (1) was obtained in the form of yellow crystals with the scent of freshly cut cedar wood.
(1E,3E)-1,4-dinitro-1,3-butadiene (1): m.p. 146.5 °C; UV-Vis (CH3OH): λmax [nm] 281; FT-IR (ATR): υ [cm−1] 3106 and 3059 (~C–H stretch, alkene, medium), 1749 (stretch, trans alkene, weak); 1606 (>C=C< stretch, conjugated alkene, medium), 1502 (~N–O stretch, asymmetrical, nitro group, strong), 1342 (~N–O stretch, symmetrical, nitro group, strong), 985 (>C=C< bend, trans alkene, strong); 1H NMR (400 MHz, CDCl3): δ [ppm] 7.63 (d, 1H, CH–NO2, J = 9.6 Hz), 7.63 (dd, 1H, =CH–, J1 = 3.2 Hz, J1 = 9.6 Hz), 7.48 (dd, 1H, =CH–, J1 = 3.2 Hz, J1 = 9.7 Hz), 7.47 (d, 1H, =CH–NO2, J = 9.7 Hz); 13C NMR (100 MHz, CDCl3): δ [ppm] 146.60 (C1 and C4); 129.50 (C2 and C3).
3.5. Analytical Techniques
For reaction progress testing, thin layer chromatography (TLC) was performed using an aluminum plates with silica (unmodified layers) as the standard procedure in a case of nitrogen-contaning organic compounds [69,70,71]. In the role of eluent cyclohexane–ethyl acetate Cy:EtOAc (80:20 v/v) was applied. The plates were developed by iodine treatment. Melting points were determined with the Boetius PHMK 05 apparatus and were not corrected. FT-IR spectra were derived from the FTS Nicolet IS 10 spectrophotometer with Attenuated Total Reflectance (ATR).1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded with a Bruker AVANCE NMR spectrometer. All spectra were obtained in CDCl3 (visible at 7.27 ppm for 1H NMR and at 77.00 ppm for 13C NMR) solution and the chemical shifts (δ) are expressed in ppm, while the J-couplings (J) are giving in Hz. TMS was used as an internal standard. UV-Vis spectra were determined in methanolic solution for the 190–500 nm range through usage of spectrometer UV-5100 Biosens. The maximum of absorption was detected below 1 AU of absorbance and adheres to Beer-Lambert Law.
3.6. Computational Details
All computations were performed using the Gaussian 16 package [72] in the Ares computer cluster of the CYFRONET regional computer centre in Cracow. DFT calculations were performed using the functional B3LYP [73] together with 6-31G(d) basis set [74]. This computational level is widely used in optimization and evaluation of processes employing nitroalkenes [75,76,77]. Calculations of all critical structures were performed at temperature T = 298 K and pressure p = 1 atm in a gas phase. All localized stationary points were characterized using vibrational analysis. It was found that starting molecules as well as products had positive Hessian matrices.
The electronic structures of (1E,3E)-1,4-dinitro-1,3-butadiene (1) and S-trans-1,3-butadiene (1’) were characterized by the Electron Localization Function (ELF) [41], the Natural Population Analysis (NPA) [42,43], the Molecular Electrostatic Potential (MEP) [44] and Non-Covalent Interactions (NCI) [52,53]. Global electronic properties of reactants were performed according to Domingo’s recommendations [45,46,47]. Electrophilic Parr functions Pk+ and nucleophilic Parr functions Pk⁻ were obtained from the Atomic Spin Density (ASD) of the reagents’ radical ions [48,49]. The physicochemical properties were evaluated by online server SwissADME [54]. In order to assess a Drug-Likeness, models based on rules of Lipinski et al. [57], Ghose et al. [58], Veber et al. [59], Egan et al. [60] and Muegge et al. [61] were applied. In turn, the analysis of prediction of activity spectra for substances were prepared by online server PASS [66].
The NPA, the ELF as well as the NCI studies were performed with TopMod [78] and Multiwfn [79] software. For visualization of the molecular geometries of all structures, and 3D representations of the radical anions and the radical cations GaussView software [80] was used. In turn, the ELF localization domains were represented by using the Paraview software [81,82] at an isovalue of 0.75 a.u. Calculations of NCI analysis were performed by using VMD [83] software and visualized as an isosurface by value of 0.5.
4. Conclusions and Future Perspective
In this research a comprehensive study on the synthesis and spectral characteristics of (1E,3E)-1,4-dinitro-1,3-butadiene (1) was presented. Additionally, a computational analysis of electronic chemical parameters based on MEDT has been made. At the end, the forecasting study of biological properties of nitrodiene 1 using ADME and PASS simulation was performed.
Among the available alternatives for synthesis of (1E,3E)-1,4-dinitro-1,3-butadiene (1), a three-step method from nitromethane (4) and glyoxal (5) was selected in the presented study. Modifications of synthesis protocol were carried out in order to increase efficiency of obtaining the final nitrodiene 1, and overall process safety. In particular, the modification of extraction method for the synthesis of 1,4-dinitrobutane-2,3-diol (3), led to greater yields of extracted product to ca. 40 % as compared to 17 % when the extraction protocol proposed by Novikov et al. [37] was applied. Furthermore, the new method uses less expensive diethyl ether instead of nitromethane. What is worth noting, the nitromethane is classified in the category 2B as possibly carcinogenic to humans by the International Agency for Research on Cancer [84], whereas diethyl ether , among other volatile anaesthetics, is classified in category 3, which means not classifiable as to its carcinogenicity to humans [85]. According to NIOSH the permissible exposure limits for diethyl ether and nitromethane are respectively: 400 ppm and 100 ppm [86]. Thus the diethyl ether can be treated as a more favoured solvent compared to nitromethane.
The structure of obtained (1E,3E)-1,4-dinitro-1,3-butadiene (1) has been verified based on measured melting point and data available in the literature [33,36,37,40]. In order to further confirm the structure of the nitrodiene 1 as well as to supplement the information missing in the literature, spectral analyses such as UV-Vis, IR, 1H NMR, 13C NMR and 2D 1H-13C HMQC NMR were performed.
A comprehensive MEDT analysis of (1E,3E)-1,4-dinitro-1,3-butadiene (1) in comparison to S-trans-1,3-butadiene (1’) shows that the introduction of two nitro groups into the molecule causes changes in the electron density and its distribution parameters, but not to all studied aspects. The ELF analysis showed practically identical distribution of electron population for molecules 1 and 1', but the presence of nitro groups had a significant effect on charge distribution. In particular, the NPA and MEP analyses indicate drastic increase of natural atomic charges on terminal carbon atoms. Based on the analysis of CDFT it can be concluded that nitrodiene 1 will play a role of a super strong electrophile and marginal nucleophile in polar reactions. The most electrophilic and nucleophilic centre for nitrodiene 1 will be located on the terminal carbon atoms in direct neighbourhood of the nitro groups. Finally, the NCI analysis presented that in the space between nitrogen atom, the terminal carbon atom and the centric carbon atom (O2N−CH=CH− fragment) an intramolecular interaction, related to two types of non-covalent effects is observed. Its first component is a steric interaction while the second component is connected with van der Waals interactions between the oxygen atom of the nitro group and the hydrogen atom in the diene part.
Based on physicochemical descriptors as well as predicted ADME and PASS parameters it can be concluded that (1E,3E)-1,4-dinitro-1,3-butadiene (1) as system can possess potential biological activity but compound 1 is not a good candidate on a drug. In particular, nitrodiene 1 has too few atoms in its structure, therefore, nitrodiene 1 also has insufficient molecular weight. After all, according to the PASS analysis the (1E,3E)-1,4-dinitro-1,3-butadiene (1) can be active, among others, as an inhibitor of Saccharopepsin, Acrocylindropepsin, and Polyporopepsin as well as a treatment for pancreatic disorders.
Presented computational study shows that the future perspective for (1E,3E)-1,4-dinitro-1,3-butadiene (1) is application of this compound as a building block in synthesis of carbo- and heterocyclic systems. For this purpose, the protocol of cycloaddition is one of the most significant reactions. Thanks to the protocol it is possible to obtain mostly five and six-membered ring. Future directions for compounds thus obtained might be determined through conversion of the nitro group to other functional groups to obtain complex cyclic systems the desired structure and properties as well as 6π-electrocyclization reaction to create bigger polycyclic systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Experimental details: pp. S2–S4, Computational data: pp. S5, Comprehensive PASS details: pp. S6–S7.
Author Contributions
Conceptualization, K.K.; methodology, K.K., B.S.M. and M.S.; software, K.K., B.S.M. and M.S.; validation, K.K. and M.S.; formal analysis, K.K., B.S.M. and M.S.; investigation, K.K. and M.S.; resources, K.K. and M.S.; data curation, K.K. and M.S.; writing—original draft preparation, K.K. and M.S.; writing—review and editing, K.K., B.S.M. and M.S.; visualization, K.K., B.S.M. and M.S.; supervision, K.K.; project administration, K.K.; funding acquisition, K.K. and M.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
This research was supported in part by PL-Grid Infrastructure. All calculations reported in this paper were performed on the “Ares” supercomputer cluster in the CYFRONET computational centre in Cracow.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the corresponding author.
References
- Ballini, R.; Petrini, M.; Rosini, G. Nitroalkanes as Central Reagents in the Synthesis of Spiroketals. Molecules 2008, 13, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, N. A Walk through Recent Nitro Chemistry Advances. Molecules 2020, 25, 3680. [Google Scholar] [CrossRef]
- Ballini, R.; Palmieri, A. Nitroalkanes: Synthesis, Reactivity, and Applications; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Zawadzińska, K.; Gaurav, G.K.; Jasiński, R. Preparation of Conjugated Nitroalkenes: Short Review. Sci. Rad. 2022, 1, 69–83. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Łapczuk-Krygier, A.; Rykała, K.; Biernasiuk, A.; Wnorowski, A.; Popiolek, Ł.; Maziarka, A.; Hordyjewska, A.; Jasiński, R. Novel Synthesis Scheme and In Vitro Antimicrobial Evaluation of a Panel of (E)-2-aryl-1-cyano-1-nitroethenes. J. Enzym. Inhib. Med. Chem. 2016, 31, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Boguszewska-Czubara, A.; Kula, K.; Wnorowski, A.; Biernasiuk, A.; Popiolek, Ł.; Miodowski, D.; Demchuk, O.M.; Jasiński, R. Novel Functionalized β-nitrostyrenes: Promising Candidates for New Antibacterial Drugs. Saudi Pharm. J. 2019, 27, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Latif, N.; Girgis, N.S.; Assad, F.M.; Grant, N. (Nitroethenyl) Salicylic Acid Anilides and Related Substances. A New Group of Molluscicidal and Microbicidal Compounds. Liebigs Ann. 1985, 6, 1202–1209. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes. Molecules 2017, 22, 895. [Google Scholar] [CrossRef] [PubMed]
- Al-Najjar, H.J.; Barakat, A.; Al-Majid, A.M.; Mabkhot, Y.N.; Weber, M.; Ghabbour, H.A.; Fun, H.-K. A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium. Molecules 2014, 19, 1150–1162. [Google Scholar] [CrossRef]
- Kras, J.; Sadowski, M.; Zawadzińska, K.; Nagatsky, R.; Woliński, P.; Kula, K.; Łapczuk, A. Thermal [3+2] Cycloaddition Reactions as Most Universal Way for the Effective Preparation of Five-Membered Nitrogen Containing Heterocycles. Sci. Rad. 2023, 2, 247–267. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzińska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the Question of the Formation of Nitro-Functionalized 2,4-Pyrazole Analogs on the Basis of Nitrylimine Molecular Systems and 3,3,3-Trichloro-1-Nitroprop-1-Ene. Molecules 2022, 27, 8409. [Google Scholar] [CrossRef]
- Kula, K.; Dobosz, J.; Jasiński, R.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Mirosław, B.; Demchuk, O.M. [3+2] Cycloaddition of Diaryldiazomethanes with (E)-3,3,3-trichloro-1-nitroprop-1-ene: An Experimental, Theoretical and Structural study. J. Mol. Struct. 2020, 1203, 127473. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. The Participation of 3,3,3-Trichloro-1-nitroprop-1-ene in the [3+2] Cycloaddition Reaction with Selected Nitrile N-Oxides in the Light of the Experimental and MEDT Quantum Chemical Study. Molecules 2021, 26, 6774. [Google Scholar] [CrossRef]
- Kras, J.; Woliński, P.; Nagatsky, R.; Demchuk, O.M.; Jasiński, R. Full Regio- and Stereoselective Protocol for the Synthesis of New Nicotinoids via Cycloaddition Processes with the Participation of Trans-Substituted Nitroethenes: Comprehensive Experimental and MEDT Study. Molecules 2023, 28, 3535–10. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R.; Żmigrodzka, M.; Dresler, E.; Kula, K. A Full Regio- and Stereoselective Synthesis of 4-Nitroisoxazolidines via Stepwise [3+2] Cycloaddition Reactions between (Z)-C-(9-anthryl)-N-arylnitrones and (E)-3,3,3-trichloro-1-nitroprop-1-ene: Comprehensive Experimental and Theoretical Study. J. Heterocyclic. Chem. 2017, 54, 3314–3320. [Google Scholar] [CrossRef]
- Fryźlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O.M.; Dresler, E.; Jasiński, R. Regio- and Stereoselective Synthesis of Nitrofunctionalized 1,2-Oxazolidine Analogs of Nicotine. Chem. Heterocycl. Comp. 2020, 56, 120–122. [Google Scholar] [CrossRef]
- García-Mingüens, E.; Ferrándiz-Saperas, M.; de Gracia Retamosa, M.; Nájera, C.; Yus, M.; Sansano, J.M. Enantioselective 1,3-Dipolar Cycloaddition Using (Z)-α-Amidonitroalkenes as a Key Step to the Access to Chiral cis-3,4-Diaminopyrrolidines. Molecules 2022, 27, 4579. [Google Scholar] [CrossRef] [PubMed]
- Żmigrodzka, M.; Sadowski, M.; Kras, J.; Dresler, E.; Demchuk, O.M.; Kula, K. Polar [3+2] Cycloaddition between N-Methyl Azomethine Ylide and Trans-3, 3, 3-trichloro-1-nitroprop-1-ene. Sci. Rad. 2022, 1, 26–35. [Google Scholar] [CrossRef]
- Hamada, T.; Iwai, K.; Nishiwaki, N. Synthesis and Characterization of Multiple Functionalized Cyclohexanone Using Diels–Alder Reaction of α-Nitrocinnamate. Reactions 2022, 3, 615–624. [Google Scholar] [CrossRef]
- Woliński, P.; Kącka-Zych, A.; Wróblewska, A.; Wielgus, E.; Dolot, R.; Jasiński, R. Fully Selective Synthesis of Spirocyclic-1,2-oxazine N-Oxides via Non-Catalysed Hetero Diels-Alder Reactions with the Participation of Cyanofunctionalysed Conjugated Nitroalkenes. Molecules 2023, 28, 4586. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules 2023, 28, 5289. [Google Scholar] [CrossRef]
- Ballini, R.; Araújo, N.; Gil, M.V.; Román, E.; Serrano, J.A. Conjugated Nitrodienes. Synthesis and Reactivity. Chem. Rev. 2013, 113, 3493–3515. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Kula, K. Nitro-functionalized Analogues of 1,3-Butadiene: An Overview of Characteristic, Synthesis, Chemical Transformations and Biological Activity. Curr. Chem. Lett. 2024, 13, 15–30. [Google Scholar] [CrossRef]
- Zapol’skii, V.A.; Bilitewski, U.; Kupiec, S.R.; Ramming, I.; Kaufmann, D.E. Polyhalonitrobutadienes as Versatile Building Blocks for the Biotargeted Synthesis of Substituted N-Heterocyclic Compounds. Molecules 2020, 25, 2863. [Google Scholar] [CrossRef] [PubMed]
- Kaberdin, R.V.; Potkin, V.I.; Zapol’skii, V.A. Nitrobutadienes and their halogen derivatives: Synthesis and reactions. Russ. Chem. Rev. 1997, 66, 827–842. [Google Scholar] [CrossRef]
- Petrillo, G.; Benzi, A.; Bianchi, L.; Maccagno, M.; Pagano, A.; Tavani, C.; Spinelli, D. Recent advances in the use of conjugated nitro or dinitro-1,3-butadienes as building-blocks for the synthesis of heterocycles. Tetrahedron Lett. 2020, 61, 152297–152309. [Google Scholar] [CrossRef]
- Al-Jumaili, M.H.A.; Hamad, A.A.; Hashem, H.E.; Hussein, A.D.; Muhaidi, M.J.; Ahmed, M.A.; Albanaa, A.H.A.; Siddique, F.; Bakr, E.A. Comprehensive Review on the Bis-heterocyclic Compounds and Their Anticancer Efficacy. J. Mol. Struct. 2023; 1271, 133970. [Google Scholar] [CrossRef]
- Iftikhar, R.; Khan, F.Z.; Naeem, N. Recent Synthetic Strategies of Small Heterocyclic Organic Molecules with Optoelectronic Applications: A Review. Mol. Divers. 2023, 27, 1–37. [Google Scholar] [CrossRef]
- Synkiewicz-Musialska, B.; Szwagierczak, D.; Kulawik, J.; Pałka, N.; Piasecki, P. Structural, Thermal and Dielectric Properties of Low Dielectric Permittivity Cordierite-Mullite-Glass Substrates at Terahertz Frequencies. Materials 2021, 14, 4030. [Google Scholar] [CrossRef] [PubMed]
- Synkiewicz, B.; Szwagierczak, D.; Kulawik, J. Multilayer LTCC Structures based on Glass-Cordierite Layers with Different Porosity. Microelectron. Int. 2017, 34, 110–115. [Google Scholar] [CrossRef]
- Synkiewicz-Musialska, B. LTCC Glass-Ceramics based on Diopside/Cordierite/Al2O3 for Ultra-High Frequency Applications. Sci. Rad. 2022, 2, 190–201. [Google Scholar] [CrossRef]
- Koc, E. Synthesis of Novel Nitroso Acetal Derivatives via Tandem 6 pi-electrocyclization/[3+ 2]-cycloaddition of 1-nitro-2-methyl-1,3-butadiene. Org. Commun. 2017, 10, 298–303. [Google Scholar] [CrossRef]
- Durden, J.A.; Heywood, D.L.; Sousa, A.A.; Spurr, H.W. Synthesis and Microbial Toxicity of Dinitrobutadienes and Related Compounds. J. Agric. Food Chem. 1970, 18, 50–56. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Rowley, G.L.; Frankel, M.B. Synthesis of Aliphatic Dinitrodienes. J. Org. Chem. 1969, 34, 1512–1513. [Google Scholar] [CrossRef]
- Perekalin, V.V.; Lerner, O.M. Synthesis of Conjugated Dinitrodiene. Dokl. Akad. Nauk SSSR 1959, 129, 1303–1305. [Google Scholar]
- Novikov, S.S.; Korsakova, I.S.; Babievskii, K.K. Synthesis of 1,4-dinitro-1,3-butadiene. Russ. Chem. Bull. 1960, 9, 882–884. [Google Scholar] [CrossRef]
- Carroll, F.I. Structure of the Isomers of 1,4-dinitro-2,3-butanediol. J. Org. Chem. 1966, 31, 366–368. [Google Scholar] [CrossRef]
- Plaut, H. Dinitrodiols and their alkali and alkaline earth metal salts, and method of preparation thereof. U.S. Patent Publication No. US2616923A, 04 November 1952. [Google Scholar]
- Carroll, F.I.; Kerbow, S.C.; Wall, M.E. The Synthesis of 1,4-dichloro-1,4-dinitro-1,3-butadiene. Can. J. Chem. 1966, 44, 2115–2117. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Leboeuf, M.; Koster, A.M.; Jug, K. Topological analysis of the molecular electrostatic potential. J. Chem. Phys. 1999, 111, 4893–4905. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Parr, R.G.; Gadre, S.R.; Bartolotti, L.J. Local Density Functional Theory of Atoms and Molecules. Proc. Natl. Acad. Sci. 1979, 76, 2522–2526. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L. v.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Aurell, M.J.; Domingo, L.R.; Pérez, P.; Contreras, R. A Theoretical Study on the Regioselectivity Of 1,3-Dipolar Cycloadditions Using DFT-based Reactivity Indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Non-covalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- SwissADME. Swiss Institute of Bioinformatics. Available online: http://www.swissadme.ch/ (accessed on 05 December 2023).
- Di, L.; Kerns, E. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Wishart, D.S. Improving early drug discovery through ADME modelling: An overview. Drugs R D 2007, 8, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Darvas, F.; Keseru, G.; Papp, A.; Dormán, G.; Urge, L.; Krajcsi, P. In Silico and Ex silico ADME approaches for drug discovery. Curr. Top. Med. Chem. 2002, 2, 1287–1304. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Way2Drug, PASS Online. Available online: http://www.way2drug.com/passonline/ (accessed on 06 December 2023).
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Poroikov, V.; Filimonov, D. Computer-Aided Prediction of Biological Activity Spectra. Application for Finding and Optimization of New Leads. In Rational Approaches to Drug Design, 1st ed.; Holtje, H.-D., Sippl, W., Eds.; Prous Science: Barcelona, Spain, 2001; pp. 403–407. [Google Scholar]
- Jasiński, R.; Mirosław, B.; Demchuk, O.M.; Babyuk, D.; Łapczuk-Krygier, A. In the search for experimental and quantumchemical evidence for zwitterionic nature of (2E)-3-[4-(dimethylamino)phenyl]-2-nitroprop-2-enenitrile—An extreme example of donor-p-acceptor push-pull molecule. J. Mol. Struct. 2016, 1108, 689–697. [Google Scholar] [CrossRef]
- Kula, K.; Dresler, E.; Demchuk, O.M.; Jasiński, R. New aldimine N-oxides as precursors for preparation of heterocycles with potential biological activity. Przem. Chem. 2015, 94, 1385–1387. [Google Scholar] [CrossRef]
- Jasiński, R.; Kula, K.; Kącka, A.; Mirosław, B. Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: Why is there no 1,3-dipolar cycloaddition? Monatsh. Chem. 2017, 148, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. GAUSSIAN 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Atoms. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Kula, K. Application of β-phosphorylated nitroethenes in [3+2] cycloaddition reactions involving benzonitrile N-oxide in the light of DFT computational study. Organics 2021, 2, 26–37. [Google Scholar] [CrossRef]
- Sadowski, M.; Utnicka, J.; Wójtowicz, A.; Kula, K. The Global and Local Reactivity of C,N-diarylnitryle Imines in [3+2] Cycloaddition Processes with Trans-β-nitrostyrene according to Molecular Electron Density Theory: A computational study. Curr. Chem. Lett. 2023, 12, 421–430. [Google Scholar] [CrossRef]
- Kula, K.; Sadowski, M. Regio- and stereoselectivity of [3 + 2] cycloaddition reactions between (Z)-C-(9-anthryl)-N-methylnitrone and analogues of trans- -nitrostyrene in the light of MEDT computational study. Chem. Heterocycl. Compd. 2023, 59, 138–144. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization, Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware Inc.: New York, NY, USA, 2015. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans “Some Industrial Chemicals"; International Agency for Research on Cancer: Lyon, France,, 2000; pp. 487–501. [Google Scholar]
- World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans “Cadmium, Nickel, Some Epoxides, Miscellaneous Industrial Chemicals and General Considerations on Volatile Anaesthetics”; International Agency for Research on Cancer: Lyon, France, 1976; pp. 487–501. [Google Scholar]
- Barson, M. NIOSH-Pocket Guide to Chemical Hazards; U.S Department of Health & Human Services: Pittsburgh, PA, USA, 2007. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



