
Submitted:
18 December 2023
Posted:
28 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Long Covid or post-acute sequelae of SARS-CoV-2 infection (PASC) can manifest as long-term symptoms in multiple organ systems, including respiratory, cardiovascular, neurological, and metabolic systems. In patients with severe COVID-19, immune dysregulation is significant, and the relationship between metabolic regulation and immune response is of great interest in determining the pathophysiological mechanisms. We aimed to characterize the metabolomic footprint for recovering severe COVID-19 patients in three consecutive time points and compare metabolite levels to controls. Our findings add proof of dysregulated amino acid metabolism in the acute phase, and dyslipidemia, glycoprotein level alterations, and energy metabolism disturbances in severe COVID-19 patients 3-4 months post hospitalization.
Keywords:
COVID-19
; SARS-CoV-2
; Post-acute sequelae of SARS-CoV-2
; metabolomics
; dyslipdemia
; Nuclear magnetic resonance
1. Introduction
During the COVID-19 pandemic, millions of people tested positive for the SARS-CoV-2 virus, hundreds of thousands of patients were hospitalized, and in the middle of year 2020, the medical and academic community were already noting Long COVID as an emerging issue in rehabilitation [1,2,3]. Long Covid or post-acute sequelae of SARS-CoV-2 infection (PASC) is a heterogeneous entity with possible long-term symptoms in multiple organ systems, including respiratory, cardiovascular, neurological, and metabolic systems, among others [4,5,6,7]. Zang and colleagues aimed to characterize PASC using Electronic Health Records (EHR) by comparing ~58 thousand patient data to ~503 thousand control EHR data. They found a significantly increased risk for new-onset diagnoses in COVID-19 patients in 12 months after the acute disease phase when compared to non-COVID-19 patients, including myopathy, dementia, cognitive problems, skin symptoms, pulmonary fibrosis, dyspnea, pulmonary embolism, thromboembolism, diabetes, cystitis, malaise and fatigue, dizziness, joint pain, and others [4].
Studies worldwide have estimated a high prevalence of long-lasting complications after the COVID-19 acute phase [6,7,8]. A systematic review that included 194 studies (735 006 participants) assessed that 45% of COVID-19 survivors, regardless of hospitalization status, were experiencing unresolved complications for ~4 months [6]. The high prevalence and unclear disease mechanisms lead to situations where healthcare systems are ill-equipped to deal with these patients without clear evidence-based rehabilitation and therapeutic guidelines for PASC patients.
In patients with severe COVID-19, immune dysregulation is significant, and the relationship between metabolic regulation and immune response is of great interest in determining the pathophysiological mechanisms of PASC [9,10]. Quantitative omics approaches, including metabolomics, have been used to understand metabolite profile differences between COVID-19 severity groups and controls, but most of them do not evaluate middle and long-term metabolic changes [11,12,13]. The dysregulation of pathways related to energy production and amino acid metabolism has been observed in several studies [11,13]. Changes in blood metabolite levels reflect more complex systemic disturbances induced by SARS-CoV-2, which affect the liver, kidneys, and other tissues. Several studies have presented evidence of plausible metabolic reprogramming of the urea cycle and/or the TCA cycle [11,13,14]. COVID-19 is associated with the development of cardiovascular diseases, including dyslipidemia and, specifically, high-density lipoprotein (HDL) dysregulation [15,16]. Another proof of disturbed energy metabolism is reports of increased risk of incident diabetes and incident antihyperglycemic use [4,17].
Considering the cardiometabolic nature of PASC, we aimed to characterize the metabolomic footprint of recovering severe COVID-19 patients in three consecutive time points and compare the metabolite levels to controls. We used NMR-based metabolomics to investigate metabolic changes during recovery from severe COVID-19. The blood plasma from hospitalized COVID-19 patients was collected at three timepoints: acute phase, a month later, and three to four months later to study metabolite profiles during recovery from severe COVID-19. To see how these profiles change compared to non-covid samples, we also measured metabolite profiles for population controls (not healthy individuals because of the relatively high prevalence of comorbidities in COVID-19 cases). Our study is designed to address these questions: how do metabolite profiles change during recovery from severe COVID-19 (linear regression for time-series data)? Can we identify significantly changed metabolites for patients with PASC and without (time series + phenotype linear regression)? What metabolites significantly differ between COVID-19 patients in each timepoint and controls (t-test, fold-change analysis)? What are the significant pathways involved in recovery from severe COVID-19?
2. Results
Primarily, we wanted to identify how metabolite profiles for COVID-19 patients change over time during the recovery process after severe disease and hospitalization. We performed limma linear regression for time series data, using Subject as a covariate to adjust for repeated within-subject samples, identifying 115 significant metabolites (p-value<0.05) (Supplementary Table 1B) across all contrasts (Timepoint C as reference group), top50 are visualized in Figure 1C heatmap. As expected, the largest number (114) of significantly different (p-value<0.05) metabolites were between the Acute phase (timepoint A) and the latest Recovery phase (timepoint C), similarly between the Acute phase (timepoint A) and the Recovery phase (timepoint B) 104 significant metabolites (p-value<0.05) were identified. The severe acute phase of COVID-19 patients can explain the significant difference in these contrasts. The top 10 most significantly changed metabolites between the Acute phase and Recovery phase (timepoint B) were GlycB, Glyc, GlycA, Glyc/SPC, Threonine, Pyruvic acid, Apo-A1, TPA1, H3FC, HDA1. Interestingly, between the Recovery phase time points B and C, we identified 17 statistically significant metabolites, top 10 - Methionine, Histidine, GlycB, Acetic Acid, Pyruvic acid, Glyc, Citric acid, GlycA, Threonine, V4TH, indicating that the recovery process is still ongoing month (35 ± 14.7.65 days) after hospitalization. See Figure 1B Venn diagram showing the significant metabolite count across three contrasts. Also, Hotelling’s T-squared (Hotelling-T^2) test was performed to detect changes or differences in the multivariate mean of the data over time and to determine if the overall pattern or distribution of these variables changes significantly across the studied time points (Supplementary Table 1C), the top Top 3 (by Hotelling-T^2 value) are visualized Figure 1D, they are GlycA, Glyc, GlycB

Table 1.
Characteristics of the study participants.
| Variable, mean (SD) or n (%) | COVID-19 patients | Population controls | |||
| Male/ Female (n, %) | 17 (41.46%)/ 24 (58.54%) | 17 (41.46%)/ 24 (58.54%) | |||
| Age, average ± SD (years ± SD) | 56.63 ± 13.16 | 53.10 ± 11.41 | |||
| BML, average (kg/m2 ± SD) | 34.40 ± 22.95 | 27.08 ± 4.60 | |||
| Time in Hospital (days ± SD) | 9.18 ± 3.25 | - | |||
| Smoker/ non-smoker (n, %) | 3 (7.32%)/ 38 (92.68%) | 3 (7.32%)/ 38 (92.68%) | |||
| Comorbidities* | |||||
| Number of patients with comorbidities (yes/no) | 30 (73.17%)/ 11 (26.83%) | 23 (56.10%)/ 18 (43.90%) | |||
| Hypertension (n, %) | 17 (41.46%) | 9 (21.95%) | |||
| Type 2 Diabetes Mellitus (n, %) | 3 (7.32%) | 3 (7.32%) | |||
| Other cardiovascular disease (n, %) | 10 (24.39%) | 13 (31.71%) | |||
| Oncological (n, %) | 2 (4.88%) | 3 (7.32%) | |||
| Clinical measurements ** | |||||
| Average (SD) | Acute COVID-19 | Recovery phase(1 month) | Recovery phase(3-4 months) | ||
| Leukocytes (μL) | 5.71 (2.15) | 6.19 (1.37) | 5.67 (1.67) | ||
| Hemoglobin (g/dL) | 13.24 (1.45) | 137.87 (11.20) | 137.88 (28.15) | ||
| Hematocrit (%) | 39.91 (4.07) | 41.47 (3.08) | 41.26 (8.05) | ||
| Platelets (μL) | 173.13 (99.07) | 267.10 (49.20) | 238.17 (59.95) | ||
| Neutrophils (μL) | 1.51 (1.50) | 52.48 (9.58) | 50.92 (13.72) | ||
| Lymphocytes (μL) | 4.86 (12.89) | 32.74 (10.36) | 32.54 (12.67) | ||
| Monocytes (μL) | 27.99 (87.84) | 9.83 (2.42) | 8.42 (2.21) | ||
| Eosinophils (μL) | 0.03 (0.05) | 3.03 (1.70) | 3.05 (1.62) | ||
| ALT (U/l) | 23.82 (18.49) | 39.23 (28.53) | 30.95 (17.54) | ||
| AST (U/l) | 28.75 (13.74) | 28.45 (13.68) | 26.75 (12.91) | ||
| GGT (U/l) | 78.33 (99.33) | 44.43 (43.10) | 28.31 (30.13) | ||
| Bilirubin (μmol/l) | 6.64 (3.22) | 13.30 (5.31) | 11.26 (4.60) | ||
| LDH (U/L) | 295.00 (168.87) | 215.70 (38.10) | 189.67 (71.51) | ||
| Creatinine ((μmol/L) | 72.45 (19.87) | 68.65 (11.53) | 70.57 (18.49) | ||
| CRP (mg/L) | 35.05 (42.05) | 3.69 (2.80) | 3.39 (4.28) | ||
| D-dimer (mg/ml) | 0.60 (0.12) | 0.48 (0.31) | 0.25 (0.15) | ||
*Comorbidities for population controls were self-reported. **Clinical measurements in Acute COVID-19 phase were performed at an in-house hospital clinical laboratory, but Recovery phase measurements in the largest local network of certified clinical laboratories outside hospitals. SD: standard deviation; BMI: body mass index; ALT: alanine aminotransferase; AST: aspartate aminotransferase, GGT: gamma-glutamyl transferase; LDH: lactate dehydrogenase; CRP: C-reactive protein.
We recognize that our two subgroups, Long Covid and Recovered, each consist of relatively small sample sizes (31 and 10 patients (x3 timepoints), respectively). However, the strength of our investigation lies in the longitudinal nature, which offers unique insights into the trajectory of COVID-19 patients over time. Metabolome data in blood samples collected at three distinct timepoints allows us to track the disease’s progression and recovery in patients after the acute phase. Therefore, we used the COVID-19 subgroups to test whether there is a significant difference between the Long Covid (n=31) and Recovered (n=10) groups. We performed limma linear regression analysis (Metaboanalyst [18]) with three experimental factors (Phenotype, Subject, and Time) and compared the phenotype groups with Time and Subject as covariates. When phenotype groups were compared, 27 metabolites were identified as significant (p-adjusted<0.05) (Supplementary Table 1D). The top 10 most significant by adjusted p-value are H2CH, H3FC, H2A2, H3A2, Choline, H2PL, H3CH, Succinic acid, Acetone, H3A1, Glucose.
Next, we set out to identify the significantly different metabolites between COVID-19 patients and population controls in each of the timepoints (A, B, C) and determine whether we see consistent changes across all contrasts that are involved in the pathogenesis of post-acute sequelae of SARS-CoV-2 infection(PASC)/ recovery from acute COVID-19, and identify the significant pathways. In contrast COVID-19 Acute phase (timepoint A) vs Population Controls, we performed a t-test and identified 116 statistically significant metabolites (FDR<0.05) (Supplementary Table 2A) and calculated Fold Change(FC) (Supplementary Table 2B), 44 metabolites were significant with FC>1.5. The summary of both analyses is visualized in the Volcano plot (Figure 2A), resulting in 13 strongly significant metabolites Fold Change treshold=2 & FDR<0,05., 3 of them downregulated (Acetic acid, Methionine, Proline), 10 upregulated (Threonine, Pyruvic acid, V5FC, Ornithine, V5CH, Acetoacetic acid, 2-Oxoglutaric acid, Sacrosine, Ethanol, 2-Hydroxybutyric acid). Figure 2B shows pathway significance (obtained by Global Test pathway enrichment analysis) and pathway impact values (from pathway topology analysis) (Supplementary Table 2C). Pathway analysis in this contrast revealed 32 significantly enriched pathways (FDR < 0.05), including (top10 by Impact values) Phenylalanine, tyrosine and tryptophan biosynthesis, Synthesis and degradation of ketone bodies, D-Glutamine and D-glutamate metabolism, Glycine, serine and threonine metabolism, Alanine, aspartate and glutamate metabolism, Phenylalanine metabolism, Arginine and proline metabolism, Pyruvate metabolism, Glycerolipid metabolism, Citrate cycle (TCA cycle). We also identified some of these pathways as significant in the acute phase vs. controls in our previous targeted metabolomics experiment (LC-MS) [12]. Using liquid chromatography-mass spectrometry (LC-MS) in blood sera, we identified tryptophan (tryptophan, kynurenine, and 3-hydroxy-DL-kynurenine) and arginine (citrulline and ornithine) metabolism, glutamine depletion, altered metabolism of several amino acids as contributing pathways in the immune response to SARS-CoV-2 in severe COVID-19 [12].
In the present study, we explored how Acute phase (timepoint A) metabolites correlate with hematological and biochemical analyses performed in a clinical lab in the recovery phase (Timepoint B and Timepoint C). Interestingly, the highest correlations (by R values) in Timepoint A metabolites vs Timepoint B Blood test contrast were Ornithine and AST (0,85), Proline and alanine aminotransferase ALT (0,83), Asparagine and C-reactive protein (CRP) (0,72). Interestingly, the highest significant correlations in Timepoint A metabolites vs Timepoint C Blood test were Tyrosine & D-dimer (-0,98), Glycerol & ALT (0,83), Ornithine & D-dimer (0,79). Figure 2 shows Timepoint A metabolite correlations with blood analysis results at Timepoints B (2C) and C (2D), correlation coefficients for the significant comparisons can be found in supplementary Table 2D,E.
In contrast: the COVID-19 Recovery phase/month post-hospitalization (timepoint B) vs Population Controls t-test identified 21 statistically significant metabolites (FDR<0.05) (Supplementary Table 3a) and calculated Fold Change(FC) (Supplementary Table 3b), 44 metabolites were FC>1.5. The summary of both analyses is visualized in the Volcano plot (Figure 3A), Acetic acid, V5FC, V5CH, V5PL, and Sacrosine were significantly different (FDR<0.05) in the analyzed contrast with Fold Change ± 1.5. Figure 3B shows pathway significance (obtained by Global Test pathway enrichment analysis) and pathway impact values (from pathway topology analysis) (Supplementary Table 3c). Pathway analysis in this contrast revealed three significantly enriched pathways (FDR < 0.05): beta-alanine metabolism, Glycolysis/Gluconeogenesis, and Pyruvate metabolism. Figure 3C visualizes the results of correlation analysis, all statistically significant R values present color between the pair. We identified three correlations as the strongest (by R-value): Succinic acid and Erythrocytes (0.89), Succinic acid and GFR (0.89), Succinic acid and Hemoglobin (0.86).
The same analysis strategy was performed in the COVID-19 Recovery phase 3-4 months post-hospitalization (timepoint C) vs Population Controls contrast, summarised in Figure 4A–C. T-test identified 10 statistically significant metabolites (FDR<0.05) (Supplementary Table 4A), and 4 metabolites were FC>1.5 (Supplementary Table 4B). Volcano plot (Figure 4A) shows two metabolites (V5FC, Ornithine) as significantly different (FDR<0.05, Fold Change ± 1.5) in the analyzed contrast. Interestingly, pathway analysis (Figure 4B) in this contrast revealed 16 significantly enriched pathways (FDR < 0.05) compared to only 3 in TimepointB vs Population controls. In Acute phase (timepoint C) vs Population Controls, the top three pathways by impact values (among statistically significant) are: D-glutamine and D-glutamate metabolism, Alanine, aspartate and glutamate metabolism, Glycine, serine and threonine metabolism. Figure 4C heatmap visualizes the results of correlation analysis between Timepoint C metabolites and Timepoint C blood test, all statistically significant R values present color between pair. We identified these three correlations as strongest (by R-value): Cholesterol-Cholesterol (0,91), Triglycerides – Triglycerides (TG) (0,89), HDL-Cholesterol-HDL-Cholesterol (0,86) which are the same molecules only measured with different methods and labs, but next strong R values identified are: Apo-B100- Cholesterol (0,83), Isoleucine-D-dimer (-0,81), Apo-B100- Non-HDL cholesterol (0,80) (Supplementary Table 4D).
3. Discussion
In this study involving 41 hospitalized COVID-19 patients (measuring metabolites in three timepoints) and 41 population controls, we found proof of dysregulated amino acid metabolism, dyslipidemia, glycoprotein level alterations and energy metabolism disturbances that we discuss below. Given the high heterogeneity of acute COVID-19 and PASC, the main limitation of our study is the relatively small sample size. However, the application of a longitudinal study design provides greater statistical power and minimizes potential interference with variable complexity at individual levels.
3.1. Dysregulations in amino acid metabolism
In our previous metabolomic investigation, we focused on the patients in the acute phase of COVID-19, uncovering significant alterations in amino acid metabolism. Using liquid chromatography-mass spectrometry (LC-MS) on blood sera, we identified tryptophan (tryptophan, kynurenine, and 3-hydroxy-DL-kynurenine) and arginine (citrulline and ornithine) metabolism, glutamine depletion as contributing pathways in the acute phase immune response to SARS-CoV-2 in severe COVID-19. We also reported altered metabolism of several amino acids (Alanine, Leucine, Histidine, Tyrosine, Methionine, Phenylalanine, Asparagine, Glutamine and others) [12]. The current study used NMR on a different cohort of COVID-19 patients also extending the observation period and including samples from the timepoint of 3-4 months post-hospitalization. Limma linear regression identified Histidine, Methionine, Phenylalanine, Threonine, Glutamine, Tyrosine, and others as significant for COVID-19 patient time-series metabolic profiles, but Choline and Valine were significantly different between Long Covid/Recovered subgroups. The amino acid concentrations tend to normalize to similar levels of controls for most patients 3-4 months post-hospitalization. The extensive dysregulation of amino acids during the acute phase indicates significant immune system activation, as recently, they are proposed to be immunometabolites and immunotrasmitters and mark immune cell activity pathways [9,10]. For example, glutamine is important as respiratory fuel for macrophage function, survival, and proliferation of T and B cells [10,19,20,21]. Interestingly, Ornithine, an amino acid involved in the urea cycle, was found to be significantly changed in the present study in time series data linear regression analysis, and we observed that it is higher in the COVID-19 acute phase. For most patients, it lowers to normal levels during the first month of recovery, but for some, it is still significantly higher than levels in controls 3-4 months after hospitalization. We also noted that Ornithine significantly positively correlated with alanine aminotransferase (ALT) and ALT (aspartate aminotransferase) in the 3-4 months post-hospitalisation recovery phase. We discuss liver involvement further in next paragraphs.
3.2. Dyslipidemia in COVID-19
Linear regression analysis for time-series data during recovery from severe COVID-19 identified Apolipoprotein-A1 (ApoA-1) and high-density lipoprotein-cholesterol (HDL-Chol) as significantly altered, and when compared to population controls, the Apo-A1 and HDL-Chol levels in blood plasma were still lower month after hospitalization, and for several patients also remained low 3-4 months after hospitalization. Importantly, HDL-Chol was among the significant metabolites when linear regression was used to investigate differences among Long Covid and Recovered patient groups. This observation is consistent with other studies, which have associated low HDL-Chol levels with poor outcomes [22,23]. HDL-Chol is considered the ‘’good’’ cholesterol because it enhances endothelial function and promotes endothelial cell integrity, Apo-A1 forms the largest part of HDL cholesterol. Adding to that, it has been previously proven that the activation of the inflammatory cascade can cause a decrease in HDL-Chol and changes in apolipoprotein profiles, which manifests as impairment in the reverse cholesterol transport mechanism by which the body removes excess cholesterol from peripheral tissues and delivers them to the liver, consequently leading to an increased accumulation of cholesterol in cells [15,24,25]. To note, we also observed a positive correlation between acute phase ApoA-1 and HDL-chol levels measured by NMR and the recovery phase (1 month and 3-4 months post-hospitalization) HDL-cholesterol levels measured in the clinical lab, indicating that patients are exposed to an abnormal level of lipoproteins for a prolonged time, and it could contribute to endothelial damage observed in COVID-19 patients [26]. We also identified changes in other lipoprotein and triglyceride fractions, indicating dyslipidemia that has been observed in COVID-19 patients before, increasing the risk of cardiovascular complications for patients recovering from COVID-19 [22,23]. Our study provides further evidence that dyslipidemia is characteristic of recovery from COVID-19 in hospitalized patients; taken together with other literature evidence, it is advisable to monitor blood lipid levels for hospitalized COVID-19 patients and consider lipid-lowering medication to avoid poor cardiovascular outcomes.
3.3. Glycoprotein level alterations
GlycA originates from a subset of glycan N-acetylglucosamine residues, GlycB (branched-chain N-acetyl signals), Glyc (N- acetylneuraminic acid) on enzymatically glycosylated acute-phase proteins (levels rise or fall in response to inflammation, modification carried out by glycosidases and glycosyltransferases) [27,28,29,30]. Inflammation-induced alterations of N-linked acute-phase glycoproteins result primarily from the addition or removal of sialic acid, galactose, or fucose residues [27,30]. Otvos et al showed that GlycA can be a robust systematic inflammation marker; Duprez et al identified it as a marker for inflammation and a strong predictor of cardiovascular events [29,31]. In the present study, GlycA, GlycB, and Glyc significantly changed during recovery from severe COVID-19, as identified by linear regression analysis (the highest levels are in the acute phase). Additionally, GlycA, GlycB, and Glyc showed the highest Hotteling-T2 values across time series data in COVID-19 recovery, indicating the most constant change pattern across patients. When compared to population controls, GlycA, Glyc, and GlycB were still elevated one month after hospitalization, and for most patients, it stayed elevated also 3-4 months after hospitalisation when compared to population controls. Importantly, GlycA is a novel marker for chronic, long-term inflammation. Leveraging population-based omics data in a 10-year-long study, increased GlycA levels were found to be chronic within individuals for up to a decade, increasing the risk of severe respiratory infections, which can lead to septicemia and pneumonia [27]. Elevated GlycA positively corresponded to inflammatory cytokines and increased neutrophil activity, suggesting chronic inflammation [27]. We also note that GlycA, GlycB, and Glyc significantly correlated with clinical blood test parameters in our cohort (plasma glucose, LDH (lactate dehydrogenase), GGT (gamma-glutamyl transferase), CRP(C-reactive protein), HDL and LDL cholesterol, neutrophil count, triglycerides). The chronic inflammation should be studied in large, prospective recovered COVID-19 and Long COVID patient cohorts to determine the best anti-inflammation therapeutic strategies for patients with PASC.
3.4. Energy metabolism disturbances
Several studies have presented evidence of disturbed energy metabolism by SARS-CoV-2 virus infection, and our results add proof in support of these proposed mechanisms as important in recovery after hospitalization [11,32,33]. In our study, two important overlapping pathways of energy metabolism, glycolysis/gluconeogenesis and pyruvate metabolism, were identified as significantly different between COVID-19 patients (in all time points) and population controls. Adding to that, the TCA cycle was identified as significantly different between COVID-19 patients in the acute phase and population controls and between COVID-19 patients in the latest recovery phase (3-4 months after hospitalization) and controls. This indicates serious alterations in energy metabolism. Caterino et al. compared metabolomes and cytokine profiles among different clinical severity patients and controls [11]. They proposed a strong connection between the hypoxemia state and the subsequent oxidative stress, which may have a major effect on the mitochondrial energy metabolism and detoxification processes in the hepatocytes, noting that the hepatic urea cycle is the main metabolic pathway involved in the detoxification processes [11]. The urea cycle metabolizes ammonia to urea with a fumarate shunt connecting the urea and TCA cycles within the liver [34]. In a hypoxia state, there is a burden of glucose production (gluconeogenesis) and urea elimination (urea cycle) to the liver. Although analyzing drug metabolites was out of this study’s scope, a mentionable fact is that corticosteroids, especially dexamethasone, were used for the treatment of severe COVID-19, and corticosteroids increase hepatic gluconeogenesis, reduce peripheral use of glucose and increase insulin levels contributing to hyperglycemia in patients. Acetic acid, Lactic acid, Pyruvic acid, Citric acid, and Succinic acid are involved in energy metabolism pathways, and we found them to be significantly changed in COVID-19 patients (linear regression for time-series data). The optimistic result in our relatively small cohort study is that 3-4 months post-hospitalization, the levels of these metabolites normalized and were similar to population controls.
Liver tissue is not the only one that changes rapidly in an oxygen deficiency environment caused by pneumonia and lung damage. Marchuenda-Egea & Narro-Serrano described how energy metabolism changes in muscles, liver, and adipose tissue, which leads to COVID-19 signature blood metabolome in severe COVID-19 patients [32]. Hypoxia caused by pneumonia impacts muscle tissue significantly because oxygen is crucial for the efficient functioning of mitochondria, where it acts as the final electron acceptor in oxidative phosphorylation (the main cellular ATP production process) [32,35]. The muscle tissue adapts to this stress by breaking down muscle proteins to generate energy, one possible pathway for that is the glucose-alanine cycle that allows myocytes to obtain energy continuously from muscle proteins by oxidizing amino acids [32,36]. The glucose-alanine cycle generates alanine that can be transported to the liver, where it can donate amino groups to pyruvate (to be used in gluconeogenesis) or to oxaloacetate, producing glutamate and initiating the urea cycle [36]. In our study, pathway analysis revealed several significantly different alanine metabolism pathways between COVID-19 patients during recovery phases and population controls, but alanine itself was not significantly altered. Adding to that, during hypoxia, the liver obtains its energy from the oxidation of lipids through β-oxidation, generating ketone bodies [37]. We also measured ketone bodies, 3-Hydroxybutyric acid was higher for some COVID-19 patients at hospitalization but was the same level as controls in both recovery phases, Acetoacetic acid was high for most patients in the acute phase and for some patients also month later, but for all patients returned to normal level 3-4 months post-acute phase. An increase in ketone bodies and plasma lipoproteins during the acute phase indicates a greater mobilization of energy source from adipose tissue, and this contributes to the disturbed lipid metabolism discussed previously.
Finally, our metabolomics study measured glucose, which was identified as statistically significant by both performed limma linear regression analysis- for COVID-19 time series data and Long Covid/ Recovered subgroups. The highest levels of glucose were measured during the acute phase. For most patients, glucose tends to go back to normal after 3-4 months of recovery (same levels as controls), but for several patients, it stays higher. It should be noted that one patient had new-onset diabetes, and another had increased blood glucose in EHR data during the 12-month post-acute period. In the scientific literature, there is an increasing amount of evidence that some patients develop diabetes as post-acute sequelae of COVID-19 and need glucose-lowering therapies [4,17,38,39]. Literature and our results show that attention (blood tests) to possible hyperglycemia in recovering COVID-19 patients is advisable and should be adequately treated to avoid the development of hyperglycemia-induced complications.
4. Materials and Methods
Study design. Our study cohort consists of 41 hospitalized COVID-19 patients from whom blood was collected at three timepoints: (1) Acute phase (Timepoint A) samples were collected on 1st or 2nd day of hospitalization, (2) Recovery phase (Timepoint B) samples were collected 35 ± 14.7.65 days later, and (3) later Recovery phase (Timepoint C) samples were collected 99 ± 16.79 days after the first sample. Hematological and biochemical analyses in the acute phase were performed at the hospital’s clinical lab, but during recovery in a certified clinical laboratory in an ambulatory setting. Health Registry data was obtained for the patients, and they were divided into two subgroups (Recovered (n=10)/Long Covid (n=31), by these criteria: if they have new post-acute sequelae of SARS-CoV-2 infection (PASC) diagnosis in 12 months following COVID-19 acute phase (1-month acute phase, 12 month period as a post-acute period). We used the PASC diagnosis selection from the recently published work of Zang et al [4]. We also compared COVID-19 patients to population controls (n=41) without acute infection at sample collection. All samples were collected during the same period (years 2020-2021). The study design is visualized in Figure 1A, but cohort characteristics are summarised in Table 1. Written informed consent was obtained from every participant before their inclusion in the study, and the study protocol was approved by the Central Medical Ethics Committee of Latvia (No. 01-29.1.2/928).
Sample preparation and instrumental analysis. The blood samples were collected in EDTA blood collection tubes and centrifuged to separate blood plasma, it was aliquoted, frozen, and stored according to standard operating procedures SOPs of the Genome Database of Latvian Population (National Biobank [40]). The raw NMR spectrum in thawed blood plasma samples was recorded using the Bruker IVDr (B.I.) method package, following SOPs. Together, three modules of analytes were measured: B.I. QUANT-PS™, B.I. LISA™, B.I. BioBank QC™ using Bruker AVNEO 600 MHz IVDr NMR-Solution (Bruker BioSpin GmbH, Ettlingen, Germany). The annotation and quantification of serum spectra were provided automatically via Bruker IVDr Quantification in Plasma/Serum, B.I. Quant-PS™ analysis package (Bruker BioSpin GmbH, Ettlingen, Germany).
Statistical analysis. For statistical analysis of metabolites, we used a merged table of all quantified metabolites, including free metabolites (no protein denaturing done) across different chemical classes (Alcohols and derivatives, Amines and derivatives, Amino acids and derivatives, Carboxylic acids, Essential nutrients, Keto acids and derivatives, Sugars and derivatives, Sulfones, Technical additives), N-acetylated- glycoproteins (GlycA, GlycB), Glyc (N-acetylneuraminic acid), SPC (supramolecular phospholipids composite), quantified lipids and lipoproteins (Triglycerides, Cholesterol, LDL-Chol, HDL-Chol, LDL-Phos, HDL-Phos, Apo-A1, Apo-B100) and concentrations of lipoprotein VLDL, IDL, LDL HDL classes and subclasses. We also included biologically relevant proportion calculations: Apo-B100/Apo-A1, Glyc/SPC, LDL-cholesterol (LDCH) / HDL-cholesterol (HDCH) as possible biomarkers across contrasts. Together, we analyzed 169 quantified features for 164 samples. See quantified input features for all samples in Supplementary Table 1A.
For statistical analysis, we used web-based Metaboanalyst software 5.0 [18]. To normalize metabolite data, we applied log transformation (base 10) and Pareto scaling (mean-centered and divided by the square root of the standard deviation of each variable). Linear Regression for Time-Series data analysis in COVID-19 patients. The underlying method is based on limma [41]. We analyzed two models: (1) time series data and (2) Time series + Phenotype (Recovered/Long Covid). For the (1) model, the reference group was set to be Timepoint C when compared to Timepoints A and B, but Timepoint B as a reference group to compare with Timepoint A; for the (2) model the direction of comparison was Long Covid vs Recovered. In both analyses, Subject, indicating one patient, was used as a covariate to adjust for repeated within-subject samples. For time-pattern analysis, we used multivariate empirical Bayes statistical time-series analysis (MEBA) at Metaboanalyst, a method to rank the features by calculating Hotelling’s T2. It can be applied to determine if these variables’ overall pattern or distribution changes significantly across timepoints.
Univariate analysis for two group comparisons. To identify the significantly different metabolites between COVID-19 patients and population controls in each of the timepoints (A, B, C) and determine whether we see consistent changes across all contrasts that are involved in the pathogenesis of/ recovery from acute COVID-19, we also performed the univariate analysis in each of the interesting contrasts with Metaboanalyst: t-test to determine significance and fold change analysis calculation to determine the level of changed metabolite concentration, to visualize the significance and fold change analysis results in volcano plot -log10(p-value) and log2FoldChange were calculated in Metaboanalyst and visualized with Prism Graphpad 9.
Pathway analysis. We also used Metaboanalyst to determine significant pathways in each of the contrasts, for that, a set of features (quantified metabolites) was used (only the ones that could be annotated). Log transformation, Pareto scaling was applied for data normalization. The reference pathway library was selected for Homo Sapiens (KEGG). The enrichment method (Global Test) was used to calculate adjusted p-values and -log10(p-value) for the plot. the reference pathway library was selected Homo Sapiens (KEGG).
Biochemical analysis and metabolite correlations. We also analyzed correlations of the metabolites with the hematological and biochemical markers from laboratory analyses at different timepoints. In each pairwise correlation, we excluded any patient if the respective laboratory analysis was not performed or the metabolite/marker was below detection level, and the correlation coefficient was only calculated if data were available for at least five patients. The calculation was done using the non-parametric Kendall’s Tau.
5. Conclusions
While currently there are no regulator-approved therapies that directly target the root causes of Long COVID symptoms, and the causes themselves are not widely understood, metabolomics data from longitudinal studies of recovering patients can identify the molecules behind observed complications to advise possible risks in recovery from severe COVID-19. From our data and literature evidence, we recommend checking recovering COVID-19 patients for hyperglycemia and dyslipidemia with widely available markers in blood and consider chronic inflammation as a potential risk of developing complications over time.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table 1 consists of table A: quantified input features in all samples (used for statistical analysis), B: limma linear regression results for time series data, C: Hotelling’s T-squared (Hotelling-T^2) test results, D: limma linear regression for Long Covid/Recovered subgroups; Supplementary Table 2 consists of table A: t-test results for Timepoint A vs Controls, B: fold-change analysis for Timepoint A vs Controls, C: pathway analysis results for Timepoint A vs Controls, D: correlation analysis results for Timepoint A metabolites vs Timepoint B blood test results, E: correlation analysis results for Timepoint A metabolites vs Timepoint C blood test results; Supplementary Table 3 consists of table A: t-test results for Timepoint B vs Controls, B: fold-change analysis for Timepoint B vs Controls, C: pathway analysis results for Timepoint B vs Controls, D: correlation analysis results for Timepoint B metabolites vs Timepoint B blood test results; Supplementary Table 4 consists of table A: t-test results for Timepoint C vs Controls, B: fold-change analysis for Timepoint C vs Controls, C: pathway analysis results for Timepoint C vs Controls, D: correlation analysis results for Timepoint C metabolites vs Timepoint C blood test results
Author Contributions
Writing-original draft preparation, statistical analysis: L.A. Correlation analysis, visualization: L.J. Supervision, conceptualization, project administration, writing- review and editing: V.R., J.K., L.A., M.B. Data curation: D.B., L.P. Metabolomic mehod: A.T, R.L. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the European Regional Development Fund under the project “Identification of molecular determinants associated with the risk for various COVID-19 long-term effects: a comprehensive cohort-based study in Latvian population. (POST-COVID-TRACK)” (Project No. 1.1.1.1/21/A/029).
Institutional Review Board Statement
The study was carried out in accordance with the principles of the Declaration of Helsinki and approved by the Central Medical Ethics Committee (No. 01-29.1.2/928).
Informed Consent Statement
Informed consent was obtained from all participants at the beginning of the study.
Data Availability Statement
The data presented in this study are available upon request from the corresponding authors due to privacy or ethical restrictions.
Acknowledgments
We acknowledge the Genome Database of Latvian Population and Latvian Biomedical Research and Study Centre for organizing participants’ recruitment and bio-sample processing and data storage.
Conflicts of Interest
A.T. and R.L. are employed by Bruker BioSpin GmbH but were not involved in the study design and analysis of the present data. Their contribution consisted of providing AVNEO 600 MHz IVDr NMR-Solution for sample analysis. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265–1273. [Google Scholar] [CrossRef]
- Wade, D.T. Rehabilitation after COVID-19: an evidence-based approach. Clin. Med. 2020, 20, 359–365. [Google Scholar] [CrossRef]
- Zang, C.; Zhang, Y.; Xu, J.; Bian, J.; Morozyuk, D.; Schenck, E.J.; Khullar, D.; Nordvig, A.S.; Shenkman, E.A.; Rothman, R.L.; et al. Data-driven analysis to understand long COVID using electronic health records from the RECOVER initiative. Nat. Commun. 2023, 14, 1–14. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: a systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- O’Mahoney, L.L.; Routen, A.; Gillies, C.; Ekezie, W.; Welford, A.; Zhang, A.; Karamchandani, U.; Simms-Williams, N.; Cassambai, S.; Ardavani, A.; et al. The prevalence and long-term health effects of Long Covid among hospitalised and non-hospitalised populations: A systematic review and meta-analysis. EClinicalMedicine, 2022, 55, 101762. [Google Scholar] [CrossRef]
- Han, Q.; Zheng, B.; Daines, L.; Sheikh, A. Long-Term Sequelae of COVID-19: A Systematic Review and Meta-Analysis of One-Year Follow-Up Studies on Post-COVID Symptoms. Pathogens 2022, 11, 269. [Google Scholar] [CrossRef]
- Chen, C.; Haupert, S.R.; Zimmermann, L.; Shi, X.; Fritsche, L.G.; Mukherjee, B. Global Prevalence of Post-Coronavirus Disease 2019 (COVID-19) Condition or Long COVID: A Meta-Analysis and Systematic Review. J. Infect. Dis. 2022, 226, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, M. Amino acids: key sources for immunometabolites and immunotransmitters. Int. Immunol. 2020, 32, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Costanzo, M.; Fedele, R.; Cevenini, A.; Gelzo, M.; Di Minno, A.; Andolfo, I.; Capasso, M.; Russo, R.; Annunziata, A.; et al. The Serum Metabolome of Moderate and Severe COVID-19 Patients Reflects Possible Liver Alterations Involving Carbon and Nitrogen Metabolism. Int. J. Mol. Sci. 2021, 22, 9548. [Google Scholar] [CrossRef]
- Ansone, L.; Briviba, M.; Silamikelis, I.; Terentjeva, A.; Perkons, I.; Birzniece, L.; Rovite, V.; Rozentale, B.; Viksna, L.; Kolesova, O.; et al. Amino Acid Metabolism is Significantly Altered at the Time of Admission in Hospital for Severe COVID-19 Patients: Findings from Longitudinal Targeted Metabolomics Analysis. Microbiol. Spectr. 2021, 9, e0033821. [Google Scholar] [CrossRef] [PubMed]
- Liptak, P.; Baranovicova, E.; Rosolanka, R.; Simekova, K.; Bobcakova, A.; Vysehradsky, R.; Duricek, M.; Dankova, Z.; Kapinova, A.; Dvorska, D.; et al. Persistence of Metabolomic Changes in Patients during Post-COVID Phase: A Prospective, Observational Study. Metabolites 2022, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, G.; Bissinger, R.; Liu, A.; Cannet, C.; Schäfer, H.; Kienzle, K.; Bitzer, M.; Häberle, H.; Göpel, S.; Trautwein, C.; et al. Maintained imbalance of triglycerides, apolipoproteins, energy metabolites and cytokines in long-term COVID-19 syndrome patients. Front. Immunol. 2023, 14, 1144224. [Google Scholar] [CrossRef] [PubMed]
- Kluck, G.E.G.; Yoo, J.-A.; Sakarya, E.H.; Trigatti, B.L. Good Cholesterol Gone Bad? HDL and COVID-19. Int. J. Mol. Sci. 2021, 22, 10182. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Hussien, N.R.; Al-Niemi, M.S.; Fahad, E.H.; Al-Buhadily, A.K.; Al-Gareeb, A.I.; Al-Hamash, S.M.; Tsagkaris, C.; Papadakis, M.; Alexiou, A.; et al. SARS-CoV-2 induced HDL dysfunction may affect the host’s response to and recovery from COVID-19. Immunity, Inflamm. Dis. 2023, 11, e861. [Google Scholar] [CrossRef]
- Xie, Y.; Al-Aly, Z. Risks and burdens of incident diabetes in long COVID: a cohort study. Lancet Diabetes Endocrinol. 2022, 10, 311–321. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef]
- Cengiz, M.; Uysal, B.B.; Ikitimur, H.; Ozcan, E.; Islamoğlu, M.S.; Aktepe, E.; Yavuzer, H.; Yavuzer, S. Effect of oral l-Glutamine supplementation on Covid-19 treatment. Clin. Nutr. Exp. 2020, 33, 24–31. [Google Scholar] [CrossRef]
- Oldani, M.; Sandini, M.; Nespoli, L.; Coppola, S.; Bernasconi, D.P.; Gianotti, L. Glutamine Supplementation in Intensive Care Patients. Medicine 2015, 94, e1319. [Google Scholar] [CrossRef]
- Obayan, A.O.E. Overview of the Rationale for L-Glutamine Treatment in Moderate-Severe COVID-19 Infection. J. Infect. Dis. Epidemiology 2021, 7. [Google Scholar] [CrossRef]
- Henry, B.M.; Szergyuk, I.; de Oliveira, M.H.S.; Abosamak, M.F.; Benoit, S.W.; Benoit, J.L.; Lippi, G. Alterations in the lipid profile associate with a dysregulated inflammatory, prothrombotic, anti-fibrinolytic state and development of severe acute kidney injury in coronavirus disease 2019 (COVID-19): A study from Cincinnati, USA. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 863–868. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Risks and burdens of incident dyslipidaemia in long COVID: a cohort study. Lancet Diabetes Endocrinol. 2023, 11, 120–128. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Memon, R.A.; Feingold, K.R.; Grunfeld, C. Infection and Inflammation-Induced Proatherogenic Changes of Lipoproteins. J. Infect. Dis. 2000, 181, S462–S472. [Google Scholar] [CrossRef]
- Esteve, E.; Ricart, W.; Fernández-Real, J.M. Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin. Nutr. 2005, 24, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Calcaterra, I.L.; Mosella, M.; Formisano, R.; D’anna, S.E.; Bachetti, T.; Marcuccio, G.; Galloway, B.; Mancini, F.P.; Papa, A.; et al. Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target. Biomedicines 2022, 10, 812. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, S.C.; Würtz, P.; Nath, A.P.; Abraham, G.; Havulinna, A.S.; Fearnley, L.G.; Sarin, A.-P.; Kangas, A.J.; Soininen, P.; Aalto, K.; et al. The Biomarker GlycA Is Associated with Chronic Inflammation and Predicts Long-Term Risk of Severe Infection. Cell Syst. 2015, 1, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, J.; Berezhnoy, G.; Cannet, C.; Schäfer, H.; Geisler, T.; Rohlfing, A.-K.; Gawaz, M.; Merle, U.; Trautwein, C. Stratification of hypertension and SARS-CoV-2 infection by quantitative NMR spectroscopy of human blood serum. Commun. Med. 2023, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Otvos, J.D.; Shalaurova, I.; Wolak-Dinsmore, J.; Connelly, M.A.; Mackey, R.H.; Stein, J.H.; Tracy, R.P. GlycA: A Composite Nuclear Magnetic Resonance Biomarker of Systemic Inflammation. Clin. Chem. 2015, 61, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Gornik, O.; Lauc, G. Glycosylation of Serum Proteins in Inflammatory Diseases. Dis. Markers 2008, 25, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Duprez, D.; Neuhaus, J.; Otvos, J.; Neaton, J.D.; Lundgren, J.D. Abstract 14731: Glyc A, a Novel Marker of Inflammation, Predicts Cardiovascular Events in HIV-Positive Patients: Results of SMART Study. Circulation 2014, 130. [Google Scholar] [CrossRef]
- Marhuenda-Egea, F.; Narro-Serrano, J. Evaluation of risk factors for COVID-19 severity or death and their relationship to metabolic pathways. Heliyon 2023, 9, e14161. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, Y.; Monárrez-Espino, J.; López, D.A.G.; Zheng, J.; Borrego, J.C.; Torres-Calzada, C.; Elizalde-Díaz, J.P.; Mandal, R.; Berjanskii, M.; Martínez-Martínez, E.; et al. The plasma metabolome of long COVID patients two years after infection. Sci. Rep. 2023, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Lamers, W.H.; Chamuleau, R.A.; Le Plénier, S.; Goron, A.; Sotiropoulos, A.; Archambault, E.; Guihenneuc, C.; Walrand, S.; Salles, J.; et al. Nitrogen metabolism and ornithine cycle function. Physiol. Rev. 1990, 70, 701–748. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. et Biophys. Acta (BBA)—Bioenerg. 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; De Sousa-Coelho, A.L.; Harata, I.; Aoun, C.; Weimer, S.; Shi, X.; Herrera, K.N.G.; Takahashi, H.; Doherty, C.; Noguchi, Y.; et al. l-Alanine activates hepatic AMP-activated protein kinase and modulates systemic glucose metabolism. Mol. Metab. 2018, 17, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- McKeigue, P.M.; McGurnaghan, S.; Blackbourn, L.; Bath, L.E.; McAllister, D.A.; Caparrotta, T.M.; Wild, S.H.; Wood, S.N.; Stockton, D.; Colhoun, H.M. Relation of Incident Type 1 Diabetes to Recent COVID-19 Infection: Cohort Study Using e-Health Record Linkage in Scotland. Diabetes Care 2023, 46, 921–928. [Google Scholar] [CrossRef]
- Wander, P.L.; Lowy, E.; Beste, L.A.; Tulloch-Palomino, L.; Korpak, A.; Peterson, A.C.; Kahn, S.E.; Boyko, E.J. The Incidence of Diabetes Among 2,808,106 Veterans with and Without Recent SARS-CoV-2 Infection. Diabetes Care 2022, 45, 782–788. [Google Scholar] [CrossRef]
- Rovite, V.; Wolff-Sagi, Y.; Zaharenko, L.; Nikitina-Zake, L.; Grens, E.; Klovins, J. Genome Database of the Latvian Population (LGDB): Design, Goals, and Primary Results. J. Epidemiology 2018, 28, 353–360. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Study design. The study cohort consists of 41 hospitalized COVID-19 patients for whom samples were collected at three timepoints (1) Acute phase (Timepoint A) on 1st or 2nd day of hospitalization, (2) Recovery phase (Timepoint B) samples were collected 35 ± 14.7.65 days later, and (3) later Recovery phase (Timepoint C) samples were collected 99 ± 16.79 days after the first sample. (B) Venn diagram shows the count of statistically significant metabolites between three timepoints A, B, and C. (C) Top50 significantly changed features (metabolites) visualized in the heatmap, samples arranged by timepoint, distance measure: euclidean, clustering algorithm: ward. (D) Time-course profiles for top 3 features.
Figure 1.
(A) Study design. The study cohort consists of 41 hospitalized COVID-19 patients for whom samples were collected at three timepoints (1) Acute phase (Timepoint A) on 1st or 2nd day of hospitalization, (2) Recovery phase (Timepoint B) samples were collected 35 ± 14.7.65 days later, and (3) later Recovery phase (Timepoint C) samples were collected 99 ± 16.79 days after the first sample. (B) Venn diagram shows the count of statistically significant metabolites between three timepoints A, B, and C. (C) Top50 significantly changed features (metabolites) visualized in the heatmap, samples arranged by timepoint, distance measure: euclidean, clustering algorithm: ward. (D) Time-course profiles for top 3 features.
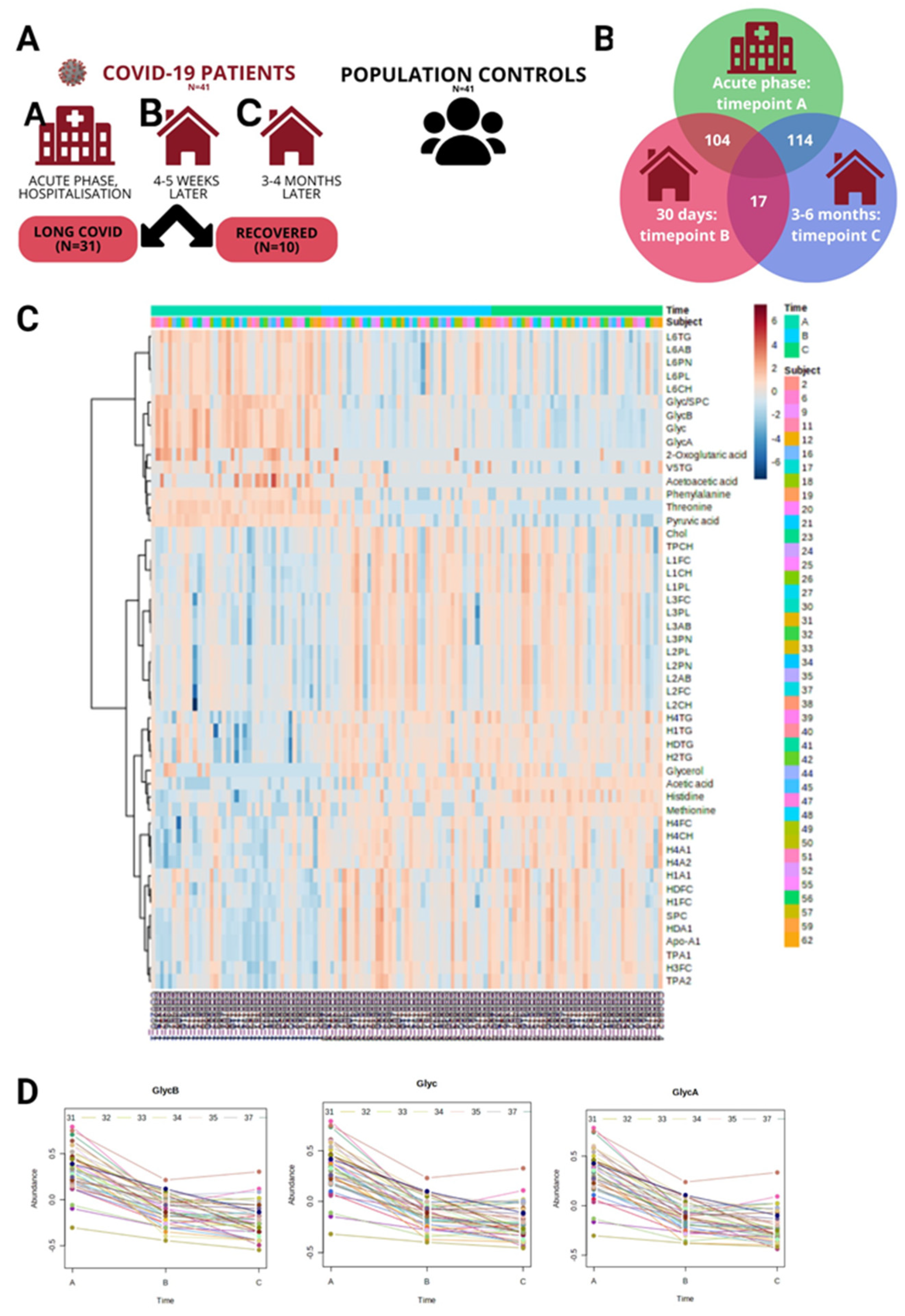
Figure 2.
Timepoint A vs Population controls. (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively, resulting in 13 strongly significant metabolites (Fold Change>1.5 and FDR<0,05) indicated in red in the plot. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint A metabolites (x axis) vs Timepoint B blood test (y axis) in COVID-19 patients. Heatmap with clinically most relevant metabolite (timepoint A) correlations with biochemical and hematological analysis results from the clinical lab at recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown. (D) Timepoint A metabolites (x axis) vs Timepoint C blood test (y axis). Heatmap visualizing metabolite (Acute phase) correlations with biochemical and hematological analysis results in recovery phase timepoint C (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
Figure 2.
Timepoint A vs Population controls. (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively, resulting in 13 strongly significant metabolites (Fold Change>1.5 and FDR<0,05) indicated in red in the plot. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint A metabolites (x axis) vs Timepoint B blood test (y axis) in COVID-19 patients. Heatmap with clinically most relevant metabolite (timepoint A) correlations with biochemical and hematological analysis results from the clinical lab at recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown. (D) Timepoint A metabolites (x axis) vs Timepoint C blood test (y axis). Heatmap visualizing metabolite (Acute phase) correlations with biochemical and hematological analysis results in recovery phase timepoint C (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
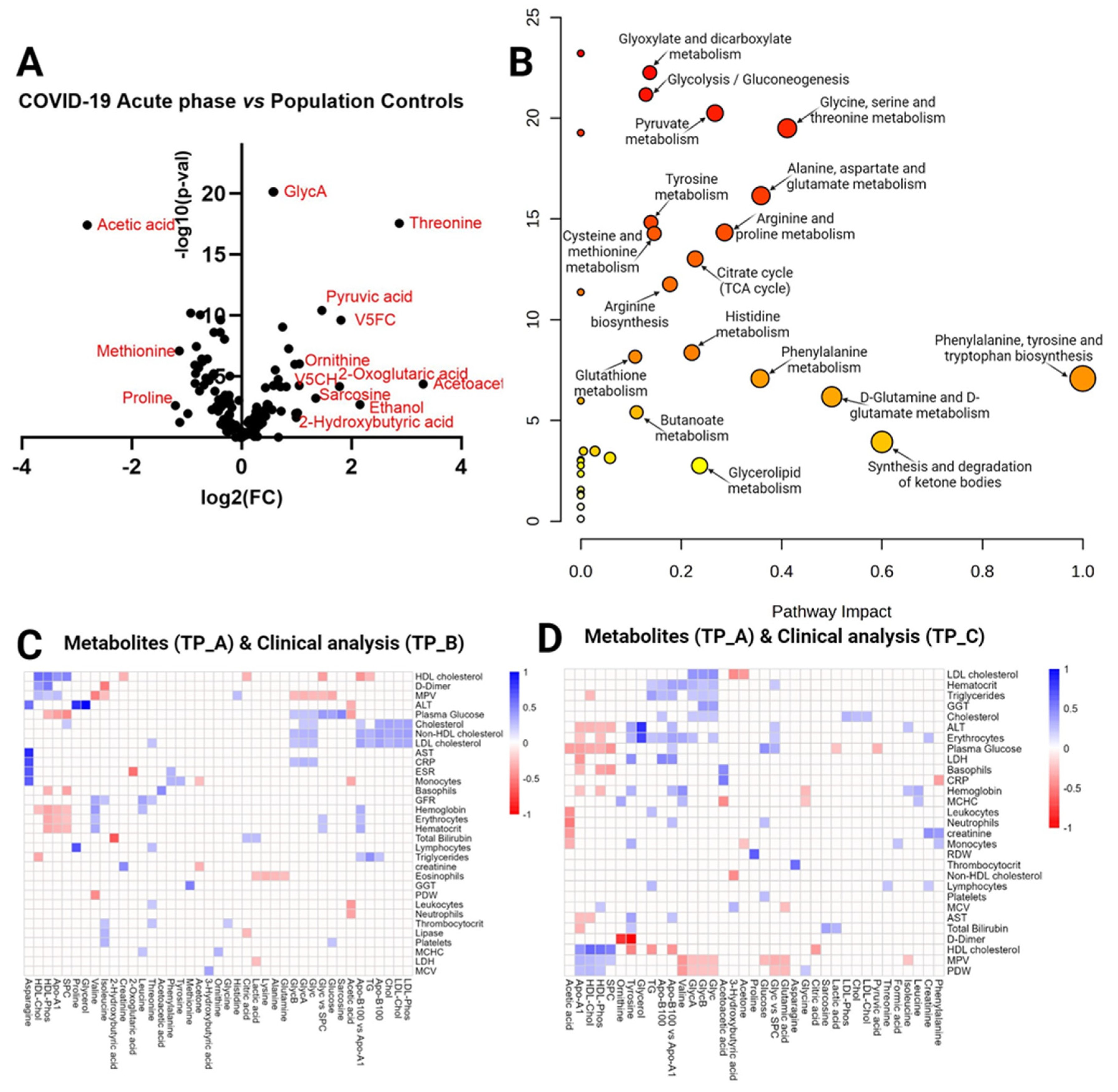
Figure 3.
Timepoint B vs Population controls (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint B metabolites (x axis) vs Timepoint B blood test (y axis) in Covid-19 patients. Heatmap visualizing statistically significant metabolite correlations with biochemical and hematological analysis results in recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
Figure 3.
Timepoint B vs Population controls (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint B metabolites (x axis) vs Timepoint B blood test (y axis) in Covid-19 patients. Heatmap visualizing statistically significant metabolite correlations with biochemical and hematological analysis results in recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
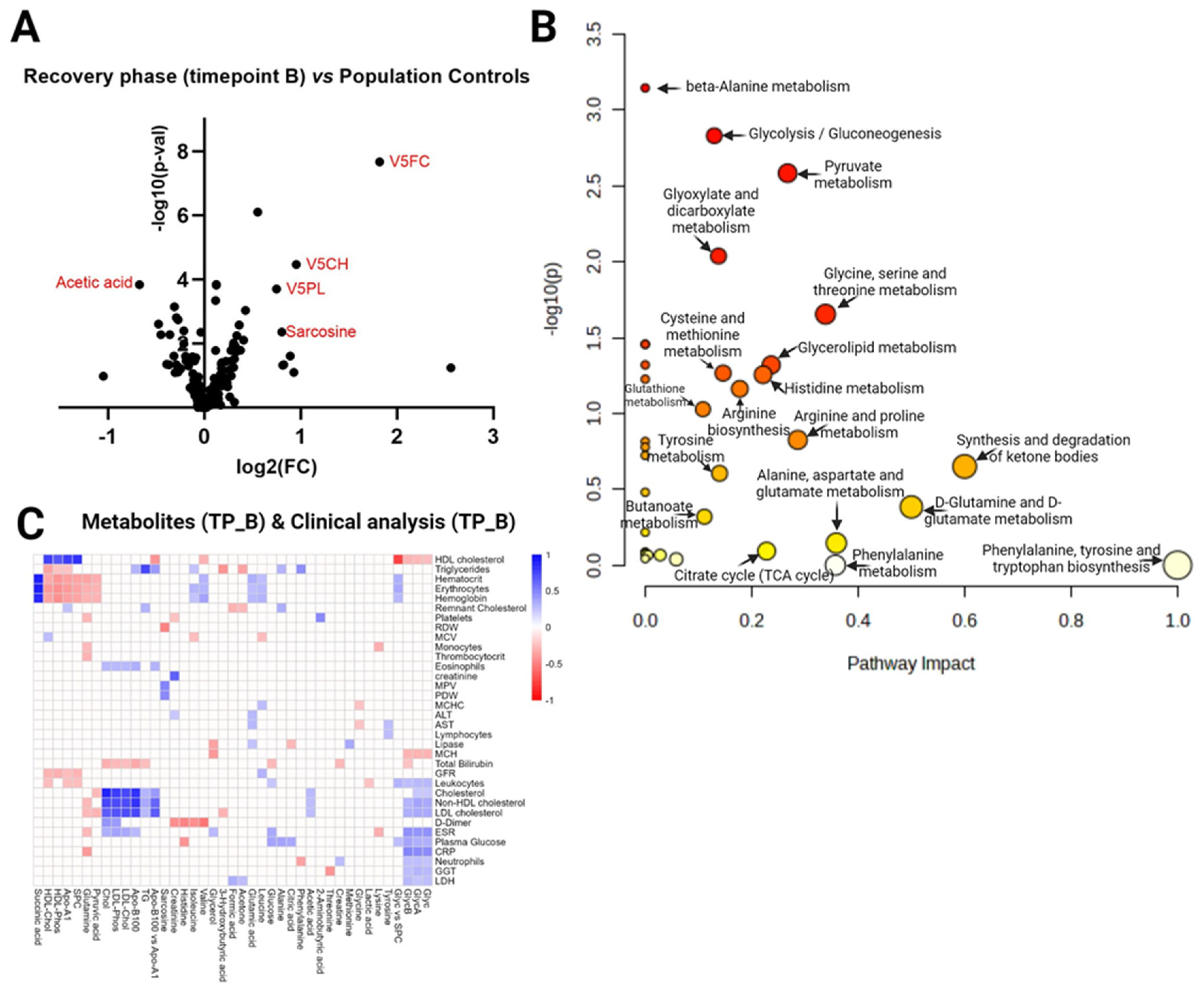
Figure 4.
Timepoint C vs Population controls. (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint C metabolites (x axis) vs Timepoint C blood test (y axis) in Covid-19 patients. Heatmap visualizing statistically significant metabolite correlations with biochemical and hematological analysis results in recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
Figure 4.
Timepoint C vs Population controls. (A) Volcano plot visualizes the distribution of metabolites in the analyzed contrast, significance (-log10(p-value)) versus log2 fold change is plotted on the y and x axes, respectively. (B) Scatterplot representing the most relevant metabolic pathways from KEGG library arranged by adjusted P values (obtained by Global Test pathway enrichment analysis) on the y-axis, and pathway impact values (from pathway topology analysis) on the x-axis. The node color is based on its P value, and the node radius is determined based on pathway impact values. (C) Timepoint C metabolites (x axis) vs Timepoint C blood test (y axis) in Covid-19 patients. Heatmap visualizing statistically significant metabolite correlations with biochemical and hematological analysis results in recovery phase timepoint B (~month after admission at the hospital), color represents the strength of relationship and its direct or inverse nature; only the pairs with sufficient data and significant correlations (p-value > 0.05) are shown.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



