
Submitted:
27 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
Serine proteases regulate cell functions through G protein-coupled protease-activated receptors (PARs). Cleavage of one peptide bond of the receptor amino terminus results in the formation of a new N-terminus ("tethered ligand") that can specifically interact with the second extracellular loop of the PAR receptor and activate it. Activation of PAR1 by thrombin (canonical agonist) and activated protein C (APC, noncanonical agonist) was described as biased agonism. Here we have supposed that synthetic peptide analogues to the PAR1 tethered ligand liberated by APC could have neuroprotective effects like APC. To verify this hypothesis a model of the ischemic brain impairment based on glutamate (Glu) excitotoxicity in primary neuronal cultures of neonatal rats has been used. It was shown that nanopeptide NPNDKYEPF-NH2 (AP9) effectively reduced the neuronal death induced by Glu. The influence of AP9 on cell survival was comparable to that of APC. Both APC and AP9 reduced the dysregulation of intracellular calcium homeostasis in cultured neurons induced by excitotoxic Glu (100 µM) or NMDA (200 µM) concentrations. PAR1 agonist synthetic peptides might be a noncanonial PAR1-agonist and a basis of novel neuroprotective drugs for disorders related to Glu excitotoxicity such as brain ischemia, trauma and some neurodegenerative diseases.
Keywords:
activated protein C
; neuroprotection
; protease-activated receptors
; neurons
; glutamate
; intracellular calcium
1. Introduction
The cerebral blood circulation decrease in ischemic stroke is one of the main pathological factors that leads to oxygen-glucose deprivation and glutamate excitotoxicity followed by neuronal cell death [1,2], inflammation [3] and BBB disruption that exacerbate brain injury [4].
A classic paradigm for G protein-coupled receptors (GPCR) activation was based on the understanding that receptor-bound agonist triggers or stabilizes receptor-related changes until it reaches an active conformation. In the last decade, it is dominantly viewed that activation with different ligands may result in distinct active receptor conformations with unique divergent signaling profiles [5,6,7]. A recognition of these biased ligands resulted in deeper understanding of mechanisms underlying biased agonism, improved assessment of ligand efficiency, and advanced search and synthesis of novel ligands for clinical use.
Acute ischemic stroke and brain injury are accompanied with the initiation of blood coagulation and the activation of hemostatic serine proteases, in particular, thrombin [8] and activated protein C (APC) [9]. Moreover, special type of receptors – protease-activated receptors (PARs) that mediate thrombin and APC-dependent regulation of cell functions can be involved in brain ischemia [10,11]. PARs are a family of highly conserved GPCR activated by proteolytic cleavage. Currently, a role played by a biased agonism for APC and other hemostatic proteases acting on PARs has been extensively investigated and discussed [12,13,14].Proteases cleave one peptide bond of the receptor amino terminus, which results in the formation of a new N-terminus ("tethered ligand") capable of specific interaction with the second extracellular loop and activate the PAR [15]. Thrombin cleaves peptide bond Arg41–Ser42 in the extracellular N-terminal sequence LDPR41S42FLLRN of PAR1, thereby disclosing a new N-terminal peptide [15,16]. However, in contrast to thrombin APC hydrolyzes peptide bond Arg46–Asn47 in the extracellular N-terminal sequence LDPRS FLLR46N47PNDKYEP of PAR1, disclosing a new N-terminal peptide NPNDKYEP - “tethered ligand” [12], responsible for cytoprotective activity of APC on endothelial cells and neurons [17]. Effects of APC can be mimicked by synthetic peptide analogues of the tethered ligands that are created after PAR1 cleavage at Arg46. By examining human endothelial cell line EA.hy926, it is found that 30 min of exposure with peptide TR47 resembling PAR1 residues 47-66 results in phosphorylation (inhibition) of glycogen synthase kinase 3β (GSK3β) at Ser9. TR47-induced GSK3β phosphorylation is inhibited by PAR1 antagonist SCH79797, suggesting that PAR1 is necessary for TR47-induced signaling [12]. However, cleavage of PAR1 by thrombin at Arg41 induces phosphorylation of extracellular regulated kinase (ERK1/2). The canonical PAR1 agonist peptide TRAP liberated by thrombin (TFLLRNPNDK) rapidly induces ERK1/2 phosphorylation, whereas TR47 does not [12]. Thrombin and АРС, interacting with the same PAR1 receptor, exert multidirectional effects during excitotoxicity and inflammation [18,19]. Thrombin increases the expression of proinflammatory and proapoptotic factors together with the procoagulant effect [19]. APC is a neuroprotector in stressed neurons and in hypoxic brain endothelium [18,20]. Recently we have reported about the anti-apoptotic effects of APC on hippocampal neurons at glutamate excitotoxicity [21]. APC (10 nM) was shown to prevent the development of apoptosis induced in cortical neurons by NMDA and staurosporine [22]. By activating PAR1 APC controls the gene expression of proinflammatory and proapoptotic factors, stabilizes endothelial cells and neurons and protects them from death [18,19,20].
It has been shown recently that the multidirectional effects of thrombin and APC may be due to the biased agonism under the action of thrombin and APC on the PAR1 in endothelial cells [12,23]. Cleavage of PAR1 by APC at Arg46 results in GSK3β phosphorylation at Ser9, whereas PAR1 hydrolyzation by thrombin at Arg41 results in phosphorylation of ERK1/2 [24]. Activation of PAR1 by thrombin can lead to concomitant activation of the Gi/o, Gq, and G12/13 families of G proteins leading to various signaling pathways that ultimately result in transient endothelial barrier disruption. This transient endothelial barrier disruption is an important physiological response promoted by the activation of PAR1 receptors that couple to multiple heterotrimeric G protein subtypes including Gq/11 and G12/13 proteins. Biased agonism leads to the induction of distinct signaling mechanisms, such as the activation of PI3K, Akt, and Rac1 by APC, which results in neuroprotection [25]. APC or TR47-induced activation of PAR1 stabilizes different conformers of PAR1 that preferentially interact with β-arrestin-2. It is known that β-arrestins play a key role in desensitizing PARs [26]. By activating caveolar PAR1 bound to β-arrestins, APC results in dissociation of receptor and adaptor protein [23]. β-Arrestin is required for activation of small GTPase Rac1, which is a key mechanism in the accomplishment of APC-related cytoprotective effect. The PAR1-dependent β-arrestin-2 signaling via Dvl-2 involves the activation of downstream signaling pathways such as PI3K/Akt and Rac1 and inhibits NF-kB, which promote cell survival and enhance barrier integrity [27]. Overall, the interaction between PAR1, EPCR, and S1P1 signaling pathways plays a critical role in mediating the cytoprotective effects of APC and thrombin in various physiological and pathological conditions [28]. Examining functions of proteases and PAR1 peptide agonists as well as analogs of tethered ligands liberated by thrombin or APC will increase understanding of the essence of biased agonism and outline ways for controlling PAR1-induced signaling mechanisms and cellular responses in inflammation, proliferation, tissue regeneration, and neurotoxicity.
We have supposed that new synthetic nanopeptide NPNDKYEPF-NH2 (AP9) analogues of the PAR1 tethered ligand liberated by APC may also have a neuroprotective effect similar to that of APC. We used the model of glutamate excitotoxicity to simulate ischemic brain damage. In case of brain ischemia, glutamate (Glu) is released massively into the intercellular space, which leads to the hyperactivation of pre- and postsynaptic glutamate receptors. The subsequent increase in intracellular free Ca2+ concentration ([Ca2+]i) may result in mitochondrial dysfunction, generation of reactive oxygen species, and activation of proteases, phosphatases and endonucleases [29]. Massive influx of Ca2+ into the nerve cells through the channels of ionotropic Glu receptors disturbs the intracellular Ca2+ homeostasis, triggers the cascade of intracellular reactions which end up with rapid or delayed cell death via the mechanisms of necrosis or apoptosis [30,31]. The prolonged exposure of primary neuronal cultures to excitotoxic Glu concentrations results eventually in a secondary rise of [Ca2+]i (delayed calcium dysregulation, DCD) followed by high [Ca2+]i plateau [32]. The DCD is accompanied by the synchronous profound mitochondrial depolarization (MD) and the secondary decrease in mitochondrial NADH and pH [33,34]. These injurious processes include Ca2+ overload of mitochondria, reactive oxygen and nitrogen species formation, activation of caspases and release of apoptosis-inducing factor [35,36]. Neurons having DCD die in a few hours by necrosis or apoptosis [37,38].
The search for agents that help the cells to resist excitotoxicity and studies of the mechanisms triggered by these agents are necessary for optimization of therapy for acute neurological diseases. Although protective effects of APC against Glu excitotoxicity are well documented [18,20,21,22] the data about function of hemostatic proteinases and their receptors in the central nervous system are still contradictory [19,39,40,41,42,43,44,45,46]. The goal of the present work was to identify the possible neuroprotective properties of a new synthetic peptide AP9 and to compare them with the effects of APC in primary neuronal cultures subjected to glutamate excitotoxicity.
2. Results
2.1. The toxic effects of glutamate and NMDA on the cultured cortical neurons
The exposure of cultured rat cortical neurons to glutamate (Glu, 100 µM, 40 min) led to the significant decrease in cell survival. With MTT assay a 40% decrease in cell survival in 24 h after treatment with Glu was shown (Figure 1). Glutamate excitotoxicity is mostly due to calcium overload of cytoplasm and mitochondrial dysfunction caused by calcium and sodium influx through the NMDA-subtype of glutamate receptors [47,48,49]. Blockage of NMDARs by MK-801 abolished glutamate-induced cell death (Figure 1). These results confirm that glutamate cell death in neurons is mediated by NMDARs [50].
2.2. The effects of APC and AP9 on survival of cultured neurons at glutamate excitotoxicity
In present work, preincubation of cultured cortical neurons in the presence of 10 nM APC resulted in a more than 1,4-fold increase in the number of living cells (Figure 2) at glutamate excitotoxicity. These data are in accordance with previous observations that APC has neuroprotective properties at staurosporine- or NMDA-induced toxicity [18,20].
In order to estimate the neuroprotective effects of a new nanopeptide (AP9) liberated by APC from the N-terminal sequences of PAR1 exodomain we examined the survival of neurons during glutamate-induced excitotoxicity in the presence of different concentrations of AP9. It was found that AP9 at concentration of 20 and 200 µM significantly reduces cell death at Glu-induced toxicity (Figure 2). Thus, the protective effects of AP9 were similar to APC impact on neurons. It has been shown that the number of living neurons in the presence of peptide does not significantly differ from their number in the presence of APC (Figure 2). These results confirm that AP9 nanopeptide at low concentrations (2, 200 μM) has similar neuroprotective effects as APC, consistent with previous reports [51].
2.3. PAR1 is required for protective effects of AP9 and Glu at glutamate induced excitotoxicity
Previously it was shown that PAR1 or the PAR/EPCR complex is required for the realization of the cytoprotective effects of APC [52,53,54]. Taking into consideration the similarities in the neuroprotective effects between APC and AP9, which is an analogue to tethered ligand liberated by APC, we suggested that the AP9 might also realize its protective effects through the same type of receptor as APC – PAR1. In the next series of experiments the estimation of cell survival was made using the MTT-test during Glu excitotoxicity in the presence of AP9 and / or in the presence of a PAR1-inhibitor - SCH79797 (Figure 3). Preincubation with APC (10 nM) or AP9 (20 μM) significantly increased the neuron survival at Glu excitotoxicity. The blockage of PAR1 by SCH79797 abolished the neuroprotective effect of both APC and AP9 demonstrating the necessity of PAR1 for the protective effects of these substances.
2.4. The effects of APC and AP9 on the intracellular free Ca2+ concentration ([Ca2+]i) dysregulated by Glu and NMDA
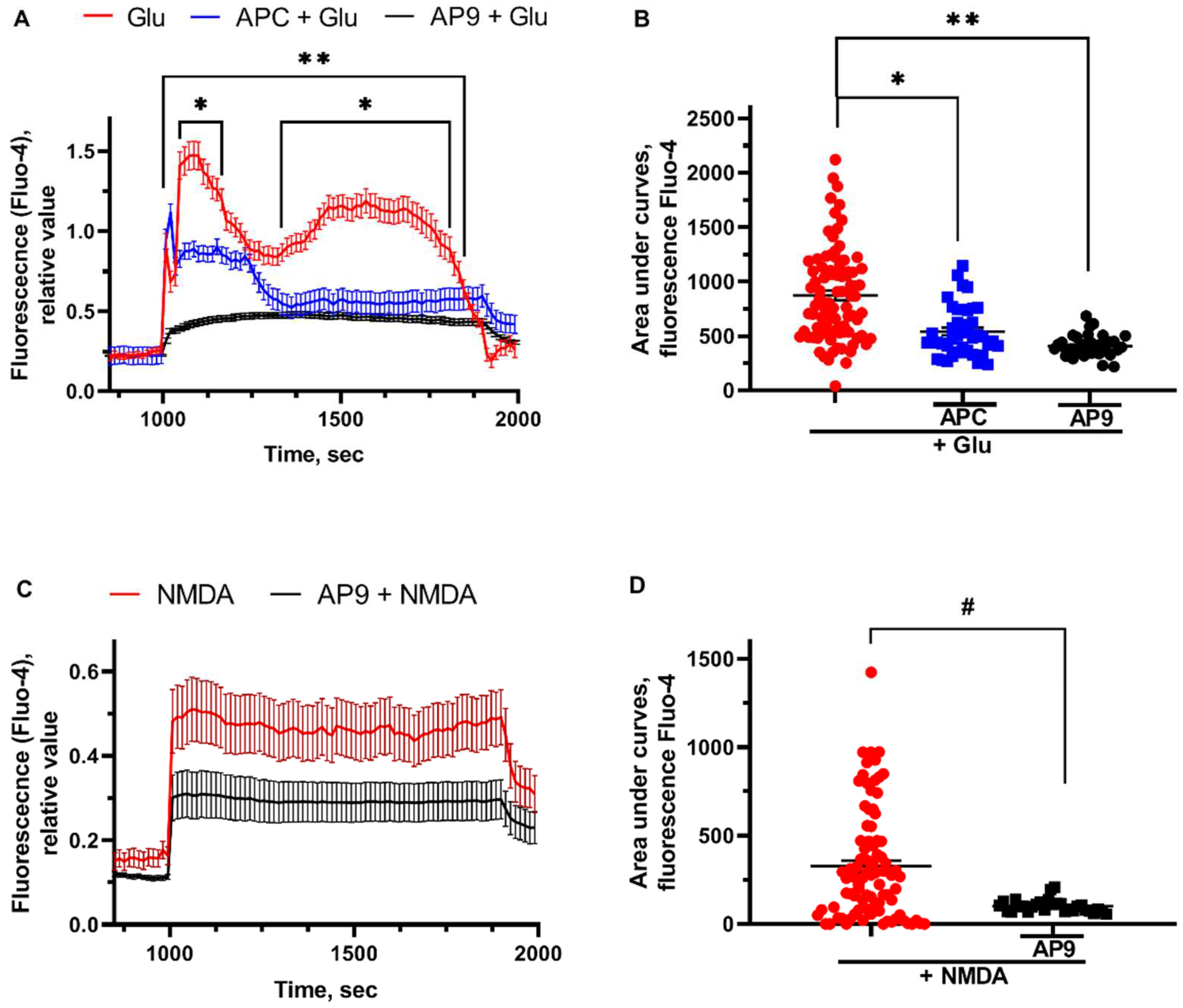
Glu (100 µM, 15 min) induced delayed calcium dysregulation (DCD) was followed by sustained 2 sec plateau. [Ca2+]i was altered in neurons by NMDA (300µM, 15 min) application similarly to the glutamate effects (Figure 4A). The pretreatment of the cells with PAR1-agonists - APC and AP9 - significantly decreased the maximum response of [Ca2+]i to toxic Glu concentration. Moreover, pretreatment of neurons with AP9 (20 μM) before the application of NMDA (300 μM) leads to the stabilization of calcium homeostasis that is expressed in a significant decrease of the area under the curve (Figure 4B). As the glutamate-induced calcium flow triggers calcium overload and neurotoxicity [55], the reduction of initial calcium influx may illustrate the protective effects of APC. AP9 was also shown to restore the Glu- and NMDA-induced [Ca2+]i dysregulation, however, the exact mechanisms of its regulatory effects are under debates.
3. Discussion
In the present work, we have studied the protective effects of PAR1 biased activation by APC and a new syntetic peptide AP9 at glutamate-induced neurotoxicity. Previously we have shown the protective effect of APC on the survival of hippocampal neurons exposed to toxic dose of glutamate [21]. Here, we demonstrated that APC prevents Glu-excitotoxicity via PAR1 in primary cortical neurons (Figure 3). Recently, it was shown that thrombin and APC have distinctly different properties because each is able to stabilize a different subsent of the dynamic conformational ensembles of PAR1 [12]. While trombin and thrombin receptor activating peptide (TRAP) promote PAR1 signaling via different G-proteins, APC-induced activation promote signaling via β-arrestin and dishevelled-2 [12,23]. Compared to thrombin, APC allosterically modulates PAR1 [14,53]. APC-cleaved PAR1 is localized in caveolae, plasma membrane microdomains, lipid rafts enriched in cholesterol and caveolin-1. Herein, APC-activated PAR1 is colocalized on the endothelial membrane with EPCR bound to caveolin-1 and necessary for cytoprotective functions accomplished by APC [56,57]. To demonstrate that PAR1-dependent signaling by APC involves a novel cleavage of the receptor’s N-terminal domain, differing from that of thrombin, we used a new synthetic peptide analogue of the tethered ligand liberated by APC - AP9 (NPNDKYEPF-amide).We studied the effect of the AP9 on the neuronal survival under the influence of glutamate in comparison with APC and we measured changes of [Ca2+]i under the influences of glutamate and NMDA as an indicator of high neuronal sensibility to used cytotoxic glutamate concentrations [47,58,59,60].
In our work, we proved that the presence of APC and AP9 protects neurons and restores the basal level of calcium, significantly increased during glutamate-induced excitotoxicity (Figure 4). PAR1 is required for neuroprotective action of APC and AP9 (Figure 3). Earlier we have shown that APC prevents neuronal death by decrease in translocation NF-kBp65 into the nucleous and abolishes increase in proapoptotic proteins at excitotoxicity [52]. Here, we show for the first time in hippocampal and cortical neurons that AP9 demonstrates a protective effect similar to APC. The novel peptide-agonist of PAR1 increases survival of neurons in culture after glutamate-induced toxicity (Figure 2, 3). Thus, our data support the hypothesis that APC’s cleavage of PAR1 of hippocampal and cortical neurons occurs at Arg46 and agrees with another recent report, which advances the paradigm that APC’s PAR1-dependent protective actions are based on Arg46 cleavage. These studies showed that the TR47 peptide representing the sequence of the novel N-terminus that is generated by cleavage at Arg46 exerts remarkable biological activities on endothelial cells and HEK [61]. Our present results corroborate well and extend another our report [62] that demonstrates the β-arrestin-2-dependent protective properties of a PAR1 agonist peptide, AP9, in vivo on a mouse model of photothrombosis-induced brain ischemia.
Excessive entry of Ca2+ through the NMDA receptor is thought to be the major cause of glutamate toxicity in brain neurons [63]. Here we have shown that NMDARs are involved in Glu-toxicity in cultural neurons, consistent with previous reports [48]. Moreover, we have demonstrated the significant impact of APC and AP9 on Glu- and NMDA-induced dysregulation of calcium hemostasis (Figure 4).
Our interest to NMDAR was induced by the data of previous studies that pointed to the possibility of PAR1-mediated NMDAR potentiation by thrombin [64]. Thrombin and other serine proteases can entry into brain parenchyma during intracerebral hemorrhage or extravasation of plasma proteins during blood–brain barrier breakdown may exacerbate glutamate-mediated cell death and possibly participate in post-traumatic ischemic injuries. For years, PAR1 has been regarded as positive modulator of NMDAR potentiation as an important mechanism for seizure initiation and subsequent neurodegeneration. We have suggested the possibility of similar PAR1-mediated effects of APC and AP9 on NMDAR that, in contrast to thrombin lead to the stabilization of intracellular calcium homeostasis and neuroprotection. The present data revealed that biased agonists of PAR1 may be new candidates for neuroprotective drugs, and PAR1-dependent signaling has a wider variety of pathways than we have known. Thus, understanding the molecular mechanisms of the protective action and the peptide structure of non-canonical PAR1 agonists may facilitate the development of new therapeutic pathways for neurodegenerative brain damage.
4. Materials and Methods
4.1. Reagents
Human APC, NaCl, KCl, CaCl2, MgCl2, KH2PO4, Hepes, glucose, glutamate, NMDA, Ara C, PAR1 inhibitor SCH79797 were from Sigma-Aldrich (Taufkirchen, Germany). Neurobasal medium A (NBM), Supplement B27 and L-GlutaMax were from Gibco (Darmstadt, Germany). AP9 (NPNDKYEPF-amide) was synthesized at the Laboratory of peptide synthesis of the Russian Cardiology Research and Production Complex by the standard technology of solid phase peptide synthesis using Fmoc (9-fluorenemethoxycarbonyl) strategy. The structure and homogeneity of the peptide were confirmed by H-NMR spectroscopy and analytical HPLC.
4.2. Preparation of cell cultures
Experiments with animals were performed in accordance with the ethical principles and regulatory documents recommended by the European Convention on the Protection of Vertebrate Animals used for experiments [65], as well as in accordance with the “Good Laboratory Rules practice”, approved by order of the Ministry of Health of the Russian Federation No. 199n of 04/01/2016.
Primary cultures of rat brain hippocampal and cortical neurons were prepared from 1-2-day old Wistar rats as described earlier [34,66]. Briefly, the rats were anesthetized, decapitated, and the hippocampus or cortex was removed and separated from the meninges. The extracted tissues were washed in a Ca2+- and Mg2+-free Hanks solution, crushed, and placed in a papain solution for 15 min at 36oC, washed with standard Hanks solution and Minimal Essential Medium (MEM), and dispersed in fresh MEM. A homogeneous suspension was precipitated in a centrifuge at 200 g for 2x5 min. The precipitated cells were resuspended to concentration of 106 cells/ml in neurobasal medium (NBM), supplemented with 2% B-27, 1% GlutaMAX, and 1% penicillin/streptomycin (NBM+). The suspension (200 µl) was transferred onto coverslips attached to the wells of 35 mm plastic glass-bottom Petri dishes (MatTek, Ashland, MA) or into wells (400 µl/well) of 24-well plastic plates (Corning costar 3338, USA). In 1 hour, 1.5 ml of NBM+ was added in each dish. The dishes and plates were pre-coated with 10 mg/ml of polyethyleneimine. The cells were kept in an incubator at 37oC, 95% air C 5% CO2, and a relative humidity of 100% until use at 9-10 day in vitro (DIV). Cytosine arabinoside (AraC, 5 µM) was added to the medium for two or three days to prevent the proliferation of glial cells. Every three days, the cells were fed by replacing 1/3 of the old medium with a fresh NBM+.
4.3. Cytotoxicity assays
A biochemical MTT assay and morphological method employing fluorescence vital dyes were used to estimate cell viability. A colorimetric MTT assay is based on the reduction of the yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) by mostly mitochondria of living cells to dark-blue formazan [67,68].
Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) or N-methyl-D-aspartate (NMDA, 200 µM) for 40 min at 37oC. Control HBSS included (mM): NaCl, 145; KCl, 5; CaCl2, 1.8; MgCl2, 1.0 HEPES, 20; glucose, 5 (pH 7.4). Glycine (10 µM) was added and MgCl2 omitted in HBSS containing Glu or NMDA. APC (10 nM) or AP9 (2, 20, 200 µM) were added alone to HBSS or 15 min prior to Glu and NMDA. NBM+ was aspirated from the cells seeded in a 24-well plates, then HBSS buffers containing Glu, NMDA, APC and AP9 in appropriate combinations were added to the corresponding wells. Next, cells were washed with saline, NBM+ was returned to the wells, and the cells were put for 24 h back to the CO2 incubator.
After 24 h, NBM+ was removed from the all 24 wells of the plate and 0.5 ml of MTT in HBSS (4 mg/ml) was added to each well. In 30 min, MTT-containing buffer was aspirated, and formazan wwas dissolved in 300 ml of DMSO. The light absorbance of formazan solution was measured at 550 nm (A550) using a plate reader (ClarioStar BMG LABTECH, Germany). Absorbance at 650 nm was subtracted from A550 to compensate light absorbance and scattering by the bottom of the plastic plate. The optical density of the control group and cell-free wells were considered as 100 and 0% survival, respectively.
4.4. Intracellular free Ca2+ ([Ca2+]i ) measurements
The experimental setup included an ZEISS LSM 700 confocal microscopy. To measure [Ca2+]i the cells were loaded with high-affinity Ca2+ indicator Fluo-4 in the form of the acetoxymethyl (AM) ester (1-2 μM Fluo-4, 40 min, 37°C). Fluo-4 fluorescence was excited at 488 nm and monitored at 505-535 nm. All measurements were carried out at 27-29°C in HBSS. Changes of [Ca2+]i induced by Glu or NMDA were measured in Mg2+-free glycine containing buffers as mentioned above. Glu or NMDA were wash out by a nominally calcium-free solutions containing 0.1 mM EGTA instead of CaCl2 and 2 mM MgCl2. Replacement of solutions was performed by quick (<13 s, 2 × 200 mkl) suck of the previous buffer out and addition of a new one into the dish with cells. To compare relative changes in [Ca2+]i induced by Glu or NMDA in experiments performed at different days we calibrated fluorescence signals of Fluo-4. To this end at the final part of experiments a Ca2+ ionophore ionomycin (2 μM) was applied in the presence of 5 mM Ca2+ to saturate indicator with Ca2+ and measure its maximum signals.
4.5. Data processing
The data of 4-6 independent experiments were analyzed using the GraphPad Prism 8. The data were processed in paired samples using Student’s t-test. Cytosolic calcium levels were compared using Two-way ANOVA (Dunnett’s test). The differences were considered significant at p < 0.05; n was the number of independent experiments. The results are presented as the mean with the standard error of the mean.
5. Conclusions
For the first time, the neuroprotective effect of the new nanopeptide analogue to the "tethered ligand" liberated by APC from PAR1 was demonstrated in this research under excitotoxicity. These findings lead to the conclusion that the protective effects of this peptide are comparable with the protective effect of APC under glutamate-induced excitotoxicity. The application of low-molecular-mass compounds in contrast to large proteins decreases the possibility of immunological reactions and increases the success of the solution to the problem of "addressed" drug delivery. The study of the mechanisms of PAR1 agonist-peptide action, as well as the development of new modifications of noncanonical PAR1-agonists with high neuroprotective activity, may be an important and relevant trend in the search for novel neuroprotective agents for treating neurodegenerative diseases and stroke.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure 1: Activation of NMDA glutamate receptors decreases cultured cortical neurons survival. Cells were cultured in NBM+ during 9-10 day, Figure 2: AP9 increases survival of cultured cortical neurons under excitotoxicity more efficiently than APC, Figure 3: Effect of PAR1 blockage on the neuronal survival at glutamate toxicity and APC/AP9 treatment, Figure 4: APC and AP9 restore the Glu- and NMDA-induced [Ca2+]i dysregulation.
Author Contributions
Conceptualization, L.G.; experimental procedures, I.B., I.S., T.M., A.S. and L.G.; AP9 (NPNDKYEPF-amide) synthesis, M.S., data analysis, I.B., T.M. and L.G.; writing—original draft preparation, review and editing, I.B., I.S. and L.G. . All authors have read and agreed to the published version of the manuscript.
Funding
The reported study was funded by Russian Science Foundation, grant No. 23-74-01144.
Institutional Review Board Statement
The study was approved by the animal ethics committee of Federal State Autonomous Educational Institution of Higher Education «N.I. Pirogov Russian National Research Medical University» of the Ministry of Health of the Russian Federation: protocol code 23/2021 from 13 December 2021.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
References
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Kaplan-Arabaci, O.; Acari, A.; Ciftci, P.; Gozuacik, D. Glutamate Scavenging as a Neuroreparative Strategy in Ischemic Stroke. Front. Pharmacol. 2022, 13, 866738. [Google Scholar] [CrossRef]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Nian, K.; Harding, I.C.; Herman, I.M.; Ebong, E.E. Blood-Brain Barrier Damage in Ischemic Stroke and Its Regulation by Endothelial Mechanotransduction. Front. Physiol. 2020, 11, 605398. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends in Pharmacological Sciences 2018, 39, 367–386. [Google Scholar] [CrossRef]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity Determinants of GPCR–G-Protein Binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G Protein–Coupled Receptor (GPCR) Assays From Traditional to a State-of-the-Art Biosensor Assay. J Pharmacol Sci 2014, 126, 302–309. [Google Scholar] [CrossRef]
- Bushi, D.; Ben Shimon, M.; Shavit Stein, E.; Chapman, J.; Maggio, N.; Tanne, D. Increased Thrombin Activity Following Reperfusion after Ischemic Stroke Alters Synaptic Transmission in the Hippocampus. Journal of Neurochemistry 2015, 135, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.H.; Fernández, J.A.; Lyden, P.D.; Zlokovic, B.V. Activated Protein C Promotes Neuroprotection: Mechanisms and Translation to the Clinic. Thrombosis Research 2016, 141, S62–S64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jin, H.; Hua, Y.; Keep, R.F.; Xi, G. Role of Protease-Activated Receptor-1 in Brain Injury After Experimental Global Cerebral Ischemia. Stroke 2012, 43, 2476–2482. [Google Scholar] [CrossRef]
- Shavit-Stein, E.; Mindel, E.; Gofrit, S.G.; Chapman, J.; Maggio, N. Ischemic Stroke in PAR1 KO Mice: Decreased Brain Plasmin and Thrombin Activity along with Decreased Infarct Volume. PLoS ONE 2021, 16, e0248431. [Google Scholar] [CrossRef]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased Agonism of Protease-Activated Receptor 1 by Activated Protein C Caused by Noncanonical Cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar] [CrossRef]
- Sinha, R.K.; Wang, Y.; Zhao, Z.; Xu, X.; Burnier, L.; Gupta, N.; Fernández, J.A.; Martin, G.; Kupriyanov, S.; Mosnier, L.O.; et al. PAR1 Biased Signaling Is Required for Activated Protein C in Vivo Benefits in Sepsis and Stroke. Blood 2018, 131, 1163–1171. [Google Scholar] [CrossRef]
- Künze, G.; Isermann, B. Targeting Biased Signaling by PAR1: Function and Molecular Mechanism of Parmodulins. Blood 2023, blood.2023019775. [CrossRef]
- Coughlin, S.R. Protease-activated Receptors in Hemostasis, Thrombosis and Vascular Biology. Journal of Thrombosis and Haemostasis 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin Signalling and Protease-Activated Receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Schuepbach, R.A.; Madon, J.; Ender, M.; Galli, P.; Riewald, M. Protease-activated Receptor-1 Cleaved at R46 Mediates Cytoprotective Effects. Journal of Thrombosis and Haemostasis 2012, 10, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The Cytoprotective Protein C Pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Riewald, M.; Ruf, W. Protease-Activated Receptor-1 Signaling by Activated Protein C in Cytokine-Perturbed Endothelial Cells Is Distinct from Thrombin Signaling. Journal of Biological Chemistry 2005, 280, 19808–19814. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Griffin, J.H. Cytoprotective Protein C Pathways and Implications for Stroke and Neurological Disorders. Trends in Neurosciences 2011, 34, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Gorbacheva, L.R.; Storozhevykh, T.P.; Pinelis, V.G.; Davydova, O.N.; Ishiwata, S.; Strukova, S.M. Activated Protein C via PAR1 Receptor Regulates Survival of Neurons under Conditions of Glutamate Excitotoxicity. Biochemistry Moscow 2008, 73, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, D.; Gelbard, H.; Cheng, T.; Insalaco, R.; Griffin, J.H.; Zlokovic, B.V. Activated Protein C Prevents Neuronal Apoptosis via Protease Activated Receptors 1 and 3. Neuron 2004, 41, 563–572. [Google Scholar] [CrossRef]
- Soh, U.J.K.; Trejo, J. Activated Protein C Promotes Protease-Activated Receptor-1 Cytoprotective Signaling through β-Arrestin and Dishevelled-2 Scaffolds. Proc. Natl. Acad. Sci. U.S.A. 2011, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ubl, J.J.; Stricker, R.; Reiser, G. Thrombin (PAR-1)-Induced Proliferation in Astrocytes via MAPK Involves Multiple Signaling Pathways. American Journal of Physiology-Cell Physiology 2002, 283, C1351–C1364. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated Protein C, Protease Activated Receptor 1, and Neuroprotection. Blood 2018, 132, 159–169. [Google Scholar] [CrossRef]
- Barsi-Rhyne, B.; Manglik, A.; Von Zastrow, M. Discrete GPCR-Triggered Endocytic Modes Enable β-Arrestins to Flexibly Regulate Cell Signaling. eLife 2022, 11, e81563. [Google Scholar] [CrossRef]
- Willis Fox, O.; Preston, R.J.S. Molecular Basis of Protease-activated Receptor 1 Signaling Diversity. Journal of Thrombosis and Haemostasis 2020, 18, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.V.; Ardeshirylajimi, A.; Dinarvand, P.; Yang, L.; Rezaie, A.R. Occupancy of Human EPCR by Protein C Induces β-Arrestin-2 Biased PAR1 Signaling by Both APC and Thrombin. Blood 2016, 128, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A. Excitotoxic Mechanisms in Stroke: An Update of Concepts and Treatment Strategies. Neurochemistry International 2007, 50, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Nicotera, P. Ca2+ Signals and Neuronal Death in Brain Ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef]
- Berliocchi, L.; Bano, D.; Nicotera, P. Ca2+ Signals and Death Programmes in Neurons. Phil. Trans. R. Soc. B 2005, 360, 2255–2258. [Google Scholar] [CrossRef]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed Calcium Dysregulation in Neurons Requires Both the NMDA Receptor and the Reverse Na+/Ca2+ Exchanger. Neurobiology of Disease 2012, 46, 109–117. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. Mechanisms Underlying the Loss of Mitochondrial Membrane Potential in Glutamate Excitotoxicity. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2008, 1777, 953–964. [Google Scholar] [CrossRef]
- Surin, A.M.; Gorbacheva, L.R.; Savinkova, I.G.; Sharipov, R.R.; Khodorov, B.I.; Pinelis, V.G. Study on ATP Concentration Changes in Cytosol of Individual Cultured Neurons during Glutamate-Induced Deregulation of Calcium Homeostasis. Biochemistry Moscow 2014, 79, 146–157. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm Shift in Neuroprotection by NMDA Receptor Blockade: Memantine and Beyond. Nat Rev Drug Discov 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Adam-Vizi, V. Calcium, Mitochondria and Oxidative Stress in Neuronal Pathology: Novel Aspects of an Enduring Theme. The FEBS Journal 2006, 273, 433–450. [Google Scholar] [CrossRef]
- Li, V.; Wang, Y. Molecular Mechanisms of NMDA Receptor-Mediated Excitotoxicity: Implications for Neuroprotective Therapeutics for Stroke. Neural Regen Res 2016, 11, 1752. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Fernandes, J.; Burgeiro, A.; Thomas, G.M.; Huganir, R.L.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. Excitotoxicity through Ca2+-Permeable AMPA Receptors Requires Ca2+-Dependent JNK Activation. Neurobiology of Disease 2010, 40, 645–655. [Google Scholar] [CrossRef]
- Hollenberg, M.D. Physiology and Pathophysiology of Proteinase-Activated Receptors (PARs): Proteinases as Hormone-Like Signal Messengers: PARs and More. J Pharmacol Sci 2005, 97, 8–13. [Google Scholar] [CrossRef]
- Suo, Z.; Citron, B.A.; Festoff, B.W. Thrombin: A Potential Proinflammatory Mediator in Neurotrauma and Neurodegenerative Disorders. Current Drug Targets - Inflammation & Allergy 2004, 3, 105–114. [Google Scholar] [CrossRef]
- Ruf, W. Is APC Activation of Endothelial Cell PAR1 Important in Severe Sepsis? : Yes. Journal of Thrombosis and Haemostasis 2005, 3, 1912–1914. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Reiser, G.; Keep, R.F. The Role of Thrombin and Thrombin Receptors in Ischemic, Hemorrhagic and Traumatic Brain Injury: Deleterious or Protective? Journal of Neurochemistry 2003, 84, 3–9. [Google Scholar] [CrossRef]
- Rohatgi, T.; Sedehizade, F.; Reymann, K.G.; Reiser, G. Protease-Activated Receptors in Neuronal Development, Neurodegeneration, and Neuroprotection: Thrombin as Signaling Molecule in the Brain. Neuroscientist 2004, 10, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Vergnolle, N.; McArthur, J.C.; Silva, C.; Vodjgani, M.; Andrade-Gordon, P.; Hollenberg, M.D.; Power, C. Proteinase-Activated Receptor-2 Induction by Neuroinflammation Prevents Neuronal Death during HIV Infection1. The Journal of Immunology 2005, 174, 7320–7329. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.E.; Lyuboslavsky, P.; Traynelis, S.F.; McKeon, R.J. PAR-1 Deficiency Protects against Neuronal Damage and Neurologic Deficits after Unilateral Cerebral Hypoxia/Ischemia. J Cereb Blood Flow Metab 2004, 24, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.-Q.; Lim, C.S.; Hwang, J.K.; Ha, I.; Han, J.-S. Anti-Oxidant and Anti-Inflammatory Activities of Macelignan in Murine Hippocampal Cell Line and Primary Culture of Rat Microglial Cells. Biochemical and Biophysical Research Communications 2005, 331, 1264–1269. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and Neuronal Survival. Physiological Reviews 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Khodorov, B. Glutamate-Induced Deregulation of Calcium Homeostasis and Mitochondrial Dysfunction in Mammalian Central Neurones. Progress in Biophysics and Molecular Biology 2004, 86, 279–351. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA Receptor-Dependent Glutamate Excitotoxicity in Human Embryonic Stem Cell-Derived Neurons. Neuroscience Letters 2013, 543, 95–100. [Google Scholar] [CrossRef]
- Simões, A.P.; Silva, C.G.; Marques, J.M.; Pochmann, D.; Porciúncula, L.O.; Ferreira, S.; Oses, J.P.; Beleza, R.O.; Real, J.I.; Köfalvi, A.; et al. Glutamate-Induced and NMDA Receptor-Mediated Neurodegeneration Entails P2Y1 Receptor Activation. Cell Death Dis 2018, 9, 297. [Google Scholar] [CrossRef]
- Savinkova, I.G.; Gorbacheva, L.R.; Bespalova, Z.D.; Pinelis, V.G.; Strukova, S.M. Peptides Analogous to Tethered Ligands Liberated by Activated Protein C Exert Neuroprotective Effects in Glutamate-Induced Excitotoxicity. Biochem. Moscow Suppl. Ser. A 2014, 8, 116–120. [Google Scholar] [CrossRef]
- Gorbacheva, L.; Pinelis, V.; Ishiwata, S.; Strukova, S.; Reiser, G. Activated Protein C Prevents Glutamate- and Thrombin-Induced Activation of Nuclear Factor-κB in Cultured Hippocampal Neurons. Neuroscience 2010, 165, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated Protein C: Biased for Translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Pendurthi, U.R.; Rao, L.V.M. Endothelial Cell Protein C Receptor-Dependent Signaling. Current Opinion in Hematology 2018, 25, 219–226. [Google Scholar] [CrossRef]
- Randall, R.; Thayer, S. Glutamate-Induced Calcium Transient Triggers Delayed Calcium Overload and Neurotoxicity in Rat Hippocampal Neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Canto, I. ; J. K. Soh, U.; Trejo, J. Allosteric Modulation of Protease-Activated Receptor Signaling. MRMC 2012, 12, 804–811. [Google Scholar] [CrossRef]
- Bae, J.-S.; Yang, L.; Rezaie, A.R. Lipid Raft Localization Regulates the Cleavage Specificity of Protease Activated Receptor 1 in Endothelial Cells. Journal of Thrombosis and Haemostasis 2008, 6, 954–961. [Google Scholar] [CrossRef]
- Blandini, F.; Porter, R.H.P.; Greenamyre, J.T. Glutamate and Parkinson’s Disease. Molecular Neurobiology 1996, 12, 73–94. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidative Damage in Neurodegenerative Diseases. Neuroscientist 1997, 3, 21–27. [Google Scholar] [CrossRef]
- Krieger, C.; Duchen, M.R. Mitochondria, Ca2+ and Neurodegenerative Disease. European Journal of Pharmacology 2002, 447, 177–188. [Google Scholar] [CrossRef]
- Burnier, L.; Mosnier, L.O. Novel Mechanisms for Activated Protein C Cytoprotective Activities Involving Noncanonical Activation of Protease-Activated Receptor 3. Blood 2013, 122, 807–816. [Google Scholar] [CrossRef]
- Galkov, M.; Kiseleva, E.; Gulyaev, M.; Sidorova, M.; Gorbacheva, L. New PAR1 Agonist Peptide Demonstrates Protective Action in a Mouse Model of Photothrombosis-Induced Brain Ischemia. Front. Neurosci. 2020, 14, 335. [Google Scholar] [CrossRef]
- Konradi, C.; Heckers, S. Molecular Aspects of Glutamate Dysregulation: Implications for Schizophrenia and Its Treatment. Pharmacology & Therapeutics 2003, 97, 153–179. [Google Scholar] [CrossRef]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA Receptor Function by the Serine Protease Thrombin. J. Neurosci. 2000, 20, 4582–4595. [Google Scholar] [CrossRef] [PubMed]
- National Research Council; Division on Earth and Life Studies; Institute for Laboratory Animal Research; Committee for the Update of the Guide for the Care and Use of Laboratory Animals Guide for the Care and Use of Laboratory Animals: Eighth Edition; National Academies Press, 2010; ISBN 0-309-18663-3.
- Krasil’nikova, I.; Surin, A.; Sorokina, E.; Fisenko, A.; Boyarkin, D.; Balyasin, M.; Demchenko, A.; Pomytkin, I.; Pinelis, V. Insulin Protects Cortical Neurons Against Glutamate Excitotoxicity. Front. Neurosci. 2019, 13, 1027. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. Journal of Immunological Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid Colorimetric Assay for Cell Growth and Survival. Journal of Immunological Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
Figure 1.
Activation of NMDA glutamate receptors decreases cultured cortical neurons survival. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. NMDAR blocker MK-801 (10 µM) was added alone to HBSS or 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4), МК – with МК-801).
Figure 1.
Activation of NMDA glutamate receptors decreases cultured cortical neurons survival. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. NMDAR blocker MK-801 (10 µM) was added alone to HBSS or 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4), МК – with МК-801).
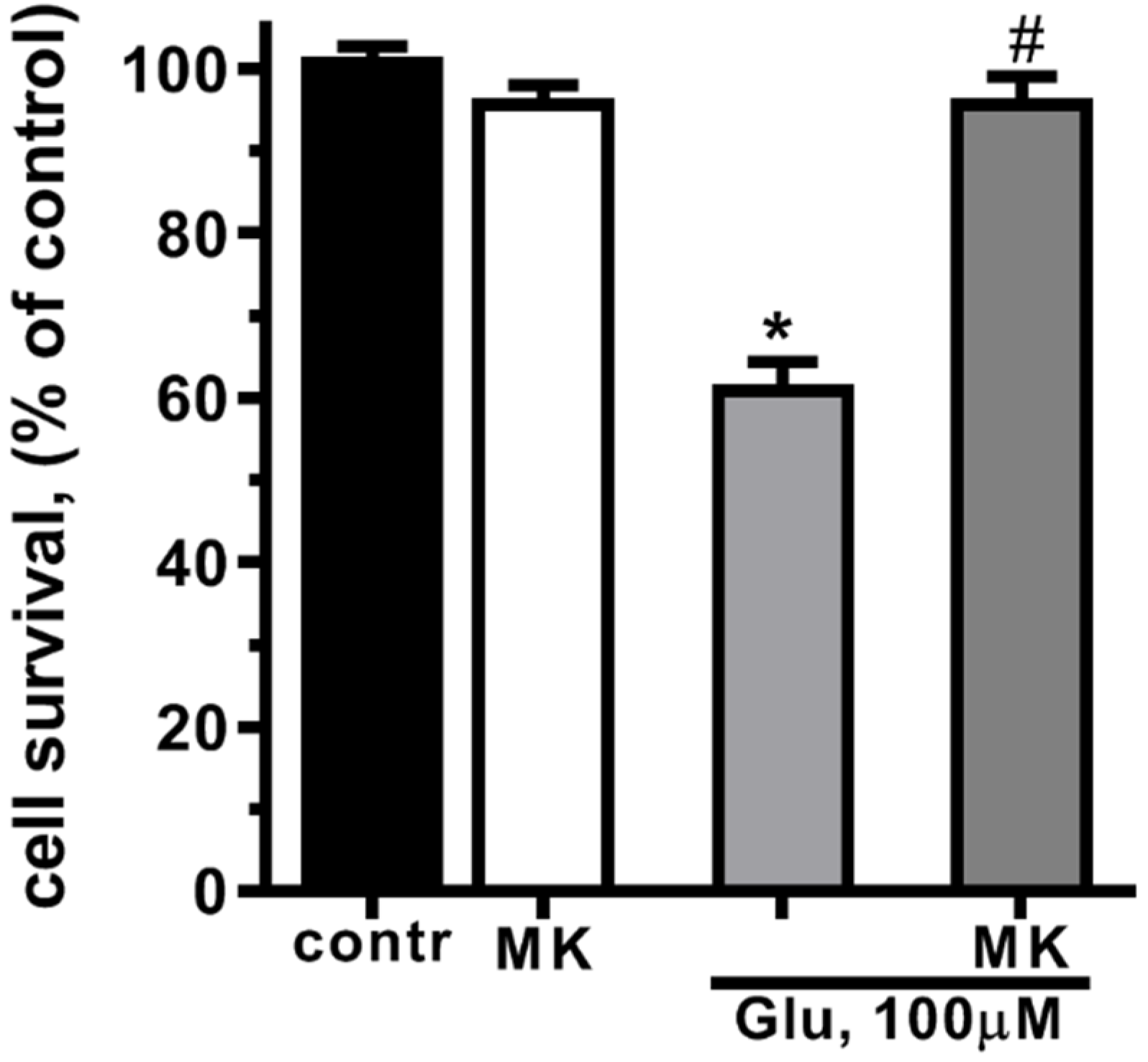
Figure 2.
AP9 increases survival of cultured cortical neurons under excitotoxicity more efficiently than APC. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. APC (10 nM) or AP9 (2 or 200 µM) were added alone to HBSS or 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4)).
Figure 2.
AP9 increases survival of cultured cortical neurons under excitotoxicity more efficiently than APC. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. APC (10 nM) or AP9 (2 or 200 µM) were added alone to HBSS or 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4)).
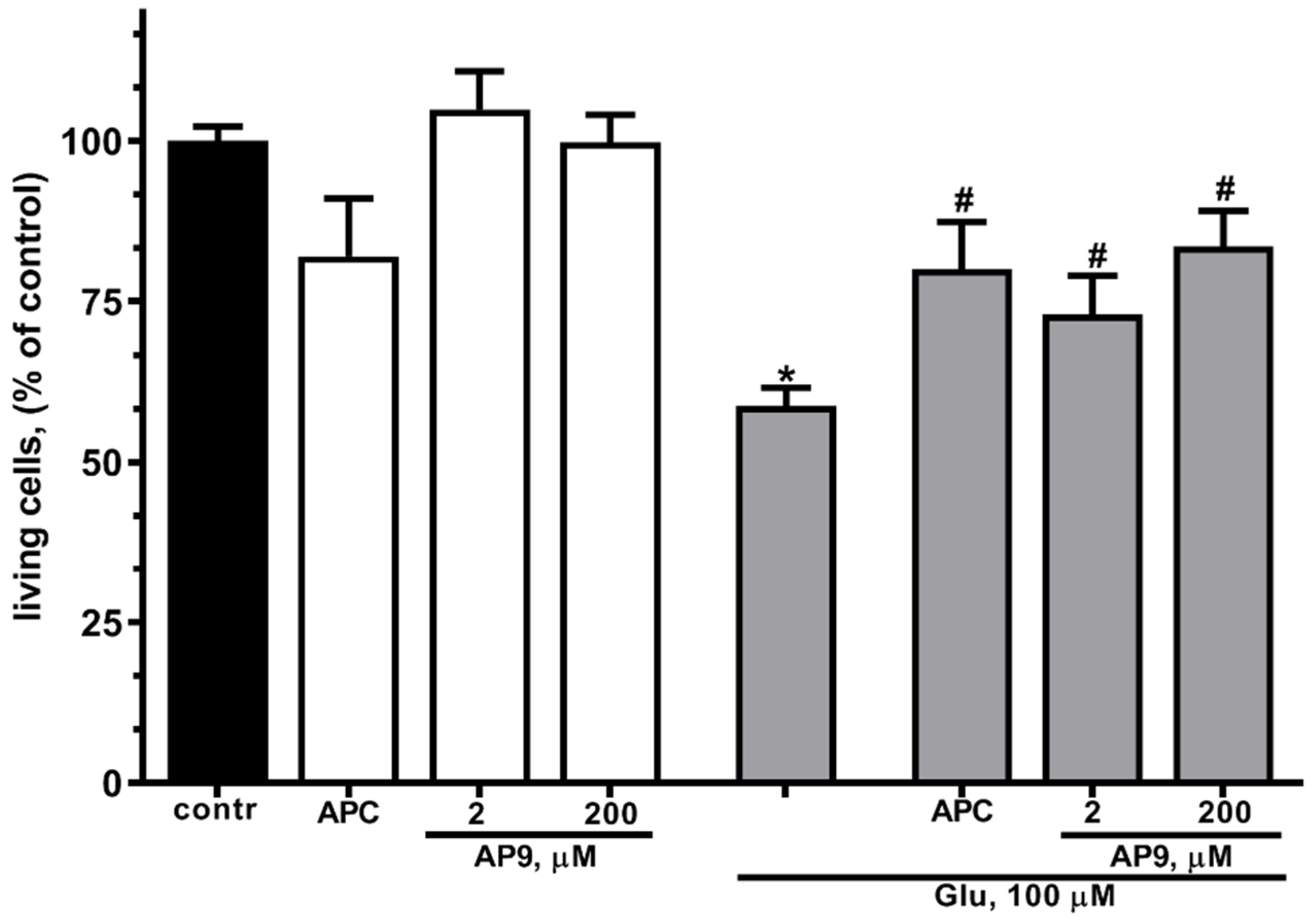
Figure 3.
Effect of PAR1 blockage on the neuronal survival at glutamate toxicity and APC/AP9 treatment. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. PAR1 blocker SCH79797 (50 nM) was added to HBSS 30 min prior to Glu, APC (10 nM) or AP9 (20 µM) were added to HBSS 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4), SCH – SCH79797).
Figure 3.
Effect of PAR1 blockage on the neuronal survival at glutamate toxicity and APC/AP9 treatment. Cells were cultured in NBM+ during 9-10 day. Excitotoxicity was reached by substitution of NBM+ for buffered saline solutions (HBSS) containing glutamate (100 µM) for 40 min at 37oC. PAR1 blocker SCH79797 (50 nM) was added to HBSS 30 min prior to Glu, APC (10 nM) or AP9 (20 µM) were added to HBSS 15 min prior to Glu. After 24 h, NBM+ was removed and cell viability was detected with the MTT assay kit as describes in chapter 4.3. Values are expressed as the mean mean±S.D. of triplicate cultures.* - p<0,05 compared to control group, # - p<0,05 compared to group with glutamate (contr – with control HBSS containing 145 mM NaCl; 5 mM KCl; 1,8 mM CaCl2; 1,0 mM MgCl2; 20 mM HEPES; 5 mM glucose (pH 7.4), SCH – SCH79797).
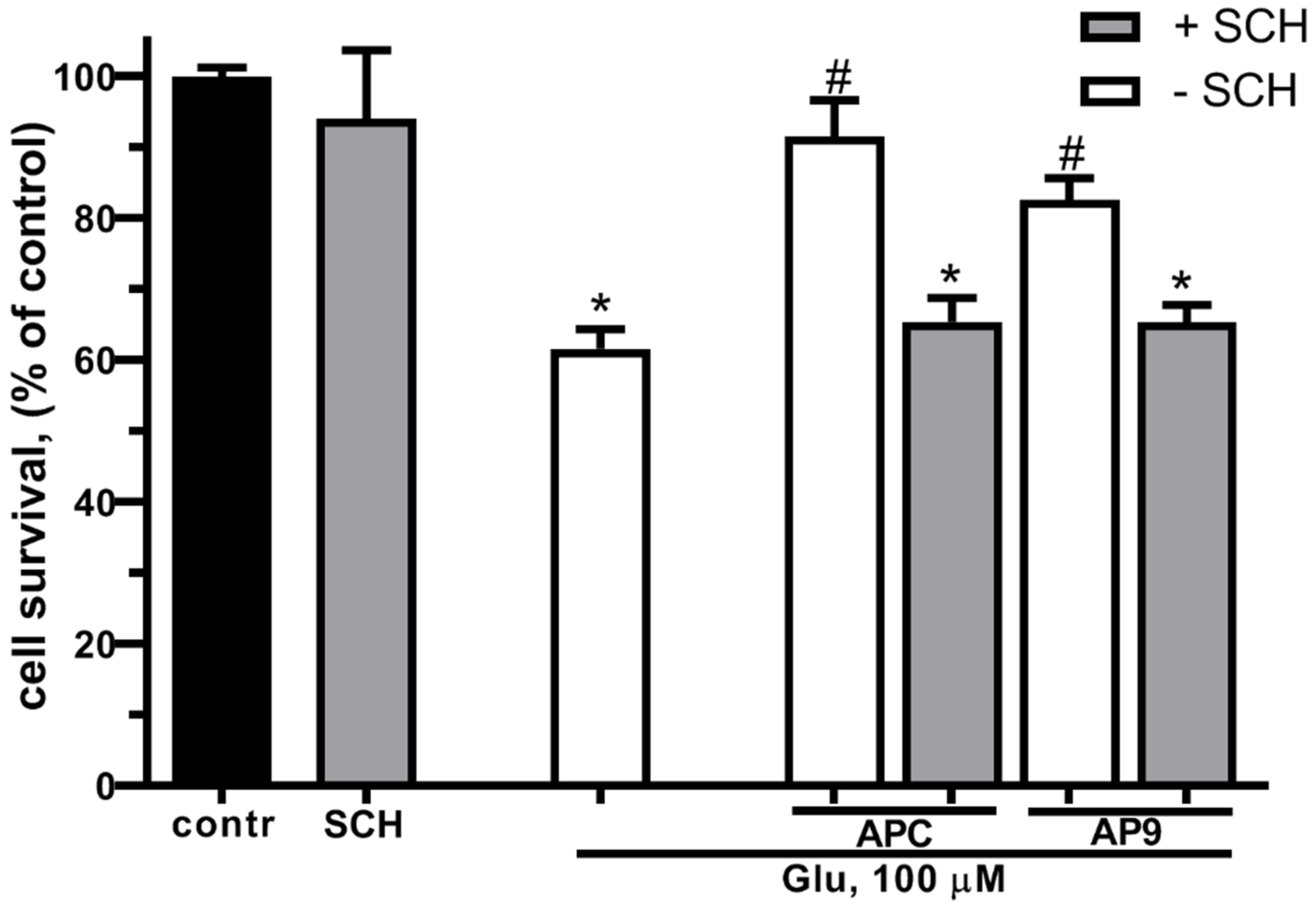
Figure 4.
APC and AP9 restore the Glu- and NMDA-induced [Ca2+]i dysregulation. (A) Glutamate-induced [Ca2+]i alterations in cultured neurons and (B) changes of the areas under curves measured with Ca-depended fluorescence Fluo-4 (effects of glutamate and PAR1-agonists), (C) NMDA-induced [Ca2+]i alterations in cultured neurons and (D) changes of the areas under curves measured with Ca-depended fluorescence Fluo-4 (effects of NMDA and PAR1-agonists). For [Ca2+]i measurement the cells were loaded with high-affinity Ca2+ indicator Fluo-4 in the form of the acetoxymethyl (AM) ester (1-2 μM Fluo-4, 40 min, 37°C). Fluo-4 fluorescence was excited at 488 nm and monitored at 505-535 nm. All measurements were carried out at 27-29°C in HBSS. Glu or NMDA were wash out by a nominally calcium-free solutions containing 0.1 mM EGTA instead of CaCl2 and 2 mM MgCl2. For the measurement of the maximal signal at the final part of experiments a Ca2+ ionophore ionomycin (2 μM) was applied in the presence of 5 mM Ca2+ to saturate indicator with Ca2+. * - p<0,05 APC pretreatment compared to glutamate, **- p<0,05 AP9 pretreatment compared to glutamate, # — p<0,05 AP9 pretreatment compared to NMDA.
Figure 4.
APC and AP9 restore the Glu- and NMDA-induced [Ca2+]i dysregulation. (A) Glutamate-induced [Ca2+]i alterations in cultured neurons and (B) changes of the areas under curves measured with Ca-depended fluorescence Fluo-4 (effects of glutamate and PAR1-agonists), (C) NMDA-induced [Ca2+]i alterations in cultured neurons and (D) changes of the areas under curves measured with Ca-depended fluorescence Fluo-4 (effects of NMDA and PAR1-agonists). For [Ca2+]i measurement the cells were loaded with high-affinity Ca2+ indicator Fluo-4 in the form of the acetoxymethyl (AM) ester (1-2 μM Fluo-4, 40 min, 37°C). Fluo-4 fluorescence was excited at 488 nm and monitored at 505-535 nm. All measurements were carried out at 27-29°C in HBSS. Glu or NMDA were wash out by a nominally calcium-free solutions containing 0.1 mM EGTA instead of CaCl2 and 2 mM MgCl2. For the measurement of the maximal signal at the final part of experiments a Ca2+ ionophore ionomycin (2 μM) was applied in the presence of 5 mM Ca2+ to saturate indicator with Ca2+. * - p<0,05 APC pretreatment compared to glutamate, **- p<0,05 AP9 pretreatment compared to glutamate, # — p<0,05 AP9 pretreatment compared to NMDA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



