
Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
There is a continuous interest in cannabinoids like Δ9-tetrahydro cannabinol (Δ9-THC) and cannabidiol (CBD). A patent of Webster et al., described the conversion of CBD to either D8-THC or D9-THC, depending on the acid catalyst applied. The use of para-toluene sulfonic acid (pTSA) led to the formation of D8-THC, while boron trifluoride etherate (BF3·Et2O) yielded mainly D9-THC. The enormous difference in product selectivity between these two catalysts was investigated with Molecular Modeling. It was found that pTSA leads to fast isomerization of Δ9-CBD to Δ8-CBD and subsequent ring closure to Δ8-THC. BF3·Et2O catalysis leads to the formation of tertiary carbenium ions in the Transition States which yield D9-THC and some iso D8-THC. Under dry conditions in refluxing toluene, it was found that pTSA is predominantly present as a dimer, and only a small fraction is available as monomeric catalyst. Applying the computationally derived activation barriers in Transition State theory yielded reaction rates predicting amounts of cannabinoids that are in close agreement with the experimental findings.
Keywords:
cannabinoids
; density functional theory
; transition state theory
1. Introduction
There is a continuous interest in cannabinoids like
Δ9-tetrahydro cannabinol (Δ9-THC) and cannabidiol (CBD)
to mention the two most important ones [1,2].
CBD shows a different medical and pharmacological profile than Δ9-THC.
CBD is a promising candidate drug which shows analgesic, anticonvulsant, muscle
relaxant, anxiolytic, antipsychotic, neuroprotective, anti-inflammatory, and
antioxidant activity [3,4,5]. At the same time,
it does not show drug induced intoxication like Δ9-THC.
However, the interest in CBD is not only explained
by its own favorable medical and pharmacological profile but also by its
potential to be converted to Δ9-THC [2,6]
which has caused a hot controversy in the scientific literature. The latter can
be easily understood as CBD can account for 40% of the cannabis plant extract [3].
Still, it is strongly advised to use any
cannabinoid on explicit medical prescription only. In addition, most products
formed by the conversion of CBD are THC variants which fall under the statutory
definition of a Schedule I controlled substance pursuant to Ind. Code §
35-48-2-4(d)(31) [2,7].
In 2004 a patent application by Webster et al.,
was published, in 2008 followed by the granted one, describing the catalytic
conversion of CBD to either Δ8-THC
or Δ9-THC, depending on the
acid catalyst applied [8,9]. The use of the Brønsted
acid para-toluene sulfonic acid (pTSA) led to the formation of Δ8-THC, while the use of the Lewis
acid boron trifluoride etherate (BF3·Et2O) yielded mainly
Δ9-THC and some Δ8-iso THC. The enormous
difference in product selectivity between these two catalysts is intriguing and
worthwhile investigating. In this Molecular Modeling study, the mechanisms of
the Brønsted and Lewis acid catalyzed conversion of CBD into other cannabinoids
are investigated, and the computational results are compared with the
experimental data of Webster et al.
2. Materials and Methods
The Spartan’20 suite of Wavefunction [10] was used for all Molecular Simulations. It
contains a large variety of academic Quantum Mechanical and Molecular Mechanics
codes. The Merck Forcefield (MMFF) is one of the most widely used Molecular
Mechanics methods. A description of MMFF can be found in a series of five
closely related articles by T.A. Halgren et al., [11,12,13,14,15].
The general computational approach can be described in three steps:
- Finding suitable geometries.
- Full geometry optimization.
- Determination of properties.
- 1.
- Finding suitable geometries.
MMFF was used to explore the conformational
behavior of the various cannabinoids by generating Conformer Distributions
(CD), including their Boltzmann weights. Until now Molecular Mechanics MMFF
usually does a better job with respect to the relative energies of possible
conformers and computational time than QM-methods.
- 2.
- Full geometry optimization.
Selected results of Molecular Mechanics
calculations were used as input for quantum chemical density functional theory
(DFT) calculations. All structures underwent a full geometry optimization,
applying B3LYP/6-31G*. This DFT-code can be considered as one of the most
validated codes. Other codes like ωB97X-D or MO6-2X whether or not combined
with higher basis sets yield similar results.
- 3.
- Determination of properties.
Finally, reaction and activation enthalpies were calculated based upon total energies and enthalpy corrections obtained from the fully optimized structures on the B3LYP/6-31G* level. The latter does require a frequency calculation. Entropy corrections were not made due to the huge simplifications made in the systems already. Transition States were characterized by their unique imaginary vibrational frequency. The concept of isodesmic reactions [16] was used to obtain high quantitative accuracy (error < ±5 kJ/mol) by gross cancelation of methodological or code errors. Transition State Theory [17] was used to estimate reaction rate constants, using the simplified expression:
k = kB*T/h*e-ΔHa/RT originating from k = κ*kB*T/h*e-ΔGa/RT
To arrive at the simplified expression, additional approximations are that the transmission coefficient κ = 1, and ΔGa ~ ΔHa, the latter in line with the fact that enthalpy corrections were made only.
In the case of catalysis with the Lewis acid BF3·Et2O, the pKa value of ArOH-BF3 was estimated using the relation between Maximum Electrostatic Potential (MEP) at the isodensity surface: 0.002 e/au3 and pKa [18]. The accuracy of the estimate was increased using structurally similar acids with experimentally determined pKa values to calibrate the MEP’s.
3. Results
3.1. Experimental Results
Webster et al., describe in their patents [8,9] two examples only, which both are of crucial interest for this study.
Example 1 describes the conversion of Δ9-CBD to Δ8-THC using dry pTSA as a catalyst in refluxing toluene (110.6 0C). Progress of the reaction was monitored with HPLC. After 15 minutes 79.3% of Δ8-THC was formed, after 30 minutes 81.7%, after 60 minutes 86.0%, and finally after 120 minutes 84.6%. No other products are mentioned explicitly but column chromatography yielded 81% Δ8-THC with a purity of 98.6%. So, the byproduct most likely is Δ9-THC because it is known that Δ8-THC and Δ9-THC are very difficult to separate completely [2]. It is mentioned that toluene is the best solvent and that dry pTSA was used. It seems that conversion is essentially almost complete after 60 minutes.
Example 2 describes the conversion of CBD to Δ9-THC using BF3.Et2O as a catalyst in dichloromethane at 0 0C. According to HPLC 27.0% of iso Δ8-THC and 66.7% of Δ9-THC was formed after 60 minutes. Contrary to example 1, the progress of the reaction was not specified further. But as the sum of the products accounts for almost 94%, conversion must be almost complete.
3.2. Computational Results
3.1.1. Cannabinoid Structures
To avoid confusion, the structures of all relevant cannabinoids are shown in Figure 1 and shortly discussed below.
The structures of Δ9-CBD, Δ8-CBD , Δ9-THC, and Δ8-THC have been discussed before [19] and originate from CD calculations as described in 2. Materials and Methods. The same approach was adopted for iso Δ8-THC, using a model wherein the C5-chain was reduced to a methyl group too. Like Δ9-THC and Δ8-THC, the conformational freedom of iso Δ8-THC is restricted by the ring closure of the phenolic group to the Δ8-C of the (former) cyclohexenyl substituent and shows 13 conformers only, all of them with the newly formed cyclohexane ring in a chair position with three axial substituents: the C-C(Ar)-, the C-O(Ar)-, and the isopropenyl substituent. Only the C-CH3 at the Δ8-position is in an equatorial position. The main differences between the conformers are in the position of the remaining phenolic group and the orientation of the isopropenyl substituent. The best conformer accounts for 84.6% in the Boltzmann weights, and the second-best conformer, showing a phenol group pointing to the isopropenyl cyclohexane substituent, accounts for 6.6 % in the Boltzmann weights.
3.1.1. The Conversion of Δ9-CBD to Δ8-THC, with pTSA as Brønsted Acid Catalyst
The conversion of Δ9-CBD to Δ8-THC, with pTSA as Brønsted acid catalyst, requires two steps: a ring closure and an isomerization of the double bond from the Δ9 to the Δ8-position. Ring closure reactions catalyzed by pTSA are quite rare. A recent example can be found in the pTSA condensation polymerization of dicarboxylic acids and polyols like sorbitol, where it appears as a side reaction on a secondary carbon atom [20]. Ring closure by a phenol on a tertiary carbon seems possible only because of the intramolecular presence of the phenolic group, the absence of more suitable nucleophiles, and activation of the alkene by pTSA. These conditions are fulfilled by the reaction conditions described above: dry pTSA as a catalyst in refluxing toluene. Figure 2 shows the Transition States of the ring closure of Δ9-CBD to Δ9-THC and Δ8-CBD to Δ8-THC catalyzed by pTSA.
The two Transition States look very similar. There is almost complete proton transfer from pTSA to the CH2 of the isopropenyl group: the CH2-HOSO2Ar-pCH3 distance is 1.237 Å and 1.212 Å respectively. This is also visible from the reduced electron density plot. The phenolic O-atom is poised to form a C-O bond with the tertiary C of the isopropenyl group despite the relative long distance between them: 2.758 Å and 3.204 Å respectively. The electrostatic potentials on the reduced electron density surface of the tertiary C’s are +1594 and +1544 kJ/mol respectively, a clear indication of their carbocation character. Animation of both imaginary frequencies show the right movement of all atoms involved. The H-atom of the phenol group is H-bridged to an O-atom of pTSA with distances of 1.723 Å and 1.746 Å respectively. The activation barriers are 70.5 and 57.5 kJ/mol respectively, the latter a reflection of the relative stability of Δ9-CBD compared to Δ8-CBD, which is 11.8 kJ/mol in favor of Δ9-CBD.
For the isomerization reaction two mechanisms were considered:
- a)
- a concerted process with simultaneous proton transfers from pTSA to D9-C and D7-C to pTSA, and
- b)
- a two-step process, starting with proton transfer from pTSA to D9-C, followed by proton transfer from D7-C to pTSA.
These mechanistic proposals for acid catalyzed isomerization are not new but go back to 1932 [21,22] and evidence has been obtained for both cases, depending on specific reaction conditions and catalysts. Figure 3 shows the Transition States of the concerted and the two-step process of the isomerization of Δ9-CBD to Δ8-CBD with pTSA.
On first sight the two Transition States look very similar, however appearances are deceiving. In the concerted process proton transfer from pCH3ArSO3H to Δ9-C is almost complete with an SO-H distance of 1.911 Å and a Δ9 C-H distance of 1.126 Å, while the SO-H Δ7 C distance is 1.720 Å and a Δ7 C-H distance is 1.169 Å. In the two-step process, proton transfer from pCH3ArSO3H to Δ9-C is less complete with an SO-H distance of 1.621 Å and a Δ9 C-H distance of 1.199 Å. Note that in this case the SO-H Δ7 C distance is 2.175 Å and a Δ7 C-H distance is 1.112 Å, also quite different from the concerted case. Once more the imaginary frequencies are low, the one for the two-step process even very low, but show the correct movement in both cases. The activation barriers are 75.3 and 67.2 kJ/mol respectively. The two-step process is clearly favored over the concerted process. The starting complex for both processes is the same. The activation barriers for the reverse process, the isomerization of Δ8->Δ9 CBD, show significantly higher activation barriers of 93.4 and 81.8 kJ/mol. The reason for that is the higher stabilization enthalpy of the starting complex caused by the H-bridge of the phenol group of Δ8 CBD to an O=S of pTSA, compared to the H-bridge of that phenol group of Δ9 CBD to an HO-S of pTSA.
For the isomerization of Δ9 THC and Δ8 THC again the two previous mentioned options were considered. However, it turned out that for the concerted option only a true Transition State could be established, despite many attempts to locate a Transition State for the two-step process. These attempts either led to the Transition State of the concerted process or to non converged structures which can be described best as intimate ion pairs, consisting of the Δ8 THC cation and the pTSA anion with no good imaginary frequency left, and a total energy of 1-3 kJ/mol higher only than the energy of the Transition State of the concerted process. In the concerted process proton transfer from pCH3ArSO3H to Δ9-C is almost complete with an SO-H distance of 1.836 Å and a Δ9 C-H distance of 1.146 Å, while the SO-H Δ7 C distance is 1.755 Å and a Δ7 C-H distance is 1.162 Å. The imaginary frequency is i152 cm-1 and its animation shows the correct movement of the proton transfers and the corresponding skeletal adaptation. The activation barriers for the TS Δ9->Δ8 THC and Δ8->Δ9 THC are 95.7 and 103.9 kJ/mol, reflecting the relative stabilities of Δ9 and Δ8 THC.
3.1.2. The Conversion of Δ9-CBD to Δ8-THC, with BF3·Et2O as Lewis Acid Catalyst
Though BF3·Et2O is a well know catalyst, it is difficult to find examples wherein ring closure of an O-nucleophile or alkene isomerization are described. Closest to the actual BF3·Et2O catalyzed ring closure are the inverse reactions, the B(C6F5)3 catalyzed ring opening of an 2,2-disubstituted oxetane to a homoallylic alcohol, and the BF3·Et2O catalyzed decomposition of a t-butyldimethylsilyl ether of a tertiary alcohol [23,24]. The latter is very close to the previously described Example 2 with respect to the reaction conditions applied. In 2022 an example of an alkene isomerization with B(C6F5)3 was published, including a mechanistic description [25]. They found experimental and computational evidence that the isomerization of 2-propenyl benzene occurs via a direct hydride abstraction by B(C6F5)3 leading to a mixed allylic-benzylic carbenium ion and a 1,2-hydride shift to the terminal alkene- B(C6F5)3 complex. They used the M06-2X DFT functional with an extended basis set and solvent model. Unfortunately, the computational data are not available in their Supplementary Material. Attempts to locate similar Transition States with BF3, using either the M06-2X or the B3LYP functional, were not successful.
As the electron deficient BF3 wants to interact with electron rich systems, the complexation of BF3 with the alkene and phenol groups of Δ9 CBD was investigated to get an impression of the complexation energy. Results are listed in Table 1. As a reference BF3·Et2O is listed too.
From Table 1 it is clear that complexation of BF3 with a phenol group is favored over complexation with an alkene like the 2-propenyl substituent on the cyclohexene ring, or ethers like in BF3·Et2O and BF3· Δ9 THC. It was realized that complexation of the Lewis acid BF3 with a phenol transforms the weakly acidic phenol into a strong Brønsted acid. Figure 4 shows the BF3·phenol complex. There is a clear blue spot at the surface of the phenolic group, indicative for a positive electrostatic potential. The Maximum Electrostatic Potential (MEP) shows a value of +314.7 kJ/mol. Using an earlier derived linear relation [26] between MEP and pKa yielded -2.17 as an estimate for the pKa of the BF3·phenol complex which is more acidic than pTSA (MEP = +261.6 kJ/mol); pKa = -1.34) and slightly less acidic than H2SO4 (MEP = +327.0 kJ/mol; pKa = -2.49).
Thus, reactions starting from a BF3-phenolic cannabinoid complex were investigated. Figure 5 shows the Transition States of the ring closure to Δ9-THC and iso Δ8-THC.
Both Transition States show nearly complete proton transfer from the phenol to the CH2 of the 2-propenyl substituent and the Δ9-C of the cyclohexenyl substituent respectively. Their O-H, H2C-H and O-H, Δ9-C distances are: 1.850 Å, 1.142 Å and 1.743 Å, 1.157 Å and their activation barriers are 80.9 and 82.1 kJ/mol respectively. The animation of their imaginary frequencies clearly shows the proton transfer process and some skeletal adaptation to the formation of the tertiary carbenium ion. No movement of the phenolic O to the tertiary is visible, and no sign of double bond isomerization is visible. Therefore, so called Energy Profiles (EP) were constructed, starting from the Transition States, and leading to either the starting BF3·Δ9-CBD complexes or the BF3·Δ9-THC, BF3· iso Δ8-THC, or the double bond isomerized products. In an EP a constraint is applied and next this constraint is varied in regular small steps from the starting to the final situation in a series of full geometry optimizations. A plot of the (relative) energy versus the steps provides an impression of the feasibility of such a pathway. Figure 6 shows the case for the formation of iso Δ8-THC, starting from BF3·Δ9-CBD.
In Figure 6 two EPs are combined which both start from the TS ring closure iso Δ8-THC as shown in Figure 5. The first EP is the reverse of the proton transfer from BF3·Δ9-CBD to the Δ9-C of the cyclohexane ring. This EP ranges from molecule 8-1, and the ArO-H distance changes in steps of ~0.1 Å from 1.743 Å to 0.993 Å, the ArO-H equilibrium distance (red line). The second EP is the movement of the phenolic-O to the Δ8-C of the cyclohexane ring. This EP ranges from molecule 8-22, and the ArO- Δ8-C distance changes in steps of ~0.1 Å from 2.857 Å to 1.475 Å, the ArO- Δ8-C equilibrium distance (green line). The energy curve (blue) does not show additional local maxima or minima. The ΔE from the TS ring closure iso Δ8-THC to iso Δ8-THC is -133.2 kJ/mol. A very similar EP was determined showing the formation of Δ8-CBD, the isomerized double bond product, from the TS ring closure iso Δ8-THC. That energy curve does not show additional local maxima or minima too. However, the ΔE from the TS ring closure iso Δ8-THC to iso Δ8-CBD is -92.0 kJ/mol. So, in principle fully reversible isomerization is very well possible but at sufficiently high reaction rates, the thermodynamic product, iso Δ8-THC, will be formed exclusively.
3.1.3. Kinetic Models: Comparison of Computational and Experimental Results
Table 2 gives an overview of all activation barriers related to the conversion of Δ9-CBD to Δ8-THC, with pTSA and BF3·Et2O.
As discussed in 2. Materials and Methods, reaction rates were calculated using:
k = k0*[catalyst]/ [substrate]*e-Ha/RT (s-1)
Thus, k represents a pseudo first order rate constant. As all Transition State structures contain the catalyst, pTSA or BF3, the pseudo first order rate constant was corrected for the [catalyst]/ [substrate] ratio, as this the maximum amount of substrate that can react in time. The activation barriers for pTSA catalyzed ring closure do lead to pseudo first order rate constants which are far too high. It took some time before it was realized that pTSA in an apolar solvent under dry conditions actually is predominantly present as a dimer with a ΔH = -77.5 kJ/mol. The corresponding equilibrium constant K= 3.31*10+10, and the fraction pTSA-monomer is 5.50*10-6 only. Correction for this low amount of monomeric pTSA led to rate constants kc, listed in the last column of Table 2.
For the case of catalysis with pTSA a kinetic model was developed using the kc values for ring closure Δ9 THC pTSA, ring closure Δ8-THC pTSA and two-step isomerization Δ9->Δ8-CBD pTSA only. The concerted isomerization of Δ9-CBD is not operative as discussed above, and all other values for kc are too low to play a role. Using the exact values of Table 2, a yield of 73.7% Δ8-THC at a total conversion of Δ9-CBD of 99.9% was predicted. It seems that overall selectivity to Δ8-THC is slightly too low, while the overall conversion is slightly too high. Probably more important is the apparent absence of Δ8-CBD in the reaction mixture after less than 15 minutes, because the presence of Δ8-CBD was not observed experimentally. Furthermore, the high selectivity to Δ8-THC instead of Δ9-THC is explained by the lower activation barriers for two-step isomerization from Δ9-CBD to Δ8-CBD and ring closure to Δ8-THC. Adaptations of +2 kJ/mol) in activation barriers of ring closure of Δ9 THC and ring closure of Δ8-THC, lead to an almost perfect fit. The fitted selectivity is 84% compared to 86% experimentally. Alternatively, the ΔH of dimerization of pTSA could be adapted, yielding a very similar result. It should be realized that such small adaptations are within the error limit of the calculations.
For the case of catalysis with BF3·Et2O a kinetic model was developed using the k values for ring closure Δ9 THC BF3·Et2O, and ring closure iso Δ8-THC BF3·Et2O. Using the exact values of Table 2, a yield of 61.9 % Δ9-THC and 36.5 % iso Δ8-THC at a total conversion of Δ9-CBD of 98.5% was predicted. It seems that overall selectivity to Δ9-THC is slightly too low, while the overall conversion is slightly too high. An adaptation from 80.9 to 81.5 kJ/mol of the activation barrier for ring closure to Δ9 THC BF3·Et2O and an adaptation from 82.1 to 83.5 kJ/mol of the activation barrier for ring closure to iso Δ8-THC BF3·Et2O leads to a perfect fit with 66.6 % Δ9-THC and 27.6 % iso Δ8-THC and a total conversion of 94.2%.
4. Discussion and Conclusions
The basic research question of this study was the origin of the enormous difference in product selectivity between the catalysts pTSA and BF3·Et2O in the conversion of Δ9-CBD. Molecular Modeling was able to elucidate the mechanisms of pTSA and BF3·Et2O catalyzed ring closure and isomerization reactions of Δ9-CBD.
In the pTSA case, the two mechanistic options for isomerization of an alkene with a sulfonic acid like pTSA are not new but goes back to 1932. The two-step mechanism for isomerization is more favorable in this case because a tertiary carbenium ion is formed, and the pTSA-anion is stabilized by the phenolic OH-group. The activation barriers for ring closure of Δ9-CBD and Δ8-CBD to Δ9-THC and Δ8-THC are 70.5 and 57.5 kJ/mol respectively, reflecting the relative stability of Δ9-CBD over Δ8-CBD, which is 11.8 kJ/mol. Using Transition State theory and the earlier described expression for the reaction rate, led initially too much to high reaction rates in the kinetic. Later it was realized that pTSA predominantly is present as a dimer in an apolar dry solvent. Correction for the correct amount of pTSA-monomer led to reaction rates that predict amounts of Δ9-THC and Δ8-THC that a very close to the experimentally observed ones.
In the BF3·Et2O case it turned out that ring closure to Δ9-THC and iso Δ8-THC had to be taken into consideration only. Δ9-THC and iso Δ8-THC is formed exclusively because they are the thermodynamically favored products. Complexation with the Lewis acid BF3 has turned the weakly acidic phenol into a powerful Brønsted acid, capable of protonating the alkene groups completely. Using the computational activation barriers to calculate pseudo first order reaction rates, led to an almost perfect fit of the kinetic model with the experimental data. Summarizing it can be said that the application of Molecular Modeling, using standard DFT quantum chemical calculations and Transition State theory to estimate reaction rates, led to a qualitative and quantitative understanding of the underlying reaction mechanisms and various reaction products formed experimentally.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Google Trends: https://trends.google.de/trends/explore?q=CBD&geo=DE&date=2013-01-01%202020-03-11#TIMESERIES (accessed on 27 December 2023).
- Golombek, P., Müller, M., Bartholt, I., Sproll, C., Lachenmeier, D.W. Conversion of cannabidiol (CBD) into psychotropic cannabinoids including tetrahydrocannabinol (THC): a controversy in the scientific literature, Toxics, 2020, 8, 41. [CrossRef]
- PubChem, National Library of Medicine, Cannabidiol, PubChem CID 644019. https://pubchem.ncbi.nlm.nih.gov/compound/644019. (accessed on 27 December 2023).
- Healthline: https://www.healthline.com/health/cbd-vs-thc#_noHeaderPrefixedContent (accessed on 27 December 2023).
- Nelson, K.M., Bisson, J., Singh, G., Graham, J.G., Chen, S-N., Friesen, J.B., Dahlin, J.J., Niemitz, M.,. Walters, M.A., Pauli, G.F. The essential medicinal chemistry of cannabidiol (CBD), J. Med. Chem. 2020, 63, 12137–12155. [CrossRef]
- Buijs, W; The scientific controversy on the conversion of CBD into THC in the human stomach: Molecular modelling and experimental results compared. Forensic Chemistry 2023, 32, 100467. [CrossRef]
- OFFICIAL OPINION 2023-1 Tetrahydrocannabinol Variants and Other Designer Cannabinoid Products: https://www.in.gov/attorneygeneral/files/Official-Opinion-2023-1.pdf (accessed on 27 December 2023).
- Webster, G.B.; Sarna, L.P.; Mechoulam, R. Conversion of CBD to Δ8-THC and Δ9-THC. US 2004/0143126A1, 2004, July 22.
- Webster, G.B.; Sarna, L.P.; Mechoulam, R. Conversion of CBD to Δ8-THC and Δ9-THC. US 20040143126A1, 2008, July 15.
- Wavefunction, Inc., 18401 Von Karman Avenue, Suite 435, Irvine, CA 92612 U.S.A. www.wavefun.com (accessed on 27 December 2023).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem., 1996, 17, 490-519.
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comp. Chem., 1996, 17, 520-552.
- Halgren, T.A. Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94. J. Comp. Chem., 1996, 17, 553-586.
- Halgren, T.A., Nachbar, R.B. Merck molecular force field. IV. conformational energies and geometries for MMFF94. J. Comp. Chem., 1996, 17, 587-615.
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comp. Chem., 1996, 17, 616-641.
- Hehre, W.J., Ditchfield, R., Radom, L., Pople, J.A. Molecular orbital theory of the electronic structure of organic compounds. V. Molecular theory of bond separation, J. Am. Chem. Soc. 1970, 92, 4796. [CrossRef]
- Pt´aˇcek, P., ˇSoukal, F., Opravi, T. Introduction to the Transition State Theory, Chapter 2, Open Research. [CrossRef]
- Virant, M., Drvaric Talian, S., Podlipnik, C., Hribar-Lee, B. Modelling the correlation between molecular electrostatic potential and pKa on sets of carboxylic acids, phenols and anilines, Acta Chim. Slov. 2017 64, 560–563. [CrossRef]
- Buijs, W. The scientific controversy on the conversion of CBD into THC in the human stomach: Molecular modelling and experimental results compared. Forensic Chemistry 2023 32, 10046. [CrossRef]
- Goddard A. R., Apebende E. A., Lentz J. C., Carmichael K., Taresco V., Irvine D. J., Howdle S. M. Synthesis of water-soluble surfactants using catalysed condensation polymerisation in green reaction media. Polym Chem. 2021, 20, 2992-3003. [CrossRef]
- Whitmore, F.C.. The common basis of intramolecular rearrangements J. Amer. Chem. Soc. 1932, 54, 3274.
- Ipatieff, V. N., Corson, B.B. Catalytic polymerization of gaseous olefins by liquid phosphoric acid. Butylene. Ind. Eng. Chem., 1935, 27, 1069.
- Cabré, A., Rafael, S., Sciortino, G., Ujaque, G., Verdaguer, X.,. Lledós, A, Rier, A. Catalytic Regioselective Isomerization of 2,2-Disubstituted Oxetanes to Homoallylic Alcohols. Angew. Chem. Int. Ed. 2020, 59, 7521.
- Posner, G. H.,. Shulman-Roskes, E. M, Oh, C. H,. Carry, J-C,. Green, J. V, Brian-Clark, A.,. Dai, H,. Anjeh, T. E.N. BF3-OEt2 promotes fast, mild, clean and regioselective dehydration of tertiary alcohols. Tetrahedron Letters, 1991, 32, 5, 6489-6492.
- Kustiana, B. A, Elsherbeni, S. A., Linford-Wood, T. G., Melen, R. L.,. Grayson, M. N., Morrill, L. C. B(C6F5)3-Catalyzed E-Selective Isomerization of Alkenes. Chem. Eur. J. 2022, 28, e20220245. [CrossRef]
- Buijs, W. Role of Fe Complexes as Initiators in the Oxidative Degradation of Amine Resins for CO2 Capture: Molecular Modeling and Experimental Results Compared. 2023. [CrossRef]
Figure 1.
The structure of Δ9-CBD, Δ8-CBD , Δ9-THC, Δ8-THC and iso Δ8-THC. Display: ball and spoke; C: black, H: white, O: red.
Figure 1.
The structure of Δ9-CBD, Δ8-CBD , Δ9-THC, Δ8-THC and iso Δ8-THC. Display: ball and spoke; C: black, H: white, O: red.
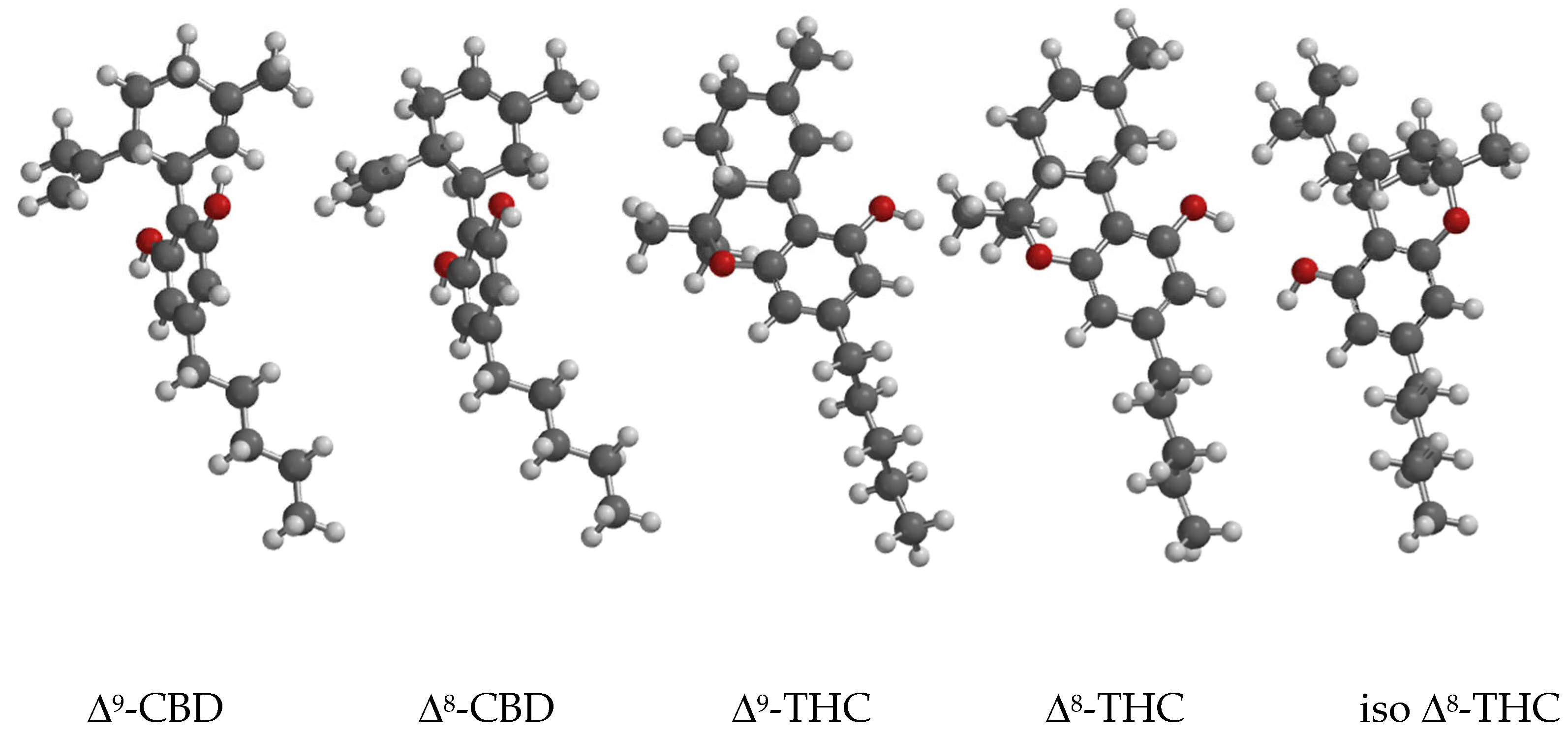
Figure 2.
Transition States ring closure of Δ9-CBD to Δ9-THC and Δ8-CBD to Δ8-THC. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
Figure 2.
Transition States ring closure of Δ9-CBD to Δ9-THC and Δ8-CBD to Δ8-THC. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
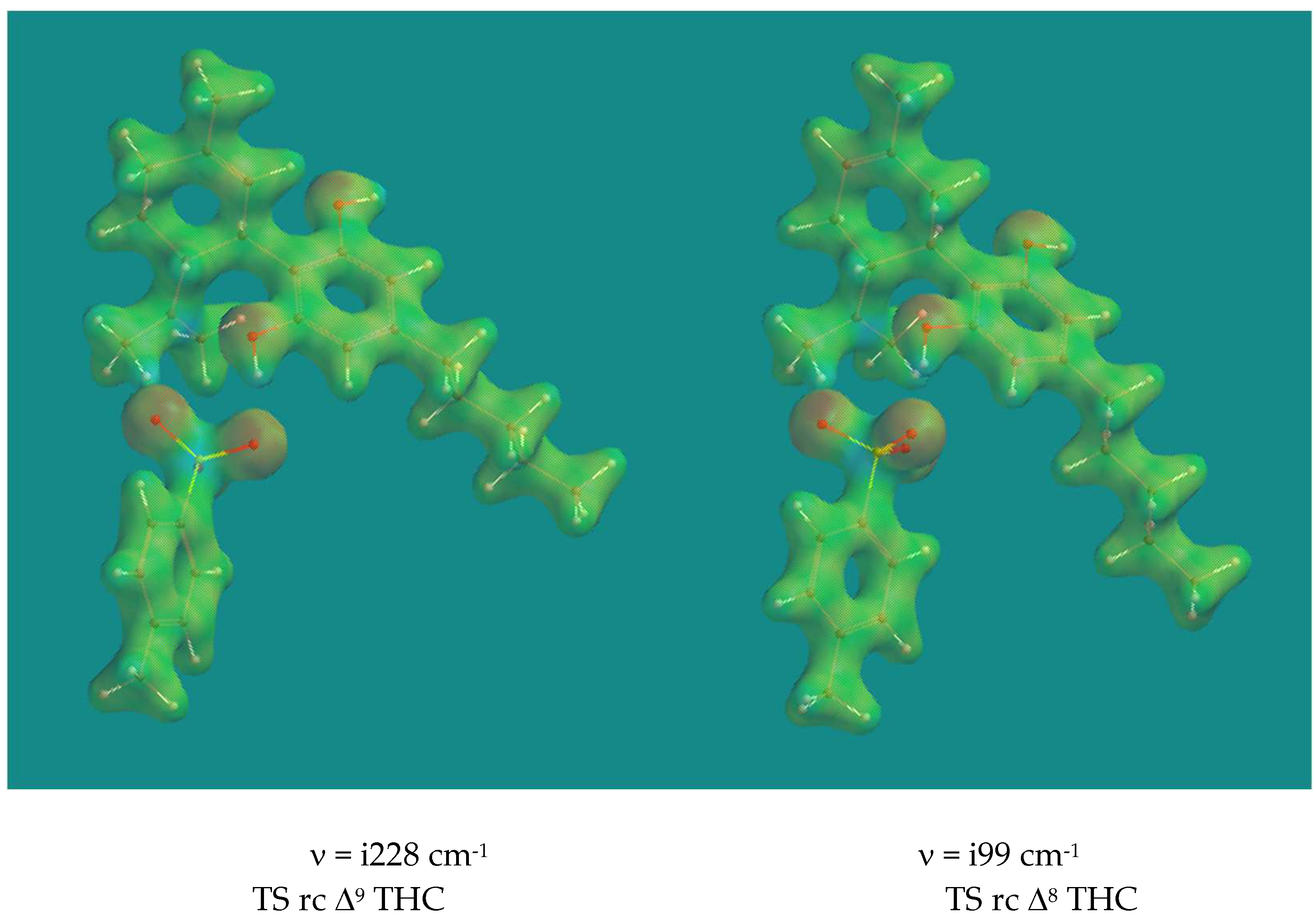
Figure 3.
Transition States isomerization of Δ9-CBD to Δ8-CBD via a concerted and a two-step mechanism. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
Figure 3.
Transition States isomerization of Δ9-CBD to Δ8-CBD via a concerted and a two-step mechanism. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
Figure 4.
BF3·phenol complex. B3LYP/6-31G* Surface: electron density = 0.002 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +300 (blue) kJ/mol.
Figure 4.
BF3·phenol complex. B3LYP/6-31G* Surface: electron density = 0.002 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +300 (blue) kJ/mol.
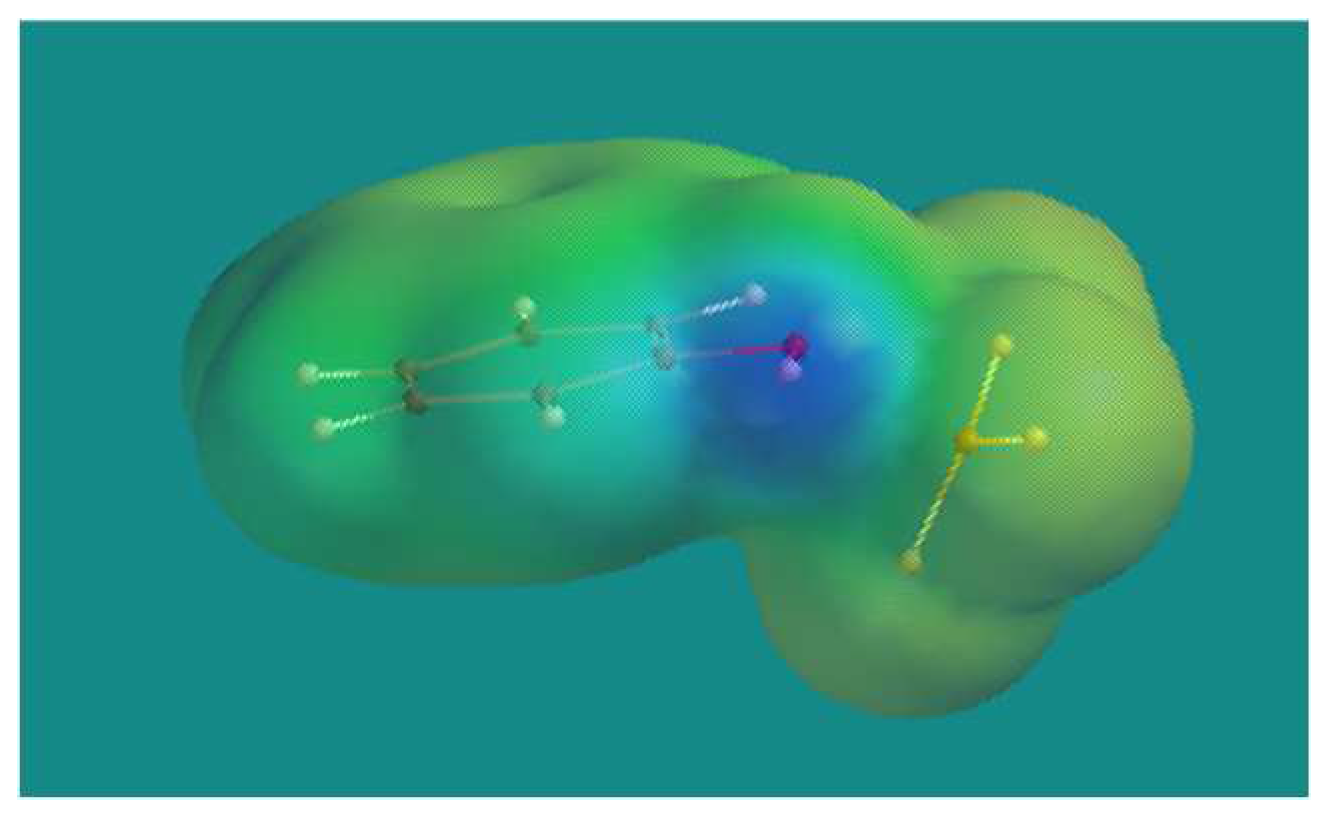
Figure 5.
Transition States ring closure of BF3·Δ9-CBD to Δ9-THC and BF3·Δ9-CBD to iso Δ8-THC. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
Figure 5.
Transition States ring closure of BF3·Δ9-CBD to Δ9-THC and BF3·Δ9-CBD to iso Δ8-THC. B3LYP/6-31G* Surface: reduced electron density = 0.08 e/au3, projected property on electron density: electrostatic potential, range: -200 (red)- +2000 (blue) kJ/mol.
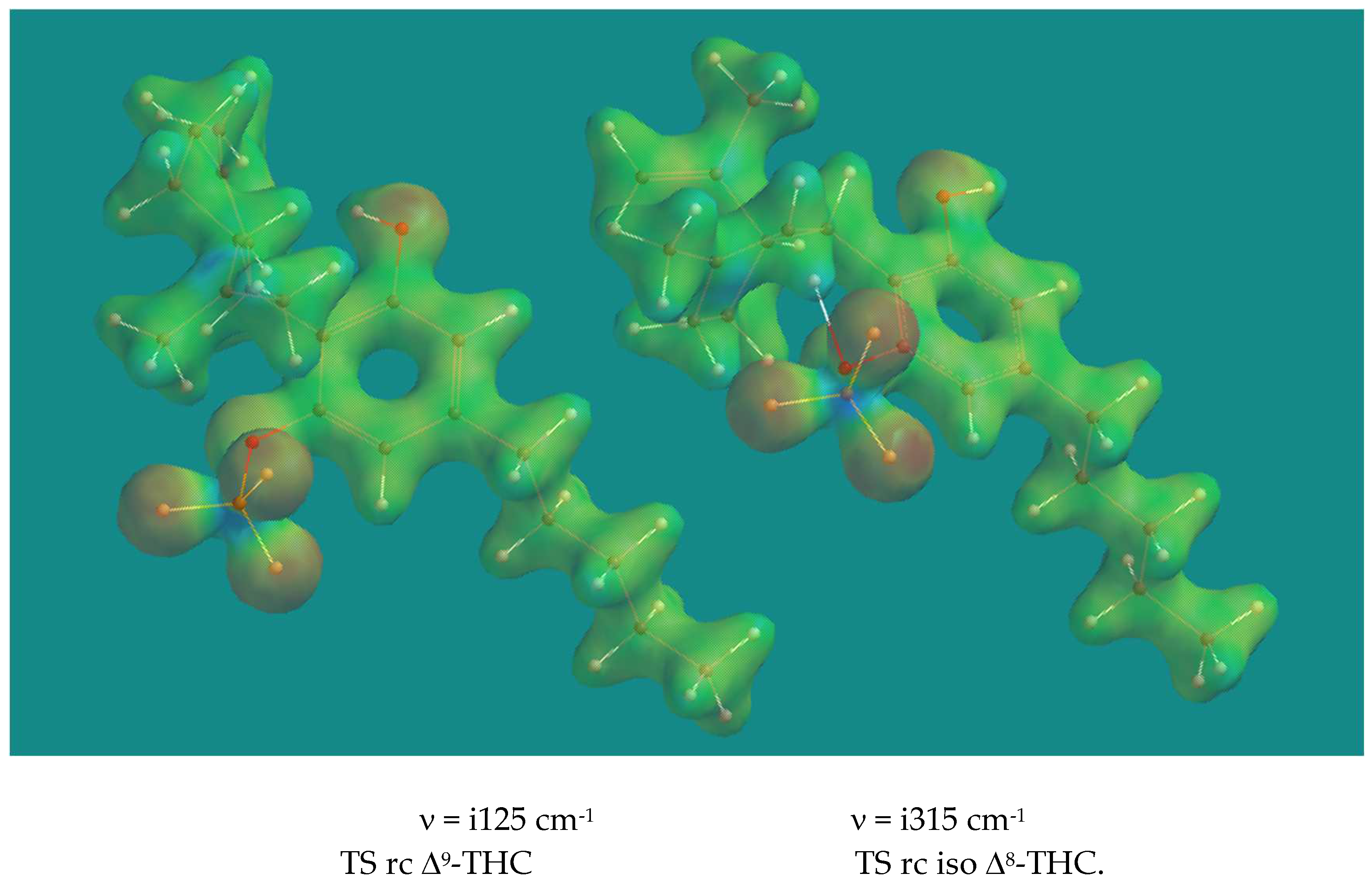
Figure 6.
Combined Energy Profiles (B3LYP/6-31G*) for the formation of iso Δ8-THC from BF3·Δ9-CBD.
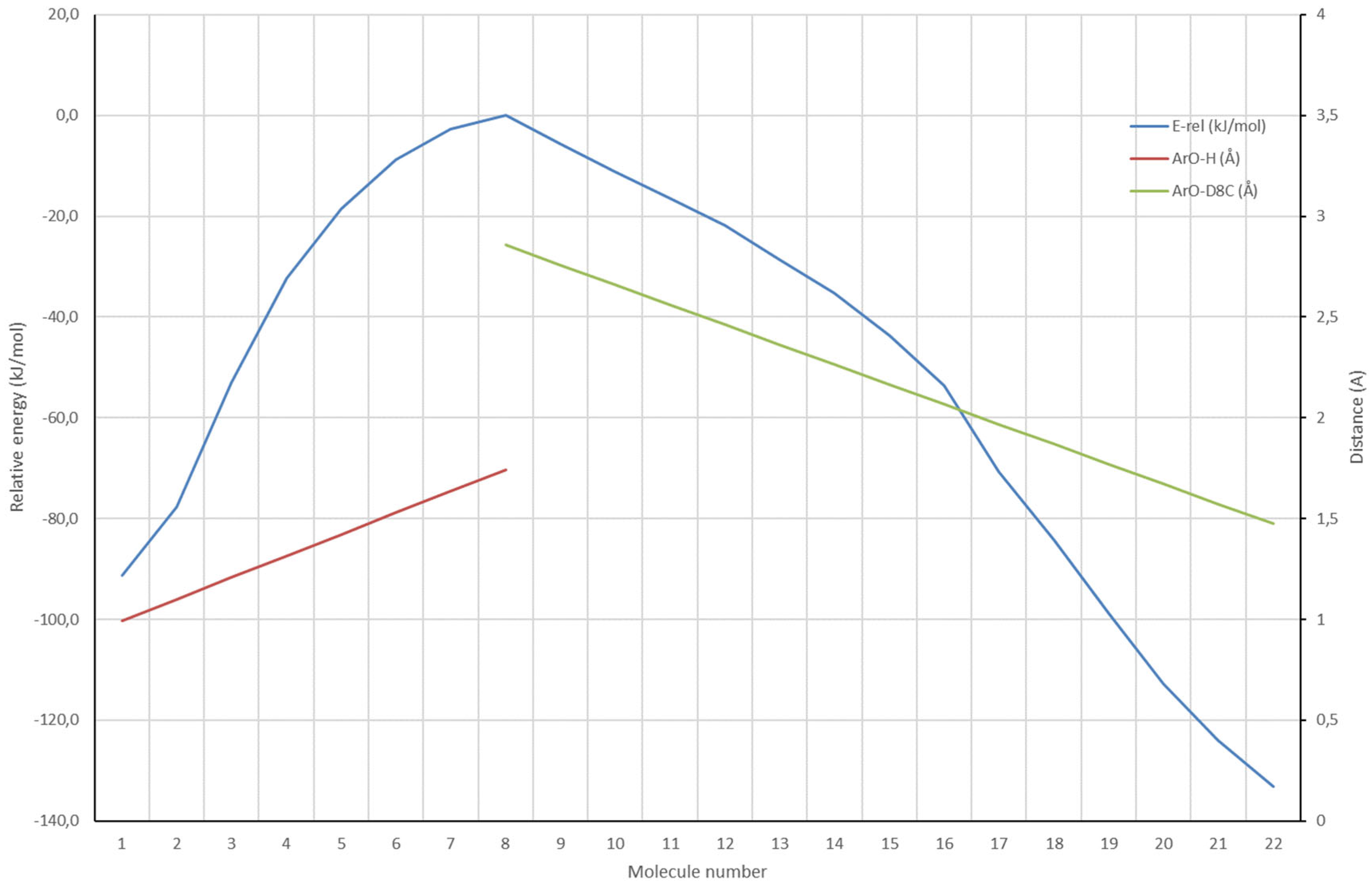
Figure 7.
Fit kinetic model for formation of Δ9 THC and Δ8-THC, catalyzed by pTSA via two-step isomerization of Δ9-CBD to Δ8-CBD.
Figure 7.
Fit kinetic model for formation of Δ9 THC and Δ8-THC, catalyzed by pTSA via two-step isomerization of Δ9-CBD to Δ8-CBD.
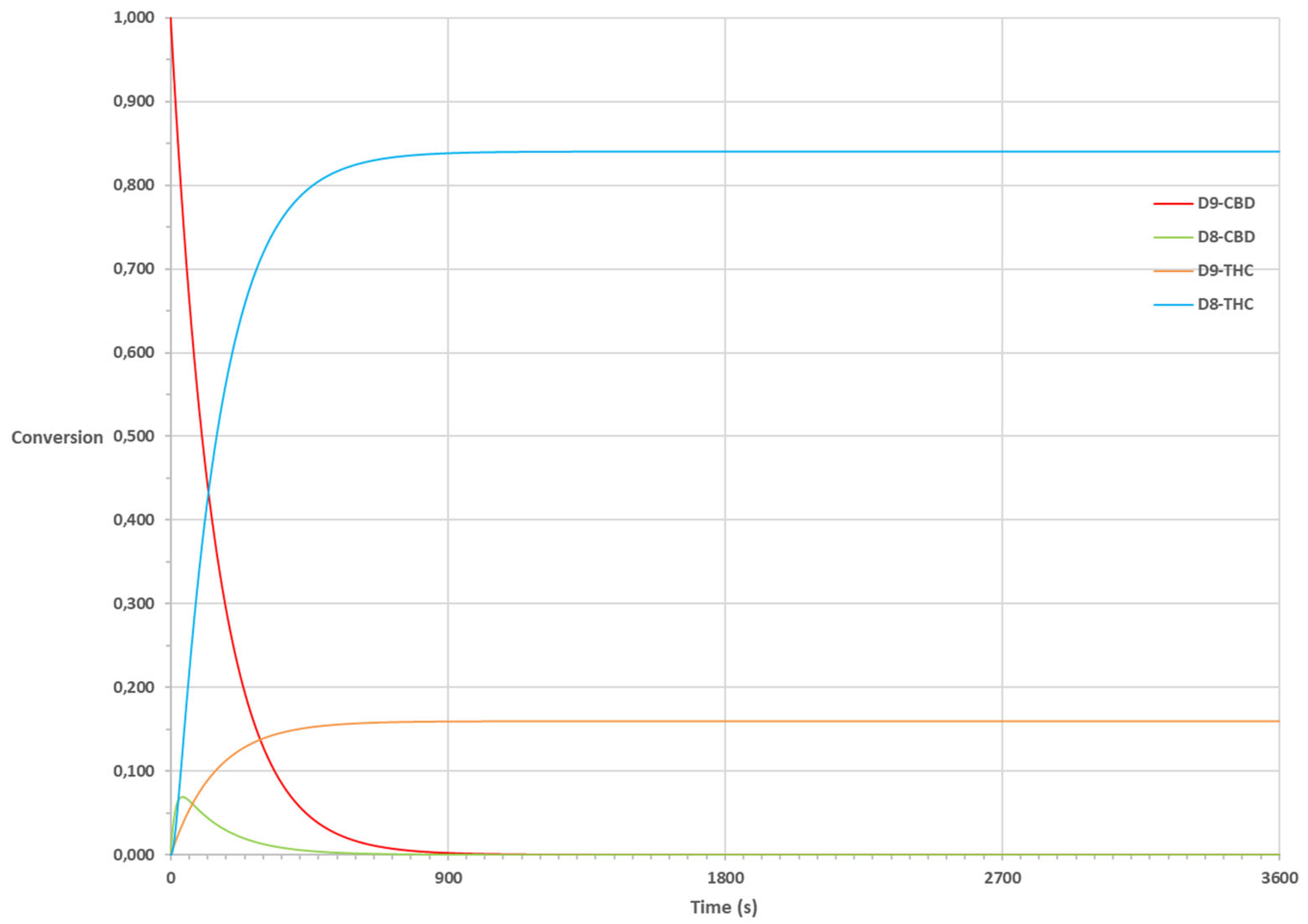

Table 1.
Overview Interaction Energy (B3LYP/6-31G*; kJ/mol) of various BF3 complexes.
| Complex | Interaction energy (kJ/mol) |
|---|---|
| BF3·Et2O | -23,1 |
| BF3· 2-propenyl Δ9 CBD | -18.8 |
| BF3· phenol Δ9 CBD to Δ9 THC | -39,1 |
| BF3· Δ9 THC | -24,3 |
Table 2.
B3LYP/6-31G* activation barriers and pseudo first order reaction rate constants related to the conversion of Δ9-CBD to Δ8-THC, with pTSA and BF3·Et2O. In the pTSA case, T = 383.8 K (110.6 0C), In the BF3·Et2O case T = 273.2 K (0.0 0C).
Table 2.
B3LYP/6-31G* activation barriers and pseudo first order reaction rate constants related to the conversion of Δ9-CBD to Δ8-THC, with pTSA and BF3·Et2O. In the pTSA case, T = 383.8 K (110.6 0C), In the BF3·Et2O case T = 273.2 K (0.0 0C).
| Molecular system | ν (cm-1) | ΔHa (kJ/mol) | k (s-1) | kc (s-1) |
|---|---|---|---|---|
| TS ring closure Δ9 THC pTSA | i228 | 70.5 | 3.69*10+2 | 2.03*10-3 |
| TS ring closure Δ8-THC pTSA | i99 | 57.5 | 2.19*10+4 | 1.21*10-1 |
| TS concerted isomerization Δ9->Δ8-CBD pTSA | i100 | 75.3 | 8.21*10+1 | 4.51*10-4 |
| TS concerted isomerization Δ8->Δ9-CBD pTSA | i100 | 86.8 | 2.23*10+0 | 1.23*10-5 |
| TS two-step isomerization Δ9->Δ8-CBD pTSA | i31 | 67.2 | 1.04*10+3 | 5.71*10-3 |
| TS two-step isomerization Δ8->Δ9-CBD pTSA | i91 | 81.8 | 1.07*10+1 | 5.88*10-5 |
| TS concerted isomerization Δ9->Δ8-THC pTSA | i152 | 95.7 | 1.37*10-1 | 7.54*10-7 |
| TS concerted isomerization Δ8->Δ9-THC pTSA | i152 | 103.9 | 1.05*10-2 | 5.77*10- |
| TS ring closure Δ9-THC BF3·Et2O | i125 | 80.9 | 7.28*10-4 | |
| TS ring closure iso Δ8-THC BF3·Et2O | i315 | 82.1 | 2.89*10-4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



