
Submitted:
29 December 2023
Posted:
29 December 2023
You are already at the latest version
Abstract
An efficient approach to the previously unknown furo[2',3':2,3]pyrrolo[2,1-a]isoquinoline deriv-atives from readily available 1-R-1-ethynyl-2-vinylisoquinolines is described. The reaction fea-tures a simple procedure, occurs in hexaflouroisopropanol and does not require elevated tem-peratures. It has been found that the addition of glacial acetic acid significantly increases the yields of the target spirolactone products. Using trifluoroethanol instead of hexaflouroisopro-panol results into formation of pyrido[2,1-a]isoquinolines.
Keywords:
hexaflouroisopropanol
; lactonic pyrrolo[2
; 1-a]isoquinolines
; pyrido[2
; [1
; 2]-sigmatropic rearrangement
; trifluoroethanol
1. Introduction
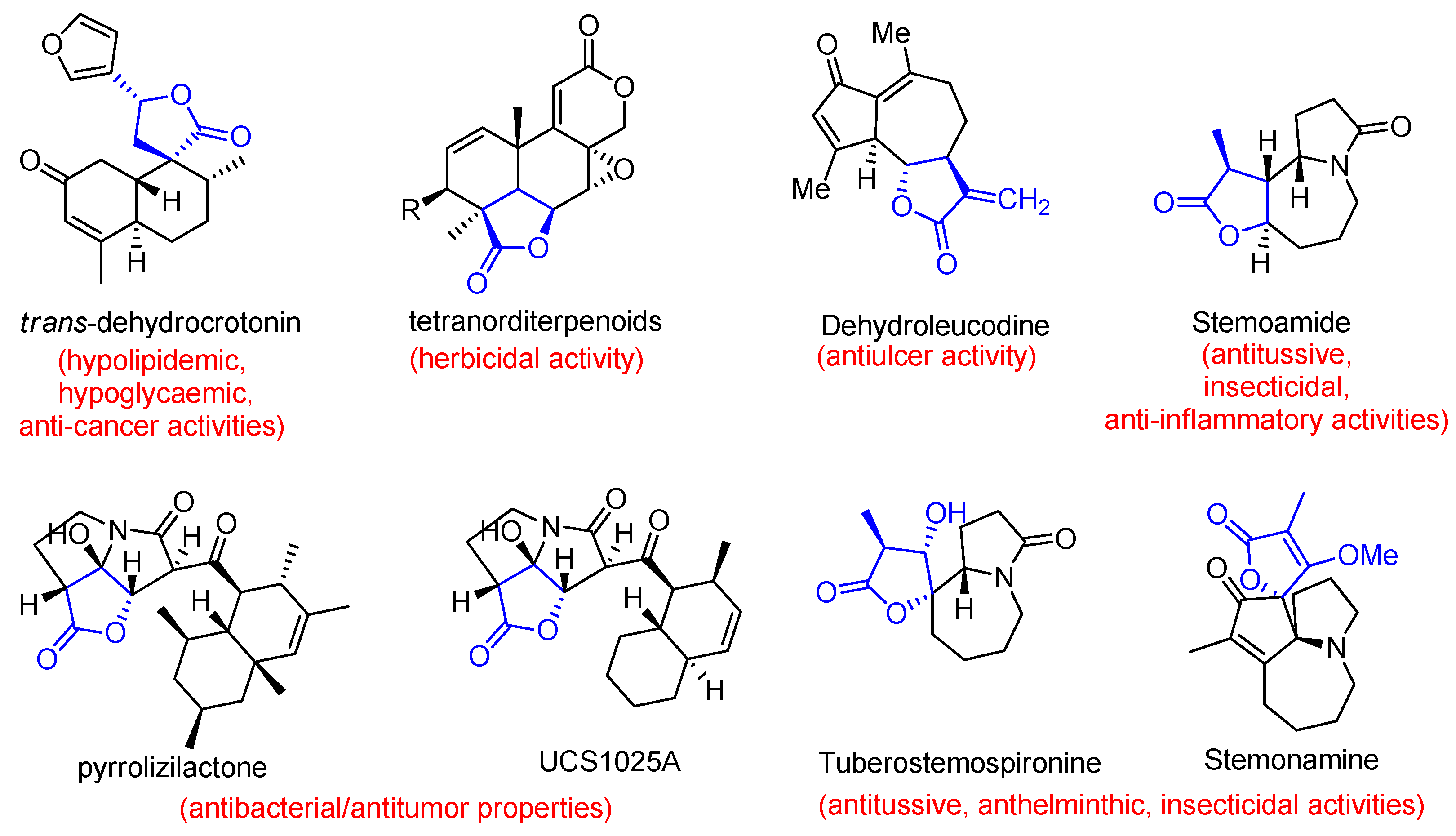
The γ-lactone moiety is present in many bioactive natural products isolated from various plants and fungal metabolites [1,2,3]. Compounds with lactone and spirolactone fragments are characterized by a broad range of bioactivities and find their application in the field of medicine and agriculture. Thus, trans-dehydrocrotonin exhibits hypolipidemic and hypoglycaemic properties, anti-cancer activity [4,5,6,7], tetranorditerpenoids can be used as herbicides [8], dehydroleucodine has anti-inflammatory and antiulcer activities [9], Stemoamide, Stemonamine and Tuberostemospironine, being Stemona alkaloids, possess anti-inflammatory, insecticidal, antitussive activities [2,10,11]. Lactonic pyrrolizidinone alkaloids - pyrrolizilactone and UCS1025A demonstrate potent antibacterial and antitumor effects [3,12].
Figure 1.
Biologically active natural lactone and spirolactone molecules.
Due to a wide profile of pharmaceutical activities spirolactones attract considerable attention of scientists and forward both the development of simple and effective synthetic routes to such structures and the further study of their properties. Recently numerous methods for the synthesis of spirolactones have been described in literature [13,14,15,16]. Among a variety of the known approaches, those, that are based on mild, free-metal and step-ecomonic reactions, start from readily available materials and meet the requirements of modern and “advantageous” synthetic chemistry, deserve a special attention. Domino processes, incorporating rearrangements and reconstructions of carbon skeleton and leading to quick complexity of a molecule structure in one step, can be considered as an eligible candidate fitting all claims of such “advantageous chemistry” [17].
2. Results and Discussion
Herein we report on a study devoted to divergent transformations of 1-R-1-ethynyl-2-vinyl substituted 1,2,3,4-tetrahydroisoquinolines 1a-g occurring in protic fluorinated solvents. One of the observed transformation proceeds via a 1,2-rearrangement step in the presence of AcOH/HFIP and opens an access to previously unexplored furo[2′,3′:2,3]pyrrolo[2,1-a]isoquinoline derivatives 3.
Previously we have described the chemical behavior of 1-R-1-ethynyl-2-vinyl substituted 1,2,3,4-tetrahydroisoquinolines in aprotic solvents [18]. It has been shown that the route of the MW-stimulated rearrangements deeply depends on the type of the solvent. The use of toluene favored the formation of pyrrolo[2,1-b][3]benzazepines while switching to acetonitrile afforded pyrido[2,1-a]isoquinolines in good yields. Encouraged by the unusual results we decided to examine the influence of protic solvents, particularly fluorinated alcohols – trifluoroethanol and hexafluoroisopropanol (HFIP), on the disclosed rearrangements. Fluorinated alcohols are characterized by low nucleophilicity, high ionizing and solvating power, increased Brønsted acidity of the hydroxyl proton, high polarity as well as the ability to affect the regio- and chemoselectivity of a reaction and its process rate [19,20]. In other words they could open new directions of the well-known transformations.
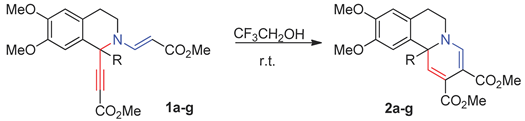
The starting 1-R-1-ethynyl-2-vinyl substituted 1,2,3,4-tetrahydroisoquinolines 1a-g were obtained according to the previously described procedure from the corresponding 3,4-dihydroisoquinolines and methyl propiolate [18]. We initiated our study with transformations of tetrahydroisoquinolines 1a-g to arise in less acidic trifluoroethanol (pKa = 12.4) [19]. To our delight, the conversions did not require elevated temperatures and proceeded smoothly at 20 oС to give to pyrido[2,1-a]isoquinolines in 55-95% yields as sole products (Table 1). To understand what caused the change in the transformation route, the effect of the fluorinated alcohol or simply the presence of a protic solvent we carried out a reaction of isoquinoline 1a in non-fluorinated ethanol (pKa = 15.9) [19]. Substrate 1a was transformed into product 2a but the use of ethanol as a solvent slowed down the process three times, besides, the yield of the target compound decreased to 78%. We have already reported on the synthesis of pyrido[2,1-a]isoquinolines from isoquinolines 1a-f in acetonitrile in the presence of triphenylphosphine [18]. In that case the conversions required more severe conditions, that makes it less attractive compared to the present protocol.
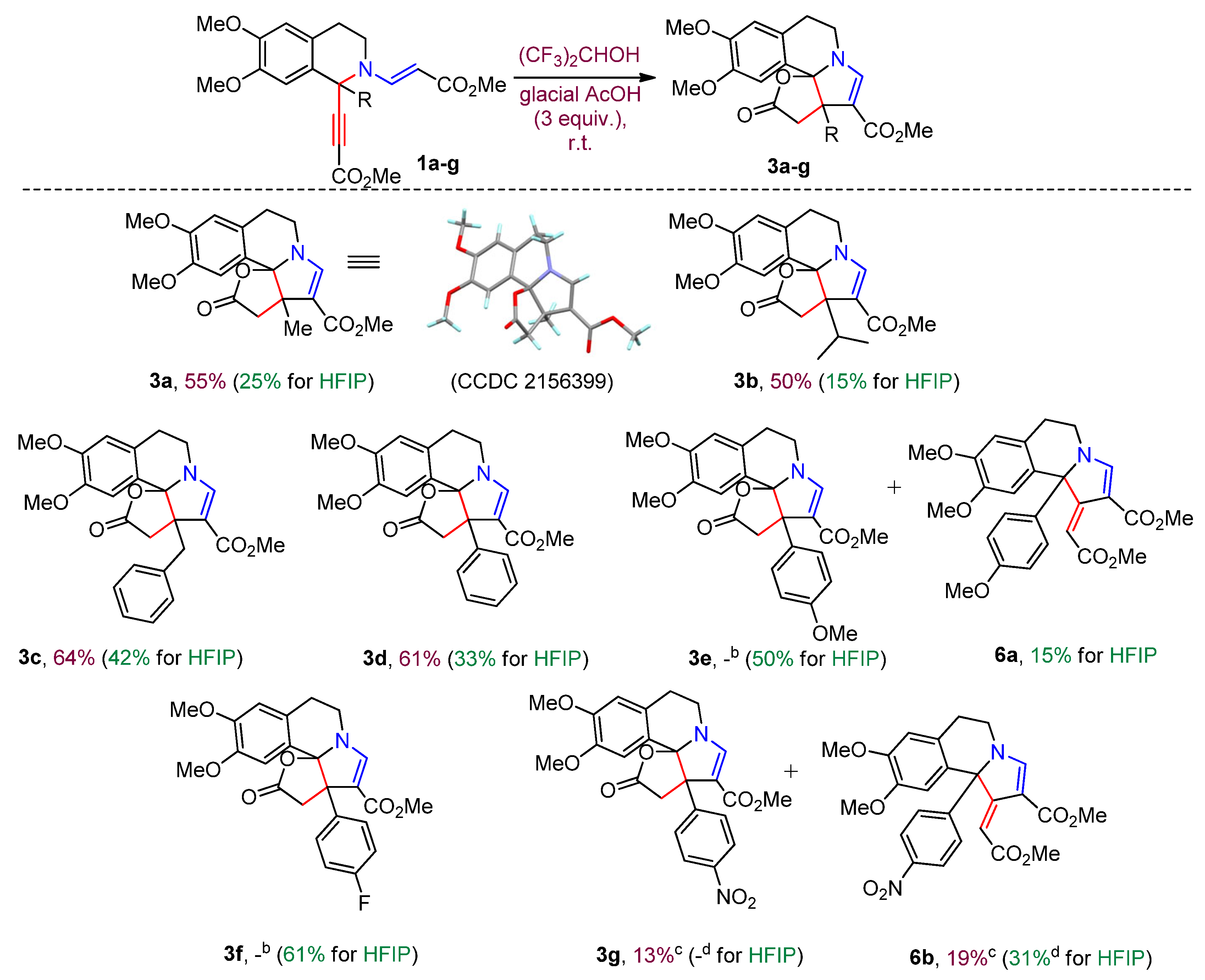
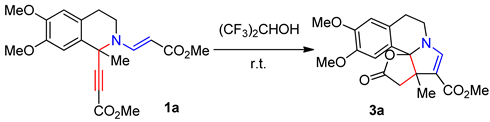
Inspired by the results obtained in trifluoroethanol, we decided to explore the intramolecular changes of starting tetrahydroisoquinolines 1a-g in more acidic hexafluoroisopropanol (HFIP) (pKa = 9.3) [19]. Using isoquinoline 1a as a model substrate we performed a reaction at 20 °C. The transformation proceeded smoothly but, to our surprise, led to a reaction mixture which consists of lactonic pyrrolo[2,1-a]isoquinoline 3a (25%) and pyrido[2,1-a]isoquinoline 2a (71%) (Table 2, entry 1). The formation of 3a was absolutely unexpected. The literature survey has not revealed the analogous structures, we have succeeded only in finding isomeric one [21]. It was obvious that the acidity of the solvent played a key role. Given our earlier published studies demonstrating that the use of more acidic solvents HFIP and AcOH can alter the routes of transformation of 1-R-ethynyl decorated tetrahydroisoquinolines in reaction with activated alkynes towards more thermodynamically stable products, we considered that increasing in the acidity of the medium with acetic acid would promote the construction of product 3a [22,23]. Indeed, the yield of desired 3a was improved to 43% by adding 0.5 equiv of glacial acetic acid; however the formation of compound 2a was still observed (Table 2, entry 2). The best result was achieved with 3.0 equiv of AcOH to furnish lactone 3a in 55% yield. It is noteworthy, that a further increase of acetic acid did not have any significant effect on the yield of the target compound 3a (Table 2, entries 3 and 4).
Having the optimized conditions in hand we investigated the scope of the discovered transformation. To estimate the effect of the substituents attached at C-1 intramolecular changes of tetrahydroisoquinolines 1b-g with different alkyl and aryl substituents were carried out. Isoquinolines 1b-d with isopropyl, benzyl and phenyl groups proved to be good substrates for the transformation producing lactonic pyrrolo[2,1-a]isoquinolines 3b-d in 50-64% yields (Scheme 1). However, the presence of substituents in phenyl radical at C-1 affected both the composition and the ration of reaction mixtures. Thus, isoquinolines 1e-f containing electron-donating substituents (-OMe and -F) in para-position of phenyl ring provided pyrrolo[2,1-b][3]benzazepines 4 and 5, no traces of lactones were observed (Scheme 2). We have already published a paper describing the construction of pyrrolo[2,1-b][3]azepines scaffold via [3,3]-sigmatropic rearrangement in vinyl-, ethynyl substituted di(tetra)hydroisoquinolines [18], but again the present version of the reaction stood out with its simplicity and mild reaction conditions. para-Nitrophenyl substituted isoquinoline 1g gave a mixture of products consisting of pyrido[2,1-a]isoquinoline 2g (47%), 1-ylidene pyrrolo[2,1-a]isoquinoline 6b (19%) and lactone 3g (13%) (Scheme 1).
The structure of 1-ylidene pyrrolo[2,1-a]isoquinoline 6b was assigned on the basis of NOESY, HMQC, and HMBC spectra (Figure 2). The NOESY spectrum has correlations of H-1 to H-3 of the pyrrole cycle as well as its correlation to H-5 and H-10 of the isoquinoline moiety. In the HMBC spectrum there are correlations of H-1 to C-1, C-3, C-10b of pyrrole cycle; to C-5, C-2 of the ester group and to C-6 of the aryl substituent.
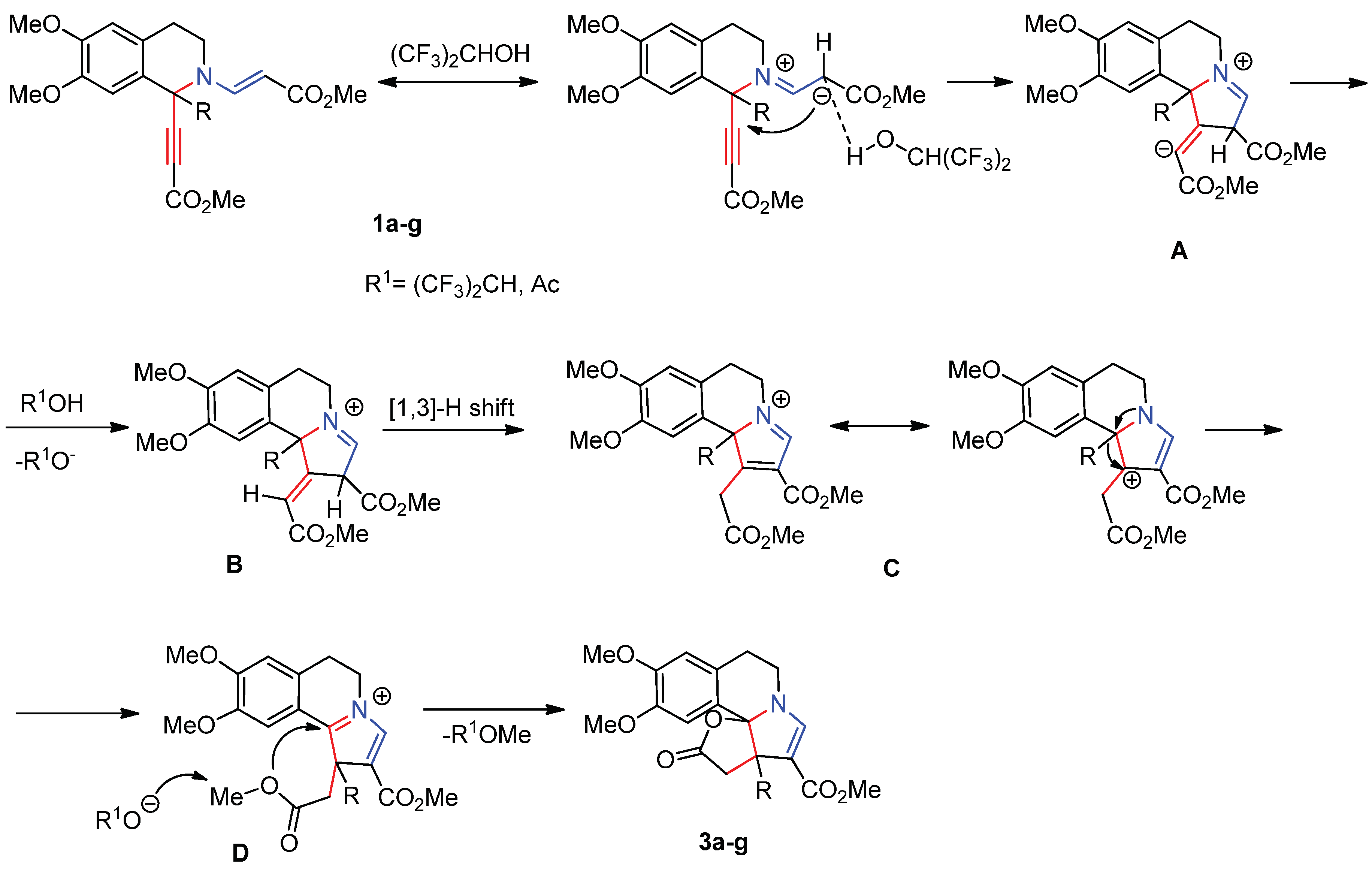
Catalytic routes towards lactones where HFIP facilitates the formation of the product [24,25] are known in the literature. We believe that the transformation commences with the HFIP-assisted polarization of the enamine moiety. The subsequent formation of the pyrrole ring (A) followed by migration of a proton from the solvent to the anionic center of ylidene fragment results into intermediate B. The following [1,3]-shift gives cation C in which a Wagner-Meerwein rearrangement occurs to furnish intermediate D. The final lactonization of the latter leads to target products 3.
Scheme 3.
Plausible Reaction Mechanism for the Formation of furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinolines 3a-g from 1a-g.
Scheme 3.
Plausible Reaction Mechanism for the Formation of furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinolines 3a-g from 1a-g.
An ambiguous behavior of isoquinolines in HFIP in the presence of 3.0 equiv of acetic acid returned us to the idea of carrying out these reactions without any additives. At 20 °C in HFIP isoquinolines 1a-g formed multicomponent mixtures, from which products were isolated using column chromatography. As it was expected in the case of starting compounds 1b-d with isopropyl, benzyl and phenyl substituents the yields of lactones 3 decreased (Scheme 1). But again isoquinolines 1e-g decorated with para-OMe, para-F and para-NO2 phenyl radicals at C-1 stood out of the general scheme. Now isoquinolines 1e-f having electron-donating groups demonstrated the highest yields of desired lactone 3. The formation of lactone 3e was accompanied by formation of product 6a - 1-ylidene substituted pyrrolo[2,1-a]isoquinolines - in 15 % yield (Scheme 1). In the case of isoquinoline 1g with para-NO2 phenyl radical we did not fix the corresponding lactone 3g, from the obtained reaction mixture pyrido[2,1-a]isoquinoline 2g and 1-ylidene-pyrrolo[2,1-a]isoquinoline 6b were isolated in 43% and 31% yields, respectively (Scheme 1).
3. Materials and Methods
3.1. General Information
IR spectra were recorded on an Infralum FT-801 FTIR spectrometer in KBr tablets for crystalline compounds or in a film for amorphous compounds (ISP SB RAS, Novosibirsk, Russia). 1H and 13C NMR spectra were acquired on 600-MHz NMR spectrometer (JEOL Ltd., Tokyo, Japan) in CDCl3 for compounds with a solvent signal as internal standard (7.27 ppm for 1Н nuclei, 77.2 ppm for 13С nuclei); peak positions were given in parts per million (ppm, δ). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), m (multiplet). Coupling constants, J, are reported in Hertz. HRMS spectra were recorded on AB SCIEX TripleTOF 5600+ mass-spectrometer (Singapore) using electrospray ionization (ESI). The measurements were conducted in a positive ion mode mass range from m/z 100 to 1000. A syringe injection was used for solutions in MeOH (concentration 100 ng/mL, flow rate 100 μL/min). Melting points were determined on a SMP-10 apparatus (Bibby Sterilin Ltd., Stone, UK) in open capillary tubes. Sorbfil PTH-AF-A-UF plates (Imid Ltd., Krasnodar, Russia) were used for TLC, visualization in an iodine chamber or using KMnO4 and H2SO4 solutions. Silica gel (40–60 μm, 60 Å) Macherey-Nagel GmbH&Co (Loughborough, UK) was used for column chromatography. All reagents (Sigma-Aldrich, St. Louis, MO, USA; Merck, Darmstadt, Germany; J.T. Baker, Phillipsburg, NJ, USA) were used without additional purification. Compounds 1a-f, 2a-f and 4 were also prepared earlier according to the described procedures [18].
3.2. General Procedure for the Synthesis of Compound 1g
Methyl propiolate (3.0 mmol) was added to the solution of corresponding isoquinoline (1.0 mmol) in 7 ml of CH2Cl2. The reaction was carried out at room temperature. The progress of the reaction was monitored by TLC (Sorbfil, EtOAc-hexane, 1:1). The solvent was removed under reduced pressure, in case of compound 1g the residue was purified by column chromatography on silica gel (1:5 EtOAc – hexane).
Methyl (2E)-3-[6,7-dimethoxy-1-(3-methoxy-3-oxoprop-1-yn-1-yl)-1-(4-nitrophenyl)-3,4-dihydroisoquinolin-2(1H)-yl]prop-2-enoate (1g). Yield 0.397 g (83%), yellow oil. IR spectrum (KBr), υ/сm-1: 2231 (СС), 1717 (C=O), 1519, 1349 (NO2) . 1H NMR (600 MHz, CDCl3) δ 8.23–8.21 (m, 2H, H-Ar), 7.68–7.66 (m, 2H, H-Ar), 7.36 (d, J = 13.6 Hz, 1H, -CHCH-CO2Me), 6.65 (s, 1H, 8-CH), 6.39 (s, 1H, 5-CH), 4.94 (d, J = 13.6 Hz, 1H, -CHCH-CO2Me), 3.88 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 3.66–3.63 (m, 1Н, 3-CH2), 3.62 (s, 3H, OCH3), 3.49–3.46 (m, 1Н, 3-CH2), 3.09–3.05 (m, 1H, 4-CH2), 2.95–2.92 (m, 1H, 4-CH2). 13C NMR (150 MHz, CDCl3) δ 168.9, 153.3, 149.2, 148.8, 148.6, 148.4, 147.9, 128.5 (2С), 127.1, 125.4, 124.2 (2C), 111.1, 111.0, 92.7, 84.6, 80.4, 64.2, 56.1, 56.0, 53.1, 51.0, 42.6, 27.8. HRMS (ESI) m/z calc’d for C25H24N2O8 [M+Н]+ 481.1605, found: 481.1605 (0.0 ppm).
3.3. General Procedure for the Synthesis of Compounds 2a-g
Isoquinolines 1a-g (0.3 mmol) was dissolved in 2,2,2-trifluoroethanol (7 ml). The reaction was carried at room temperature. The progress of the reaction was monitored by TLC (Sorbfil, EtOAc-hexane, 1:1). The solvent was removed under reduced pressure, the residue crystallized from Et2O to give compounds 2a, 2c-f, in case of compounds 2b and 2g the residue was purified by column chromatography on silica gel (1:5 EtOAc – hexane). Yields of 2a-f in 2,2,2-trifluoroethanol: 2a (95%), 2b (55%), 2c (56%), 2d (71%), 2e (79%), 2f (80%). The spectral data of compounds 2a-f are similar to those previously obtained and reported in [18].
Dimethyl 11b-(4-nitrophenyl)-9,10-dimethoxy-7,11b-dihydro-6H-pyrido[2,1-a]isoquinoline-2,3-dicarboxylate (2g). Yield 0.098 g (68%), light yellow oil. IR spectrum (KBr), υ/сm-1: 1688 (C=O), 1519, 1347 (NO2). 1H NMR (600 MHz, CDCl3) δ 8.10 (d, J = 8.8 Hz, 2H, H-Ar), 7.81 (s, 1H, 4-CH), 7.54 (s, 1H, 1-CH), 7.27–7.25 (m, 2H, H-Ar), 7.01 (s, 1H, 11-CH), 6.67 (s, 1H, 8-CH), 3.90 (s, 3H, OCH3), 3.76 (s, 6H, 2*OCH3), 3.60–3.56 (m, 1H, 6-CH2), 3.39 (s, 3H, OCH3), 3.34–3.30 (m, 1H, 6-CH2), 3.00–2.96 (m, 1H, 7-CH2), 2.80–2.77 (m, 1H, 7-CH2). 13C NMR (150 MHz, CDCl3) δ 166.8, 164.2, 161.1, 155.8, 148.9, 147.7, 147.4, 147.1, 129.3 (2C), 126.1, 126.0, 122.7 (2C), 112.2, 111.2, 105.6, 104.0, 78.6, 56.0, 55.6, 51.0, 50.8, 42.2, 29.0. HRMS (ESI) m/z calc’d for C25H24N2O8 [M+Na]+ 503.1425, found: 503.1421 (-0.8 ppm).
3.4. General Procedure for the Synthesis of Compounds 3a-g, 4, 5a,b and 6a,b
A) Isoquinoline 1 (0.3 mmol) was dissolved in 7 ml HFIP. The reaction was carried out at room temperature. The progress of the reaction was monitored by TLC (Sorbfil, EtOAc-hexane, 1:1). The solvent was removed under reduced pressure, the residue were chromatographed on silica gel (1:3 EtOAc – hexane) to obtained compounds 3a-g and 6a,b.
B) To a solution of isoquinoline 1 (0.3 mmol) in 7 ml HFIP glacial AcOH (0.9 mmol) was added. The reaction was carried at room temperature. The progress of the reaction was monitored by TLC (Sorbfil, EtOAc-hexane, 1:1). The solvent was removed under reduced pressure, compounds 3a-g, 4, 5a,b and 6b were chromatographed on silica gel (1:5 EtOAc – hexane (for 4 and 6a,b), 1:3 EtOAc – hexane (for 3a-g, 5a,b)).
Methyl 10,11-dimethoxy-3a-methyl-2-oxo-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3a). Yield 0.059 g (55%), white solid, mp 210-212°C. IR spectrum (KBr), υ/сm-1: 1764, 1680 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.19 (s, 1H, 5-CH), 6.69 (s, 1H, H-Ar), 6.60 (s, 1H, H-Ar), 3.90 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.70 (s, 3H, OCH3), 3.68–3.60 (m, 2H, 7-CH2), 3.52 (d, J = 18.2 Hz, 1H, 3-CH2), 2.90 (d, J = 18.2 Hz, 1H, 3-CH2), 2.90–2.85 (m, 1H, 8-CH2), 2.74–2.70 (m, 1H, 8-CH2), 1.03 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3) δ 174.8, 164.4, 149.9, 148.1, 146.4, 128.9, 122.2, 111.4, 109.1, 108.3, 104.8, 56.2, 55.9, 53.9, 50.6, 42.6, 40.4, 29.6, 21.5. HRMS (ESI) m/z calc’d for C19H21NO6 [M+Н]+ 360.1442, found: 360.1451 (2.5 ppm).
Methyl 10,11-dimethoxy-2-oxo-3a-(propan-2-yl)-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3b). Yield 0.081 g (50%), white solid, mp 237-239°C. IR spectrum (KBr), υ/сm-1: 1751, 1675 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.35 (s, 1H, 5-CH), 6.67 (s, 1H, H-Ar), 6.62 (s, 1H, H-Ar), 3.90 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.79 (d, J = 17.9 Hz, 1H, 3-CH2), 3.68 (s, 3H, OCH3), 3.66–3.64 (m, 2H, 7-CH2), 2.98–2.93 (m, 1H, 8-CH2), 2.95 (d, J = 17.9 Hz, 1H, 3-CH2), 2.75–2.71 (m, 1H, 8-CH2), 1.85–1.79 (m, 1H, CH(CH3)2), 0.98 (d, J = 6.7 Hz, 3H, CH(CH3)2), 0.43 (d, J = 6.7 Hz, 3H, CH(CH3)2). 13C NMR (150 MHz, CDCl3) δ 174.4, 165.1, 150.0, 148.5, 147.9, 128.8, 122.2, 111.3, 109.6, 105.0, 102.0, 60.7, 56.3, 55.9, 50.6, 42.2, 39.5, 34.2, 29.2, 20.3, 16.3. HRMS (ESI) m/z calc’d for C21H25NO6 [M+H]+ 388.1755, found: 388.1765 (2.6 ppm).
Methyl 3a-benzyl-10,11-dimethoxy-2-oxo-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3c). Yield 0.083 g (64%), white solid, mp 218-220°C. IR spectrum (KBr), υ/сm-1: 1762, 1676 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.07 (t, J = 7.6 Hz, 1H, H-Ph), 7.04 (s, 1H, 5-CH), 6.95 (t, J = 7.6 Hz, 2H, H-Ph), 6.71 (s, 1H, H-Ar), 6.59 (s, 1H, H-Ar), 6.25 (d, J = 7.6 Hz, 2H, H-Ph), 3.95 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.65 (d, J = 18.2 Hz, 1H, 3-CH2), 3.46–3.42 (m, 1H, 7-CH2), 3.31 (d, J = 14.1 Hz, 1H, -CH2-Ph), 3.30–3.27 (m, 1H, 7-CH2), 3.07 (d, J = 18.2 Hz, 1H, 3-CH2), 2.65 (d, J = 14.1 Hz, 1H, -CH2-Ph), 2.38–2.34 (m, 1H, 8-CH2), 1.85–1.80 (m, 1H, 8-CH2). 13C NMR (150 MHz, CDCl3) δ 174.2, 164.9, 150.4, 148.5, 147.7, 135.4, 130.2, 130.1 (2C), 127.1 (2C), 126.4, 122.3, 111.4, 109.3, 104.6, 104.0, 57.9, 56.4, 56.2, 50.9, 42.2, 41.1, 39.2, 28.7. HRMS (ESI) m/z calc’d for C25H25NO6 [M+H]+ 436.1755, found: 436.1757 (0.5 ppm).
Methyl 10,11-dimethoxy-2-oxo-3a-phenyl-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3d). Yield 0.077 g (61%), white solid, mp 212-214°C. IR spectrum (KBr), υ/сm-1: 1759, 1679 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H, 5-CH), 7.10–7.08 (m, 2H, H-Ph), 7.06–7.04 (m, 1H, H-Ph), 7.02 (d, J = 7.6 Hz, 2H, H-Ph), 6.53 (s, 1H, H-Ar), 6.14 (s, 1H, H-Ar), 3.85–3.81 (m, 1H, 7-CH2), 3.79 (s, 3H, OCH3), 3.78 (br. d, J = 5.0 Hz, 2H, 3-CH2), 3.75–3.73 (m, 1H, 7-CH2), 3.57 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.04–3.00 (m, 1H, 8-CH2), 2.82–2.79 (m, 1H, 8-CH2). 13C NMR (150 MHz, CDCl3) δ 174.0, 164.0, 149.4, 147.6, 146.7, 138.5, 128.1 (3C), 127.4, 126.3 (2C), 122.5, 110.8, 110.4, 109.3, 105.9, 60.7, 55.8, 55.7, 50.7, 42.2, 37.9, 28.9. HRMS (ESI) m/z calc’d for C24H23NO6 [M+H]+ 422.1598, found: 422.1604 (1.4 ppm).
Methyl 10,11-dimethoxy-3a-(4-methoxyphenyl)-2-oxo-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3e). Yield 0.067 g (50%), light yellow solid, mp 196-198°C. IR spectrum (KBr), υ/сm-1: 1760, 1675 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.34 (s, 1H, 5-CH), 6.92 (d, J = 8.6 Hz, 2H, H-Ar), 6.62 (d, J = 8.6 Hz, 2H, H-Ar), 6.53 (s, 1H, H-Ar), 6.18 (s, 1H, H-Ar), 3.84–3.80 (m, 1H, 7-CH2), 3.81 (s, 3H, OCH3), 3.75 (br. s, 2H, 3-CH2), 3.73-3.71 (m, 1H, 7-CH2), 3.69 (s, 3H, OCH3), 3.58 (s, 6H, 2*OCH3), 3.03–2.98 (m, 1H, 8-CH2), 2.81–2.78 (m, 1H, 8-CH2). 13C NMR (150 MHz, CDCl3) δ 174.1, 164.1, 158.5, 149.4, 147.6, 146.5, 130.5, 128.1, 127.4 (2C), 122.6, 113.5 (2C), 110.8, 110.4, 109.3, 105.8, 60.2, 55.8, 55.7, 55.0, 50.6, 42.3, 38.1, 29.0. HRMS (ESI) m/z calc’d for C25H25NO7 [M+H]+ 452.1704, found: 452.1714 (2.2 ppm).
Methyl 3a-(4-fluorophenyl)-10,11-dimethoxy-2-oxo-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3f). Yield 0.080 g (61%), white solid, mp 206-208°C. IR spectrum (KBr), υ/сm-1: 1771, 1683 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.35 (s, 1H, 5-CH), 6.99–6.97 (m, 2H, H-Ar), 6.80–6.77 (m, 2H, H-Ar), 6.54 (s, 1H, H-Ar), 6.14 (s, 1H, H-Ar), 3.83–3.80 (m, 1H, 7-CH2), 3.81 (s, 3H, OCH3), 3.76 (d, J = 15.7 Hz, 2H, 3-CH2), 3.75–3.71 (m, 1H, 7-CH2), 3.58 (s, 6H, 2*OCH3), 3.03–2.98 (m, 1H, 8-CH2), 2.82–2.79 (m, 1H, 8-CH2). 13C NMR (150 MHz, CDCl3) δ 173.7, 163.9, 161.7 (d, J = 247.1 Hz, 1C), 149.6, 147.7, 146.7, 134.5, 128.2, 128.0 (d, J = 8.1 Hz, 2C), 122.3, 115.1 (d, J = 21.6 Hz, 2C), 110.9, 110.2, 109.2, 105.7, 60.3, 55.9, 55.8, 50.7, 42.2, 38.1, 29.0. HRMS (ESI) m/z calc’d for C24H22FNO6 [M+H]+ 440.1504, found: 440.1500 (-0.9 ppm).
Methyl 10,11-dimethoxy-3a-(4-nitrophenyl)-2-oxo-3,3a,7,8-tetrahydro-2H-furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinoline-4-carboxylate (3g). Yield 0.018 g (13%), yellow solid, mp 147-149°C. IR spectrum (KBr), υ/сm-1: 1774, 1682 (C=O), 1519, 1347 (NO2). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 8.8 Hz, 2H, H-Ar), 7.43 (s, 1H, 5-CH), 7.22 (d, J = 8.8 Hz, 2H, H-Ar), 6.58 (s, 1H, H-Ar), 6.12 (s, 1H, H-Ar), 3.90–3.83 (m, 3H, 3-CH2, 7-CH2), 3.81 (s, 3H, OCH3), 3.78 (d, J = 17.4 Hz, 1H, 3-CH2), 3.58 (s, 3H, OCH3), 3.56 (s, 3H, OCH3), 3.09–3.04 (m, 1H, 8-CH2), 2.89–2.85 (m, 1H, 8-CH2). 13C NMR (150 MHz, CDCl3) δ 172.7, 163.6, 150.3, 148.0, 147.0, 146.9, 145.9, 128.7, 127.4 (2C), 123.4 (2C), 121.4, 111.1 (2С), 108.7 (2С), 60.7, 56.0, 55.8, 50.9, 42.4, 37.8, 28.9. HRMS (ESI) m/z calc’d for C24H22N2O8 [M+H]+ 467.1449, found: 467.1455 (1.3 ppm).
Dimethyl 11-hydroxy-8,9-dimethoxy-11-(4-methoxyphenyl)-6,11-dihydro-5H-pyrrolo[2,1-b][3]benzazepine-1,2-dicarboxylate (5a). Yield 0.079 g (55%), orange oil. IR spectrum (KBr), υ/сm-1: 3521 (OH), 1723, 1709 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.61 (s, 1H, 3-CH), 7.17 (s, 1H, 10-CH), 6.98 (d, J = 8.9 Hz, 2H, H-Ar), 6.77 (d, J = 8.9 Hz, 2H, H-Ar), 6.60 (s, 1H, 7-CH), 4.03–3.99 (m, 1H, 5-CH2), 3.93 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.88–3.84 (m, 1H, 5-CH2), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.64 (s, 1H, OH), 2.98–2.94 (m, 1H, 6-CH2), 2.86–2.82 (m, 1H, 6-CH2). 13C NMR (150 MHz, CDCl3) δ 169.5, 164.0, 159.4, 148.2, 147.5, 138.4, 137.0, 133.9, 128.3 (2C), 127.3, 127.1, 116.8, 113.9 (2C), 113.3, 113.2, 110.6, 77.6, 56.1, 56.0, 55.3, 52.7, 51.4, 48.1, 33.1. HRMS (ESI) m/z calc’d for C26H27NO8 [M+Na]+ 504.1629, found: 504.1641 (2.4 ppm).
Dimethyl 11-(4-fluorophenyl)-11-hydroxy-8,9-dimethoxy-6,11-dihydro-5H-pyrrolo[2,1-b][3]benzazepine-1,2-dicarboxylate (5b). Yield 0.062 g (44%), orange oil. IR spectrum (KBr), υ/сm-1: 3449 (OH), 1715 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.59 (s, 1H, 3-CH), 7.17 (s, 1H, 10-CH), 7.06–7.03 (m, 2H, H-Ar), 6.94–6.91 (m, 2H, H-Ar), 6.61 (s, 1H, 7-CH), 4.02–3.98 (m, 1H, 5-CH2), 3.93 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.87–3.85 (m, 1H, 5-CH2), 3.80 (s, 3H, OCH3), 3.75 (s, 1H, OH), 2.95–2.91 (m, 1H, 6-CH2), 2.87–2.83 (m, 1H, 6-CH2). 13C NMR (150 MHz, CDCl3) δ 169.4, 163.9, 162.2 (d, J = 248.5 Hz, 1C), 148.4, 147.6, 142.0, 136.7, 133.5, 128.9 (d, J = 8.1 Hz, 2C), 127.4, 127.3, 116.9, 115.4 (d, J = 21.6 Hz, 2C), 113.5, 113.3, 110.5, 77.5, 56.1, 56.0, 52.8, 51.5, 48.2, 33.1. HRMS (ESI) m/z calc’d for C25H24FNO7 [M+Na]+ 492.1429, found: 492.1434 (1.0 ppm).
Methyl (1E)-8,9-dimethoxy-1-(2-methoxy-2-oxoethylidene)-10b-(4-methoxyphenyl)-1,5,6,10b-tetrahydropyrrolo[2,1-a]isoquinoline-2-carboxylate (6a). Yield 0.021 g (15%), beige solid, mp 227-229°C. IR spectrum (KBr), υ/сm-1: 1721, 1679 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.31 (s, 1H, 3-CH), 7.21 (d, J = 8.8 Hz, 2H, H-Ar), 6.84 (d, J = 8.8 Hz, 2H, H-Ar), 6.60 (s, 1H, H-Ar), 6.42 (s, 1H, H-Ar), 5.60 (s, 1H, =CH-CO2Me), 3.90 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.74–3.70 (m, 1H, 5-CH2), 3.69 (s, 3H, OCH3), 3.33–3.30 (m, 1H, 5-CH2), 3.12–3.08 (m, 1H, 6-CH2), 2.69–2.66 (m, 1H, 6-CH2). 13C NMR (150 MHz, CDCl3) δ 169.4, 165.6, 159.3, 148.3, 148.2, 146.6, 136.7, 130.5, 129.7 (2C), 129.2, 126.1, 119.8, 113.5 (2C), 111.3, 109.2, 94.3, 64.6, 56.1, 55.9, 55.3, 52.3, 50.9, 47.8, 28.6. HRMS (ESI) m/z calc’d for C26H27NO7 [M+H]+ 466.1860, found: 466.1861 (0.2 ppm).
Methyl (1E)-8,9-dimethoxy-1-(2-methoxy-2-oxoethylidene)-10b-(4-nitrophenyl)-1,5,6,10b-tetrahydropyrrolo[2,1-a]isoquinoline-2-carboxylate (6b). Yield 0.045 g (31%), orange oil. IR spectrum (KBr), υ/сm-1: 1733, 1699 (С=O), 1518, 1349 (NO2). 1H NMR (700 MHz, CDCl3) δ 8.19–8.17 (m, 2H, H-Ar), 7.51–7.49 (m, 2H, H-Ar), 7.32 (s, 1H, 3-CH), 6.65 (s, 1H, H-Ar), 6.35 (s, 1H, H-Ar), 5.59 (s, 1H, =CH-CO2Me), 3.91 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.68–3.64 (m, 1H, 5-CH2), 3.41–3.39 (m, 1H, 5-CH2), 3.16–3.11 (m, 1H, 6-CH2), 2.73–2.70 (m, 1H, 6-CH2). 13C NMR (176 MHz, CDCl3) δ 168.9, 165.2, 150.0, 148.8, 148.6, 147.5, 146.7, 130.6, 129.1 (2C), 128.9, 126.3, 123.6 (2C), 118.7, 111.5, 108.9, 95.7, 64.5, 56.2, 56.0, 52.4, 51.1, 48.0, 28.4. HRMS (ESI) m/z calc’d for C25H24N2O8 [M+H]+ 481.1605, found: 481.1605 (0.0 ppm).
4. Conclusions
In summary, we have described a novel procedure for the synthesis of lactonic pyrrolo[2,1-a]isoquinolines and pyrido[2,1-a]isoquinolines through the rearrangements of 1-R-1-ethynyl-2-vinyl-1,2,3,4-tetrahydroisoquinolines in fluorinated alcohols. It has been demonstrated that the rearrangements depend on the acidity of the used solvents. In some cases the addition of 3 equiv of AcOH increased the yields of the target lactones. The substituent at C-1 in the starting isoquinolines affects the composition and the ration of the products in the transformation occurring in HFIP both with and without AcOH.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.V.V. and A.A.T.; methodology, A.A.T; investigation, A.Y.O; resources, V.B.R.; writing—original draft preparation, A.A.T., A.V.L. and A.Y.O.; writing—review and editing, T.N.B. and A.V.V.; visualization, A.Y.O; supervision, L.G.V. and A.V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and in the Supplementary Materials.
Acknowledgments
This paper has been supported by the RUDN University Strategic Academic Leadership Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hitotsuyanagi, Y.; Fukaya, H.; Takeda, E.; Matsuda, S.; Saishu, Y.; Zhu, S.; Komatsu, K.; Takeya, K. Structures of stemona-amine B and stemona-lactams M–R. Tetrahedron 2013, 69, 6297–6304. [Google Scholar] [CrossRef]
- Xu, Y.; Xiong, L.; Yan, Y.; Sun, D.; Duan, Y.; Li, H.; Chen, L. Alkaloids From Stemona Tuberosa and Their Anti-Inflammatory Activity. Front. Chem. 2022, 10, 847595. [Google Scholar] [CrossRef] [PubMed]
- Nogawa, T.; Kawatani, M.; Uramoto, M.; Okano, A.; Aono, H.; Futamura, Y.; Takahashi, S.; Osada, H. Pyrrolizilactone, a new pyrrolizidinone metabolite produced by a fungus. J. Antibiot. 2013, 66, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Salatino, A.; Salatino, M. L. F.; Negri, G. Traditional uses, chemistry and pharmacology of Croton species (Euphorbiaceae). J. Braz. Chem. Soc. 2007, 18, 11–33. [Google Scholar] [CrossRef]
- Farias, R.; Rao, V.; Viana, G.; Silveira, E.; Maciel, M.; Pino, A. Hypoglycemic Effect of Trans-Dehydrocrotonin, a Nor-Clerodane Diterpene from Croton Cajucara. Planta Med. 1997, 63, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Haun, M. Cytotoxicity of Trans-Dehydrocrotonin from Croton Cajucara on V79 Cells and Rat Hepatocytes. Planta Med. 1999, 65, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Aleman, J.; del Solar, V.; Martin-Santos, C.; Cubo, L.; Ranninger, C. N. Tandem Cyclization–Michael Reaction by Combination of Metal-and Organocatalysis. J. Org. Chem. 2011, 76, 7287–7293. [Google Scholar] [CrossRef] [PubMed]
- Herath, H. B.; Herath, W. H.; Carvalho, P.; Khan, S. I.; Tekwani, B. L.; Duke, S. O.; Tomaso-Peterson, M.; Nanayakkara, N. D. Biologically active tetranorditerpenoids from the fungus Sclerotinia homoeocarpa causal agent of dollar spot in turfgrass. J. Nat. Prod. 2009, 72, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Ivanescu, B.; Miron, A.; Corciova, A. Sesquiterpene lactones from Artemisia genus: Biological activities and methods of analysis. J. Anal. Methods Chem. 2015, 2015. [Google Scholar] [CrossRef]
- Greger, H. Structural classification and biological activities of Stemona alkaloids. Phytochem. Rev. 2019, 18, 463–493. [Google Scholar] [CrossRef]
- Pilli, R. A.; Rosso, G. B.; de Oliveira, M. D. C. F. The chemistry of Stemona alkaloids: An update. Nat. Prod. Rep. 2010, 27, 1908–1937. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tang, M. C.; Tang, S.; Gao, S.; Soliman, S.; Hang, L.; Xu, W.; Ye, T.; Watanabe, K.; Tang, Y. Genome mining and assembly-line biosynthesis of the UCS1025A pyrrolizidinone family of fungal alkaloids. J. Am. Chem. Soc. 2018, 140, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, A.; Rodier, F.; Commeiras, L.; Parrain, J. L.; Chouraqui, G. Construction of spirolactones with concomitant formation of the fused quaternary centre–application to the synthesis of natural products. Nat. Prod. Rep. 2011, 28, 763–782. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, Z.; Li, P.; Miao, T.; Wang, L. Synthesis of Spirolactones via a BF3·Et2O-Promoted Cascade Annulation of α-Keto Acids and 1,3-Enynes. Org. Lett. 2021, 23, 5698–5702. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, M.; Zhu, J.P.; Zhang, X. F.; Liu, Z.J.; Li, H.R.; Chen, Y.; Chen, H.-P.; Zhao, J.; Pu, J.-X.; Yu, M.; Liu, J.-K.; Wu, B. An unexpected photoinduced cyclization to synthesize fully substituted γ-spirolactones via intramolecular hydrogen abstraction with allyl acrylates. Org. Chem. Front. 2022, 9, 2316–2321. [Google Scholar] [CrossRef]
- Nair, D.; Basu, P.; Pati, S.; Baseshankar, K.; Sankara, C. S.; Namboothiri, I. N. Synthesis of Spirolactones and Functionalized Benzofurans via Addition of 3-Sulfonylphthalides to 2-Formylaryl Triflates and Conversion to Benzofuroisocoumarins. J. Org. Chem. 2023, 88, 4519–4527. [Google Scholar] [CrossRef] [PubMed]
- Delayre, B.; Wang, Q.; Zhu, J. Natural product synthesis enabled by domino processes incorporating a 1,2-rearrangement step. ACS Cent. Sci. 2021, 7, 559–569. [Google Scholar] [CrossRef]
- Obydennik, A.Y.; Titov, A.A.; Listratova, A.V.; Borisova, T.N.; Sokolova, I.L.; Rybakov, V.B.; Van der Eycken, E.V.; Voskressensky, L.G.; Varlamov, A.V. Divergent and Nucleophile-Assisted Rearrangement in the Construction of Pyrrolo [2,1-b][3]benzazepine and Pyrido[2,1-a]isoquinoline Scaffolds. Chem.– Eur. J. 2023, e202302919. [Google Scholar] [CrossRef]
- Motiwala, H.F.; Armaly, A.M.; Cacioppo, J.G.; Coombs, T.C.; Koehn, K.R.; Norwood IV, V.M.; Aube, J. HFIP in organic synthesis. Chem. Rev. 2022, 122, 12544–12747. [Google Scholar] [CrossRef]
- Listratova, A. V.; Titov, A. A.; Obydennik, A. Y.; Varlamov, A. V. N-propargyl aza-Claisen rearrangement in the synthesis of heterocycles. Tetrahedron 2022, 121, 132914. [Google Scholar] [CrossRef]
- Lee, J. Y.; Lee, Y. S.; Chung, B. Y.; Park, H. Asymmetric synthesis of both enantiomers of novel tetracyclic heterocycle, furo [3′,2′:2,3]pyrrolo[2,1-a] isoquinoline derivative via a diastereoselective N-acyliminium ion cyclization. Tetrahedron 1997, 53, 2449–2458. [Google Scholar] [CrossRef]
- Titov, A.A.; Kobzev, M.S.; Borisova, T.N.; Listratova, A.V.; Evenko, T.V.; Varlamov, A.V.; Voskressensky, L.G. Facile Methods for the Synthesis of 8-Ylidene-1,2,3,8-tetrahydrobenzazecines. Eur. J. Org. Chem. 2020, 2020, 3041–3049. [Google Scholar] [CrossRef]
- Titov, A.A.; Purgatorio, R.; Obydennik, A.Y.; Listratova, A.V.; Borisova, T.N.; De Candia, M.; Catto, M.; Altomare, C.D.; Varlamov, A.V.; Voskressensky, L.G. Synthesis of Isomeric 3-Benzazecines Decorated with Endocyclic Allene Moiety and Exocyclic Conjugated Double Bond and Evaluation of Their Anticholinesterase Activity. Molecules 2022, 27, 6276. [Google Scholar] [CrossRef] [PubMed]
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Call, A.; Cianfanelli, M.; Besalú-Sala, P.; Olivo, G.; Palone, A.; Vicens, L.; Ribas, X.; Luis, J. M.; Bietti, M.; Costas, M. Carboxylic Acid Directed γ-Lactonization of Unactivated Primary C–H Bonds Catalyzed by Mn Complexes: Application to Stereoselective Natural Product Diversification. J. Am. Chem. Soc. 2022, 144, 19542–19558. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 3a-g in HFIP in the presence of glacial AcOH. a Reaction conditions: A mixture of 1a-g (0.3 mmol), glacial AcOH (0.9 mmol, 3.0 equiv) in HFIP (7.0 ml) was stirred at rt. b Formation of pyrrolo[2,1-b][3]benzazepines 4, 5 instead furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinolines 3e-f. c Pyrido[2,1-a]isoquinoline 2g (47%) and 1-ylidene-pyrrolo[2,1-a]isoquinoline 6b (19%) were isolated in addition of 3g. d Formation of mixture pyrido[2,1-a]isoquinoline 2g (43%) and 1-ylidene- pyrrolo[2,1-a]isoquinoline 6b (31%).
Scheme 1.
Synthesis of 3a-g in HFIP in the presence of glacial AcOH. a Reaction conditions: A mixture of 1a-g (0.3 mmol), glacial AcOH (0.9 mmol, 3.0 equiv) in HFIP (7.0 ml) was stirred at rt. b Formation of pyrrolo[2,1-b][3]benzazepines 4, 5 instead furo[2’,3’:2,3]pyrrolo[2,1-a]isoquinolines 3e-f. c Pyrido[2,1-a]isoquinoline 2g (47%) and 1-ylidene-pyrrolo[2,1-a]isoquinoline 6b (19%) were isolated in addition of 3g. d Formation of mixture pyrido[2,1-a]isoquinoline 2g (43%) and 1-ylidene- pyrrolo[2,1-a]isoquinoline 6b (31%).
Scheme 2.
Transformations of isoquinolines 1e-f.
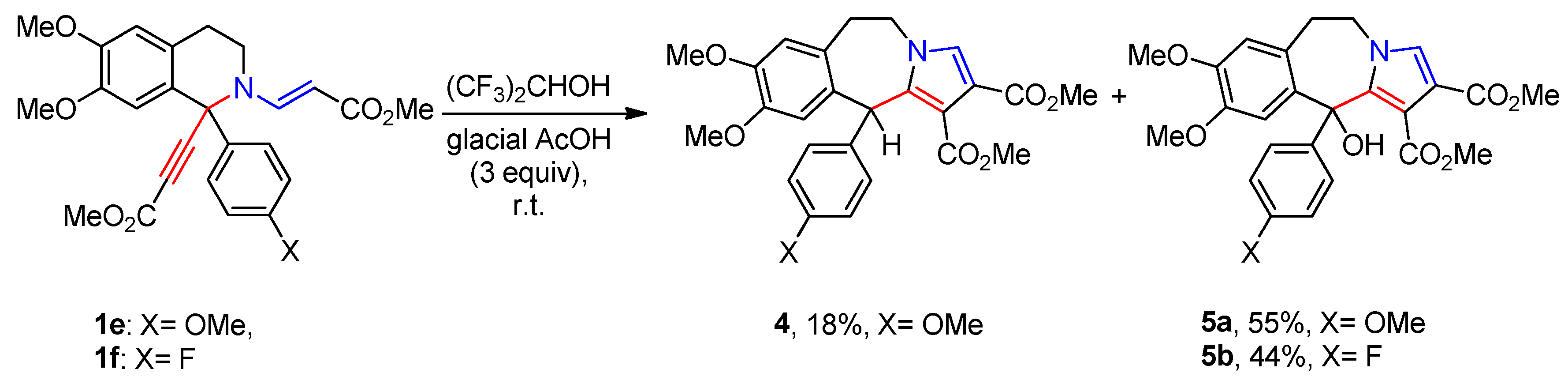
Figure 2.
Results of correlation spectra of compound 6b.
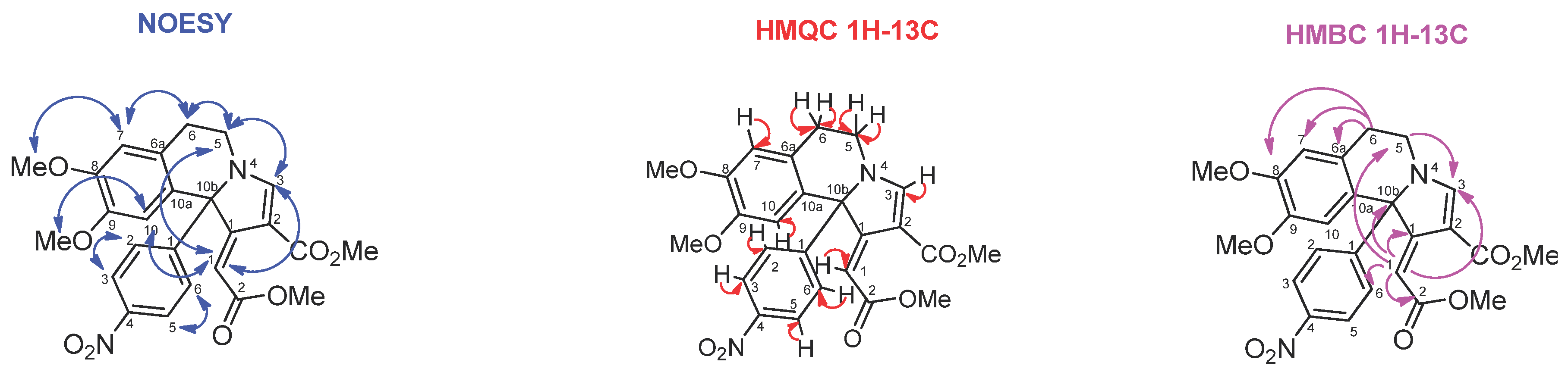

Table 1.
Synthesis of pyrido[2,1-a]isoquinolines 2a-g.
| Entry | R | Product | Yield, % |
| 1 | Me | 2a | 95 a |
| 2 | i-Pr | 2b | 55 |
| 3 | Bn | 2c | 56 |
| 4 | Ph | 2d | 71 |
| 5 | 4-OMe-C6H4- | 2e | 79 |
| 6 | 4-F-C6H4- | 2f | 80 |
| 7 | 4-NO2-C6H4- | 2g | 68 |
a 78% for reaction in C2H5OH.
Table 2.
Optimization of the Reaction Conditions.
| Entry | glacial AcOH (equiv.) | Yield 3a, % | Yield 2a, % |
| 1 | - | 25 | 71 |
| 2 | 0.5 | 43 | 43 |
| 3 | 3.0 | 55 | - a |
| 4 | 5.0 | 56 | - a |
a no traces of pyridoisoquinoline.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



