Submitted:
29 December 2023
Posted:
30 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
COVID-19 was an inflammatory pneumonia caused by a respiratory infection with a coronavirus named SARS-CoV-2. Severely ill COVID-19 patients could die of acute pulmonary and systemic inflammation. Thus, there was a theory that long COVID-19 shared many similarities with systemic autoimmune diseases. There was also a theory that the SARS-CoV-2 virus could infect many cell- and tissue-types distributed throughout multiple organ systems of the body. I aim to review the above theories using unbiased high-throughput datasets cataloguing gene expression. Studies reviewed used the technology of next generation sequencing, either as bulk tissue RNA sequencing, or as single-cell RNAs sequencing; ingenious methods for signal deconvolution were then used to identify individual cell types. Datasets reviewed suggested that severe COVID-19 induced expression of genes associated with pro-inflammatory signaling, tissue development or remodeling, scarring, or cell death. The datasets also provided evidence for several inflammatory syndromes associated with COVID-19, such as neuronal COVID, acute respiratory disease syndrome, vascular inflammation, or multisystem inflammatory syndrome. Indeed, inflammatory signal during severe COVID-19 was reported in many cell types, for example in structural cells, in infiltrating immune cells, in the vascular system, or in circulating cells in the blood. There was also frequently a change in proportions of immune cell-types in the blood, epithelial cells in the lungs, or among infiltrating immune cells.
Keywords:
COVID-19
; SARS-CoV-2
; transcriptomics
; RNA-seq
; scRNA-seq
; inflammation
; pneumonia
; neuronal COVID
; acute respiratory disease syndrome
; multisystem inflammatory syndrome
1. Introduction
Coronavirus Disease 2019 — COVID-19 — was a novel type of pneumonia caused by severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2). The disease rapidly emerged in Wuhan in the year 2019, and it soon spread throughout the world. This unusual disease was described one year later in 2020. In particular, its respiratory component — SARS and acute respiratory disease syndrome (ARDS) — were then characterized [1,2,3,4,5]. It was also established that the infectious agent for COVID-19 was SARS-CoV-2 — a coronavirus of probable zoonotic origin [6]. Confusingly, COVID-19 manifested an extremely wide spectrum of clinical symptoms. While most patients have mild or moderate illness, around 15% of patients progress in severity and length of the disease so much that they required hospitalization. The fatality rate in such hospitalized patients was many times higher than that of influenza [7]. Long, or severe, COVID-19 [8] was a severe and prolonged form of the disease [9]. Long COVID-19 probably led to inflammatory deterioration in function of multiple organs, including lungs, heart, gastrointestinal tract, neurological system, blood vessels, and the reproductive system. In most severe cases, patients developed ARDS, which required mechanical ventilation. Respiratory failure, sepsis, acute cardiac injury, or heart failure tended to be direct causes of death from ARDS. Thus, etiology of COVID-19 was complex, but it could be elucidated herein using unbiased genomics datasets.
Natural history of severe COVID-19 should be introduced now. The SARS-CoV-2 virus would first infect upper airways replicating in ciliated epithelial cells and inducing inflammation therein. This tended to cause sore throat, fever, and coughing. Next, the virus would strike at lungs causing pneumonia. There was severe pulmonary and systemic inflammation, which tended to cause gross damage to lung tissue. For example, alveoli would harden, inflame and fail at their role in gas exchange. Indeed, there was fibrotic scarring, lack of respiratory surfactant, and a leaky state in the lung. Moreover, coagulopathy, endotheliopathy, and vasculitis could result from the damage to the EC layer in the lungs and then spread throughout the body [10]. Thus, SARS-CoV-2 impacted negatively on many cell- and tissue-types throughout all physiological systems of the body [11]. Most importantly, the virus appeared to infect type II alveolar cells (AT2 cells) located in tiny air sacs at the ends of air tubes of the lungs. In fact, coronavirus particles could be observed in these cells using electron microscopy, and it was obvious that infected AT2 cells were damaged through senescence [12]. However, there was also ample evidence that SARS-CoV-2 attacked ECs. For example, the coronavirus could infect and damage ECs in vivo [13]. Moreover, it was shown that SARS-CoV-2 could infect and damage human umbilical vein ECs — HUVECs [14], as well as ECs in an aortic ring assay [15]. However, only single-cell genomics could provide unbiased evidence about cell types affected by SARS-CoV-2.
Based on traditional biochemical and molecular markers, it was suggested that long COVID-19 appeared related to autoimmune diseases [16]. Indeed, patients with pre-existing inflammatory or autoimmune conditions such as cancer, obesity, hypertension, cardiovascular disease, type 2 diabetes, or connective tissue disorders were at a higher risk of a fatal outcome if autoantibodies were present. One could hypothesize that the body produced high concentrations of autoantibodies to many host proteins in response to a significant viral load in many tissues. The antibodies then attacked host proteins distributed across many tissues, which resulted in a state of systemic inflammation. Indeed, presence of autoantibodies, vascular inflammation, and an inflammatory state of endothelial cells (ECs) were typical systemic symptoms of long COVID-19 [17,18]. As these changes occur over several weeks — that is times-scales suggesting transcriptional and epigenetic changes, I will review these hypotheses using unbiased genomic datasets.
Note that the genome of SARS-CoV-2 was published early in 2020 [3]. It was deposited in GenBank with a RefSeq accession number NC_045512.2. It was a single-strand ribonucleic acid (RNA) genome consisting of 29,903 base pairs. There were 11 protein-coding genes in the genome. For example, there was a multiunit replicase and a multiunit protease, and four structural proteins. Among these, protein S was a viral surface glycoprotein, which could bind to a mammalian receptor for the angiotensin-converting enzyme 2 (ACE2). Note that ACE2 could be found attached to cell membranes in the lungs, the small intestine, arteries or veins [19], but it is not generally a very abundant protein. Protein E was an envelope protein. Protein M was a membrane glycoprotein. Protein N was a phosphoprotein of the nucleocapsid. There may be additional open reading frames (ORFs), probably non-coding, which could have a regulatory role [20]. There were also at least 41 RNA modification sites on the viral RNAs [21]. Viral transcripts were typically expressed at very high levels, dominating the expression of host genes [20]. Somewhat surprisingly, the genome of the virus also encoded a furin cleavage site, which was absent from its closest known phylogenetic relatives. The furin site might enhance the tropism of the virus towards a greater number of tissue- and cell-types [22,23]. Therefore, we had sufficient genomic information about SARS-CoV-2 to investigate COVID-19 using functional genomics as early as 2020.
Herein, I aimed to review etiology of COVID-19 [24] using unbiased high-throughput functional genomics datasets. I was aided in my task by a high number and high quality of relevant genomics datasets in public databases. For example, there are hundreds of gene expression datasets relevant to COVID-19 in Gene Expression Omnibus (see Table 1). Next generation sequencing (NGS) was typically performed on messenger RNAs (mRNAs) isolated from either in vitro cell cultures or from bulk tissue samples.
2. Unbiased evidence from functional genomic studies of tissues and cells focusing on the etiology of COVID-19.
To start with, I discuss 11 whole-tissue RNA-sequencing (RNA-seq) studies focusing on the etiology of COVID-19 listed in Table 2. Bulk-sample examples were discussed below and listed in Table 2, focusing on inflammatory changes induced in peripheral blood mononuclear cells — PBMCs [25,26,27,28] during severe COVID-19, as well as within lung cells / airways [29,30,31], or in pancreatic islets [32], or within cerebrospinal fluid — CSF [33], or in conjunctival epithelium [34].
Next, I introduce 16 single-cell RNA-seq (scRNA-seq) studies, listed in Table 3, which also aim at elucidating the etiology of COVID-19 (but discriminating between individual cells). These scRNA-seq studies focused on PBMCs [35,36,37,38,39,40,41,42,43], the broncho-alveolar lavage fluid — BALF [44,45], brain [46], lungs and lung epithelium [32,47,48], heart [47], liver [47], vascular endothelium [48], or kidneys [47].
Additionally, eight meta-analyses were reviewed (see Table 4). Five of these focused on gene expression of ACE2 in scRNA-seq datasets from the lungs or airways [49,50,51,52], or kidneys [53]. Another meta-analysis built a reference library for immune cells in normal body and in different diseases [54]. Other authors looked at similarities in gene expression between COVID-19 and cancer [55]. Finally, there was also a meta-analysis of 9 scRNA-seq studies focusing on PBMCs [56].
2.1. An in vitro study in a cell line infected with SARS-CoV-2.
As early as 2020, Blanco-Melo et al. profiled transcriptomes of immortalized human cancer cell lines infected with SARS-CoV-2 and few other common respiratory viruses (such as a strain of influenza). Note that the cancer cell lines were isolated from lung tumors [29]. In short, the cells responded to SARS-CoV-2 with an inflammatory gene expression program. For example, levels of expression of chemokines were relatively high in the SARS-CoV-2 group, while levels of interferon type I and III were relatively low. Thus, these studies suggested that suboptimal transcription of antiviral countermeasures (such as interferons, inflammatory cytokines, and chemokines) played a role in the etiology of COVID-19.
In another in vitro study, Müller et al. [57] investigated islet cells from four deceased COVID-19 patients. The authors were prompted by the discovery that proteins of the virus could be detected in pancreatic tissue in the vicinity of the islets of Langerhans. Their goal was to establish whether SARS-CoV-2 could infect pancreas causing long-term damage to β-cells (potentially resulting in diabetes). An answer obtained was generally positive.
2.2. Examples of in vivo transcriptional responses in bulk tissue samples exposed to SARS-CoV-2.
The in vitro studies described above were deficient in evidence about organismal context in which a viral infection occurred. For example, status of the immune system could not be known. Neither could we know interactions between the virus and hundreds of cell types in many tissues of the body. Instead, Daamen et al. [31] followed an alternative and advantageous approach, in which they focused on in vivo activities of immune cells. Specifically, they profiled gene expression in blood samples from hospitalized patients with COVID-19. To be precise, the authors performed bulk RNA-seq on clinical samples of PBMCs. Additionally, postmortem lung tissue and postmortem samples from diseased airways were processed. Daamen et al. interpreted resulting COVID-19 datasets by discussing the importance of the expression of immunological genes. For example, many genes associated with innate immune responses were increased in expression in infected tissues. Specifically, type I interferon and other genes important for anti-viral immunity were increased in their mRNA levels. In contrast, gene expression signatures of adaptive immune response tended to be decreased.
Interestingly, not only structural cell types like epithelia, but also infiltrating immune cells were the source of transcriptional signal related to COVID-19. For example, Daamen et al. detected populations of myeloid-like cells with high inflammatory transcriptional signatures. Daamen et al. also observed that there were insufficient numbers of activated natural killer (NK) cells in diseased samples. This deficiency could prevent efficient clearing of diseased virus-laden cells. Note also that there were also insufficient numbers of regulatory, that is cluster of differentiation 8 positive (CD8+) T cells in tissues from COVID-19 patients (these cells normally mediate adaptive immunity).
2.3. Examples of RNA-seq studies of individual cells in COVID-19.
A significant advantage of single-cell analysis — scRNA-seq [58] — was in ability to identify changes in cell proportions as well as DEGs gene-by-gene and cell-by-cell. Another strategic advantage was that one could describe heterogeneity of gene expression across many cells. Moreover, viruses generally attack tissues such that infected cells are mixed with healthy ones. Recall also that there were many different cell types in each infected tissue (such as ECs, stromal cells, infiltrating immune cells, or epithelial cells). Moreover, recall that the infection was a dynamic process, and it took place in the context of co-existing pathologies (such as cancer, atherosclerosis, dementia, or chronic autoimmune disorders).
2.3.1. Expression profiling of single cells in the immune system.
Individual cells of the immune system, either circulating in the blood or present in body fluids, were analyzed to verify whether and how COVID-19 was a disease of systemic inflammation (see studies listed in Table 3). In the first of examples, Chua et al. [59] performed scRNA-seq of nasopharyngeal and bronchial samples from 19 well-characterized COVID-19 patients with either moderate or critical disease (there were also 5 healthy controls). Major epithelial cell types were identified including basal, secretory and ciliated cells, as well as FOXN4+ cells and ionocytes. Moreover, there was a subpopulation of epithelial cells with a strong interferon gamma response. There were also 13 different cell types or states of immune cells, including macrophages, dendritic cells, mast cells, neutrophils, B cells, T cells, or NK cells. In critical cases of COVID-19, there were stronger inflammatory interactions between immune and epithelial cells in comparison to moderate cases. The inflammatory interactions were identified through expression profiling of ligand–receptor pairs in both epithelial and immune cells (these were inferred from the CellPhoneDB database). These inflammatory interactions led to damage in respiratory tissues and correlated with COVID-19 severity. In other words, even more lung injury, respiratory failure, and inflammatory tissue damage was observed in critical cases. Additionally, epithelial cells from COVID-19 patients had three times greater expression of ACE2: an entry receptor for the SARS-CoV-2 virus, which correlated with interferon signalling among immune cells. In comparison to moderate cases, critical cases had inflammatory expression profiles of ligand-receptor pairs, for example inflammatory macrophages expressing many potent chemokines, chemokine ligands, interleukin 8 (IL8), interleukin -1 beta (IL1B), and tumor necrosis factor (TNF).
Similar conclusions were reached by Zhu et al. who extensively sequenced individual transcriptomes of PBMCs [60]. Five COVID-19 patients and three healthy people donated their blood for isolation of PBMCs. An atlas of single-cell gene expression was generated in both COVID-19 and influenza patients. For example, three signaling pathways were turned on in COVID-19: the apoptosis pathway, signal transducer and activator of transcription 1 (STAT1), and interferon regulatory factor 3 (IRF3) pathway. Specifically, three different mechanisms of apoptosis were more active in T cells in COVID-19: X-linked inhibitor of apoptosis (XIAP)-associated factor 1 (XAF1) pathway, TNF, and Fas receptor pathway. In influenza, signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa-light-chain enhancer of activated B cells (NF-kappa-beta) tended to be active instead of STAT1 / IRF3. According to the results of the authors, there was also an increase in the number of fully differentiated, i.e. producing specific antibodies, B cells among the PBMCs in COVID-19. There was also a decrease in the number of lymphocytes, and this effect was mediated through the apoptosis pathway. Furthermore, individual up-regulated DEGs tended to encode pro-inflammatory cytokines, cytokine receptors, or interferon-responsive transcription factors. Overall, the study suggested that the transcriptional response in immune cells during COVID-19 was both diminished in magnitude and pro-inflammatory.
Note that a similar study was performed by Wilk et al. [27] who sequenced transcriptomes of PBMCs from 7 hospitalized patients with COVID-19, four of whom had ARDS, as well as six health controls. 44,721 cells were sequenced with an average cell number of 3,194 per sample. Uniform manifold approximation and projection (UMAP) identified 30 cell clusters and DEGs were calculated for each cluster. Large phenotypic differences were identified between COVID-19 patients and controls, in cell populations of monocytes, T cells and NK cells. There were also COVID-19-related changes in cell proportions. There were also several cell types that seemed depleted in COVID-19, namely a subtype of T-cells, conventional dendritic cells (DCs), plasmacytoid dendritic cells (pDCs), CD16+ monocytes and NK cells. Note that in patients with ARDS only DCs, CD16+ monocytes and NKs were only significantly depleted. An expanding population of plasmablasts and developing neutrophils was also detected: levels of these cells were most elevated in patients with ARDS.
Another set of authors applied scRNA-seq of PBMCs to compare immune cells in the blood in even further detail. There were 57,669 high-quality transcriptomes sequenced and assembled for 7 COVID-19 patients compared against 5 healthy controls [25]. As many as 25 immune cell subsets were identified using UMAP in resulting NGS data. A few subsets of PBMCs were primarily identified in patients with COVID-19, for example monocytes and T cells with high expression of interferon-stimulated genes including interferon-alpha inducible protein 27 (IFI27), IFITM3, or ISG15. Moreover, DEGs were determined between analogous subsets in the COVID-19 group versus control. In these DEGs, there were antiviral pathways, which were induced in the COVID-19 group in monocytes and dendritic cells. Special attention was paid to the expression of interferons, however, only modest levels of interferon-gamma expression in T and NK cells were detected. Thus, a comprehensive atlas of transcriptional immune modulations in PBMCs in COVID-19 was presented. It was also confirmed that very high levels of inflammatory cytokines in COVID-19 led to severe autoimmune disease (resulting in shock, multiple organ failure, or respiratory failure).
Bernardes et al. also profiled immune responses in PBMCs, but they explored disease trajectories chronologically in a longitudinal approach [26]. The analysis included scRNA-seq data for 358,930 cells with 10,900 cells on average per sample. There were up to four longitudinal samples per patient. The goal was to identify dynamic changes in expression, which could be correlated with disease severity in COVID-19. This goal was achieved by extensive bioinformatics analyses of whole-blood scRNA-seq data along five different time points. Following sequencing, DEGs, functional trends, networks of transcription factors, and co-expression modules were identified. Generally, the observed set of DEGs suggested that there was a transcription factor activity related to inflammation and a signature of interferon signaling in COVID-19. However, interferon response seemed to be defective in critical cases. Moreover, severe COVID-19 was associated with hypoxia and hypoxic signaling (strongly increasing erythropoiesis), as well as genome-wide hypomethylation. There was an increase in the expression of genes for a pathway linked with platelet production when survivors were compared with non-survivors. There was also an induction of pro-inflammatory cytokines in the non-survivor group. Finally, increased numbers of plasmablasts, erythroid cells, and interferon-activated megakaryocytes were found to be characteristic of severe disease. When survivors were compared with non-survivors, there were 16 TFs that were differentially regulated in non-survivors only, and 7 TFs in the opposing group. Pathway analysis using the Reactome database suggested significant enrichment for the following two terms in the non-survivor group: megakaryocytes development and platelet production, and TRAF6-mediated induction of pro-inflammatory cytokines.
Another study compared cytotoxic T-cells in a severely ill group with those in a moderately affected group [61]. In the first step, RNA libraries were constructed each representing individual cytotoxic T-cells. In the second step, single-cell sequencing was performed. As a result of the sequencing, the M protein of SARS-CoV-2 was identified as a frequent target of the cytotoxic T-cell receptor (TCR). Specifically, the M198–206 amino-acid sequence was identified as an important epitope. In fact, 81.1% of the libraries, i.e. 30 out of 37, were identified as responding to this epitope. Further downstream in their analysis, Ogura et al. identified DEGs in a comparison between populations of mature cytotoxic T cells with those that were naive. For example, granzyme A and B (GZMA and GZMB) were serine proteases, which were crucial for induction of apoptosis in a cell that was attacked with cytotoxicity. Overall, the authors concluded that T cells in after severe COVID were less capable of a cytotoxic response than those in moderate COVID. However, a similar study by Meckiff et al. [39] found more cytotoxic T cells among PBMCs in hospitalized patients with COVID-19.
In another example [28], NGS sequencing was performed on single-cell transcriptomes of PBMCs from ten moderate, six critical, and five fatal cases of SARS-CoV-2. To be precise, 57,049 single-cell transcriptomes of PBMCs were sequenced. The authors identified eight functional clusters of cells among the PBMCs: (1) CD4+ T cells, (2) CD8+ T cells, (3) B cells, (4) plasma cells, (5) NKs, (6) conventional dendritic cells, (7) canonical monocytes, and (8) non-canonical monocytes. Significantly, there were greater proportions of transcriptomes derived from myeloid cells in PBMCs of fatal COVID-19 cases (in comparison with moderate cases). These myeloid cells (i.e. proliferating bone marrow cells) were characterized by biased up-regulation of a platelet-activating signature. The authors also concluded that endothelial injury and pathological thrombotic events were common in COVID-19 (and that these events were positively correlated in frequency with activity of myeloid cells). Note that two additional and related studies of PBMCs also suggested that subtle changes in proportions of myeloid cells correlated with severity of COVID-19 [62,63].
Next, Sinha et al. [64] looked at the effects of the administration of dexamethasone on circulating neutrophils during severe COVID-19. At that time, dexamethasone became a standard treatment for COVID-19, and it was given either oral or intravenous at a dose of 6 mg once daily. A total of 15 thousand single cells, which contained an equal proportion of leukocytes and lymphocytes, were analyzed. Dexamethasone expanded immunosuppressive properties of neutrophils and changed them functionally from information receivers into information providers.
In another example focusing on immune cells within the respiratory system, Bost et al. [44] performed scRNA-seq of immune cells isolated from BALF. The patients had either severe or mild COVID-19. In parallel, the authors developed a method called Viral-Track to computationally interpret their datasets. Viral-Track was a robust, unsupervised, bioinformatics method that could detect viral RNAs in large scRNA-seq datasets. The method was benchmarked on NGS datasets from virus-infected tissues. Using Viral-Track, Bost et al. compared immune cells from severe cases with those from mild cases of COVID-19. Deep impact of the virus on the immune system was detected in severe cases. For example, the authors detected marked differences in proportions of different subtypes of immune cell: either myeloid, or lymphoid, or epithelial. There was also an increase in alveolar macrophages and plasmacytoid dendritic cells (pDCs) in BALF isolated from bronchia in mild cases of COVID-19. However, tissue-resident alveolar macrophages were replaced with recruited macrophages, monocytes, or neutrophils in severe cases.
Another study by Heming et al. [33] focused on immune cells from CSF of patients who experienced heterogenous neurological symptoms during COVID-19 (this was called neuro-COVID). For example, patients with neuro-COVID could develop headache, dizziness, cognitive abnormalities, seizures, encephalitis, stroke, or brain hemorrhage. There was also an increased number of de-differentiated monocytes and exhausted cluster of differentiation 4 positive (CD4+) T cells in neuro-COVID. (Note that exhausted T cells arose because of chronic over-stimulation in a site of active inflammation.) Significantly, the exhausted T cells had diminished effector functions, and expressed co-inhibitory receptors. Finally, there was a reduced interferon response in neuro-COVID in comparison to viral encephalitis.
Another study focused on young individuals with multisystem inflammatory syndrome (MIS-C) following COVID-19 [41]. Note that MIS-C was a very dangerous inflammatory condition in which internal and external body parts became inflamed. This inflammation could be so pervasive throughout the body that it could include the lungs, heart, brain, eyes, kidneys, or the tissues of the gastrointestinal tract. Inflammatory symptoms of MIS-C could also include a cytokine storm or fever. Following scRNA-seq, transcriptomes of PBMCs were fractioned computationally using UMAP into 30 different subpopulations of immune cells. Subsequently, DEGs were identified for each of the subpopulations of PBMCs (in a comparison between cases of MIS-C and healthy controls). The goal was to identify gene markers allowing us to better diagnose and predict severity of MIS-C. For example, MIS-C patients had higher expression of S100A-family alarmins, as well as decreased gene expression signatures characteristic of antigen presentation. Finally, there was also an elevated expression level of cytotoxicity genes in NKs and CD8+ T cells.
On the other hand, Su et al. [65] investigated the etiology of post-acute sequelae of COVID-19 (PASC), using scRNA-seq and several other multi-omics technologies. This study focused on PBMCs, and it had a long follow-up period. The study found that there were four risk factors associated with the diagnosis of PASC: (1) type 2 diabetes, (2) high SARS-CoV-2 load in the plasma, (3) Epstein-Barr viremia, (4) and the presence of specific autoantibodies.
2.3.2. Expression profiling of the lung tissue.
As patients with severe COVID-19 die of pulmonary inflammation and ARDS, scRNA-seq on samples from diseased lungs were also necessary for the understanding of the etiology of COVID-19. For example, Melms et al. [30] provided a comprehensive census of lung cellular signaling and differentiation states in lethal COVID-19. They noted that individuals with COVID-19 had highly inflamed lungs with marked infiltration of activated macrophages, but also with diminished T cell responses. Impaired activities of T cells were observed, and these were likely to have contributed to fatal outcomes. Myeloid cells were also an important source of dysregulated inflammation, being more prevalent in diseased lungs in comparison to lungs from the control group. Moreover, there was significant lung fibrosis in COVID-19, and it was correlated with the length of the duration of the disease. Indeed, numerous pathological fibroblasts were observed in disorganized tissue regions. A reduction in the epithelial cell compartment was also observed, because there were fewer alveolar type I (AT1) and alveolar type II (AT2) cells in inflamed lungs, impairing their regeneration.
Another study of multiple organs led to similar conclusions about inflammation in COVID-19. Specifically, Delorey et al. generated an extensive single-cell RNA atlas from the lungs, kidneys, livers, and hearts of dead COVID-19 patients. Following transcriptional profiling, integrated computational analysis suggested that diseased lungs were characterized by extensive inflammatory damage and failed regeneration. There was evidence of such damage in cells from patients, with and without viral RNAs. Epithelial progenitors failed to regenerate sufficiently to prevent a reduction in the numbers of AT2 cells. Moreover, gene pathways that were increased in transcriptional output in COVID-19 included: apoptosis linked with oxidative stress in pericytes, various immune pathways, cell adhesion pathways, as well as the pathway for fibroblast differentiation.
2.3.3. Expression profiling of the brain, ocular epithelia, and the vasculature.
The NGS or scRNA-seq approaches were already applied in 2021 to profile brain tissues infected with SARS-CoV-2 [46]. Cells from brains of eight patients ill with COVID-19 were compared with cells obtained from 14 healthy controls. The cause of death for nearly all patients was pneumonia with extreme inflammation, which followed more than fourteen days of mechanical ventilation. The authors discovered that microglia and astrocyte subpopulations in the brain resembled those found in neurodegenerative disease. Moreover, the study was successful in linking genetic susceptibility to COVID-19 with genetic susceptibility to neurological degeneration (for example, with genes for impaired cognition, schizophrenia, or depression). In conclusion, the study found a considerable amount of evidence that long-term inflammation is a part of brain-linked COVID-19, as if inflammation from the lung tissue was relayed to the brain [46].
In another example, Jackson et al. [34] applied scRNA-seq to an in vitro model of human ocular epithelia infected with the virus. In their experiment, ocular epithelial cells were isolated from eyes donated for research from COVID-19 patients. Specifically, adult human eyes from three female donors aged 52, 78, and 80 years old were donated. The ocular epithelial cells were grown in vitro on mitotically inactivated 3T3 feeder cells. Discussing their results, Jackson et al. suggested that there was evidence for the detection of the coronavirus in the ocular epithelial cells. Moreover, conjunctival epithelium was permissive to the entry of the SARS-CoV-2 virion. However, there was no evidence of productive replication of the virus within such epithelial cells [34]. In other words, the virion of SARS-CoV-2 could probably infect more cell types than those in which it could replicate.
Another set of authors [48] characterized the vascular system in COVID-19. Before this study, ECs were already known to contribute significantly to inflammation in COVID-19, as well as to the invasion of immune cells, vascular leakage, thrombosis, and hypoxia [66]. Motivated by such observations, de Rooij et al. performed NGS on mRNAs from ECs in cases of lethal COVID-19. In the first step, single-cell transcriptomes from more than 175,000 cell nuclei were sequenced. Next, UMAP analysis of the resulting expression profiles allowed the differentiation of the nuclei into four major classes: epithelial, stromal, endothelial, or from the immune system. In the following step, a more detailed UMAP analysis identified as many as 14 different endothelial subtypes among 35,000 EC nuclei. For example, there were arterial, pulmonary vein, large vessel, systemic vein, proliferating, capillary, or lymphatic ECs. In their conclusions, the authors showed that pulmonary ECs of deceased COVID-19 patients were enriched in genes involved in cellular stress. Moreover, there were gene expression signatures in ECs that were suggestive of failed immunomodulation and impaired vessel integrity. When proportions of cells were quantified, there were more capillary and venous ECs in COVID-19 than in controls.
3. Meta-analyses of datasets related to the etiology of COVID-19.
As there are so many datasets of gene expression available for COVID-19, it was also possible and interesting to merge multiple datasets in a meta-analysis (see Table 4). Goals of a typical meta-analysis include re-analysis, recalculation of tests of significance, statistical tests of replicability and of robustness of conclusions.
In the first example, Ziegler et al. [49] focused on identifying lung cells that expressed two major receptors for the SARS-CoV-2 virus: namely ACE2 and transmembrane serine protease 2 (TMPRSS2). The authors first focused on identifying cellular targets for SARS-CoV-2 in the lungs of humans and non-human primates, then in the gastrointestinal tract, then in the upper airways. ACE2 was identified as the main target. Next, it was established that ACE2 expression was enhanced by interferons. Similarly, a meta-analysis reported by Aguiar et al. (Aguiar, Tremblay et al. 2020) focused on ACE2, and reported evidence for a mechanism dynamically regulating ACE2 expression in response to an infection with SARS-CoV-2. Another related meta-analysis looked at ACE2 levels in kidney disease associated with COVID-19 [53].
In another example, a major re-analysis and meta-analysis published by Garg et al. re-evaluated 20 published hypotheses about the nature of the immune response in COVID-19 by integrating 9 PBMC-focused datasets [27,36,56,67,68,69,70]. The cells were classified into 5 major populations: lymphoid cells, myeloid cells, B cells, epithelial cells, and platelets. However, this meta-analysis focused on the characteristics of a single cellular compartment (i.e., immune cells) in COVID-19, rather than trying to compare responses across different cell- and tissue-types. T cells tended to decrease in number with increasing severity of COVID-19. Interferon type 1 responses tended to increase in COVID-19, and B cells tended to expand in selected clones in response to antigens from SARS-CoV-2.
Furthermore, Chen et al. [55] set out to compare transcriptional responses observed in human cell lines during infection with SARS-CoV-2 with those observed in cancer samples processed by The Cancer Genome Atlas (TCGA). The following cell lines were infected with the virus: lung adenocarcinoma cell lines A549 and Calu-3, normal human bronchial epithelial cells. Many similarities were detected, suggesting that the immune cell responses and inflammatory programmes induced in host cells were similar in both diseases.
Note that smokers are strongly susceptible to severe COVID-19. To investigate the impact of smoking on COVID-19, Smith et al. [32] performed an extensive meta-analysis of expression profiles measured for lung and airway epithelial samples. Another goal of this meta-analysis was to identify cell and tissue types that have high levels of the SARS-CoV-2 receptor ACE2.
In contrast, the goal of Muus et al. [52] was to identify cell type specific associations of age, sex, or smoking, with expression levels of ACE2 and other SARS-CoV-2 receptor molecules. To this end, publicly available scRNA-seq datasets were downloaded from GEO. These datasets were searched using the following requirement: (1) that unnormalized count data were provided; (2) that data were generated using the 10X Genomics’s Chromium platform; and (3) that human samples were profiled. In effect, the authors performed a meta-analysis of 31 lung scRNA-seq studies with over 1 million 300 thousand cells, from 377 airway and lung samples, from 228 individuals. It was confirmed that cell-specific expression patterns play a role in the etiology of COVID-19.
Finally, Zhang et al. [54] set out to identify cellular phenotypes, which were shared across many inflammatory diseases including COVID-19. Through a meta-analysis and an integration pipeline, similarities in gene expression and signalling between COVID-19 and other inflammatory diseases were to be uncovered. This meta-analysis modelled also the effects of technology, tissue of origin, and donor. The meta-analysis built a reference library for > 300,000 cells in normal body and in different inflammatory diseases as well as in COVID-19. This major cross-disease study suggested that interferon-gamma and TNA-alpha signalling in macrophages were essential for the inflammatory phenotype seen in severe COVID-19. In particular, the CXCL10+ CCL2+ inflammatory macrophage state was very abundant in severe COVID-19.
4. The etiology of severe COVID-19 in the light of gene expression data.
Based on 27 datasets reviewed together with 8 meta-analyses, we arrive at mature understanding of the etiology of severe COVID-19 (see Cover Figure). Note that I considered mostly high-impact examples of RNA-seq studies of COVID-19 (the average number of citations was 372). These studies were well-interpreted through quality publications in a peer-reviewed journal with an impact factor (IF). The advantage of RNA-seq is that changes in gene expression were likely to be longer term and stable, perhaps mediated through epigenetic reprogramming, and are likely to better characterize etiology of COVID-19 than biochemical markers. Moreover, there were several inflammatory syndromes complicating long COVID-10 and described by gene expression datasets from various tissues and organs, for example neuro-COVID [33,46], ARDS, or MIS-C [41,43].
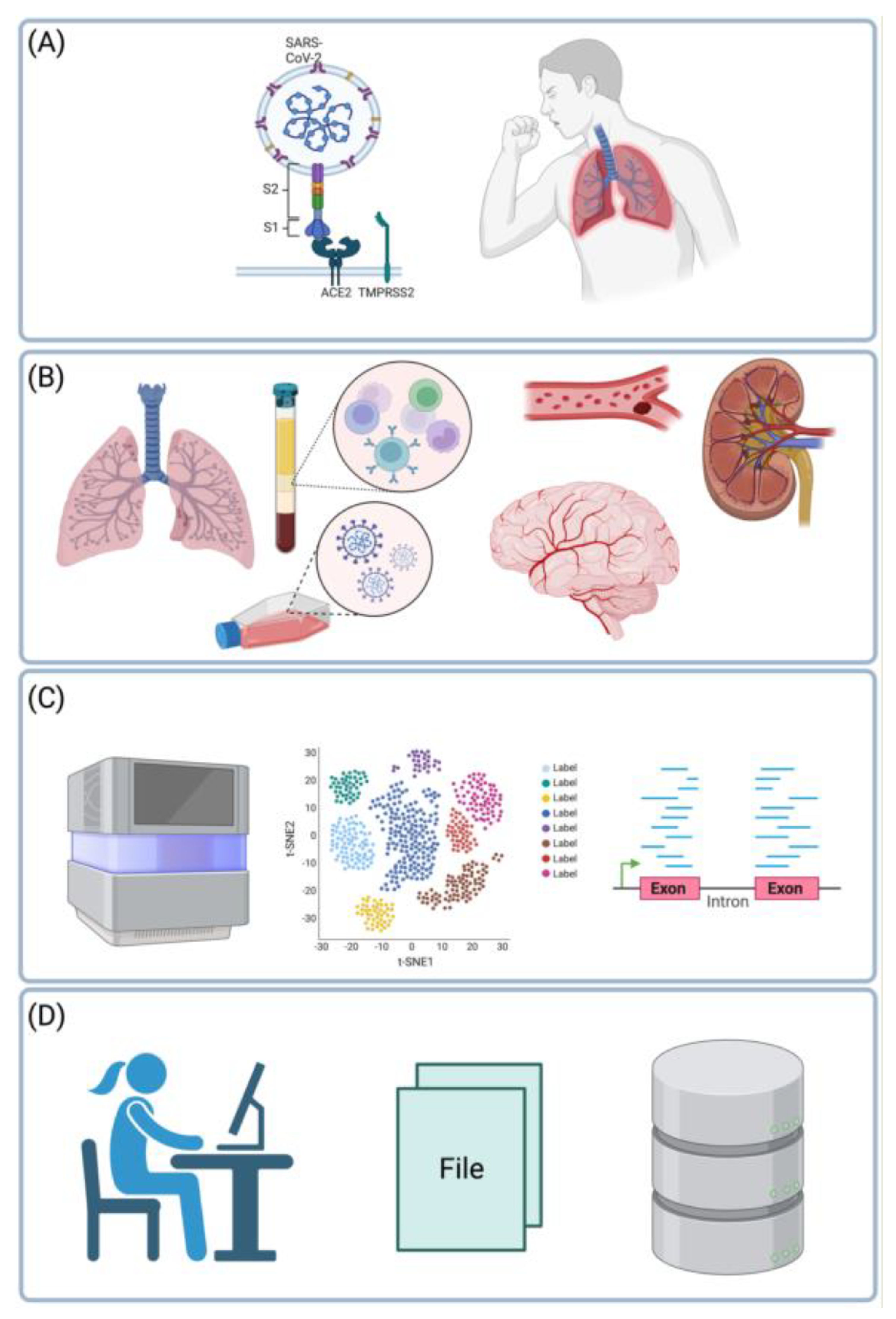
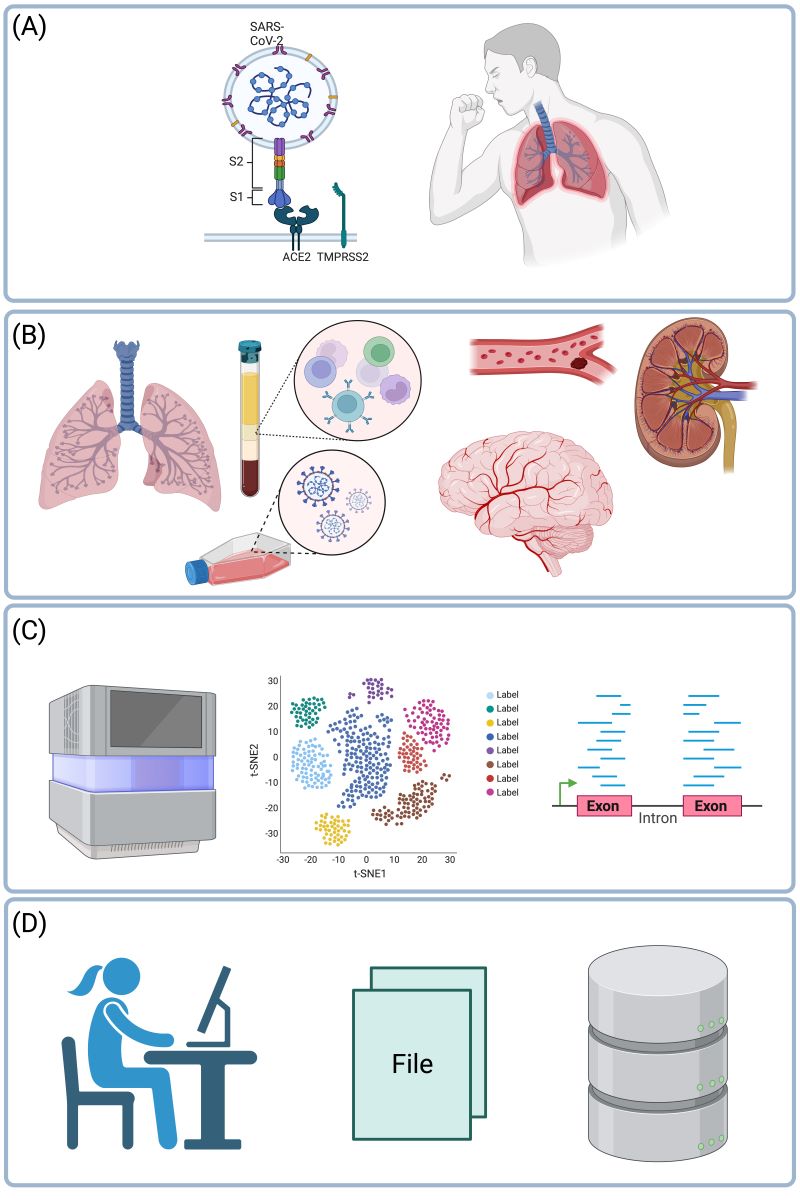
Cover Figure.
Main themes of the review. In severe COVID-19, the SARS-CoV-2 virus affects the lungs and the bronchi (A). However, not only the respiratory system is affected, but also the blood, or blood vessels, or endothelial cells, the brain, or kidneys (B). Changes in gene expression in these organs can be characterized in unbiased fashion using RNA-seq or single-cell RNA-seq (C). Resulting datasets are processed using bioinformatics tools or databases and analyzed statistically (D). Based on 27 such datasets reviewed herein together with 8 meta-analyses, we arrive at mature understanding of the etiology of severe COVID-19. Created with BioRender.com.
Cover Figure.
Main themes of the review. In severe COVID-19, the SARS-CoV-2 virus affects the lungs and the bronchi (A). However, not only the respiratory system is affected, but also the blood, or blood vessels, or endothelial cells, the brain, or kidneys (B). Changes in gene expression in these organs can be characterized in unbiased fashion using RNA-seq or single-cell RNA-seq (C). Resulting datasets are processed using bioinformatics tools or databases and analyzed statistically (D). Based on 27 such datasets reviewed herein together with 8 meta-analyses, we arrive at mature understanding of the etiology of severe COVID-19. Created with BioRender.com.
In general, lists of DEGs and differentially expressed pathways strongly suggested that inflammatory mechanisms were responsible for the development of severe COVID-19 (see Table 5). In particular, six pathways were differentially expressed (Table 6). There was frequently increased interferon response, interferon signaling, or interferon-responsive transcription factors (TFs). Moreover, there was a tendency for increased immune or inflammatory responses. Expression of cytokines, chemokines, or their receptors also tended to be increased. Increased interleukin-1 family signaling. There was also Increased interleukin-1 family signaling and increased hypoxic signalling. These increases were accompanied by diminished immune system regulation, diminished angiogenesis, and diminished vessel integrity.
Not only DEGs and host-viral interactions were studied, but also cellular composition of tissues and organs. For example, changes in proportions of immune or epithelial cells detected through scRNA-seq (Table 7) gave clues as to why severe COVID-19 becomes an out-of-control immune disease affecting many organs and tissue-types. Indeed, there were frequent quantitative changes in proportions of immune cells [25,31,33,44,60,61] especially among PBMCs or among infiltrating immune cells or among NKs. For example, several studies suggested that inflammatory effector cells such as antibody-producing B-cells (plasmablasts and plasma cells), or neutrophils, or cytotoxic T cells, or activated macrophages increase in proportions in severe COVID-19 (see Figure 1 and Table 7). In contrast, regulatory immune cells such as regulatory T-cells, or dendritic cells, or antigen-presenting cells decrease in proportions. These observations suggest a hypothesis according to which a powerful immune stimulation over 7-10 days at the beginning of COVID-19 gives rise to a dysregulated immune system, which attacks the lungs leading to their scaring and fibrosis (Figure 1).
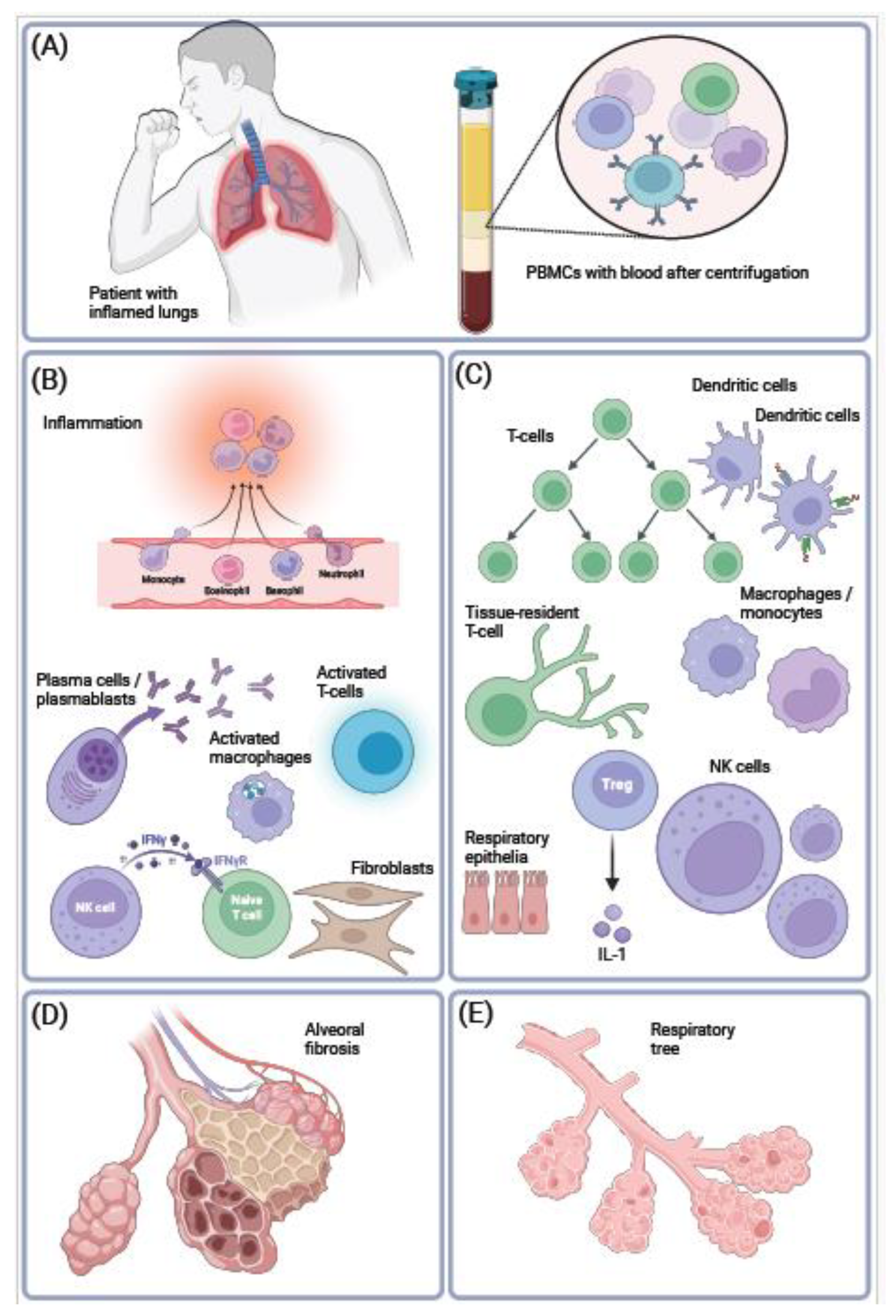
Figure 1.
Inflammatory changes in proportions of immune cells in COVID-19. SARS-CoV-2 replicates in the lungs in epithelial cells, macrophages, lymphocytes, or in neutrophils (A). This leads to inflammatory syndromes in various organs and tissues in case of severe COVID-19. To investigate this, PBMCs were frequently harvested from blood samples for gene expression profiling using scRNA-seq. In response to COVID-19-induced inflammation, there were changes in proportions of cell types, which resembled auto-immune diseases or immune aging (B). The following inflammatory cell types were found to increase in proportion: neutrophils, recruited monocytes, plasma cells and plasmablasts, or activated NK cells (C). In contrast, immune cells involved in modulation of immune response tended to decrease in proportion, for example regulatory T cells, or naive NK cells. Furthermore, lung’s alveoli (E) harden, inflame, and undergo scaring as a result of severe pulmonary COVID-19 (D). Created with BioRender.com.
Figure 1.
Inflammatory changes in proportions of immune cells in COVID-19. SARS-CoV-2 replicates in the lungs in epithelial cells, macrophages, lymphocytes, or in neutrophils (A). This leads to inflammatory syndromes in various organs and tissues in case of severe COVID-19. To investigate this, PBMCs were frequently harvested from blood samples for gene expression profiling using scRNA-seq. In response to COVID-19-induced inflammation, there were changes in proportions of cell types, which resembled auto-immune diseases or immune aging (B). The following inflammatory cell types were found to increase in proportion: neutrophils, recruited monocytes, plasma cells and plasmablasts, or activated NK cells (C). In contrast, immune cells involved in modulation of immune response tended to decrease in proportion, for example regulatory T cells, or naive NK cells. Furthermore, lung’s alveoli (E) harden, inflame, and undergo scaring as a result of severe pulmonary COVID-19 (D). Created with BioRender.com.
Finally, there was also transcriptional evidence for endothelial injury and pathological thrombotic events [28,48].
Note also that the transcription of the SARS-CoV-2 virus itself could be verified using RNA-seq and specialized bioinformatics pipelines, for example in epithelial cells and in lymphocytes, or macrophages, or neutrophils in bronchi or lungs of COVID-19 samples (see Table 8).
5. Conclusions.
A mean time from diagnosis to death in severe COVID-19 was estimated to be between 17 and 19 days [71], suggesting sufficient time for changes in gene expression or epigenetic regulation. Thus, severe COVID-19 is not only a viral infection with SARS-CoV-2, but also a weeks-long inflammatory transition occurring throughout the body. This transition is accompanied by global changes in gene expression patterns, particularly among PBMCs. Many tissues also undergo inflammatory transformation in cellular composition, which is accompanied by fibrosis or loss of cells that can regulate immunological reactions.
Abbreviations.
ACE2 angiotensin-converting enzyme 2.
ARDS acute respiratory disease syndrome.
AT1 alveolar type I.
AT2 alveolar type II.
BALF broncho-alveolar lavage fluid.
BioC Bioconductor.
CD4 cluster of differentiation 4.
CD8 cluster of differentiation 8.
COVID-19 Coronavirus Disease 2019.
CSF cerebrospinal fluid.
ChIP-seq chromatin immunoprecipitation.
DEG differentially expressed gene.
EC endothelial cell.
FastQC Quality Control tool for High Throughput Sequence Data.
FDR false discovery rate.
GEO Gene Expression Omnibus.
GO gene ontology.
GSEA gene set enrichment.
GZMA granzyme A.
GZMB granzyme B.
HPIV3 human parainfluenza virus 3.
HUVEC human umbilical vein EC.
IAV H1N1 influenza A virus.
ID identifier.
IPF idiopathic pulmonary fibrosis.
IRF7 interferon regulatory factor 7.
ISG15 interferon-stimulated gene 15.
KEGG Kyoto Encyclopedia of Genes and Genomes.
LUCAT1 lung cancer associated transcript 1.
MGI Mouse Genome Informatics.
MIS-C multisystem inflammatory syndrome.
NGS next generation sequencing.
NK natural killer.
ORFs open reading frames.
PBMCs peripheral blood mononuclear cells.
PMID PubMed unique identifier.
RNA-seq RNA sequencing.
SARS severe acute respiratory syndrome.
SARS-CoV-2 SARS coronavirus 2.
STRING protein-protein interaction networks functional enrichment analysis.
Scopus Elsevier's abstract and citation database.
TCR T-cell receptor.
TCGA The Cancer Genome Atlas.
UMAP uniform manifold approximation and projection.
hACE2 human angiotensin-converting enzyme 2.
pDCs plasmacytoid dendritic cells.
scRNA-seq single-cell RNA sequencing.
ssGSVA single-sample gene-set variation analysis.
References
- Wiersinga, W. J.; Rhodes, A.; Cheng, A. C.; Peacock, S. J.; Prescott, H. C., Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, (8), 782-793.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; Niu, P.; Zhan, F.; Ma, X.; Wang, D.; Xu, W.; Wu, G.; Gao, G. F.; Tan, W., A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 2020, 382, (8), 727-733.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y. M.; Wang, W.; Song, Z. G.; Hu, Y.; Tao, Z. W.; Tian, J. H.; Pei, Y. Y.; Yuan, M. L.; Zhang, Y. L.; Dai, F. H.; Liu, Y.; Wang, Q. M.; Zheng, J. J.; Xu, L.; Holmes, E. C.; Zhang, Y. Z., A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, (7798), 265-269.
- Gostin, L. O.; Gronvall, G. K., The Origins of Covid-19 — Why It Matters (and Why It Doesn't). New England Journal of Medicine 2023.
- Guan, W.-j.; Ni, Z.-y.; Hu, Y.; Liang, W.-h.; Ou, C.-q.; He, J.-x.; Liu, L.; Shan, H.; Lei, C.-l.; Hui, D. S. C.; Du, B.; Li, L.-j.; Zeng, G.; Yuen, K.-Y.; Chen, R.-c.; Tang, C.-l.; Wang, T.; Chen, P.-y.; Xiang, J.; Li, S.-y.; Wang, J.-l.; Liang, Z.-j.; Peng, Y.-x.; Wei, L.; Liu, Y.; Hu, Y.-h.; Peng, P.; Wang, J.-m.; Liu, J.-y.; Chen, Z.; Li, G.; Zheng, Z.-j.; Qiu, S.-q.; Luo, J.; Ye, C.-j.; Zhu, S.-y.; Zhong, N.-s., Clinical Characteristics of Coronavirus Disease 2019 in China. New England Journal of Medicine 2020, 382, (18), 1708-1720.
- Holmes, E. C.; Goldstein, S. A.; Rasmussen, A. L.; Robertson, D. L.; Crits-Christoph, A.; Wertheim, J. O.; Anthony, S. J.; Barclay, W. S.; Boni, M. F.; Doherty, P. C.; Farrar, J.; Geoghegan, J. L.; Jiang, X.; Leibowitz, J. L.; Neil, S. J. D.; Skern, T.; Weiss, S. R.; Worobey, M.; Andersen, K. G.; Garry, R. F.; Rambaut, A., The origins of SARS-CoV-2: A critical review. Cell 2021, 184, (19), 4848-4856.
- Wu, Z.; McGoogan, J. M., Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72‚ÄØ314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, (13), 1239-1242.
- Davis, H. E.; McCorkell, L.; Vogel, J. M.; Topol, E. J., Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023, 21, (3), 133-146.
- Sherif, Z. A.; Gomez, C. R.; Connors, T. J.; Henrich, T. J.; Reeves, W. B., Pathogenic mechanisms of post-acute sequelae of SARS-CoV-2 infection (PASC). Elife 2023, 12.
- Iba, T.; Connors, J. M.; Levy, J. H., The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm Res 2020, 69, (12), 1181-1189.
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W. V.; Gerlo, S.; Vandekerckhove, L., On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog 2020, 16, (10), e1009037.
- Konstantinos, E.; Dimitris, V.; Koralia, P.; Periklis, G. F.; Nefeli, L.; Marios, D.; Angelos, P.; Bindu, K.; Orsalia, H.; Aikaterini, P.; Sophia, H.; Athanassios, K.; Christos, K.; Athanasios, G. T.; Laurence de, L.; Demetris, V.; Sotirios, T.; Barry, R. S.; Argyris, P.; Giovanni, B.; Ioannis, K.; Peter, J. B.; Vassilis, G. G., Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis. European Respiratory Journal 2022, 60, (2), 2102951.
- Ackermann, M.; Verleden, S. E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; Li, W. W.; Li, V. W.; Mentzer, S. J.; Jonigk, D., Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. New England Journal of Medicine 2020, 383, (2), 120-128.
- Costa, T. J.; Potje, S. R.; Fraga-Silva, T. F. C.; da Silva-Neto, J. A.; Barros, P. R.; Rodrigues, D.; Machado, M. R.; Martins, R. B.; Santos-Eichler, R. A.; Benatti, M. N.; de Sá, K. S. G.; Almado, C. E. L.; Castro Í, A.; Pontelli, M. C.; Serra, L.; Carneiro, F. S.; Becari, C.; Louzada-Junior, P.; Oliveira, R. D. R.; Zamboni, D. S.; Arruda, E.; Auxiliadora-Martins, M.; Giachini, F. R. C.; Bonato, V. L. D.; Zachara, N. E.; Bomfim, G. F.; Tostes, R. C., Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascul Pharmacol 2022, 142, 106946.
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wrsdrfer, P.; Midkiff, C. C.; Mudd, J.; Belyaeva, E.; Milligan, N. S.; Rorison, T. D.; Wagner, N.; Bodem, J.; Dlken, L.; Aktas, B. H.; Vander Heide, R. S.; Yin, X. M.; Kolls, J. K.; Roy, C. J.; Rappaport, J.; Ergn, S.; Qin, X., SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front Cell Infect Microbiol 2021, 11, 701278.
- Stjepanovic, M. I.; Stojanovic, M. R.; Stankovic, S.; Cvejic, J.; Dimic-Janjic, S.; Popevic, S.; Buha, I.; Belic, S.; Djurdjevic, N.; Stjepanovic, M. M.; Jovanovic, D.; Stojkovic-Laloševic, M.; Soldatovic, I.; Bonaci-Nikolic, B.; Miskovic, R., Autoimmune and immunoserological markers of COVID-19 pneumonia: Can they help in the assessment of disease severity. Front Med (Lausanne) 2022, 9, 934270.
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y., The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun Rev 2021, 20, (4), 102792.
- Damoiseaux, J.; Dotan, A.; Fritzler, M. J.; Bogdanos, D. P.; Meroni, P. L.; Roggenbuck, D.; Goldman, M.; Landegren, N.; Bastard, P.; Shoenfeld, Y.; Conrad, K., Autoantibodies and SARS-CoV2 infection: The spectrum from association to clinical implication: Report of the 15th Dresden Symposium on Autoantibodies. Autoimmun Rev 2022, 21, (3), 103012.
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C., The protein expression profile of ACE2 in human tissues. Mol Syst Biol 2020, 16, (7), e9610.
- Finkel Y., M. O., Nachshon A., Weingarten-Gabbay S., Morgenstern D., Yahalom-Ronen Y., Tamir H., Achdout H., Stein D., Israeli O., Beth-Din A., Melamed S., Weiss S., Israely T., Paran N., Schwartz M. & Stern-Ginossar N., The coding capacity of SARS-CoV-2. Nature 2020, 589, 125-130.
- Kim, D.; Lee, J. Y.; Yang, J. S.; Kim, J. W.; Kim, V. N.; Chang, H., The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, (4), 914-921 e10.
- Papa, G.; Mallery, D. L.; Albecka, A.; Welch, L. G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H. T.; Goodfellow, I. G.; Carter, A.; Munro, S.; James, L. C., Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog 2021, 17, (1), e1009246.
- Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., Auerbach, A., Li, F., Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences of the United States of America 2020, 117, (21), 11727-11734.
- Lamers, M. M. H., B. L., SARS-CoV-2 pathogenesis. Nat Rev Microbiol 2022, 20, (5), 270-284.
- Arunachalam, P. S.; Wimmers, F.; Mok, C. K. P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; Wagh, D.; Coller, J.; Pellegrini, K. L.; Kazmin, D.; Alaaeddine, G.; Leung, W. S.; Chan, J. M. C.; Chik, T. S. H.; Choi, C. Y. C.; Huerta, C.; Paine McCullough, M.; Lv, H.; Anderson, E.; Edupuganti, S.; Upadhyay, A. A.; Bosinger, S. E.; Maecker, H. T.; Khatri, P.; Rouphael, N.; Peiris, M.; Pulendran, B., Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, (6508), 1210-1220.
- Bernardes, J. P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J. I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; Köhler, P.; Künstner, A.; Rosati, E.; Aschenbrenner, A. C.; Bacher, P.; Baran, N.; Boysen, T.; Brandt, B.; Bruse, N.; Dörr, J.; Dräger, A.; Elke, G.; Ellinghaus, D.; Fischer, J.; Forster, M.; Franke, A.; Franzenburg, S.; Frey, N.; Friedrichs, A.; Fuß, J.; Glück, A.; Hamm, J.; Hinrichsen, F.; Hoeppner, M. P.; Imm, S.; Junker, R.; Kaiser, S.; Kan, Y. H.; Knoll, R.; Lange, C.; Laue, G.; Lier, C.; Lindner, M.; Marinos, G.; Markewitz, R.; Nattermann, J.; Noth, R.; Pickkers, P.; Rabe, K. F.; Renz, A.; Röcken, C.; Rupp, J.; Schaffarzyk, A.; Scheffold, A.; Schulte-Schrepping, J.; Schunk, D.; Skowasch, D.; Ulas, T.; Wandinger, K. P.; Wittig, M.; Zimmermann, J.; Busch, H.; Hoyer, B. F.; Kaleta, C.; Heyckendorf, J.; Kox, M.; Rybniker, J.; Schreiber, S.; Schultze, J. L.; Rosenstiel, P., Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, (6), 1296-1314 e9.
- Wilk, A. J.; Rustagi, A.; Zhao, N. Q.; Roque, J.; Martínez-Colón, G. J.; McKechnie, J. L.; Ivison, G. T.; Ranganath, T.; Vergara, R.; Hollis, T.; Simpson, L. J.; Grant, P.; Subramanian, A.; Rogers, A. J.; Blish, C. A., A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 2020, 26, (7), 1070-1076.
- Iwamura, C.; Hirahara, K.; Kiuchi, M.; Ikehara, S.; Azuma, K.; Shimada, T.; Kuriyama, S.; Ohki, S.; Yamamoto, E.; Inaba, Y.; Shiko, Y.; Aoki, A.; Kokubo, K.; Hirasawa, R.; Hishiya, T.; Tsuji, K.; Nagaoka, T.; Ishikawa, S.; Kojima, A.; Mito, H.; Hase, R.; Kasahara, Y.; Kuriyama, N.; Tsukamoto, T.; Nakamura, S.; Urushibara, T.; Kaneda, S.; Sakao, S.; Tobiume, M.; Suzuki, Y.; Tsujiwaki, M.; Kubo, T.; Hasegawa, T.; Nakase, H.; Nishida, O.; Takahashi, K.; Baba, K.; Iizumi, Y.; Okazaki, T.; Kimura, M. Y.; Yoshino, I.; Igari, H.; Nakajima, H.; Suzuki, T.; Hanaoka, H.; Nakada, T.-A.; Ikehara, Y.; Yokote, K.; Nakayama, T., Elevated Myl9 reflects the Myl9-containing microthrombi in SARS-CoV-2-induced lung exudative vasculitis and predicts COVID-19 severity. Proceedings of the National Academy of Sciences of the United States of America 2022, 119, (33), e2203437119-e2203437119.
- Blanco-Melo, D.; Nilsson-Payant, B. E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Muller, R.; Jordan, T. X.; Oishi, K.; Panis, M.; Sachs, D.; Wang, T. T.; Schwartz, R. E.; Lim, J. K.; Albrecht, R. A.; tenOever, B. R., Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, (5), 1036-1045.e9.
- Melms, J. C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A. F.; Amin, A. D.; Schapiro, D.; Frangieh, C. J.; Luoma, A. M.; Filliol, A.; Fang, Y.; Ravichandran, H.; Clausi, M. G.; Alba, G. A.; Rogava, M.; Chen, S. W.; Ho, P.; Montoro, D. T.; Kornberg, A. E.; Han, A. S.; Bakhoum, M. F.; Anandasabapathy, N.; Suárez-Fariñas, M.; Bakhoum, S. F.; Bram, Y.; Borczuk, A.; Guo, X. V.; Lefkowitch, J. H.; Marboe, C.; Lagana, S. M.; Del Portillo, A.; Tsai, E. J.; Zorn, E.; Markowitz, G. S.; Schwabe, R. F.; Schwartz, R. E.; Elemento, O.; Saqi, A.; Hibshoosh, H.; Que, J.; Izar, B., A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, (7865), 114-119.
- Daamen, A. R.; Bachali, P.; Owen, K. A.; Kingsmore, K. M.; Hubbard, E. L.; Labonte, A. C.; Robl, R.; Shrotri, S.; Grammer, A. C.; Lipsky, P. E., Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Scientific reports 2021, 11, (1), 7052-7052.
- Assou, S.; Ahmed, E.; Morichon, L.; Nasri, A.; Foisset, F.; Bourdais, C.; Gros, N.; Tieo, S.; Petit, A.; Vachier, I.; Muriaux, D.; Bourdin, A.; De Vos, J., The Transcriptome Landscape of the In Vitro Human Airway Epithelium Response to SARS-CoV-2. Int J Mol Sci 2023, 24, (15).
- Heming, M.; Li, X.; Ruber, S.; Mausberg, A. K.; Brsch, A. L.; Hartlehnert, M.; Singhal, A.; Lu, I. N.; Fleischer, M.; Szepanowski, F.; Witzke, O.; Brenner, T.; Dittmer, U.; Yosef, N.; Kleinschnitz, C.; Wiendl, H.; Stettner, M.; Meyer Zu Hrste, G., Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity 2021, 54, (1), 164-175 e6.
- Jackson, R. M.; Hatton, C. F.; Spegarova, J. S.; Georgiou, M.; Collin, J.; Stephenson, E.; Verdon, B.; Haq, I. J.; Hussain, R.; Coxhead, J. M.; Mudhar, H.-S.; Wagner, B.; Hasoon, M.; Davey, T.; Rooney, P.; Khan, C. M. A.; Ward, C.; Brodlie, M.; Haniffa, M.; Hambleton, S.; Armstrong, L.; Figueiredo, F.; Queen, R.; Duncan, C. J. A.; Lako, M., Conjunctival epithelial cells resist productive SARS-CoV-2 infection. Stem Cell Reports 2022, 17, (7), 1699-1713.
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; Wei, X.; Sun, H. X.; Ye, B.; Wu, Y.; Zhang, N.; Lei, G.; Yu, L.; Yan, J.; Diao, G.; Meng, F.; Bai, C.; Mao, P.; Yu, Y.; Wang, M.; Yuan, Y.; Deng, Q.; Li, Z.; Huang, Y.; Hu, G.; Liu, Y.; Wang, X.; Xu, Z.; Liu, P.; Bi, Y.; Shi, Y.; Zhang, S.; Chen, Z.; Wang, J.; Xu, X.; Wu, G.; Wang, F. S.; Gao, G. F.; Liu, L.; Liu, W. J., Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity 2020, 53, (3), 685-696.e3.
- Zhang, J. Y.; Wang, X. M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J. W.; Fan, X.; Xia, P.; Fu, J. L.; Wang, S. Y.; Xu, R. N.; Dai, X. P.; Shi, L.; Huang, L.; Jiang, T. J.; Shi, M.; Zhang, Y.; Zumla, A.; Maeurer, M.; Bai, F.; Wang, F. S., Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol 2020, 21, (9), 1107-1118.
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A. G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; Rameau, P.; Catelain, C.; Alfaro, A.; Dussiau, C.; Friedrich, C.; Sourdeau, E.; Marin, N.; Szwebel, T. A.; Cantin, D.; Mouthon, L.; Borderie, D.; Deloger, M.; Bredel, D.; Mouraud, S.; Drubay, D.; Andrieu, M.; Lhonneur, A. S.; Saada, V.; Stoclin, A.; Willekens, C.; Pommeret, F.; Griscelli, F.; Ng, L. G.; Zhang, Z.; Bost, P.; Amit, I.; Barlesi, F.; Marabelle, A.; Pène, F.; Gachot, B.; André, F.; Zitvogel, L.; Ginhoux, F.; Fontenay, M.; Solary, E., Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, (6), 1401-1418.e18.
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; De Domenico, E.; Wendisch, D.; Grasshoff, M.; Kapellos, T. S.; Beckstette, M.; Pecht, T.; Saglam, A.; Dietrich, O.; Mei, H. E.; Schulz, A. R.; Conrad, C.; Kunkel, D.; Vafadarnejad, E.; Xu, C. J.; Horne, A.; Herbert, M.; Drews, A.; Thibeault, C.; Pfeiffer, M.; Hippenstiel, S.; Hocke, A.; Müller-Redetzky, H.; Heim, K. M.; Machleidt, F.; Uhrig, A.; Bosquillon de Jarcy, L.; Jürgens, L.; Stegemann, M.; Glösenkamp, C. R.; Volk, H. D.; Goffinet, C.; Landthaler, M.; Wyler, E.; Georg, P.; Schneider, M.; Dang-Heine, C.; Neuwinger, N.; Kappert, K.; Tauber, R.; Corman, V.; Raabe, J.; Kaiser, K. M.; Vinh, M. T.; Rieke, G.; Meisel, C.; Ulas, T.; Becker, M.; Geffers, R.; Witzenrath, M.; Drosten, C.; Suttorp, N.; von Kalle, C.; Kurth, F.; Händler, K.; Schultze, J. L.; Aschenbrenner, A. C.; Li, Y.; Nattermann, J.; Sawitzki, B.; Saliba, A. E.; Sander, L. E., Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, (6), 1419-1440 e23.
- Meckiff, B. J.; Ramirez-Sustegui, C.; Fajardo, V.; Chee, S. J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; Sette, A.; Ay, F.; Seumois, G.; Ottensmeier, C. H.; Vijayanand, P., Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 2020, 183, (5), 1340-1353 e16.
- Witkowski, M.; Tizian, C.; Ferreira-Gomes, M.; Niemeyer, D.; Jones, T. C.; Heinrich, F.; Frischbutter, S.; Angermair, S.; Hohnstein, T.; Mattiola, I.; Nawrath, P.; McEwen, S.; Zocche, S.; Viviano, E.; Heinz, G. A.; Maurer, M.; Kölsch, U.; Chua, R. L.; Aschman, T.; Meisel, C.; Radke, J.; Sawitzki, B.; Roehmel, J.; Allers, K.; Moos, V.; Schneider, T.; Hanitsch, L.; Mall, M. A.; Conrad, C.; Radbruch, H.; Duerr, C. U.; Trapani, J. A.; Marcenaro, E.; Kallinich, T.; Corman, V. M.; Kurth, F.; Sander, L. E.; Drosten, C.; Treskatsch, S.; Durek, P.; Kruglov, A.; Radbruch, A.; Mashreghi, M. F.; Diefenbach, A., Untimely TGF-beta responses in COVID-19 limit antiviral functions of NK cells. Nature 2021, 600, (7888), 295-301.
- Ramaswamy, A.; Brodsky, N. N.; Sumida, T. S.; Comi, M.; Asashima, H.; Hoehn, K. B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N. G.; Bishai, J.; Khan, A.; Lau, W.; Sellers, B.; Bansal, N.; Guerrerio, P.; Unterman, A.; Habet, V.; Rice, A. J.; Catanzaro, J.; Chandnani, H.; Lopez, M.; Kaminski, N.; Dela Cruz, C. S.; Tsang, J. S.; Wang, Z.; Yan, X.; Kleinstein, S. H.; van Dijk, D.; Pierce, R. W.; Hafler, D. A.; Lucas, C. L., Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, (5), 1083-1095 e7.
- You, M.; Chen, L.; Zhang, D.; Zhao, P.; Chen, Z.; Qin, E. Q.; Gao, Y.; Davis, M. M.; Yang, P., Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat Cell Biol 2021, 23, (6), 620-630.
- Aznaourova, M.; Schmerer, N.; Janga, H.; Zhang, Z.; Pauck, K.; Bushe, J.; Volkers, S. M.; Wendisch, D.; Georg, P.; Ntini, E.; Aillaud, M.; Gendisch, M.; Mack, E.; Skevaki, C.; Keller, C.; Bauer, C.; Bertrams, W.; Marsico, A.; Nist, A.; Stiewe, T.; Gruber, A. D.; Ruppert, C.; Li, Y.; Garn, H.; Sander, L. E.; Schmeck, B.; Schulte, L. N., Single-cell RNA sequencing uncovers the nuclear decoy lincRNA PIRAT as a regulator of systemic monocyte immunity during COVID-19. Proceedings of the National Academy of Sciences of the United States of America 2022, 119, (36), e2120680119.
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; Deczkowska, A.; Zhang, S.; Schwikowski, B.; Zhang, Z.; Amit, I., Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, (7), 1475-1488 e12.
- Chua, R. L.; Lukassen, S.; Trump, S.; Hennig, B. P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M. T.; Kazmierski, J.; Timmermann, B.; Twardziok, S.; Schneider, S.; Machleidt, F.; Muller-Redetzky, H.; Maier, M.; Krannich, A.; Schmidt, S.; Balzer, F.; Liebig, J.; Loske, J.; Suttorp, N.; Eils, J.; Ishaque, N.; Liebert, U. G.; von Kalle, C.; Hocke, A.; Witzenrath, M.; Goffinet, C.; Drosten, C.; Laudi, S.; Lehmann, I.; Conrad, C.; Sander, L. E.; Eils, R., COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol 2020, 38, (8), 970-979.
- Yang, A. C.; Kern, F.; Losada, P. M.; Agam, M. R.; Maat, C. A.; Schmartz, G. P.; Fehlmann, T.; Stein, J. A.; Schaum, N.; Lee, D. P.; Calcuttawala, K.; Vest, R. T.; Berdnik, D.; Lu, N.; Hahn, O.; Gate, D.; McNerney, M. W.; Channappa, D.; Cobos, I.; Ludwig, N.; Schulz-Schaeffer, W. J.; Keller, A.; Wyss-Coray, T., Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, (7868), 565-571.
- Delorey, T. M.; Ziegler, C. G. K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Ö.; Abbondanza, D.; Fleming, S. J.; Subramanian, A.; Montoro, D. T.; Jagadeesh, K. A.; Dey, K. K.; Sen, P.; Slyper, M.; Pita-Ju√°rez, Y. H.; Phillips, D.; Biermann, J.; Bloom-Ackermann, Z.; Barkas, N.; Ganna, A.; Gomez, J.; Melms, J. C.; Katsyv, I.; Normandin, E.; Naderi, P.; Popov, Y. V.; Raju, S. S.; Niezen, S.; Tsai, L. T. Y.; Siddle, K. J.; Sud, M.; Tran, V. M.; Vellarikkal, S. K.; Wang, Y.; Amir-Zilberstein, L.; Atri, D. S.; Beechem, J.; Brook, O. R.; Chen, J.; Divakar, P.; Dorceus, P.; Engreitz, J. M.; Essene, A.; Fitzgerald, D. M.; Fropf, R.; Gazal, S.; Gould, J.; Grzyb, J.; Harvey, T.; Hecht, J.; Hether, T.; Jan√©-Valbuena, J.; Leney-Greene, M.; Ma, H.; McCabe, C.; McLoughlin, D. E.; Miller, E. M.; Muus, C.; Niemi, M.; Padera, R.; Pan, L.; Pant, D.; Pe‚Äôer, C.; Pfiffner-Borges, J.; Pinto, C. J.; Plaisted, J.; Reeves, J.; Ross, M.; Rudy, M.; Rueckert, E. H.; Siciliano, M.; Sturm, A.; Todres, E.; Waghray, A.; Warren, S.; Zhang, S.; Zollinger, D. R.; Cosimi, L.; Gupta, R. M.; Hacohen, N.; Hibshoosh, H.; Hide, W.; Price, A. L.; Rajagopal, J.; Tata, P. R.; Riedel, S.; Szabo, G.; Tickle, T. L.; Ellinor, P. T.; Hung, D.; Sabeti, P. C.; Novak, R.; Rogers, R.; Ingber, D. E.; Jiang, Z. G.; Juric, D.; Babadi, M.; Farhi, S. L.; Izar, B.; Stone, J. R.; Vlachos, I. S.; Solomon, I. H.; Ashenberg, O.; Porter, C. B. M.; Li, B.; Shalek, A. K.; Villani, A.-C.; Rozenblatt-Rosen, O.; Regev, A., COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, (7865), 107-113.
- de Rooij, L.; Becker, L. M.; Teuwen, L. A.; Boeckx, B.; Jansen, S.; Feys, S.; Verleden, S.; Liesenborghs, L.; Stalder, A. K.; Libbrecht, S.; Van Buyten, T.; Philips, G.; Subramanian, A.; Dumas, S. J.; Meta, E.; Borri, M.; Sokol, L.; Dendooven, A.; Truong, A. K.; Gunst, J.; Van Mol, P.; Haslbauer, J. D.; Rohlenova, K.; Menter, T.; Boudewijns, R.; Geldhof, V.; Vinckier, S.; Amersfoort, J.; Wuyts, W.; Van Raemdonck, D.; Jacobs, W.; Ceulemans, L. J.; Weynand, B.; Thienpont, B.; Lammens, M.; Kuehnel, M.; Eelen, G.; Dewerchin, M.; Schoonjans, L.; Jonigk, D.; van Dorpe, J.; Tzankov, A.; Wauters, E.; Mazzone, M.; Neyts, J.; Wauters, J.; Lambrechts, D.; Carmeliet, P., The pulmonary vasculature in lethal COVID-19 and idiopathic pulmonary fibrosis at single-cell resolution. Cardiovasc Res 2023, 119, (2), 520-535.
- Ziegler, C. G. K.; Allon, S. J.; Nyquist, S. K.; Mbano, I. M.; Miao, V. N.; Tzouanas, C. N.; Cao, Y.; Yousif, A. S.; Bals, J.; Hauser, B. M.; Feldman, J.; Muus, C.; Wadsworth, M. H., 2nd; Kazer, S. W.; Hughes, T. K.; Doran, B.; Gatter, G. J.; Vukovic, M.; Taliaferro, F.; Mead, B. E.; Guo, Z.; Wang, J. P.; Gras, D.; Plaisant, M.; Ansari, M.; Angelidis, I.; Adler, H.; Sucre, J. M. S.; Taylor, C. J.; Lin, B.; Waghray, A.; Mitsialis, V.; Dwyer, D. F.; Buchheit, K. M.; Boyce, J. A.; Barrett, N. A.; Laidlaw, T. M.; Carroll, S. L.; Colonna, L.; Tkachev, V.; Peterson, C. W.; Yu, A.; Zheng, H. B.; Gideon, H. P.; Winchell, C. G.; Lin, P. L.; Bingle, C. D.; Snapper, S. B.; Kropski, J. A.; Theis, F. J.; Schiller, H. B.; Zaragosi, L. E.; Barbry, P.; Leslie, A.; Kiem, H. P.; Flynn, J. L.; Fortune, S. M.; Berger, B.; Finberg, R. W.; Kean, L. S.; Garber, M.; Schmidt, A. G.; Lingwood, D.; Shalek, A. K.; Ordovas-Montanes, J., SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, (5), 1016-1035 e19.
- Smith, J. C.; Sausville, E. L.; Girish, V.; Yuan, M. L.; Vasudevan, A.; John, K. M.; Sheltzer, J. M., Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev Cell 2020, 53, (5), 514-529.e3.
- Aguiar, J. A.; Tremblay, B. J.; Mansfield, M. J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A.; Revill, S.; Miller, M. S.; Carlsten, C.; Organ, L.; Joseph, C.; John, A.; Hanson, P.; Austin, R. C.; McManus, B. M.; Jenkins, G.; Mossman, K.; Ask, K.; Doxey, A. C.; Hirota, J. A., Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur Respir J 2020, 56, (3).
- Muus, C.; Luecken, M. D.; Eraslan, G.; Sikkema, L.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E. D.; Subramanian, A.; Smillie, C.; Jagadeesh, K. A.; Duong, E. T.; Fiskin, E.; Torlai Triglia, E.; Ansari, M.; Cai, P.; Lin, B.; Buchanan, J.; Chen, S.; Shu, J.; Haber, A. L.; Chung, H.; Montoro, D. T.; Adams, T.; Aliee, H.; Allon, S. J.; Andrusivova, Z.; Angelidis, I.; Ashenberg, O.; Bassler, K.; Bécavin, C.; Benhar, I.; Bergenstråhle, J.; Bergenstråhle, L.; Bolt, L.; Braun, E.; Bui, L. T.; Callori, S.; Chaffin, M.; Chichelnitskiy, E.; Chiou, J.; Conlon, T. M.; Cuoco, M. S.; Cuomo, A. S. E.; Deprez, M.; Duclos, G.; Fine, D.; Fischer, D. S.; Ghazanfar, S.; Gillich, A.; Giotti, B.; Gould, J.; Guo, M.; Gutierrez, A. J.; Habermann, A. C.; Harvey, T.; He, P.; Hou, X.; Hu, L.; Hu, Y.; Jaiswal, A.; Ji, L.; Jiang, P.; Kapellos, T. S.; Kuo, C. S.; Larsson, L.; Leney-Greene, M. A.; Lim, K.; Litviňuková, M.; Ludwig, L. S.; Lukassen, S.; Luo, W.; Maatz, H.; Madissoon, E.; Mamanova, L.; Manakongtreecheep, K.; Leroy, S.; Mayr, C. H.; Mbano, I. M.; McAdams, A. M.; Nabhan, A. N.; Nyquist, S. K.; Penland, L.; Poirion, O. B.; Poli, S.; Qi, C.; Queen, R.; Reichart, D.; Rosas, I.; Schupp, J. C.; Shea, C. V.; Shi, X.; Sinha, R.; Sit, R. V.; Slowikowski, K.; Slyper, M.; Smith, N. P.; Sountoulidis, A.; Strunz, M.; Sullivan, T. B.; Sun, D.; Talavera-López, C.; Tan, P.; Tantivit, J.; Travaglini, K. J.; Tucker, N. R.; Vernon, K. A.; Wadsworth, M. H.; Waldman, J.; Wang, X.; Xu, K.; Yan, W.; Zhao, W.; Ziegler, C. G. K., Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med 2021, 27, (3), 546-559.
- Menon, R.; Otto, E. A.; Sealfon, R.; Nair, V.; Wong, A. K.; Theesfeld, C. L.; Chen, X.; Wang, Y.; Boppana, A. S.; Luo, J.; Yang, Y.; Kasson, P. M.; Schaub, J. A.; Berthier, C. C.; Eddy, S.; Lienczewski, C. C.; Godfrey, B.; Dagenais, S. L.; Sohaney, R.; Hartman, J.; Fermin, D.; Subramanian, L.; Looker, H. C.; Harder, J. L.; Mariani, L. H.; Hodgin, J. B.; Sexton, J. Z.; Wobus, C. E.; Naik, A. S.; Nelson, R. G.; Troyanskaya, O. G.; Kretzler, M., SARS-CoV-2 receptor networks in diabetic and COVID-19-associated kidney disease. Kidney Int 2020, 98, (6), 1502-1518.
- Zhang, F.; Mears, J. R.; Shakib, L.; Beynor, J. I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Donlin, L. T.; Raychaudhuri, S., IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med 2021, 13, (1), 64.
- Chen, F.; Zhang, Y.; Sucgang, R.; Ramani, S.; Corry, D.; Kheradmand, F.; Creighton, C. J., Meta-analysis of host transcriptional responses to SARS-CoV-2 infection reveals their manifestation in human tumors. Scientific reports 2021, 11, (1), 2459.
- Garg, M.; Li, X.; Moreno, P.; Papatheodorou, I.; Shu, Y.; Brazma, A.; Miao, Z., Meta-analysis of COVID-19 single-cell studies confirms eight key immune responses. Scientific reports 2021, 11, (1), 20833.
- Müller, J. A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C. P.; Read, C.; Fois, G.; Eiseler, T.; Gehrmann, J.; van Vuuren, J.; Wessbecher, I. M.; Frick, M.; Costa, I. G.; Breunig, M.; Grüner, B.; Peters, L.; Schuster, M.; Liebau, S.; Seufferlein, T.; Stenger, S.; Stenzinger, A.; MacDonald, P. E.; Kirchhoff, F.; Sparrer, K. M. J.; Walther, P.; Lickert, H.; Barth, T. F. E.; Wagner, M.; Münch, J.; Heller, S.; Kleger, A., SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nature Metabolism 2021, 3, (2), 149-165.
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y., Single-cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med 2022, 12, (3), e694.
- Chua, R. L.; Lukassen, S.; Trump, S.; Hennig, B. P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M. T.; Kazmierski, J.; Timmermann, B.; Twardziok, S.; Schneider, S.; Machleidt, F.; Müller-Redetzky, H.; Maier, M.; Krannich, A.; Schmidt, S.; Balzer, F.; Liebig, J.; Loske, J.; Suttorp, N.; Eils, J.; Ishaque, N.; Liebert, U. G.; von Kalle, C.; Hocke, A.; Witzenrath, M.; Goffinet, C.; Drosten, C.; Laudi, S.; Lehmann, I.; Conrad, C.; Sander, L.-E.; Eils, R., COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nature Biotechnology 2020, 38, (8), 970-979.
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; Wei, X.; Sun, H. X.; Ye, B.; Wu, Y.; Zhang, N.; Lei, G.; Yu, L.; Yan, J.; Diao, G.; Meng, F.; Bai, C.; Mao, P.; Yu, Y.; Wang, M.; Yuan, Y.; Deng, Q.; Li, Z.; Huang, Y.; Hu, G.; Liu, Y.; Wang, X.; Xu, Z.; Liu, P.; Bi, Y.; Shi, Y.; Zhang, S.; Chen, Z.; Wang, J.; Xu, X.; Wu, G.; Wang, F. S.; Gao, G. F.; Liu, L.; Liu, W. J., Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity 2020, 53, (3), 685-696 e3.
- Ogura, H.; Gohda, J.; Lu, X.; Yamamoto, M.; Takesue, Y.; Son, A.; Doi, S.; Matsushita, K.; Isobe, F.; Fukuda, Y.; Huang, T.-P.; Ueno, T.; Mambo, N.; Murakami, H.; Kawaguchi, Y.; Inoue, J.-i.; Shirai, K.; Yamasaki, S.; Hirata, J.-I.; Ishido, S., Dysfunctional Sars-CoV-2-M protein-specific cytotoxic T lymphocytes in patients recovering from severe COVID-19. Nature communications 2022, 13, (1), 7063.
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A. G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; Rameau, P.; Catelain, C.; Alfaro, A.; Dussiau, C.; Friedrich, C.; Sourdeau, E.; Marin, N.; Szwebel, T. A.; Cantin, D.; Mouthon, L.; Borderie, D.; Deloger, M.; Bredel, D.; Mouraud, S.; Drubay, D.; Andrieu, M.; Lhonneur, A. S.; Saada, V.; Stoclin, A.; Willekens, C.; Pommeret, F.; Griscelli, F.; Ng, L. G.; Zhang, Z.; Bost, P.; Amit, I.; Barlesi, F.; Marabelle, A.; Pène, F.; Gachot, B.; André, F.; Zitvogel, L.; Ginhoux, F.; Fontenay, M.; Solary, E., Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, (6), 1401-1418 e18.
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; De Domenico, E.; Wendisch, D.; Grasshoff, M.; Kapellos, T. S.; Beckstette, M.; Pecht, T.; Saglam, A.; Dietrich, O.; Mei, H. E.; Schulz, A. R.; Conrad, C.; Kunkel, D.; Vafadarnejad, E.; Xu, C. J.; Horne, A.; Herbert, M.; Drews, A.; Thibeault, C.; Pfeiffer, M.; Hippenstiel, S.; Hocke, A.; Mller-Redetzky, H.; Heim, K. M.; Machleidt, F.; Uhrig, A.; Bosquillon de Jarcy, L.; Jrgens, L.; Stegemann, M.; Glsenkamp, C. R.; Volk, H. D.; Goffinet, C.; Landthaler, M.; Wyler, E.; Georg, P.; Schneider, M.; Dang-Heine, C.; Neuwinger, N.; Kappert, K.; Tauber, R.; Corman, V.; Raabe, J.; Kaiser, K. M.; Vinh, M. T.; Rieke, G.; Meisel, C.; Ulas, T.; Becker, M.; Geffers, R.; Witzenrath, M.; Drosten, C.; Suttorp, N.; von Kalle, C.; Kurth, F.; Hndler, K.; Schultze, J. L.; Aschenbrenner, A. C.; Li, Y.; Nattermann, J.; Sawitzki, B.; Saliba, A. E.; Sander, L. E., Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, (6), 1419-1440 e23.
- Sinha, S.; Rosin, N. L.; Arora, R.; Labit, E.; Jaffer, A.; Cao, L.; Farias, R.; Nguyen, A. P.; de Almeida, L. G. N.; Dufour, A.; Bromley, A.; McDonald, B.; Gillrie, M. R.; Fritzler, M. J.; Yipp, B. G.; Biernaskie, J., Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nature Medicine 2022, 28, (1), 201-211.
- Su, Y.; Yuan, D.; Chen, D. G.; Ng, R. H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; Kornilov, S. A.; Scherler, K.; Pavlovitch-Bedzyk, A. J.; Dong, S.; Lausted, C.; Lee, I.; Fallen, S.; Dai, C. L.; Baloni, P.; Smith, B.; Duvvuri, V. R.; Anderson, K. G.; Li, J.; Yang, F.; Duncombe, C. J.; McCulloch, D. J.; Rostomily, C.; Troisch, P.; Zhou, J.; Mackay, S.; DeGottardi, Q.; May, D. H.; Taniguchi, R.; Gittelman, R. M.; Klinger, M.; Snyder, T. M.; Roper, R.; Wojciechowska, G.; Murray, K.; Edmark, R.; Evans, S.; Jones, L.; Zhou, Y.; Rowen, L.; Liu, R.; Chour, W.; Algren, H. A.; Berrington, W. R.; Wallick, J. A.; Cochran, R. A.; Micikas, M. E.; Wrin, T.; Petropoulos, C. J.; Cole, H. R.; Fischer, T. D.; Wei, W.; Hoon, D. S. B.; Price, N. D.; Subramanian, N.; Hill, J. A.; Hadlock, J.; Magis, A. T.; Ribas, A.; Lanier, L. L.; Boyd, S. D.; Bluestone, J. A.; Chu, H.; Hood, L.; Gottardo, R.; Greenberg, P. D.; Davis, M. M.; Goldman, J. D.; Heath, J. R., Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, (5), 881-895 e20.
- Varga, Z.; Flammer, A. J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A. S.; Mehra, M. R.; Schuepbach, R. A.; Ruschitzka, F.; Moch, H., Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, (10234), 1417-1418.
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J.; Dong, L.; Cui, X.; Miao, Y.; Wang, D.; Dong, J.; Xiao, C.; Chen, W.; Wang, H., Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov 2020, 6, 31.
- Lee, J. S.; Park, S.; Jeong, H. W.; Ahn, J. Y.; Choi, S. J.; Lee, H.; Choi, B.; Nam, S. K.; Sa, M.; Kwon, J. S.; Jeong, S. J.; Lee, H. K.; Park, S. H.; Park, S. H.; Choi, J. Y.; Kim, S. H.; Jung, I.; Shin, E. C., Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol 2020, 5, (49).
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; Liu, L.; Amit, I.; Zhang, S.; Zhang, Z., Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 2020, 26, (6), 842-844.
- Chua, R. L.; Lukassen, S.; Trump, S.; Hennig, B. P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M. T.; Kazmierski, J.; Timmermann, B.; Twardziok, S.; Schneider, S.; Machleidt, F.; Müller-Redetzky, H.; Maier, M.; Krannich, A.; Schmidt, S.; Balzer, F.; Liebig, J.; Loske, J.; Suttorp, N.; Eils, J.; Ishaque, N.; Liebert, U. G.; von Kalle, C.; Hocke, A.; Witzenrath, M.; Goffinet, C.; Drosten, C.; Laudi, S.; Lehmann, I.; Conrad, C.; Sander, L. E.; Eils, R., COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol 2020, 38, (8), 970-979.
- Marschner, I. C., Estimating age-specific COVID-19 fatality risk and time to death by comparing population diagnosis and death patterns: Australian data. BMC Med Res Methodol 2021, 21, (1), 126.
- Wang, T.; Zhang, X.; Liu, Z.; Yao, T.; Zheng, D.; Gan, J.; Yu, S.; Li, L.; Chen, P.; Sun, J., Single-cell RNA sequencing reveals the sustained immune cell dysfunction in the pathogenesis of sepsis secondary to bacterial pneumonia. Genomics 2021, 113, (3), 1219-1233.
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; Cuapio, A.; Ask, E. H.; Hull, R. M.; Haroun-Izquierdo, A.; Schaffer, M.; Klingström, J.; Folkesson, E.; Buggert, M.; Sandberg, J. K.; Eriksson, L. I.; Rooyackers, O.; Ljunggren, H. G.; Malmberg, K. J.; Michaëlsson, J.; Marquardt, N.; Hammer, Q.; Strålin, K.; Björkström, N. K., Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol 2020, 5, (50).
- Zhang, W.; Xu, X.; Fu, Z.; Chen, J.; Chen, S.; Tan, Y., PathogenTrack and Yeskit: tools for identifying intracellular pathogens from single-cell RNA-sequencing datasets as illustrated by application to COVID-19. Front Med 2022, 16, (2), 251-262.
| 1 | According to the Elsevier's abstract and citation database (Scopus) database. Retrieved on 31 July 2023. |
| 2 | According to the Scopus database. Retrieved on 31 July 2023. |
| 3 | At the China National GeneBank DataBase (db.cngb.org/cnsa). |
| 4 | This is an accession identifier (ID) in the Chinese National Genomics Data Center (NGDC). |
| 5 | In the European Genome-Phenome Archive. |
| 6 | This is an accession ID in the Chinese National Genomics Data Center (NGDC). |
| 7 | This is an accession ID in the Sequence Read Archive (SRA). |
| 8 | These are libraries for single-cell T cell-receptor (TCR) sequencing (scTCR-seq), and TCR-FACS-index-ATAC sequencing (Ti-ATAC-seq). |

Table 1.
There are many high-throughput datasets related to COVID-19 in the GEO database. If one searched Gene Expression Omnibus (GEO) with the keyword SARS-CoV-2, they would find as many as 800 data sets. The GEO database was searched with two alternative keywords: SARS-CoV-2, or COVID-19. The datasets were assigned a species label, depending on the species from which tissues and sampled and profiled. Numbers of datasets were given in total, or for the following four species: H. sapiens, C. sabaeus, M. musculus, or in SARS-CoV-2. Numbers of datasets according to an experimental platform were also given (either NGS, or microarray, or chromatin immunoprecipitation — ChIP-seq). What was the most typical workflow? Firstly, polyadenylated mRNAs were typically sequenced in parallel from survey or experimental samples. Subsequently, NGS reads were aligned to a reference genome, and transcript abundance was quantified. Next, differentially expressed genes — DEGs — were identified using popular biostatistics software tools or packages. The DEGs were then categorized into functional classes using standard functional genomic tools and databases.
Table 1.
There are many high-throughput datasets related to COVID-19 in the GEO database. If one searched Gene Expression Omnibus (GEO) with the keyword SARS-CoV-2, they would find as many as 800 data sets. The GEO database was searched with two alternative keywords: SARS-CoV-2, or COVID-19. The datasets were assigned a species label, depending on the species from which tissues and sampled and profiled. Numbers of datasets were given in total, or for the following four species: H. sapiens, C. sabaeus, M. musculus, or in SARS-CoV-2. Numbers of datasets according to an experimental platform were also given (either NGS, or microarray, or chromatin immunoprecipitation — ChIP-seq). What was the most typical workflow? Firstly, polyadenylated mRNAs were typically sequenced in parallel from survey or experimental samples. Subsequently, NGS reads were aligned to a reference genome, and transcript abundance was quantified. Next, differentially expressed genes — DEGs — were identified using popular biostatistics software tools or packages. The DEGs were then categorized into functional classes using standard functional genomic tools and databases.
| Keyword | Species | Total | NGS | Microarray | ChIP-seq |
|---|---|---|---|---|---|
| SARS-CoV-2 | All | 808 | 642 | 22 | 52 |
| H. sapiens | 596 | 472 | 16 | 42 | |
| C. sabaeus | 14 | 10 | n/a | 1 | |
| M. musculus | 16 | 14 | n/a | 2 | |
| SARS-CoV-2 | 29 | 20 | n/a | n/a | |
| COVID-19 | All | 536 | 407 | 22 | 33 |
| H. sapiens | 536 | 407 | 22 | 33 | |
| C. sabaeus | 6 | 5 | n/a | 1 | |
| M. musculus | 10 | 9 | 1 | 1 | |
| SARS-CoV-2 | 19 | 14 | n/a | n/a |
Table 2.
Examples of bulk-sample RNA-seq studies of COVID-19. Specifications of biological models, sample types, and conclusions were given for transcriptional studies related to SARS-CoV-2. The following information was also given: PubMed unique identifier (PMID), data set identification number (ID), impact factor (IF) of a journal, number of citations, and technology of a high-throughput platform. The average number of citations was 472.
Table 2.
Examples of bulk-sample RNA-seq studies of COVID-19. Specifications of biological models, sample types, and conclusions were given for transcriptional studies related to SARS-CoV-2. The following information was also given: PubMed unique identifier (PMID), data set identification number (ID), impact factor (IF) of a journal, number of citations, and technology of a high-throughput platform. The average number of citations was 472.
| Year of publication and reference. | Specification of the biological model and sample type. | Main conclusions of the study. | (1) GEO ID. (2) PMID. (3) Dataset size. (4) Number of samples. |
(1) IF. (2) Citations 1. |
Platform. |
|---|---|---|---|---|---|
| 2020 [29]. | Human cell lines were infected in vitro with several common viruses. Specifically, H1N1 influenza A virus (IAV), human respiratory syncytial virus (RSV), human parainfluenza virus 3 (HPIV3), and SARS-CoV-2 were used to infect human lung cancer cell lines. | Transcriptional responses did not appear to stop the replication of SARS-CoV-2. Instead, they suggested a protracted inflammatory response. | (1) GSE147507. (2) PMID 32416070. (3) 2.7 Mb. (4) 110. |
(1) IF = 67. (2) 2771 citations. |
Illumina NextSeq 500 (H. sapiens). |
| 2020 [25]. | Immune cells isolated from life COVID-19 patients were profiled with a gene expression platform. | A comprehensive atlas of immune modulations during COVID-19 was presented. The authors identified three significant immunotypes related to disease severity. | (1) GSE152418. (2) PMID 32788292. (3) 1.6 Mb. (4) 34. |
(1) IF = 48. (2) 629 citations. |
Illumina NovaSeq 6000 (H. sapiens). |
| 2020 [26]. | PBMCs from life COVID-19 patients were profiled in a longitudinal approach. (PBMCs from thirteen patients were isolated, and there were five time points profiled for each patient.) In total, scRNA-seq data were obtained for 358,930 cells, and there were 10,900 cells per sample on average. | COVID-19 was associated with a set of dynamic changes in expression patterns in PBMCs. In particular, severe disease induced both hypoxic and pro-inflammatory signalling. There was also an impaired activation of the interferon pathway. | (1) GSE161777. (2) PMID 33296687. (3) 0.5 Mb. (4) 101. |
(1) IF = 43. (2) 189 citations. |
Illumina MiSeq andIllumina NovaSeq 6000 (H. sapiens). |
| 2020 [27]. | PBMCs from 7 hospitalized patients with COVID-19, four of whom had ARDS, as well as six health controls. | Strong downregulation of human leukocyte antigen (HLA) of class I and class II, and interferon-driven inflammatory reactions in monocytes are characteristics for infection with SARS-CoV-2. | (1) GSE150728. (2) PMID 32514174. (3) 372 Mb. (4) 13. |
(1) IF = 83. (2) 896 citations. |
Illumina NovaSeq 6000 (H. sapiens). |
| 2021 [30]. | Lung cells from dead COVID-19 patients were profiled and compared with normal cells. | Lungs from individuals with COVID-19 were very inflamed, and T cell responses were impaired. There were also fewer epithelial cells in diseased lungs. Instead, there was an increase in numbers of macrophages / monocytes, neuronal cells, and fibroblasts. | (1) GSE171524. (2) PMID 33915568. (3) 1601.4 Mb. (4) 27. |
(1) IF = 70. (2) 213 citations. |
Illumina NovaSeq 6000 (M. musculus). |
| 2021 [31]. | Blood, lungs, and airways of dead COVID-19 patients were profiled. | Results of the expression screen underlined the importance of the changes in expression of immunological genes. Note that not only structural cell-types like epithelia, but also infiltrating immune cells were the source of the signal. | (1) GSE147507. (2) PMID 33782412. (3) 1800.9 Mb. (4) 110. |
(1) IF = 5. (2) 75 citations. |
Illumina NextSeq 500 (H. sapiens). |
| 2021 [33]. | CSF was collected from 7 males and one female with neuro-COVID (aged from 53 to 82). | This study pointed at the expansion of dedifferentiated monocytes, as well as at exhausted CD4+ T cells in the CSF of patients with neuro-COVID. | (1) GSE26495. (2) PMID 33382973. (3) 83 Mb. (4) 16. |
(1) IF = 43. (2) 84 citations. |
Illumina NextSeq 500, or Illumina NovaSeq 6000 (H. sapiens). |
| 2021 [32]. | Cell culture of human pancreatic islets. | As a result of the infection of cultured pancreatic islets with the SARS-CoV-2 virus, the number of insulin-secretory granules in β-cells was reduced, and glucose dependent insulin secretion was impaired. Infection was associated with morphological, transcriptional and functional changes in β-cells of the islets. As a result, glucose-stimulated insulin secretion was impaired. | (1) GSE159717. (2) PMID 33536639. (3) 3.9 Mb. (4) 4. |
(1) IF = 21. (2) 318. |
HiSeq 4000 instrument (Illumina). |
| 2022 [28]. | The study investigated tissue samples isolated from COVID-19 patients. For example, blood samples were taken for transcriptome profiling from 21 patients. In total, 57,049 single-cell transcriptomes of PBMCs were sequenced. | Endothelial injury and pathological thrombotic events were frequently a part of clinical manifestations of COVID-19. These pathological changes were positively correlated with increased activity of myeloid cells. | (1) GSE208337. (2) PMID 35895716. (3) 573.4 Mb. (4) 105. |
(1) IF = 13. (2) 7 citations. |
Illumina NovaSeq 6000 (H. sapiens ). |
| 2022 [34]. | Conjunctival epithelium was isolated from eyes of dead patients and cultured as organotypic clusters. 15,821 cell transcriptomes from 3 infected, 3 uninfected cultures were processed. | There was evidence of the entry of the coronavirus into ocular epithelial cells. At the same time, there was no evidence of productive viral replication in this cell type. | (1) GSE191232. (2) PMID 35750043. (3) 78 Mb. (4) 7. |
(1) IF = 7. (2) 2 citations. |
Table 3.
Examples of scRNA-seq studies of gene expression related to COVID-19. Specifications of biological models, sample types, and conclusions are given for transcriptional studies related to SARS-CoV-2. The following information was given: data set identification number (ID), impact factor (IF) of a journal, number of citations, and technology of a high-throughput platform. The average number of citations was 303.
Table 3.
Examples of scRNA-seq studies of gene expression related to COVID-19. Specifications of biological models, sample types, and conclusions are given for transcriptional studies related to SARS-CoV-2. The following information was given: data set identification number (ID), impact factor (IF) of a journal, number of citations, and technology of a high-throughput platform. The average number of citations was 303.
| Year of publication and reference. | Specification of the biological model and sample type. | Main conclusions of the study. | (1) GEO ID. (2) PMID. (3) Dataset size. (4) Number of samples. |
(1) IF. (2) Citations 2. |
Platform. |
|---|---|---|---|---|---|
| 2020 [60]. | Five COVID-19 patients and three healthy people donated their blood for isolation of PBMCs. | An atlas of single-cell gene expression was generated in both COVID-19 and influenza patients. Three signaling pathways were turned on in COVID-19: apoptosis, Signal transducer and activator of transcription 1 (STAT1), and interferon regulatory factor 3 (IRF3). Plasma cells were increased in COVID-19. | (1) CNP0001102 3. (2) PMID 32783921.(3) n/a.(4) n/a. |
(1) IF = 43. (2) 206 citations. |
DNBelab C4 library and DIPSEQ T1 sequencer. |
| 2020 [36]. | PBMCs from five healthy donors and 13 patients with COVID-19 (this included moderate, severe and convalescent cases). | COVID-19 induced acute inflammatory response with a strong induction of the interferon-alpha pathway. There were also many cytotoxic effector T cells. However, there was also evidence for disorganized interferon response and immune exhaustion in severe cases of COVID-19. | (1) HRA000150 4. (2) PMID 32788748. (3) n/a. (4) 64. |
(1) IF = 31. (2) 356 citations. |
Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [44]. | This is a single-cell analysis of broncho-alveolar immune cells from patients with COVID-19. Samples from 9 patients with COVID-19 and 3 healthy controls were sequenced. | A new protocol and a computational method were developed (Viral-Track) to detect infected cells. The method showed that SARS-CoV-2 had a detrimental effect on the host immune system. | (1) GSE145926. (2) PMID 32479746. (3) 225.1 Mb. (4) 21. |
(1) IF = 67. (2) 285 citations. |
Beijing Genomics Institute MGISEQ-2000 platform, scRNA-seq. |
| 2020 [37]. | PBMCs from 10 patients (either mild, severe, or control). | Subtle changes in percentages of subtypes of neutrophils or monocytes correlate with severity of COVID-19. | (1) E-MTAB-9221. (2) 32810439 PMID.(4) 10. |
(1) IF = 67. (2) 496 citations. |
10X Chromium droplet-based platform. Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [63]. | PBMCs from 18 COVID-19 patients (8 mild and 10 severe) were collected between day 3 and day 20 after symptoms were first diagnosed. A total of 48,266 single-cell expression profiles of PBMCs were analyzed along with 50,783 control PBMCs from publicly available datasets. | There are major alterations in the myeloid compartment during a SARS-CoV-2 infection. Inflammatory monocytes are increased in mild cases. Dysfunctional monocytes are increased in severe cases, and there is rapid myelopoiesis in severe cases, resulting in the production of immature neutrophils. | (1) GSE145926. (2) PMID 32479746. (3) 225.1 Mb. (4) 21. |
(1) IF = 67. (2) 805 citations. |
10X Chromium droplet-based platform. Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [59]. | Nasopharyngeal and bronchial samples from 19 COVID-19 patients with either moderate or critical disease. (There were also five healthy controls.) There were transcriptional profiles of 160,528 cells obtained from a total of 36 samples. The study identified 22 different cell types and states within the epithelial cell and immune cell populations. | In critical cases, there were stronger inflammatory interactions between epithelial and immune cells. Even more lung injury, respiratory failure, and inflammatory tissue damage was observed in critical cases. | (1) EGAS00001004481 5. (2) 32591762 PMID. (4) 36. |
(1) IF = 59. (2) 635 citations. |
Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2020 [39]. | PBMCs from hospitalized and non-hospitalized COVID-19 patients, and healthy non-exposed subjects, as well as PBMCs from subjects before and/or after receiving flu vaccination. | scRNA-seq conducted for CD4+ T cells from 40 COVID-19 patients showed that hospitalization resulted in increased fraction of cytotoxic follicular helper cells and cytotoxic T helper cells, as well as a reduction in regulatory T cells. | (1) GSE152522. (2) PMID 33096020. (3) 518 Mb. (4) 78. |
(1) IF = 67. (2) 300 citations. |
Illumina NovaSeq6000(H. sapiens), scRNA-seq. |
| 2021 [40]. | 52 samples of PBMCs were obtained from 13 patients with severe COVID-19. There were also 5 samples from 5 healthy donors. After cell sorting, there were 80,325 NKs obtained from the 68 samples of peripheral blood. From these NKs single-cell transcriptomes were generated. UMAP representation divided the transcriptomes into several types, for example effector I and II, terminally differentiated, transitional, and proliferating. | Severe COVID-19 is associated with early high serum levels of TGF-beta. High levels of this cytokine early in the course of infection are associated with the inhibition of interferon-driven activation of NKs, and impaired immunological response. | (1) GSE184329. (2) PMID 34695836. (3) 174.8 Mb. (4) 13. |
(1) IF = 65. (2) 104 citations. |
Illumina NextSeq500 (H. sapiens), scRNA-seq. |
| 2021 [47]. | Single-cell atlases of 24 lung, 19 heart, 16 liver, and 16 kidney tissue samples from COVID-19 autopsies were generated. | COVID-19 is characterized by failed tissue regeneration, and pathological re-modeling of diseased tissues. In the lungs, there were more fibroblasts and less epithelial cells in COVID-19 samples. The RNA of SARS-CoV-2 was enriched in phagocytic and ECs. In the heart, there was a reduction in the number of cardiomyocytes and pericytes. There was also more vascular ECs in the heart. | (1) GSE163530. (2) PMID 33915569. (3) 40.4 Mb. (4) 1194. |
(1) IF = 65. (2) 320 citations. |
NextSeq 550, Illumina NovaSeq 6000, NextSeq 550, Nanostring GeoMx 2020 Broad COVID Platform, scRNA-seq. |
| 2021 [46]. | Brain and choroid plexus samples isolated from dead patients with severe COVID-19 were profiled at a single cell level. (There were 65,309 high-quality nuclei.) | Considerable amount of evidence was presented suggesting that long-term inflammation is a part of both COVID-19 and neurodegenerative diseases. | (1) GSE159812. (2) PMID 34153974. (3) 944 Mb. (4) 30. |
(1) IF = 70. (2) 270 citations. |
Illumina NovaSeq 6000 (M. musculus), scRNA-seq. |
| 2021 [41]. | The study profiled COVID-19, MIS-C, as well as healthy pediatric and adult individuals using scRNA-seq. The results of scRNA-seq were correlated with disease severity, flow cytometry, antigen receptor repertoire analysis, and serum proteomics. | The study defined gene expression signatures associated with MIS-C, which could find application in clinical diagnostics to predict inflammatory complications of COVID-19. In particular, MIS-C tissues had increased expression levels of S100A-family alarmins, a signature of decreased antigen presentation, and increased cytotoxicity in NK and CD8+ T cells. | (1) GSE166489. (2) PMID 33891889. (3) 826.1 Mb. (4) 54. |
(1) IF = 43. (2) 118 citations. |
Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2021 [42]. | PBMCs were isolated from 5 healthy donors, 7 individuals who recovered from moderate disease, and 3 donors who recovered from severe COVID-19. The resulting 97.315 epigenomes of PBMCs were divided by UMAP representation into monocytes, effector, memory, naive, plasma cells, or NKs. | This study catalogued patterns of global re-modeling of the chromatin accessibility landscape in convalescing COVID-19 patients. These patterns suggested establishment of immunity against SARS-CoV-2 through immunological memory. | (1) HRA000562 6, PRJNA7180097. (2) PMID 34108657. (3) n/a. (4) n/a. |
(1) IF = 28. (2) 43 citations. |
Libraries8 were processed on HiSeq X Ten platform, Illumina. |
| 2021 [32]. | To understand the role of neutrophils during COVID-19 and to elucidate the effects of dexamethasone. | A single-cell atlas of COVID-19 neutrophil states and molecular mechanisms of action of dexamethasone was generated. See http://biernaskielab.ca/COVID_neutrophil. | (1) GSE157789. (2) 34782790. (3) 360 Mb. (4) 31. |
(1) IF = 53. (2) 90 citations. |
Sequencing was performed using Illumina NovaSeq S2 and SP 100. |
| 2022 [43]. | PBMCs from seven children with MIS-C (plus six healthy controls). | Immune responses by SARS-CoV-2 are regulated by long non-coding RNAs (lncRNAs). For example, lncRNA PIRAT forms a negative feedback loop with the PU.1 transcription factor, which promotes transcription of alarmins — that is proteins promoting inflammation in response to SARS-CoV-2. Inflammation in COVID-19 is promoted by down-regulation of PIRAT, and up-regulation of lung cancer associated transcript 1 (LUCAT1) — that is a lncRNA promoting inflammation. | (1) GSE142503. (2) PMID 35998224. (3) 16.7 Mb. (4) 15. |
(1) IF = 10. (2) 6 citations. |
Illumina NovaSeq 6000 (H. sapiens), scRNA-seq. |
| 2022 [38]. | 209 COVID-19 patients (plus 100 patients with post-acute COVID-19) and 457 healthy controls were investigated for 2 to 3 months: from initial diagnosis to convalescence. The patients were investigated at the time of clinical diagnosis, acute disease, as well as at the time of convalescence. | Diabetes, viremia, and auto-immune conditions were risk factors associated with the diagnosis of PASC. | (1) E-MTAB-10129. (2) PMID 32810438. (3) 5.2 Mb. (4) 309. |
(1) IF = 67. (2) 802 citations. |
Illumina NovaSeq 6000. |
| 2023 [48]. | The study focused on single-nucleus RNA-seq performed on samples of frozen lungs from 7 deceased COVID-19 patients, 6 pairs of lungs from patients with idiopathic pulmonary fibrosis (IPF), and 12 individuals from a control group. There were 38,794 cell nuclei from a vascular fraction, which could be fractioned into 14 different subtypes of endothelial type. There were also 38,794 non-vascular nuclei. The goal was to focus on ECs and on a comparison between IPF and COVID-19. | There was an enrichment of genes involved in cellular stress, and a signature of diminished immunomodulation, and impaired vessel integrity. There was also a set of receptor-ligand interactions that were specifically enriched or depleted in either COVID-19 or IPF. | (1) GSE159585. (2) PMID 35998078. (3) 488 Mb. (4) 53. |
(1) IF = 13. (2) 7 citations. |
Illumina HiSeq 4000 / NovaSeq 6000 (H. sapiens), scRNA-seq |
Table 4.
Meta-analyses. This table lists high-impact meta-analyses of RNA-seq and scRNA-seq datasets focusing on the etiology of COVID-19. The average number of citations was 287[9].
Table 4.
Meta-analyses. This table lists high-impact meta-analyses of RNA-seq and scRNA-seq datasets focusing on the etiology of COVID-19. The average number of citations was 287[9].
| Year of publication and reference. | Goal. | Conclusions. | Datasets. | Computational methods. | (1) IF. (2) Citations.(3) PMID. |
|---|---|---|---|---|---|
| 2020 [49]. | A comprehensive meta-analysis of scRNA-seq datasets to identify cell subsets expressing ACE2 and, therefore, targeted by SARS-CoV-2. | ACE2 and TMPRSS2 promote cellular entry of SARS-CoV-2. Type I interferons, and to a lesser extent type II interferons, upregulate ACE2. Cells vulnerable to infection were identified in the lungs. | GSE148829, GSE135069, GSE19190, GSE22147. | Re-analysis with Drop-Seq Computational Protocol v2.0, Seurat. Meta-analysis with UMAP, PCA, GSEA, etc. | (1) IF = 65. (2) 1574 citations. (3) PMID 32413319. |
| 2020 [50]. | To investigate the impact of smoking on COVID-19 utilizing scRNA-seq gene expression data from lung and airway epithelial samples from human, mouse, or rat. | For ACE2 levels of protein and mRNA were highly correlated (r = 0.82, P-value < 0.0001) across 53 cell lines. Smokers had higher levels of gene expression of ACE2 in the lungs. ACE2 expression was uncorrelated with age or sex. However, inflammation in the lungs was linked to the induction of expression of ACE2. ACE2 was also stimulated in expression by interferon-signalling. | GSE132040, GSE53960, GSE53960, GSE34378, GSE53960, GSE44555, GSE132040, GSE6591, GSE80680, GSE1643, GSE18344, GSE13933, GSE22047, GSE64614, GSE76925, GSE79209, GSE121611, GSE122960, GSE134174, GSE75715, GSE39059, GSE135188, GSE57148, GSE103174, GSE2052, GSE47460, GSE43696, GSE16538, GSE3100, GSE11056, GSE86623, GSE41789, GSE32138, GSE32138, GSE47963, GSE100504, GSE51392, and GSE19392 plus some samples from TCGA, the GTEx portal, the Human Cell Atlas, the Human Protein Atlas, and the Single-Cell Expression Atlas. | The meta-analysis was performed using Python, Excel, and Graphpad Prism. Regressions were performed using Python using ordinary least squares and the statsmodels package. Analysis of single-cell expression data was performed using Python, Scanpy, and Multicore-TSNE packages. Variable genes were prioritized utilizing the Seurat function in Scanpy. The variable genes were then analyzed using PCA. | (1) IF = 14. (2) 275 citations. (3) PMID 32425701. |
| 2020 [51]. | To determine expression patterns for ACE2 or other receptors for SARS-CoV-2 in the respiratory mucosa using scRNA-seq data. | The levels of mRNA and protein of ACE2 are very low in the upper airways and in the lungs. However, there is a mechanism dynamically regulating ACE2 expression in response to SARS-CoV-2. | GSE19190, GSE11906, GSE4302, GSE67472, GSE37147, GSE108134, GSE135893, the FANTOM5 dataset, and few proteomics datasets including the Human Proteome Map. | The Cell Ranger pipeline, UMAP, Zenbu genome browser, R packages: pheatmap, Seurat, ggplot2. | (1) IF = 25. (2) 107 citations. (3) PMID 32675206. |
| 2020 [53]. | scRNA-seq data were used to investigate how kidney diseases or medications may alter ACE2 expression in kidneys. | ACE2 expression in proximal tubular epithelial cells of the kidney facilitated infection with SARS-CoV-2. | Nephrocell data were archived at http://nephrocell.miktmc.org. COVID-19 kidney data were archived at https://hb.flatironinstitute.org/covid-kidney. | scRNA-seq data were analyzed according to protocols of the Kidney Precision Medicine Project (see https://www.kpmp.org/for-researchers#protocols). | (1) IF = 8. (2) 52 citations. (3) PMID 32675206. |
| 2021 [54]. | scRNA-seq data were used to identify cellular phenotypes shared across disparate inflammatory diseases. | Similarities in gene expression and signalling between COVID-19 and other inflammatory diseases are uncovered. | GSE134809, GSE122960, GSE145926, GSE155249, GSE47189, GSE147507, GSE168710, phs001457.v1.p1, phs001529.v1.p1, phs001457.v1.p1, SCP259. | A meta-analysis and integration pipeline was constructed, which models and removes the effects of technology, tissue of origin, and donor. The meta-analysis built a reference library for immune cells in normal body and in different diseases. | (1) IF = 12. (2) 83 citations. (3) PMID 33879239. |
| 2021 [52]. | Muus et al. performed a meta-analysis of receptor genes for SARS-CoV-2 by looking at gene expression in a meta-analysis of 31 lung scRNA-seq studies. | An atlas of cell type-specific associations of age, sex, and smoking with expression levels of ACE2 (and other co-receptors for SARS-CoV-2). | SCP865, SCP895, SCP891, SCP903, SCP871, SCP870, SCP874, SCP878, SCP887, SCP899, SCP898, SCP902, SCP894, SCP869, SCP872, SCP866, SCP1240, SCP1241, SCP868, SCP877, SCP867, SCP897, SCP886, SCP879, SCP860, SCP881, SCP875, SCP876, SCP900, SCP892, SCP890, SCP889, SCP880, SCP882, SCP882, GSE102592, GSE103918, GSE104600, GSE107747, GSE108571, GSE109037, GSE110973, GSE111014, GSE111360, GSE112570, GSE112845, GSE113036, GSE114530, GSE114724, GSE114802, GSE115149, GSE115189, GSE117211, GSE117403, GSE117824, GSE118127, GSE119212, GSE119506, GSE119507, GSE119561, GSE119594, GSE120446, GSE121267, GSE121600, GSE122342, GSE122703, GSE122960, GSE123926, GSE124263, GSE124334, GSE124472, GSE124494, GSE124898, GSE125680, GSE127472, GSE128066, GSE128169, GSE128518, GSE128889, GSE129845, GSE130073, GSE130117, GSE130151, GSE130238, GSE130318, GSE130430, GSE130888, GSE131685, GSE132802, GSE133704, GSE134809, GSE135618, GSE135929, GSE136103, GSE136314, GSE136394, GSE139249, GSE139324, GSE98201. | Data generated by Chromium instrument 10X were integrated using the Cell Ranger pipeline. Datasets were post-processed using Python harmony-pytorch (for batch correction and leiden clustering) of Scanpy. | (1) IF = 87. (2) 183 citations. (3) PMID 33654293. |
| 2021 [55]. | Chen et al. used cell-line and bulk-tissue RNA-seq to detect overlap between host transcriptional responses observed in cancer and those observed in COVID-19 in response to SARS-CoV-2. | There were many similarities in host-disease interactions between cancer and COVID-19. In particular, immune cell infiltration and inflammation. | GSE147507, GSE148729, GSE36969, GSE59185, GSE68820, GSE119856, GSE115770, GSE147507, GSE146507, GSE156063. | All data were public and derived either from GEO or from TCGA. Pathway analysis was performed using GO, wikiPathways, SigTerms software. Standard statistical methods were used, for example two-sided p-values, log-transformed gene expression values, FDR correction for multiple testing, and heat-maps. | (1) IF = 4.3. (2) 12 citations. (3) PMID 33510359. |
| 2021 [56]. | Garg et al. set up a multi-dataset that included samples from 9 scRNA-seq studies. | The multi-dataset consisted of 159 samples, deriving from 7 medical procedures focused on PBMCs and 2 focused on BALF. 8 out of 20 evaluated hypotheses were confirmed. | PRJCA002413, PRJCA002564, GSE149689, PRJCA002579, GSE150728, GSE145926, GSE147143, and EGAS00001004481. | The SCANPY protocol was used for the meta-analysis. The following steps were included: normalization of the datasets, log-transformation of the data, selection of genes variable in expression levels, and PCA. Harmony was used to integrate data from different samples. UMAP was then used to cluster, visualize, and annotate gene expression data by cell type. | (1) IF = 4.3. (2) 12 citations. (3) PMID 34675242. |
Table 5.
DEGs and DEG sets in scRNA-seq datasets in severe COVID-19.
| Cell or tissue type | DEG genes | DEG pathways | Reference |
|---|---|---|---|
| PBMCs. | ISG15 ubiquitin like modifier (ISG15) ↑ Interferon induced protein 44 like (IFI44L) ↑ MX dynamin like GTPase 1 (MX1) ↑ XIAP associated factor 1 (XAF1) ↑ |
Pro-inflammatory cytokines ↑ Cytokine receptors ↑ Interferon-responsive TFs ↑ Response to type 1 interferon signaling ↑ Defense response to virus signaling ↑ Endoplasm and protein-unfolding ↑ Regulation of chromosome organization ↑ DNA conformation change ↑ |
[60]. |
| Thrombospondin 1 (THBS1) ↑ |
Neutrophil degranulation ↑ Plate activation, signaling and aggregation ↑ Semaphorin interactions ↑ Crosslinking of collagen fibrils ↑ Interleuking-1 family signaling ↑ Platelet degranulation ↑ |
[28]. | |
| lncRNA LUCAT1 ↑ CXCL2 ↑ IL-6 ↑ lncRNA PIRAT ↓ |
Hematopoietic cell lineage ↑ Cytokine-cytokine receptor interaction ↑ Chemokine signaling pathway ↑ |
[43]. | |
| Immunoglobulin heavy constant alpha 1 (IGHA1) ↑ Immunoglobulin heavy constant gamma 1 (IGHG1) ↑ Lactotransferrin (LTF) ↑ Interferon regulatory factor 1 (IRF1) ↑ Signal transducer and activator of transcription 3 (STAT3) ↑ Hypoxia inducible factor 1 subunit alpha (HIF1A) ↑ RAR related orphan receptor C (RORC) ↓ |
IL-1β and vasodilatory signaling ↑ IFN-related transcripts ↑ myeloid-cell-mediated immunity ↑ neutrophil degranulation ↑ erythroid cell differentiation ↑ cell differentiation pathway ↑ hypoxic signaling ↑ inflammation and IFN signaling ↑ ribosomal structural proteins ↓ |
[26]. | |
| HLA-DPB1 and HLA-DMA in monocytes ↓ |
Type I interferon-driven inflammatory signature in monocytes ↑ |
[27]. | |
| Interferon Induced Transmembrane Protein 2 (IFITM2) ↑ Interferon-induced transmembrane protein 3 (IFITM3) ↑ Interferon-induced protein 20 (ISG20) ↑ Interferon-induced protein 15 (ISG15) ↑ |
Interferon gamma response ↑ |
[72]. | |
| Interferon-ɑ response upregulation ↑ |
HLA-class II downregulation in CD14+ monocytes ↓ |
[56]. | |
| ECs. | Heat shock protein 90 alpha family class A member 1 (HSP90AA1) ↑ Heat shock protein family A (Hsp70) member 1A (HSPA1A) ↑ TIMP metallopeptidase inhibitor 1 (TIMP1) ↑ Fibrillin 1 (FBN1) ↑ Matrix metallopeptidase 16 (MMP16) ↑ Collagen type XV alpha 1 chain (COL15A1) ↑ Indoleamine 2,3-dioxygenase 1 (IDO1) ↑ Intercellular adhesion molecule 1 (ICAM1) ↓ Interferon regulatory factor 1 (IRF1) ↓ Cadherin 5 (CDH5) ↓ Integrin subunit beta 1 (ITGB1) ↓ Member of RAS oncogene family (RAP1B) ↓ Cell division cycle 42 (CDC42) ↓ Occludin (OCLN) ↓ Vinculin (VCL) ↓ Sphingosine-1-phosphate receptor 1 (S1PR1) ↓ Protein C receptor (PROCR) ↓ Thrombomodulin (THBD) ↓ |
Genes involved in cellular stress ↑ Heat shock proteins ↑ Genes involved in antigen presentation ↑ Hypoxia signalling ↑ Extracellular matrix (ECM) interactions ↑ ECM production/remodeling ↑ Immune system regulation ↓ Vessel maintenance/integrity ↓ Inflammation ↓ Angiogenesis ↓ Cell–cell adhesion ↓ Chemokines/cytokines ↓ TNF and JAK/STAT signalling ↓ |
[48]. |
| Lungs. | ACE2 ↑ |
Interferon response ↑ |
[49,50,51]. |
| ACE2 ↑ TMPRSS2 ↑ CTSL ↑ |
Interferon response ↑ |
[52]. | |
| Lung cancer cell lines. | Interferon Gamma Receptor 1 (IFNGR1) ↑ Interferon Gamma Receptor 2 (IFNGR2) ↑ Colony Stimulating Factor 2 (CSF2) ↑ Colony Stimulating Factor 3 (CSF3) ↑ C-X-C Motif Chemokine Ligand 1 (CXCL1) ↑ C-X-C Motif Chemokine Ligand 2 (CXCL2) ↑ Interleukin 1 Alpha (IL1A) ↑ Interleukin 1 Beta (IL1B) ↑ Interleukin 6 (IL6) ↑ Tumor Necrosis Factor Superfamily Member 14 (TNFSF14) ↑ |
Type II interferon signaling ↑ Immune response ↑ Response to stress ↑ Cytokine-mediated signaling ↑ Inflammatory response ↑ Cytokine activity ↑ Extracellular space ↑ Growth factor receptor binding ↑ Response to virus ↑ |
[55]. |
| Kidney. | ACE2 ↑ |
Interferon response ↑ |
[53]. |
Table 6.
Gene expression effects on gene sets in severe COVID-19.
| Effects on DEGs. | References. |
|---|---|
| Increased interferon response, interferon signaling, interferon-responsive TFs. | [26,27,49,50,51,52,53,55,60,72]. |
| Increased immune or inflammatory responses. | [27,55,60]. |
| Increased expression of cytokines, chemokines, or receptors. | [43,48,55,60]. |
| Increased interleukin-1 family signaling. | [28,55]. |
| Increased hypoxic signalling. | [26,48]. |
| Diminished immune system regulation, angiogenesis, and vessel integrity. | [48,56]. |
Table 7.
Changes in proportions of cell types detected by scRNA-seq in case of inflammatory COVID-19.
Table 7.
Changes in proportions of cell types detected by scRNA-seq in case of inflammatory COVID-19.
| Tissue type. | Reference. | Cell type increasing in frequency in severe COVID-19. | Less common cell type. |
|---|---|---|---|
| PBMCs. | [56]. | Plasma cells. Increased B-cell clonal expansion. |
Regulatory T cells. |
| [60]. | Plasmablasts. | Lymphocytes. | |
| [72]. | Plasmablasts. | n/a | |
| [28]. | Myeloid cells. | n/a | |
| [27]. | Developing neutrophils.CD14+ monocytes.Plasmablasts. | CD16+ monocytes.Plasmacytoid dendritic cells, conventional dendritic cells, and NK cells. | |
| [39]. | Cytotoxic follicular helper cells, and cytotoxic T helper cells. | Regulatory T cells. | |
| [73]. | Highly cytotoxic NK cells containing high levels of cytotoxic proteins such as perforin. | Unarmed NK cells. | |
| [70]. | Activated macrophages. | n/a | |
| BALF. | [44]. | Recruited macrophages, monocytes, or neutrophils. | Alveolar macrophages. |
| [70]. | Neutrophils. | Basal epithelial cells. | |
| CSF. | [33]. | De-differentiated monocytes, and CD4+ T cells. | n/a |
| Lungs. | [30]. | Fibroblasts, myeloid and neuronal cells. | Antigen presenting cells, epithelia. |
Table 8.
The transcription of SARS-CoV-2 was confirmed in several studies.
| Tissue type. | Positive cell types. | Type of evidence. | Computational method. | Reference. |
|---|---|---|---|---|
| If detected, transcription of the viral genome in swabs or brush specimens was used to confirm the diagnosis of COVID-19. | n/a | The entire SARS-CoV-2 genome sequence was annotated as one viral ‘gene’. The viral gene was appended to the hg19 annotation gtf file. All reads aligning to the SARS-CoV-2 genome per sample were aggregated and divided by the total number of reads in that sample. | Transcripts were aligned to a customized reference genome in which the SARS-CoV-2 genome (Refseq-ID: NC_045512) was added as an additional chromosome to the human reference genome hg19. Viral load was calculated on the raw data matrices output by CellRanger. | [59]. |
| BALF from severe and mild COVID-19 patients. | Epithelial cells and macrophages. | Viral mRNAs were identified among scRNA-seq reads that did not map to the human genome. | Viral-Track was an R-based pipeline that utilized the STAR algorithm to align reads to the SARS-CoV-2 genomes. | [44]. |
| BALF from two severe COVID-19 patients [69]. | Epithelial cells, lymphocytes, macrophages, neutrophils. | Yeskit integrates host gene expression profiles with virus detection. | R and Python-based packages utilizing the STAR algorithm. Yeskit was an R package for data integration, clustering, identification of DEGs, functional annotation, and visualization. | [74]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



