
Submitted:
31 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
Chemo-resistance is a substantial challenge in the realm of cancer treatment that requires exploring new therapeutic approaches for effective mitigation. Achieving this goal requires examination of the molecular mechanisms involved in both tumor growth and therapeutic interventions. The potential of NRF2 (Nuclear factor E2-related factor 2) in addressing resistance to chemotherapy across diverse cancer types highlights its value as a promising therapeutic approach based on cancer characteristics. Manipulating the NRF2 signaling pathway has a dual impact, offering promise for both preventing and treating cancer, as well as inhibiting carcinogenesis. The influence of the NRF2/KEAP1 pathway on the progression of tumor formation and resistance to drugs has been well-documented. The interplay between the NRF2 signaling pathway and processes such as endoplasmic reticulum (ER) stress, unfolded protein response (UPR), and autophagy plays a crucial protective role. A deeper understanding of NRF2's role in the modulating these pathways is necessary to develop novel approaches for improving chemotherapeutic efficacy. This article discusses the significance of the NRF2-KEAP1 pathway in preventing/promoting cancer and resistance mechanisms to various chemotherapeutic agents, with a focus on the complementary effects of antioxidants via NRF2-mediated signaling pathways. This study aims to provide a molecular basis for targeting NRF2 via inhibitors/activators as promising therapeutic strategies to overcome chemo-resistance.
Keywords:
NF-E2-related factor 2
; Drug resistance
; Reactive oxygen species
; Autophagy
; Unfolded protein response
1. Introduction
Cytotoxic chemotherapy (CTX) is repeatedly employed for early-stage cancers; primarily serving to monitor further spread [1]. In reality, adjuvant CTX is the principal approach in anti-cancer therapy. It involves a wide spectrum of medications with influential cytotoxic outputs that beneficially, though not exclusively, target the rapidly dividing tumor cells [2,3]. Despite remarkable achievements in cancer treatment over the last decade, failure of cancer chemotherapy and/or resistance to new anticancer agents persists, leading to tumor recurrence and metastasis, poorer prognosis, and emerging as a primary obstacle in cancer treatment[4].
The chemo-resistance can be pre-existent (primary resistance) or acquired (secondary resistance), governed by the molecular characteristics of an individual cancer. Primary resistance may be identified early at diagnosis when tumor cells do not initially respond to classical chemotherapeutic agents [5]. On the contrary, secondary resistance may emerge following chemotherapy [6]. Consequently, it is crucial to comprehend molecular systems of cancer, carcinogenesis, and chemo-resistance mechanisms to develop efficient and appropriate care procedures for anticancer treatment. Based on current knowledge, the molecular patterns of chemotherapy insensitivity are associated with DNA repair, proto-oncogenes, anti-oncogene, genes, autophagy, epithelial mesenchymal transition (EMT), tumor cell survival, transporter pumps, mitochondrial alteration, redox controlling complex, and exosomes [7,8,9,10] , as outlined in (Table 1).
Solid tumors are the most common type of tumor affected by cancer hypoxia-induced responses associated with genomic imbalances, which results in increased production of reactive oxygen species (ROS) and abnormalities in damaged DNA re-synthesis pathways [11]. The ROS imbalance under these circumstances, can activate the autophagy pathway through either endoplasmic reticulum (ER) stress or unfolded response protein (UPR) approach, and induces chemoresistance associated with cell cycle arrest and intensifying EMT or cancer stem-like cells [12]. Essentially, autophagy plays an underlying role in cellular viability and survival by eliminating aggregated or misfolded proteins and damaged cellular organelles [13]. The recruitment of autophagy in cancer therapy can play a dual role. Initially, it plays a pro-death role by eliminating transformed cells and damaged cell compartments. However, in in later stages, it plays a pro-survival role by providing protection against hypoxic stress, energy deficits, and chemotherapeutic medications associated with chemo resistance [14].
Autophagy as a multi-step process involved in scavenging damaged cellular components (such as proteins and organelles), engages approximately 40 proteins called autophagy-related proteins (ATGs) [15]. These ATGs are responsible for the formation of autophagosomes, which are double membrane structures that remove their contents upon fusion with lysosomes [16,17]. Initiation, nucleation, elongation, maturation, and fusion are the four phases of the autophagy process [18,19].
In normal biological processes, autophagy maintains cellular health by recycling damaged organelles and proteins, fostering cellular homeostasis, and adapting to stress.[20,21]. Conversely, dysregulation of autophagy is implicated in various pathological conditions. For instance, neurodegenerative diseases such as Alzheimer's and Parkinson's arise from aggregated proteins due to flawed autophagy. Cancer displays a complex relationship with autophagy—promoting tumor survival under stress while also preventing tumor initiation. Recognizing autophagy's role in both physiological balance and disease progression unveils potential avenues for therapeutic interventions and disease management[22].
Both in vitro and in vivo studies reveal that oncogenic activation, intrinsic stresses of tumor cells, and extrinsic pressures from the tumor microenvironment (TME), collectively contribute to an increase in misfolded protein levels in the ER and subsequent activation of the UPR pathway. The Unfolded Protein Response (UPR) is coordinated by three proteins located within the endoplasmic reticulum (ER) membrane: activated transcription factor 6 (ATF6), protein kinase RNA-like ER kinase (PERK), and serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1α (IRE1α) [22]. Once initiated, UPR signaling can lead to either cellular adaptation to stress or cell death, depending on variables such as cell type, the specific stress trigger, and the duration and intensity of cellular stress[23]. Proliferating cancer cells might utilize different aspects of the UPR to enhance their existing oncogenic mechanisms for resisting chemotherapy[24]. The UPR has a dual function in the progression of cancer. Initially, it acts as a survival mechanism, enabling cancer cells to maneuver the challenging tumor microenvironment by managing issues like misfolded proteins and limited nutrients. This adaptive reaction supports the survival and adjustment of tumor cells[25]. Conversely, prolonged UPR activation can drive cancer progression. Excessive UPR signaling has the potential to stimulate tumor growth, angiogenesis, and resistance to treatments. Furthermore, UPR-induced inflammation and microenvironment changes can facilitate tumor invasion and metastasis, contributing to aggressive behavior. The paradoxical nature of the UPR underscores the intricate interplay between its safeguarding and harmful effects in the context of cancer[26]. The UPR has emerged as a therapeutic target for the treatment of cancer, given its over-activation in cancer compared to healthy, non-proliferative cells.
Still, cancer-originated hypoxic niches are able to additionally stimulate a large proportion of leader antioxidant gene directors, including the nuclear factor erythroid 2-related factor 2 (NRF2), a family of transcription factors [27,28], existing in various tumors with chemo-resistant phenotypes [29,30,31]. Previous studies focused on the activators of NRF2 members and their chemo-preventive functions suggest that NRF2 has both have a dark/ negative and light/positive side. The positive aspect shields cells against external stress factors and is deliberately activated to safeguard organisms from diverse diseases. In a similar manner to how NRF2 guards’ healthy cells against injury, it can also shield malignant cells. As cells undergo transformation, they encounter numerous stressors, and excessive activation of NRF2 can aid this transformation, supporting cancer cells' growth, dissemination, and resistance to treatment [32,33]. Consequently, research indicates that inhibiting NRF2 may sensitize cancer cells to chemical treatments[34].
Given the inconclusive findings on the role of NRF2 in cancer, a clear picture of NRF2 is necessary to assist researchers in clarifying this intricate arrangement of antioxidant regulation pathways in tumor progression. In addition, it is also essential to define when NRF2 is triggered or repressed in different environments or how it can be affected by the influence of diverse stimuli [35]. In the following parts of the current review, we will explain the complementary effects of antioxidants via NRF2-mediated signaling pathways in more detail, in addition to considering the interplay between oxidative stress/redox regulatory networks, autophagy, and UPR-dependent chemo-resistance pathways.
1.1. The Family of Nuclear Factor (Erythroid 2)-Like (NRF) Transcription Factors
Among redox-responsive transcription factors (TFs), NRF2 stands out as one of the most crucial controllers of the cellular defense mechanisms against xenobiotics and oxidative stress [36]. The NF-E2-like BZIP Transcription Factor 2 (NFE2L2) gene encodes the NRF 2, which belongs to the cap 'n' collar basic region leucine zipper (CNC-bZIP) family with four closely related members, including NRF 1, NRF 2, NRF 3, and p45 nuclear factor erythroid-derived 2 (NFE2) protein [37]. These protein members feature seven functional NRF2-ECH homology (Neh) domains with high evolutionary conservation, each playing specific roles in modulating its transcriptional activity. The Neh1 region, which holds a bZIP binding sequence, engages with members of the small musculoaponeurotic fibrosarcoma (sMAFs) family (MAFF, MAFG, and MAFK), along with other bZIP motifs. This interaction enables the binding to functional antioxidant response elements (AREs), prompting the initiation of transcriptional gene expression. On the other hand, the Neh2 domain comprises two distinct binding sequences, namely DLG and ETGE. These sequences independently form dimers with the Kelch domains of Kelch-like-ECH-associated protein 1 (KEAP1), ultimately facilitating the degradation of NRF2 through UPR-mediated processes.[38,39]. The Neh3-5 transcription activation domains interact with different elements of the core-transcriptional machinery.
Two highly conserved redox-insensitive motifs in Neh6-domain, namely DSGIS and DSAPGS, form dimers with β-transducin repeat-containing E3 ubiquitin protein ligase (β-TrCP), inducing NRF2 degradation in oxidative stressed cells [40]. Neh7 domain of NRF2 characteristically heterodimerizes with retinoid X receptor alpha (RXRα) and suppresses the NRF2 function [41]. These domains regulate NRF2 integrity and the trans-activation of the downstream target genes across transcriptional and post-transcriptional modifications along with post-translational directive pathways against different lesions [42]. All four members of the NRF family are characterized by a unique N-terminal 43-aa CNC domain and play crucial roles during embryo development and in response to environmental stresses [37,43]. However, recent studies have identified novel NRF2 target genes with a number of additional features of NRF2 beyond its redox-managing roles, including regulation of inflammatory responses, cell metabolism, autophagy, proteostasis, ER stress, and the UPR, especially in tumorigenesis [44,45,46,47]. Recognizing the diverese features and functions of NRF2 and its emerging activities will pose additional challenges beyond exploring its potential in NRF2-targeted anti-cancer drugs.
The expression profiles of NRF transcription factors vary significantly based to tissue specificity. While NRF1 and NRF2 exhibit widespread expression, NRF3 is notably confined to the placenta and liver. Additionally, NF-E2 is specifically limited to megakaryocytes, mast cells, erythrocytes, and hematopoietic progenitors. [48,49,50]. It appears NRF1 assists in the the proteasome transcriptional bounce-back response to proteasome blocker processing [51]. The Activated NRF1 accumulates, migrates across the ER membrane, and then acts as a nuclear transcription factors following de-glycosylation and partial proteolytic cleavage processing in the nucleus [52]. However, its proteasome-mediated degradation activity and transcriptional capabilities have not been fully elucidated [53].
It is now established that NRF1 plays an important role in the development of resistance to cancer treatments[54,55]. Apparently, NRF1 protects tumor cells from proteotoxicity, which is enhanced by antitumor proteasome blockers [56]. Amongst the NRF transcription factor members, recent studies on NRF3 protein have demonstrated that it drives key functions of 20S in tumor proliferation and progression of malignant tumors by down- modulating the tumor suppressor proteins p53 and retinoblastoma-associated protein (pRB) over driving the 20S proteasome in different tumor cell types [57,58,59,60]. Moreover, these actions directly contribute to the subsequent metastasis and induction of angiogenesis in malignant tumors [61].
1.2. Deciphering the transcriptional NRF2 -regulated target genes
The NRF2 function is meticulously regulated. Despite the binding of activated NRF2 to DNA, it has been observed through DNA transcription profiling that not all genes in close proximity to the activated NRF2 are transcriptionally controlled by NRF2 attachment [62]. These NRF2 target genes need the cooperative recruitment of NRF2 and NRF2 interactive co-activators, such as cofactors and transcription factors or mediator proteins for a full stimulation [63]. The small MAF-TFs, namely MAF-F, MAF-G, and MAF-K, interact with transcriptional NRF2 across the associated bZIP motifs to make NRF2/ MAF heterodimer identify the ARE and trigger transcriptional regulatory function of NRF2 about genes encoding detoxification enzymes [64].
Transcriptional modulator BTB and CNC homology 1 (BACH1) is required to repress heme oxygenase (HO)-1 gene transcription, which goes against NRF2/ sMAFs interplay in the upstream promotor region and target NRF2-dependent transcription NAD(P)H: quinone oxidoreductase1 (NQO1) [65,66]. The upregulation of the NRF2/HO-1 binding results in HO-1 expression and requires the deactivation of BACH1[67,68]. Intrestingly, NRF2- activating transcription factor 4 (ATF4) dimers bind with NRF2 at ARE sites to activate HO-1 transcription as well. Physical Interaction of the Activator protein 1 (AP-1) subunit c-Jun with NRF2 stimulates NRF2-driven transcriptional inducers, although another AP-1 subunit c-FOS can inhibit favorable NRF2 activity [69]. The collaboration between the cAMP-response element binding protein (CREB)-binding protein (CBP) and its co-activator p300 is utilized to jointly bind to the antioxidant response element (ARE) by employing the transactivation domains Neh4/5 of NRF2. This interaction plays a role in facilitating the activation of gene transcription. Both histone acetyltransferases CBP and p300 cause chromatin decompaction to be conducive to the employment of the transcription apparatus [68,70].
Nonetheless, ATF3 mediator can interact with CBP, inhibiting the binding of NRF2 and repressing transcription arising from the NRF2–CBP complex. Remarkably, the removal of Atf3 increases NRF2 destruction via overexpression of the KEAP1 gene and the loss of human DJ-1 in the H157 NSCLC squamous cell line [71]. Hence, ATF3 functions as a potential regulator, either positively or negatively, involved in the modulation of NRF2 activity [72,73].
Additional transcription complex co-regulators, including SIRT6, an NAD+-dependent histone deacetylase, the ATPase subunit of the chromatin-remodeling complex SWI/SNF, RAC3, a co-activator linked to receptors, CHD6, a chromodomain helicase DNA-binding protein, BRG-1, a gene associated with Brahma, and subunit 16 of the mediator involved in RNA polymerase II transcription, have the capability to activate NRF2, thereby influencing the transactivation process of genes that are targeted by NRF2 [39]. Nevertheless, the actual implication of such interactions are not yet fully understood. The nuclear receptors estrogen receptors α and peroxisome proliferator-activated receptor gamma (PPARγ) can selectively bind to NRF2, inhibiting its transcriptional activity [74]. Notably, gene expression profiles from the livers of Keap1 knockout/knockdown and Nfe2l2-null mice relative to the corresponding control wild-type mice in a gene dose response experiment, demonstrated that Nrf2 activity is correlated with the expression level of Nrf2[75]. A comprehensive understanding of the gene transactivation orchestrated by NRF2 necessitates a concentrated examination of the synergistic interplay and potential rivalry between NRF2 and various other categories of transcription factors and co-regulators that are intricately linked to both ARE sequences and similar ARE-like regions.
1.3. NRF2- driven response to oxidative stress and drug metabolizing
Antioxidant detoxification and drug metabolizing control via transcriptional activation of ARE-mediated β-globin genes are recognized as significant emerging activities of NRF2 as a well-known NC-bZIP TF [76]. Recent studies have shown that the activation of antioxidant cytoprotective genes over the Nrf2/antioxidant response component in response to cellular oxidative stress results in a complex of collaborating enzymes complicated in phase I, II, and III biotransformation reactions and the removal of oxidative inducers to maintain homeostasis [77,78,79,80]. Phase I reactions of xenobiotic metabolism correspond to oxidoreductase and hydrolysis, catalyzed by NQO1, aldo-keto reductases (AKRs), adenine dinucleotide phosphate (NADPH)–oxidoreductases cytochrome P450s (CYPs), and carbonyl reductases (CBRs) as well as aldehyde dehydrogenase 1 (ALDH1) enzymes [81]. Enzymatic conjugation, UDP-glucuronic acid production enzymes UDP-glucuronosyltransferase (UGT), glutathione S-transferase (GST), and heme oxygenase 1 (HO-1) are all included in Phase II. Phase III mechanisms of xenobiotic conjugation and transport primarily revolve around the accumulation of non-toxic or conjugated metabolites subsequent to phase II reactions. These mechanisms predominantly involve drug efflux pumps such as the breast cancer resistance-related protein (BCRP/ABCG2), multidrug-resistance-associated-proteins (MDR), and ATP-binding cassette G8 (ABCG8) [82,83].
Conventional antioxidant defense systems triggered by NRF2-driven enzymes participate in the biosynthesis of reduced glutathione (GSH) (glutathione synthetase (GSS)) and its usage and recycling (glutamate-cysteine ligase catalytic (GCLC) and modulator (GCLM) subunits)[84]. In addition, several other antioxidant enzymes, involved in the removal of reactive oxygen and nitrogen products (ROS/RNS) (glutathione s-transferases, peroxiredoxins, superoxide dismutase, thioredoxins, and thioredoxins reductases) are all known target genes of NRF2 [85]. play a crucial role in maintaining normal homeostasis disturbed by redox signaling. They are instrumental in addressing various disorders characterized by oxidative stress, including neurodegenerative and cardiovascular diseases, autoimmune disorders with metabolic syndrome, and cancer. [86].
Studies suggest that the induction of the NRF2 signaling pathway is a powerful approach in tumor suppression, and a feasible strategy in anticancer therapy [87,88]. Furthermore, pharmaceutical induction of NRF2, can reduce carcinogenesis and perform a protective function compared to tumor initiation in normal cells. The absence of NRF2 decreases prompt GST expression, leading to an increases in ROS level, resulting in DNA damage and a predisposition to carcinogenesis [89]. Besides, other investigations suggest that NRF2 may also support cancer development. For example, there is a focus on developing NRF2 inhibitors to reverse the resistance of cancer cells to chemotherapy. Simultaneously, researchers are exploring Nrf2 activators as potential safeguards against the harmful impacts or undesirable outcomes of chemotherapy treatments [90].
1.4. NRF2 signaling pathway in cancer
The significance of NRF2 in cancer is widely recognized. It is crucial to understand its negative regulator, KEAP1, which modulates NRF2 to decrease its cellular expression and effectively control metabolic balance [91]. The KEAP1 activity is closely correlated with cellular levels of NRF2 protein after normal or low/moderate responses to stressful situations to monitor cellular antioxidant responses, as well as detoxification responses involved in cancer prevention and treatment [92].Understanding these regulators would enable researchers to channel NRF2's footprint into a more targeted approach aiming to completely eradicate tumors, while also addressingits pro-oncogenic effects, most of which are associated with the "dark side" of NRF2, clearly linked to the metastatic behavior of tumor cells [93].
Numerous studies have shown that the activation of NRF2 preserves the health of cells exposed to diverse toxic components and illnesses; it has also been observed that the over-activation of NRF2 promotes tumor development and protects tumor cells from oxidative injury, which can further induce chemo-resistance [94]. The elevated levels of NRF2 observed in cancer contributes to an increased expression of key metabolic enzymes including transketolase (TKT), phosphogluconate dehydrogenase (PGD), glucose 6-phosphate dehydrogenase (G6PD), and several others [95]. This heightened activity of glucose metabolic enzymes promotes the production of purine and amino acids, along with the regeneration of the NADPH pool through the pentose phosphate pathway (PPP). Consequently,, there is a reconfiguration of metabolic pathways to faciliate cellular growth and enhance antioxidant capacity [96].
As a key regulator in carcinogenesis, the cell cycle is closely linked to Nrf2 over-activation, as Nrf2 deficiency leads to arrest in the G2/M phase of the cell cycle. It appears that Nrf2 is a controller in the regulation of the cell cycle through the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling [97]. Some studies have indicated that the over-expression of NRF2 induces the phosphorylation of AKT, glycogen synthase kinase-3 (GSK3), PPP, and KRAS activation[98,99]. In addition, cooperation between the PI3K/AKT and KRAS/MAPK pathways can lead to an increase in the anabolic pathways efficiency, the inhibition of the apoptosis, and induce survival and self-renewal inducement in cancer stem cells (CSCs) by anti-apoptotic factor B-cell lymphoma 2 (Bcl-2) [100]. These mechanisms contribute to the development and progression of cancer[101].
Furthermore, the activation of Nrf2 is associated with the inhibition of chronic inflammation, a factor linked to cancer[102]. Inthe absence of Nrf2, certain pro-inflammatory factors such as inducible nitric oxide synthase (iNOS), tumor necrosis factor-alpha (TNF-α), and cyclooxygenase-2 (COX-2), are increased significantly [103]. The iNOS can produce nitric oxide, which contributes in inflammation and tumor development[104]. It also can cause mutagenesis and DNA degradation [105]. The TNF-α is one of the main cytokines regulating the progression of inflammation through different signaling pathways [105,106]. Additionally, Cox-2 serves as another regulator of inflammatory diseases [106] (Figure 1).
Furthermore, Nrf2 regulates the fundamental expression of mouse double minute 2 homolog (Mdm2), a potential repressor of tumor suppressor p53 [107]. Similarly, the overexpression of the transcription factor Nrf2 secondarily down-modulates TP53 and enhances cancer cell self-renewal by inhibiting p53-linked apoptotic death signals [108]. In an investigation involving human refractory ovarian cancer cells, the elevated expression of NRF2 was observed compared to the non-resistant cell line, and NRF2 knockout by siRNA reintroduced drug response. In addition, chemical NRF2 induction delivered a survivability nature to immortalize neuroblastoma cell lines responding to tumor medications such as etoposide, cisplatin, and doxorubicin [109,110]. Based on these findings,, Cho and his team confirmed that the knockdown of NRF2 through siRNA treatment enhanced the cisplatin sensitivity in ovarian cancer cells [109]. Likewise, Long-term stimulation of NRF2 has been found to impair efficacy of combined drug and radiation treatment in human respiratory tract cancer cells. Conversely, lower levels of NRF2 increased the cellular response to ionizing radiation and cytotoxic drugs [111]. The above results suggest that Nrf2 function alone or in combination with complementary drugs can be considered a productive method to improve the response of metastatic cells to chemotherapy. Moreover, recent investigations highlight the role of Nrf2 in malignancy, where functional Nrf2 promotes lung cancer metastasis by preventing the degradation of the heme-binding Bach1 transcription regulator [112].
The overproduction of NRF2 triggers cell growth and metastasis in breast cancer by activating the RhoA gene expression along with its downstream effectors [113]. The significant role of NRF2 in proliferation and invasion is also observed in hepatocellular carcinoma, where it regulates the post-transcriptional expression of target genes such as metalloproteinase-9 (MMP-9) and BCL-XL transcripts [93]. Recent findings have highlighted that, beyond facilitating tumor advancement, UPR activation can also play a role in the development of chemo resistance (as depicted in Table 1) [114,115,116,117]. Silencing all three branches of the UPR has been linked to the restoration of sensitivity in previously resistant cancer cells [116,118,119]. Rapidly dividing tumor cells might exploit distinct UPR branches to complement their pre-existing mechanisms of chemo-resistance. Furthermore, the targeted activation of a specific UPR arm may be intertwined with other inherent mechanisms of chemo-resistance [24].
Antioxidants play a complex role in cancer therapy, resembling a double-edged sword. While they can potentially protect healthy cells from the damaging effects of treatments like chemotherapy and radiation by neutralizing harmful free radicals, they might also inadvertently shield cancer cells from these therapies, reducing their effectiveness. This duality underscores the need for a balanced approach when considering the use of antioxidants in cancer treatment strategies [120]. This regulation exhibits the dual character of ROS: at low concentrations, it acts as a critical second intracellular messenger in various signal transduction pathways, whereas at high concentrations, through a change in gene expression, cells try to confront stress by converting to antioxidant response [27].
As mentioned earlier, the activation of NRF2 signaling has gained attention for its potential to mitigate the adverse effects of chemotherapy. This pathway plays a protective role by shielding cells from oxidative harm. However, inhibiting oxidative-induced cell death through NRF2 activation can lead to the development of chemo-resistance in cancer cells. Evidently, NRF2 might contribute to the promotion of tumor-initiating cell lineage, consequently giving rise to chemo resistance. [35]. Increased NRF2 expression has been studied in various cancer types, including head and neck, lung, epithelial, gastric, and pancreatic cancer [93,121,122]. It has been found that elevated NRF2 expression can lead to resistance to radiation, 5-fluorouracil, and cisplatin, possibly through the induction of antioxidants [123]. Therefore, there may be a need to consider inhibiting the NRF2 pathway during chemotherapy [34].

Table 1.
Molecular Mechanisms involved in chemo-resistance.
| Underlying molecular mechanism | Cellular effect | Reference |
|---|---|---|
| Transporter pumps (ABC proteins, SERCA, V-ATPase) |
These proteins exhibit elevated expression levels in chemo-resistant cancer cells and play a role in the development of drug resistance. | [124,125,126] |
| Oncogenes EGFR |
The overexpression of EGFR triggers the activation of NF-κB and STAT3, which subsequently leads to the development of chemo-resistance and unfavorable treatment outcomes. |
[127] |
| KRAS | Oncogenic KRAS promotes drug resistance via upregulation of the cell protective stress response gene, NRF-2, at the transcriptional level. | [128] |
| (PI3K)/Akt | AKT involves in apoptosis, migration, and proliferation. | [129] |
| NF-кB | Following activation, NF-κB translocates to the nucleus, elevating the expression of BCL-2, BCL-XL, XIAP, survivin, and AKT, thereby contributing to accelerated tumorigenesis, increased aggressiveness, drug resistance, and induction of EMT. | [130] |
| ERKs |
ERKs are recognized for their role as activators of various transcription factors, including ETS Like1, along with downstream protein kinases. These factors are closely linked to processes such as cell proliferation, drug resistance, and apoptosis. | [131] |
| Oncogenic Viruses |
Viral onco-proteins contribute to chemo-resistance through multiple mechanisms, including the regulation of cellular transporters and drug targets, modulation of signaling pathways involved in drug-induced cell death responses, and activation of pathways that counteract the effects of drugs. | [120] |
| Rb |
Oncogenic p53 causes chemo-resistance of cancer cells by increasing the expression of MDR-1. | [132,133,134,135,136,137] |
| CKIs |
These mechanisms involve inducing cell cycle arrest and activating DNA repair processes | [138,139,140] |
| PTEN |
Increase apoptosis, regulating cell cycle progression. | [141,142] |
| BRCA1 |
Reduction of cell proliferation, migration, survival and cell size, Regulating transcription, cell cycle checkpoint, DNA repair, and apoptosis. |
[143,144,145,146] |
|
Mitochondrial alteration SERCA |
Bcl-2 and Bcl-xL contribute to heightened drug resistance, while reducing their expression enhances the cytotoxic impact of cisplatin and gemcitabine. Moreover, the level of survivin expression was found to be linked to the degree of cisplatin resistance in gasteric cancer cells. |
[147] |
| V-ATPase |
Somatic mutations occurring in the mitochondrial genome (mtDNA) of cancer cells lead to impaired mitochondrial function, which in turn contributes to the development of chemo-resistance. | [148] |
| DNA repair |
BER and NER can confer the resistance to chemo drugs that target DNA. RAD51, a crucial participant in homologous recombination during double-strand break (DSB) repair, being overexpressed, serves as a marker for resistance to Cisplatin (CDDP) in non-small cell lung cancer (NSCLC). Similarly, elevated expression of ERCC1, a component of the nucleotide excision repair (NER) pathway, is associated with resistance to CDDP in both human hepatocellular carcinoma (HCC) cell lines and specimens. | [149,150,151] |
|
Autophagy |
In tamoxifen-resistance breast cancer cells, SAHA, as a HDAC inhibitor, can induce autophagic cell death and reduce tumor growth. Despite the challenges about the anticancer and pro-survival function of autophagy, in vitro and in vivo research has been more confirmed that autophagy could be considered as a facilitator of cancer chemo-resistance. In NSCLC cells, autophagy inhibition using Chloroquine, before paclitaxel treatment, prevents drug resistance. |
[14,152,153] |
| UPR |
CSCs and actively dividing tumor cells might exploit distinct branches of the UPR to reinforce their pre-existing mechanisms of chemo-resistance. Notably, the suppression of all three UPR branches—GRp78, ATF6, ATF4, and XBP1s—has shown a correlation with the restoration of sensitivity in chemotherapy-resistant cancer cells. | [24,116,117,118,119] |
| EMT |
EMT has been identified as a promoter of chemo-resistance against the DNA alkylating agent cyclophosphamide and the DNA synthesis inhibitor gemcitabine. Specifically, the attenuation of Snail or Twist has been linked to increased sensitivity to chemotherapy. | [154] |
| Cancer stemness |
Cancer stem cells (CSCs) resist chemotherapy by increasing the levels of P-glycoprotein, ABCG2, BCL-2, and survivin. Recent findings highlight NRF2's role in preserving stemness, intensifying tumorigenicity, and initiating chemo-resistance within CSCs. |
[155,156] |
| Regulatory redox network |
The mechanisms of ROS-mediated acquired chemo-resistance include autophagy, ER stress, overcoming cell cycle arrest, and enhancing epithelial to mesenchymal transition or cancer stem-like cells. Numerous chemotherapy agents, including cisplatin, doxorubicin, etoposide, paclitaxel, and bortezomib, induce cancer cell death by elevating ROS levels. Adjusting intracellular antioxidant levels holds potential therapeutic benefits but can be complex. While antioxidants may impede chemotherapy efficacy by scavenging ROS, they can also trigger chemotherapy-related toxicity, highlighting a delicate balance. | [12,120,157] |
2. Redox regulatory network involved in the induction of autophagy/UPR and tumor chemo-resistance
The activation of UPR relieves ER stress by reducing protein translation to lessen protein load in the ER, increasing the translation of chaperones to facilitate the ER protein folding, and removing misfolded proteins [158]. Sustained UPR activation induces apoptosis, but tumor cells can bypass the apoptosis and use the UPR for tumor proliferation. Recent findings reveal that UPR is also involved in the chemo-resistance of the tumor cells [24]. Based on several findings, multiple elements of the UPR are associated with advanced tumor stage and resistance to chemotherapy [24,159,160]. In this context, the overexpression of XBP1 is a significant event associated with chemo-resistance and short-term survival in lymphoma [161]. In the case of ABCs, as major contributors to chemo-resistance, ABC activity can be diminished by inhibiting GRP78 which also reduces the antioxidant response due to ROS accumulation. Recently, it has been shown that IT-139, a small inhibitor of the GRP78 molecule, can sensitize chemically resistant PDAC cells to gemcitabine [162]. GRP78-mediated ER homeostasis is associated with the activity of specificity protein 1 (SP1). SP1 inhibits homeostasis, negatively affecting the UPR and inducing cancer cell death. This modulation occurs through the regulation of NRF2 antioxidant responses and ABC transporter activity by inhibiting GRP78-mediated ER homeostasis. [163].
The PERK pathway also exerts its adaptive effect(s), which includes transient inhibition of eIF2α and antioxidant response by inducing the transcription factor NRF2 [164,165,166]. NRF2, which governs the response to oxidative stress, functions downstream of PERK. This regulatory role can extend to influencing the expression of the ABCC subfamily. While NRF2 predominantly functions as a tumor suppressor, heightened activity of its antioxidant response elements can enhance the survival of both normal and cancerous cells (Figure 2) [167] .
Additionally, studies have shown that cells experience an increase in mitochondrial ROS, metabolic changes, and the accumulation of free radicals during hypoxia, leading to metabolic stress. Under the hypoxic conditions, HIF-1α protein accumulation, the central regulator of the cellular response to hypoxia, activates the UPR pathway as a mechanism of adaptation of tumor cells, causing tumor growth and resistance to chemo and radiation therapy [168]. Indeed, hypoxia induces the PERK/eIF2α/ATF4 axis, with PERK ultimately leading to phosphorylation and subsequent activation of the FOXO-1 (anti-apoptotic forkhead box O-1) as well as pro-autophagic components. The up-regulation of autophagy, along with the suppressed apoptosis, make cells resistant to chemotherapeutic drugs (Figure 2) [169]. In contrast, prolonged activation of UPR pathway, triggers cell death mechanisms once environmental ER stress is not relieved. Under these conditions, different pathways that lead to cell death will be initiated, limiting the progression of cancer. There is substantial evidence that ROS-induced autophagic cell death may play an important role in these pathways; as ROS can regulate the expression of ATGs, such as ATG4 and Beclin1 [157].
Nonetheless, ROS exhibit a dual nature in the context of cancer. The increased ROS levels contribute to the enhanced growth of cancer cells by triggering signaling pathways, such as the PI3K/AKT and up-regulation of their antioxidant components, which consequently leads to the development of drug resistance within cancer cells [11]. Furthermore, high levels of ROS may guide tumor cells toward different pathways of cell death and restrict their expansion [170]. Accordingly, anti-cancer therapies that increase ROS or inhibit antioxidant levels are considered as novel treatment strategies in this context.
The utility of chemo-drugs, such as cisplatin, doxorubicin, and paclitaxel with the ability of inducing ROS levels inside the cancer cells, is considered an appropriate option for curing cancers by triggering cell death-related pathways. Amongst these pathways, autophagy ROS-induced cell death plays a considerable role [171]. Studies have shown that antioxidants can inhibit the autophagy flux and indicate a direct involvement of ROS in inducing autophagy. Autophagy has been proven as a significant degradation system for eliminating harmful protein components from the endoplasmic reticulum (ER) and is capable of breaking down a broader array of substrates [172]. Recent studies have shown that ROS is necessary for autophagy induction. ROS oxidizes the cysteine protease ATG4, which results in ATG8 lipidation and the formation of autophagosomes. These reactive species also regulate autophagy by the activation of TFEB-nuclear translocation. High levels of ROS activate adenosine monophosphate-activated protein kinase (AMPK) and mitogen-activated protein kinase (MAPK), inducing autophagy [173]. Autophagy is thus a strategy exploited by tumor cells to get adopted to tumor environment, resulting in chemo-resistance development [174].
Regarding this matter, a study conducted on breast cancer cell lines revealed that the application of carnosol polyphenols resulted in cell death through a process involving ROS-triggered autophagy followed by late-stage apoptosis[175]. New evidence suggests that autophagy inhibitors and antioxidants may be able to prevent cancer cell death [17,176,177]. Consistently, data obtained about utility of resveratrol or psoralen showed that human colon cancer (COLO 201, HT-29) and human lung cancer (A549) cells die by increasing ROS accumulation and autophagy levels, respectively. However, the use of 3-MA or antioxidants reverse these effects [178].
This hypothesis can also be exemplified by studies conducted on glioma cells, as demonstrated that after treatment with polycyclic ammonium ion sanguinarine, H2O2 could increase the autophagic cell death. In addition, ROS can induce the expression of SQSTM1/p62 and Beclin1/ATG6 genes via the NF-κB, and thus can regulate autophagy inside the malignant cells [178] . Although autophagy can play a suppressive role durig the early stages of cancer progression, it can also increase cell survival and metastasis, thus protecting tumor cells against environmental and drug stresses [179].
Conclusively, ROS act as a double-edged sword, with both sides being used therapeutically. There is still much debate about the use of antioxidant supplementation or its inhibition to modulate ROS levels in cancer therapy. Due to the effective role of the UPR pathway and autophagy in maintaining cellular homeostasis, resistance and survival of cancer cells, further studies are strictly needed to target these two pathways in modifying ROS content during cancer.
2.1. The cross talk between different arms of UPR-autophagy in drug resistance and cancer cell survival
Despite the significance of autophagy and ER stress in multiple human illnesses [180,181,182], the interplay between autophagy and the UPR is still unclear. Recently it was shown that UPR’s signaling arms strongly correlate with autophagy. We discussed in detail the interplay between UPR signaling arms with autophagy, principal molecular mechanisms, and their role in drug resistance in the following section.
2.1.1. Interaction among IRE1/XBP1s, IRE1/TRAF2/ASK1/JNK, and Autophagy
Activating the IRE1/XBP1s signaling arm of the UPR can be a reliable method to correct impairments of ER proteostasis indicated in different diseases [183]. IRE1 contains two major domains, namely a serine/threonine kinase domain, an endoribonuclease domain [184]. Unconventional splicing of X-box-binding protein-1 (XBP1) and regulated IRE1 dependent decay (RIDD) are two downstream mechanisms through which IRE1 primarily exerts its pro-survival effects[185,186]. It should be noted that the IRE1 pathway is actually the most evolutionarily conserved arm of the UPR, as it is identified in almost all eukaryotes [187]. Spliced XBP1 (XBP1s), which is a key transcription factor produced by the IRE1’s endoribonuclease activity [188], can bind to the UPR-targeted genes inside the nucleus (e.g., GRP78) to accelerate the protein folding capacity and restore cellular homeostasis [185].
As depicted in (Figure 3), the IRE1/XBP1s pathway can induce autophagy in three main phases: (i) XBP1s indirectly induces autophagy by operating the expression of Bcl-2 [189,190,191], XBP1s may trigger autophagy by facilitating the LC3βI to LC3β II conversion, and the overexpression of Beclin-1(BECN) [192,193], and (iii) XBP1s can form a homo- or heterodimer and attach to the BECN1 gene promoter to up-regulate the Beclin -1(BECN) expression [194]. Accordingly, IRE1/XBP1s axis is believed to be a positive modulator of autophagy with pro-survival effects. Nonetheless, IRE1/XBP1s deficiency has been reported to cause an increase in autophagy and cell survival in a group of amyotrophic lateral sclerosis patients [195,196]. Xbp1s deficiency may also lead to the overexpression of Forkhead box O1 (FoxO1) to stimulate neuronal autophagy [197].
Furthermore, C-Jun NH2-terminal kinase (JNK) is known as a stress-associated protein involved in a vast array of cellular processes [198]. Once the UPR becomes initiated, the adaptor protein tumor necrosis factor receptor-associated factor-2 (Traf2) is recruited by the activated IRE1 and creates the Ire1-Traf2 complex. Afterward, the apoptosis-signal regulating kinase 1 (Ask1) arranges the formation of the Ire1-Traf2-Ask1 complex [199]. Interestingly, the IRE1/TRAF-2/JNK1 axis can operate ER stress-triggered autophagy, as mouse embryonic fibroblast cells deficient in IRE1/TRAF-2 have revealed a significant decrease in the formation of autophagosomes [191]. Pharmacological inhibitors of JNK1 can also suppress autophagosome formation; SP600125 is a good example in this regard [200].
The JNK1 pathway also has a central role in the transcriptional modulation of Beclin-1(BECN) expression. It has been demonstrated that JNK1 is involved in starvation-triggered autophagy by phosphorylating BCL-2, localized in ER, resulting in separation of Beclin-1(BECN) from BCL-2 and subsequent initiation of autophagic flux [201]. In sum, the IRE1/JNK1/c-JUN axis is a pivotal mechanism to induce autophagy. IRE1/XBP1s and IRE1/JNK1 stimulated-autophagy pathways join at BECN. Hence, BECN can be a promising therapeutic target to mediate the ER stress-triggered autophagy in drastic pathological conditions, such as malignancies, cancer drug resistance, and cancer cell survival.
2.1.2. The role of IRE1/XBP1s arm of UPR and autophagy in drug resistance and cancer cell survival
IRE1 modulates irregular splicing of XBP1 mRNA, as well as the expression of cyclin A1, supporting the IRE1-induced cancer cell growth [202]. High levels of XBP1s, which results from increased XBP1 splicing, have been reported in multiple cancers and represent a poor prognosis [203,204]. For instance, a high ratio of XBP1s/XBP1 has been demonstrated to be strongly correlated with poor prognosis and a shortened relapse period in myeloma patients [205]. Treating multiple myeloma cells with MKC-3946, as an inhibiting agent of IRE1α endoribonuclease activity, desirably results in the repression of XBP1 splicing and induction of ER-regulated apoptosis in these cells when simultaneously treated with bortezomib. It can be concluded that the IRE1-XBP1 axis is central to the viability of (multiple myeloma) cells, and directing this pathway may lead to anti-tumor effects [205]. Furthermore, we recently presented evidence that simvastatin (Simva), through the activation of the IRE-1 arm of UPR, enhances temozolomide (TMZ)-induced cell death in U87, U251 glioblastoma cells. Moreover, our result revealed that IRE-1 regulated Simva-TMZ mediated autophagy flux inhibition and improved TMZ efficacy[22] .
The involvement of XBP1s in triggering relapse of triple-negative breast cancer (TNBC) tumors in vivo has also been observed. After doxorubicin therapy, MDA-MB-231 xenografts initially experience a decrease in tumor size, followed by tumor regrowth once the therapy is stopped [24,206]. When XBP1 is suppressed in MDA-MB-231 xenografts, the regrowth of tumors following doxorubicin withdrawal is inhibited. Additionally, the use of the MKC8866 inhibitor to reduce IRE1 RNase activity in MDA-MB-231 cells prevents regrowth after paclitaxel withdrawal, suggesting a correlation between XBP1s signaling and tumor regrowth [207].
In addition to XBP1s, GRP78 overexpression has been identified to be strongly associated with chemotherapy impairment through certain molecular mechanisms [208,209]. Interestingly, during ER stress, GRP78 is capable of becoming anchored as a cell surface receptor [210]. This novel receptor can activate the PI3K/AKT signaling pathway and inhibit the transforming growth factor (TGF-β) pathway, to induce cell survival and growth [211]. Previous studies have suggested that upregulation of GRP78 may contribute to chemo resistance of a group of cancers, such as breast cancer, glioblastoma, and other aggressive gliomas [212,213]. This finding proposed that GPR78 could be considered as a promising link between metabolic alterations and tumor survival. Therefore, targeting GRP78 emerges as a potential approach to overcome chemotherapeutic failure in the coming years.
2.1.3. Cross-talk between the PERK/eukaryotic translation initial factor 2α (eIF2α)/ ATF4/CHOP axis and autophagy
PERK/eIF2α/ATF4/CHOP pathway constitutes another branch of the UPR. PERK is a serine/threonine kinase is activated through both autophosphorylation and homodimerization upon releasing from GRP78 [214]. Once activated, PERK inhibits protein synthesis by phosphorylating eIF2α, disrupting the attachment of methionyl-tRNA and ribosomes [215]. Subsequently, phosphorylated eIF2α promotes ATF4 translation, aiding the ER in protein folding[216]. Moreover, the overexpressed ATF4 induces the translation of CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP), which contributes to ER stress-mediated apoptosis and is considered as a marker to evaluate the stimulation of the UPR [217].
In association with autophagy, ATF4 plays a central role inin upregulating ATG12, an essential component of the ATG5-ATG12-ATG16L complex (figure 3), and critical for autophagosome elongation [218]. It has also been reported that ATF4 can directly bind to the cAMP response element binding site located in the light chain 3B (LC3B) promoter, inducing the expression of LC3B to trigger the autophagic flux [219]. Furthermore, in the context of melanoma, B-RAF proto-oncogene (BRAF)-induced phosphorylation of PERK is pivotal for autophagosome generation[220]. Pharmacological suppression of PERK using GSK2606414, or siRNA-induced blockade of this serine/threonine kinase, significantly reduces the Lc3B II/Lc3 BI ratio [221]. Regarding CHOP, as a powerful transcription factor involved in autophagy, UPR and some other cellular processes, it has been reported that its overexpression induces the expression of ATG5 and BH3-only proteins like Bim and Puma.
Furthermore, CHOP downregulates the expression of Bcl-2,facilitating the release of Beclin-1 from this anti-apoptotic protein [217,222,223]. When CHOP collaborates with PERK in the form of PERK-CHOP pathway, it promotes the expression of tribbles-related protein3 (TRB3), an AKT blocker [224]. Upon AKT inactivation, it represses the phosphorylation of tuberous sclerosis complex 2 (TSC2), resulting in inactivation of the mammalian target of rapamycin complex1 (mTORC1). The inactivated mTORC1 then dephosphorylates ATG13 and the ULK1/2 complex to provoke the formation of autophagosomes [225]. Moreover, the eIF2α/ATF4/CHOP pathway positively affects the expression of p62 to induce autophagic flux [226].
2.1.4. The role of PERK arm of UPR and autophagy in drug resistance and cancer cell survival
As mentioned earlier, the activated PERK phosphorylates eIF2α and NRF2 after dissociating from GRP78 [227]. Phosphorylated NRF2, in turn, activates ROS-scavenging enzymes, making cells resistant to hypoxia [228]. Hence, the inhibition of PERK enhances chemosensitivity by increasing ROS accumulation [160,229]. GSK2656157 is a well-known PERK inhibitor that blocks the phosphorylation of eIF2α and the expression of ATF4 and CHOP, following the suppression of PERK autophosphorylation. GSK2656157 also refuses angiogenesis as a pivotal process in tumor cell growth and development [230]. More interestingly, PERK-induced up-modulation of cellular inhibitors of apoptosis (i.e., cIPA1 and cIPA2) can protect cells against tunicamycin-induced death [231].
The PERK-eIF2α pathway has also been demonstrated to be up-regulated in chronic myeloid leukemia (CML) cells with high expression levels of BCR-ABL [232]. An intriguing study revealed that transfecting CML cells with dominant-negative mutants of PERK or dominant-negative eIF2α-S51A mutant, significantly increases the apoptotic pathway activity in these cells when treated with imatinib [233]. Recent findings suggested that PERK arm of UPR is involved in the crucial effects of Simva-TMZ combination therapy, enhancing cell death and improving TMZ effectiveness in GBM cells. Surprisingly, Simva-TMZ, through PERK, causes the p62 accumulation and regulates autophagy flux inhibition in U87 and U251[22] .
PERK activation has also been implicated in cervical cancer stem cells (CSCs), which exhibit resistance to ER stress-triggered apoptosis [234]. Consequently, pharmacological suppression of PERK and not IRE1 makes CSCs sensitive to ER-stress induced apoptosis. However, when these corresponding CSCs undergo cisplatin therapy, they become dependent on IRE1 rather than PERK. It can be concluded that CSCs defeat tumor progression-related stresses by triggering the PERK. Switching from PERK to IRE1 due to tolerating extra chemotherapeutic stress is actually a potential mechanism to protect cells from CHOP-induced cell death [24,234]. Consistent with these facts, targeting the PERK-eIF2α axis emerges as another effective approach to overcome challenges associated with cancer therapy.
2.1.5. Interplay between ATF6 and autophagy
When ATF6, a third component of the Unfolded Protein Response (UPR), detects an elevation in the levels of misfolded proteins, it exposes Golgi localization signals to ensure smooth transport to the Golgi apparatus (GA). Subsequently, Site 1 and Site 2 proteases initiate the cleavage of ATF6, resulting in its activated form[235]. This activated ATF6 then translocates to the nucleus, where it binds to elements associated with endoplasmic reticulum (ER) stress. Upon binding, ATF6 enhances the expression of essential factors involved in proper protein folding, namely GRP78, XBP1, CHOP, and PDI (protein disulfide isomerase) [236].
Atf6 has been reported to play an essential role in autophagy induction by assisting the death-associated kinase 1 (Dapk1) [237]. The underlying mechanism of this type of autophagy induction is an interactive connection between ATF6 and C/EBP-β to generate a transcriptional complex to promote the expression of DAPK1 through binding to CRE/ATF elements located on the DAPK1 promoter [238]. Along with this finding, silencing of ATF6 with specific small hairpin RNAs (shRNAs) can significantly decrease the expression of DAPK1 and subsequent formation of autophagosomes. Indeed, DAPK1 is a major contributor to the formation of autophagosomes through phosphorylating of BECN. Upregulation of CHOP, XBP1, and GRP78 also contributes to ATF6-triggered autophagy [239]. This axis has transformed ER stress-stimulated autophagy into a more complex process.
2.1.6. The role of ATF6 arm of UPR/ autophagy in drug resistance and cancer cell survival
Although ATF6’s role in cancer drug resistance is not fully understood, it has been revealed as a crucial contributor to chemoresistance. A great model has been described for ATF6-induced imatinib resistance in leukemia [240]. In this study, PDIA5 was identified as responsible for the activation of ATF6 and export of ER proteins in a way that PDIA5 impairment could mitigate the expression levels of ATF6-specific target genes. In addition, the down-regulation of ATF6 promoted chemosensitivity in imatinib-resistant leukemia cell line (K562R cells) [240]. ATF6 activation has also been demonstrated in tunicamycin or thapsigargin-treated melanoma, suggesting the essential role of ATF6 in protecting melanoma cells against ER stress-induced death [241].
Beyond its role in drug resistance and cell survival, ATF6 has been reported to be involved in cancer recurrence [242]. The activation of ATF6α induced by p38 signaling has been shown in D-HEp3 cell line. More interestingly, the number of viable D-HEp3 cells with silenced ATF6α expression significantly decreases after doxorubicin treatment. It is noteworthy that ATF6α exerts its anti-chemotherapeutic effects through the activation of the mammalian target of rapamycin (mTOR) [243]. Therefore, targeting the ATF6 arm of the UPR presents another effective approach to overcome challenges associated with cancer therapy (Figure 3).
2.1.6. Other pathways involved in ER stress-induced autophagy
Based on previous studies, proper ER function positively supports autophagic flux, which must be initiated and elongated accurately. Hence, inhibiting a ER key regulator and/or molecular chaperone, e.g., GRP78, can disrupt ER function, leading to the repression of ER stress-triggered autophagy [244,245]. In this regard, Cook et al. reported that activating AMPK and TSC2 by GRP78 could stimulate autophagy in breast cancer by suppressing the mTOR [246]. P38 MAPK signaling cascade is another central pathway to the mentioned phenomenon; the accumulation of misfolded acid α-glucosidase (GAA) can stimulate ER stress, which enhances LC3 II levels in Pompe disease. Interestingly, a significant decrease in p38 phosphorylation, achieved by employing a pharmacological chaperone for misfolded GAA and/or a particular inhibitor of p38 MAPK, can markedly mitigate p38-correlated ER stress [247]. SB203580 is one of these inhibitors that can suppress ER stress-triggered autophagy. It is important to highlight that the IRE1/ASK1 pathway leads to the phosphorylation of p38. Additionally, within the IRE1/ASK1 axis, JNK is a commonly targeted component, often involving TRAF2. However, it's noteworthy that no alterations in the levels of phosphorylated JNK and ERK have been documented in cells exposed to ER stress. As a result, the exact pathway among the three MAPK pathways that serves as an autophagy inducer during ER stress conditions appears to remain uncertain. [214].
ER stress promotes the translocation of misfolded proteins to the cytoplasm, where they undergo ubiquitination and subsequent removal by the by the ubiquitin-proteasome system, a process known as ER-associated degradation (ERAD) [248,249]. When the ERAD is insufficient for degradation, ER stress-induced autophagy serves as an alternative process to eliminate misfolded proteins in order to maintain proteostasis [250]. Autophagy may also neutralize ER expansion by sequestering the ER into double membrane-bounded and autophagosomal-like structures [251]. ER stress caused by hypoxia/ischemia can be reduced in vivo by the powerful autophagy activator rapamycin [169] , but it can be completely restored by the pharmacological autophagy inhibitor 3-methyladenine (3-MA).[252].
In general, chemoresistance can occur due to multiple factors such as amplified drug efflux, changes in drug targets, drug inactivation, all of which contribute to accelerating drug removal from cancerous cells. The potential role of UPR and the UPR-autophagy network in cancer drug resistance is now evident. Cancer cells typically employ the adaptive power of the UPR arms to ensure survival when exposed to chemotherapeutics. Therefore, suppressing XBP1, GRP78, ATF6, and ATF4 is believed to contribute to the re-sensitization of chemoresistant cells, suggesting that all UPR’s arms and their downstream factors can be considered as potential targets to overcome drug resistance. Multiple processes, such as oxidative stress and ROS generation, occur upstream or downstream of the UPR, and they may be directly or indirectly involved in chemoresistance. Since it is crucial to understand the interplay between these up-/downstream events and UPR, the following section will provide comprehensive information in this regard.
3. NRF2 controls UPR and proteostasis
As mentioned before, ER stress resulting from the aggregation of misfolded proteins induces UPR by stimulating three signaling branches equipped with the IRE1-XBP1, PERK -eIF2a, ATF4, and ATF6 [253]. The deposition and aggregation of misfolded proteins trigger the unrestricted generation of ROS from mitochondria, ER, and other sources, which could activate NRF2 [254]. The NRF2-KEAP1-ARE pathway is a flexible cellular response that safeguards against oxidative and xenobiotic stress. ROS or electrophile-induced changes in Keap1 cysteine sites prevent NRF2 degradation, leading to its accumulation in the cytosol. NRF2 then enters the nucleus, partnering with MAF proteins to activate genes with ARE sequences in their regulatory regions. This process enhances the cell's ability to counter stress (Figure 4A, canonical pathway) [255].
During cellular stress, NRF2 triggers the activation of cytoprotective genes, which encode a network of enzymes collaborating to detoxify drugs through phases I, II, and III, and to eliminate pro-oxidants, maintaining cellular hemostasis. Notably, SQSTM1/p62, a protein acting as an autophagy adapter, is instrumental in orchestrating the formation of protein aggregates marked for autophagic turnover. This suggests that p62 plays a role in selectively removing protein burdens via autophagy. In SQSTM1/p62, the 349-DPSTGE-354 motif located in its KEAP1-interacting region (KIR) domain is pivotal. This motif establishes a direct interaction between p62 and KEAP1 [256]. Consequently, p62 sequesters KEAP1 within autophagosomes, hindering the ubiquitination of NRF2. As a result, the NRF2 signaling pathway becomes activated, contributing to cellular defense against stress-induced damage[255] (Figure 4B, noncanonical pathway).
Moreover, stress-induced PERK activity initiates the phosphorylation of NRF2, resulting in the dissociation of the NRF2/KEAP1 system and subsequent activation of NRF2 [257]. NRF2 acts as a pivotal central hub for sensing critical signals arising from the accumulation of misfolded proteins, orchestrating a coordinated transcriptional response. This function mirrors that of SKN-1, the C. elegans homolog of NRF2, which triggers certain aspects of UPR genes, such as XBP1 and ATF6, thereby promoting a UPR strategy to preserve endoplasmic reticulum integrity and protein homeostasis [258]. Notably, the activation of UPR target genes by SKN-1 may be mediated through NRF1[259]. Additionally, NRF2 contributes to the induction of ATF4 expression, a protein closely associated with amino acid metabolism and the ability to withstand oxidative stress [260]. NRF2 and ATF4 form a heterocomplex to activate the expression of target genes to, enabling the cell to withstand proteotoxic insults [39].
Furthermore, NRF2 binds to the promoters of genes coding proteasome maturation protein (POMP), an intermediate in proteasome assembly and activates its expression [261]. The heightened activation of NRF2 in cancers is strongly associated with increased proteasome activity and resistance to the proteasome blocker bortezomib [262]. In conclusion, NRF2 not only enhances proteasome activity but also upregulates the expression of antioxidant genes, facilitating cellular adaptation to stress. [39].
3.1. NRF2 modulates autophagy
Autophagy relies on a coordinated collaboration among a group of proteins that collectively assemble autophagosomes and autolysosomes [263,264]. Additionally, selective cargo-recognizing proteins identify specific targets and guide them toward degradation[265,266]. Nrf2 contributes to the upregulation of mRNA levels for autophagy-related genes like Sqstm1/p62, calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2/Ndp52), unc-51-like kinase 1 (Ulk1), autophagy protein 5 (Atg5), and gamma-aminobutyric acid receptor-associated protein-like 1 (Gabarapl-1). These genes collectively contribute to facilitating the autophagy process [267]. Surprisingly, in conditions induced by NRF2, a therapeutic focus on promoting autophagy might prove ineffective. Paradoxically, insufficient autophagy results in the accumulation of oxidized proteins or organelles, triggering NRF2 activation. Notably, a deficiency in autophagy results in the buildup of p62, a multifunctional cargo receptor that can sequester KEAP1 and stabilize NRF2, ultimately leading to NRF2 induction [268]. Hence, a reciprocal loop is formed between p62 and NRF2, establishing a positive feedback mechanism that governs a multitude of cellular processes. [39,265].
The function of autophagy in cancer is slightly paradoxical; it can contribute to eliminating tumor cells in some instances, while in others, cancer cells may suppress autophagy as a defense against nutrient deficiency, oxidative stress, and other stressors [269,270,271,272]. Autophagy defects can potentially promote cancer through Nrf2 induction. In a study, the inhibition of the critical autophagy gene Atg7 in the liver of mice induced accretion of p62, Nrf2 stimulation, and the development of hepatocellular carcinoma (HCC) [273]. In addition, ectopic expression of p62 was sufficient to activate NRF2 and promote development of HCC [265], highlighting the fundamental role for p62 elevation in HCC generation downstream of autophagy deficit. In pancreatic ductal adenocarcinoma, perturbation of inflammation- induced autophagy also triggered p62-mediated stimulation of NRF2, contributing to neoplasm progression through NRF2-intervened MDM2 induction [274].
In cancers, not only NRF2 stimulation through p62 is pro-tumorigenic, but it also performs a vital role in the cellular response to autophagy defects in normal cells. NRF2 preserves small intestine damage and animal death after entire body deletion of ATG7 and TP53 [20], implying that NRF2 induction by p62 is crucial for cellular adaptation and homeostatic management. NRF2 may regulate the expression of specific proteasome subunits, which could clarify why it is protective in the background of autophagy deficiency [265]. Furthermore, in response to proteasome defects, NRF2 is also activated and subsequently collaborates with autophagy to respond to the stress[275]. In summary, these studies emphasize the critical role for NRF2 in responding to autophagic defect and/or proteasomal strain [265]. Unresolved ER stress can trigger programmed cell death, but certain tumor cells evade ER stress signaling to promote their growth, and NRF2 activation plays a role in this evasion[276]. For instance, the PERK enzyme inhibits cap-dependent translation to alleviate proteotoxic stress while promoting ATF4 expression[277]. ATF4 is associated with resistance against oxidative stress, enhanced amino acid metabolism, and autophagy induction, possibly through interaction with NRF2. NRF2 and ATF4 mutually induce each other's expression, forming a positive feedback loop[253] . NRF2 and ATF4 prevent ER stress-induced cell death, enabling cancer cells to endure proteotoxic stress. Furthermore, unresolved ER stress triggers the UPR and activates the ER-associated degradation (ERAD) pathway. NRF2-induced proteasome genes aid in ERAD, thus reducing proteotoxic stress [253,257].
4. Evidence show overexpression of NRF2 promotes post-initiation stages of cancer
Although the overexpression of NRF2 is not sufficient to launch tumorigenesis, its upregulation has been established in certain types of cancers. NRF2 overexpression contributes to carcinogenesis by amending oxidative stress and promoting cell growth in different ways, such as over activation of pentose phosphate pathway (PPP), serine synthesis, autophagy, and weakening the immune system [76]. In a murine Kras oncogenic pancreatic cancer model, research about the suppressive action of oxidative stress on carcinogenesis has demonstrated that Nrf2 is needed for Kras-guided pre-invasive pancreatic intraepithelial neoplastic lesions. Likewise, through a CRISPR-Cas9 approach, deletion of Keap1 has been shown to hasten Kras-guided lung adenocarcinoma [76,278]. Since cancer cells generate higher levels of ROS to sustain growth and must tolerate oxidative stress within metastasis, it is highly possible that overexpression of NRF2 gives advantages to the tumor, so the subsequent upregulation of antioxidant genes inhibits ROS-induced cell apoptosis [279]. In addition to the increased activity of antioxidant enzymes that eliminate ROS, NRF2 also handles the transcription of PPP genes in both the oxidative and non-oxidative arms. The overexpression of this pathway could contribute to the existence and proliferation of cancer cells by enhancing the production of NADPH and ribonucleotides [280].
In addition to the elevation of antioxidant enzymes regulated by NRF2, there are alternative strategies available to boost glutathione (GSH) levels during advanced cancer stages. One effective approach involves increasing the production of NADPH through the folate pathway. Other techniques encompass activating mTOR signaling, raising mitochondrial metabolism, and facilitating glutamine flux through estrogen-related receptor α (ERRα) pathways [281]. Furthermore, the indirect influence of NRF2 over ATF4 governs serine synthesis by controlling the transcription of genes encoding phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase-1 (PSAT1), and serine hydroxymethyltransferase-2 (SHMT2). While serine serves as a vital intermediate for glutathione and nucleotide synthesis, excessive activation of NRF2 can stimulate cancer cell growth [260]. In conjunction with antioxidant systems and the pentose phosphate pathway (PPP), NRF2 also impacts the transcription of proteasome subunits and components within the autophagy complex. The hyperexpression of NRF2 activates this system, potentially providing support for cancer cell viability and growth [76]. Notably, autophagy plays a crucial role in suppressing liver carcinogenesis by preventing the buildup of defective mitochondria and mitigating oxidative stress. This is exemplified through studies involving mice with mosaic deletion of Atg5 and hepatocyte-specific deletion of Atg7, where hepatic adenomas developed [282]. However, before tumor stabilization occurs, autophagy typically stimulates cancer cell proliferation by recycling unnecessary cellular components to fuel oxidative phosphorylation, thus enabling these cells to overcome nutrient stress [76].
Hence, the role of autophagy in cancer is context-dependent and might be linked to the timing of NRF2 stimulation via an unconventional mechanism [253]. Autophagic flux intensifies in response to oxidative, proteotoxic, and metabolic stresses, aiming to restore homeostasis and impede genome instability, inflammation, and overall tissue damage [283]. Notably, regulated and effective autophagy in normal cells or tissues acts to suppress the initiation of cancer [253]. However, many cancer cells rely on autophagy to withstand heightened levels of proteotoxic, metabolic, oxidative, and hypoxic stress. Notably, cancers carrying KRAS mutations heavily depend on extensive autophagy for proliferation and invasion [253,284]. Disrupting autophagy in NSCLC through the activation of KrasG12D and BrafV600E, either alone or combined with Trp53 deletion, halts cancer progression and leads to less severe damages[285]. That is why autophagy inhibitors are utilized in cancer treatment [253,286].
Yet, in cases where these agents fail to effectively initiate cellular death, they may inadvertently trigger NRF2 via non-canonical pathways, resulting in chemo-resistance and prolonged cell survival. Moreover, given NRF2's role in governing the transcription of autophagy-related genes like Sqstm1/p62, Calcoco2, Ulk1, Atg5, and Gabarapl1, non-canonical activation of NRF2 could undermine the effectiveness of treatments targeting autophagy [253,267]. Nonetheless, a combined therapeutic strategy addressing both autophagy and Nrf2 could potentially overcome this resistance. On the other hand, impaired autophagy, achieved through genetic disruption of components like ATG5, ATG7, or BECN1, has been shown to trigger liver cancer [282]. The impairment of autophagy (via deletion of ATG5, ATG7, or BECN1) leads to an accumulation of SQSTM1/p62, resulting in subsequent non-canonical sustained activation of NRF2 [253]. Interestingly, studies have illustrated that the elimination of Sqstm1/p62 reinstates the carcinogenicity linked to malfunctioning autophagy in mice, which corresponds to a reduction in Nrf2 levels [282]. Correspondingly, Nrf2 deletion inverses the impacts of malfunctioned autophagy made by deletion of Atg5 in mouse liver and lessens carcinogenesis [77,253].
4.1. NRF2 as double-edged sword in cancers
Cancer chemopreventive agents serve to safeguard normal tissues from the initiation of carcinogenesis by activating NRF2-targeted genes. These genes encode enzymes that mitigate the genotoxic and cytotoxic effects of carcinogens. However, extended activation of NRF2 in cancer cells can predictably result in resistance to both drug and radiotherapy treatments. Thus, NRF2 demonstrates a dual nature, acting as a "double-edged sword" by facilitating both cancer chemoprevention and the promotion or progression of tumors [76]. The potential adverse consequences arising from the overstimulation of NRF2 in pre-neoplastic lesions and cancer cells have been termed the "dark side" of this transcription factor by Donna Zhang's research group [287]. This aspect is underscored by clinical observations linking elevated NRF2 expression to unfavorable prognosis and decreased overall survival rates among patients with lung, head and neck, esophageal, gastric, liver, and colorectal cancers. [76].
Tumor cells are theoretically more sensitive to oxidative stress than normal cells due to their high levels of ROS induced by activation of oncogenes. Therefore, therapeutic approaches aiming to increase ROS production or decrease their antioxidant capacity have been considered as a means of generating selective toxicity in cancers [288].
4.2. Targeting of Nrf2 Signaling to fight Chemo-resistance: NRF2 inhibitors
Regarding the distinct NRF2 levels between cancer and normal cells, inhibiting NRF2 activity with small molecular inhibitors might be a safe and promising strategy to overcome multidrug resistance in cancers. For example, sequential therapy of breast cancer cells with vitamin C and quercetin has been found to reduce the expression of NRF2 at both the mRNA and protein levels [289]. Additionally, combination therapy with vitamin C and quercetin has been reported to enhance the cytotoxic feature of chemotherapeutic drugs in breast cancer cells as compared with the drug treatment alone [290].Developing novel and potent NRF2 inhibitors is undoubtedly a challenging task, although only a small number of NRF2 inhibitors have been proposed for further preclinical experiments. [291].
There is a large number of synthetic molecules and flavonoids that inhibit Nrf2, but in this review, we only focused on NRF2 inhibitors studied based on data from various preclinical and experimental models. Studies have frequently pointed to the inhibition of the NRFrf2 pathway as a promising therapeutic choice for the treatment of cancer, which requires more investigation and authentication in the clinical settings. The inhibitors we have discussed, as outlined in (Table 2), exhibit a range of inhibitory mechanisms. These mechanisms include the inhibition of overall protein synthesis (brusatol, halofuginone, camptothecin/CPT), disruption of NRF2 nuclear translocation (trigonelline, ATRA), inhibition of DNA binding (ML385), suppression of associated kinase pathways (clobetasol propionate, flavonoids), initiation of NRF2 degradation (clobetasol propionate), or an effect that remains undisclosed (triptolide, IM3829, AEM1).
The use of this approach is exemplified by well-known instances such as the quassinoid brusatol [292], as well as other compounds like retinoic acid [261] and natural flavonoids such as luteolin, apigenin, chrysin, and wogonin [293,294,295,296]. Brusatol, derived from the natural product of Brucea javanica, functions as an NRF2 inhibitor, validated through a stable ARE-luciferase reporter gene cell line known as MDA-MB-231-ARE-Luc [228]. Recent studies have shown that it effectively reduces NRF2 protein levels across various cell lines, including MDA-MB-231, Hela, Ishikawa, and SPEC-2, while NRF2 mRNA levels remain unaffected. Furthermore, brusatol does not impact KEAP1 protein and mRNA levels. Notably, it sensitizes A549 cells and xenografts to a range of chemotherapeutic drugs including carboplatin, 5-fluorouracil, etoposide, and paclitaxel [292]. Histological investigation showed that brusatol treatment decreased the expression of antioxidant genes such as NRF2, solute carrier 7A11 (SLC7A11), GCLC, and GCLM in a xenografted glioma model [297].
Table 2.
Research Summary on Nrf2-ARE Inhibitors in Preclinical and Experimental Models.
| Classification and origins |
Compound | Structure | Dose and time | Mechanism | Model and effect | Refs. |
|---|---|---|---|---|---|---|
|
Brucea javanica Plant |
Brusatol (Bru) | 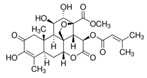 |
2 mg/kg, five times for 16 days via intraperitoneally | ↓ NRF2/GSH axis | ↓tumor mass ↓NRF2, SLC7A11, GCLC, and GCLM expression Six-to-eight-week-old NSG mice |
[298] |
|
Flavonoid |
Luteolin (Lut) |  |
40 mg/kg BW/day; 14 days |
Unclear | ↓NRF2 protein levels in mouse liver and intestine (C57BL/6, Male, 6 weak old) | [293] |
| Flavonoid | Apigenin (Api) |  |
In vivo: 50 mg/kg BW/day; every 3 days for 7 times |
PI3K/Akt pathway: ↓p-Akt | ↓tumor size in male BALB/c nude mice (aged 5 weeks) that were implanted with BEL-7402 cells; ↓level of NRF2 protein | [294] |
| Flavonoid | Chrysin (Chry) |  |
40 and 80 mg/kg/day by oral gavage; once a day, 5 times per week | ↓ERK/NRF2 signaling pathway | ↓Tumor size of mice (male BALB/c athymic nude mice, 4–6 weeks) ↓translocation of NRF2 into the nucleus and ↓ expression of (HO-1) and NQO-1 |
[295] |
| Flavonoid | Wogonin (Wog) | 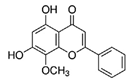 |
40 mg/kg intravenously, once every other day for 30 days | ↓NF-κB/NRF2 pathway | NOD/SCID immunodeficient mice (aged 5–6 weeks) ↓ nuclear NF-κB p65, p-Stat3 and NRF2 expression and ↓phosphorylation of IKKα and IκBα ↓NF-κB p65 and NRF2 expression in spleen |
[296] |
| A traditional Chinese medicine | triptolide | 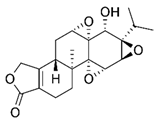 |
0.25 mg/kg, by intraperitoneal every other day for 10 days | transcriptional regulation of NRF2 |
↓Tumor growth and weight C57BL/6 mice (6–8 weeks, male) ↓NRF2 and downstream genes Gclc and Gclm increases the chemosensitivity of xenograft tumors to epirubicin |
[299] |
| An alkaloid | Trigonelline (Trig) | 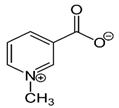 |
0.02 mg/kg intraperitoneally for 21 days | ↓ a nuclear level of activated NRF2 protein |
↓tumor growth and weight 8-week-old female SCID–beige mice ↑responsiveness of various cell lines to both anticancer drugs and apoptosis induced by TRAIL. |
[300] |
| Vitamin derivative | all-trans-retinoic acid |  |
10 µM, 48h 40 mg/kg, three times weekly via intraperitoneally |
↓NRF2/POMP axis | ↓cell viability, purified CD138+ plasma cells from patients with myeloma ↓tumor growth 6-week-old non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice |
[261] |
|
Aniline-based compound |
IM3829 (4-(2-Cyclohex-ylethoxy) aniline) | 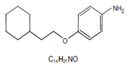 |
5mg/kg/day), intraperitoneally for 4 days | ↓NRF2-binding activity and expression of NRF2 target genes, increase ROS accumulation in irradiated cell |
↓tumor growth, without changes in body weight, Five-week, female, athymic BALB/c nude mice |
[301] |
| A probe molecule that binds to Nrf2 | ML385 | 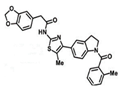 |
30 mg/kg daily Monday to Friday), intraperitoneally for 3 weeks | blocks NRF2 transcriptional activity | ↓tumor growth, athymic nude mice, ↓NRF2, NQO1, and ABCG2 expression | [302] |
| A chemical substance | ARE expression modulator 1 (AEM1) | 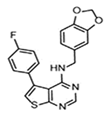 |
50 mg/kg, BW; twice a day for 10 days |
Unclear | ↓Tumor growth of mice which were implanted with A549 cells (Nude mice, Male, 6 wk old) | [303] |
| A febrifugine derivatives | halofuginone | 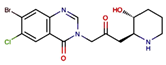 |
0.25 mg/kg, every day intraperitoneally | Inducing a cellular amino acid starvation response that repressed protein level of NRF2 |
↓tumor growth, 6–8-week-old male nude mice, enhances the anticancer effects of cisplatin. without severe toxicity |
[304] |
| Corticosteroid drug | Clobetasol propionate | 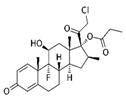 |
CP (0.5 or 1 mg/kg, n = 5 per group) were intraperitoneally injected every 2 days (3 days per week) for 40 days. |
↓NRF2 in a GR and GSK3-dependent manner | Balb/c-nu mice (6–8 weeks). ↓NRF2 and the expression of its key targets in KEAP1 mutant NSCLCs and induction of oxidative stress. ↓tumor growth and shrinkage of tumor size |
[305] |
| Purified from Streptomyces sp. 3728-17 strain |
K-563 |  |
100 mg/kg, subcutaneously to the mice twice a day for 1 day |
↓Keap1/NRF2 pathway | Male severe combined immunodeficient (SCID) mice, 5 weeks’ old ↓expressions of Keap1/NRF2 pathway Targeted genes (HMOX1, GCLC, GCLM,AKR1C1, ME1, NQO1, and TXNRD1) |
[306] |
| Anti-tumor drug | Camptothecin |  |
CPT (3 mg/ kg body weight) were intraperitoneally (IP) injected twice a week for a total of three times. | ↓NRF2–ARE pathway activity | ↓tumor growth BALB/Cnu/nu mice (4–6 weeks, male). Sensitization of a variety of cancer cells and a xenograft hepatocellular carcinoma model to chemotherapeutic drugs | [307] |
| Type 2 diabetes drug | Metformin |  |
200 μg/mL, diluted in drinking water and administered for day 22 | ↓NRF2, HO-1, KI-67 and PCNA expression |
↓tumor growth, Female BALB/C nude mice (6-8 weeks of age) enhancing the anti-cancer effect of EGCG on NSCLC xenografts |
[308] |
| Cardiac glycoside drug | Digoxin |  |
0.1 mg/kg, daily, i.g for 24 days | ↓Activity of NRF2 through suppressing PI3k/Akt signaling pathway |
↓tumor growth In female BALB/c nude mice (aged 6 weeks, weighing 18 ± 2 g), the resistance to gemcitabine was effectively reversed by inhibiting NRF2 in SW1990/Gem and Pac-1/Gem cells. |
[30] |
These compounds possess drawbacks like lack of specificity and potential off-target effects. For instance, brusatol exhibits a global protein translation suppression effect [228]. Luteolin (3′,4′,5′,7-tetrahydroxyflavone) reduces both NRF2 mRNA and protein levels in A549 cells by promoting NRF2 mRNA degradation, sensitizing them to antitumor drugs. In mice, luteolin treatment decreases NRF2 protein levels and reduces tumor size in the liver and intestine [293]. Apigenin (4′, 5, 7-trihydroxyflavone) and chrysin (5, 7-dihydroxyflavone) also decrease NRF2 mRNA and protein levels, rendering hepatocellular carcinoma BEL-7402 cells more responsive to the anti-tumor drug doxorubicin. Recent research highlights that apigenin and chrysin's NRF2-suppressive effects stem from inhibiting the PI3K/AKT pathway. Notably, apigenin treatment induces tumor size reduction in mice implanted with hepatocellular carcinoma cells. [294].
Notably, apigenin and chrysin exhibit direct inhibition of the PI3K/AKT pathway, a critical regulator of cancer cell proliferation [228,294]. Their distinct actions on cancer cells compared to normal cells, along with their low toxicity, make them promising candidates for cancer treatment. Another compound, wogonin (5, 7-dihydroxy-8-methoxyflavone), derived from the traditional Chinese herb Scutellariae radix, has emerged as an NRF2 inhibitor methoxyflavone) [228]. Wogonin effectively counteracts drug resistance in various cell lines, including human breast adenocarcinoma MCF-7/DOX, human myelogenous leukemia K562/A02, and HepG2 cells by suppressing NRF2 mRNA and protein expression. Notably, it enhances the anticancer effects of Adriamycin in leukemia by inhibiting pY705-Stat3 and NRF2 signaling pathways[296].
Triptolide, an agent from traditional Chinese medicine, has been proven to effectively inhibit the expression and transcriptional activity of NRF2 in various cancer cell types, including non-small cell lung cancer (NSCLC) and liver cancer cells. This suppression of NRF2 activity leads to enhanced chemosensitivity of cancer cells to antitumor drugs both in laboratory settings and in a xenograft tumor model of lung carcinoma cells. [299].
Trigonelline, an alkaloid found in various plants and coffee, has the potential to enhance cell susceptibility to apoptosis by inhibiting the nuclear translocation of NRF2. This action is accompanied by a reduction in proteasomal gene expression and proteasome activity[228]. The favorable safety profile of this natural coffee constituent in both regular consumption and therapeutic applications suggests its viability for combination therapy in pancreatic and other types of cancers.[300]. All-trans-retinoic acid (ATRA), also identified as tretinoin, is a vitamin in the retinoid family of medicines [228]. As an NRF2 inhibitor, ATRA diminishes cellular levels of NRF2 and boosts the anti-prolife\rative and pro-apoptotic actions of bortezomib in resistant cells while reducing proteasome activity. The combination therapy with all-trans-retinoic acid plus bortezomib exhibited great activity versus primary patient samples and in a mice-bearing bortezomib-resistant myeloma model [261]. A number of research teams have purified several chemical libraries for small molecules that hinder NRF2 activity with various cell-dependent reporter analyses [228]. Employing ARE-directed luciferase assays, researchers found compounds such as 4-(2-cyclohexylethoxy) aniline (IM3829) [301], ML385 [302], halofuginone [304], and clobetasol propionate [305] were powerful suppressors of NRF2 activity.
IM3829 stands as the first synthetic NRF2 inhibitor, as revealed by Song et al. Their pursuit involved screening a synthetic compound library of 8000 substances through a HEK293 cell-based ARE-luciferase reporter assay, leading them to identify IM3829 [4-(2-cyclohexylethoxy) aniline] as a small-molecule NRF2 blocker [228]. This compound was found to decrease the mRNA levels of NRF2 and its target genes, HO-1 and NQO1, in both H1229 and A549 cells. In tandem with irradiation (IR) therapy, IM3829 inhibited the nuclear translocation of NRF2, increased ROS levels in lung cancer cells, and significantly delayed tumor growth in H1299 and A549 xenograft models, surpassing the effects of IR-only treatment or Dimethyl sulfoxide (DMSO). Overall, IM3829 holds potential as a radiosensitizer for non-small cell lung cancer (NSCLC) [301]. Moving on, ML385 selectively suppresses the growth of KEAP1-mutated NSCLC A549 and H460 cells while leaving normal lung epithelial BEAS2B cells, possessing wild-type NRF2 and KEAP1, unaffected. Additionally, ML385 enhances the sensitivity of NSCLC cells to the chemotherapeutic drug carboplatin both in vitro and in vivo. [302].
In a recent study, a compound called ARE expression modulator 1 (AEM1) has been identified as a potent suppressor of antioxidant response element (ARE) activity. It effectively reduces ARE-luciferase activity in 3T3 cells and downregulates HO-1 expression in A549 cells with an IC50 of 650 nM. AEM1 also enhances the susceptibility of A549 cells to the cytotoxic effects of chemotherapy drugs like etoposide, 5-fluorouracil, and doxorubicin. In vivo experiments have shown that AEM1 can inhibit tumor growth in an A549 xenograft-model. [303].
Halofuginone, derived from febrifugine and known for its low toxicity, has been employed as an animal antibiotic [309]. It also demonstrates a dose-dependent repression of NRF2 function and enhances the effectiveness of cisplatin in fighting cancer in vivo. Similar to brusatol, halofuginone blocks NRF2's protein translation [304]. Another instance is clobetasol propionate (CP), which notably reduces NRF2 activity to 40% at a concentration of 1 nM. Treating of KEAP1 mutant cells (H2228 and A549) with CP results in decreased NRF2 target gene expression and increased accumulation of reactive oxygen species (ROS). Furthermore, the combination of CP and rapamycin has been observed to effectively hinder the growth of KEAP1-mutated tumors both in vitro and in vivo [305].
Another study investigated a newly discovered inhibitor of the KEAP1/NRF2 pathway, K563, obtained from Streptomyces sp [228]. K563 was found to hinder the expression of downstream genes and proteins related to this pathway, leading to reduced glutathione production and increased reactive oxygen species in A549 cells. It also exhibited similar effects in cancer cells with mutated KEAP1 or NRF2 genes. In vivo experiments on xenograft mice with A549 cells showed that K563 effectively suppressed the KEAP1/NRF2 signaling pathway within lung cancer tumors. [306].
CPT, known for its role as a topoisomerase inhibitor in the treatment of gastrointestinal and head and neck cancers, has also emerged as a potential NRF2 inhibitor for anticancer purposes. Chen and colleagues introduced CPT as a novel compound that can block NRF2 activity. Through this action, CPT sensitized a range of cancer cells (HepG2, SMMC-7721, A549) and even a xenograft model of hepatocellular carcinoma to various chemotherapeutic agents like As2O3, epirubicin, fluorouracil, and cisplatin. The inhibition of NRF2 by CPT specifically contributed to enhanced chemosensitivity in HepG2, A549, and SMMC-7721 cells[307].
Metformin, a traditional medication for diabetes management, has been found to impede the advancement of cancer. It inhibits the expression of HO-1 and amplifies the anticancer effects of EGCG by augmenting levels of reactive oxygen species. In a particular investigation, the size of transplanted tumors subjected to the combined treatment of metformin and EGCG exhibited reduction in comparison to the control groups. Metformin heightens the susceptibility of non-small cell lung cancer (NSCLC) cells to EGCG treatment by obstructing the NRF2/HO-1 signaling pathway. [308].
In an experimental study, the potent cardiac glucoside digoxin was found to effectively counteract drug resistance to gemcitabine in SW1990/Gem and Pac-1/Gem cells by inhibiting NRF2. Digoxin achieved this by blocking NRF2 through the suppression of the PI3K/AKT signaling pathway[30]. Given NRF2's pivotal role in cancer hallmarks, targeting this transcription factor appears to be a promising therapeutic strategy. While NRF2 activators could potentially prevent chemical carcinogenesis, inhibiting NRF2 holds promise for cancer treatment. Despite challenges in developing safe, potent, and specific NRF2 inhibitors due to NRF2's dual nature, extensive efforts have been dedicated to this area with limited success thus far. It is anticipated that upcoming years will clarify NRF2's potential as both a prognostic biomarker and a therapeutic target within cancer therapy.
4.3. Clinical Overview
Studies have indicated that inducing the NRF2 signaling pathway is a potent approach for tumor suppression and a viable strategy in anticancer therapy[87,88]. The transcription factor NRF2 plays a pivotal role in maintaining cellular redox and hemostasis, as well as in proliferation, differentiation, and the regulation of inflammation through its influence on a wide array of target genes [310,311,312]. Therefore, it emerges as a promising target for clinical interventions where such pathways contribute to the underlying pathophysiology. Over the past few decades, ongoing efforts have been dedicated to targeting NRF2 signaling as a therapeutic strategy. Numerous preclinical studies have investigated the targeting NRF2 pathways in diseases where inflammatory and oxidative processes constitute critical components, like autoimmune diseases. Particularly, four main NRF2 inducers are of particular interest: dimethyl fumarate, bardoxolone methyl, oltipraz and sulforaphane. These agents disruptNRF2 signaling through KEAP1, a key cytoplasmic receptor for NRF2 [312,313]. However, use of such agents in cancer treatment remains a topic of controversy, as knocking down NRF2 may increase susceptibility to carcinogenesis [314]. Although, NRF2 overexpression has been observed in numerous tumors, particularly in advanced stages, helping tumors in adapting to their microenvironment and inducing resistance to therapeutic strategies [315,316].
Therefore, their application in clinical setting for cancer is still in its early stages. Various compounds, such as glucocorticoid agonists [305,317] and retinoic acid receptor-alpha, which inhibit NRF2 activity [318,319] may exert NRF2-blocking activities, although they lack specificity.. However, certain compounds, such as ML385 have been identified through in silico studies, exhibiting a degree of specific interactions with NRF2 activities. ML385 inhibits the transcriptional function of NRF2 by interacting with its DNA binding domain, thereby increasing chemosensitivity of KEAP1-deficient cells [320]. AEM1 is another compound that downregulate NRF2-conrolled genes and increase chemosensitivity[303]. Additionally, a compound named 4f, a pyrazolyl hydroxamic acid, has demonstrated inhibitory effects on NRF2 activities. It increases apoptosis and reduces proliferation in acute myeloid leukemia cells [321]. A novel aziridonin, YD0514, derived from medical herb Rabdosia rubescens, targets the NRF2/ Ras homolog family member A (RHOA)/Rho-associated protein kinase (ROCK) pathway to suppress metastatic growth of breast cancer in vitro and in vivo[322]. The main component of coffee, Trigonelline, has been introduced as a potential treatment candidate for lung adenocarcinoma. It targets multiple pathways, including NRF2, cyclin D1, Nuclear Factor Kappa B (NF-кB), and BAX/Bcl2, inducing cell cycle arrest and apoptosis [323]. An alkaloid of black pepper, piperine, is another compound that suppresses NF-kB by inducing NRF-2 and has therefore found to be effective for prophylactic treatment of colorectal cancer in vivo[324].
Available evidence indicated that phytochemical antioxidants such as sulforaphane and curcumin target the NRF2/NF-kB and Androgen Receptor pathways, making them proper candidates for chemoprevention in prostate cancer[325]. As a polyphenolic composition of soy-based plants, daidzein modulates immune responses and antioxidant function in mice with Benzo(a)pyrene -Induced Lung Cancer by targeting Proliferating Cell Nuclear Antigen (PCNA)/ NF-κB, Cytochrome P450 Family 1 Subfamily A Member 1 (CYP1A1), and NRF[326]. Another in vitro and vivo study indicated that downregulation of NRF2/Heme oxygenase (HO-1) by miR-144-3p increased the anticancer effect of curcumin in non-small cell lung cancer (NSCLC)[327]. It has also been shown that L-carnosine reduces the peripheral neuropathy induced by oxaliplatin in colorectal cancer patients by targeting Nrf-2/ NF-κB pathways[328]. Taken together, NRF2 may be considered as a promising signaling target for cancer treatment. Further studies will contribute to identifying the exact clinical effect of NRF2 in cancer therapy. Understanding the intricate connections between NRF1, NRF2, redox homeostasis, PERK, autophagy, and chemo sensitivity opens doors to novel therapeutic interventions. Targeting specific components of these pathways could enhance the efficacy of chemotherapy in a patient-tailored manner. Developing biomarkers to assess NRF1/NRF2 activity, redox status, and autophagy levels could guide treatment decisions, optimizing outcomes in diverse cancer contexts.
5. Conclusion
NRF2 plays a crucial role in the hallmarks of cancer, acting as a double-edged sword in both the promotion and prevention of different tumors. Activation of UPR and autophagy by NRF2 may result in cancer cell survival and chemoresistance or cancer cell death during the progression of malignancies, depending on the stage and cellular origin. Therefore, proper targeting of Nrf2 may be a promising strategy in overcoming chemoresistance,a major obstacle in cancer therapy. Nrf2-targeting agents can be employed to tackle chemo-resistance in a variety of cancers. The crosstalk between ER-stress, UPR, autophagy, and NRF2 signaling pathways has recently received considerable attention. the use of antioxidant supplements and activation of the NRF/KEAP1 pathways in individuals undergoing chemotherapy are topics of controversy. Adjusting the internal levels of antioxidants, which hold therapeutic promise , poses a dual challenge. While these agents have the potential to diminish the effectiveness of chemotherapy by eliminating reactive oxygen species (ROS), they can also contribute to the toxicity induced by chemotherapy. These conflicting outcomes suggest that the impact of supplementing antioxidants during chemotherapy varies based on the specific cellular environment of the tumor. Consequently, achieving an optimal balance between the cancer-preventive and cancer-promoting functions of NRF2 may offer clinical benefits to cancer patients.
Author Contributions
Conceptualization, S.G (Saeid Ghavami).; P.M (Pooneh Mokarram) and ZM (Zohreh Mostafavi-pour); methodology, S.M.SH (Seyed Mohammad Shafiee).; Software, M.S (Morvarid Siri).; Validation, S.D (Sanaz Dastghaib).; N.A (Niloufar Ashtari) and M.Z (Mozhdeh Zamani).; Investigation, F.R (Fatemeh Ramezani).;F.T (Farhad Tabasi).; and J.S.Ch (Javad Safari Chaleshtori).; Resources, O.V (Omid Vakili).; Data curation, M.M.N (Mahshid Moballegh Nasery), S.I (Somayeh Igder), F.K (Fariba Kokabi), and E.W (Emilia Wiechec).; Writing—original draft preparation, F.R , S.D, and J.S.Ch.; Writing—review and editing, P.M., S.D, M.Z, M.M.N and F.K.; Visualization, M.S.; Supervision, S.G., P.M. and Z.M.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3-MA | 3-Methyladenine |
| 5-FU | 5-Fluorouracil |
| ABC | Advanced Breast Cancer |
| ABCCs | Multidrug resistance (MDR)-associated proteins |
| ABCG2 | Breast cancer resistance protein |
| AEM | Are Expression Modulator |
| AKRs | Aldo-Keto Reductases |
| AKT | Protein kinase B |
| ALDH1 | Aldehyde Dehydrogenase 1 |
| AMPK | Adenosine monophosphate-activated protein kinase |
| AP-1 | Activator protein 1 |
| ARE | Antioxidant Response Element |
| ARS | Antioxidants and redox signaling |
| ASK1 | Apoptosis-signal regulating kinase 1 |
| ATF | Activating Transcription Factor |
| ATG | Autophagy-related protein |
| ATRA | All-Trans-Retinoic Acid |
| BACH1 | BTB and CNC homology 1 |
| BCRP/ABCG2 Breast cancer resistance protein | |
| BECN | Encoding beclin |
| BRAF | B-Raf proto-oncogene |
| BRG-1 | Brahma-Related Gene 1 |
| bZIP | Basic Region/Leucine Zipper |
| CALCOCO2 | Calcium-Binding and Coiled-Coil Domain-Containing Protein 2 |
| CBP | cAMP -binding protein |
| CBRs | Carbonyl Reductases |
| CDDP | Cisplatin |
| CHD6 | Chromodomain Helicase Dna-Binding Protein 6 |
| CHOP | Homologous protein |
| cIPA | Cellular inhibitors of apoptosis |
| CML | Chronic Myeloid Leukemia |
| CNC | Cap ‘N’ Collar |
| CNC-bZIP | Collar Basic Region Leucine Zipper |
| COX-2 | Cyclooxygenase-2 |
| CP | Clobetasol Propionate |
| CPT | Camptothecin |
| CQ | Chloroquine |
| CREB | cAMP-response element binding protein |
| CSCs | Cancer Steam Cells |
| CTX | Cytotoxic Chemotherapy |
| CYPs | Cytochrome P450s |
| DAPK1 | Death-Associated Kinase 1 |
| DMSO | Dimethyl sulfoxide |
| EBP | Enhancer Binding Protein |
| EGCG | Epigallocatechin-3-gallate |
| eIF2a | Initiation Factor 2-Alpha |
| EMT | Epithelial-Mesenchymal Transition |
| ER | Endoplasmic Reticulum |
| ERAD | ER-associated degradation |
| ERRα | Estrogen-Related Receptor A |
| FOXO-1 | Anti-apoptotic forkhead box O-1 |
| G6PD | Glucose 6-phosphate dehydrogenase |
| GA | Golgi apparatus |
| GAA | Acid α-glucosidase |
| GABARAPL1Gamma-aminobutyric acid receptor-associated protein-like 1 | |
| GCL | Glutamate-Cysteine Ligase |
| GCLC | Glutamate-Cysteine Ligase Catalytic |
| GCLM | Glutamate-Cysteine Ligase Modulator |
| GR | Glutathione reductase |
| GRP78 | Glucose-Regulated Protein 78 |
| GSCs | Glioma Stem Cells |
| GSH | Glutathione |
| GSK3 | Glycogen synthase kinase-3 |
| GSS | Glutathione synthetize |
| GSSG | Oxidized glutathione |
| GST | Glutathione S-Transferase |
| HCC | Hepatocellular Carcinoma |
| HO | Heme Oxygenase |
| IL | Interleukin |
| iNOS | Induced Nitric Oxide Synthase |
| IR | Irradiation |
| IRE1 | Inositol-requiring enzyme1 |
| JNK | Jun NH2-terminal kinase |
| KEAP1Kelch-Like-Ech-Associated Protein 1 | |
| KIR | Keap1-Interacting Region |
| LC3B | light chain 3B |
| MAPK | Mitogen-activated protein kinase |
| MDR | Multidrug resistance-associated proteins |
| MMP-9 | Metalloproteinase-9 |
| MRP1 | Multidrug-resistance-associated protein-1 |
| MSCs | Mesenchymal Stem Cells |
| mTOR | Mammalian target of rapamycin |
| mTORC | Mammalian target of rapamycin complex |
| NADPH | Adenine Dinucleotide Phosphate |
| Neh | Nrf2-ECH homology |
| NFE2 | Nuclear factor erythroid-derived 2 |
| NF-E2 | Nuclear Factor-Erythroid 2 |
| NFE2L2 | NFE2 like BZIP Transcription Factor 2 |
| NQO1 | Nad(P)H:Quinine Oxidoreductase 1 |
| Nrf2 | Nuclear Related Factor 2 |
| OX | Electrophiles |
| PDI | Protein Disulfide Isomerase |
| PERK | Protein kinase RNA-like ER kinase |
| PGD | Phosphogluconate Dehydrogenase |
| PHGDH | Phosphoglycerate Dehydrogenase |
| PI3K | Phosphoinositide 3-Kinase |
| POMP | Proteasome Maturation Protein |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| PPP | Pentose Phosphate Pathway |
| PSAT1 | Phosphoserine Aminotransferase-1 |
| RAC3 | Receptor-Associated Co-Activator 3 |
| Rb | Retinoblastoma |
| RIDD | Regulated Ire1 Dependent Decay |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| ROS/RS | Reactive Oxygen and Nitrogen Products |
| RXRα | Retinoid X receptor alpha |
| SERCA | Calcium transport ATPase |
| SHMT2 | Serine Hydroxymethyltransferase-2 |
| shRNAs | Small hairpin RNA |
| Simva | Simvastatin |
| Sirt6 | Sirtuin 6 |
| SLC7A11 | Solute carrier 7A11 |
| sMAF | Small musculoaponeurotic fibrosarcoma |
| SMRT | Silencing mediator for retinoid and thyroid hormone receptor |
| SOD | Superoxide Dismutase |
| SP1 | Specificity Protein 1 |
| TFs | Transcription Factors |
| TGF-β | Transforming growth factor |
| TKT | Transketolase |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| TNBC | Triple-Negative Breast Cancer |
| TNF-α | Tumor Necrosis Factor |
| TRAF2 | Tumor Necrosis Factor Receptor-Associated Factor-2 |
| TRB3 | Tribbles-Related Protein3 |
| Trx | Thioredoxin |
| TSC2 | Tuberous Sclerosis Complex 2 |
| UGT | Udp-Glucuronosyltransferase |
| ULK | Unc-51-Like Kinase |
| UPR | Unfolded Protein Response |
| VEGF | Vascular Endothelial Growth Factor |
| XBP1 | X-Box-Binding Protein-1 |
| β-TrCP | β-transducin repeat-containing E3 ubiquitin protein ligase |
References
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death & Differentiation 2014, 21, 15–25. [Google Scholar]
- Bailly, C.; Thuru, X.; Quesnel, B.J.N.c. Combined cytotoxic chemotherapy and immunotherapy of cancer: modern times. NAR cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Malone, E.; Maltese, M.; Coady, L.; Hammond, L.; Silva, N.; Gullo, G.; Crown, J.J.A.o.O. Use and clinical impact of conventional cytotoxic chemotherapy (CTx) subsequent to immunotherapy in metastatic melanoma. Annals of Oncology 2016, 27, vi396. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: a brief review. Advanced pharmaceutical bulletin 2017, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Davodabadi, F.; Sajjadi, S.F.; Sarhadi, M.; Mirghasemi, S.; Hezaveh, M.N.; Khosravi, S.; Andani, M.K.; Cordani, M.; Basiri, M.; Ghavami, S. Cancer chemotherapy resistance: Mechanisms and recent breakthrough in targeted drug delivery. European Journal of Pharmacology 2023, 958, 176013. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008. [Google Scholar] [CrossRef]
- Lu, C.; Shervington, A. Chemoresistance in gliomas. Molecular and cellular biochemistry 2008, 312, 71–80. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.r.; Albokashy, M.; Butterfield, Y. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. The FEBS journal 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.M.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: a new approach to combat temozolomide chemoresistance in glioblastoma. Journal of Investigative Medicine 2018, 66, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive oxygen species: a key constituent in cancer survival. Biomarker insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Kim, E.-K.; Jang, M.; Song, M.-J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-mediated mechanism of chemoresistance in cancer cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef]
- Mishra, J.; Bhatti, G.K.; Sehrawat, A.; Singh, C.; Singh, A.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Modulating autophagy and mitophagy as a promising therapeutic approach in neurodegenerative disorders. Life Sciences 2022, 121153. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Choudhury, D.; Das, A.; Mukherjee, D.D.; Dasgupta, M.; Bandopadhyay, S.; Chakrabarti, G. Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human nonsmall lung adenocarcinoma A549 cells via ROS mediated modulation of β-catenin pathway. Apoptosis 2019, 24, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Kochan, M.M.; Stewart, V.D.; Drewnik, D.A.; Hannila, S.S.; Ghavami, S. Inhibition of Autophagy Flux Promotes Secretion of Chondroitin Sulfate Proteoglycans in Primary Rat Astrocytes. Mol Neurobiol 2021, 58, 6077–6091. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R. New frontiers in the treatment of colorectal cancer: autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Siri, M.; Behrouj, H.; Dastghaib, S.; Zamani, M.; Likus, W.; Rezaie, S.; Hudecki, J.; Khazayel, S.; Los, M.J.; Mokarram, P.; et al. Casein Kinase-1-Alpha Inhibitor (D4476) Sensitizes Microsatellite Instable Colorectal Cancer Cells to 5-Fluorouracil via Authophagy Flux Inhibition. Arch Immunol Ther Exp (Warsz) 2021, 69, 26. [Google Scholar] [CrossRef]
- Peixoto, P.; Grandvallet, C.; Feugeas, J.-P.; Guittaut, M.; Hervouet, E. Epigenetic control of autophagy in cancer cells: a key process for cancer-related phenotypes. Cells 2019, 8, 1656. [Google Scholar] [CrossRef]
- da Silva Rosa, S.C.; Martens, M.D.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; Hai, Y.; Chapman, D.; Diehl-Jones, W.; Aliani, M.; West, A.R.; et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021, 17, 2257–2272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karsli-Uzunbas, G.; Poillet-Perez, L.; Sawant, A.; Hu, Z.S.; Zhao, Y.; Moore, D.; Hu, W.; White, E. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes & development 2020, 34, 688–700. [Google Scholar]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life sciences 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin induces unfolded protein response and enhances temozolomide-induced cell death in glioblastoma cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jäger, R.; Gorman, A.M.; Samali, A. The unfolded protein response in breast cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biology of the Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. International journal of molecular sciences 2019, 20, 4354. [Google Scholar] [CrossRef]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Letters 2020, 490, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Vrishni, S.; Singh, B.K.; Rahman, I.; Kakkar, P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free radical research 2010, 44, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Eguchi, S.; Alam, A.; Ma, D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017, 312, L155–L162. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radical Biology and Medicine 2013, 57, 119–131. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Yang, M.; Wang, K.; Liu, Y.; Zhang, M.; Yang, Y.; Jin, C.; Wang, R.; Hu, R. Digoxin sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine via inhibiting Nrf2 signaling pathway. Redox biology 2019, 22, 101131. [Google Scholar] [CrossRef]
- Tazehkand, A.P.; Akbarzadeh, M.; Velaie, K.; Sadeghi, M.R.; Samadi, N. The role of Her2-Nrf2 axis in induction of oxaliplatin resistance in colon cancer cells. Biomedicine & Pharmacotherapy 2018, 103, 755–766. [Google Scholar]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Dodson, M.; Chapman, E.; Zhang, D.D. NRF2-targeted therapeutics: New targets and modes of NRF2 regulation. Current opinion in toxicology 2016, 1, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacological research 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.V.; Hushmandi, K.; Ashrafizadeh, M. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, S.; Ehmer, U.; Freiberg, N.; Manns, M.P.; Strassburg, C.P. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. Journal of Biological Chemistry 2010, 285, 5993–6002. [Google Scholar] [CrossRef]
- Shelton, P.; Jaiswal, A.K. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? The FASEB Journal 2013, 27, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-Y.; Song, M.-Y.; Kim, E.-H. Role of Oxidative Stress and Nrf2/KEAP1 Signaling in Colorectal Cancer: Mechanisms and Therapeutic Perspectives with Phytochemicals. Antioxidants 2021, 10, 743. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. International journal of molecular sciences 2020, 21, 4777. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radical Biology and Medicine 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer research 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Marchev, A.S.; Dimitrova, P.A.; Burns, A.J.; Kostov, R.V.; Dinkova-Kostova, A.T.; Georgiev, M.I.J.A.o.t.N.Y.A.o.S. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Annals of the New York Academy of Sciences 2017, 1401, 114–135. [Google Scholar] [CrossRef]
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. Journal of Biological Chemistry 2019, 294, 18131–18149. [Google Scholar] [CrossRef]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 pathway in age-related neurological disorders: insights into MicroRNAs. Cellular Physiology and Biochemistry 2018, 47, 1951–1976. [Google Scholar] [CrossRef]
- Wang, R.; Liang, L.; Matsumoto, M.; Iwata, K.; Umemura, A.; He, F. Reactive Oxygen Species and NRF2 Signaling, Friends or Foes in Cancer? Biomolecules 2023, 13, 353. [Google Scholar] [CrossRef]
- Bauer, A.K.; Cho, H.-Y.; Miller-DeGraff, L.; Walker, C.; Helms, K.; Fostel, J.; Yamamoto, M.; Kleeberger, S.R. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PloS one 2011, 6, e26590. [Google Scholar] [CrossRef]
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.-W.; Gustafson, D.L.; Thorburn, A. Cancer cells upregulate NRF2 signaling to adapt to autophagy inhibition. Developmental cell 2019, 50, 690–703. e696. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; O'Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap'n'collar transcription factors in aging and human disease. Science signaling 2010, 3, re3–re3. [Google Scholar] [CrossRef] [PubMed]
- Luk, A.D.W.; Yang, X.; Alcasabas, A.P.; Hao, R.C.; Chan, K.W.; Lee, P.P.; Yang, J.; Chan, G.C.F.; So, J.C.C.; Yang, W.J.B.j.o.h. NF-E2 mutation as a novel cause for inherited thrombocytopenia. British Journal of Haematology 2020, 189, e41. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Molecular cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear factor erythroid-2 like 1 (NFE2L1): structure, function and regulation. Gene 2016, 584, 17–25. [Google Scholar] [CrossRef]
- Ibrahim, L.; Mesgarzadeh, J.; Xu, I.; Powers, E.T.; Wiseman, R.L.; Bollong, M.J. Defining the Functional Targets of Cap ‘n’collar Transcription Factors NRF1, NRF2, and NRF3. Antioxidants 2020, 9, 1025. [Google Scholar] [CrossRef]
- Bhawe, K.; Roy, D. Interplay between NRF1, E2F4 and MYC transcription factors regulating common target genes contributes to cancer development and progression. Cellular Oncology 2018, 41, 465–484. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, T.; Zhou, W.; Zhang, Y.; Xu, G.; Xu, Q.; Li, S.; Gao, Y.; Wang, Z.; Xu, J. Long noncoding RNA LINC01132 enhances immunosuppression and therapy resistance via NRF1/DPP4 axis in hepatocellular carcinoma. Journal of Experimental & Clinical Cancer Research 2022, 41, 270. [Google Scholar]
- Sekine, H.; Motohashi, H. Roles of CNC Transcription Factors NRF1 and NRF2 in Cancer. Cancers 2021, 13, 541. [Google Scholar] [CrossRef]
- Chowdhury, A.M.A.; Katoh, H.; Hatanaka, A.; Iwanari, H.; Nakamura, N.; Hamakubo, T.; Natsume, T.; Waku, T.; Kobayashi, A. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Scientific reports 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Immonen, A.; Haapasaari, K.-M.; Skarp, S.; Karihtala, P.; Teppo, H.-R. NRF3 Decreases during Melanoma Carcinogenesis and Is an Independent Prognostic Marker in Melanoma. Oxidative medicine and cellular longevity 2022, 2022. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Z.; Chen, Q.; Pan, Y.; Lu, H.; Zhang, H.; Yu, Y.; Dai, Y. NRF3 suppresses breast cancer cell metastasis and cell proliferation and is a favorable predictor of survival in breast cancer. OncoTargets and therapy 2019, 12, 3019. [Google Scholar] [CrossRef]
- Kobayashi, A.; Waku, T. New addiction to the NRF2-related factor NRF3 in cancer cells: ubiquitin-independent proteolysis through the 20S proteasome. Cancer Science 2020, 111, 6–14. [Google Scholar] [CrossRef]
- Kobayashi, A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers 2020, 12, 2681. [Google Scholar] [CrossRef]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 activation in cancer: from DNA to protein. Cancer research 2019, 79, 889–898. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxidants & redox signaling 2018, 29, 1727–1745. [Google Scholar]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): history, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef]
- Inouye, S.; Kubo, T.; Miyamoto, T.; Iyoda, T.; Okita, N.; Akagi, R. Heat shock-induced heme oxygenase-1 expression in a mouse hepatoma cell line is dependent on HSF1 and modified by NRF2 and BACH1. Genes to Cells 2022, 27, 719–730. [Google Scholar] [CrossRef]
- Mafra, D.; Alvarenga, L.; Cardozo, L.F.; Stockler-Pinto, M.B.; Nakao, L.S.; Stenvinkel, P.; Shiels, P.G. Inhibiting BTB domain and CNC homolog 1 (Bach1) as an alternative to increase Nrf2 activation in chronic diseases. Biochimica et Biophysica Acta (BBA)-General Subjects 2022, 1866, 130129. [Google Scholar] [CrossRef]
- Warnatz, H.-J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. Journal of Biological Chemistry 2011, 286, 23521–23532. [Google Scholar] [CrossRef]
- Paramasivan, P.; Kankia, I.H.; Langdon, S.P.; Deeni, Y.Y. Emerging role of nuclear factor erythroid 2-related factor 2 in the mechanism of action and resistance to anticancer therapies. Cancer Drug Resistance 2019, 2, 490–515. [Google Scholar] [CrossRef]
- Sarcinelli, C.; Dragic, H.; Piecyk, M.; Barbet, V.; Duret, C.; Barthelaix, A.; Ferraro-Peyret, C.; Fauvre, J.; Renno, T.; Chaveroux, C. ATF4-dependent NRF2 transcriptional regulation promotes antioxidant protection during endoplasmic reticulum stress. Cancers 2020, 12, 569. [Google Scholar] [CrossRef]
- Chung, S.; Kim, S.; Son, M.; Kim, M.; Koh, E.S.; Shin, S.J.; Park, C.W.; Kim, H.-S. Inhibition of p300/CBP-associated factor attenuates renal tubulointerstitial fibrosis through modulation of NF-kB and Nrf2. International journal of molecular sciences 2019, 20, 1554. [Google Scholar] [CrossRef]
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Rao, J.; Qian, X.; Li, G.; Pan, X.; Zhang, C.; Zhang, F.; Zhai, Y.; Wang, X.; Lu, L. ATF3-Mediated NRF2/HO-1 Signaling regulates TLR4 innate immune responses in mouse liver ischemia/reperfusion injury. American Journal of Transplantation 2015, 15, 76–87. [Google Scholar] [CrossRef]
- Kratschmar, D.V.; Calabrese, D.; Walsh, J.; Lister, A.; Birk, J.; Appenzeller-Herzog, C.; Moulin, P.; Goldring, C.E.; Odermatt, A. Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11β-HSD1-mediated glucocorticoid activation in hepatic cells. PloS one 2012, 7, e36774. [Google Scholar] [CrossRef]
- Namani, A.; Li, Y.; Wang, X.J.; Tang, X. Modulation of NRF2 signaling pathway by nuclear receptors: implications for cancer. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2014, 1843, 1875–1885. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiological Genomics 2018, 50, 77–97. [Google Scholar] [CrossRef]
- Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. Nrf2 and the ambiguous consequences of its activation during initiation and the subsequent stages of tumourigenesis. Cancers 2020, 12, 3609. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded Nrf2 activation on phase-I and-II drug metabolizing enzymes and transporters in mouse liver. PloS one 2012, 7, e39006. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. Journal of Biological Chemistry 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Shen, G.; Kong, A.N. Nrf2 plays an important role in coordinated regulation of Phase II drug metabolism enzymes and Phase III drug transporters. Biopharmaceutics & drug disposition 2009, 30, 345–355. [Google Scholar]
- Wang, X.J.; Li, Y.; Luo, L.; Wang, H.; Chi, Z.; Xin, A.; Li, X.; Wu, J.; Tang, X. Oxaliplatin activates the Keap1/Nrf2 antioxidant system conferring protection against the cytotoxicity of anticancer drugs. Free Radical Biology and Medicine 2014, 70, 68–77. [Google Scholar] [CrossRef]
- Fu, Z.D.; Selwyn, F.P.; Cui, J.Y.; Klaassen, C.D. RNA sequencing quantification of xenobiotic-processing genes in various sections of the intestine in comparison to the liver of male mice. Drug Metabolism and Disposition 2016, 44, 842–856. [Google Scholar] [CrossRef]
- Kohalmy, K.; Vrzal, R. Regulation of phase II biotransformation enzymes by steroid hormones. Current Drug Metabolism 2011, 12, 104–123. [Google Scholar] [CrossRef]
- Jaramillo, A.C.; Saig, F.A.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resistance 2018, 1, 6–29. [Google Scholar] [CrossRef]
- Mani, M.; Khaghani, S.; Mohammadi, T.G.; Zamani, Z.; Azadmanesh, K.; Meshkani, R.; Pasalar, P.; Mostafavi, E. Activation of Nrf2-antioxidant response element mediated glutamate cysteine ligase expression in hepatoma cell line by homocysteine. Hepatitis monthly 2013, 13. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Meierjohann, S. NRF2-dependent stress defense in tumor antioxidant control and immune evasion. Pigment Cell & Melanoma Research 2021, 34, 268–279. [Google Scholar]
- Dovinova, I.; Kvandova, M.; Balis, P.; Gresova, L.; Majzunova, M.; Horakova, L.; Chan, J.Y.; Barancik, M. The Role of Nrf2 and PPARγ in the Improvement of Oxidative Stress in Hypertension and Cardiovascular Diseases. Physiological Research 2020, 69. [Google Scholar] [CrossRef]
- Giudice, A.; Montella, M. Activation of the Nrf2–ARE signaling pathway: a promising strategy in cancer prevention. Bioessays 2006, 28, 169–181. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacological reports 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Kamiya, T.; Courtney, M.; Laukkanen, M.O. redox-activated signal transduction pathways mediating cellular functions in inflammation, Differentiation, Degeneration, Transformation, and Death. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, J.; Cao, M.; Zhao, Z.; Cao, B.; Yu, S. The potential roles of Nrf2/Keap1 signaling in anticancer drug interactions. Current Research in Pharmacology and Drug Discovery 2021, 2, 100028. [Google Scholar] [CrossRef]
- Gallorini, M.; Carradori, S.; Panieri, E.; Sova, M.; Saso, L. Modulation of NRF2: biological dualism in cancer, targets and possible therapeutic applications. Antioxidants and Redox Signaling 2023. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-J.; Cheng, X.-D.; Zhang, J.; Zhang, W.-D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: a systematic review. Cell Communication and Signaling 2019, 17, 1–15. [Google Scholar] [CrossRef]
- Telkoparan-Akillilar, P.; Panieri, E.; Cevik, D.; Suzen, S.; Saso, L. Therapeutic targeting of the NRF2 signaling pathway in cancer. Molecules 2021, 26, 1417. [Google Scholar] [CrossRef]
- Hu, T.; Pan, C.; Zhang, T.; Ni, M.; Wang, W.; Zhang, S.; Chen, Y.; Wang, J.; Fang, Q. Nrf2 overexpression increases the resistance of acute myeloid leukemia to cytarabine by inhibiting replication factor C4. Cancer Gene Therapy 2022, 29, 1773–1790. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Wakabayashi, N.; Kensler, T.W. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism. Biochemical Society Transactions 2015, 43, 639–644. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Kleeberger, S.; Bream, J.; Fallon, P.; Kensler, T.; Yamamoto, M.; Reddy, S. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Chi, Z.; Zhou, X.; Ren, G.; Zhou, R.; Li, Y.; Tang, X.; Wang, X.J. Interplay of MKP-1 and Nrf2 drives tumor growth and drug resistance in non-small cell lung cancer. Aging (Albany NY) 2019, 11, 11329. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. Journal of Biological Chemistry 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Yun, S.-M.; Song, M.-Y.; Jung, K.; Kim, E.-H. Cyanidin chloride induces apoptosis by inhibiting NF-κB signaling through activation of Nrf2 in colorectal cancer cells. Antioxidants 2020, 9, 285. [Google Scholar] [CrossRef]
- Mas, G.; Man, N.; Nakata, Y.; Martinez-Caja, C.; Karl, D.; Beckedorff, F.; Tamiro, F.; Chen, C.; Duffort, S.; Itonaga, H. The SWI/SNF chromatin-remodeling subunit DPF2 facilitates NRF2-dependent antiinflammatory and antioxidant gene expression. The Journal of clinical investigation 2023, 133. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radical Biology and Medicine 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Belgorosky, D.; Girouard, J.; Langle, Y.V.; Hamelin-Morrissete, J.; Marino, L.; Agüero, E.I.; Malagrino, H.; Reyes-Moreno, C.; Eiján, A.M. Relevance of iNOS expression in tumor growth and maintenance of cancer stem cells in a bladder cancer model. Journal of Molecular Medicine 2020, 98, 1615–1627. [Google Scholar] [CrossRef]
- Umesalma, S.; Sudhandiran, G. Differential inhibitory effects of the polyphenol ellagic acid on inflammatory mediators NF-κB, iNOS, COX-2, TNF-α, and IL-6 in 1, 2-dimethylhydrazine-induced rat colon carcinogenesis. Basic & clinical pharmacology & toxicology 2010, 107, 650–655. [Google Scholar]
- Paul, S.; Modak, D.; Chattaraj, S.; Nandi, D.; Sarkar, A.; Roy, J.; Chaudhuri, T.K.; Bhattacharjee, S. Aloe vera gel homogenate shows anti-inflammatory activity through lysosomal membrane stabilization and downregulation of TNF-α and Cox-2 gene expressions in inflammatory arthritic animals. Future Journal of Pharmaceutical Sciences 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Z.; Tang, M.; Xing, C.; Chen, H.; Zheng, K.; Zhao, Z.; Zhou, S.; Zhao, A.Z.; Li, F. Endogenous production of ω-3 polyunsaturated fatty acids mitigates cisplatin-induced myelosuppression by regulating NRF2-MDM2-p53 signaling pathway. Free Radical Biology and Medicine 2023, 201, 14–25. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Nam, C.-w.; Wakabayashi, N.; Yamamoto, M.; Kensler, T.W.; Kwak, M.-K. Transcription factor Nrf2 maintains the basal expression of Mdm2: an implication of the regulation of p53 signaling by Nrf2. Archives of biochemistry and biophysics 2011, 507, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Wu, J.; Dodson, M.; Rojo de la Vega, E.M.; Ning, Y.; Zhang, Z.; Yao, M.; Zhang, D.D.; Xu, C.; Yi, X. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Molecular carcinogenesis 2017, 56, 1543–1553. [Google Scholar] [CrossRef]
- Orozco-Morales, M.; Hernández-Pedro, N.Y.; Barrios-Bernal, P.; Arrieta, O.; Ruiz-Godoy, L.M.; Aschner, M.; Santamaría, A.; Colín-González, A.L.J.A.-C.D. S-allylcysteine induces cytotoxic effects in two human lung cancer cell lines via induction of oxidative damage, downregulation of Nrf2 and NF-κB, and apoptosis. Anti-Cancer Drugs 2021, 32, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Murakami, K.; Nawamaki, M.; Kashiwakura, I. Effects of Nrf2 knockdown on the properties of irradiated cell conditioned medium from A549 human lung cancer cells. Biomedical reports 2018, 8, 461–465. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 2019, 178, 330–345. e322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, H.-J.; Bao, Q.-C.; Wang, L.; Guo, T.-K.; Chen, W.-L.; Xu, L.-L.; Zhou, H.-S.; Bian, J.-L.; Yang, Y.-R. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Yang, F.; Thorne, R.F.; Zhu, B.K.; Hersey, P.; Zhang, X.D. Human melanoma cells under endoplasmic reticulum stress acquire resistance to microtubule-targeting drugs through XBP-1-mediated activation of Akt. Neoplasia 2009, 11, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Zhai, W.L.; Yang, H.Y.; Jin, H.; Zhang, Q.X. Induction of ER stress protects gastric cancer cells against apoptosis induced by cisplatin and doxorubicin through activation of p38 MAPK. Biochemical and biophysical research communications 2011, 406, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 2017, 8, 51164. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Molecular cancer 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. Journal of cellular and molecular medicine 2009, 13, 3888–3897. [Google Scholar] [CrossRef]
- Kim, J.K.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Han, X.; Fernando, P.M.D.J.; Oh, M.C.; Park, J.E.; Shilnikova, K.; Boo, S.J. Endoplasmic reticulum stress induces 5-fluorouracil resistance in human colon cancer cells. Environmental toxicology and pharmacology 2016, 44, 128–133. [Google Scholar] [CrossRef]
- Nahand, J.S.; Rabiei, N.; Fathazam, R.; Taghizadieh, M.; Ebrahimi, M.S.; Mahjoubin-Tehran, M.; Baghi, H.B.; Khatami, A.; Abbasi-Kolli, M.; Mirzaei, H.R. Oncogenic viruses and chemoresistance: What do we know? Pharmacological Research 2021, 105730. [Google Scholar] [CrossRef]
- Tao, W.; Wang, N.; Ruan, J.; Cheng, X.; Fan, L.; Zhang, P.; Lu, C.; Hu, Y.; Che, C.; Sun, D. Enhanced ROS-boosted phototherapy against pancreatic cancer via Nrf2-mediated stress-defense pathway suppression and ferroptosis induction. ACS Applied Materials & Interfaces 2022, 14, 6404–6416. [Google Scholar]
- Fu, D.; Wang, C.; Yu, L.; Yu, R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cellular & molecular biology letters 2021, 26, 26. [Google Scholar]
- Moon, E.J.; Giaccia, A. Dual roles of NRF2 in tumor prevention and progression: possible implications in cancer treatment. Free Radical Biology and Medicine 2015, 79, 292–299. [Google Scholar] [CrossRef]
- Almasi, S.; El Hiani, Y.J.C. Exploring the therapeutic potential of membrane transport proteins: Focus on cancer and chemoresistance. Cancers 2020, 12, 1624. [Google Scholar] [CrossRef]
- Cole, S.; Bhardwaj, G.; Gerlach, J.; Mackie, J.; Grant, C.; Almquist, K.; Stewart, A.; Kurz, E.; Duncan, A.; Deeley, R.G.J.S. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science signaling 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Deeley, R.G.; Cole, S.P.J.F.l. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS letters 2006, 580, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Fatemian, T.; Hoque Chowdhury, E. Targeting oncogenes and tumor suppressors genes to mitigate chemoresistance. Current cancer drug targets 2014, 14, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer research 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-S.; Du, J.; Fan, Y.-J.; Liu, F.-J.; Cao, L.-L.; Liang, N.; Xu, D.-G.; Zhang, J.-D. Activation of endoplasmic reticulum stress promotes autophagy and apoptosis and reverses chemoresistance of human small cell lung cancer cells by inhibiting the PI3K/AKT/mTOR signaling pathway. Oncotarget 2016, 7, 76827. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guan, Z.; Liang, L.; Cheng, Y.; Zhou, J.; Li, J.; Xu, Y. NF-κB signaling plays irreplaceable roles in cisplatin-induced bladder cancer chemoresistance and tumor progression. International journal of oncology 2016, 48, 225–234. [Google Scholar] [CrossRef]
- Dong, Q.; Fu, L.; Zhao, Y.; Tan, S.; Wang, E. Derlin-1 overexpression confers poor prognosis in muscle invasive bladder cancer and contributes to chemoresistance and invasion through PI3K/AKT and ERK/MMP signaling. Oncotarget 2017, 8, 17059. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Yin, Y.; Kelly, R.E.; Lee, E.; Bradley, A.; Li, W.; Bertino, J.R.; Wahl, G.M. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proceedings of the National Academy of Sciences 1995, 92, 5436–5440. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Booth, D.; Naderi, S.; Sever-Chroneos, Z.; Fribourg, A.F.; Hunton, I.C.; Feramisco, J.R.; Wang, J.Y.; Knudsen, E.S. RB-dependent S-phase response to DNA damage. Molecular and cellular biology 2000, 20, 7751–7763. [Google Scholar] [CrossRef]
- Sharma, A.; Comstock, C.E.; Knudsen, E.S.; Cao, K.H.; Hess-Wilson, J.K.; Morey, L.M.; Barrera, J.; Knudsen, K.E. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer research 2007, 67, 6192–6203. [Google Scholar] [CrossRef] [PubMed]
- Bosco, E.E.; Wang, Y.; Xu, H.; Zilfou, J.T.; Knudsen, K.E.; Aronow, B.J.; Lowe, S.W.; Knudsen, E.S. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. The Journal of clinical investigation 2007, 117, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Donati, G.; Mazzini, G.; Montanaro, L.; Vici, M.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Treré, D. Loss of retinoblastoma tumor suppressor protein makes human breast cancer cells more sensitive to antimetabolite exposure. Clinical cancer research 2008, 14, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.R.; Dean, J.L.; Seeley, S.L.; Mayhew, C.N.; Knudsen, E.S. RB status governs differential sensitivity to cytotoxic and molecularly-targeted therapeutic agents. Cell cycle 2008, 7, 1095–1103. [Google Scholar] [CrossRef]
- Chow, L.; Wang, X.; Kwong, D.; Sham, J.; Tsao, S.; Nicholls, J.M. Effect of p16INK4a on chemosensitivity in nasopharyngeal carcinoma cells. International journal of oncology 2000, 17, 135–175. [Google Scholar] [CrossRef]
- Barboule, N.; Chadebech, P.; Baldin, V.; Vidal, S.; Valette, A. Involvement of p21 in mitotic exit after paclitaxel treatment in MCF-7 breast adenocarcinoma cell line. Oncogene 1997, 15, 2867–2875. [Google Scholar] [CrossRef]
- Yu, D.; Jing, T.; Liu, B.; Yao, J.; Tan, M.; McDonnell, T.J.; Hung, M.-C. Overexpression of ErbB2 blocks Taxol-induced apoptosis by upregulation of p21Cip1, which inhibits p34Cdc2 kinase. Molecular cell 1998, 2, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.J.; Han, J.; Kim, S.J.; Lee, M.J.; Ju, X.; Lee, Y.L.; Son, J.H.; Cui, J.; Jang, Y.; Chung, W.J.O.R. PTEN/AKT signaling mediates chemoresistance in refractory acute myeloid leukemia through enhanced glycolysis. Oncology Reports 2019, 42, 2149–2158. [Google Scholar] [CrossRef]
- Singh, M.; Chaudhry, P.; Fabi, F.; Asselin, E.J.B.c. Cisplatin-induced caspase activation mediates PTEN cleavage in ovarian cancer cells: a potential mechanism of chemoresistance. BMC cancer 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nature Reviews Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Dapic, V.; Monteiro, A.N. Functional implications of BRCA1 for early detection, prevention, and treatment of breast cancer. Critical Reviews™ in Eukaryotic Gene Expression 2006, 16. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic acids research 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Yu, H.; Deng, C.-X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proceedings of the National Academy of Sciences 2004, 101, 17108–17113. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.; Qian, X.; Ding, Y.; Yu, L.; Liu, B. Gambogic acid, a potent inhibitor of survivin, reverses docetaxel resistance in gastric cancer cells. Cancer letters 2008, 262, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jones, A.; Fassone, E.; Sweeney, M.; Lebiedzinska, M.; Suski, J.; Wieckowski, M.; Tajeddine, N.; Hargreaves, I.; Yasukawa, T. PGC-1β mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 2013, 32, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liang, W.; Liu, J.; Zhang, L.; Wei, J.; Yang, J.; Zhang, Y.; Huang, Z. Autophagy-mediating microRNAs in cancer chemoresistance. Cell biology and toxicology 2020, 1–20. [Google Scholar] [CrossRef]
- Takenaka, T.; Yoshino, I.; Kouso, H.; Ohba, T.; Yohena, T.; Osoegawa, A.; Shoji, F.; Maehara, Y. Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non-small cell lung cancer. International journal of cancer 2007, 121, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Shirabe, K.; Morita, K.; Umeda, K.; Kayashima, H.; Uchiyama, H.; Soejima, Y.; Taketomi, A.; Maehara, Y. Evaluation of ERCC1 expression for cisplatin sensitivity in human hepatocellular carcinoma. Annals of surgical oncology 2011, 18, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Hajiahmadi, S.; Seyfoori, A.; Amereh, M.; Zamani, M.; Shahsavari, Z.; Shojaei, S.; Akbari, M.; Mokarram, P.; Ghavami, S. Role of apoptosis, autophagy, and the unfolded protein response in glioblastoma chemoresistance. In Glioblastoma Resistance to Chemotherapy: Molecular Mechanisms and Innovative Reversal Strategies; Elsevier: 2021; pp. 201-242.
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. International journal of medical sciences 2012, 9, 881. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; del Río Hernández, A. Chemoresistance and the self-maintaining tumor microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950. [Google Scholar] [CrossRef] [PubMed]
- Kahroba, H.; Shirmohamadi, M.; Hejazi, M.S.; Samadi, N. The Role of Nrf2 signaling in cancer stem cells: From stemness and self-renewal to tumorigenesis and chemoresistance. Life sciences 2019, 239, 116986. [Google Scholar] [CrossRef]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews Molecular cell biology 2012, 13, 89–102. [Google Scholar] [CrossRef]
- McMellen, A.; Yamamoto, T.M.; Qamar, L.; Sanders, B.E.; Nguyen, L.L.; Ortiz Chavez, D.; Bapat, J.; Berning, A.; Post, M.D.; Johnson, J. ATF6-mediated signaling contributes to PARP Inhibitor Resistance in Ovarian Cancer. Molecular Cancer Research 2023, 21, 3–13. [Google Scholar] [CrossRef]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Scientific reports 2019, 9, 3210. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, H.; Yang, Y.; Zhao, D.; Wen, Y.; Lv, C.; Qiu, H.; Wang, C. The regulation of miR-320a/XBP1 axis through LINC00963 for endoplasmic reticulum stress and autophagy in diffuse large B-cell lymphoma. Cancer cell international 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Molecular cancer therapeutics 2016, 15, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Molecular oncology 2018, 12, 1498–1512. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Zuliani, I.; Tramutola, A.; Barone, E.; Blarzino, C.; Folgiero, V.; Caforio, M.; Valentini, D.; Villani, A.; Locatelli, F. Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention. Progress in Neurobiology 2021, 196, 101892. [Google Scholar] [CrossRef]
- Wei, R.; Zhao, Y.; Wang, J.; Yang, X.; Li, S.; Wang, Y.; Yang, X.; Fei, J.; Hao, X.; Zhao, Y. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. International journal of biological sciences 2021, 17, 2703. [Google Scholar] [CrossRef] [PubMed]
- Küper, A.; Baumann, J.; Göpelt, K.; Baumann, M.; Sänger, C.; Metzen, E.; Kranz, P.; Brockmeier, U. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death & Disease 2021, 12, 82. [Google Scholar]
- Lanzillotta, C.; Zuliani, I.; Tramutola, A.; Abisambra, J.F.; Barone, E.; Perluigi, M.; Domenico, F.D. PERK inhibition promotes the rescue of protein translation and Nrf2-related antioxidant response: Molecular and cell biology/oxidative stress. Alzheimer's & Dementia 2020, 16, e041867. [Google Scholar]
- Ivanova, I.G.; Park, C.V.; Yemm, A.I.; Kenneth, N.S. PERK/eIF2α signaling inhibits HIF-induced gene expression during the unfolded protein response via YB1-dependent regulation of HIF1α translation. Nucleic acids research 2018, 46, 3878–3890. [Google Scholar] [CrossRef]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: dangerous liaisons. Journal of Experimental & Clinical Cancer Research 2021, 40, 28. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxidative medicine and cellular longevity 2016, 2016, 3164734–3164734. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: the bright side of the moon. Experimental & Molecular Medicine 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cellular and Molecular Life Sciences 2013, 70, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: further developments. Journal of hematology & oncology 2020, 13, 1–18. [Google Scholar]
- Eskelinen, E.-L. The dual role of autophagy in cancer. Current opinion in pharmacology 2011, 11, 294–300. [Google Scholar] [CrossRef]
- Al Dhaheri, Y.; Attoub, S.; Ramadan, G.; Arafat, K.; Bajbouj, K.; Karuvantevida, N.; AbuQamar, S.; Eid, A.; Iratni, R. Carnosol induces ROS-mediated beclin1-independent autophagy and apoptosis in triple negative breast cancer. PloS one 2014, 9, e109630. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.-L. RITA plus 3-MA overcomes chemoresistance of head and neck cancer cells via dual inhibition of autophagy and antioxidant systems. Redox Biology 2017, 13, 219–227. [Google Scholar] [CrossRef]
- Du, F.; Feng, Y.; Fang, J.; Yang, M. MicroRNA-143 enhances chemosensitivity of Quercetin through autophagy inhibition via target GABARAPL1 in gastric cancer cells. Biomedicine & Pharmacotherapy 2015, 74, 169–177. [Google Scholar]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biology 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Frontiers in oncology 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Li, J.; Chen, J.; Hu, Q.; Gu, C.; Lin, W.; Chen, G. Endoplasmic reticulum stress is associated with neuroprotection against apoptosis via autophagy activation in a rat model of subarachnoid hemorrhage. Neuroscience letters 2014, 563, 160–165. [Google Scholar] [CrossRef]
- Shimodaira, Y.; Takahashi, S.; Kinouchi, Y.; Endo, K.; Shiga, H.; Kakuta, Y.; Kuroha, M.; Shimosegawa, T. Modulation of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1α (IRE1α) in human colon cancer cells. Biochemical and biophysical research communications 2014, 445, 524–533. [Google Scholar] [CrossRef]
- Rubiolo, J.; López-Alonso, H.; Martínez, P.; Millán, A.; Cagide, E.; Vieytes, M.; Vega, F.; Botana, L. Yessotoxin induces ER-stress followed by autophagic cell death in glioma cells mediated by mTOR and BNIP3. Cellular signalling 2014, 26, 419–432. [Google Scholar] [CrossRef]
- Grandjean, J.M.; Madhavan, A.; Cech, L.; Seguinot, B.O.; Paxman, R.J.; Smith, E.; Scampavia, L.; Powers, E.T.; Cooley, C.B.; Plate, L. Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nature chemical biology 2020, 16, 1052–1061. [Google Scholar] [CrossRef]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiological reviews 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, J.; Ma, Y.; Lu, L.; Ma, C.; Qin, P.; Gao, E.; Zuo, M.; Yang, J.; Yang, L. Electroacupuncture Pretreatment Mitigates Myocardial Ischemia/Reperfusion Injury via XBP1/GRP78/Akt Pathway. Frontiers in Cardiovascular Medicine 2021, 8. [Google Scholar] [CrossRef]
- Xin, W.; Zhang, M.; Yu, Y.; Li, S.; Ma, C.; Zhang, J.; Jiang, Y.; Li, Y.; Zheng, X.; Zhang, L. BCAT1 binds the RNA-binding protein ZNF423 to activate autophagy via the IRE1-XBP-1-RIDD axis in hypoxic PASMCs. Cell death & disease 2020, 11, 1–16. [Google Scholar]
- Zhang, L.; Zhang, C.; Wang, A. Divergence and conservation of the major UPR branch IRE1-bZIP signaling pathway across eukaryotes. Scientific reports 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. The FASEB Journal 2007, 21, 4013–4027. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell death & disease 2019, 10, 1–15. [Google Scholar]
- Ogata, M.; Hino, S.-i.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K. Autophagy is activated for cell survival after endoplasmic ReticulumStress. Molecular and cellular biology 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.-G.; Jiang, Z.-X.; Li, J.-H.; Zhou, Z.; Zhang, Q.-H. Spliced XBP1 promotes macrophage survival and autophagy by interacting with Beclin-1. Biochemical and biophysical research communications 2015, 463, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. Journal of Biological Chemistry 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. XBP1: a pivotal transcriptional regulator of glucose and lipid metabolism. Trends in Endocrinology & Metabolism 2016, 27, 119–122. [Google Scholar]
- Suzuki, H.; Kanekura, K.; Levine, T.P.; Kohno, K.; Olkkonen, V.M.; Aiso, S.; Matsuoka, M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. Journal of neurochemistry 2009, 108, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. International journal of molecular sciences 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Human molecular genetics 2012, 21, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lei, H.; Zhang, B.-G.; Xu, Z.-C.; Dong, C.; Hao, Y.-Q. c-Jun NH2-terminal kinase (JNK)/stress-activated protein kinase-associated protein 1 is a critical regulator for arthritis progression by meditating inflammation in mice model. International immunopharmacology 2020, 81, 106272. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, D.; Li, Y.; Qiao, H.; Shan, Z. Ketamine enhances autophagy and endoplasmic reticulum stress in rats and SV-HUC-1 cells via activating IRE1-TRAF2-ASK1-JNK pathway. Cell Cycle 2021, 1–16. [Google Scholar] [CrossRef]
- Wang, L.; Wang, P.; Dong, H.; Wang, S.; Chu, H.; Yan, W.; Zhang, X. Ulk1/FUNDC1 prevents nerve cells from hypoxia-induced apoptosis by promoting cell autophagy. Neurochemical research 2018, 43, 1539–1548. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Li, H.; Wei, C.; Mao, A.; Liu, W.; Pan, G. Polyphyllin D induces apoptosis and protective autophagy in breast cancer cells through JNK1-Bcl-2 pathway. Journal of Ethnopharmacology 2021, 114591. [Google Scholar] [CrossRef]
- Thorpe, J.A.; Schwarze, S.R. IRE1α controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress and Chaperones 2010, 15, 497–508. [Google Scholar] [CrossRef]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omedè, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14. [Google Scholar] [CrossRef]
- Yang, W.; Xu, X.; Xu, M.; Zhou, J.; Xi, Z.; Guo, H.; Ming, J.; Huang, T. XBP1s Acts as a Tumor Suppressor to Inhibit the EMT Process and Metastasis of Papillary Thyroid Cancer. OncoTargets and therapy 2021, 14, 2339. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.-T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood, The Journal of the American Society of Hematology 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nature communications 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-A.; Fang, S.-U.; Su, C.-L.; Hsiao, C.-J.; Chang, C.-C.; Lin, Y.-F.; Cheng, C.-W. Silencing glucose-regulated protein 78 induced renal cell carcinoma cell line G1 cell-cycle arrest and resistance to conventional chemotherapy. In Proceedings of the Urologic Oncology: Seminars and Original Investigations, 2014; pp. 29. e21–29. e11. [Google Scholar]
- Raiter, A.; Lipovetsky, J.; Hyman, L.; Mugami, S.; Ben-Zur, T.; Yerushalmi, R. Chemotherapy Controls Metastasis Through Stimulatory Effects on GRP78 and Its Transcription Factor CREB3L1. Frontiers in oncology 2020, 10, 1500. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Gopal, U.; Austin, R.C.; Pizzo, S.V. Glucose-regulated protein (GRP78) is an important cell surface receptor for viral invasion, cancers and neurological disorders. IUBMB life 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-y.; Yu, J.-y.; Leng, Y.-l.; Zhu, R.-r.; Liu, H.-x.; Wang, X.-y.; Yang, T.-t.; Guo, Y.-n.; Tang, J.-l.; Zhang, X.-c. MiR-181c sensitizes ovarian cancer cells to paclitaxel by targeting GRP78 through the PI3K/Akt pathway. Cancer Gene Therapy 2021, 1–14. [Google Scholar] [CrossRef]
- Suyama, K.; Watanabe, M.; Sakabe, K.; Okada, Y.; Matsuyama, D.; Kuroiwa, M.; Mochida, J. Overexpression of GRP78 protects glial cells from endoplasmic reticulum stress. Neuroscience letters 2011, 504, 271–276. [Google Scholar] [CrossRef]
- Elfiky, A.A.; Baghdady, A.M.; Ali, S.A.; Ahmed, M.I. GRP78 targeting: Hitting two birds with a stone. Life sciences 2020, 118317. [Google Scholar] [CrossRef]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncology letters 2015, 10, 1959–1969. [Google Scholar] [CrossRef]
- Wang, Y.-c.; Li, X.; Shen, Y.; Lyu, J.; Sheng, H.; Paschen, W.; Yang, W. PERK (protein kinase RNA-like ER kinase) branch of the unfolded protein response confers neuroprotection in ischemic stroke by suppressing protein synthesis. Stroke 2020, 51, 1570–1577. [Google Scholar] [CrossRef]
- Teske, B.F.; Baird, T.D.; Wek, R.C. Methods for analyzing eIF2 kinases and translational control in the unfolded protein response. Methods in enzymology 2011, 490, 333–356. [Google Scholar]
- B'chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.-C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cellular signalling 2014, 26, 1385–1391. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Cheon, C.; Ko, S.-G. SH003 activates autophagic cell death by activating ATF4 and inhibiting G9a under hypoxia in gastric cancer cells. Cell death & disease 2020, 11, 1–14. [Google Scholar]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; Mcafee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.J.T.J.o.c.i. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. The Journal of clinical investigation 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Sun, Z.; Zhang, Q. Intermittent-hypoxia-induced autophagy activation through the ER-stress-related PERK/eIF2α/ATF4 pathway is a protective response to pancreatic β-cell apoptosis. Cellular Physiology and Biochemistry 2018, 51, 2955–2971. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacology & therapeutics 2012, 134, 306–316. [Google Scholar]
- Wu, M.-Z.; Fu, T.; Chen, J.-X.; Lin, Y.-Y.; Yang, J.-E.; Zhuang, S.-M. LncRNA GOLGA2P10 is induced by PERK/ATF4/CHOP signaling and protects tumor cells from ER stress-induced apoptosis by regulating Bcl-2 family members. Cell death & disease 2020, 11, 1–12. [Google Scholar]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. The EMBO journal 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B.J.M.; biology, c. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Molecular and cellular biology 2012, 32, 2–11. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic acids research 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Chung, H.; Kim, Y.R.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. Journal of Experimental & Clinical Cancer Research 2020, 39, 1–14. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. Journal of Biological Chemistry 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Krishnamoorthy, J.; Rajesh, K.; Mirzajani, F.; Kesoglidou, P.; Papadakis, A.; Koromilas, A.E. Evidence for eIF2α phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications. Cell Cycle 2014, 13, 801–806. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Ji, X.; Liebhaber, S.A.; Diehl, J.A. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2009, 28, 910–920. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Cmoch, A.; Wolczyk, M.; Bugajski, L.; Tkaczyk, M.; Dadlez, M.; Nieborowska-Skorska, M.; Koromilas, A.E.; Skorski, T.; Piwocka, K. Increased phosphorylation of eIF2α in chronic myeloid leukemia cells stimulates secretion of matrix modifying enzymes. Oncotarget 2016, 7, 79706. [Google Scholar] [CrossRef]
- Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Peidis, P.; Glodkowska-Mrowka, E.; Wolanin, K.; Leszak, G.; Seferynska, I.; Stoklosa, T.; Koromilas, A.E.; Piwocka, K. The PERK-eIF2α phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 2012, 11, 4069–4078. [Google Scholar] [CrossRef]
- Fujimoto, A.; Kawana, K.; Taguchi, A.; Adachi, K.; Sato, M.; Nakamura, H.; Ogishima, J.; Yoshida, M.; Inoue, T.; Nishida, H. Inhibition of endoplasmic reticulum (ER) stress sensors sensitizes cancer stem-like cells to ER stress-mediated apoptosis. Oncotarget 2016, 7, 51854. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Hong, M.-Y.; Pei, J.; Gao, Y.-H.; Zheng, Y.; Xu, X. ER stress mediated-autophagy contributes to neurological dysfunction in traumatic brain injury via the ATF6 UPR signaling pathway. Molecular Medicine Reports 2021, 23, 1–1. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. Journal of cell science 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Gade, P.; Ramachandran, G.; Maachani, U.B.; Rizzo, M.A.; Okada, T.; Prywes, R.; Cross, A.S.; Mori, K.; Kalvakolanu, D.V. An IFN-γ–stimulated ATF6–C/EBP-β–signaling pathway critical for the expression of Death Associated Protein Kinase 1 and induction of autophagy. Proceedings of the National Academy of Sciences 2012, 109, 10316–10321. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, S.; Dai, C.; Tang, S.; Yang, X.; Li, D.; Zhao, K.; Xiao, X. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell biology and toxicology 2016, 32, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J.J.O.L. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncology Letters 2015, 10, 1959–1969. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.-M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Molecular and cellular biology 2014, 34, 1839–1849. [Google Scholar] [CrossRef]
- Tay, K.H.; Luan, Q.; Croft, A.; Jiang, C.C.; Jin, L.; Zhang, X.D.; Tseng, H.-Y. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cellular signalling 2014, 26, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Min, H.-Y.; Pei, H.; Wei, X.; Sim, J.Y.; Park, S.-H.; Hwang, S.J.; Lee, H.-J.; Hong, S.; Shin, Y.K. The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis. Cancers 2020, 12, 1772. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proceedings of the National Academy of Sciences 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.; Lee, A. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death & Differentiation 2008, 15, 1460–1471. [Google Scholar]
- Rashid, M.-u.; Lorzadeh, S.; Gao, A.; Ghavami, S.; Coombs, K.M. PSMA2 knockdown impacts expression of proteins involved in immune and cellular stress responses in human lung cells. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2023, 1869, 166617. [Google Scholar] [CrossRef]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer research 2012, 72, 3337–3349. [Google Scholar] [CrossRef]
- Shimada, Y.; Kobayashi, H.; Kawagoe, S.; Aoki, K.; Kaneshiro, E.; Shimizu, H.; Eto, Y.; Ida, H.; Ohashi, T. Endoplasmic reticulum stress induces autophagy through activation of p38 MAPK in fibroblasts from Pompe disease patients carrying c. 546G> T mutation. Molecular genetics and metabolism 2011, 104, 566–573. [Google Scholar] [CrossRef]
- Verchot, J. The ER quality control and ER associated degradation machineries are vital for viral pathogenesis. Frontiers in plant science 2014, 5, 66. [Google Scholar] [CrossRef]
- Lemberg, M.K.; Strisovsky, K. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Molecular Cell 2021. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature Reviews Nephrology 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. ER-Phagy, ER homeostasis, and ER quality control: implications for disease. Trends in Biochemical Sciences 2021. [Google Scholar] [CrossRef]
- Peng, Y.; Guo, L.; Gu, A.; Shi, B.; Ren, Y.; Cong, J.; Yang, X. Electroacupuncture alleviates polycystic ovary syndrome-like symptoms through improving insulin resistance, mitochondrial dysfunction, and endoplasmic reticulum stress via enhancing autophagy in rats. Molecular Medicine 2020, 26, 1–13. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox biology 2017, 11, 543–553. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; De La Vega, M.R.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radical Biology and Medicine 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Molecular and cellular biology 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and cellular biology 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS genetics 2013, 9, e1003701. [Google Scholar] [CrossRef]
- Zhu, Y.-p.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of opposites between two antioxidant transcription factors Nrf1 and Nrf2 in mediating distinct cellular responses to the endoplasmic reticulum stressor tunicamycin. Antioxidants 2020, 9, 4. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E. Erratum: NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nature genetics 2016, 48, 473. [Google Scholar] [CrossRef]
- Li, B.; Fu, J.; Chen, P.; Ge, X.; Li, Y.; Kuiatse, I.; Wang, H.; Wang, H.; Zhang, X.; Orlowski, R.Z. The nuclear factor (erythroid-derived 2)-like 2 and proteasome maturation protein axis mediate bortezomib resistance in multiple myeloma. Journal of Biological Chemistry 2015, 290, 29854–29868. [Google Scholar] [CrossRef]
- Srivastava, R.; Fernández-Ginés, R.; Encinar, J.A.; Cuadrado, A.; Wells, G. The current status and future prospects for therapeutic targeting of KEAP1-NRF2 and β-TrCP-NRF2 interactions in cancer chemoresistance. Free Radical Biology and Medicine 2022. [Google Scholar] [CrossRef]
- Hajiahmadi, S.; Lorzadeh, S.; Iranpour, R.; Karima, S.; Rajabibazl, M.; Shahsavari, Z.; Ghavami, S. Temozolomide, simvastatin and acetylshikonin combination induces mitochondrial-dependent apoptosis in GBM cells, which is regulated by autophagy. Biology 2023, 12, 302. [Google Scholar] [CrossRef]
- Alizadeh, J.; da Silva Rosa, S.C.; Weng, X.; Jacobs, J.; Lorzadeh, S.; Ravandi, A.; Vitorino, R.; Pecic, S.; Zivkovic, A.; Stark, H. Ceramides and Ceramide Synthases in Cancer: Focus on Apoptosis and Autophagy. European Journal of Cell Biology 2023, 151337. [Google Scholar] [CrossRef]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the crosstalk between NRF2 signaling and metabolic processes in cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Cordani, M.; Donadelli, M.; Ghavami, S. Metastatic outgrowth via the two-way interplay of autophagy and metabolism. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2023, 166824. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature cell biology 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes & development 2019, 33, 150–165. [Google Scholar]
- Barzegar Behrooz, A.; Latifi-Navid, H.; da Silva Rosa, S.C.; Swiat, M.; Wiechec, E.; Vitorino, C.; Vitorino, R.; Jamalpoor, Z.; Ghavami, S. Integrating Multi-Omics Analysis for Enhanced Diagnosis and Treatment of Glioblastoma: A Comprehensive Data-Driven Approach. Cancers 2023, 15, 3158. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Kavoosi, M.; Singh, N.; Lorzadeh, S.; Ravandi, A.; Kidane, B.; Ahmed, N.; Mraiche, F.; Mowat, M.R.; Ghavami, S. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers 2023, 15, 2195. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Lu, W.; Paul, M.K.; Jha, N.K.; Gupta, P.K.; Ojha, S.; Chattopadhyay, I.; Rao, P.V.; Ghavami, S. Autophagy and EMT in cancer and metastasis: Who controls whom? Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2022, 1868, 166431. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. Journal of Cell Biology 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer cell 2017, 32, 824–839. e828. [Google Scholar] [CrossRef]
- Kageyama, S.; Sou, Y.-s.; Uemura, T.; Kametaka, S.; Saito, T.; Ishimura, R.; Kouno, T.; Bedford, L.; Mayer, R.J.; Lee, M.-S. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. Journal of Biological Chemistry 2014, 289, 24944–24955. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic reticulum stress and cancer. Journal of cancer prevention 2014, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-N.; Kavianpour, S.; Zhang, T.; Zhang, X.; Nguyen, D.; Thombre, R.; He, L.; Wang, J. MARK2 phosphorylates eIF2α in response to proteotoxic stress. PLoS biology 2021, 19, e3001096. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nature medicine 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends in biochemical sciences 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.-P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nature communications 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes & development 2011, 25, 795–800. [Google Scholar]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Molecular cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell'Antonio, G. Pancreatic cancers require autophagy for tumor growth. Genes & development 2011, 25, 717–729. [Google Scholar]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer discovery 2014, 4, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, K.; Wang, H.; Dai, Y. Inhibition of autophagy by chloroquine enhances the antitumor efficacy of sorafenib in glioblastoma. Cellular and molecular neurobiology 2016, 36, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Natural product reports 2020, 37, 797–826. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochemical pharmacology 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Ramezani, F.; Keshavarzi, F.; Samadi, N. The role of quercetin and vitamin C in Nrf2-dependent oxidative stress production in breast cancer cells. Oncology letters 2017, 13, 1965–1973. [Google Scholar] [CrossRef]
- Ramezani, F.; Samadi, N.; Mostafavi-Pour, Z. Sequential therapy of breast cancer cell lines with vitamin C and quercetin improves the efficacy of chemotherapeutic drugs. Nutrition and cancer 2017, 69, 881–891. [Google Scholar] [CrossRef]
- Lin, H.; Qiao, Y.; Yang, H.; Nan, Q.; Qu, W.; Feng, F.; Liu, W.; Chen, Y.; Sun, H. Small molecular Nrf2 inhibitors as chemosensitizers for cancer therapy. Future medicinal chemistry.
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Chian, S.; Thapa, R.; Chi, Z.; Wang, X.J.; Tang, X. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochemical and Biophysical Research Communications 2014, 447, 602–608. [Google Scholar] [CrossRef]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Sun, K.; Wang, X.; Pan, H.; Zhu, J.; Ji, X.; Li, X. Chrysin suppresses proliferation, migration, and invasion in glioblastoma cell lines via mediating the ERK/Nrf2 signaling pathway. Drug design, development and therapy 2018, 12, 721. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, X.; Zhang, Y.; Yang, L.; Liu, Y.; Huang, S.; Lu, L.; Kong, L.; Li, Z.; Guo, Q. Wogonin reversed resistant human myelogenous leukemia cells via inhibiting Nrf2 signaling by Stat3/NF-κB inactivation. Scientific reports 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol Cancer Ther 2020, 19, 221–230. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, Z.; Guo, R.; Ren, F. Nrf2 inhibitor, brusatol in combination with trastuzumab exerts synergistic antitumor activity in HER2-positive cancers by inhibiting Nrf2/HO-1 and HER2-AKT/ERK1/2 pathways. Oxidative medicine and cellular longevity 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Chen, F.; Lv, H.; Xu, Z.; Fu, J.; Hou, Y.; Xu, Y.; Pi, J. Triptolide enhances chemotherapeutic efficacy of antitumor drugs in non-small-cell lung cancer cells by inhibiting Nrf2-ARE activity. Toxicology and Applied Pharmacology 2018, 358, 1–9. [Google Scholar] [CrossRef]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Lee, S.; Lim, M.J.; Kim, M.H.; Yu, C.H.; Yun, Y.S.; Ahn, J.; Song, J.Y. An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic Biol Med 2012, 53, 807–816. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem Biol 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Bollong, M.J.; Yun, H.; Sherwood, L.; Woods, A.K.; Lairson, L.L.; Schultz, P.G. A small molecule inhibits deregulated NRF2 transcriptional activity in cancer. ACS chemical biology 2015, 10, 2193–2198. [Google Scholar] [CrossRef]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radical Biology and Medicine 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Jung, B.; Lee, S.; Yoo, H.; Shin, E.; Ko, H.; Chang, S.; Kim, S.; Jeon, S. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef] [PubMed]
- Hori, R.; Yamaguchi, K.; Sato, H.; Watanabe, M.; Tsutsumi, K.; Iwamoto, S.; Abe, M.; Onodera, H.; Nakamura, S.; Nakai, R. The discovery and characterization of K-563, a novel inhibitor of the Keap1/Nrf2 pathway produced by Streptomyces sp. Cancer medicine 2019, 8, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, H.; Zhu, J.; Zhao, R.; Xue, P.; Zhang, Q.; Bud Nelson, M.; Qu, W.; Feng, B.; Pi, J. Camptothecin suppresses NRF2–ARE activity and sensitises hepatocellular carcinoma cells to anticancer drugs. British journal of cancer 2017, 117, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Jiao, Y.; Xue, J.; Zhang, Q.; Yang, H.; Xing, L.; Chen, G.; Wu, J.; Zhang, S.; Zhu, W. Metformin sensitizes non-small cell lung cancer cells to an epigallocatechin-3-gallate (EGCG) treatment by suppressing the Nrf2/HO-1 signaling pathway. International journal of biological sciences 2017, 13, 1560. [Google Scholar] [CrossRef]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 2012, 8, 311–317. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who's listening? Antioxid Redox Signal 2010, 13, 1649–1663. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol 2005, 25, 4150–4165. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Su, L.; Ye, Q.; Zhang, S.; Kung, H.; Jiang, F.; Jiang, G.; Miao, J.; Zhao, B. Discovery of a novel Nrf2 inhibitor that induces apoptosis of human acute myeloid leukemia cells. Oncotarget 2017, 8, 7625–7636. [Google Scholar] [CrossRef]
- Li, D.; Wang, H.; Ding, Y.; Zhang, Z.; Zheng, Z.; Dong, J.; Kim, H.; Meng, X.; Zhou, Q.; Zhou, J. Targeting the NRF-2/RHOA/ROCK signaling pathway with a novel aziridonin, YD0514, to suppress breast cancer progression and lung metastasis. Cancer letters 2018, 424, 97–108. [Google Scholar] [CrossRef]
- Hamzawy, M.; Abo-Youssef, A.; Malak, M.; Khalaf, M. Multiple targets of Nrf 2 inhibitor; trigonelline in combating urethane-induced lung cancer by caspase-executioner apoptosis, cGMP and limitation of cyclin D1 and Bcl2. European Review for Medical & Pharmacological Sciences 2022, 26. [Google Scholar]
- Rehman, M.U.; Rashid, S.; Arafah, A.; Qamar, W.; Alsaffar, R.M.; Ahmad, A.; Almatroudi, N.M.; Alqahtani, S.M.; Rashid, S.M.; Ahmad, S.B. Piperine regulates Nrf-2/Keap-1 signalling and exhibits anticancer effect in experimental colon carcinogenesis in Wistar rats. Biology 2020, 9, 302. [Google Scholar] [CrossRef]
- Khurana, N.; Sikka, S.C. Targeting crosstalk between Nrf-2, NF-κB and androgen receptor signaling in prostate cancer. Cancers 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Meng, W.; Liu, Y.; Li, C.; Zhang, C.; Wang, P.; Almoallim, H.S.; Manikandan, V.; Guan, H. Elucidating the immunomodulatory effect of daidzein in Benzo (a) pyrene-Induced lung cancer mice model through modulation of proliferating cell nuclear antigen, NF-κB, CYP1A1, and NRF. Pharmacognosy Magazine 2022, 18, 193. [Google Scholar]
- Chen, P.; Fang, X.; Mao, H.; Lin, Y.; Li, B. Curcumin combination with miR-144-3p suppresses non-small cell lung cancer by regulating Nrf-2/HO-1 in vitro and vivo study. Journal of Biomaterials and Tissue Engineering 2019, 9, 476–484. [Google Scholar] [CrossRef]
- Yehia, R.; Saleh, S.; El Abhar, H.; Saad, A.S.; Schaalan, M. L-Carnosine protects against Oxaliplatin-induced peripheral neuropathy in colorectal cancer patients: A perspective on targeting Nrf-2 and NF-κB pathways. Toxicology and applied pharmacology 2019, 365, 41–50. [Google Scholar] [CrossRef]
Figure 1.
NRF-affected pathways. A perspective of the pathways affecting chemical resistance.
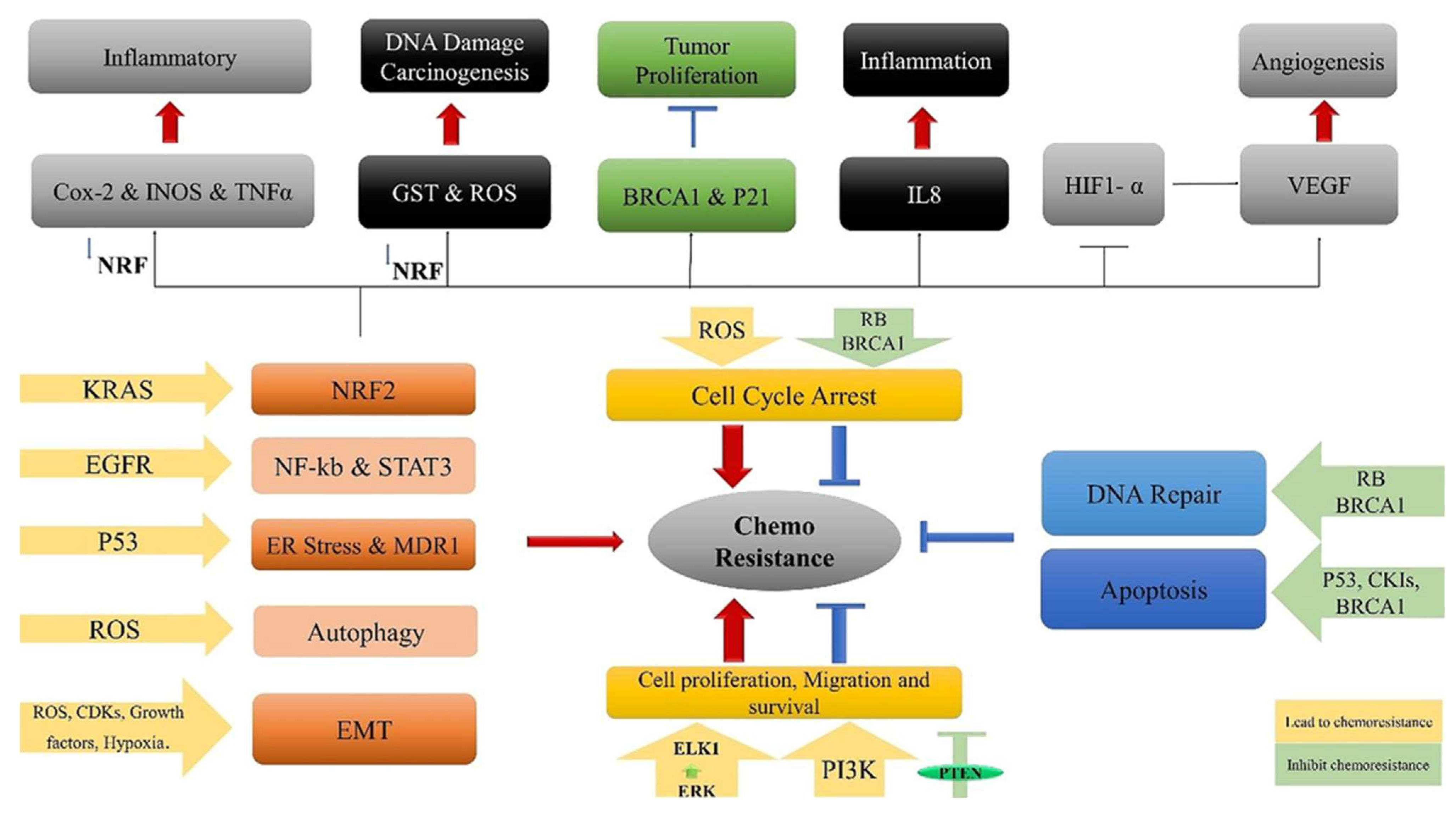
Figure 2.
Schematic view of oxidative stress, UPR sensors, autophagy, and drug resistance. In oxidative stress, the UPR sensors cooperate in chemo-resistance induction. PERK/ eIF2α/ATF4 axis leads to activation of the anti-apoptotic factor Fork-head box O-1 (FOXO-1) and transcription factor NRF2, which regulates the expression of ABC transporters. Increased autophagy flux and the inhibition of apoptosis pathway makes cells resistant to chemotherapy agents.
Figure 2.
Schematic view of oxidative stress, UPR sensors, autophagy, and drug resistance. In oxidative stress, the UPR sensors cooperate in chemo-resistance induction. PERK/ eIF2α/ATF4 axis leads to activation of the anti-apoptotic factor Fork-head box O-1 (FOXO-1) and transcription factor NRF2, which regulates the expression of ABC transporters. Increased autophagy flux and the inhibition of apoptosis pathway makes cells resistant to chemotherapy agents.
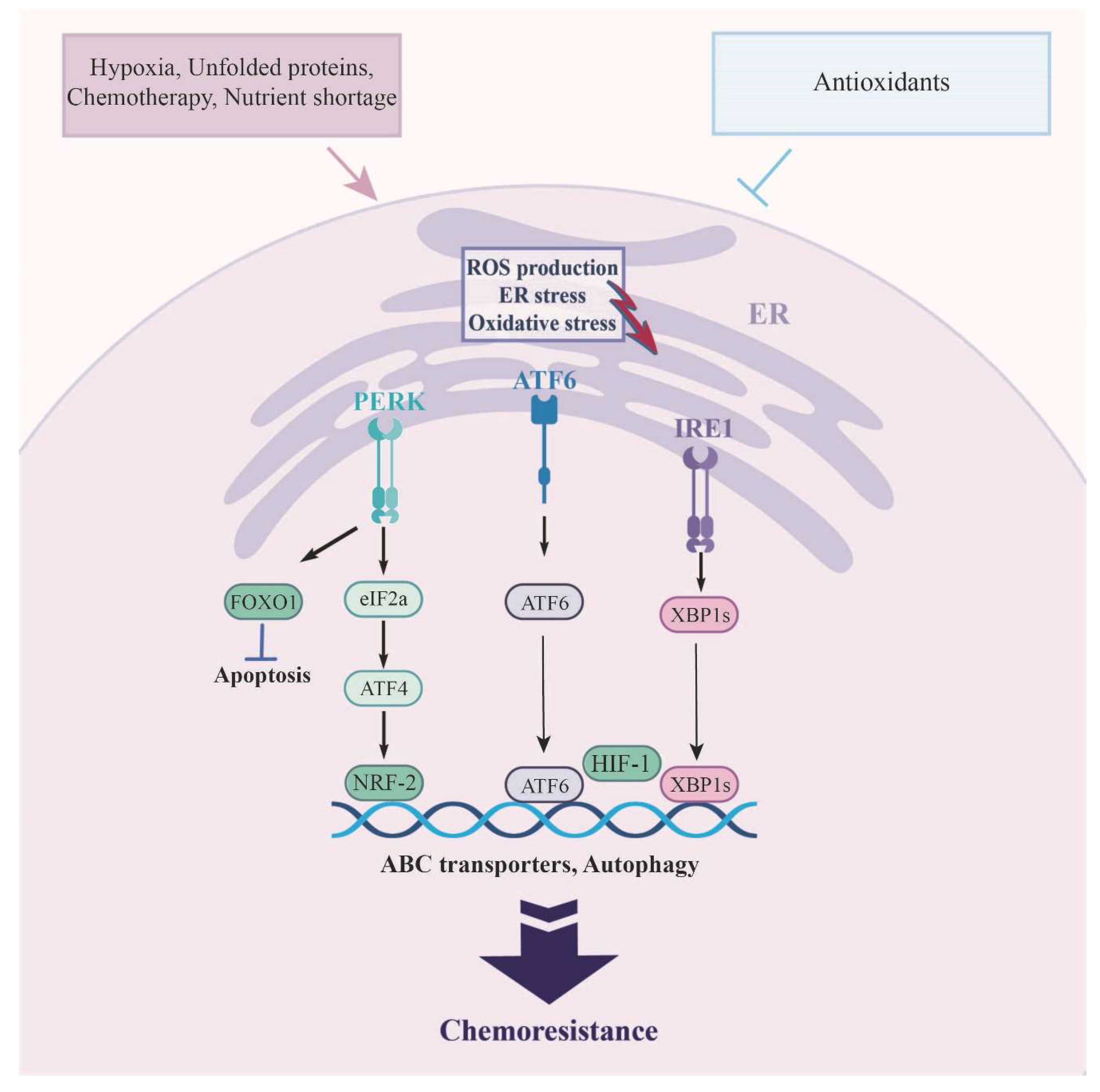
Figure 3.
Schematic views of crosstalk between the UPR, autophagy, tumor survival, and drug resistance. The presence of unfolded proteins inside the ER lumen, accompanied by the ER stress, promotes the dissociation of GRP78 from IRE1 and PERK, leading to dimerization and auto-phosphorylation of these two proteins and subsequent activation of XBP1 and eIF2α, respectively. The activated XBP1 and eIF2α (i.e., XBP1s and phosphorylated eIF2α) then trigger their downstream targets to modulate autophagic flux, as well as tumor survival/drug resistance. On the other hand, ATF6, the UPR’s third arm, activates ATF6-N to target autophagy and tumor survival/drug resistance. GRP78, by itself, regulates particular pathways in order to stimulate and/or suppress tumor survival and chemoresistance. Follow the arrows for more detailed information. AKT, Protein kinase B; ASK1, Apoptosis signal-regulating kinase 1; ATF6, Activating transcription factor 6; ATG, Autophagy-related protein; BCL2, B-cell lymphoma apoptosis regulator 2; BIK, BCL-2 interacting killer; Ca, Calcium; CDK, Cyclin-dependent kinase; CHOP, C/EBP homologous protein; DDIT3, DNA damage inducible transcript 3; eIF2α, Eukaryotic initiation factor-2α; ER, Endoplasmic reticulum; FIP200, Focal adhesion kinase family-interacting protein of 200 kDa; FOXO1, Forkhead box O1 ; GA, Golgi apparatus; Gln, Glutamine; GRP78, Glucose-regulated protein of 78 kDa; HIF1α, Hypoxia inducible factor 1α; HSPA5, Heat shock protein family A member 5; IRE1, Inositol-requiring enzyme 1; LC3, Light chain 3; mTOR, Mammalian target of rapamycin; NRF2, Nuclear factor-erythroid factor 2-related factor 2; p, Phosphate; PDIA5, Protein disulfide isomerase family A member 5; PERK, Protein kinase RNA-like endoplasmic reticulum kinase; PI3K, Phosphoinositide 3-kinase; pJNK1, Phosphorylated c-Jun N-terminal kinase 1; RheB, Ras homolog enriched in brain; ROS, Reactive oxygen species; TGF-β, Transforming growth factor-βeta; TRAF, Tumor necrosis factor receptor-associated factor; TRIB3, Tribbles pseudokinase 3; TSC1/2, Tuberous sclerosis complex 1/2; ULK1/2, Unc-51 like autophagy activating kinase 1/2; XBP1, X-box-binding protein 1; XBP1s, spliced XBP1.
Figure 3.
Schematic views of crosstalk between the UPR, autophagy, tumor survival, and drug resistance. The presence of unfolded proteins inside the ER lumen, accompanied by the ER stress, promotes the dissociation of GRP78 from IRE1 and PERK, leading to dimerization and auto-phosphorylation of these two proteins and subsequent activation of XBP1 and eIF2α, respectively. The activated XBP1 and eIF2α (i.e., XBP1s and phosphorylated eIF2α) then trigger their downstream targets to modulate autophagic flux, as well as tumor survival/drug resistance. On the other hand, ATF6, the UPR’s third arm, activates ATF6-N to target autophagy and tumor survival/drug resistance. GRP78, by itself, regulates particular pathways in order to stimulate and/or suppress tumor survival and chemoresistance. Follow the arrows for more detailed information. AKT, Protein kinase B; ASK1, Apoptosis signal-regulating kinase 1; ATF6, Activating transcription factor 6; ATG, Autophagy-related protein; BCL2, B-cell lymphoma apoptosis regulator 2; BIK, BCL-2 interacting killer; Ca, Calcium; CDK, Cyclin-dependent kinase; CHOP, C/EBP homologous protein; DDIT3, DNA damage inducible transcript 3; eIF2α, Eukaryotic initiation factor-2α; ER, Endoplasmic reticulum; FIP200, Focal adhesion kinase family-interacting protein of 200 kDa; FOXO1, Forkhead box O1 ; GA, Golgi apparatus; Gln, Glutamine; GRP78, Glucose-regulated protein of 78 kDa; HIF1α, Hypoxia inducible factor 1α; HSPA5, Heat shock protein family A member 5; IRE1, Inositol-requiring enzyme 1; LC3, Light chain 3; mTOR, Mammalian target of rapamycin; NRF2, Nuclear factor-erythroid factor 2-related factor 2; p, Phosphate; PDIA5, Protein disulfide isomerase family A member 5; PERK, Protein kinase RNA-like endoplasmic reticulum kinase; PI3K, Phosphoinositide 3-kinase; pJNK1, Phosphorylated c-Jun N-terminal kinase 1; RheB, Ras homolog enriched in brain; ROS, Reactive oxygen species; TGF-β, Transforming growth factor-βeta; TRAF, Tumor necrosis factor receptor-associated factor; TRIB3, Tribbles pseudokinase 3; TSC1/2, Tuberous sclerosis complex 1/2; ULK1/2, Unc-51 like autophagy activating kinase 1/2; XBP1, X-box-binding protein 1; XBP1s, spliced XBP1.
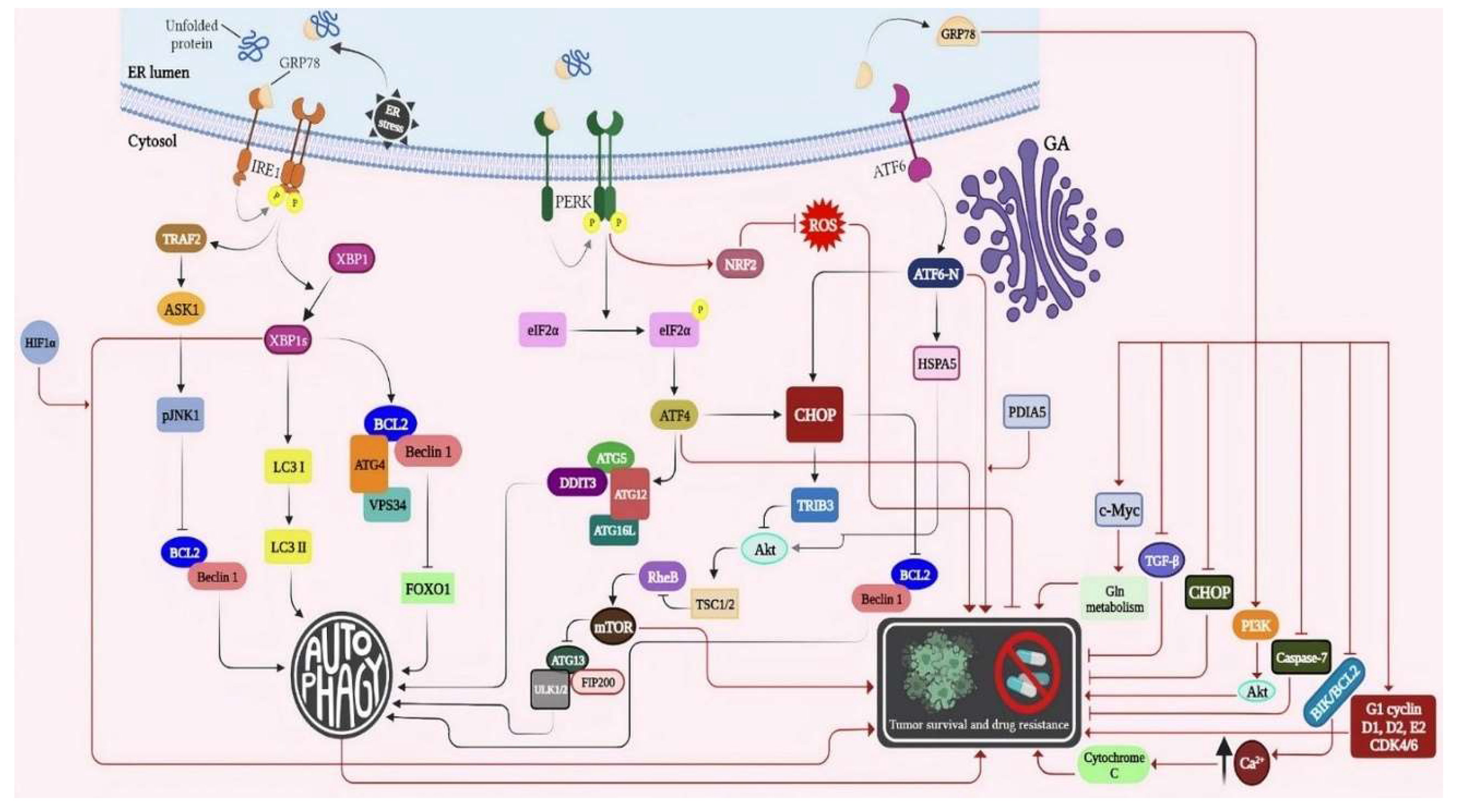
Figure 4.
The KEAP1–NRF2 pathway. (A)Canonical KEAP1-NRF2 Pathway:(1) The KEAP1 homodimer functions as an adaptor protein for the Cul3-based Rbx1 ubiquitin ligase complex. It recognizes NRF2 by binding to its ETGE motif and DLGex motif, crucial for proper ubiquitination by Rbx1. Continuous ubiquitination tags NRF2 for degradation by the proteasome. (2) Oxidative stress or electrophilic exposure causes specific cysteine residues on KEAP1 to be modified by reactive oxygen species and electrophiles (OX). This results in NRF2 dissociating from KEAP1. (3) Released NRF2 translocates to the nucleus, collaborating with transcription factors sMAFs, to activate target genes. (B) P62-Mediated KEAP1-NRF2 Pathway: (1) Similar to the canonical pathway, the KEAP1-Cul3-Rbx1 complex interacts with and ubiquitinates NRF2, leading to its proteasomal degradation. P62, too, is targeted for autophagic degradation through its ubiquitination by the ubiquitin ligase. (2) Selective autophagy triggers P62-mediated pathway in response to factors like defective proteostasis, mitochondrial dysfunction, and invasive microbes. Phosphorylation of P62's Ser349 by mTORC1, CK1 (CSNK1A1), TAK1, and PKCδ hampers KEAP1-DLGex motif interaction, preventing new NRF2 from binding to KEAP1. (3) Stabilized NRF2 translocates to the nucleus, inducing the transcription of its target genes, including P62. (4) P62's ubiquitination on lysine 420 by KEAP1-Cul3-Rbx1 complex augments its autophagic degradation, whether bound to KEAP1 or not. ARE, antioxidant response element.
Figure 4.
The KEAP1–NRF2 pathway. (A)Canonical KEAP1-NRF2 Pathway:(1) The KEAP1 homodimer functions as an adaptor protein for the Cul3-based Rbx1 ubiquitin ligase complex. It recognizes NRF2 by binding to its ETGE motif and DLGex motif, crucial for proper ubiquitination by Rbx1. Continuous ubiquitination tags NRF2 for degradation by the proteasome. (2) Oxidative stress or electrophilic exposure causes specific cysteine residues on KEAP1 to be modified by reactive oxygen species and electrophiles (OX). This results in NRF2 dissociating from KEAP1. (3) Released NRF2 translocates to the nucleus, collaborating with transcription factors sMAFs, to activate target genes. (B) P62-Mediated KEAP1-NRF2 Pathway: (1) Similar to the canonical pathway, the KEAP1-Cul3-Rbx1 complex interacts with and ubiquitinates NRF2, leading to its proteasomal degradation. P62, too, is targeted for autophagic degradation through its ubiquitination by the ubiquitin ligase. (2) Selective autophagy triggers P62-mediated pathway in response to factors like defective proteostasis, mitochondrial dysfunction, and invasive microbes. Phosphorylation of P62's Ser349 by mTORC1, CK1 (CSNK1A1), TAK1, and PKCδ hampers KEAP1-DLGex motif interaction, preventing new NRF2 from binding to KEAP1. (3) Stabilized NRF2 translocates to the nucleus, inducing the transcription of its target genes, including P62. (4) P62's ubiquitination on lysine 420 by KEAP1-Cul3-Rbx1 complex augments its autophagic degradation, whether bound to KEAP1 or not. ARE, antioxidant response element.
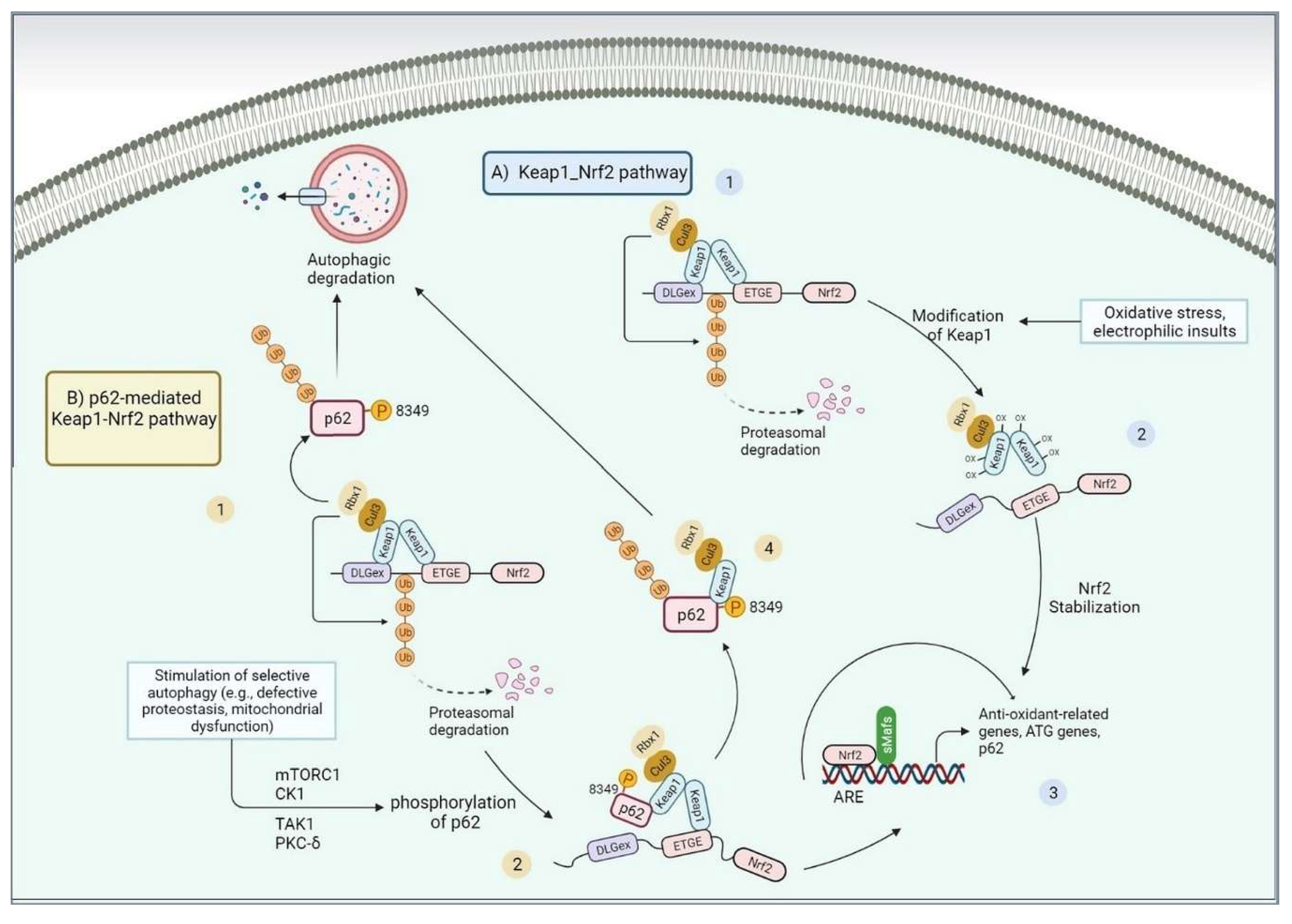
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



