Submitted:
01 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
The field of nephrology has recently directed a considerable amount of attention towards the stimulator of interferon genes (STING) molecule since it appears to be a potent driver of chronic kidney disease (CKD). STING and its activator, the cyclic GMP-AMP synthase (cGAS) are among the most relevant inducers that promote the expression of type I interferons (IFN-Is). These cytokines have been long recognized as part of the mechanism used by the innate immune system to battle viral infections; however, their involvement in sterile inflammation remains unclear. Mounting evidence pointing to the involvement of the IFN-I pathway in sterile kidney inflammation provides potential insights into the complex interplay between the innate immune system and damage to the most sensitive segment of the nephron, the glomerulus. The STING pathway is often cited as the cause of renal disease originating from non-infection related caused by the induction of IFN-I expression. However, other receptors have been implicated in this process, mainly by recognizing host-derived nucleic acids. This review explores the main endogenous inducers of IFN-I in glomerular cells, discusses what exposure to the main autocrine and paracrine cytokines has on these cells, and identifies the pathways that are implicated in the development of glomerular damage.
Keywords:
Glomerulonephritis
; IFN-I pathway
; Intracellular Pattern-Recognition Receptors
; STING
; sterile inflammation
1. Introduction
Over one hundred years ago, Paul Ehrlich introduced the term “horror autotoxicus” to the field of immunopathology. He used this term to refer to the process whereby the immune system directs its attention to attack itself. This term finds increased relevance today and can be used to describe many types of hypersensitivity reactions, including autoimmune diseases (types II and III) and disorders stemming from a hyperinflammatory response, including cytokine storm syndrome and other autoinflammatory diseases.
Recently, molecules typically associated with an antiviral immune response, cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING), have been linked to the development of chronic kidney diseases (CKD) without involving infection. Instead, activation is through host-derived endogenous nucleic acids. Most relevant to potential therapeutic intervention, STING blockade results in reduced kidney damage,1 especially at the level of the glomerulus.2,3 Interestingly, although the expression of type I interferons (IFN-I) is one of the expected outcomes after the activation of cGAS/STING and other intracellular pattern-recognition receptors (PRRs), it is still not clear if the renoprotective effects of cGAS/STING blockade rely on the subsequent abrogation of IFN-I production more than other involved mechanisms.
Glomerulonephritis (GN) encompasses a group of immune-mediated diseases characterized by inflammation and fibrosis within the glomerulus causing well-described histopathological lesions that are used for clinical classification. Seven major categories are included in GN and include: 1) membranoproliferative GN (MPGN), 2) minimal change disease (MCD), 3) focal segmental glomerulosclerosis (FSGS), 4) membranous nephropathy (MN), 5) IgA nephropathy (IgAN), 6) crescentic GN (CGN), and 7) lupus nephritis (LN).4 Although the current classification is helpful for diagnosis, several and often different etiologies are involved in the development of each. Indeed, among the different etiologies, infections are responsible for only a fraction of GN cases while most are caused by a) endogenous factors often involving auto-antibodies against a broad spectrum of glomerular cell-specific molecules, b) genetic defects leading to autoinflammatory disorders, and c) as a consequence of other non-communicable diseases such as diabetes.5 Taken together, this group of diseases is part of the above mentioned “horror autotoxicus”.
Among the different molecular mechanisms involved in the pathogenesis of GN, there is accumulating evidence that the pathways that induce IFN-I expression along with the pathways activated downstream of IFN-I recognition (hereafter both referred to as the IFN-I pathway) participate in the progression of glomerular damage.
Much evidence stems from patient data derived from kidney biopsies, identifying the upregulation of gene products involved in the IFN-I pathway. For example, patients diagnosed with C3 glomerulopathy MPGN show an up-regulation of IFI44L, IFIT1, MX1 and OAS2 genes,6 and STING was observed to be over-expressed in MCD and IgAN.7 Using a weighted gene co-expression network analysis (WGCNA), FSGS patients show an enrichment in the IFN-I signaling pathway in glomerular endothelial cells,8 while anti-PLA2R MN patients show a similar enrichment in the IFN-I pathway using gene ontology (GO) analysis in mesangial cells. Specifically, these patients show increased expression of IFI6, a gene stimulated after IFN-I recognition.9 In LN, the IFN-I pathway is consistently recognized as a key marker of the disease in kidney and other tissues, with immune cells being the main contributors of the activated IFN-I pathway.10
The most compelling evidence suggesting the direct involvement of the IFN-I pathway in the development and progression of GN comes from patients receiving recombinant IFN therapy or were discovered to have genetic alterations that cause enhanced IFN-I production. Of note, patients diagnosed with multiple sclerosis, hepatitis C, some types of cancer, and other diseases developed glomerular lesions after received recombinant IFN-I (rIFN-I) therapy.11-14 Among the inborn errors causing enhanced IFN-I production are Aicardi-Goutières syndrome,15 STING-associated vasculopathy with onset in infancy (SAVI),16,17 and systemic lupus erythematosus (SLE).18 These genetic diseases all present with glomerular injury.
Results from experiments using animal models have provided additional support for the direct effect of IFN-I on the glomerulus. A murine model of nephritis generated by the injection of anti-glomerular basement membrane (GBM) antibodies showed an increase in IFNα in renal tissue and correlated with renal dysfunction demonstrated by increased levels of blood urea nitrogen (BUN) and proteinuria. Moreover, the exogenous overexpression of IFNα using an adenoviral vector increased proteinuria and lead to a higher GN score in treated animals.19 In a similar approach, Migliorini et al. combined a model of adriamycin nephropathy with the administration of IFNα and IFNβ and showed enhanced kidney dysfunction through the perturbation of parietal epithelial cells (PEC) and podocyte loss.20 In testing a novel model of murine SLE, Nacionales et al. showed that blocking IFN-I activity by knocking out an IFN-I receptor (IFNAR), kidney dysfunction and glomerular injury could be ameliorated.21 Taken together, results from these experiments suggest that the different causes of GN drive a diverse set of complex immunopathologies, including the stimulation of the IFN-I pathway which, in turn, participates in further aggravating the disease.
The goal of this review is to 1) present a general overview of the IFN- pathway, 2) explore how the IFN-I pathway is induced, focusing on endogenous stress activators such as host-derived self-nucleic acids, and 3) outline how kidney specific cells are affected by these cytokines.
2. IFN-I Induction In Sterile Inflammation
An early immune response to foreign pathogens, such as viral nucleic acids, is the activation of intracellular pattern-recognition receptors (PRRs), leading to the induction of IFN-I expression. These responses can also be triggered by endogenous danger signals, including self-nucleic acids detected in the cytosol22 by proteosome dysfunction leading to proteotoxic stress,23 and by elevated cellular oxidative stress.24 In physiological homeostatic conditions, mechanisms are in place to avoid aberrant PRR activation by self-nucleic acids. This is achieved by their quick degradation using DNA and RNA nucleases, protective epigenetic and posttranscriptional modifications, and by physically compartmentalizing nucleic acids to the nucleus and mitochondria. Certain stress-inducing conditions, however, can cause nucleic acid damage and drive their release into the cytoplasm. These conditions include the presence of xenobiotics, metabolic stress, elevated reactive oxygen species (ROS), and the unfolded protein response (UPR).25,26
In many cases of CKD, including several types of GN, patient biopsies show evidence of DNA damage.27 In a retrospective study using human biopsy samples, Ott et al. observed DNA fragmentation across a spectrum of different types of GN.28 More specifically, it was shown in mice that double stranded DNA breaks in podocytes caused by the endonuclease I-Ppol correlated with proteinuria, glomerulosclerosis, and tubulointerstitial fibrosis.29 This study also identified portions of the immune system as being responsible for the development of disease. Similarly, Dhillon et al. show that the expression of DNA damage-causing transposable elements (TE) and endogenous retroviruses (ERVs) could be detected in diseased samples from human patients and mouse models of renal fibrosis.30 Interestingly, Dhillon et al. link the expression of ERVs to the IFN-I pathway. In support of these findings, De Cecco et al. show an association between retrotransposable elements and IFN-I pathway signaling trough the detection of cytoplasmic cDNA.31 As previously mentioned, the first response is to degrade nucleic acids. Mechanisms to guard against immune activation caused by self-nucleic acid detection exist in the kidney. For example, case-study data report that the nucleases TREX1 and RNaseH215,32 are key enzymes involved in cytoplasmic nucleic acid degradation and when mutated have been linked to kidney-predominant thrombotic microangiopathy (TMA) and Aicardi-Goutières syndrome,15,32 respectively.
The second response involves the immune system, and more specifically the activation of intracellular PRRs. There is a vast variety of PRRs able to recognize nucleic acids, among these are the cGAS-like receptors (cGLR), RIG-I-like receptors (RLRs), and toll-like receptors (TLRs).22 Table 1 provides a general summary of the distinct types of intracellular PRRs and their cognate ligands.
Although different nucleic acid receptors activate different signal transduction pathways, most converge at the nexus of activating TANK-binding kinase 1 (TBK1) and, in turn, the phosphorylation of interferon regulatory factor (IRF)3 and IRF7, transcription factors that allow IFN-I expression. Once IFN-Is are released, they act in an autocrine/paracrine manner through their recognition by IFN-I receptors (IFNAR1/2). IFN-I ligand-receptor binding activates a signal transduction pathway that allows for the formation of an interferon stimulated gene factor 3 (ISGF3) complex, leading to the expression of hundreds of interferon-stimulated genes (ISGs).33,34
Figure 1 provides a graphical representation of the main molecular interactions which occur to initiate and propagate signaling through the IFN-I pathway. The remainder of this review will highlight certain aspects of these interactions as they relate to the available data concerning glomerulonephritis and other prominent kidney diseases.
3. cGAS/STING Activation In The Kidney
cGAS and its analogs, cGAS-like receptors (cGLR), are evolutionary conserved receptors in metazoans that detect cytosolic double stranded (ds) DNA.35 Once these receptors recognize their ligands, they have the ability to enzymatically convert triphosphate nucleotides into signaling linear or cyclic dinucleotides. For example, cGAS can generate 2’3’-cGAMP, a second messenger that binds and activates STING in the endoplasmic reticulum (ER).36-38 STING then oligomerizes and translocates to the Golgi, leading to the activation of TBK1 and IRF3, which in turn translocates to the nucleus and induces IFN-I expression.
Primarily due to its cellular localization and inability to discriminate between host and foreign dsDNA, the cGAS/STING system is among the first mechanisms to respond to nuclear and mitochondrially (mt)-derived DNA present in cytoplasm. Interest in the cGAS/STING pathway has recently increased within the field of nephrology with the discovery that key glomerular cells express components of the pathway at the early stage of kidney dysfunction. Zang et al. show that podocyte injury can be triggered in a mouse model of diabetic kidney disease (DKD) through lipotoxicity-induced mitochondrial damage and mtDNA leakage leading to the activation of the cGAS/STING pathway.3 Mitrofanova et al. observed similar results in cultured human and mouse podocytes as well as in mouse models of DKD and Alport syndrome that were exposed to a STING-specific agonist.2 In an attempt to identify molecular pathways responsible for apolipoprotein 1 (APOL1)-induced kidney dysfunction, Wu et al. identified the expression of STING as an early event leading to podocyte cytotoxicity and cell death.39 In all cases, pharmacologic inhibition or genetic deletion of the STING pathway reduced glomerular damage in these mouse models of disease. A more detailed exploration of the involvement of the cGAS/STING pathway in the progression to Lupus Nephritis End Stage Renal Disease (LN-ESRD) using cultured human podocytes was recently reported. Davis et al. show that if podocytes are treated with nucleosome-associated dsDNA fragments, a common marker found in the blood of Lupus patients, IFNγ-inducible protein 16 (IFI16) triggers the expression of both IFNβ and APOL1 and the activation of the cGAS/STING pathway.40 More directly, this group shows that exposing podocytes to IFNβ promotes the expression of IFI16, suggesting a positive feedback loop between dsDNA recognition and cGAS/STING pathway activation. Additional evidence implicating the cGAS/STING pathway in lupus-mediated podocyte damage was provided by Li et al., who treated mouse podocytes with LN patient serum. It was shown that in these podocytes, the cGAS/STING pathway gets activated, IFN-I expression is stimulated, and ER stress is induced leading to apoptosis.41 Podocytes from diabetic mice also show increased levels of cGAS and STING as well as activation of TBK1. Palmitic acid, a fatty acid associated with DKD, can elicit mitochondrial damage and leakage of mtDNA into the cytoplasm to activate the STING pathway. As a result, several inflammatory markers are induced and levels of apoptosis increase. Interestingly, IFN-I expression does not seem to be elevated in this mouse model.3
The bulk of research linking the activation of the cGAS/STING pathway to glomerular disease has focused on the podocyte, however, some evidence exists showing that other cells within glomerulus are affected by this pathway and could contribute to renal dysfunction. There are several reports that implicate the STING pathway, either directly or indirectly, in glomerular endothelial cell damage. In diabetic mice, Qi et al. present evidence that mtDNA contributes to glomerular damage indirectly through GECs by inducing podocyte loss leading to proteinuria,42 while Caselena et al. show that GECs display severe mitochondrial damage when treated with high glucose media.43 Mitochondrial dysfunction is also seen in GECs from a transgenic mouse model of FSGS. This study shows the crosstalk between glomerular cells where the activation of tumor growth factor β receptor 1(TGFβR1) in podocytes is associated with an increase in mitochondrial stress and release of mtDNA in GECs, leading to a subsequent loss of podocytes.44 More indirect evidence comes from studies using mice expressing an APOL1 renal risk variants (RSK). The endothelial cells of these mice display high levels of mitophagy with mtDNA leakage into the cytoplasm and IFN-I pathway activation.45 Although mostly indirect, there is evidence that the cGAS/STING pathway gets activated in GECs and that this activation leads to glomerular damage. It is interesting to note that in 2009, Hagele et al. reported that GECs could be activated by dsDNA in a TLR-independent manner, showing increased IFN-I pathway expression;46 the report being published in years close to the cGAS/STING pathway was discovered.47,48
Activation of the cGAS/STING pathway in mesangial cells (MCs) is still somewhat unexplored. There is some evidence, however, that mitochondrial damage leading to leakage of mtDNA occurs in cultured human mesangial cells (HMCs). This group showed that culturing HMCs with galactose deficient IgA from IgAN patients leads to mitochondrial damage due to the reduction in peroxisome proliferator-activated receptor alpha (PPARα) expression.49 Curiously, MCs grown in hyperglycemic conditions also shown markers of mitochondrial damage, suggesting that leakage of mtDNA and the subsequent activation of the cGAS/STING pathway could also be operating in some instances of DN.
It should be including here that there are other cell types not typically associated with the glomerulus that could also contribute to glomerular injury through the STING pathway. As an example, plasmacytoid dendritic cells (pDCs) are a special type of immune cell characterized, in part, by their high expression of IFN-I. Indeed, Deng et al. show that pDCs rapidly infiltrate the kidney after AKI and produce IFNα, leading to kidney damage.50 These cells have also been implicated in lupus nephritis,51,52 where it has been reported that STING activation promotes the maturation of pDCs with their subsequent participation in glomerular injury.53 However, it must be emphasized that it is still not clear if pDCs’ contribution to renal disease is through their involvement systemically or at the level of the kidney.
4. RLRs Activation in The Kidney
Melanoma differentiation-associated gene 5 (MDA5) and retinoic acid inducible gene I (RIG-I), are two RLR family members which reside in the cytoplasm and act as receptors for dsRNA, and in a manner similar to cGAS, activate the mitochondrial antiviral-signaling protein (MAVS), TBK1, and IRF3 to elicit IFN-I expression. Although cytoplasmic endogenous dsRNA is a less common indicator of cellular stress as compared to cytoplasmic dsDNA, sources include ERE, mt-dsRNA and other secondary RNA structures that arise from epigenetic alterations, mitochondrial damage, or defects in RNA processing.54-59 Murine models of autoimmune disease have been instrumental in showing that mt-dsRNA plays a relevant role in driving sterile inflammation through IFN-I pathway activation.60,61
The activation of RLRs has been documented in a variety of kidney diseases, both in human patient samples and animal models of AKI and several different types of GN. A number of studies have shown that tubule-specific damage is linked to active RLRs. For example, Zhu et al. showed that renal tubule injury in mice caused by ischemia-reperfusion-injury (IRI) or unilateral ureteral obstruction (UUO) was closely tied to an accumulation of mt-dsRNA.62 Similar results were reported by Doke et al. also using the murine IRI method.63 Exploring the mechanisms responsible for AKI caused by Crush Syndrome (CS), Wang et al. utilized rats and were able to show a significant increase in RIG-I associated signaling.64 A study using whole exome sequencing on patients with LN identified a gain-of-function mutation in RIG-I leading to its constitutive activation.65 RLR activation and associated inflammation was observed in kidney samples from CKD patients showing an increase in ERE expression.30
Cells within the glomerulus also express the major sensors for dsRNA and can activate the associated immune signaling pathways. Yamashita et al. used cultured human and mouse podocytes to show that regardless of the source of dsRNA, be it from mitochondria, ERE or secondary structures spontaneously formed, RLR activation in podocytes leads to the expression of IFN-I, IL-6, and cytoskeleton alterations,66 the latter being instrumental in causing effacement and eventual cell loss.67 The remaining bulk of evidence linking RLRs to podocyte dysfunction comes from investigations into the mechanisms whereby APOL1 causes kidney damage. Fang et al. used long-term injections of recombinant APOL1 in mice and observed increased expression of RIG-I in podocytes. Similar findings were seen in cultured human podocytes engineered to express APOL1 RRV. Knock-down of RIG-I, either using siRNA against RIG-I in in vitro experiments or AAV-shRIG-I in in vivo mouse experiments, blunted the expression of several pro-inflammatory genes and attenuated podocyte and glomerular damage, respectively.68 One caveat emerging from this study is that it remains unclear what the direct activator of RIG-I is, and if RIG-I drives the expression of IFN-I along with other proinflammatory genes. Further confounding the involvement of RLRS in APOL1-mediated podocyte damage is the discovery that APOL1 mRNA itself possesses structural characteristics that allows for the formation of dsRNA secondary structures,69 which indeed have been shown to be recognized by MDA5 in podocytes.70 However, as just mentioned, it is yet to be determined if these APOL1 secondary structures can be also recognized by RIG-I.
There is a dearth of evidence for the expression of RLRs in other glomerular cells. Results from in vitro experiments support the notion that RLRs can contribute to glomerular injury. Hagele et al. used a synthetic agonist of RLR to treat cultured murine GECs. These experiments revealed that there is an increase in the expression of IFN-I, IL-6, and intracellular adhesion molecule 1 (ICAM-I). It can be extrapolated that these changes in GEC could lead to increased albumin permeability.71 The same synthetic agonist was used to in vitro treat human mesangial cells. The result was an increase in the expression of RIG-I chemokine ligand 5 (CCL5) and the C-X-C motif chemokine ligand 10 (CXCL10).72,73 Taken together, these results suggest that if GECs and MCs are under stress and are exposed to RLR ligands, they are able to promote inflammation favoring chemotaxis and the adhesion of leukocytes. However, since the synthetic ligand for RLRs can also activate TLR3, signaling through TLR3-mediated pathways cannot be ruled out.
5. TLRs Activation in The Kidney
TLRs are comprised of a large family of PRRs that are expressed on the plasma membrane and intracellularly, the latter location being activated by nucleic acids and highly associated with sterile inflammation.74 Endosomally-restricted TLRs include: TLR3 (which recognizes dsRNA), TLR7/8 (which recognizes ssRNA), and TLR9 (which recognizes dsDNA). It must be highlighted that intracellular TLRs cannot participate in the first response to self-derived nucleic acids in the cytoplasm as the other RLRs mentioned above can since they are physically sequestered within endosomes. Instead, these TLRs are only activated in the event that extracellular DNA or RNA from damaged or dead bystander cells has been endocytosed and the nucleic acid-containing endosome fuses with an acidified endosome containing the TLRs.75 Indeed, there are several reports in the literature where extracellular mRNA or mitochondrial RNA derived from damaged cells has been engulfed by neighboring cells resulting in TLR3 activation and sterile inflammation.60,76,77 Highlighting a route for entry for nucleic acids, Bertheloot et al. show that RNA released from damaged cells can be detected by the receptor for advanced glycation-end products (RAGE) to elicit their internalization and activate TLR7 and TLR8. 78 It is thought that a similar mechanism exists for the detection and internalization of mtDNA from damaged mitochondria, leading the activation of TLR9.79 Extracellular nucleic acids are shielded from degradation if they are bound to other molecules. These shielding molecules include antibodies, antimicrobial peptides, neutrophil extracellular traps (NETS), and microvesicles; all of which have been detected in patients with lupus.80-83 Once formed, these nucleic acid-protein complexes can be taken up through endocytosis by specialized cells, including macrophages and dendritic cells, but also by podocytes, MCs and GECs.84-86 However, we should consider that the activation of this class of TLRs could be the consequence of previous IFN-I or other pathway activation and/or as a result of actions carried out by any number of immune cell types, rather than directly by cells within the glomerulus.
In the context of CKD and GN, the function of TLRs has been extensively reviewed in previous publications.87,88 Therefore, this review will narrowly focus on the involvement of TLR signaling in podocytes, GECs, and MCs.
Experiments using cultured human podocytes confirm that these cells highly express TLR3 and upon its activation, were shown to induce the expression of IFNβ, several chemokines (IL-6, MCP-1, CCL5), the costimulatory molecule CD80, and APOL1, but not IFNα.89-91 TLR7 agonists have been widely used to generate mouse models of lupus which display varying degrees of podocyte injury; however, podocyte damage can be traced to the action of other cell types, including those that belong to the immune system.92,93 A more direct involvement of TLR8 in podocyte damage has been seen in mice models of lupus and in rodents that have undergone UUO. In these experimental models, TLR8 expression is increased in podocytes and is associated with the elevation of micro-RNA 21 (miR21),94,95 an endogenous ligand for TLR8.96 Interestingly, it has been reported that miR21 is increased in the urine and renal tissue of CKD patients, including those diagnosed with DN and IgAN.97-99 In a rat model of nephrotic syndrome and in a mouse model of autoimmune GN, TLR9 expression is increased and associated with podocyte injury.100,101 Moreover, mtDNA and/or dsDNA seems to be the ligand for this receptor in podocytes, inducing their apoptosis in vitro. Seemingly contradictory results were reported by Bossaller et al., who show that TLR9 deficiency aggravates LN induced by TLR7 agonists in mice,102 suggesting a role for TLR9 as a negative regulator to limit glomerular damage.
Only the involvement of TLR3 in GECs has been reported. A number of publications originating from the same group using cultured human cells, show that the in vitro activation of TLR3 elicits the expression IFN-I, RLRs, chemokines and plasminogen activator inhibitor-1 (PAI-1), a negative regulator of fibrinolysis.103-108 Taken together, this body of work suggests that the activation of TLR3 in GECs may contribute to inflammatory and fibrotic responses, events that could also be a result of a secondary response to the IFN-I pathway signaling.
Similar to reports for GECs, much of the work exploring the action of TLRs in MCs was accomplished using cultured human mesangial cells. These studies show that the in vitro activation of TLR3 in MCs results in the expression of several different chemokines, adhesion molecules and matrix metalloproteinase 9 (MMP9).109-112 It is worth mentioning that MMP9 is an enzyme relevant to the inflammatory response and is detected in the biopsies of patients diagnosed with active and chronic GN,113 as well as in the urine of patients with MN.114 A report by Shen et al. revealed that TLR9 was significantly elevated in MCs grown in vitro under hyperglycemic conditions and that knockdown of TLR9 under these same conditions reduces the expression of several inflammatory markers, and the incidence of apoptosis.115 In the same study using a mouse model of diabetes, silencing TLR9 reduced glomerular matrix cell expansion.
Taken together, these various reports suggest that under conditions that result in cellular stress, nucleic acids may be released into the extracellular space where they can be taken up by any number of different cells, including cells within the glomerulus. This, in turn, results in the activation of the IFN-I pathway and may include other signal pathways such those that induce nuclear factor κB (NF-κB) activation. The end result is to prolong and propagate sterile inflammation in the kidney.
6. Effect of IFN-I in Glomerular Cells
The activation of intracellular PRRs leads to the synthesis of IFN-I. The term ‘IFN’ encompasses a group of cytokines divided into three family types referred as type I, type II (also named g), and type III (also named l). Interferons were initially discovered for their essential role in the anti-viral and anti-cancer immune response, and more recently, for their participation in autoimmune and autoinflammatory disorders.116-119 Among the IFN-I family members, several subtypes have been described in mammals and include a, b, d, e, k, t, and w. In humans, IFN-I contains 13 isoforms of IFNα and one isoform of IFNb, and are primarily recognized by their biological function.116,120
IFN-I are recognized by a heterodimeric receptor composed by interferon a and b receptor subunits 1 and 2 (IFNAR1 and IFNAR2). Although both subunits are necessary for IFN-recognition, the signal transduction is initiated by IFNAR1. Attached to the cytoplasmic tail of IFNAR1 is tyrosine kinase 2 (TYK2) which is initially activated, followed by the phosphorylation of Janus kinase 1 (JAK1), the phosphorylation of signal transduction and activator of transcription (STAT)1 and STAT2, which are associated with the cytoplasmic tail of IFNAR2. Next is the formation of ISGF3, a heterotrimeric transcriptional factor made up of STAT1, STAT2, and IRF9 which functions to promote the expression of interferon signature genes (ISGs). Cellular stress, including ribosomal stress, drives crosstalk between the above-mentioned canonical IFN-I pathway and other signaling pathways to boost the expression of hundreds of ISGs. The end result is the alteration of several cellular processes such as transcription, translation and metabolism.121-123
Recently, Manoharan et al. report that the cytokine tissue factor (TF) exhibits substantial homology with IFNAR2 and can physically interact with IFNAR1 to restrict its activation in glomerular cells under steady state conditions. Moreover, TF deletion in podocytes promotes IFN-I pathway activation and the development of GN. Interestingly, biopsies from patients diagnosed with different forms of GN show a reduction in TF levels within the glomerulus.124
The effects of IFNα signaling in podocytes includes the induction of autophagy and the inhibition of the mammalian target of rapamycin complex 1 (mTORC1) pathway.125 These observations provide an additional link between IFN-I signaling and the development of GN since the loss of mTORC1 in podocytes causes proteinuria associated with glomerular damage.126 Furthermore, IFNβ induces the expression of APOL191 and apoptosis in mature podocytes, along with the reduction of nephrin expression during podocyte differentiation in vitro.20 Li et al. also show that IFN-I activation leads to the induction of viperin (also known as RSAD2) a common ISG reported to be a negative regulator of podocyte differentiation.127 Similar effects have been described in other kidney-specific cells, such as parietal epithelial cells (PECs) and tubule epithelial cells (TECs), where IFN-I signaling disrupts the cell cycle and promote cell death.20,128 It is becoming clear that elevated IFN-I expression in podocytes can drive autophagy, impair cellular differentiation, and promote podocyte apoptosis, events that can exacerbate glomerular disease. On the other hand, Lee et al. showed that by targeting IFNAR1/2 signals using bariticinib, a JAK inhibitor, podocyte damage in a mouse LN model could be reduced, along with a general reduction in inflammation pointing to a systemic effect of the inhibitor.129
There is little direct evidence of what effect IFN-I has on glomerular endothelial cells. A series of publications from Hiroshi Tanaka’s group using cultured human GECs identified a set of ISGs with the potential to be used as biomarkers for LN.103,130,131 Instead, we might draw on work accomplished using other types of endothelial cells to extrapolate IFN-I signaling effects on GECs. For example, work using brain, liver, and lung endothelial cells indicates that IFN-I cytokines, mainly IFNβ, affects tight junctions through the down-regulation of VE-cadherin, reducing the synthesis of nitric oxide (NO), the induction of PAI-1 secretion, and the reduction in caveolin 1 expression.132-134 Combined, these effects could promote endothelial permeability and stiffness. Additionally, interferon-stimulated gene 15 (ISG15), one of the byproducts of IFNAR1/2 activation, has been linked to the development of vascular stiffness in cases of hypertension.135
In mesangial cells, it has been demonstrated that IFNβ promotes the expression of IL-6 and participates in regulating proliferation.136 Using primary human mesangial cells, Zhang et al. discovered a feed-forward regulatory pathway where IFN-I stimulates the expression of miR744, which boosts the activation of JAK1/STAT1/STAT2 to promote the expression of several chemokines, including CXCL10.137 More recently, Gao et al. show that CXCL10 can stimulate mesangial cell proliferation and migration in vitro, and participates in mesangial cell expansion in a mouse model of glomerulonephritis.138
7. Concluding Remarks and Future Perspectives
The IFN-I pathway has been extensively studied as part of the innate immune system’s response against viral infections. This pathway originates with the recognition of viral nucleic acids by intracellular PRRs, followed by the transduction of signals that induce IFN-I production and release, and terminates with the recognition of IFN-I by specific receptors to induce the expression of genes that reduces transcription and translation rates, as well as alter nucleotide acid and lipid metabolism to block the viral replication cycle.139 However, the function of this pathway in sterile inflammation is not yet fully understood. With the description of interferonopathies more than a decade ago, much attention turned to focus on the relationship between these cytokines and autoinflammatory effects.119,140 Soon to follow was the discovery and description of a new cytoplasmic nucleic acid receptor system, cGAS/STING which also was linked to IFN-I expression.47,48 Henceforth, new light was shed on how endogenous stress, through self-nucleic acid recognition, drives sterile inflammation via cGAS/STING and other PRR signaling pathways.
Although there is ample evidence supporting the hypothesis that blocking intracellular receptors blunts sterile inflammation, there still exists a substantial gap in knowledge regarding the exact mechanisms responsible. This is especially true if the protective effects are due to limiting IFN-I expression or if other pathways are the primary stimulators of the inflammation since these intracellular receptors can also participate in crosstalk with other signaling pathways that activate NF-kB.
This review explored and described the endogenous activators of intracellular PRRs that drive IFN-I expression, as well the specific effects these cytokines have on kidney and glomerular cells that, when considered together, could indicate a direct involvement of this pathway in GN progression. Figure 2 summarizes the key findings of this review with respect to the effect of the IFN-I pathway on glomerular health. Many questions, however, remain unanswered. For example, is it possible that IFN-I pathway activation is merely an indirect reaction to an initial insult rather than the main cause of GN? Since GN is a group of diseases with each group displaying complex pathophysiology, it may prove difficult to accurately uncover the responsible mechanisms for each since in vivo and in vitro models seldom fully resemble the pathology displayed by human patients. Nonetheless, and despite the complexity of the molecular mechanisms involved, it is clear that the IFN-I pathway is participating in the development and progression of GN. The evidence of IFN-I pathway function in glomerular cells presents an opportunity to explore the potential exploitation of this pathway for therapeutic purposes. For example, manipulation of the IFN-I pathway and related cytokines could boost the production of antibodies for vaccine development141 directed at autoimmune driven GN. Another avenue to explore is the effect that IFN-I has on the cellular metabolism of glomerular cells, including glycolysis, oxidative phosphorylation, and lipid synthesis, as has been reported for macrophages and T cells.142-144 The relationship the IFN-I pathway has with other cellular stress responses, such as the activation of inflammasomes and the unfolded protein response (UPR)23,145 could be investigated further, specifically in glomerular cells to inform new treatment options. Finally, new insights into renal fibrosis, glomerular cell senescence, and aging could be gained by exploring IFN-I pathway crosstalk with tumor growth factor β (TGF-β) pathway,146 DNA damage and epigenetic changes,147 or inflammaging process,148 respectively.
References
- Chung, K. W. et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab 30, 784-799 e785. [CrossRef]
- Mitrofanova, A. et al. Activation of Stimulator of IFN Genes (STING) Causes Proteinuria and Contributes to Glomerular Diseases. J Am Soc Nephrol 33, 2153-2173,. [CrossRef]
- Zang, N. et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience 25, 105145. [CrossRef]
- Tinawi, M. Update on the etiology, classification, and management of glomerular diseases. Avicenna J Med 10, 61-67. [CrossRef]
- Anders, H. J., Kitching, A. R., Leung, N. & Romagnani, P. Glomerulonephritis: immunopathogenesis and immunotherapy. Nat Rev Immunol 23, 453-471. [CrossRef]
- Koh, J. H. et al. Spatially resolved transcriptomic profiling for glomerular and tubulointerstitial gene expression in C3 glomerulopathy. medRxiv, 2023.2006.2030.23292064. [CrossRef]
- Yu, B. C. et al. Minimal Change Disease Is Associated with Mitochondrial Injury and STING Pathway Activation. J Clin Med 11. [CrossRef]
- Menon, R. et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 5. [CrossRef]
- Xu, J. et al. Single-Cell Profiling Reveals Transcriptional Signatures and Cell-Cell Crosstalk in Anti-PLA2R Positive Idiopathic Membranous Nephropathy Patients. Front Immunol 12, 683330. [CrossRef]
- Deng, Y. et al. Expression characteristics of interferon-stimulated genes and possible regulatory mechanisms in lupus patients using transcriptomics analyses. EBioMedicine 70, 103477. [CrossRef]
- Irifuku, T., Okimoto, K., Masuzawa, N. & Masaki, T. Nephrotic-range proteinuria and membranoproliferative glomerulonephritis-like pattern caused by interferon-beta1b in a patient with multiple sclerosis. CEN Case Rep 12, 275-280. [CrossRef]
- Gianassi, I., Allinovi, M., Caroti, L. & Cirami, L. C. Broad spectrum of interferon-related nephropathies-glomerulonephritis, systemic lupus erythematosus-like syndrome and thrombotic microangiopathy: A case report and review of literature. World J Nephrol 8, 109-117. [CrossRef]
- Markowitz, G. S., Nasr, S. H., Stokes, M. B. & D'Agati, V. D. Treatment with IFN-alpha, -beta, or -gamma is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5, 607-615. [CrossRef]
- Kayar, Y. et al. Interferon Induced Focal Segmental Glomerulosclerosis. Case Rep Nephrol 2016, 6967378. [CrossRef]
- He, T., Xia, Y. & Yang, J. Systemic inflammation and chronic kidney disease in a patient due to the RNASEH2B defect. Pediatr Rheumatol Online J 19, 9,. [CrossRef]
- Abid, Q. et al. APOL1-Associated Collapsing Focal Segmental Glomerulosclerosis in a Patient With Stimulator of Interferon Genes (STING)-Associated Vasculopathy With Onset in Infancy (SAVI). Am J Kidney Dis 2020 75, 287-290. [CrossRef]
- Staels, F. et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front Immunol 2020 11, 575219. [CrossRef]
- Hou, G. et al. Integrative Functional Genomics Identifies Systemic Lupus Erythematosus Causal Genetic Variant in the IRF5 Risk Locus. Arthritis Rheumatol 75, 574-585. [CrossRef]
- Fairhurst, A. M. et al. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J Immunol 2009 183, 6831-6838. [CrossRef]
- Migliorini, A. et al. The antiviral cytokines IFN-alpha and IFN-beta modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol 2013 183, 431-440. [CrossRef]
- Nacionales, D. C. et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum 56, 3770-3783. [CrossRef]
- Okude, H., Ori, D. & Kawai, T. Signaling Through Nucleic Acid Sensors and Their Roles in Inflammatory Diseases. Front Immunol 11, 625833. [CrossRef]
- Davidson, S. et al. Protein kinase R is an innate immune sensor of proteotoxic stress via accumulation of cytoplasmic IL-24. Sci Immunol 7, eabi6763. [CrossRef]
- Hwang, I. et al. Cellular stress signaling activates type-I IFN response through FOXO3-regulated lamin posttranslational modification. Nat Commun 12, 640. [CrossRef]
- Miller, K. N. et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506-5526. [CrossRef]
- Chen, Y. G. & Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol 23, 286-301. [CrossRef]
- Yoshimoto, N. et al. Significance of podocyte DNA damage and glomerular DNA methylation in CKD patients with proteinuria. Hypertens Res 46, 1000-1008. [CrossRef]
- Ott, U. et al. DNA fragmentation in chronic glomerulonephritis: an immunohistological analysis. Nephron Clin Pract 105, c18-28. [CrossRef]
- Nakamichi, R. et al. DNA-damaged podocyte-CD8 T cell crosstalk exacerbates kidney injury by altering DNA methylation. Cell Rep 2023 42, 112302. [CrossRef]
- Dhillon, P. et al. Increased levels of endogenous retroviruses trigger fibroinflammation and play a role in kidney disease development. Nat Commun 2023 14, 559. [CrossRef]
- De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019 566, 73-78. [CrossRef]
- Song, Z. R. et al. Kidney-Predominant Thrombotic Microangiopathy Associated With TREX1 Frameshift Mutation. Kidney Int Rep 2023 8, 2172-2176. [CrossRef]
- Zhou, R., Zhang, Q. & Xu, P. TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim Biophys Sin (Shanghai) 2020 52, 757-767. [CrossRef]
- Chen, H. J., Tas, S. W. & de Winther, M. P. J. Type-I interferons in atherosclerosis. J Exp Med 2020 217. [CrossRef]
- Li, Y. et al. cGLRs are a diverse family of pattern recognition receptors in innate immunity. Cell 2023 186, 3261-3276 e3220. [CrossRef]
- Hall, J. et al. The catalytic mechanism of cyclic GMP-AMP synthase (cGAS) and implications for innate immunity and inhibition. Protein Sci 2017, 26, 2367-2380. [CrossRef]
- Shang, G., Zhang, C., Chen, Z. J., Bai, X. C. & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389-393. [CrossRef]
- Motani, K. et al. The Golgi-resident protein ACBD3 concentrates STING at ER-Golgi contact sites to drive export from the ER. Cell Rep2022 41, 111868. [CrossRef]
- Wu, J. et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest 2021 131. [CrossRef]
- Davis, S. E., Khatua, A. K. & Popik, W. Nucleosomal dsDNA Stimulates APOL1 Expression in Human Cultured Podocytes by Activating the cGAS/IFI16-STING Signaling Pathway. Sci Rep2019, 9, 15485. [CrossRef]
- Li, X. et al. SMURF1 activates the cGAS/STING/IFN-1 signal axis by mediating YY1 ubiquitination to accelerate the progression of lupus nephritis. Autoimmunity2023 56, 2281235. [CrossRef]
- Qi, H. et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 66, 763-778. [CrossRef]
- Casalena, G. A. et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal 18, 105, doi:10.1186/s12964-020-00605-x. [CrossRef]
- Daehn, I. et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124, 1608-1621. [CrossRef]
- Wu, J. et al. APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity 54, 2632-2649 e2636. [CrossRef]
- Hagele, H. et al. Double-stranded DNA activates glomerular endothelial cells and enhances albumin permeability via a toll-like receptor-independent cytosolic DNA recognition pathway. Am J Pathol 175, 1896-1904. [CrossRef]
- Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788-792. [CrossRef]
- Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826-830. [CrossRef]
- Wu, J. et al. Downregulation of PPARalpha mediates FABP1 expression, contributing to IgA nephropathy by stimulating ferroptosis in human mesangial cells. Int J Biol Sci 18, 5438-5458,. [CrossRef]
- Deng, B. et al. Plasmacytoid dendritic cells promote acute kidney injury by producing interferon-alpha. Cell Mol Immunol 18, 219-229,. [CrossRef]
- Nehar-Belaid, D. et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat Immunol 21, 1094-1106. [CrossRef]
- Iwamoto, T. et al. High Systemic Type I Interferon Activity Is Associated With Active Class III/IV Lupus Nephritis. J Rheumatol 49, 388-397. [CrossRef]
- Thim-Uam, A. et al. STING Mediates Lupus via the Activation of Conventional Dendritic Cell Maturation and Plasmacytoid Dendritic Cell Differentiation. iScience 23, 101530. [CrossRef]
- Mehdipour, P. et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169-173. [CrossRef]
- Tunbak, H. et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat Commun 11, 5387. [CrossRef]
- Silva, S., Camino, L. P. & Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc Natl Acad Sci U S A 115, 11024-11029. [CrossRef]
- Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238-242. [CrossRef]
- Tigano, M., Vargas, D. C., Tremblay-Belzile, S., Fu, Y. & Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477-481,. [CrossRef]
- Garcia-Gonzalez, C. et al. ADAR1 Prevents Autoinflammatory Processes in the Heart Mediated by IRF7. Circ Res 131, 580-597. [CrossRef]
- Kim, S. et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep 40, 111178. [CrossRef]
- Yoon, J. et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren's syndrome. Mol Ther Nucleic Acids 30, 257-269. [CrossRef]
- Zhu, Y. et al. Polynucleotide phosphorylase protects against renal tubular injury via blocking mt-dsRNA-PKR-eIF2alpha axis. Nat Commun 14, 1223. [CrossRef]
- Doke, T. et al. NAD(+) precursor supplementation prevents mtRNA/RIG-I-dependent inflammation during kidney injury. Nat Metab 5, 414-430. [CrossRef]
- Wang, P. T. et al. RIG-I, a novel DAMPs sensor for myoglobin activates NF-kappaB/caspase-3 signaling in CS-AKI model. Mil Med Res 8, 37,. [CrossRef]
- Peng, J. et al. Clinical Implications of a New DDX58 Pathogenic Variant That Causes Lupus Nephritis due to RIG-I Hyperactivation. J Am Soc Nephrol 34, 258-272,. [CrossRef]
- Yamashita, M. et al. Antiviral innate immunity disturbs podocyte cell function. J Innate Immun 5, 231-241. [CrossRef]
- Schell, C. & Huber, T. B. The Evolving Complexity of the Podocyte Cytoskeleton. J Am Soc Nephrol 28, 3166-3174,. [CrossRef]
- Fang, J. et al. ApoL1 induces kidney inflammation through RIG-I/NF-kappaB activation. Biochem Biophys Res Commun 527, 466-473. [CrossRef]
- Okamoto, K. et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol 1, 188. [CrossRef]
- Riella, C. V. et al. ADAR regulates APOL1 via A-to-I RNA editing by inhibition of MDA5 activation in a paradoxical biological circuit. Proc Natl Acad Sci U S A 119, e2210150119. [CrossRef]
- Hagele, H., Allam, R., Pawar, R. D. & Anders, H. J. Double-stranded RNA activates type I interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol Dial Transplant 24, 3312-3318. [CrossRef]
- Imaizumi, T. et al. Retinoic acid-inducible gene-I is induced by double-stranded RNA and regulates the expression of CC chemokine ligand (CCL) 5 in human mesangial cells. Nephrol Dial Transplant 25, 3534-3539. [CrossRef]
- Imaizumi, T. et al. Melanoma differentiation-associated gene 5 regulates the expression of a chemokine CXCL10 in human mesangial cells: implications for chronic inflammatory renal diseases. Tohoku J Exp Med 228, 17-26. [CrossRef]
- Patra, M. C. et al. A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models. Cells 9. [CrossRef]
- Lind, N. A., Rael, V. E., Pestal, K., Liu, B. & Barton, G. M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat Rev Immunol 22, 224-235. [CrossRef]
- Gollmann-Tepekoylu, C. et al. Toll-like receptor 3 mediates ischaemia/reperfusion injury after cardiac transplantation. Eur J Cardiothorac Surg 57, 826-835,. [CrossRef]
- Stolberg-Stolberg, J. et al. Toll-like receptor 3 activation promotes joint degeneration in osteoarthritis. Cell Death Dis 13, 224,. [CrossRef]
- Bertheloot, D. et al. RAGE Enhances TLR Responses through Binding and Internalization of RNA. J Immunol 197, 4118-4126. [CrossRef]
- Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251-255. [CrossRef]
- Lande, R. et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3, 73ra19. [CrossRef]
- Bruschi, M. et al. Neutrophil Extracellular Traps Profiles in Patients with Incident Systemic Lupus Erythematosus and Lupus Nephritis. J Rheumatol 47, 377-386. [CrossRef]
- Jorgensen, M. H., Rekvig, O. P., Jacobsen, R. S., Jacobsen, S. & Fenton, K. A. Circulating levels of chromatin fragments are inversely correlated with anti-dsDNA antibody levels in human and murine systemic lupus erythematosus. Immunol Lett 138, 179-186. [CrossRef]
- Rasmussen, N. S., Nielsen, C. T., Nielsen, C. H. & Jacobsen, S. Microvesicles in active lupus nephritis show Toll-like receptor 9-dependent co-expression of galectin-3 binding protein and double-stranded DNA. Clin Exp Immunol 204, 64-77. [CrossRef]
- Liu, Y. et al. IQGAP1 mediates podocyte injury in diabetic kidney disease by regulating nephrin endocytosis. Cell Signal 59, 13-23,. [CrossRef]
- Bryniarski, M. A. et al. Megalin-mediated albumin endocytosis in cultured murine mesangial cells. Biochem Biophys Res Commun 529, 740-746,. [CrossRef]
- Moriyama, T., Karasawa, K., Hasegawa, F., Uchida, K. & Nitta, K. Sertraline Reduces Albuminuria by Interfering with Caveolae-Mediated Endocytosis through Glomerular Endothelial and Epithelial Cells. Am J Nephrol 50, 444-453. [CrossRef]
- Liu, F., Chen, H., Cao, C., Liang, Y. & Zhou, Y. The role of toll-like receptors (TLRs) and their therapeutic applications in glomerulonephritis. Int Urol Nephrol 55, 2845-2856. [CrossRef]
- Liu, M. & Zen, K. Toll-Like Receptors Regulate the Development and Progression of Renal Diseases. Kidney Dis (Basel) 7, 14-23. [CrossRef]
- Umetsu, H. et al. Interleukin-6 via Toll-Like Receptor 3 Signaling Attenuates the Expression of Proinflammatory Chemokines in Human Podocytes. Kidney Blood Press Res 46, 207-218. [CrossRef]
- Shimada, M. et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappaB-dependent pathway. Nephrol Dial Transplant 27, 81-89. [CrossRef]
- Nichols, B. et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 87, 332-342. [CrossRef]
- Carlucci, F. et al. C1q Modulates the Response to TLR7 Stimulation by Pristane-Primed Macrophages: Implications for Pristane-Induced Lupus. J Immunol 196, 1488-1494. [CrossRef]
- Zhang, D. et al. Myeloid-Derived Suppressor Cells Induce Podocyte Injury Through Increasing Reactive Oxygen Species in Lupus Nephritis. Front Immunol 9, 1443. [CrossRef]
- Masum, M. A., Ichii, O., Elewa, Y. H. A. & Kon, Y. Podocyte Injury Through Interaction Between Tlr8 and Its Endogenous Ligand miR-21 in Obstructed and Its Collateral Kidney. Front Immunol 11, 606488. [CrossRef]
- Kimura, J. et al. Overexpression of Toll-like receptor 8 correlates with the progression of podocyte injury in murine autoimmune glomerulonephritis. Sci Rep 4, 7290. [CrossRef]
- Zhang, Z. J. et al. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. J Exp Med 215, 3019-3037. [CrossRef]
- Lange, T. et al. MiR-21 is up-regulated in urinary exosomes of chronic kidney disease patients and after glomerular injury. J Cell Mol Med 23, 4839-4843. [CrossRef]
- Zang, J., Maxwell, A. P., Simpson, D. A. & McKay, G. J. Differential Expression of Urinary Exosomal MicroRNAs miR-21-5p and miR-30b-5p in Individuals with Diabetic Kidney Disease. Sci Rep 9, 10900. [CrossRef]
- Szeto, C. C. et al. Kidney microRNA-21 Expression and Kidney Function in IgA Nephropathy. Kidney Med 3, 76-82 e71. [CrossRef]
- Bao, W. et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci Rep 6, 22579. [CrossRef]
- Masum, M. A. et al. Overexpression of toll-like receptor 9 correlates with podocyte injury in a murine model of autoimmune membranoproliferative glomerulonephritis. Autoimmunity 51, 386-398. [CrossRef]
- Bossaller, L. et al. TLR9 Deficiency Leads to Accelerated Renal Disease and Myeloid Lineage Abnormalities in Pristane-Induced Murine Lupus. J Immunol 197, 1044-1053. [CrossRef]
- Karasawa, T. et al. Expression of interferon-stimulated gene 20 (ISG20), an antiviral effector protein, in glomerular endothelial cells: possible involvement of ISG20 in lupus nephritis. Ren Fail 45, 2224890. [CrossRef]
- Liu, Q. et al. Cytosolic Sensors of Viral RNA Are Involved in the Production of Interleukin-6 via Toll-Like Receptor 3 Signaling in Human Glomerular Endothelial Cells. Kidney Blood Press Res 44, 62-71. [CrossRef]
- Hirono, K. et al. Endothelial expression of fractalkine (CX3CL1) is induced by Toll-like receptor 3 signaling in cultured human glomerular endothelial cells. Mod Rheumatol 30, 1074-1081. [CrossRef]
- Sato, R. et al. Inhibitory effect of anti-malarial agents on the expression of proinflammatory chemokines via Toll-like receptor 3 signaling in human glomerular endothelial cells. Ren Fail 43, 643-650. [CrossRef]
- Aizawa, T. et al. Chloroquine attenuates TLR3-mediated plasminogen activator inhibitor-1 expression in cultured human glomerular endothelial cells. Clin Exp Nephrol 23, 448-454. [CrossRef]
- Imaizumi, T. et al. IFIT Proteins Are Involved in CXCL10 Expression in Human Glomerular Endothelial Cells Treated with a Toll-Like Receptor 3 Agonist. Kidney Blood Press Res 46, 74-83. [CrossRef]
- Merkle, M. et al. TLR3-dependent immune regulatory functions of human mesangial cells. Cell Mol Immunol 9, 334-340. [CrossRef]
- Imaizumi, T. et al. Toll-like receptor 3 signaling contributes to the expression of a neutrophil chemoattractant, CXCL1 in human mesangial cells. Clin Exp Nephrol 19, 761-770. [CrossRef]
- Imaizumi, T. et al. Cylindromatosis (CYLD), a Deubiquitinase, Attenuates Inflammatory Signaling Pathways by Activating Toll-Like Receptor 3 in Human Mesangial Cells. Kidney Blood Press Res 42, 942-950. [CrossRef]
- Merkle, M., Ribeiro, A., Koppel, S. & Wornle, M. TNFalpha enhances TLR3-dependent effects on MMP-9 expression in human mesangial cells. Cell Biol Int 36, 1155-1160. [CrossRef]
- Phillips, T. M. et al. MMP2 and MMP9 associate with crescentic glomerulonephritis. Clin Kidney J 10, 215-220. [CrossRef]
- Gilbert, A., Changjuan, A., Guixue, C., Jianhua, L. & Xiaosong, Q. Urinary Matrix Metalloproteinase-9 and Nephrin in Idiopathic Membranous Nephropathy: A Cross-Sectional Study. Dis Markers 2021, 1620545. [CrossRef]
- Shen, J., Dai, Z., Li, Y., Zhu, H. & Zhao, L. TLR9 regulates NLRP3 inflammasome activation via the NF-kB signaling pathway in diabetic nephropathy. Diabetol Metab Syndr 14, 26. [CrossRef]
- Bourdon, M., Manet, C. & Montagutelli, X. Host genetic susceptibility to viral infections: the role of type I interferon induction. Genes Immun 21, 365-379. [CrossRef]
- Arico, E., Castiello, L., Capone, I., Gabriele, L. & Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) 11. [CrossRef]
- Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370. [CrossRef]
- Crow, Y. J. & Stetson, D. B. The type I interferonopathies: 10 years on. Nat Rev Immunol 22, 471-483. [CrossRef]
- Peters, S. O. et al. Evolutionary Pattern of Interferon Alpha Genes in Bovidae and Genetic Diversity of IFNAA in the Bovine Genome. Front Immunol 11, 580412. [CrossRef]
- Shemesh, M., Lochte, S., Piehler, J. & Schreiber, G. IFNAR1 and IFNAR2 play distinct roles in initiating type I interferon-induced JAK-STAT signaling and activating STATs. Sci Signal 14, eabe4627. [CrossRef]
- Boccuni, L. et al. Stress signaling boosts interferon-induced gene transcription in macrophages. Sci Signal 15, eabq5389. [CrossRef]
- Hubel, P. et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol 20, 493-502. [CrossRef]
- Manoharan, J. et al. Tissue factor binds to and inhibits interferon-alpha receptor 1 signaling. Immunity. [CrossRef]
- Qi, Y. Y. et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-alpha in lupus nephritis. Ann Rheum Dis 77, 1799-1809. [CrossRef]
- Godel, M. et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121, 2197-2209. [CrossRef]
- Li, Z. et al. miR-200 family promotes podocyte differentiation through repression of RSAD2. Sci Rep 6, 27105. [CrossRef]
- Cordoba-David, G. et al. Crosstalk between TBK1/IKKepsilon and the type I interferon pathway contributes to tubulointerstitial inflammation and kidney tubular injury. Front Pharmacol 13, 987979. [CrossRef]
- Lee, J. et al. Baricitinib Attenuates Autoimmune Phenotype and Podocyte Injury in a Murine Model of Systemic Lupus Erythematosus. Front Immunol 12, 704526. [CrossRef]
- Karasawa, T. et al. Glomerular endothelial expression of type I IFN-stimulated gene, DExD/H-Box helicase 60 via toll-like receptor 3 signaling: possible involvement in the pathogenesis of lupus nephritis. Ren Fail 44, 137-145. [CrossRef]
- Hashimoto, S. et al. Expression of IFN-induced transmembrane protein 1 in glomerular endothelial cells. Pediatr Int 63, 1075-1081. [CrossRef]
- Jana, A. et al. Increased Type I interferon signaling and brain endothelial barrier dysfunction in an experimental model of Alzheimer's disease. Sci Rep 12, 16488. [CrossRef]
- Jia, H. et al. Endothelial cell functions impaired by interferon in vitro: Insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res 163, 105-116. [CrossRef]
- Gairhe, S. et al. Type I interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension. Proc Natl Acad Sci U S A 118. [CrossRef]
- Gonzalez-Amor, M. et al. Interferon-stimulated gene 15 pathway is a novel mediator of endothelial dysfunction and aneurysms development in angiotensin II infused mice through increased oxidative stress. Cardiovasc Res 118, 3250-3268. [CrossRef]
- Flur, K. et al. Viral RNA induces type I interferon-dependent cytokine release and cell death in mesangial cells via melanoma-differentiation-associated gene-5: Implications for viral infection-associated glomerulonephritis. Am J Pathol 175, 2014-2022. [CrossRef]
- Zhang, X. et al. miR-744 enhances type I interferon signaling pathway by targeting PTP1B in primary human renal mesangial cells. Sci Rep 5, 12987. [CrossRef]
- Gao, J. et al. Knockdown of Cxcl10 Inhibits Mesangial Cell Proliferation in Murine Habu Nephritis Via ERK Signaling. Cell Physiol Biochem 42, 2118-2129. [CrossRef]
- Schoggins, J. W. Interferon-Stimulated Genes: What Do They All Do? Annu Rev Virol 6, 567-584. [CrossRef]
- Crow, Y. J., Lebon, P., Casanova, J. L. & Gresser, I. A Brief Historical Perspective on the Pathological Consequences of Excessive Type I Interferon Exposure In vivo. J Clin Immunol 38, 694-698. [CrossRef]
- Ye, L. et al. Type I and Type III Interferons Differ in Their Adjuvant Activities for Influenza Vaccines. J Virol 93. [CrossRef]
- Olson, G. S. et al. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Rep 35, 109195. 1091; 95. [CrossRef]
- Buang, N. et al. Type I interferons affect the metabolic fitness of CD8(+) T cells from patients with systemic lupus erythematosus. Nat Commun 12, 1980. [CrossRef]
- York, A. G. et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716-1729,. [CrossRef]
- Chipurupalli, S., Samavedam, U. & Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front Med (Lausanne) 8, 758311. [CrossRef]
- Ahodantin, J. et al. Type I interferons and TGF-beta cooperate to induce liver fibrosis during HIV-1 infection under antiretroviral therapy. JCI Insight 7. [CrossRef]
- Yu, Q. et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep 11, 785-797. [CrossRef]
- Rasa, S. M. M. et al. Inflammaging is driven by upregulation of innate immune receptors and systemic interferon signaling and is ameliorated by dietary restriction. Cell Rep 39, 111017. [CrossRef]
Figure 1.
Origins and intracellular sensors of self-nucleic acids promoting the IFN-I signaling pathway. During cellular stresses, self-derived nucleic acids (DNA or RNA) are released from damaged mitochondria and the nucleus, consequently accumulating in the cytosol. In addition, extracellular nucleic acids liberated from neighboring dying or damaged cells are internalized by endocytosis. These intracellular (cytosolic or endosomal) nucleic-acids are recognized by diverse intracellular nucleic acid sensors, triggering the activation of the signaling pathways that produce IFN-I and proinflammatory cytokines. Specifically, sensors for endosomal nucleic acids include TLR3 (for double-stranded DNA), TLR7 (for single-stranded RNA), and TLR9 (for single-stranded DNA). Cytosolic double-stranded RNA is detected by RIG-I or MDA5, while cytosolic double-stranded DNA is recognized by members of the cGAS-STING pathway. Activation of these intracellular nucleic acid sensors stimulates TBK1 activation, prompting the translocation of IRFs and NF-kB into the nucleus. There, they orchestrate the expression of IFN-I and proinflammatory cytokine genes. Subsequently, binding of IFN-I to IFNAR1/IFNAR2 triggers the activation of the JAK-STAT pathway, culminating in the induction of ISGs. This cascade of events illustrates the intricate molecular mechanisms involved in the recognition of self-nucleic acids, the subsequent activation of signaling pathways leading to the expression and secretion of IFN-I and proinflammatory cytokines, and the subsequent induction of ISGs via the JAK-STAT pathway. Created with BioRender.com. IFN-I, type I interferons; TLR, Toll-like receptor; RIG-I, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated protein 5; cGAS, cyclic GMP-AMP (cGAMP) synthase; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; IFNAR, type I interferon receptor; ISGs, interferon-stimulated genes.
Figure 1.
Origins and intracellular sensors of self-nucleic acids promoting the IFN-I signaling pathway. During cellular stresses, self-derived nucleic acids (DNA or RNA) are released from damaged mitochondria and the nucleus, consequently accumulating in the cytosol. In addition, extracellular nucleic acids liberated from neighboring dying or damaged cells are internalized by endocytosis. These intracellular (cytosolic or endosomal) nucleic-acids are recognized by diverse intracellular nucleic acid sensors, triggering the activation of the signaling pathways that produce IFN-I and proinflammatory cytokines. Specifically, sensors for endosomal nucleic acids include TLR3 (for double-stranded DNA), TLR7 (for single-stranded RNA), and TLR9 (for single-stranded DNA). Cytosolic double-stranded RNA is detected by RIG-I or MDA5, while cytosolic double-stranded DNA is recognized by members of the cGAS-STING pathway. Activation of these intracellular nucleic acid sensors stimulates TBK1 activation, prompting the translocation of IRFs and NF-kB into the nucleus. There, they orchestrate the expression of IFN-I and proinflammatory cytokine genes. Subsequently, binding of IFN-I to IFNAR1/IFNAR2 triggers the activation of the JAK-STAT pathway, culminating in the induction of ISGs. This cascade of events illustrates the intricate molecular mechanisms involved in the recognition of self-nucleic acids, the subsequent activation of signaling pathways leading to the expression and secretion of IFN-I and proinflammatory cytokines, and the subsequent induction of ISGs via the JAK-STAT pathway. Created with BioRender.com. IFN-I, type I interferons; TLR, Toll-like receptor; RIG-I, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated protein 5; cGAS, cyclic GMP-AMP (cGAMP) synthase; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; IFNAR, type I interferon receptor; ISGs, interferon-stimulated genes.
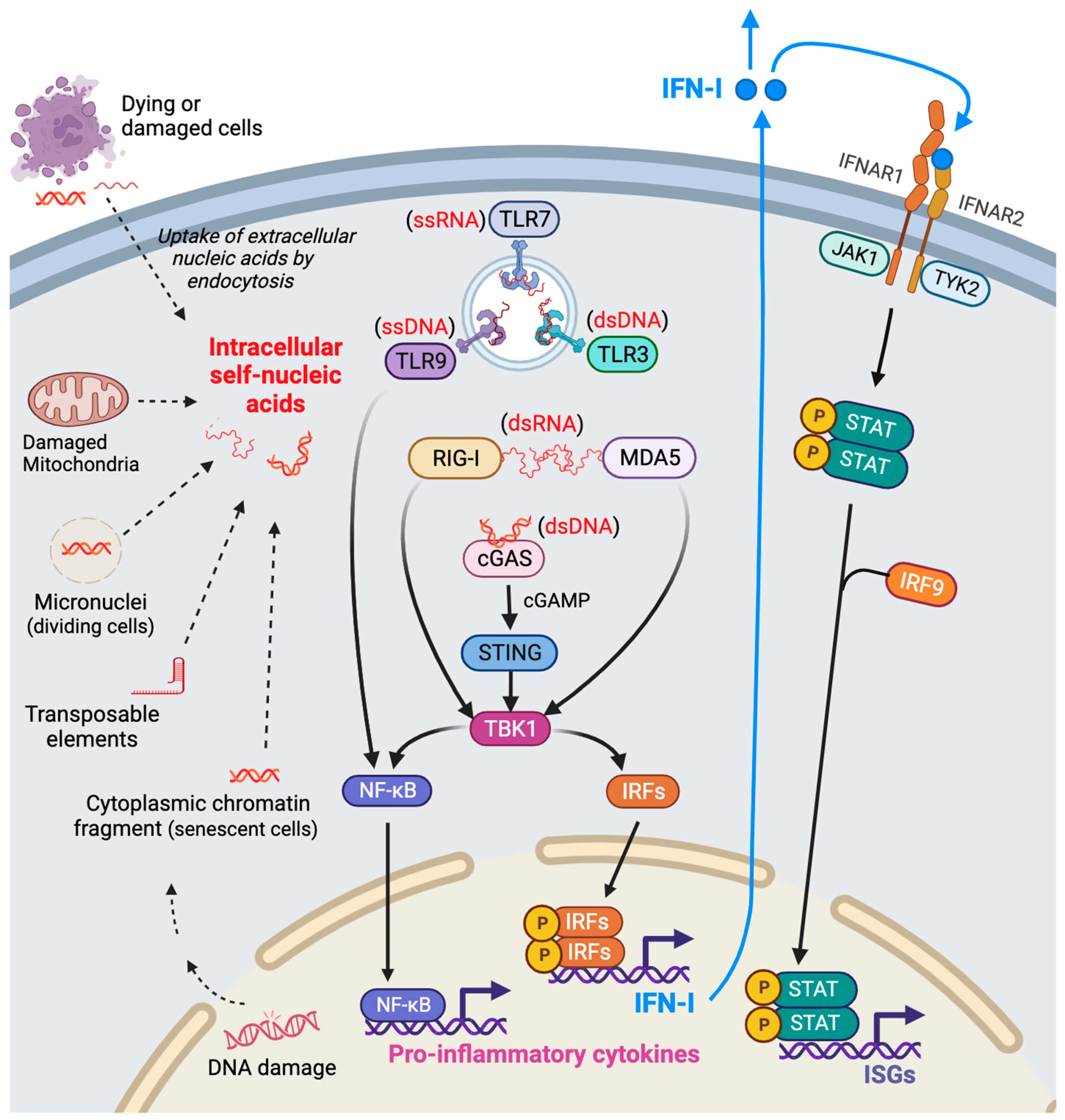
Figure 2.
The potential deleterious effects of IFN-I on different glomerular cell types. IFN-I can exhibit direct influence on diverse cell types within the glomerular compartment, altering their cellular functions. In addition, increased IFN-I levels induce the expression of interferon-stimulated genes (ISGs). This could intensify inflammatory processes by recruiting inflammatory immune cells to the kidney and increasing the production of autoantibodies. Created with BioRender.com. APOL-1, Apolipoprotein L1; VE-cadherin, Vascular endothelial-cadherin; NO, Nitric oxide.
Figure 2.
The potential deleterious effects of IFN-I on different glomerular cell types. IFN-I can exhibit direct influence on diverse cell types within the glomerular compartment, altering their cellular functions. In addition, increased IFN-I levels induce the expression of interferon-stimulated genes (ISGs). This could intensify inflammatory processes by recruiting inflammatory immune cells to the kidney and increasing the production of autoantibodies. Created with BioRender.com. APOL-1, Apolipoprotein L1; VE-cadherin, Vascular endothelial-cadherin; NO, Nitric oxide.
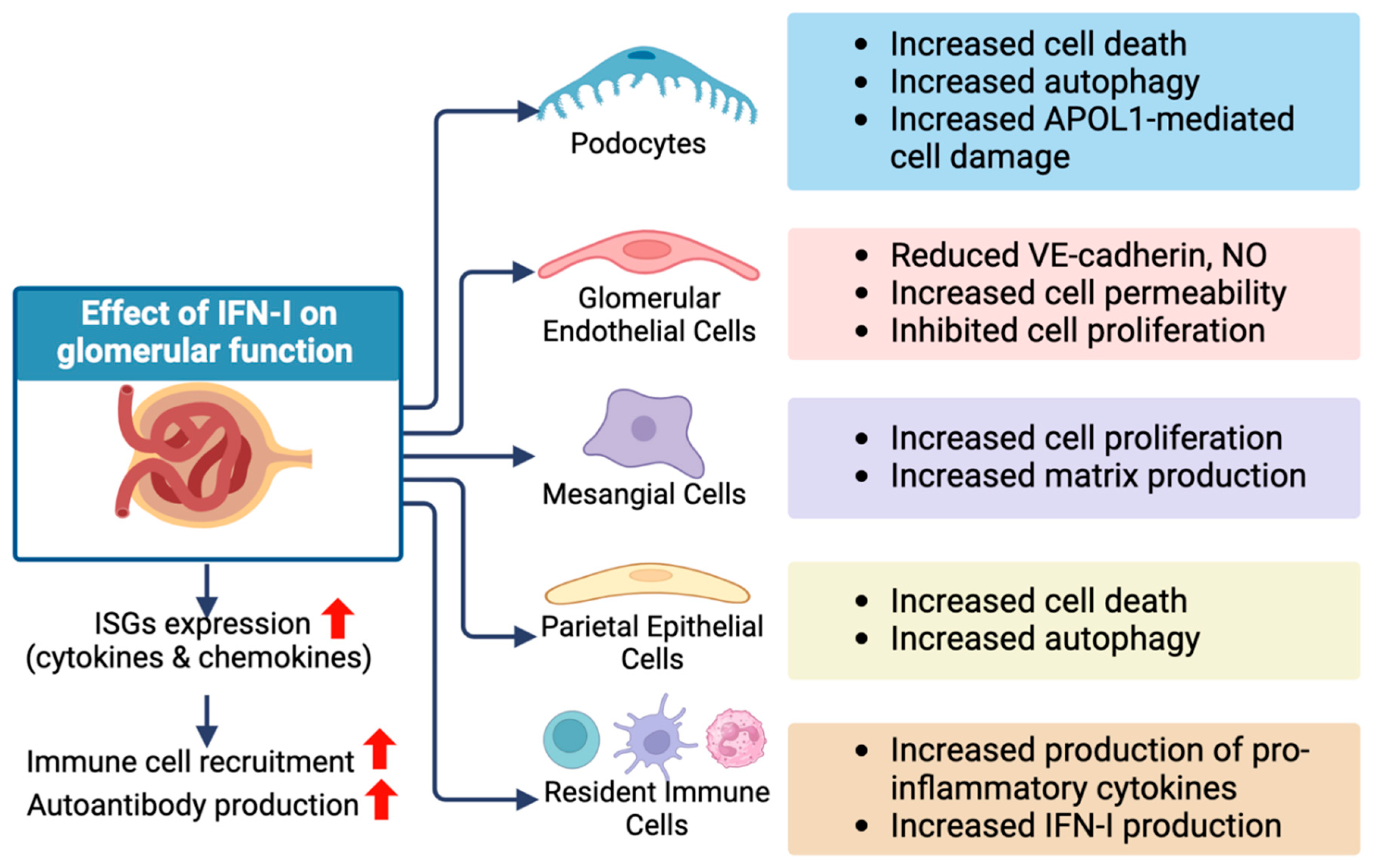

Table 1.
Intracellular pattern recognition receptors (PRRs) involved in IFN-I induction.
| Family | Name | Ligands | Cell distribution | Source of endogenous activators |
|---|---|---|---|---|
| cGLRs | cGAS | dsDNA | Cytoplasm |
|
| RLRs | RIG-I | dsRNA | Cytoplasm |
|
| MDA5 | dsRNA | Cytoplasm | ||
| TLRs | TLR3 | dsRNA | Endosomes |
|
| TLR7 | ssRNA | |||
| TLR8 | ssRNA | |||
| TLR9 | dsDNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



