
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Tobacco smoke is an environmental mixture including polycyclic aromatic hydrocarbons and may interfere in endocrine system as an EDC in aging. Thus, we performed a molecular epidemiological approach with in-depth biological monitoring of combustible cigarette smoking among adolescents and adults in South Korea (N=620) with exposure biomarkers, i.e., exhaled CO, urinary cotinine, t,t-muconic acid (TTMA), malondialdehyde (MDA), and obtained information of their lifestyle and tobacco addiction status. We also diagnosed the genetic polymorphisms on the 96 SNPs for tobacco-metabolism, -addiction, and expression differences and compared mtDNA abnormalities in buccal and blood cells. As results, there were positive associations among the above tobacco exposure biomarkers. Man or youth smokers showed high frequency in some of mtDNA alteration, such as ‘SNPs for inconsistent bases between buccal and blood cells’. Among the SNPs, the polymorphisms on SULT1A1, DRD2, and ADH1B were related to multi-exposure biomarkers. Interestingly, youth showed similar levels of urinary TTMA and MDA to adults, although their pack-year was approx. 1/6 volume of that of adults. We also observed the negative association between urinary MDA levels and the growth rate in the adolescents (p<0.05). In conclusion, the present biological monitoring provides high susceptible population, i.e., adolescents rather than adults, and reliable genetic factors affecting tobacco exposure. The inferred environment-gene-gene interaction suggests tobacco smoking accelerats aging in adolescents as an EDC.
Keywords:
tobacco smoking
; biological monitoring
; adolescent
; genetic polymorphism
; addiction
; mtDNA alteration
; aging
1. Introduction
Endocrine disrupting chemicals (EDCs) are a heterogeneous group of exogenous compounds that can restrict with several facets of endogenous hormones and accelerate aging, immune, metabolic or neurobehavioral disorders to threaten quality of life [1]. Persistent exposure to EDC can disrupt homeostasis in the body and creates oxidative stress that can lead to aging and chronic inflammation [2]. These characteristics were also found to be significant in the observation of telomere length, which is a measure of aging [3]. As aging is a complex process influenced by genetic, environmental, and lifestyle factors, changes in endogenous hormone levels are part of this intricate interplay.
Tobacco smoke is an environmental mixture with over 7,000 chemicals including polycyclic aromatic hydrocarbons (PAHs) [4] and has ability to obstruct and interfere in the function of endocrine system and has been entitled as EDCs [5]. In addition to oxidative stress or inflammasome activation by tobacco smoking, a chronic activation of aryl hydrocarbon receptor signaling can contribute to premature aging and the development of neoplasms by affecting metabolism, extracellular matrix remodeling, inflammation, pigmentation, DNA repair, and apoptosis [6]. Therefore, there is growing concern about the potential impact of tobacco smoking as an EDC, while research in this field is ongoing.
New-generation tobacco nicotine products are intended to replace combustible cigarettes to avoid tobacco smell and to consider health benefit. However, the health benefit of the new types of tobacco is still unclear [7] and only combustible cigarettes showed product use dependent exposure to PAHs [8], which are regarded as endocrine disruptors [9]. Therefore, traditional combustible cigarettes can be one kind of tobacco smoking to monitor the proper exposure to PAHs.
In addition, exposure timing and early life environment for tobacco smoking have been considered, because the response to the toxic chemicals in children or youth can be manifested into adulthood via cellular memory, such as epigenetic changes [10]. Therefore, the tobacco starting age can be the first issue for susceptibility differences. For other susceptibility biomarkers of tobacco smoking, various genome-wide association studies on tobacco smoking behavior have identified multiple loci associated with tobacco addiction [11,12] For example, genetic polymorphisms of cytochrome P450 (CYP) 2A6 and nicotinic acetylcholine receptor (nAChRs) are notable for tobacco-addiction [13,14]. We previously reported differential gene expression patterns between smokers and nonsmokers in a Korean pilot study [15].
For exposure biomarkers of tobacco smoking, parent chemicals in tobacco smoke or their specific metabolites have been used [16]. The most representative biomarkers are cotinine, a metabolite of nicotine; t,t-muconic acid (TTMA), a metabolite of benzene, which are quite attributable to their specificity and sensitivity among the various urinary metabolites of tobacco [17]. In addition, altered biological responses to tobacco smoke exposure have potential as early diagnostic biomarkers. For example, alteration in genomic and mitochondrial (mt) DNA or oxidative stress parameters, such as malondialdehyde (MDA), can be considered as popular biomarkers as responsive to tobacco smoking [15].
Therefore, we performed a molecular epidemiological approach with in-depth biological monitoring of combustible cigarette smoking among adolescents and adults in South Korea, where the current smoking rate of adolescents is 6.7% in 2019 [18]and for adults is 20.6 % in 2020 [19], with various exposure, susceptibility and response biomarkers to clarify how tobacco smoking differently affects adolescents and adults. This approach may reveal the underlying mechanisms of tobacco smoking as an EDC and provide preventive ways from EDCs.
2. Results
2.1. Characteristics of Subjects
Six hundred twenty volunteers were recruited from four sites (Table 1). We defined non- smokers, who did not smoke for the last one year. Thus, ex-smokers were included in nonsmokers and their ratio was approx. 0.8 in the non-smokers, thus, that of never-smokers reached to 0.2. The ratio of non-smokers and smokers were similar, approx. 32-37% among men. Thirty four % of the recruited male youths smoked combustible cigarettes.
Adult smokers smoked 18.49± 9.70 pack-year and started tobacco smoking at 21.75±6.23 years. There were no significant differences in age and BMI due to smoking in the adults. However, education years and alcohol drinking were negatively (p<0.05) and positively (p<0.001) related to smoking, respectively.
For youths, they smoked 2.2±1.2 pack-year and started tobacco smoking at 13.77±2.27 years. The addition scores by FTND for adults and youth were 3.23 ± 2.31 and 0.73 ± 0.88, respectively. The tobacco addiction status of most of the subjects was relatively low, however, it was higher in adults than adolescents.
2.2. Exposure Levels of Tobacco Smoking
The CO levels of exhaled gas were approx. five- fold higher in smokers than non-smokers, 13.37±8.28 ppm vs. 2.83±2.56 ppm (p<<0.001). For urinary exposure biomarkers, the ranges of urinary cotinine, TTMA and MDA were 0.015-4.35 mg/L (median 0.015 mg/L), 0.10-1324.60 ug/L (median 37.70 ug/L) and 0.06-13.26 µM (median 2.51 µM). When we compared the exposure levels by smoking, the smokers showed significantly higher in most of the biomarkers than non-smokers (Table 2), although the association somewhat decreased after creatinine modification.
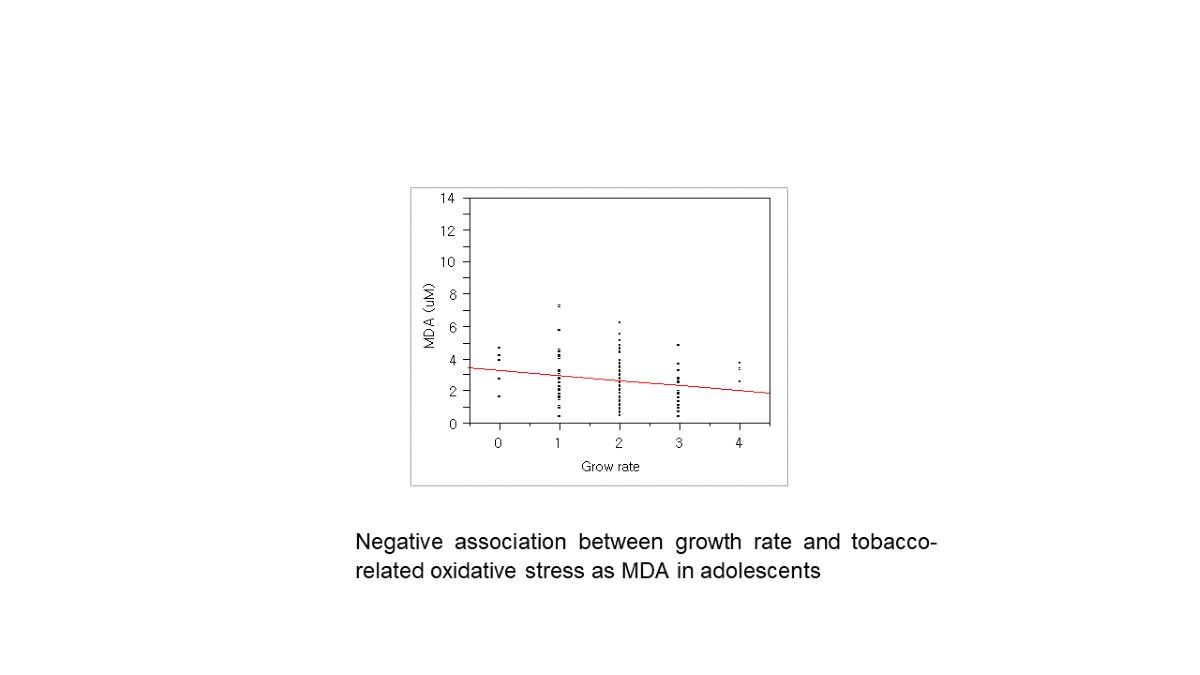
In addition, there were strong positive associations among the exposure markers. For example, there was the association between tobacco smoking and oxidative stress with urinary cotinine and MDA (Figure 1). Interestingly, the self-reported growth rate in youth was negatively related to the MDA levels (Figure 2). Due to approx. six-fold higher pack per year of smoking in adults than youth, we expected quite big differences in these exposure by smoking. As results, urinary cotinine levels were approx. three fold higher in adults than youth, i.e., 0.45± 0.88 vs. 0.14± 0.39 mg/g cre (p<0.0001). However, urinary MDA or TTMA was only 1.2-1.3 fold higher in adults than youth: For TTMA, 89.95± 116.40 vs. 74.10 ±129.99 µg/ L (p<0.05); For MDA, 2.11± 3.11 vs. 1.58± 0.86 µM/g cre (p<0.0001). Thus, these results may result from the fact that youth felt guilty for smoking and faked the exact amounts smoked. In addition, youth can have high susceptibility to bio-produced tobacco metabolites with small amounts of tobacco.
We also found the positive associations between urinary cotinine and TTMA (r=0.38, p<0.01) and between urinary MDA and TTMA (r=0.25, p<0.01).
2.3. mtDNA Alteration by Tobacco Smoking
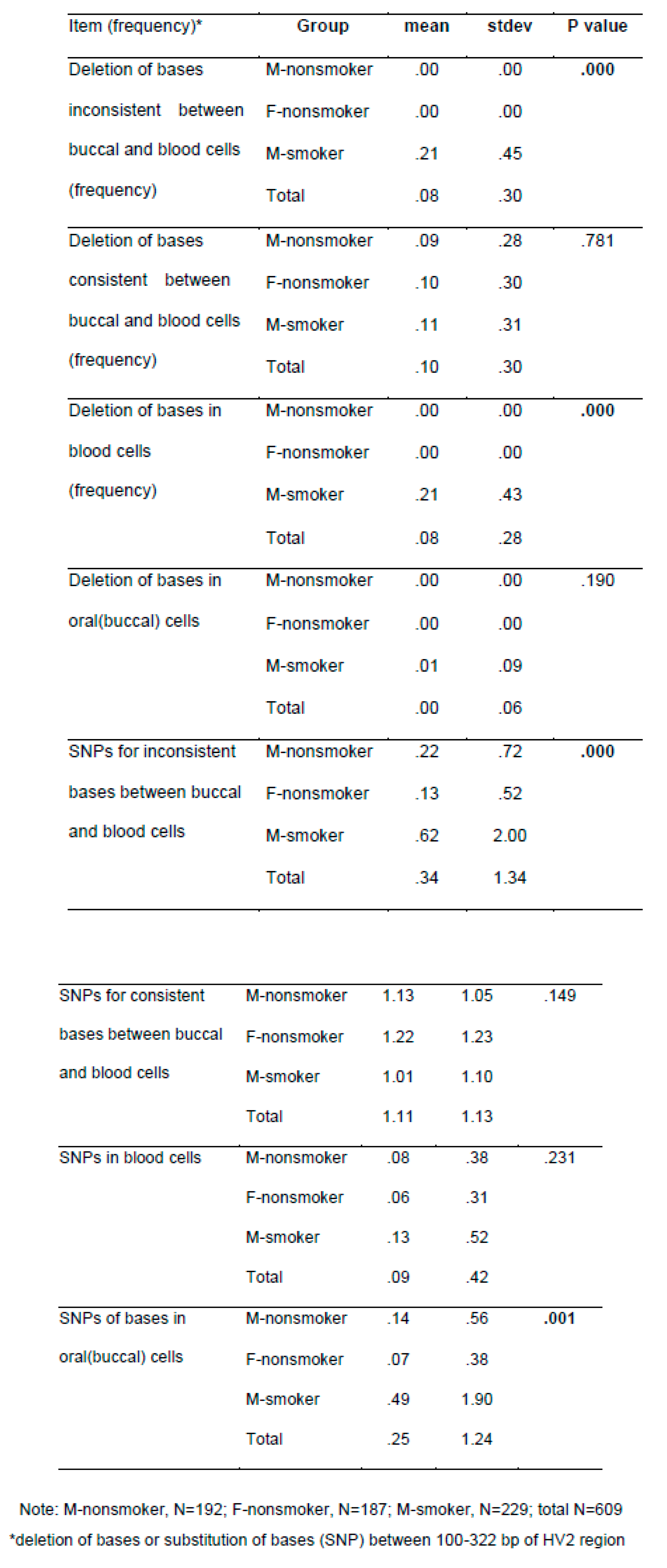
As shown in Table 3, we found smokers had high frequency in half of studied mt DNA alteration indicators, compared to nonsmokers. In detail, 45 male smokers showed inconsistent deletion mutations in blood and oral cells, deletion mutations in blood. Two male smokers showed discordant gene deletion in both blood and oral cavity. None of the non-smokers showed discordant deletion mutations. Nucleotide substitution mutations that appeared discordantly in blood and oral cells were found in 9 %, 35 out of 388 non-smokers (11 in blood, 18 in oral cavity, 6 in both blood and oral), and in 17%, 39 out of 232 smokers (11 persons in blood, 21 persons in oral cells, 7 persons in both blood and oral cells). The frequency of the SNP was significantly higher in smokers than nonsmokers (p<0.01).
2.4. Genetic Polymorphisms Affecting Exposure Biomarkers
We could diagnose the 15-86% of the gDNA samples for the 96 SNPs and found genetic polymorphisms affecting exposure biomarkers (Supplement Table S1–3 ). Six, seven, and ten genotypes among the 96 SNPs were associated with the levels of urinary cotinine, TTMA and MDA, respectively. The polymorphisms of some genes, such as SUL1A1 (rs9282861), ADH1B1 (rs1229984), and DRD2 (rs1800497), were even related to two different exposure biomarkers, i.e., cotinine and TTMA. TTMA and MD or Cotinine and MDA. Using these results, we performed pathway analyses to assess toxic mechanisms of tobacco smoking. As results, we can infer a neurobehavioral (addiction) mechanism from the interaction between SLC6A3 (dopamine transporter) and DRD2 (dopamine receptor D2) (Figure 3A). In addition, a group of metabolic enzymes for TTMA-associated polymorphisms on ADH1B, GSTM1, CYP2E1 and COMT can be involved in metabolic pathway for benzene (Figure 3B). Finally, three genes, MGMT, XRCC, and TP53, were associated with MDA to indicate the potential role of oxidative stress or aging in carcinogenesis.
3. Discussion
A molecular epidemiological approach with in-depth biological monitoring can provide reliable mechanisms and health end points of tobacco smoking as an EDC, including high susceptible population for precision prevention. For example of biomonitoring, tobacco smoking and the levels of cotinine showed association with a pronounced (~50%) reduction in fecundability, resulting in a longer time-to-pregnancy [20]Because their developmental processes amplify or magnify the toxic responses via epigenetic or cell memory [21]. From the self-reported cigarette pack-year, the present adolescents consumed over 1/6 volume of that of adults. However, urinary levels of cotinine in youth were 1/3 of adults and urinary MDA or TTMA of youth were similar to the adults. Although cotinine and TTMA are metabolites of tobacco products, MDA is an exposure and response biomarker for tobacco smoking via oxidative stress from tobacco chemicals or their combustion. If we assume that youth’s answers for tobacco consumption were not faked, youth can be highly susceptible to the bio-produced tobacco metabolites with small amounts of tobacco, compared to the adults. Around the onset of puberty, the activities of most of metabolic enzymes begin a gradual decline that continues throughout adolescence and concludes with attainment of adult capacity at the completion of pubertal development [22]. In addition, a current human liver study showed that the activities of CYP2A6 and CYP2E1, major metabolic enzymes of nicotine and benzene, were somewhat lower in adults (≤69 years) than young people (21-45 years) [23]. Thus, the same exposure to tobacco smoking can result in high levels of metabolites of tobacco components among adolescents than adults and they can be more bioactive or toxic than the parent chemicals in tobacco.
Particularly, oxidative stress and aging can be a main mechanism and a health endpoint of tobacco smoking, respectively [24]. Chronic inhalation of cigarette smoke is a prominent cause of chronic obstructive pulmonary disease (COPD) and provides an important source of exogenous oxidants [25]. In the present study, we found urinary MDA, a biomarker for oxidative stress or reactive oxygen species (ROS), was associated with tobacco smoking as urinary cotinine (Table 2; Figure 1). In addition, the growth rate of youth was negatively related to urinary MDA levels (Figure 2). To confirm the quality of the self-report, we recalled the questionnaire and confirmed the homogeneity of the answer for the growth rate with height and body weight. Thus, the present biological monitoring suggests tobacco reduces growth rate via oxidative stress in youth. As excessive ROS might react with nucleic acid, lipids, carbohydrates, and protein causing inflammation and oxidative stress that are the main causes for the development of various metabolic disorders [5]. In addition, the exposure to ROS from tobacco smoking can cause a higher rate of mutations in the mitochondrial genome that accumulate over time and reduce the efficiency of mtDNA repair systems [26,27]. The present study also shows that youth smokers, who reached to approx. 50 % of the smokers, showed high frequency of ‘SNPs for inconsistent bases between buccal and blood cells (Table 3). Thus, high susceptibility in youth to tobacco smoking was confirmed with mtDNA alteration and MDA-related growth relay.
Finally, the pathway analyses of exposure-related gene–gene interaction suggest that MDA-related genes, MGMT, XRCC, and TP53, were known to interact for carcinogenesis, a degenerative disease (Figure 3C). EDCs including tobacco smoking can increase an overall risk of ovarian aging, leading to the diminish of ovarian reserve, decline of fertility or fecundity, irregularity of the menstrual cycle and an earlier age at menopause, and/or premature ovarian insufficiency/failure in epidemiological studies [28,29]. We previously found tobacco smoking up-regulated aging genes, such as DEFA4 for hearing loss in adults [15]. In addition, others reported that tobacco smoking advanced brain aging in middle age women [30]. Moreover, tobacco smoking-related oxidative stress has been emphasized in the appearance of the clinical manifestation of skin aging [30,31]. Hexane-soluble tobacco smoke extract may induce matrix metalloproteinase-1 expression in human skin fibroblasts through the activation of the aryl hydrocarbon receptor pathway, which is pathogenetically involved in extrinsic skin aging [32]. Thus, the present susceptibility/genetic biomarkers support that oxidative stress and aging are ones of the mechanisms or health end points of tobacco smoking as an EDC.
Various kinds of tobacco attract youth and it can be a potential risk for their various future diseases [18,33] and as a reaction to that, we also consider effective tobacco control, such as raising the minimum legal age for access to tobacco [34]. The present study also can be a fresh approach to provide scientific evidences to protect adolescents from tobacco.
In conclusion, the present biological monitoring provides high susceptible population, such as adolescents rather than adults, and reliable genetic factors affecting tobacco exposure. The inferred environment-gene-gene interaction suggests accelerating aging in adolescents by tobacco smoking as an EDC.
4. Materials and Methods
4.1. Subjects and Sampling
We recruited adult volunteers, who visited Eulji University Hospital in Daejeon, South Korea for regular examination, and adolescents from high schools around Geumsan-gun near Daejeon. All subjects provided written informed consents and completed extensive questionnaires including medical and smoking history, dietary patterns, alcohol drinking, environment of residency, and smoking behaviors, such as smoking cessation, cigarettes smoker per day (CPD), duration of smoking, and smoking initiation. In addition, the Fagerstrom test of nicotine dependence (FTND) was used to assess nicotine dependence [35]. None of these subjects had any history of pulmonary, cardiovascular, endocrine, or gastrointestinal disorders. In addition, we measured carbon monoxide (CO) during exhalation with Micro CO Monitor (On-site Lab, Seoul, Korea).
Peripheral blood samples (10 ml) were collected into evacuated tubes containing sodium heparin as an anticoagulant (BD Vacutainer, Franklin Lakes, NJ, USA). In addition, spot urine specimens, the first voids of urine (40 ml) before breakfast, were collected into 50 ml of conical tubes. Both urine and blood samples were stored at -20℃ until analyses. All study protocols for this study were approved by the Institutional Review Board of Eulji University Hospital (Daejeon, Korea).
To compare mtDNA alteration in buccal cells to that in blood cells, we also collected buccal cells from the subjects with sterile cotton swabs, following our previous method [15].
4.2. Analyses of Urinary Cotinine
We analyzed urinary cotinine by our previous ion-pair HPLC/UVD method [15] with minor modifications. In brief, 900 μl of each urine sample was mixed with 100 μl of
80 μM 2-phenylimidazole as an internal standard and 330 μl of 3 M NaOH. The mixture was twice extracted with 3 ml of CH2Cl2 each time. After evaporating CH2Cl2 -extract, we dissolved the residue in 1 ml of water and injected 20 μl of its supernatant fraction to HPLC. The HPLC system consisted of dual Younglin SP930D pumps (Younglin, Seoul, Korea), an MIDAS COOL autosampler (Spark Holland, Emme, The Netherlands), an SPD-10A UV-VIS detector (Shimadzu, Kyoto, Japan), and a TSK gel ODS-80™ column (5 μm, 4.6 mm × 150 mm, Toyo Soda Co., Tokyo, Japan). Analyses were carried out with the following gradient mode: mobile phase A, a mixture of acetonitrile/water (15/85) containing 20 mM KH2PO4 and 3 mM sodium 1-octanesulfonate (pH 4.5); B, methanol; Flow rate, 0.7 ml/min; 0–20 min, ratio of A to B = 100:0; 20–25 min, ratio of A to B = 100:0 to 50:50; 25–30 min, ratio of A to B = 50:50; 30–35 min, ratio of A to B = 50:50 to 100:0; and 35-45 min, ratio of A to B = 100:0. The column was kept at 50 ℃ and the absorbance was observed at wavelength of 254 nm.
4.3. Analyses of Urinary MDA
We quantified urinary MDA as adducts of 2-thiobarbituric acid (TBA, CAS number: 504-17-6) with HPLC/UVD [36]. TBA-MDA adducts were detected at 532 nm with isocratic mode. The mobile phase was a mixture of 50 mM potassium phosphate buffer (pH 6.8) and methanol (58:42, v/v). Flow rate was set at 0.6 ml/min.
4.4. Analyses of Urinary TTMA
We analyzed urinary TTMA with UPLC-MS/MS, using the method previously described by Gagne et al. [37] with a minor modification. Briefly, TTMA standard and deuterated internal standard, d4-TTMA, were obtained from Sigma-Aldrich and CDN Isotopes Inc. (Pointe-Claire, Quebec, Canada), respectively. Standard TTMA solutions (0.025-2.5 ng/ml) were prepared in 50% methanol. In short, 950 μl of 0.1 % formic acid containing 2.38 μg/ml of d4- TTMA was mixed with 50 μl of TTMA standards or urine samples. After centrifugation at 13,000 rpm for 10 min, 5 μl of each supernatant was analyzed with UPLC-MS/MS. The UPLC-MS/MS system consisted of a Waters Acquity UPLC coupled with a Waters Xevo TQ triple quadrupole mass spectrometer (Beverly, MA), and an Acquity UPLC BEH C18 (1.7 μm, 2.1 mm × 50 mm, Waters). Mobile phases were composed of 0.1% formic acid in methanol (eluant A) and in water (eluant B). UPLC separation was achieved with a gradient from 10 to 95 % of eluant A for 1.25 min. Eluant A composition was then held constant for 0.5 min followed by a 0.5 min equilibration period at 10 % of eluant A. The flow rate was set at 0.5 mL/min and the column temperature was kept at 50ºC. The Xevo TQ was operated in negative mode. The capillary voltage was set at 2.8 kV. The source temperature was at 150 ºC. The desolvation temperature was at 500 ºC. Desolvation flow rate was at 900 L/hr and collision gas flow rate was at 0.15 mL/min. Data were acquired in multiple reaction monitoring (MRM) mode.
Urinary cotinine, MDA, and TTMA were adjusted for creatinine, measured with ion-pair HPLC/UVD method [15].
4.5. Targeted Genotyping
Genomic DNA of peripheral blood was isolated with a QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. We measured the purity and concentration of the isolated genomic DNA (gDNA) using a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). For the genotyping, we used gDNA samples with a 260:280 ratio of 1.5 or higher and concentrations of 60 ng/µl or higher.
We selected the 96 target SNPs, based on the SNPs of tobacco smoking-responsive genes, of which expression levels were altered by tobacco smoking in our previous microarray study [15], i.e., ACTG1, DEFA4, VAV3, FCGR3A, etc., as well as the SNPs related to metabolism, e.g., CYP2A6, CYP1B1, CYP2E1, and NQO1, addiction [38], e.g., CHRNA5/A3/B4, 5-HTTLPR, and DRD2 [39], risks for lung cancer, e.g., CYP1A1, GSTP1, and MPO1 [39,40], DNA repair, e.g., ERCC1, MGMT, XRCC1, etc. [41], and epigenetic modulation, e.g., HDAC1 and MTHFR [33], of tobacco smoking (supplement Table 1). For genotyping assays, the 96.96 Dynamic Array™ integrated fluidic circuits (Fluidigm Corp., South San Francisco, CA) was used. Prior to genotyping, specific target amplification (STA) was performed for each gDNA to enrich targeted SNP sequences. Briefly, 70 ng of gDNA was mixed with 50 nM of STA primer mixture, 50 nM of locus specific primer mixture, and 2.5 µL of Qiagen 2X Multiplex PCR Master Mix (Qiagen) in a final volume of 5µL. PCR reactions were performed on an Arktik Thermal Cycler (Thermo Scientific, Rockford, IL) with the following cycling conditions: 10 min at 95°C, followed by 14 cycles of a 2-step amplification profile of 15 sec at 95°C and 4 min at 60°C. STA products were 100-fold diluted in DNA suspension buffer and 2.5 µL of each product was combined with 3.0 µL of 2X Maxima® Probe/ROX qPCR Master Mix (Fermentas, St. Leon-Rot, Germany), 0.3 µL of SNPtype 20 X Sample Loading Reagent (Fluidigm), 0.1 µL of SNPtype Reagent (Fluidigm), and 0.1 µL of nuclease-free water. In parallel, 1 µL of each SNPtype assay was mixed with 2.5 µL of 2 X Assay Loading Reagent (Fluidigm) and 1.5 µL of nuclease-free water and loaded into the Fluidigm 96.96 Dynamic Genotyping Arrays. PCR and image processing were carried out on an EP1 system (Fluidigm). We analyzed data with an automated genotype calling algorithm using Fluidigm SNP Genotyping Software (v3.1.1).
4.6. Analyses of mtDNA Alteration between Blood and Buccal Cells
After extraction of DNAs from buccal cells and blood with DNeasy blood & tissue kit (Qiagen), we prepared a 96 well plate (Bioline, London, UK) to load 50 ul of DNA samples (5 ng/µl =250 ng). From the master plates, 5ng of DNA was used for a conventional PCR to amplify HV2 region. Total reaction volume was 20 µl, containing 5ng DNA and 75 µM of forward and backward primers, F015 5’-CAC CCT ATT AAC CAC TCA CG-3’ and R569 5’-GGT GTC TTT GGG GTT TGG TTG-3’, respectively. PCR conditions for amplification were performed on Primus 96 plus (MWG_Biotech, Huntsville, AL): initial denaturation at 96°C for 5 min; 35 cycles of denaturation at 96°C for 30s; annealing at 56°C for 30 s; elongation at 72°C for 1 min, with MyGenieTM 96 Gradient Thermal Block (Bioneer, Daejeon, Korea). For sequencing target mtDNA, we used BigDye Terminator v3.1 sequencing kit and ABI3730XL (Applied Biosystems, Waltham, MA ).
For data analyses, we used DNASTAR Lasergene SeqMan Pro version 7.1.0. Among the reference sequence of the mtDNA HV2 region (015-560 bp), NCBI (ref|NC_012920.1) corresponding location from100 bp to 322 bp of mtDNA HV2 region was analyzed for detecting discrepancies between blood and oral DNA on (a/g or t/c) polymorphism at the position 263 bp and polyC region (303~314 in reference C7(T)C5).
4.7. Statistical Analyses
We removed imputed SNPs with < 0.05 genotype information content, low call rates with < 0.90, and minor allele frequency (MAF) with < 0.05. The Shapiro-Wilk W test was used to test distributional normality for levels of exposure biomarkers (i.e., urinary cotinine, TTMA and MDA). ANOVA or Wilcoxon rank sum test was used to analyze differences in characteristics among never, ex-, and current smokers. Associations between SNPs and levels of exposure biomarkers were investigated with Wilcoxon Mann-Whitney Test or multi linear regression models after adjusting for age, body mass index (BMI), and alcohol consumption under a dominant model. All statistical analyses were conducted using JMP package v. 4.0.2 (SAS Institute, Cary, NC)
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This study was supported by a grant (13182MFSD765) from the Ministry of Food and Drug Safety, Korea.
Institutional Review Board Statement
All study protocols for this study were approved by the Institutional Review Board of Eulji University Hospital (Daejeon, Korea).
Acknowledgments
We deeply appreciate Sul-Kil R. Koh in Goodbeing Center, Co., Ltd., Seoul, Korea, for reviewing English.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Papalou, O.; Kandaraki, E.A.; Papadakis, G.; Diamanti-Kandarakis, E. Endocrine Disrupting Chemicals: An Occult Mediator of Metabolic Disease. Frontiers in Endocrinology 2019, 10, 112. [CrossRef]
- Ivarsson, J.; Ferrara, F.; Vallese, A.; Guiotto, A.; Colella, S.; Pecorelli, A.; Valacchi, G. Comparison of Pollutant Effects on Cutaneous Inflammasomes Activation. International Journal of Molecular Sciences 2023, 24, 16674. [CrossRef]
- Son, E.; Kwon, K.H. How Body Burden from Exposure to Endocrine Disruptors Effects Accelerated Aging? Environmental Research and Technology 2023, 6, 383-390.
- Zhou, G.; Xiao, W.; Xu, C.; Hu, Y.; Wu, X.; Huang, F.; Lu, X.; Shi, C.; Wu, X. Chemical Constituents of Tobacco Smoke Induce the Production of Interleukin-8 in Human Bronchial Epithelium, 16HBE Cells. Tobacco induced diseases 2016, 14, 1-9. [CrossRef]
- Jabeen, K.; Akash, M.S.H.; Haider, K.; Faheem, A.; Tariq, M.; Rehman, K. Tobacco Smoking as an EDC in Metabolic Disorders. Endocrine Disrupting Chemicals-induced Metabolic Disorders and Treatment Strategies 2021, 343-355.
- Vogeley, C.; Esser, C.; Tüting, T.; Krutmann, J.; Haarmann-Stemmann, T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. International Journal of Molecular Sciences 2019, 20, 6005. [CrossRef]
- Ratajczak, A.; Jankowski, P.; Strus, P.; Feleszko, W. Heat Not Burn Tobacco Product—a New Global Trend: Impact of Heat-Not-Burn Tobacco Products on Public Health, a Systematic Review. International journal of environmental research and public health 2020, 17, 409. [CrossRef]
- Scherer, G.; Scherer, M.; Rögner, N.; Pluym, N. Assessment of the Exposure to Polycyclic Aromatic Hydrocarbons in Users of various Tobacco/Nicotine Products by Suitable Urinary Biomarkers. Arch. Toxicol. 2022, 96, 3113-3126. [CrossRef]
- Zhang, Y.; Dong, S.; Wang, H.; Tao, S.; Kiyama, R. Biological Impact of Environmental Polycyclic Aromatic Hydrocarbons (ePAHs) as Endocrine Disruptors. Environmental pollution 2016, 213, 809-824. [CrossRef]
- Mitchell, C.; Schneper, L.M.; Notterman, D.A. DNA Methylation, Early Life Environment, and Health Outcomes. Pediatr. Res. 2016, 79, 212-219.
- Justice, A.E.; Winkler, T.W.; Feitosa, M.F.; Graff, M.; Fisher, V.A.; Young, K.; Barata, L.; Deng, X.; Czajkowski, J.; Hadley, D. Genome-Wide Meta-Analysis of 241,258 Adults Accounting for Smoking Behaviour Identifies Novel Loci for Obesity Traits. Nature communications 2017, 8, 14977. [CrossRef]
- Minicã, C.C.; Mbarek, H.; Pool, R.; Dolan, C.V.; Boomsma, D.I.; Vink, J.M. Pathways to Smoking Behaviours: Biological Insights from the Tobacco and Genetics Consortium Meta-Analysis. Mol. Psychiatry 2017, 22, 82-88. [CrossRef]
- Brunzell, D.H.; Stafford, A.M.; Dixon, C.I. Nicotinic Receptor Contributions to Smoking: Insights from Human Studies and Animal Models. Current addiction reports 2015, 2, 33-46. [CrossRef]
- Murphy, S.E. Nicotine Metabolism and Smoking: Ethnic Differences in the Role of P450 2A6. Chem. Res. Toxicol. 2017, 30, 410-419. [CrossRef]
- Na, H.; Kim, M.; Chang, S.; Kim, S.; Park, J.Y.; Chung, M.W.; Yang, M. Tobacco Smoking-Response Genes in Blood and Buccal Cells. Toxicol. Lett. 2015, 232, 429-437. [CrossRef]
- Haines, D.A.; Saravanabhavan, G.; Werry, K.; Khoury, C. An Overview of Human Biomonitoring of Environmental Chemicals in the Canadian Health Measures Survey: 2007–2019. Int. J. Hyg. Environ. Health 2017, 220, 13-28. [CrossRef]
- Chang, C.M.; Edwards, S.H.; Arab, A.; Del Valle-Pinero, A.Y.; Yang, L.; Hatsukami, D.K. Biomarkers of Tobacco Exposure: Summary of an FDA-Sponsored Public Workshop. Cancer Epidemiology, Biomarkers & Prevention 2017, 26, 291-302. [CrossRef]
- Kim, S.; Jo, K. Multiple Tobacco Product use among Adolescents with Asthma in Korea. International Journal of Environmental Research and Public Health 2022, 19, 9633.
- Korean Statistical Information Service, https://kosis.kr/search.
- Pollack, A.Z.; Krall, J.R.; Swan, S.H.; Louis, G.M.B. Does Older Age Modify Associations between Endocrine Disrupting Chemicals and Fecundability? International Journal of Environmental Research and Public Health 2022, 19, 8074.
- Resendiz M, Watkins DS, Öztürk NC, Zhou FC. Environmental Influence on Epigenetics in Handbook of Epigenetics, 2023; pp. 639-668.
- Kennedy, M.J. Hormonal Regulation of Hepatic Drug-metabolizing Enzyme Activity during Adolescence. Clinical Pharmacology & Therapeutics 2008, 84, 662-673. [CrossRef]
- Corton, J.C.; Lee, J.S.; Liu, J.; Ren, H.; Vallanat, B.; DeVito, M. Determinants of Gene Expression in the Human Liver: Impact of Aging and Sex on Xenobiotic Metabolism. Exp. Gerontol. 2022, 169, 111976. [CrossRef]
- Bono, R.; Squillacioti, G.; Ghelli, F.; Panizzolo, M.; Comoretto, R.I.; Dalmasso, P.; Bellisario, V. Oxidative Stress Trajectories during Lifespan: The Possible Mediation Role of Hormones in Redox Imbalance and Aging. Sustainability 2023, 15, 1814. [CrossRef]
- Nucera, F.; Mumby, S.; Paudel, K.R.; Dharwal, V.; Di Stefano, A.; Casolaro, V.; Hansbro, P.M.; Adcock, I.M.; Caramori, G. Role of Oxidative Stress in the Pathogenesis of COPD. Minerva Med. 2022. [CrossRef]
- Fetterman, J.L.; Sammy, M.J.; Ballinger, S.W. Mitochondrial Toxicity of Tobacco Smoke and Air Pollution. Toxicology 2017, 391, 18-33. [CrossRef]
- Pirini, F.; Guida, E.; Lawson, F.; Mancinelli, A.; Guerrero-Preston, R. Nuclear and Mitochondrial DNA Alterations in Newborns with Prenatal Exposure to Cigarette Smoke. International journal of environmental research and public health 2015, 12, 1135-1155. [CrossRef]
- Ding, T.; Yan, W.; Zhou, T.; Shen, W.; Wang, T.; Li, M.; Zhou, S.; Wu, M.; Dai, J.; Huang, K. Endocrine Disrupting Chemicals Impact on Ovarian Aging: Evidence from Epidemiological and Experimental Evidence. Environmental Pollution 2022, 305, 119269. [CrossRef]
- Oladipupo, I.; Ali, T.; Hein, D.W.; Pagidas, K.; Bohler, H.; Doll, M.A.; Mann, M.L.; Gentry, A.; Chiang, J.L.; Pierson, R.C. Association between Cigarette Smoking and Ovarian Reserve among Women Seeking Fertility Care. Plos one 2022, 17, e0278998. [CrossRef]
- Ning, K.; Zhao, L.; Matloff, W.; Sun, F.; Toga, A.W. Association of Relative Brain Age with Tobacco Smoking, Alcohol Consumption, and Genetic Variants. Scientific reports 2020, 10, 10. [CrossRef]
- Papaccio, F.; D′ Arino, A.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants 2022, 11, 1121.
- Morita, A.; Torii, K.; Maeda, A.; Yamaguchi, Y. Molecular Basis of Tobacco Smoke-Induced Premature Skin Aging. In Journal of Investigative Dermatology Symposium Proceedings; pp. 53-55.
- Kaur, G.; Begum, R.; Thota, S.; Batra, S. A Systematic Review of Smoking-Related Epigenetic Alterations. Arch. Toxicol. 2019, 93, 2715-2740. [CrossRef]
- Oyston, J. A Fresh Approach to Tobacco Control: Raising the Minimum Legal Age for Access. CMAJ 2017, 189, E293-E294. [CrossRef]
- Do, H.P.; Nguyen, L.H.; Nguyen, N.P.T.; Ngo, C.; Nguyen, H.L.T.; Le, G.T.; Nguyen, L.K.; Nguyen, C.T.; Tran, B.X.; Le, H.T. Factors Associated with Nicotine Dependence during Methadone Maintenance Treatment: Findings from a Multisite Survey in Vietnam. BMJ open 2017, 7, e015889. [CrossRef]
- Lee, H.; Isse, T.; Kawamoto, T.; Baik, H.W.; Park, J.Y.; Yang, M. Effect of Korean Pear (Pyruspyrifolia Cv. Shingo) Juice on Hangover Severity Following Alcohol Consumption. Food and chemical toxicology 2013, 58, 101-106. [CrossRef]
- Gagné, S. Determination of Trans, Trans-muconic Acid in Workers' Urine through Ultra-performance Liquid Chromatography Coupled to Tandem Mass Spectrometry. Biomedical Chromatography 2013, 27, 664-668. [CrossRef]
- Hasan, F.; Yadav, V.; Katiyar, T.; Yadav, S.; Pandey, R.; Mehrotra, D.; Hadi, R.; Singh, S.; Bhatt, M.L.; Parmar, D. Validation of Gene Expression Profiles of Candidate Genes using Low Density Array in Peripheral Blood of Tobacco Consuming Head and Neck Cancer Patients and Auto/Taxi Drivers with Preneoplastic Lesions. Genomics 2020, 112, 513-519. [CrossRef]
- Del Casale, A.; Paolini, M.; Gentile, G.; Borro, M.; Zocchi, C.; Fiaschè, F.; Padovano, A.; Zoppi, T.; Modesti, M.N.; De Luca, O. Dopamine DRD2 and DRD3 Polymorphisms Involvement in Nicotine Dependence in Patients with Treatment-Resistant Mental Disorders. Journal of Personalized Medicine 2022, 12, 565. [CrossRef]
- Yang, M.; Choi, Y.; Hwangbo, B.; Lee, J.S. Combined Effects of Genetic Polymorphisms in Six Selected Genes on Lung Cancer Susceptibility. Lung Cancer 2007, 57, 135-142. [CrossRef]
- Alsagaby, S.; Ahmed, A.A.; Rasheed, Z.; Althwab, S.A.; Aljohani, A.S.; Alhumaydhi, F.A.; Alhomaidan, H.T.; Alkhamiss, A.S.; Alkhowailed, M.; Alaqeel, A. Association of Genetic Polymorphisms in DNA Repair Genes ERCC2 Asp312Asn (rs1799793), ERCC2 Lys 751 Gln (rs13181), XRCC1 Arg399 Gln (rs25487) and XRCC3 Thr 241Met (rs861539) with the Susceptibility of Lung Cancer in Saudi Population. Nucleosides Nucleotides Nucleic Acids 2022, 41, 530-554.
Figure 1.
Association between tobacco smoking and oxidative stress as urinary cotinine and MDA levels: A. without creation modification, p<0.01, slope (estimate) = 0.21, r2 = 0.01 by regression analysis; B. with creation modification, p < 0.01, slope (estimate) = 0.45, r2 = 0.12 by regression analysis.
Figure 1.
Association between tobacco smoking and oxidative stress as urinary cotinine and MDA levels: A. without creation modification, p<0.01, slope (estimate) = 0.21, r2 = 0.01 by regression analysis; B. with creation modification, p < 0.01, slope (estimate) = 0.45, r2 = 0.12 by regression analysis.
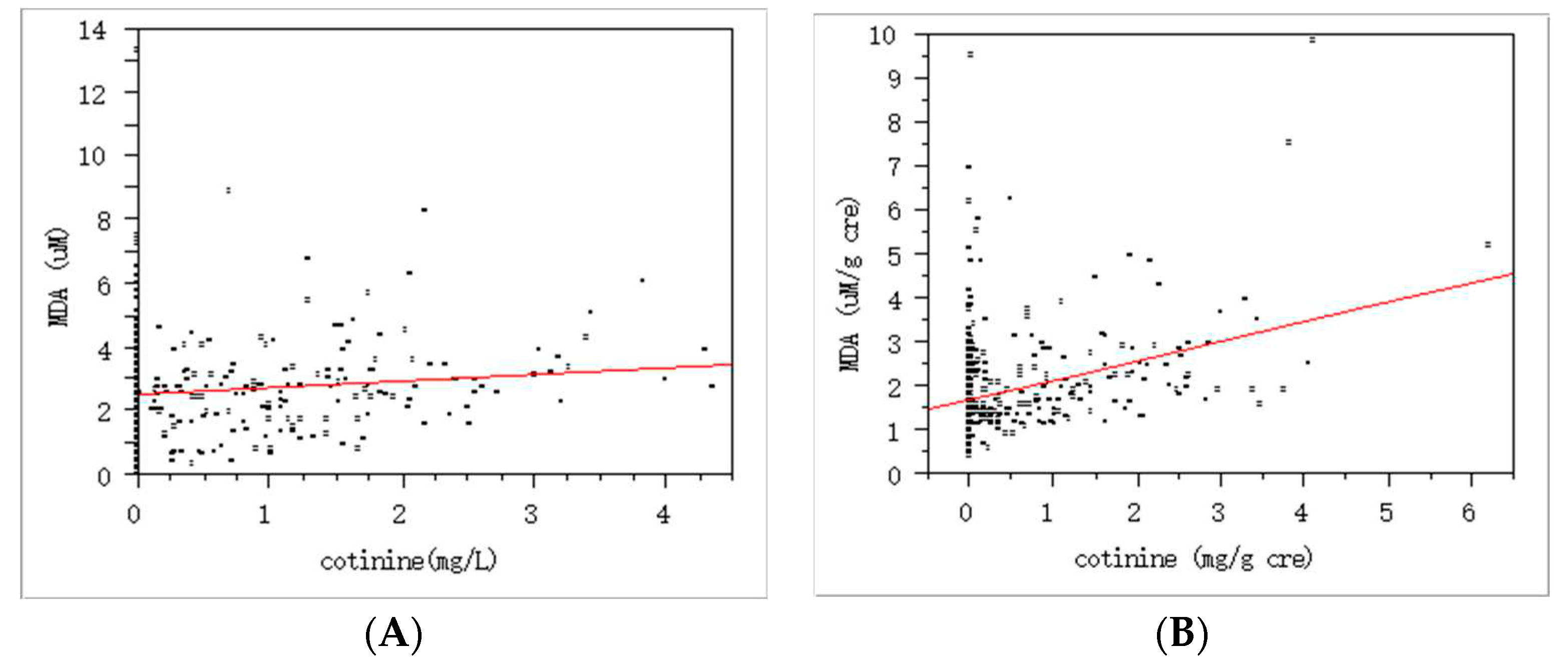
Figure 2.
Negative association between oxidative stress and growth rate in adolescents: P = 0.04, slope = -0.31, r2 = 0.18 by regression analysis (N = 129); self-reported growth rate, 0, very slow; 1, slow; 2, normal; 3, fast;, 4. very fast.
Figure 2.
Negative association between oxidative stress and growth rate in adolescents: P = 0.04, slope = -0.31, r2 = 0.18 by regression analysis (N = 129); self-reported growth rate, 0, very slow; 1, slow; 2, normal; 3, fast;, 4. very fast.
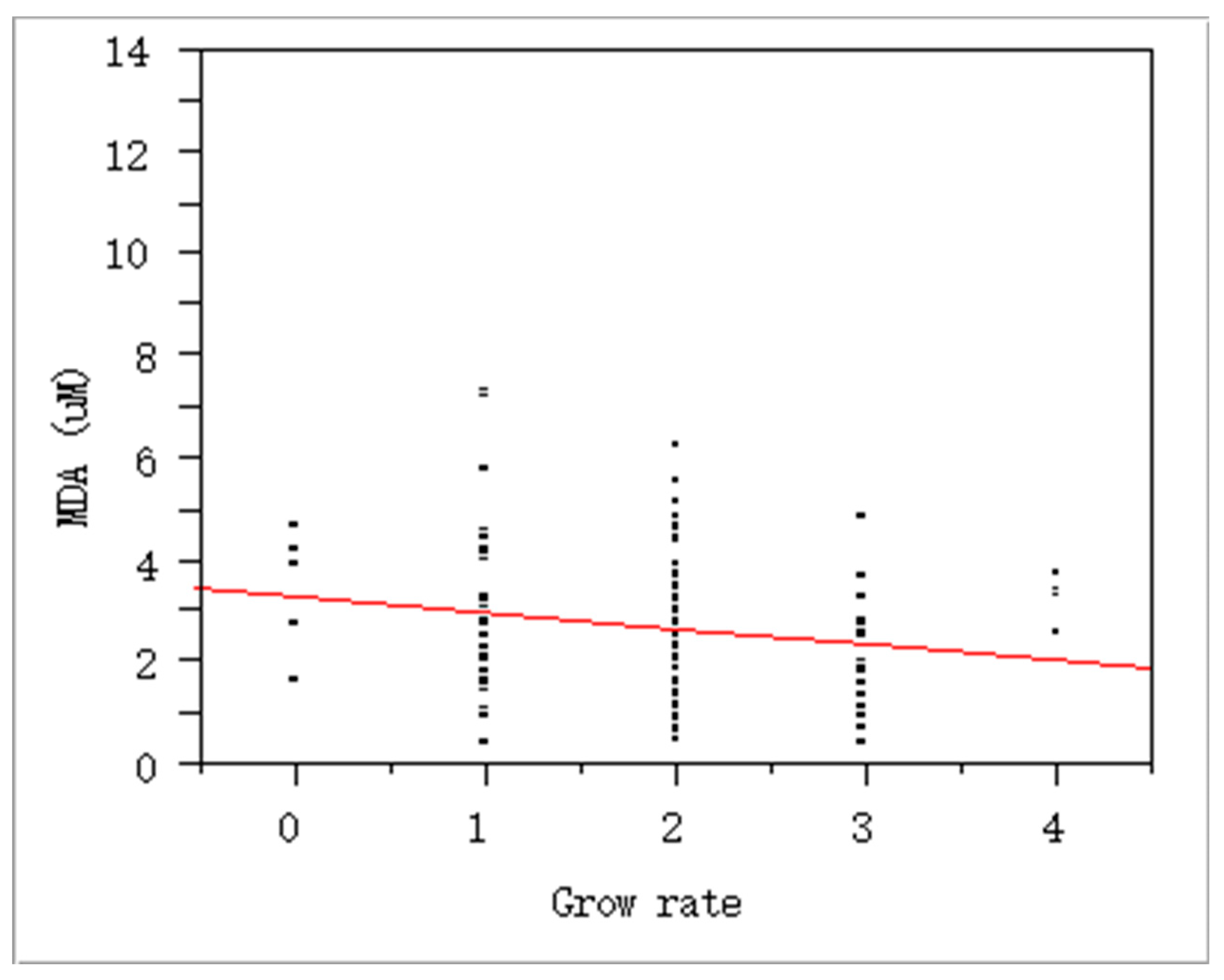
Figure 3.
Gene-gene interactions in cotinine (A), t,t-muconic acid (B) and C (MDA)-associated genetic biomarkers: A, Interactions between SLC6A3 (dopamine transporter) and DRD2 (dopamine receptor D2) to show neurobehavioral (addiction) effects of nicotine/cotinine; B. Interactions between ADH1B, GSTM1, CYP2E1 and COMT to be involved in metabolic pathway for benzene ; C. Interactions among MGMT, XRCC, and TP53 to indicate the potential role of oxidative stress in carcinogenesis (by STRING 9.05).
Figure 3.
Gene-gene interactions in cotinine (A), t,t-muconic acid (B) and C (MDA)-associated genetic biomarkers: A, Interactions between SLC6A3 (dopamine transporter) and DRD2 (dopamine receptor D2) to show neurobehavioral (addiction) effects of nicotine/cotinine; B. Interactions between ADH1B, GSTM1, CYP2E1 and COMT to be involved in metabolic pathway for benzene ; C. Interactions among MGMT, XRCC, and TP53 to indicate the potential role of oxidative stress in carcinogenesis (by STRING 9.05).
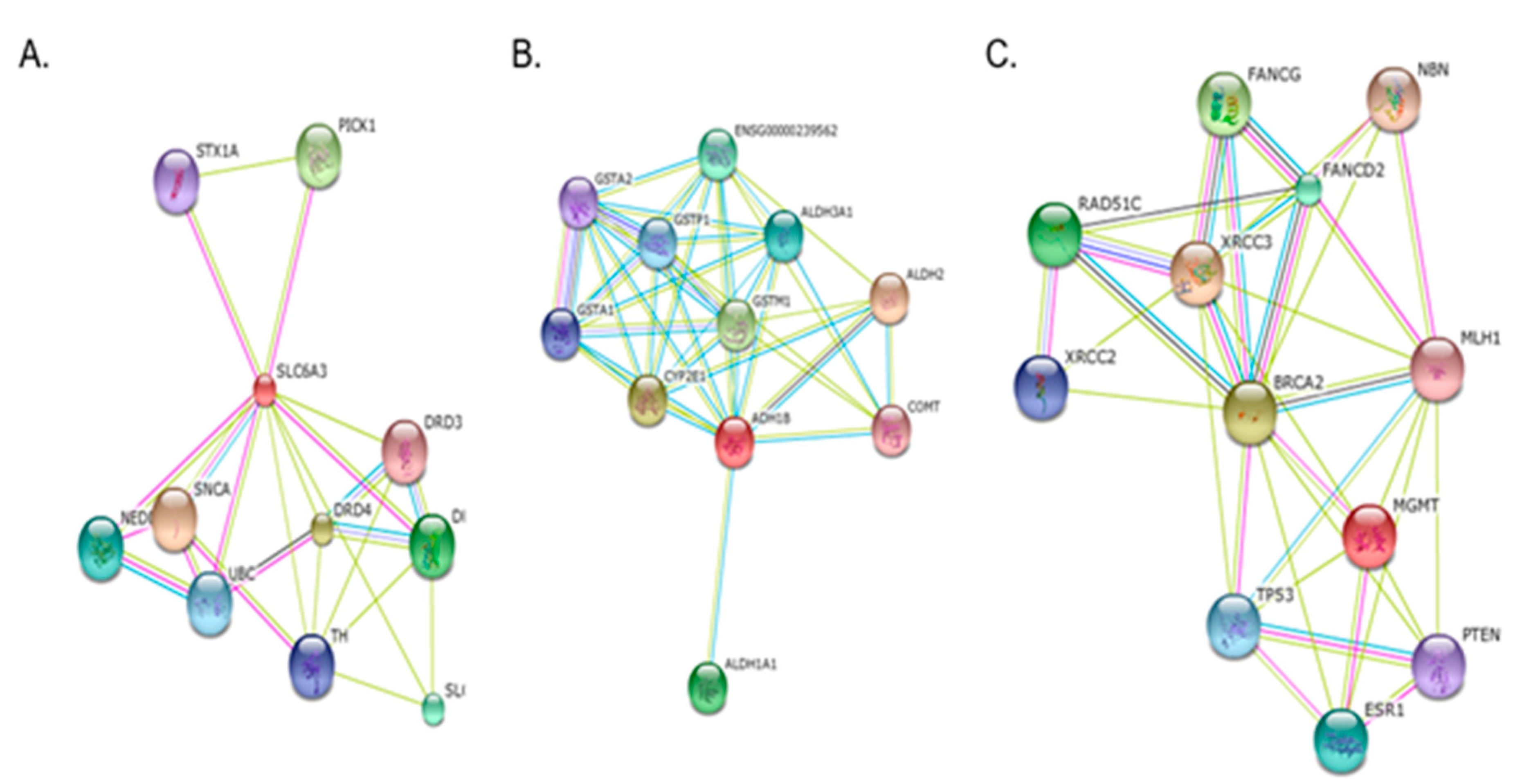

Table 1.
Composition of the subjects.
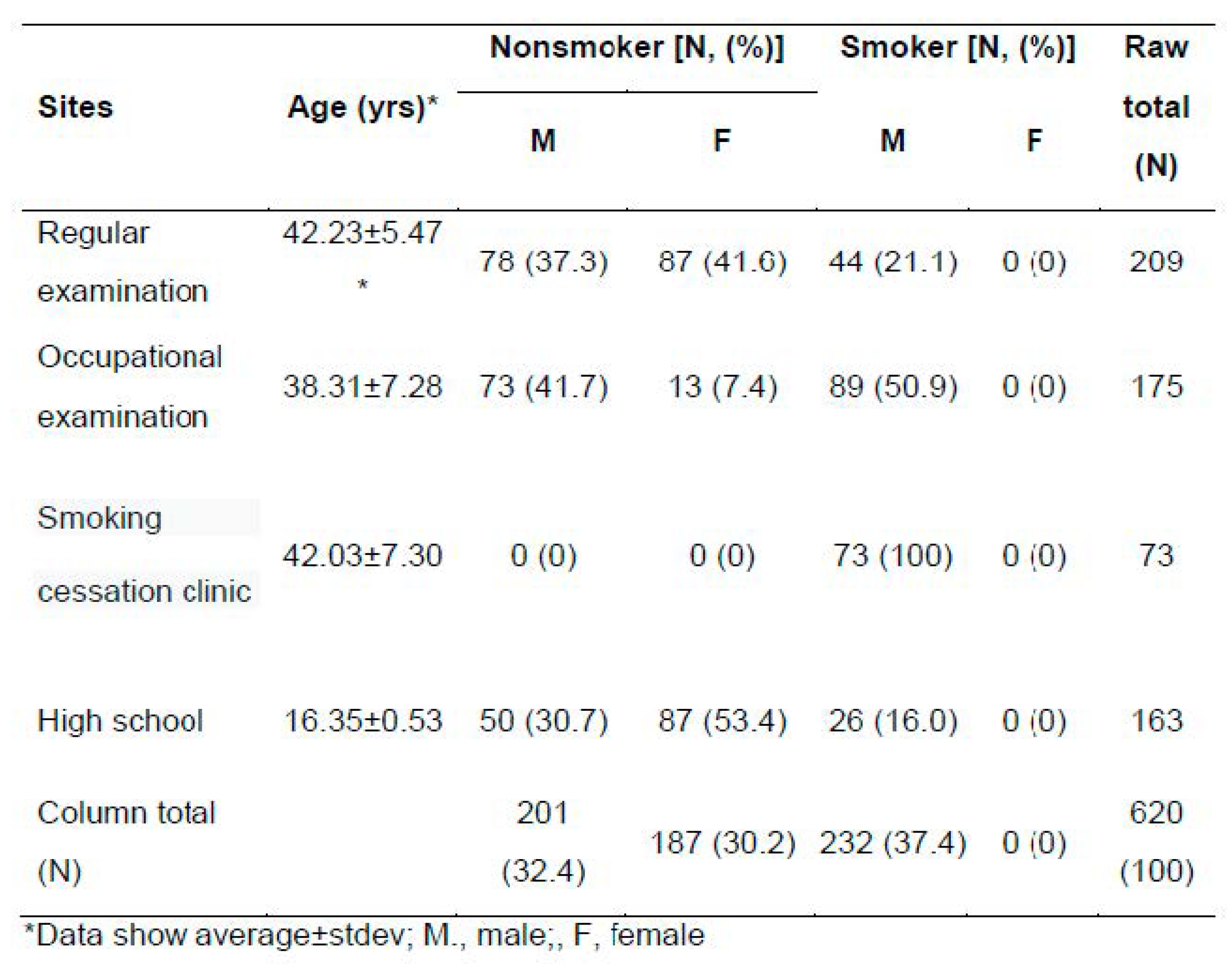 |
Table 2.
Exposure biomarker levels between somkers and nonsmokers.
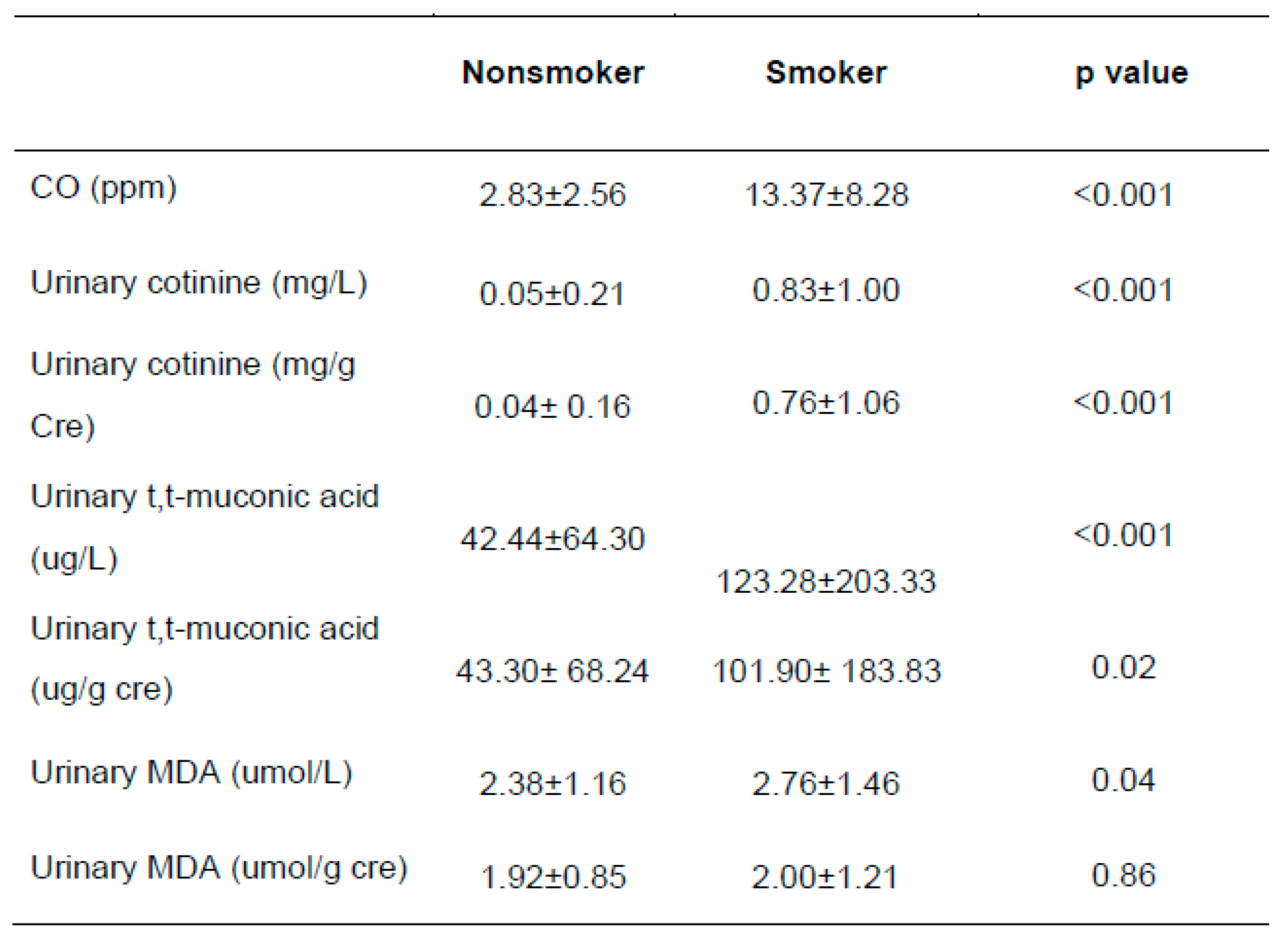 |
Table 3.
mt DNA alteration by smoking and gender.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



