Submitted:
29 December 2023
Posted:
04 January 2024
You are already at the latest version
Abstract
We present a precise and universal method for anticipating the thermodynamic characteristics of polyatomic molecules. Unlike conventional computational approaches that rely on numerous experimental spectroscopy or calorimetry data, our model only utilizes a small number of molecular constants. We accurately predicted the values of entropy, enthalpy, specific heat, and Gibbs free energy for boron trifluoride, which were in excellent accordance with the available experimental data. This investigation introduces a new approach to deal with the anharmonic vibrations of polyatomic molecules
Keywords:
Molar entropy
; Partition function
; polyatomic molecules
1. Introduction
Boron trifluoride (BF3) has been extensively utilized as a gas-filled neutron detector in place of 3He gas-filled neutron detectors, which has been unable to meet the demands for environmental security and scientific applications over the past century, specifically in the 1950s, 1960s, and 1970s. BF3 has been considered a more permanent solution to address this issue. In 1963, Fowler constructed the largest BF3 counters to date for a neutron cosmic ray observatory at Chalk River [1]. One of the main advantages of using BF3 gas in other fields is its ability to discriminate gamma-rays from neutrons, as gamma-ray noise produced in the proportional counters can affect the efficiency of the neutron detectors. Consequently, proportional counters filled with boron trifluoride gas are used as start-up detectors in the instrumentation of nuclear power reactors. Neutron spectroscopy, mixed waste monitoring, thermal neutron diffraction, and monitoring of nuclear reactors operating at low power are among the other powerful applications of neutron detectors with BF3. Despite the high cross-section for neutrons and the sensitivity to thermal neutrons of BF3 gas, it is highly toxic and corrosive. Thus, in the early 1980s, it became less popular, and 3He again became an affordable alternative. Unfortunately, 3He has become expensive, while the demand for it has increased in neutron scattering facilities. The severe shortage of 3He necessitates the re-evaluation of using BF3 as a replacement.
In the early years of this century, many researchers turned to calculate the efficiency of using BF3 counters [3-5] instead of 3He, and explored new ways to utilize BF3 gas in neutron detectors. The use of BF3 gas is highly attractive due to its easy handling procedures, particularly in terms of cost-effectiveness for multi-detector banks like cosmic neutron monitors (CNMs) and detector arrays for time-of-flight neutron scattering instruments [6]. This reconsideration of BF3 may also benefit other larger detection systems. In this study, we present efficient analytical and graphical representations of the thermodynamic properties of the BF3 polyatomic molecule.
Theoretical estimation of thermodynamic properties of molecules and atoms can be achieved by combining quantum and statistical mechanics. In the early 1930s, several researchers estimated the thermodynamic properties of organic compounds, including entropy, heat capacity, and heat content, using theoretical calculations [2,3]. At that time, Raman and infrared spectroscopy were used to estimate vibrational energy levels, which was a significant achievement in spectral analysis. Meanwhile, improvements were made in measuring thermodynamic properties, such as determining gaseous entropies, providing data that could be used to evaluate theoretical calculations successfully. The accurate determination of thermodynamic properties of polyatomic molecules is increasingly important in many fields, particularly in chemistry and physics. The partition function is the key concept behind theoretical calculations of thermodynamic properties, as it bridges quantum and statistical mechanics. It is the total sum over all bound levels of the Boltzmann factor (known as the sum-over-states), describing how atoms and molecules are distributed among various energy states at a specific temperature, where the system is in thermodynamic equilibrium.
Recently, significant progress has been made in calculating ro-vibrational states based on quantum theoretical principles of thermodynamics. However, these calculations are limited to lower degrees of freedom systems [4] and small molecules. To overcome this issue, effective Hamiltonians are fitted to experimental data to calculate the partition function and related thermodynamic data. However, the accuracy of the calculated results is lower for anharmonic systems [9] and at high temperatures [5,8].
Various methods have been proposed to study the thermodynamic properties of molecular systems by fitting experimental data using the partition function as a starting point. These approaches include analytical and direct summation methods, classical statistical mechanics methods, and Fourier path integral Monte Carlo methods [10]. Corrections such as semi-classical [12], semi-empirical [13], and quantum [11] are considered in these calculations.
Atomic and molecular partition function experimental data can be found in many several sources. For instance, Kurucz supplements Traving, Baschek, and Holweger (hereafter TBH) by providing the partition functions of neutral and ionized species of all the elements in the periodic table. However, it is a time consuming method in order to get elementary partition function data. Moreover, the evaluation of the partition using TBH requires to solve large number of exponentials and Redberg series partition functions to tabulate atomic and molecular data. In addition, due to the coarse spacing of the Kurucz tables, differentiations and interpolation are difficult to be done. Irwin [22] proposed a solution for these difficulties by using polynomials to approximate atomic and molecular partition functions. Another major sources of partition function data are Tatum [19], McBride et al (hereafter MHEG) [20], and JANAF thermodynamic tables (hereafter JANAF) [21] tabulate and calculate directly thermodynamic properties of the important molecules in astrophysics. Again, these molecular partition function tables need excessive computer storage. To overcome this nowadays, sufficient computational methods used to calculate thermodynamic properties of polyatomic molecules.
In this paper, we establish an available formulation for Boron trifluoride gas based on its molecular structure characteristics and its spectroscopic molecular constants. As such, the spectroscopic information used is of utmost importance in the developed representation to predict and calculate thermodynamic properties of BF3 such as entropies and Gibbs free energy. We compare the calulcated results with the NIST data in the temperature range 100 K and 6000 K. This study provides opportunities for better predictions of thermodynamic functions of polyatomic molecules.
2. Partition Function
The partition function serves as the link between thermodynamics and statistical mechanics, connecting these two fields through the framework of quantum mechanics. By utilizing the partition function, various thermodynamic functions like entropies, enthalpies, and heat capacities can be computed.
2.1. Overview of Methods Used to Calculate the Partition Function
This section presents a summary of various studies that use the partition function in different forms. It describes the simplifications and approximation methods employed, the molecular constants used, and the recommended temperature range. It is important to note that extrapolating data beyond the recommended range, despite its common occurrence, is not advisable. Our data and formulas, however, can be applied throughout the full temperature range of 100 K to 6000 K with minimal deviation.
In Tatum’s research [23], 14 diatomic molecules were examined, and their partition function was calculated using their dissociation equilibrium constants within a temperature range of 1000 K to 8000 K. His findings revealed that only four molecular constants were utilized to estimate the partition functions. Additionally, a cut-off was implemented at for the upper energy. These four molecular constants were (;; , ).
Other study done by Irwin [24] who used the partition function in the form of polynomials,
To calculate the partition function for numerous atomic and molecular species within the temperature range of 1000 K to 16 000 K, a least-squares fitting method was employed for each species to determine the coefficients in Equation (1). However, for other molecular data, extrapolation was used and their weight was reduced by a factor of 106 prior to fitting the coefficients. Although Irwin stated that these least-squares fits have small errors, using linear extrapolation for non-linear data generates significant errors, and the magnitude of these errors was not estimated. Bohn and Wolf [26] developed a new approximate formulation for the partition function and applied it to two diatomic molecules, H2 and CO, in the temperature range of T = 1000-6000 K. Using a limited number of molecular constants (, , , ) and accounting only for the electronic ground state, they computed specific heat capacity (cV) and internal energy (Eint) for these two molecules. Sauval and Tatum [28] evaluated the total internal partition functions for 300 diatomic molecules, 69 neutral atoms, and 19 positively charged ions. The approximated partition function polynomial for molecules and atoms is valid for a certain temperature range from 1000 K to 9000 K and has the following expression:
The molecular constants of all of these molecules, atoms, and ions are taken from [27].
In 1985, Rossi et al. [29] presented total molecular partition functions for 53 molecular species using the first few molecular constants (, , , ) which are taken from Ref. [27] in the temperature range of 1000 K to 5000 K. The authors used rigid-rotor approximation in the calculation of QJ while they claimed that they are not. However, they allow the interaction between vibration and rotation. They used polynomial approximations in order to find the “exact” specific heat which behave near the origin and more suitable to approximation schemes. Then, by integration they can obtain the partition function as follows:
where to are coefficients. Accordingly, the specific heat approximation is
Kurucz [30] commented that the approximate expressions for the partition functions used and reported in previous papers are not rigorous enough for H2 and CO. This is because they did not properly track the number of bound levels and did not include higher-order coefficients. Data for H2 were formulated and used from [31]. Kurucz summed over all the energies of the three lowest electronic states for both diatomic molecules and tabulated polynomial fits for the molecular partition functions from 1000 K to 9000 K with steps of 100 K.
Irwin [32] also studied the internal partition functions of H2 and CO in the same temperature range. He found that the errors for H2 at 4000 K are larger than for CO by approximately 2%. He used the electronic ground state of H2 and evaluated Y using the least-squares fitting method of Dunham series with the energy data from Ref. [31]. Irwin compared his evaluations with those of Kurucz [30] and Sauval [28] and found that his values of Q are higher, which he attributed to the number of electronic ground states included and the higher rotational level treatments used.
Pagano et al [33] derived internal partition functions and thermodynamic properties of Jupiter-atmosphere species in a wide range of temperatures from 50 to 50 000 K. The results are presumably self-consistent, with E(v; Jmax) for each v, in terms of maximum rotational states for each vibrational level.
In 2011, by using the methods published in 2003 and 2000 by Fischer [35] and Gamache [36] respectively, laraia et al. [34] presented molecular partition functions for certain molecules with their isotopes in the temperature range 70-3000 K. Vibrational partition functions were evaluated and determined using the approximation of the harmonic oscillator [39], however, rotational partition function were calculated as in Ref. [37,38]. In this study, all state-dependent and state-independent degeneracy factors were considered and taken into account. The molecular partition function of H2 was evaluated using direct summation and ab initio energies. In addition, for the temperature range with the intervals of 25 K, four-point Lagrange interpolation was used. Colonna et al. [40] described the statistical thermodynamics of H2 molecules in normal ortho/para mixture and determined its internal partition function on rigorous basis. Consequently, solving directly the existing ambiguity of those quantities in the definition related to the partition functions. Moreover, thermodynamic properties obtained from internal partition function for temperature range of 5 K to 10 000 K are found.
2.2. Model
The Born-Oppenheimer approximation posits that rotational, vibrational, and electronic energies of a molecule are independent of each other. As a result, the partition function of a molecule can be expressed using the standard formula of statistical mechanics by multiplying the translational, rotational, vibrational, and electronic contributions.
where , , and stand for translational, rotational, vibrational and electronic partition functions, respectively.
This total internal partition function can be written in this form by taking the assumption that the internal rotations within molecule are ignored. Moreover, Bohn and Wolf [26] illustrated that the electronic state for molecules is non-degenerate since the electronic state energy typically is higher than the dissociation energy. Therefore, .
The BF3 molecule is a molecule with trigonal planar geometry where Boron atom at the center and Fluorides are peripheral atoms at the corners, all in one plane. This molecular geometry molecule’s point group is D3h. BF3 has six possible symmetry types (six vibrations) and they are represented as follows:
B-F stretching vibrations are totally symmetric and cannot be combined with other molecular coordinates in or/and out of plane. is the only asymmetric vibration represented in . Consequently, the out-of-plane B-F bending vibrations belonging to this type which is clearly different and cannot be mixed with other coordinates. However, the vibration has two degeneracies which are in-plane bending B-F and asymmetric stretching vibrations with different energies and can be mixed partially. In the infrared (IR) spectrum, and vibration types are active, and the other two vibrations and are active in the Raman spectrum. Therefore, there are only four main vibrations of different frequencies that can occur in the vibrational spectrum of BF3 molecule. These vibrations are represented in Figure 1.
The vibrational partition function is given by
where is the vibrational partition function corresponding to the symmetric stretching mode. is the vibrational partition function corresponding to the antisymmetric stretching and bending modes.
Due to the anharmonicity effects, the symmetric stretching vibrational modes are dominant comparing to the other vibrational modes [41] and in order to reduce the computational costs, we choose the improved Tietz oscillator [43] to describe the internal symmetric stretching vibrations, and use the harmonic oscillator for the description of the antisymmetric stretching vibration and the bending vibration.
The improved Tietz oscillator is given by [43] as:
where is the dissociation energy, is the equilibrium distance, r is the internuclear separation, is a dimensionless constant which controls the range of the interaction between atoms.
Applying the vibrational partition function given in Ref. [44] for the improved Tietz oscillator, we formulate the vibrational partition function corresponding to the symmetric stretching mode for BF3 as follows,
where is the dissociation energy of BF3 into BF and F2, , is the Boltzmann’s constant, T is the temperature, , , , and .
- here h is the Planck constant, is the reduced mass of boron and fluoride atoms, denotes the equilibrium B-F bond length.
The potential parameter is given by the expression:
where c is the speed of light, denotes the symmetric stretching vibration frequency and W is the Lambert function, which satisfies [45].
When , the Tietz oscillator turns to improved Rosen-Morse potential and improved Manning-Rosen potential respectively.
- denotes the most vibrational quantum number and is given bywhere Int stands for the integer part function. The imaginary error function, denoted erfi, is defined as [47]where denotes the error function, which is a special function of sigmoid shape.
We use the harmonic oscillator to describe the antisymmetric stretching and bending vibrations of the BF3 molecule. The vibrational partition function corresponding to these three vibration modes is expressed as follows,
where , and represent the antisymmetric stretching vibrational and bending vibrational frequencies for the BF3 molecule.
We made two simplifications in this model which are the intermolecular interactions are considered to be very weak and not taking into account and the second one is using the treatment of the rigid rotors of diatomic molecules. By minimizing computation costs and keeping the accuracy requirements in engineering applications, we do not take into account the effects of the nuclear spin of protons and centrifugal distortion in the treatment of rotation. It follows that the translational and rotational partition functions for BF3 molecule can be represented as [48]:
where denotes the molecular mass of BF3 and P is the atmospheric pressure.
(i = x, y, z) denote three rotational characteristic temperatures. , and represent the rotational moments of inertia for the BF3 molecule around three main axes where is the bond angle FBF. = 6, is the number of symmetry types for the BF3 molecule.
The total partition function for BF3 is readily written as
The values of the boron trifluoride partition function can be determined from analytical expressions (17) by inputting the experimental values of , , , , , and . The partition function of boron trifluoride are the functions of pressure, temperature and several molecular constants, and do not depend on a large quantity of calorimetric data or theoretical and experimental spectroscopy data.
Applying the following basic thermodynamic relationship, we can obtain the expressions of the molar entropy, the molar enthalpy, the molar Gibbs free energy and the isobaric specific heat
where V is the spatial volume and R is the molar gas constant.
3. Application
In this section, to evaluate the efficiencies of the proposed models for thermodynamic properties predictions of boron trifluoride, the calculated values are compared with the JANAF data [21] and MHEG data [20].
MHEG tabulates the reduced Gibbs free energy, every 100 K from 100 to 6000 K. JANAF gives the value of and tabulates on the same temperature grid. and are, respectively, the Gibb’s energy and the enthalpy at temperature T. The dissociation energy, B-F bond equilibrium length and vibrational frequencies of boron trifluoride are taken from literature [ref2]: = 64700 cm−1, reOH = 1.307 Å, = 888 cm−1, = 1453 cm−1, = 480 cm−1 and = 691 cm−1.
Figure 1.a illustrates the relationship between the partition function and temperature at a pressure of 0.1 MPa for boron trifluoride, based on molecular constant values. Additionally, Figure 2.b depicts a logarithmic plot of the same data. We observe that the partition function for increases uniformly as the temperature increases. The polynomial fits of the partition function and its logarithmic are represented by the dashed lines. It is important to note that using the coefficients from the polynomial fits to compute the partition function values is not recommended because the excess rounding introduces errors.
Figure 3 presents a graphical summary of the predicted values for the molar entropy, the molar enthalpy, the reduced Gibbs energy, and the isobaric specific heat for BF3 molecules, alongside the NIST data and MHEG data. Upon inspection of Figure 3, it is clear that the predicted values for are in excellent agreement with the NIST data and MHEG data.
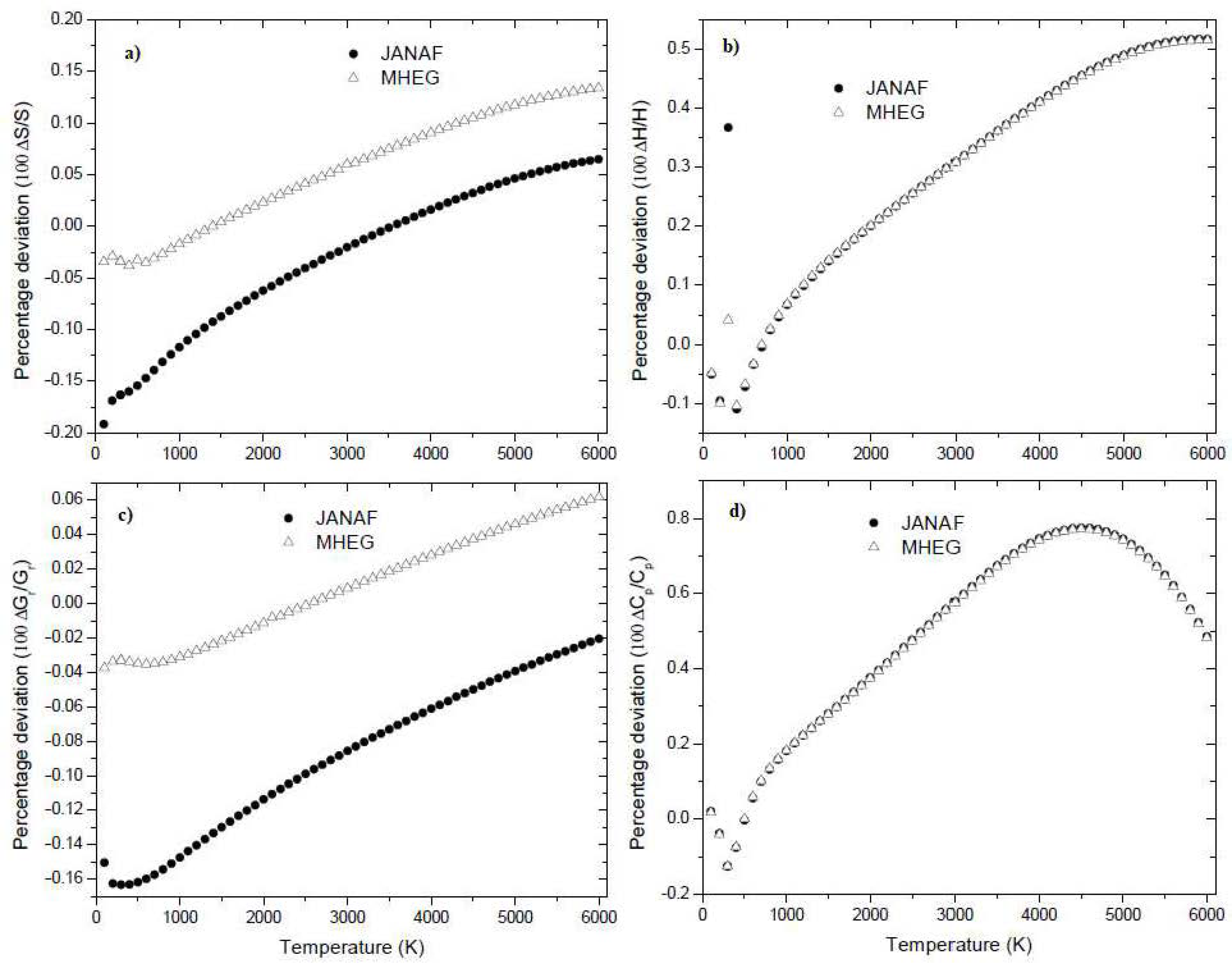
In Figure 4, the percentage deviations between the NIST values and the corresponding values predicted by the proposed model are displayed, along with the percentage deviations between the MHEG values and the calculated values. It is clear that the predicted values for BF3 align exceptionally well with the experimental data over a wide temperature range. The proposed prediction model for BF3 demonstrates a satisfactory accuracy, exhibiting an average relative deviation of 0.088 %, 0.088 %, 0.088 %, and 0.088 % for the molar entropy, the molar enthalpy, the reduced Gibbs free energy, and the specific heat, respectively, at a pressure of 0.1 MPa and in the temperature range of 100 K to 6000 K. However, Figure 4 also highlights that the present prediction models are inadequate in accurately predicting the thermodynamic properties of BF3 at lower temperatures. This shortcoming is attributed to the use of classical partition function for the vibrational partition function, which neglects quantum corrections. In the derivation of the partition function, only the lowest-order approximation was considered, and all quantum corrections were excluded.
4. Conclusions
In this article, based on the structure of the BF3 molecule, we proposed a simple and general model with independent variables of temperature and pressure for the prediction of the thermodynamic properties of BF3 molecules. The developed models require only seven experimental values of molecular constants for BF3 including the dissociation energy Des, bond length reOH, the reduced mass , vibration frequencies , , and whereas conventional analytical expressions require numerous adjustable coefficients derived from fitting numerous experimental spectroscopic or calorimetric data.
Comparison of the predicted values with NIST and MHEG data confirms that the developed analytical representations of molar entropy, molar enthalpy, reduced Gibbs energy, and isobaric specific heat for BF3 are applicable in scientific research and engineering applications.
Author Contributions
R. Horchani, N. Al Hashemi, and H. Jelassi conceived of the presented idea. O. Al Kharusi, H. Jelassi and R. Horchani developed the theory and performed the computations. U. S. Okorie and A. N. Ikot verified the analytical methods. All authors discussed the results and contributed to the final manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- I. L. Fowler (Atomic Energy of Canada Ltd., Chalk River, One.).Can. Nucl. Technol., 4: 40-4 (1965).
- Halford J O, J Chem Phys, 2 (1934) 694.
- Kemp J D, Pitzer K S. J Am Chem Soc, 59 (1937) 276.
- Prudente F V, Costa L S Acioli P H, J Phys B: At Mol Opt Phys, 33 (2000) R285.
- Harris G J, Viti S, Mussa H Y Tennyson J, J Chem Phys,109 (1998) 7197.
- Koput J, Carter S Handy N C, J Phys Chem A, 102 (1998) 6325.
- Irwin A W, Astron Astrophys Suppl, 74 (1988) 145.
- Martin J M L, Francois J P Gijbels R, J Chem Phys, 96 (1992) 7633.
- Riganelli A, Wang W Varandas A J C, J Phys Chem A, 103 (1999) 8303.
- Mielke S L, Srinivasan J Truhlar D G, J Chem Phys, 112 (2000) 8758.
- Taubmann G, Witschel W Shoendorff L, J Phys B: At Mol Opt Phys, 32 (1999) 2859.
- Messina M, Schenter G K Garrett C, J Chem Phys, 98 (1993) 4120.
- Pitzer K S Gwinn W D, J Chem Phys, 10 (1942) 428.
- Riganelli A, Prudente F V Varandas A J C, Phys Chem Chem Phys, 2 (2000) 4121.
- Taubmann G Schmatz S, Phys Chem Chem Phys, 3 (2001) 2296.
- Dahler J S, Mol Phys, 99 (2001) 1563.
- Traving, G., Baschek, B., and Holweger, H. 1966, Abh. Hamburger Sternw., 8, 3 (TBH).
- Kurucz, R. L. 1970, Smithsonian Ap. Obs. Spec. Kept., No. 309.
- Tatum, J. B. 1966, Pub. Dorn. Ap. Obs. Victoria, 13, 1.
- McBride, B. J., Heimei, S., Ehlers, J. G., and Gordon, S. 1963, Thermodynamic Properties to 6000 K for 210 Substances Involving the First 18 Elements (NASA, SP-3001) (MHEG).
- JANAF Thermochemical Tables. 1971 (2d ed.; Springfield, Va.: NBS), NSRDS-NBS-37 (JANAF).
- Irwin, A. W. 1978, Ph.D. thesis, University of Toronto.
- Tatum, J. B. 1966, Publications of the Dominion Astrophysical Observatory Victoria, 13, 1.
- Irwin, A.W. 1981, ApJS, 45, 621.
- Stull, D. R., Prophet, H. 1971, JANAF Thermochemical, 2nd ed. (Washington: USDepartment of Commerce), NSRDS-NBS37.
- Bohn, H. U., Wolf, B. E. 1984, AA, 130, 202.
- Huber, K. P., Herzberg, J. G. 1979, Molecular Spectra And Molecular Structure, Vol. IV. Constants of Diatomic Molecules (New York: Van Nostrand Reinhold).
- Sauval, A. J., Tatum, J. B. 1984, ApJS, 56, 193.
- Rossi, S. C. F., Maciel, W. J., Benevides-Soares, P. 1985, AA, 148, 93.
- Private communication, 1985.
- Dabrowski, I. 1984, Can. J. Phys., 62, 1639.
- Irwin, A.W. 1987, AA, 182, 348.
- Pagano, D., Casavola, A., Pietanza, L. D., et al. 2009, ESA Sci. Techn. Rev., 257.
- Laraia, A. L., Gamache, R. R., Lamouroux, J., Gordon, I. E., Rothman, L. S. 2011, Icarus, 215, 391.
- Fischer, J., Gamache, R. R., Goldman, A., Rothman, L. S., Perrin, A. 2003, J. Quant. Spectr. Rad. Transf., 82, 401.
- Gamache, R. R., Kennedy, S., Hawkins, R. L., Rothman, L. S. 2000, J. Mol. Struct., 517, 407.
- McDowell, R. S. 1988, J. Chem. Phys., 88, 356.
- McDowell, R. S. 1990, J. Chem. Phys., 93, 2801.
- Herzberg, J. G. 1960, Molecular Spectra and Molecular Structure, Vol. II: Infrared and Raman Spectra of Polyatomic Molecules, 2nd edn. (New York: Van Nostrand Reinhold).
- Colonna G., D’Angola A., Capitelli, M. 2012, Int. J. Hydrogen Energy, 37, 12, 9656.
- N. Elghobashi, L. González, J. Manz, J. Chem. Phys. 120 (2004) 8002–8014.
- C.S. Jia, Y.F. Diao, X.J. Liu, P.Q. Wang, J.Y. Liu, G.D. Zhang, Equivalence of the Wei potential model and Tietz potential model for diatomic molecules, J. Chem. Phys. 137 (2012), 014101.
- K.X. Fu, M. Wang, C.S. Jia, Commun. Theor. Phys. 71 (2019) 103–106.
- C.S. Jia, C.W. Wang, L.H. Zhang, X.L. Peng, R. Zeng, X.T. You, Chem. Phys. Lett. 676 (2017) 150–153.
- R.M. Corless, G.H. Gonnet, D.E.G. Hare, D.J. Jeffrey, D.E. Knuth, Adv. Comput. Math., 5 (1996), pp. 329-359.
- Bin Tang, Yi-Ting Wang; Xiao-Long Peng, Lie-Hui Zhang, Chun-Sheng Jia, Journal of Molecular Structure, Vol: 1199 (2019) Page: 126958.
- C.E. Pearson, Handbook of Applied Mathematics, Van Nostrand Reinhold, New York (1983).
- X.L. Peng, R. Jiang, C.S. Jia, L.H. Zhang, Y.L. Zhao, Gibbs free energies of gaseous phosphorus dimer, Chem. Eng. Sci. 190 (2018) 122-125.
Figure 1.
The vibrational modes of BF3.
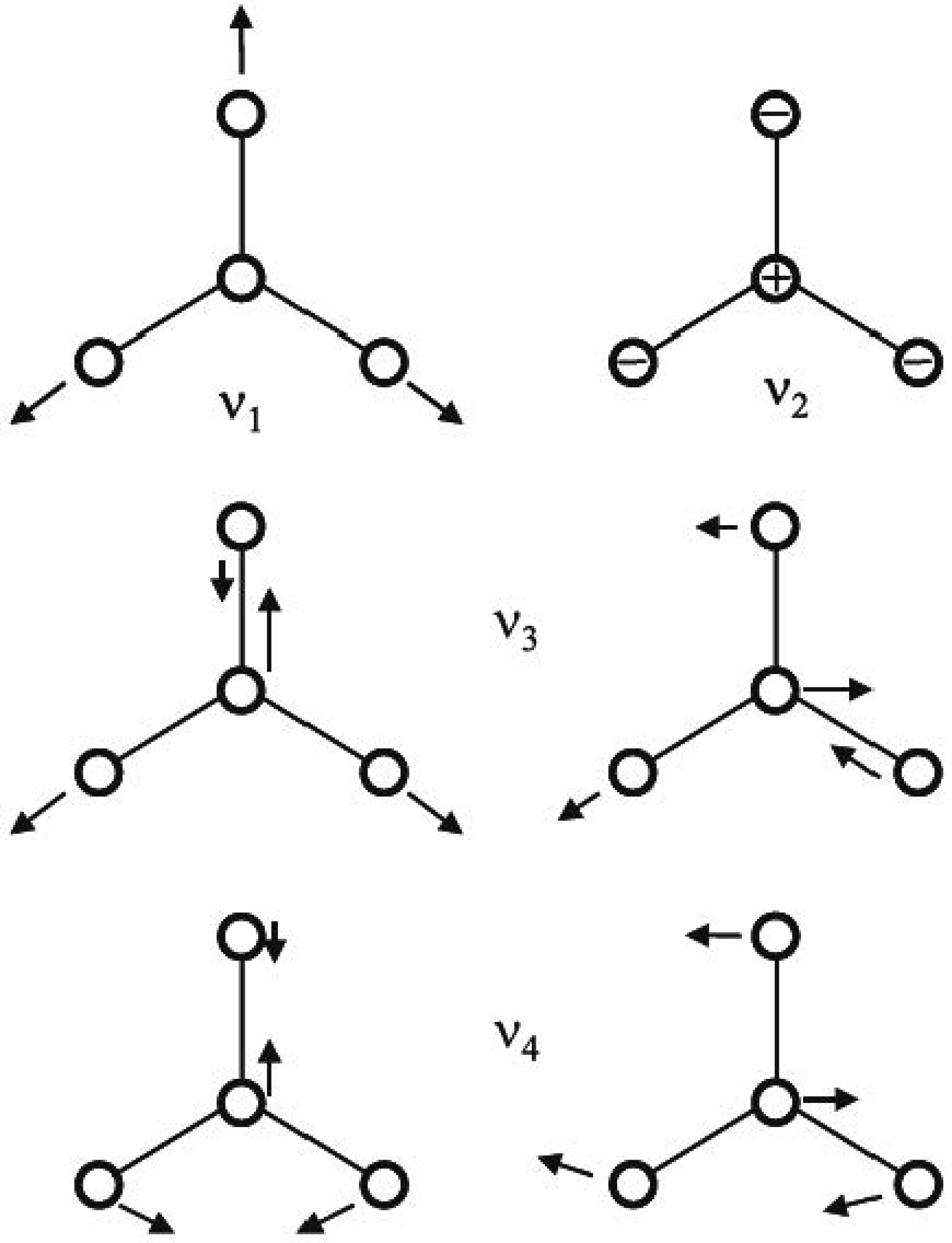
Figure 2.
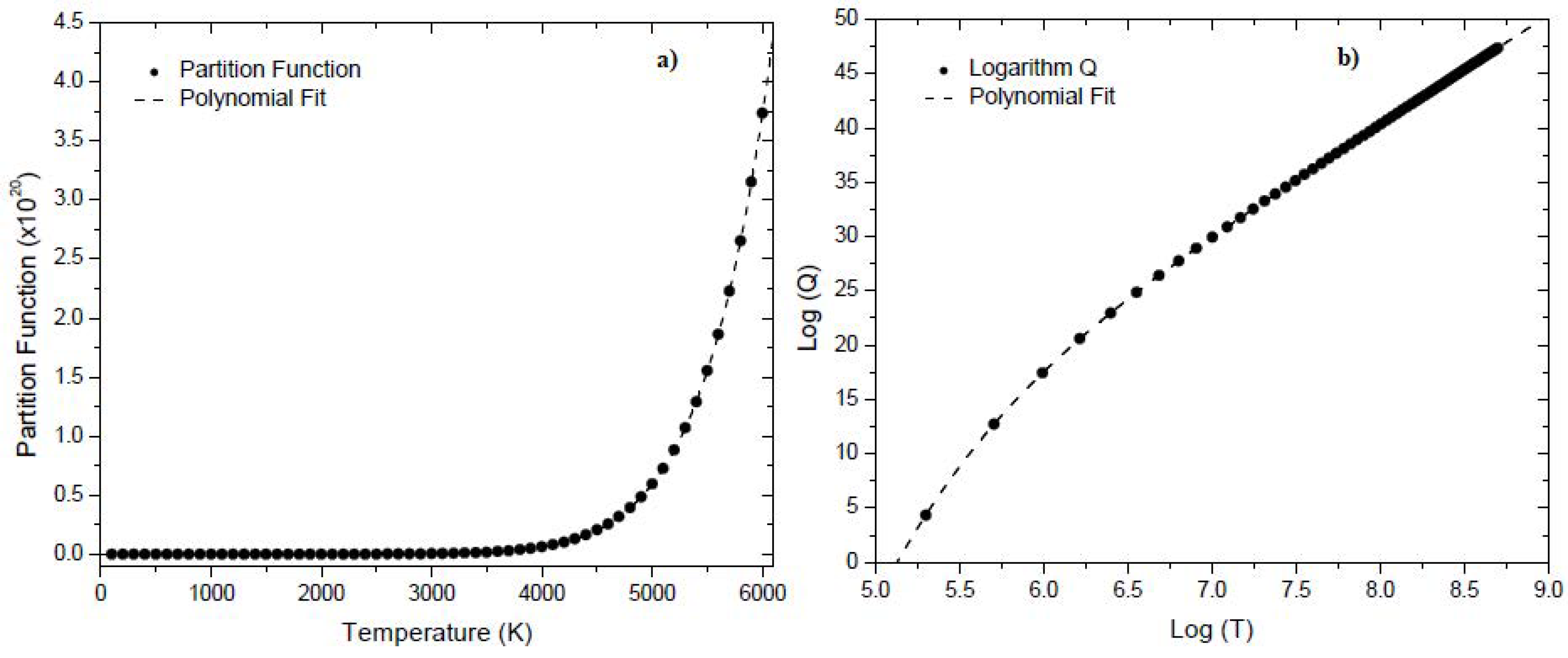
Figure 3.
Temperature variation of the thermodynamic properties of BF3 for molar entropy (a), molar enthalpy (b), reduced Gibbs energy (c) and isobaric specific heat (d).
Figure 3.
Temperature variation of the thermodynamic properties of BF3 for molar entropy (a), molar enthalpy (b), reduced Gibbs energy (c) and isobaric specific heat (d).
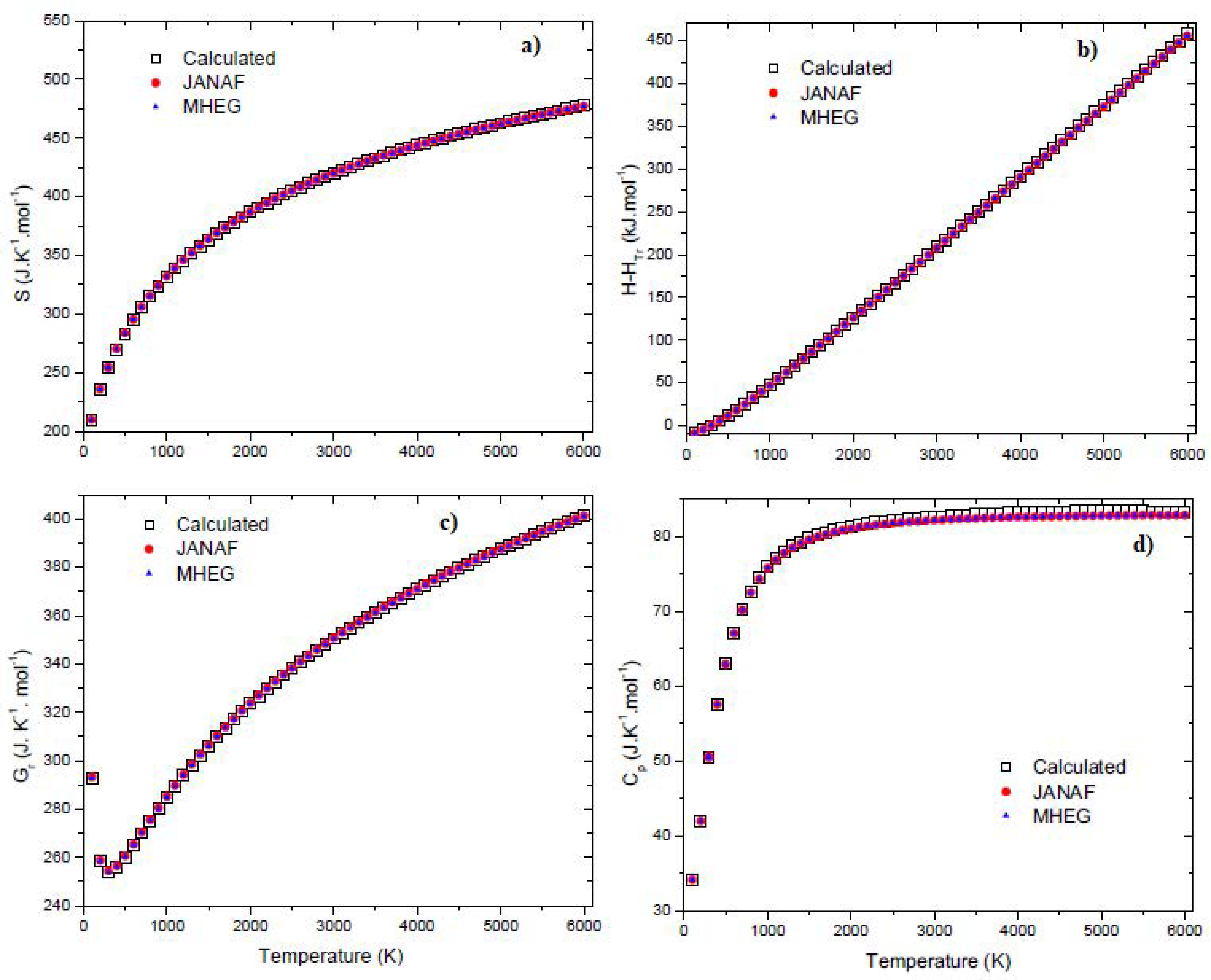
Figure 4.
Percentage deviations of the NIST data and MHEG data of BF3 for molar entropy (a), molar enthalpy (b), reduced Gibbs energy (c) and isobaric specific heat (d) from the corresponding calculated values with respect to temperature.
Figure 4.
Percentage deviations of the NIST data and MHEG data of BF3 for molar entropy (a), molar enthalpy (b), reduced Gibbs energy (c) and isobaric specific heat (d) from the corresponding calculated values with respect to temperature.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



