
Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
Ruxolitinib has been approved by FDA for the treatment of myeloproliferative neoplasms such as polycythemia vera and idiopathic myelofibrosis. Although ruxolitinib has been in the clinic for more than ten years, surprisingly, no JAK2 drug resistant mutations have been identified in patients. At the moment, it is not clear whether the absence of JAK2 mutations in MPN patients is due to a lack of JAK2 addiction or residues that mediate the drug resistance compromising the JAK2 function. To find out this, we established a Ba/F3 cell-based screening strategy to predict mutations in JAK2-V617F that cause resistance towards ruxolitinib. In this study, we have recovered seven residues in the kinase domain of JAK2 that affect ruxolitinib sensitivity. All these mutations confer cross-resistance across the panel of JAK2 kinase inhibitors except JAK2-L983F. JAK2-L983F reduces the sensitivity towards ruxolitinib. However, it is sensitive towards fedratinib indicating that our screen identifies the drug-specific resistance profiles. All the ruxolitinib resistant JAK2 variants displayed sensitivity towards Type II JAK2 inhibitor CHZ-868. Finally, our study also shows that HSP90 inhibitors are potent against ruxolitinib resistant variants through the JAK2 degradation and provides the rationale for clinical evaluation of potent HSP90 inhibitors in genetic resistance driven by JAK2 inhibitors.
Keywords:
Myeloproliferative neoplasms
; JAK2-V617F mediated resistance
; ruxolitinib
; JAK2-STAT5 signaling
; HSP90 inhibition
1. Introduction
JAK2 is an important cytoplasmic tyrosine kinase that plays a major role in the normal development of hematopoiesis and cytokine mediated signaling [1,2]. The occurrence of somatic activation mutation (valine to phenylalanine) in the pseudokinase domain (V617F) of JAK2 has been implicated in myeloproliferative neoplasms like polycythemia Vera (90%), essential thrombocythemia (50%) and Idiopathic myelofibrosis (50%) [3,4,5,6]. In addition to MPNs, JAK2-V617F mutation has also been observed at very low frequencies in myelodysplastic syndrome, chronic myelomonocytic leukemia (3-8%) and very rarely in systemic mastocytosis [7,8]. Subsets of PV patients negative to V617F mutation showed a gain of function mutations affecting the exon 12 of JAK2 [9]. Other novel mutations located in the JH2 domain are also reported in several hematological malignancies including D620E in PV patient [10], C661Y in unclassified MPN [11], L611S in ALL [12], IREED in Down syndrome [13]. Biochemical studies have shown that all these mutations lead to constitutive activation of JAK2. In addition to the point mutations, JAK2 is, also involved as a fusion protein due to chromosomal translocation. A t (9; 12) (p24: p13) leads to the generation of TEL-JAK2 fusion associated with the development of T cell childhood acute lymphoblastic leukemia [14,15]. Wild-type JAK2 signaling is also involved in some solid tumors such as breast cancer [16]. Taken together, these discoveries encouraged the development of small molecular inhibitors against JAK2. Several JAK family kinase inhibitors are developed and are currently being tested in preclinical and clinical studies [17]. Among those, ruxolitinib and fedratinib have been approved for the treatment of intermediate and high-risk myelofibrosis, while ruxolitinib was also approved for PV patients intolerant to hydroxyurea. Unlike imatinib in CML, where already 6 months of TKI treatment can result in a durable clinical response by reduction of the BCR::ABL1 transcript, JAK2 inhibitor short-term treatment does not induce a significant reduction in MPN-driving allele burden [18,19]. Nevertheless, long-term studies on ruxolitinib indicated a reduction of the mutant allele burden, improvement of bone marrow fibrosis and increase in overall survival [20,21,22,23]. Due to these benefits, ruxolitinib remains a mainstay for the treatment of MPN patients. However, it becomes evident from clinical trials that JAK2 inhibitor treatment has a limited effect on disease-driving stem cells and thus, it is unlikely that these inhibitors induce complete remission in MPN patients [24]. In addition to ruxolitinib and fedratinib, lesturtinib is also a JAK2-specific inhibitor that inhibits expanded erythroid cells in PV patient. Compared to ruxolitinib, lestaurtinib showed modest clinical recovery with improvement of spleen size and no improvement in bone marrow myelofibrosis and JAK2-V617F allele burden [25]. In CML, NSCLC and GIST, it has been demonstrated that acquired resistance to imatinib is due to the emergence of secondary resistance mutations in the target kinase [26,27,28]. In the case of BCR-ABL, Azam et al. demonstrated that more than 60 residues in the kinase domain are involved in the resistance against ABL kinase inhibitor imatinib [29]. These results led to the development of second and third generation kinase inhibitors in the CML in order to treat the disease efficiently. So far, no inhibitor resistant JAK2 mutations have been reported in patients, although ruxolitinib has been used for more than ten years in the clinic. In order to predict the drug resistant mutations against the JAK2 inhibitor ruxolitinib, we used a cell-based screening strategy. In this study, we have identified seven different exchanges in the kinase domain of JAK2 that induce strong ruxolitinib resistance. All these mutations confer cross-resistance across the panel of JAK2 kinase inhibitors except JAK2-L983F. JAK2-L983F reduces the sensitivity of JAK2 dependent cells to ruxolitinib, however, are sensitive towards fedratinib indicating that our screen identifies the compound-specific resistant mutations but not ATP-competitor specific mutations. All the ruxolitinib resistant JAK2 variants displayed sensitivity towards Type II JAK2 inhibitor CHZ-868. Finally, our study also provides that HSP90 inhibitors are also potent against ruxolitinib resistant variants.
2. Results
2.1. Frequency of drug resistant clones are decreased after increasing the ruxolitinib concentration
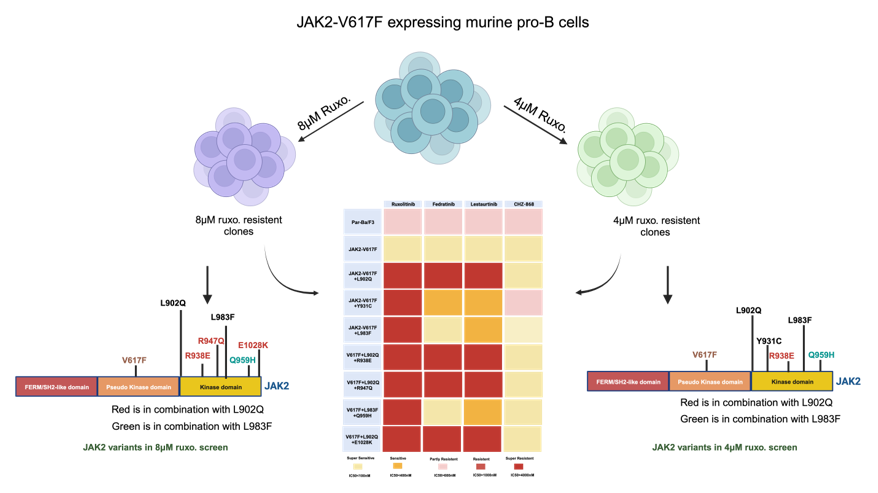
Ruxolitinib is a potent JAK2/JAK1 specific inhibitor that exhibits remarkable clinical activity against the JAK2-V617F mediated MPNs [23]. In the case of several hematological malignancies, it has been demonstrated that resistance is due to the acquisition of the point mutation in the target kinase. However, in the case of JAK2 mediated MPNs, none of the patients displayed mutations in JAK2 kinase, even though persistent to ruxolitinib therapy. The mechanism responsible for ruxolitinib persistence in MPN patients has yet to be demonstrated. In order to understand the ruxolitinib persistence, we developed a cell-based screening strategy against the various concentrations of ruxolitinib. Screening performed with 4μM and 8μM ruxolitinib concentrations did not yield any drug resistant clones; however, ENU (ethyl-nitrosourea: a chemical mutagen) pretreatment before the ruxolitinib exposure to the cells yielded resistant clones. At 4μM ruxolitinib concentration, which approximates the maximum measured plasma concentration, the frequency of resistant clones was 1.12 (Figure 1A). The frequency of resistant clones decreased in 8µM ruxolitinib concentration and was limited to 0.3 (Figure 1A).
2.1.1. L902Q, Y931C, L983F are the most frequent exchanges identified in ruxolitinib screen performed at 4 μM and 8 μM concentrations
To determine the drug resistance is due to the acquisition of point mutations in the kinase domain of JAK2, we sequenced the drug resistant clones from 4 μM and 8 μM concentrations. Resistant clones growing in the presence of 8μM ruxolitinib displayed L902Q and L983F with high abundance (44% and 18%, respectively) (Figure 1B). In addition to these two exchanges, some of the resistant clones displayed compound exchanges such as L902Q+R938E, L902Q+R947Q, L902Q+E1028K and one resistant clone displayed compound mutation together with L983F (L983F+Q959H) (Figure 1B). Analysis of 4μM drug resistant clones revealed L902Q, L983F and Y931C are the most frequent mutations with a relative frequency of 33%, 16% and 16%, respectively (Figure1 C). Similar to the 8μm resistant clones, 4μm resistant clones also displayed compound exchanges. Among those, L902Q+R938E and L983F+Q959H were also identified in 4μm resistant screen (Figure 1C and D). One clone displayed the R938E mutation alone (Figure 1C and D). Taken together, these results suggested that ruxolitinib exposure to the JAK2-V617F cells lead to the generation of JAK2 variants in the cell-based method. To determine whether these mutations are valid in the clinical situation, we checked the homologous mutations in BCR-ABL (Figure 1E). Interestingly, the most abundant mutation, L902Q, in JAK2 is homologous to M290 (C-helix) of BCR-ABL, which is involved in imatinib resistance [29,30]. JAK2-Y931 is homologous to F317 in ABL1 has also been demonstrated as associated with imatinib resistance in CML patients [29,31]. JAK2-Y931 is located in the adenine-binding region of the hinge and involved in the direct interaction with the inhibitor (Supplementary Figure S2C). Consistent with our results, previous data also suggest that JAK2-Y931C has been identified in a resistant screen generated against the JAK2-inhibitor BVB808 [32]. These results indicate that Y931 residue is ATP-competitive inhibitor specific. Finally, L983 of JAK2 is homologous to L370 of BCR-ABL. The Leucine at this position is highly conserved among the tyrosine kinases, but so far, none of the kinases have shown drug resistance by changing this residue. In addition to this, we also found that JAK2- R938 and JAK2- Q959 are homologous to the L326 and Q346 of BCR-ABL reported as hot spots in imatinib resistance [29] (Figure 1E).
2.2. Drug resistant mutations identified in the cell-based screen transform the Ba/F3 cell and showed constitutive JAK2-STAT5 activation
In the case of BCR-ABL, it has been well demonstrated that mutations in the kinase domain of the ABL not only confer the resistance but also increase the kinase activity [33]. To check this possibility in the case of JAK2, we cloned all the mutants in the V617F background and stably expressed the mutants in Ba/F3 cell line. Concurrent expression of JAK2-V617F in Ba/F3 cell line confers the IL-3 independent cell growth, as reported previously [4,34]. JAK2- L902Q, JAK2- L983F, JAK2- L902Q+R938E, JAK2-L902Q+R947Q, JAK2- L902Q+E1028K and JAK2- L983F+Q959H were able to give IL-3 independence to the Ba/F3 cell line (Figure 2A). Interestingly, JAK2-V617F+Y931C gives enhanced cell growth to the Ba/F3 cell line compared to JAK2-V617F (Figure 2A). Similarly, as expected JAK2-R938E, JAK2-R947Q, JAK2-Q959H, JAK2-E1028K in the V617F background gave IL-3 independence to the Ba/F3 cell line. Later, we analyzed the activation of the JAK2-STAT5 axis in JAK2 mutants expressing Ba/F3 cell lines and found that all the mutants could activate JAK2 and STAT5 (Figure 2B). These results suggested that all the mutants identified in ruxolitinib resistant screen promoted the competitive growth advantage to the Ba/F3 cells. Later, we hypothesized that kinase domain mutations alone could activate the JAK2 and give the cytokine independent growth to the Ba/F3 cells without V617F exchange. To check this, we cloned the L902Q and L983F mutations alone in JAK2 and compared the cytokine independent growth with JAK2-V617F+L902Q and JAK2-V617F+L983F. Interestingly, JAK2-L902Q and JAK2-L983F alone fails to transform the Ba/F3 cells (Supplementary Figure S1A and B). These results indicate that V617F exchange is indispensable for IL-3 independent growth and kinase domain mutations alone might not be able to promote the cytokine-independent growth to Ba/F3 cells.
2.3. Kinase domain mutations confers the ruxolitinib resistance and showed persistent activation of STAT5 at higher concentration of drug
To determine that these mutations can confer drug resistance, first, we checked the JAK2 mutants cell growth in the presence of increasing concentration of ruxolitinib. JAK2-V617F Ba/F3 cells were sensitive to ruxolitinib (IC 50 ~182nM), compared to parental-Ba/F3 cells (IC 50 ~ 686nM). Interestingly, all the mutants identified in the cell-based screen were able to grow in the presence of high ruxolitinib concentration. L902Q, L902Q+R938E, L902Q+R947Q, Y931C and L902Q+E1028K are totally resistant to ruxolitinib (IC 50 > 4000nM) compared to L983F and L983F+Q959H which shows even higher resistance (IC 50 > 8000nM) (Supplementary Table S1) (Figure 3A). Next, we sought to determine whether drug resistant mutants could show the persistent activation of JAK2 and STAT5 in the presence of ruxolitinib. Consistent with the cell proliferation data, western blot data also showed persistent STAT5 activation in all the mutants in higher concentrations of ruxolitinib except JAK2-V617F (Figure 3B). At 250nM ruxolitinib concentration, JAK2-V617F displayed the complete absence of STAT5 activation (Figure 3B). As shown before, JAK2 is hyperphosphorylated in the presence of ruxolitinib compared to untreated cells. These results are consistent with the previous report that activation loop phosphorylation (Tyr1007/1008) by JAK inhibitor is mode dependent and treatment with ATP-competitive inhibitors leads to JAK2 hyperphosphorylation [35] (Figure 3A). These results indicate that ruxolitinib resistant variants identified in the cell-based screen method are indeed resistant to higher concentrations of ruxolitinib and keep persistent activation of STAT5.
In order to further understand the drug resistant phenotype of the mutants, we also performed computational modeling studies using Glide package of schrodinger maestro software. Ruxolitinib forms hydrogen bond interactions with Glu 930 and Leu 932 with wildtype JAK2 domain and these interactions were consistent in all the variants except for Y931 variant where an additional carbon centered hydrogen bond was observed with Arg 980 (Supplementary Figure S2A-D). In addition to altering the shape of the protein’s binding pocket, mutations L902Q, Y931C, and L983F alleles can hinder the drug’s physical fit by disrupting crucial chemical interactions between the drug and the protein, weakening their association and making drug detachment easier (Supplementary Table S2).
2.4. Ruxolitinib resistant JAK2-L983F is sensitive towards fedratinib
Next, we analyzed for the cross-drug resistance by using another ATP-competitive inhibitor of JAK2, Fedratinib. Similar to the ruxolitinib, JAK2-V617F is sensitive towards fedratinib (IC 50 ~172nM), compared to parental-Ba/F3 cells (IC 50 ~229nM). Interestingly, L902Q, L902Q+R938E, L902Q+R947Q, L902Q+E1028K are resistant to fedratinib by increasing their cellular IC50 value to more than 1000nM. Whereas Y931C is sensitive towards fedratinib (IC 50~ 145nM). Similarly, L983F, and L983F+Q959H are inhibited at less fedratinib concentration (IC 50~88nM and 91, respectively) (Figure 4A and Supplementary Table S1). Consistent with the cell proliferation data, western blot analysis suggested that JAK2-V617F showed inhibition of STAT5 activation in the presence of 250nM concentration of fedratinib (Figure 4B) whereas JAK2-L902Q, JAK2-Y931C, JAK2-L902Q+R938E, JAK2-L902Q+R947Q, JAK2-L902Q+E1028K mutants showed persistent activation of STAT5 even at 2000nM concentration of fedratinib (Figure 4B) suggesting that these variants are completely resistant towards fedratinib (Figure 4B). Similar to the cell proliferation data, western blot data also suggested that JAK2-L983F and JAK2-L983F+Q959H showed inhibition of STAT5 at 500nM concentration of fedratinib (Figure 4B), suggesting that fedratinib might be beneficial for the ruxolitinib refractory MPN patients. Inhibition of STAT5 activation in JAK2-V617F+Y931C was observed at 1000nM concentration, suggesting that this variant might be moderately resistant towards fedratinib. Consistent with the previous data, JAK2 phosphorylation (Tyr1007/1008) was increased in the fedratinib sensitive mutants (V617F, Y931C, L983F and L983F+Q959H) but not in fedratinib insensitive mutants such as L902Q, L902Q+R938E, L902Q+R947Q and L902Q+E1028K.
To gain further understanding of fedratinib sensitivity towards the L983F variant, we performed molecular docking analysis. Fedratinib interacts with wild type JAK2, L902Q, Y931C and L983F variants by forming two hydrogen bonds with amino acid Leu 932 (Supplementary Figure S3A-D). In addition, fedratinib showed π-π interactions with Phe 983 and hydrogen bond interactions with Ser 936 and Leu 855 in the case of L983F variant suggesting stronger binding affinity in consistent with the observed low IC50. Binding energy analysis displayed both L902Q and L983 showed similar glide score, however, L902Q is completely resistant towards fedratinib (Supplementary Table S2). These results are in line with previous finding by Kesarvani et al. study where fedratinib binds substrate binding pockets with higher affinity than the ATP site in contrast to the ruxolitinib which preferentially binds to ATP site [36]. These results suggest that fedratinib might be effective in suppression of ATP-site mutations generated by ruxolitinib due to its ability to bind additional substrate binding site.
2.5. Lestauritinib is not potent against the ruxolitinib resistant variants and did not change the IC50 value
Lestaurtinib is another potent JAK2 specific inhibitor and showed inhibition of STAT5 phosphorylation in JAK2-V617F expressing cells [25]. Based on these results, we hypothesized that lestaurtinib might inhibit ruxolitinib resistant variants. Consistent to the previous reports JAK2-V617F (IC 50~89.9nM) was sensitive towards lestaurtinib compared to par-Ba/F3 cell line (IC 50~710nM) indicating that this inhibitor is specific towards JAK2-V617F, however, analysis of other JAK2 variants results suggest that L902Q, Y931C, L902Q+R938E, L902Q+R947Q, and L902Q+E1028K are resistant to lestaurtinib (IC 50 >4000nM). Even At 8μm concentration of lestaurtinib did not reduce the cell proliferation of these JAK2 variants (Figure 5A). Interestingly, lestaurtinib is potent against the L983F and L983F+Q959H (IC 50~245nM and 117nM, respectively) (Supplementary Table S1). Consistent with the MTS based cell proliferation data, biochemical data also showed inhibition of STAT5 in JAK2-V617F (Figure 5B) and increased JAK2 activation loop Tyr1007/1008 phosphorylation in the presence of lestaurtinib. Whereas, L902Q, Y931C, L902Q+R938E, L902Q+R947Q and L902Q+E1028K did not showed any decrease of STAT5 activation (Figure 5C). L983F and L983F+Q959H showed a decrease of STAT5 activation based on the dose dependent manner, suggesting that these variants are inhibited at higher concenntration of lestaurtinib. In addition to compound mutations which are in combination with L902Q or L983F, we also established the single constitutes of R938E, R947Q, Q959H and E1028K and found that these variants did not display resistance towards ruxolitinib, fedratinib, and lestaurtinib except R938E (Supplementary Figure S4A, B and C). R938E has increased 2.5 fold increase of IC50 value towards ruxolitinib and lestauritinib but no change in IC50 value against fedratinib. These results suggest that L902Q and L983F exchanges are indispensable for strong drug resistance phenotype in these variants.
Molecular docking analysis suggest that Lestaurtinib forms two carbon-centered hydrogen bonds with wild type JAK2 Arg 980. The L902Q (with Leu 855), Y931C (with Val 863) mutants showed single carbon-centered hydrogen bond interaction with lestaurtinib indicating less binding affinity which can be correlated with high IC50 values (Supplementary Figure S5A-D). In contrast, for the L983F variant, lestaurtinib showed multiple hydrogen bond interactions (with Leu 932, Leu 855, Val 863, and Glu 930) and π-π interaction (with Phe 983) indicating stronger binding affinity thus low IC50 (Supplementary Table S2). Taken together, computational data with the biochemical IC50 value data suggest that each inhibitor has distinct affinity towards the kinase conformation and alteration of these conformational states governs the drug binding state which subsequently leads to drug resistance phenotype. However, a complete three-dimensional structure of JAK2 needed to be required in order to explain the resistance phenotype of each variant involved in the modulation of JAK2 kinase confirmation.
2.6. Type II JAK2 inhibitor CHZ 868 is potent towards the ruxolitinib variants
Since type I inhibitors showed less efficiency toward the ruxolitinib resistant variants, we decided to check the efficacy of type II JAK2 inhibitor CHZ-868 towards the ruxolitinib resistant variants. Previously, it has been demonstrated that type II JAK2 inhibitor CHZ-868 showed remarkable efficacy in MPN mouse models compared to ruxolitinib [37]. Based on our previous results, that L902Q, Y931C and L983F are the crucial resistant variants as other variants such as R938E, R947Q, Q959H and E1028K without L902Q or L983F did not shift their cellular IC50 value, we focused on only L902Q, Y931C and L983F variants towards the CHZ-868. Interestingly, L902Q, which is resistant to ruxolitinib, fedratinib and lestaurtinib, is sensitive towards CHZ-868 (Figure 6A) (Supplementary Table S1). L983F is highly resistant towards ruxolitinib is also sensitive towards the CHZ-868 (Figure 6A), suggesting that fedratinib and CHZ-868 should be beneficial to ruxolitinib refractory MPN patients. Surprisingly, Y931C, which is not a strong resistant variant towards the ruxolitinib, lestaurtinib and fedratinib, is less sensitive towards CHZ-868 (Figure 6A), suggesting that type II inhibitors are more potent towards the resistant variants of type I inhibitors and also the mode of inhibitor interaction to the kinase domain determines the resistant phenotype. Analysis of JAK2 and STAT5 activation after CHZ-868 treatment results clearly showed decrease of STAT5 and JAK2 activation in V617F, L902Q and L983F (Figure 6B). Interestingly, Y931C displayed less sensitivity towards CHZ-868 by displaying the inhibition of STAT5 at 200nM concentration (Figure 6B).
Unlike type I JAK2 inhibitors bind the ATP-binding site of active JAK2, type II JAK2 inhibitor CHZ-868 binds allosteric sites of JAK2 in addition to the ATP-binding site of inactive JAK2 [38]. Molecular docking analysis with type II JAK2 inhibitor CHZ-868 revealed that numerous hydrophobic interactions hold the drug with residues Leu 855, Val 863, Ala 880, Val 911, Met 929, Leu 932 and Leu 983 that line the binding pocket (Supplementary Figure S6A-D). In addition, CHZ-868 forms hydrogen bonds with D994 and E898, and van der Waal interaction with side chains of L983 and G993.
CHZ-868 forms two hydrogen bond interactions in wild type JAK2 (with Glu 390 and Leu 932) (Supplementary Figure S6A), L902Q (with Glu 390 and Leu 932) (Supplementary Figure S6B), Y931C (with Glu 390 and Leu 932), (Supplementary Figure S6C) and L983F (with Glu 390 and Leu 932) (Supplementary Figure S6D) variants. The mutations did not alter the binding interactions of CHZ-868 signifying similar binding affinities in all the variants (Supplementary Table S2).
2.6.1. JAK1-L1010F exchange (L983F homologous JAK2) drives strong ruxolitinib resistance
In order to evaluate the role of ruxolitinib resistance in JAK1, we created the JAK2 homologous exchanges in JAK1 and measured the ruxolitinib resistance (Supplementary Figure S7A). In contrast to JAK2-L902Q which is strong resistant towards ruxolitinib, however, homologous exchange JAK1-L929Q give only moderate resistance towards ruxolitinib suggesting that structural variation among JAK-family kinases (Supplementary Figure S7B, D). Interestingly, JAK2-L983F homologous exchange JAK1-L1010F gives strong ruxolitinib resistance (Supplementary Figure S7C, E) suggesting that this exchange might pose a challenge in JAK1 mediated diseases as a resistant variant when patients treated with JAK1 inhibitors.
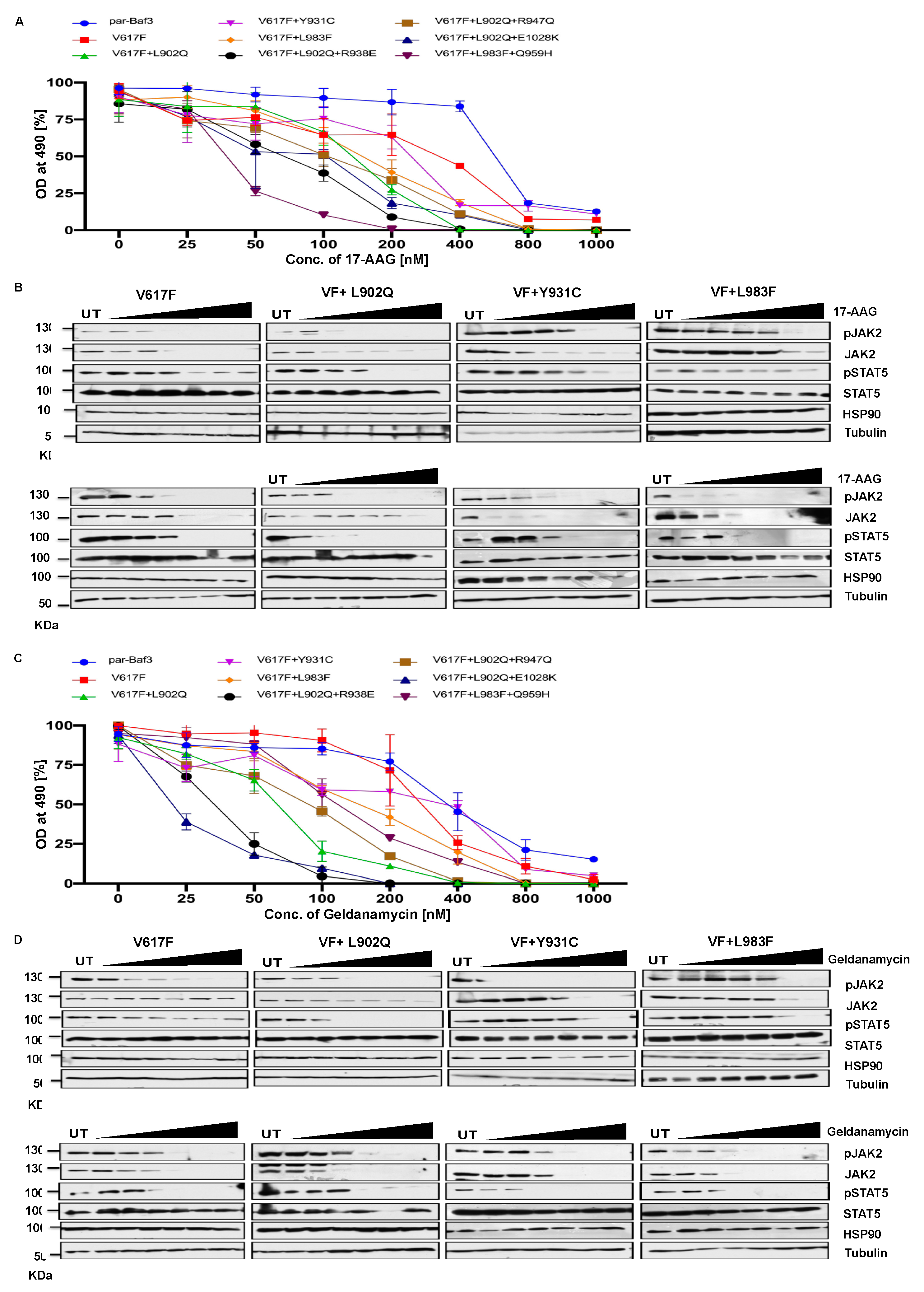
2.7. HSP 90 inhibitors are effective therapeutic agents against the ruxolitinib resistant variants
JAK2 is the known client protein of HSP90 and inhibition of HSP90 by small molecular inhibitors leads to the degradation of both JAK2 wild-type and JAK2-V617F [39]. HSP 90 inhibitors were shown to be effective in the survival of JAK2 mediated MPNs in mice models and also shown as an alternative to genetic resistance mediated by JAK2 enzymatic inhibitors [39]. We then hypothesized ruxolitinib resistant variants might also depend on the HSP90 pathway. Consistent to our hypothesis, HSP90 inhibitor 17-AAG treatment results suggest that V617F+L902Q, V617F+Y931C, V617F+L983F, V617F+L902Q+R938E, V617F+L902Q+R947Q, V617F+L902Q+E1028K and V617F+L983F+Q959H are sensitive to 17-AAG (Figure 7A). These results indicate that these mutants are dependent on the HSP90 for their folding. To know that downregulation of JAK2 protein leads to the decrease of cell proliferation, we performed biochemical analysis on these mutant JAK2 cells and found that ruxolitinib resistant variants are sensitive towards 17-AAG and treatment of the cells with 17-AAG leads to the downregulation of JAK2 protein and decrease of STAT5 activation (Figure 7B).
To confirm that HSP90 inhibition leads to the inhibition of cell proliferation of mutant JAK2 cells, we used another potent HSP90 inhibitor, geldanamycin. Similar to the 17-AAG results, geldanamycin is also very effective in the inhibition of cell growth both in JAK2-V617F cells and drug resistant mutants (Figure 7C). Western blot data also suggested that treatment of the cells with geldanamycin leads to the downregulation of the JAK2 protein more effectively in the drug resistant mutants compared to JAK2-V617F and also decreased the STAT5 activation (Figure 7D).
3. Discussion
JAK2-V617F mutation is frequently reported in myeloproliferative neoplasms (MPNs) such as polycythemia vera, essential thrombocythemia and idiopathic myelofibrosis [3,4,5,7]. Constitutive JAK2 signaling is also involved in several solid tumors and other lymphoid malignancies [15,16,40]. The occurrence of JAK2 variants in several malignancies suggests that therapeutic inhibition of JAK2 is important. Two JAK2 inhibitors, ruxolitinib and fedratinib, have been approved for patients with intermediate and high risk myelofibrosis. It has been demonstrated that in chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST), acquired resistance to imatinib is due to the emergence of secondary kinase domain mutations in target kinase. In the case of BCR-ABL, more than 50 different exchanges have been described that confer drug resistance in CML patients [41]. Results from imatinib resistance in CML suggested sequential treatment with TKIs and approval of second-generation ABL kinase inhibitors. However, in the case of JAK2, no drug resistant variants are reported in MPN patients even though they are resistant (persistent) to JAK2 inhibitors. The mechanism responsible for JAK2-inhibitor persistence in MPN patients is not understood well. One possible explanation for the persistence against the JAK2 inhibitors without acquiring the point mutations in the kinase domain of the JAK2 is due to compromisation of kinase function. Thus, it is essential to identify the critical residues that mediate the inhibitor resistance. In this study, we have identified seven different exchanges in the kinase domain of JAK2 that conferred strong resistance towards ruxolitinib. We functionally evaluated these residues in the kinase domain function and resistance towards the type I and Type II JAK2 inhibitors (Figure 2-6). All the mutations identified in our cell-based screen showed similar transformation ability and JAK2 and STAT5 activation except Y931C. JAK2-Y931C gives enhanced cell growth compared to other JAK2 variants. This result is in line with Charlotte et al. study using the ATF7IP-JAK2 model where ruxolitinib resistance was mediated by Y931C exchange, which also induced the enhanced cell proliferation and JAK/STAT signaling pathway [42]. Only seven resistant mutations identified in our screen are in line with other studies by Deshpande et al. and Kesarvani et al. study, which demonstrated the limited repertoire of JAK2 mutations against the JAK2-inhibitor resistance in contrast to ABL inhibitors [36,43].
In the case of c-Kit mediated drug resistance, the acquisition of point mutation (D816V) leads to a change in the equilibrium of the kinase towards the active confirmation that results in the activation of the c-Kit [44]. Similarly, FLT3-D835 exchange is known to cause activation of the FLT3 and resistance towards the FLT3 inhibitors [45]. In the case of JAK2, the drug resistant mutations L902Q and L983F did not activate the JAK2 and STAT5 without V617F exchange, suggesting that V617F exchange is indispensable for the activation of JAK2-STAT5 and cytokine independent growth. Similar to our results, Hornakoa et al. study identified several mutations in JAK1 by cytokine deprivation. In this study, they found F958C exchange in cytokine independent clones. Interestingly, JAK1-F958C is resistant to JAK inhibitors CMP6 and ruxolitinib [46]. JAK1-F958 is homologous to JAK2-Y931, which we observed as a drug resistant variant in our screen. In addition, two other publications used random mutagenesis screens and observed JAK2-Y931C mutation against ruxolitinib and BVB808 [32,47]. Interestingly, a more comprehensive study by Keserawani et al. detected 211 amino acid substitutions in whole JAK2 protein, which give cross resistance against the panel of JAK inhibitors by using randomly mutagenized JAK2-V617F expressed in Ba/F3 cells [36]. In line with their study, our chemical mutagenesis screen also detected JAK2 mutations at residues L902, Y931, R938, L983 and E1028, which give strong resistance against the ruxolitinib. In our study, we found only seven different exchanges in our cell-based screen. This is in contrast to BCR-ABL, where 112 distinct amino acid substitutions affect 90 residues in BCR-ABL [29]. Since we used a high concentration of ruxolitinib, such as 4µM (IC50~20 times) and 8µM (IC50~40 times) in our cell-based screen, which might lead to the escape of moderate drug resistant clones. However, ruxolitinib screen performed with low concentration did not yield any kinase domain mutations (unpublished data). One possibility for this is a heterodimeric association of JAK1 and JAK2 or TYK2, as reported by Koppiker et al. study [48]. We found a 45-kDa novel JAK2 variant that alters kinase domain structure and generates ruxolitinib resistance in Ba/F3 cell line model system without ENU treatment suggests that novel short form of JAK2 varaints need to be analyzed in ruxolitinib resistant MPN patients [49].
In this study, we identified JAK2-V617F+L902Q, JAK2-V617F+Y931C and JAK2-V617F+L983F are the most frequent ruxolitinib resistant mutation. These mutations conferred resistance to ruxolitinib and cross-resistance to lestaurtinib and fedratinib. JAK2-V617F+L983F and V617F+L983F+Q959H identified in the ruxolitinib screen conferred cross-resistance to other JAK2 inhibitors but were very sensitive towards fedratinib and CHZ-868. In line with this study, Kesarvani et al. demonstrated that fedratinib binds to both ATP and substrate binding sites. Due to the ability of ATP- binding and substrate binding, fedratinib showed a complete lack of genetic resistance [36]. Our results suggest that ruxolitinib resistant variant L983F poses a significant challenge in MPN patients. However, fedratinib and type II JAK2 inhibitor CHZ-868 could be used in these patients. In contrast to type I JAK2 inhibitors, which bind JAK2 in a phosphorylated state, type II JAK2 inhibitor CHZ-868 which binds JAK2 in unphosphorylated form and is active against the JAK2-L902Q, JAK2-Y931 and JAK2-L983. In addition, CHZ-868 did not increase the JAK2 activation loop Tyr1007/1008 phosphorylation compared to type I inhibitors. These results are in line with Charlotte et al. study, where ATF7IP-JAK2 mediated acute lymphoblastic leukemia (ALL), CHZ-868 is potent against the ruxolitinib resistant variants such as Y931C and L983F [42]. These observations suggest that clinical evaluation of type II JAK2 inhibitor CHZ-868 is essential to guide future rational drug designing strategies to overcome drug resistance in JAK2 mediated diseases. In this study, we also created the ruxolitinib resistant homologous mutations in JAK1 and identified that L1010F (L983F in JAK2) exchange in JAK1 mediates the ruxolitinib resistance. Previously, it was well established in AML, that resistance against the FLT3-TKIs is also due to the acquisition of activation mutations in JAK1 and JAK3 [50]. Based on these observations, it is important to perform combination treatment approaches such as FLT3 inhibitors (Midostaurin or Glitaritinib) in combination with JAK-family kinase inhibitors (ruxolitinib or Momelotinib) to achieve better therapeutic response in AML. It is also noteworthy that L1010F exchange in JAK1 might pose a challenge as this variant drives the resistance against JAK1 inhibitors.
Similar to our study, a study by Deshpande et al. using the random mutagenesis method also identified R938L as a ruxolitinib resistant mutation [43]. They hypothesize that the R938L mutation changes the main chain conformation that affects the receptor binding and affinity due to their proximity to the binding pocket. In our screen, we have seen R938E instead of R938L. Substitution of Asp at this position is not enough to give strong drug resistance compared to Leu with a more hydrophobic nature. Although we used JAK2- V617F Ba/F3 cells to predict the ruxolitinib resistant mutations, one must focus on other JAK2 variants such as point mutations in JAK2 other than V617F (exon 12 mutations) [9], additional JAK2 point mutations in pediatric or adult B-ALL [51,52], or active form of JAK2 include TEL-JAK2 [14], PCM-JAK2 [53] and BCR-JAK2 [54] in order to predict resistance mechanisms against the JAK inhibitors.
Computational modeling studies further provide the evidence for the ruxolitinib resistance in L902Q, Y931C and L983F. In many studies, isolated JH1 domain is used to perform inhibitor-kinase interaction to define the residues that mediate the drug resistance phenotype. However, it is also essential to consider the performance of computational modeling together with the JH2 domain, as V617F is located in this domain, affecting the conformation of the kinase domain. This could be one reason for certain kinase domain residues whose binding energies are not significantly altered in molecular docking studies, however, biochemical results showed a strong resistance phenotype.
Heat shock protein 90 (HSP90) is a molecular chaperone that plays a major role in the maturation of several client proteins, such as fusion kinases and oncogenic proteins. Inhibition of the HSP90 pathway has been proven as a therapeutic strategy for the treatment of myeloma and other cancers [55]. Recently, it has been demonstrated that JAK2 is the client protein of HSP90. Treatment of JAK2-V617F cells with HSP90 inhibitors leads to the downregulation of JAK2 protein and HSP90 inhibitors are active in mice models of JAK2-V617F and MPL mediated MPNs [39]. In this study, we also demonstrated that HSP90 inhibition overcomes the ruxolitinib resistant mutations. Both 17-AAG and geldanamycin showed inhibition of ruxolitinib resistant mutations more effectively than JAK2-V617F and Parental Ba/F3 cells. We observed that JAK2-V617F+L902Q, JAK2-V617F+Y931C and JAK2-V617F+L983F mutations are sensitive to HSP90 inhibitors compared with cells lacking resistant mutation (Figure 7 A and B). This observation suggested that drug resistant mutations depend more on the HSP90 activity than non-resistant mutations (JAK2-V617F). Taken together, our study also provides a rationale that HSP90 inhibitors are the possible therapeutic agents in the case of JAK2 kinase inhibitor resistant mutations and suggests the importance of the clinical evolution of HSP90 inhibitors in drug resistant MPN patients.
4. Materials and Methods
4.1. Inhibitors
Ruxolitinib was a kind gift from Novartis Pharma AG, Basel, Switzerland. Fedratinib was purchased from Selleckchem (Houston, USA). Lestauritinib and CHZ-868 were purchased from Calbiochem. 17-AAG and Geldanamycin were purchased from Sigma-Aldrich (Taufkirchen, Germany). All the inhibitors were dissolved in dimethyl sulfoxide to make stock solutions of 10mM and stored at –20°C.
4.2. Cell culture and DNA constructs
Ba/F3 cells were obtained from the German Resource Centre for Biological Material (DSMZ) in 2005 and authenticated by DSMZ by DNA typing, species PCR and immunophenotyping. Cells were passaged for less than 6 months and were maintained in the presence of 2ng/ml interleukin-3 (R&D, Wiesbaden, Germany). Ba/F3 cells were transfected by retroviral gene transfer and transformed upon withdrawal of interleukin-3. JAK2 mutations were introduced in MSCV-EYFP-V617FJAK2 using the QuickChnage mutagenesis kit (Stratgene, Amsterdam, The Netherlands). Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). For RT-PCR of JAK2 encompassing the kinase domain, the following primers are used. JAK2 RT–KD for 5’gaaaatgacatgttaccaaatatg-3’ and JAK2 RT-KD rev 5’-ggagtaaacaaactgttaaag-3’. For sequencing the kinase domain, the following primers were used: 5’-ctagggttttctggtgcctttgaag-3’ and 5’-gggcgttgatttacattattgttcc-3’
4.3. Generation of drug-resistant variants
The selection of ruxolitinib resistant clones was described previously [56]. Briefly, Ba/F3 MSCV-EYFP-V617FJAK2 cells were cultured in 96–well plates at a density of 4 x 10E5 cells per well in the presence of ruxolitinib at indicated concentrations. Colonies that became visible were picked, expanded and analyzed. When indicated, Ba/F3 MSCV-EYFP-JAK2-V617F cells were pretreated with chemical mutagen N-ethyl-N-nitrosourea (ethyl nitrosourea) twice for 12h at a concentration of 50ug/ml. Resulting inhibitor-resistant sublines were cultured in the presence of inhibitor at a concentration corresponding to that used during the screen.
4.4. Proliferation assay
Proliferation was measured using an MTS (3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium)-based method by absorption of formazan at 490 nm (CellTiter 96; Promega, Madison, WI). Measures were taken as triplicates after 48 and 72 hours of culture without cytokines, as described previously [34]. IC50 value is calculated using Prism Software.
4.5. Western blot
Ba/F3 cells were cultured for 2.5 hours without and in the presence of inhibitor at the indicated concentrations. Cell lysis, sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotting were done as described previously [57]. JAK2, pSTAT5 and phosphotyrosine antibodies were purchased from Upstate Biotechnology (4G10 and PY20) (Biozol, Eching, Germany). STAT5 and pJAK2 antibodies were obtained from Santacruz Biotechnology (Heidelberg, Germany). Bands were visualized using the enhanced chemiluminescence (ECL) system (Amersham, Braunschweig, Germany).
4.6. Protein and Ligand Preparation
The high-resolution 3D structure of human tyrosine-protein JAK2 kinase JH1 domain (PDB ID: 7LL4) with exceptional resolution 1.31Å, is directly retrieved from the protein data bank, selected specifically for the in silico analysis. Alongside the wild-type structure of the JAK2 JH1 domain, three mutants (L902Q, Y931C, and L983F) were engineered using Maestro within the Schrodinger suite following standard protocols. All variants of the JAK2 JH1 domain structure, both wild-type and mutant, underwent optimization and successive minimization steps until reaching a 0.30 Å convergence, employing the OPLS-3e Force Field. Simultaneously, the ligands Ruxolitinib, Fedratinib, Lestaurtinib, and CHZ-868 were retrieved from PubChem. These ligands underwent conformational generation under standard pH conditions, generating up to 32 conformations per ligand, followed by minimization using the OPLS-3e force field.
4.7. Molecular Docking Simulation
The prepared wild-type and mutant proteins undergo active site prediction using the sitemap module, and the predicted sites are crosslinked with co-crystal-bound ligand structure. Subsequently, the predicted sites are manually picked for the Glide-based grid generation for positioning the ligands to be docked, including co-crystal ligands. The grid box is set at 2Å from the center radii. Successful grids for wild-type and mutant proteins enable docking with prepared ligands utilizing the eXtra Precision mode (XP) docking in the Glide module. The final binding positions are validated through MM/GBSA calculations. The best scoring pose, indicating optimal bonding, is visualized using the Maestro visualizer, and the corresponding scores are documented.
5. Conclusions
Using cell-based screening methodology, we were able to detect seven residues in the JAK2 kinase domain, which mediates the strong resistance towards ruxolitinib. Among seven variants, L902Q, Y931C and L983F were identified as frequent variants in JAK2-V617F. L983F exchange mediates cross-resistance across other JAK2 inhibitors and is sensitive towards the fedratinib and CHZ-868. Type II JAK2 inhibitor displayed more potency against the ruxolitinib variants. Finally, HSP90 inhibitors target the ruxolitinib resistant variants by JAK2 degradation. Thus, developing potent HSP90 inhibitors is important to overcome drug resistance mechanisms in ruxolitinib treated MPN patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
S.P.G. designed the study, S.P.G., G.P., J.O. and V.B. performed experiments and analyzed data; D.C.D performed and analyzed structural studies; N.v.B. and J.D. provided critical materials, suggested experiments and reviewed the manuscript; S.P.G wrote the manuscript.
Funding
S.P.G; O.J. and N.v.B. are supported by DFG (3554/1-3). G.P. is funded by DAAD (91865598). V.B is funded by LeoPharma.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable for this study.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Acknowledgments
The authors would like to thank their funding agencies. S.P.G. and N.v.B. are supported by DFG (3554/1-3). G.P. was funded by DAAD fellowship. Graphical abstract was created using Biorender software (Biorender.com). We are thankful for Chandrabose SelvaRaj (C.S.) and Kumar Reddy Kakularam for structural analysis and discussion. C.S thankfully acknowledged the professor Sanjeev Kumar Singh, Alagappa university, for providing the technical support in Schrodinger, maestro.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
JAK2 - Janus Kinase 2; HSP90-Heat Shock Protein 90; FDA-Food and Drug Administration; MPN-Myeloproliferative Neoplasms; PV-Polycythemia Vera; JH2-pseudokinase domain; ALL-Acute Lymphocytic Leukemia; AML-Acute Myeloid Leukemia; CML-Chronic Myelogenous Leukemia; TKI-Tyrosine Kinase Inhibitor; BCR-B Cell Receptor; NSCLC-Non-Small-Cell Lung Cancer; GIST-Gastrointestinal Stromal Tumors; ENU-Ethyl-Nitrosourea; ATP-Adenosine Triphosphate; IL-3-Interleukin-3; STAT5-Signal Transducer and Activator of Transcription 5; IC50-Half-Maximal Inhibitory Concentration; WT-Wild Type; MTS-3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; 17-AAG-17-allylamino-17-demethoxy- geldanamycin; FLT3-FMS-like Tyrosine Kinase 3; TYK2-Tyrosine Kinase 2; MPL-Myeloproliferative Leukemia.
References
- Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U & Pfeffer K (1998) Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93, 397-409. [CrossRef]
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G & Ihle JN (1998) Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93, 385-395. [CrossRef]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN & Green AR (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054-1061. [CrossRef]
- James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N & Vainchenker W (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144-1148. [CrossRef]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M & Skoda RC (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352, 1779-1790. [CrossRef]
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ & Gilliland DG (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387-397. [CrossRef]
- Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, Berger R, Clark JJ, Willis SG, Nguyen KT, Flores NJ, Estey E, Gattermann N, Armstrong S, Look AT, Griffin JD, Bernard OA, Heinrich MC, Gilliland DG, Druker B & Deininger MW (2005) The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 106, 3377-3379. [CrossRef]
- Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG & Tefferi A (2005) The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both "atypical" myeloproliferative disorders and myelodysplastic syndromes. Blood 106, 1207-1209. [CrossRef]
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF & Green AR (2007) JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 356, 459-468. [CrossRef]
- Grunebach F, Bross-Bach U, Kanz L & Brossart P (2006) Detection of a new JAK2 D620E mutation in addition to V617F in a patient with polycythemia vera. Leukemia 20, 2210-2211. [CrossRef]
- Schnittger S, Bacher U, Kern W, Schröder M, Haferlach T & Schoch C (2006) Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia 20, 2195-2197. [CrossRef]
- Kratz CP, Böll S, Kontny U, Schrappe M, Niemeyer CM & Stanulla M (2006) Mutational screen reveals a novel JAK2 mutation, L611S, in a child with acute lymphoblastic leukemia. Leukemia 20, 381-383. [CrossRef]
- Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, Macintyre EA, Villeval JL, Vainchenker W, Berger R, Bernard OA, Delabesse E & Penard-Lacronique V (2007) Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood 109, 2202-2204. [CrossRef]
- Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, Berthou C, Lessard M, Berger R, Ghysdael J & Bernard OA (1997) A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 278, 1309-1312. [CrossRef]
- Najfeld V, Cozza A, Berkofsy-Fessler W, Prchal J & Scalise A (2007) Numerical gain and structural rearrangements of JAK2, identified by FISH, characterize both JAK2617V>F-positive and -negative patients with Ph-negative MPD, myelodysplasia, and B-lymphoid neoplasms. Exp Hematol 35, 1668-1676. [CrossRef]
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gönen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA & Polyak K (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J Clin Invest 121, 2723-2735. [CrossRef]
- Sonbol MB, Firwana B, Zarzour A, Morad M, Rana V & Tiu RV (2013) Comprehensive review of JAK inhibitors in myeloproliferative neoplasms. Ther Adv Hematol 4, 15-35. [CrossRef]
- Pardanani A, Tefferi A, Jamieson C, Gabrail NY, Lebedinsky C, Gao G, Liu F, Xu C, Cao H & Talpaz M (2015) A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J 5, e335. [CrossRef]
- Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R & Tefferi A (2010) Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 363, 1117-1127. [CrossRef]
- Bose P & Verstovsek S (2020) JAK Inhibition for the Treatment of Myelofibrosis: Limitations and Future Perspectives. Hemasphere 4, e424. [CrossRef]
- Kvasnicka HM, Thiele J, Bueso-Ramos CE, Sun W, Cortes J, Kantarjian HM & Verstovsek S (2018) Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J Hematol Oncol 11, 42. [CrossRef]
- Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, Harrison CN, Paquette R, Sun W, Naim A, Langmuir P, Dong T, Gopalakrishna P & Gupta V (2017) Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 10, 156. [CrossRef]
- Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH, Jr., Arcasoy MO, Hexner EO, Lyons RM, Paquette R, Raza A, Jones M, Kornacki D, Sun K & Kantarjian H (2017) Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 10, 55. [CrossRef]
- Pandey G, Kuykendall AT & Reuther GW (2022) JAK2 inhibitor persistence in MPN: uncovering a central role of ERK activation. Blood Cancer J 12, 13. [CrossRef]
- Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, Kennedy D, Estrov Z, Cortes J & Verstovsek S (2010) Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 115, 1131-1136. [CrossRef]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN & Sawyers CL (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876-880. [CrossRef]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG & Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2, e73. [CrossRef]
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, Bertulli R, Colecchia M, Casali PG, Pierotti MA & Pilotti S (2004) A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 127, 294-299. [CrossRef]
- Azam M, Latek RR & Daley GQ (2003) Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831-843. [CrossRef]
- Lee TS, Potts SJ & Albitar M (2009) Basis for resistance to imatinib in 16 BCR-ABL mutants as determined using molecular dynamics. Recent Pat Anticancer Drug Discov 4, 164-173. [CrossRef]
- von Bubnoff N, Schneller F, Peschel C & Duyster J (2002) BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet 359, 487-491. [CrossRef]
- Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, Toms AV, Marubayashi S, Christie AL, McKeown M, Paranal RM, Bradner JE, Yoda A, Gaul C, Vangrevelinghe E, Romanet V, Murakami M, Tiedt R, Ebel N, Evrot E, De Pover A, Regnier CH, Erdmann D, Hofmann F, Eck MJ, Sallan SE, Levine RL, Kung AL, Baffert F, Radimerski T & Weinstock DM (2012) Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med 209, 259-273. [CrossRef]
- Griswold IJ, MacPartlin M, Bumm T, Goss VL, O’Hare T, Lee KA, Corbin AS, Stoffregen EP, Smith C, Johnson K, Moseson EM, Wood LJ, Polakiewicz RD, Druker BJ & Deininger MW (2006) Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol 26, 6082-6093. [CrossRef]
- Gorantla SP, Dechow TN, Grundler R, Illert AL, Zum Buschenfelde CM, Kremer M, Peschel C & Duyster J (2010) Oncogenic JAK2V617F requires an intact SH2-like domain for constitutive activation and induction of a myeloproliferative disease in mice. Blood 116, 4600-4611. [CrossRef]
- Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, Marque F, Régnier CH, De Pover A, Ryckelynck H, Bhagwat N, Koppikar P, Goel A, Wyder L, Tavares G, Baffert F, Pissot-Soldermann C, Manley PW, Gaul C, Voshol H, Levine RL, Sellers WR, Hofmann F & Radimerski T (2012) Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov 2, 512-523. [CrossRef]
- Kesarwani M, Huber E, Kincaid Z, Evelyn CR, Biesiada J, Rance M, Thapa MB, Shah NP, Meller J, Zheng Y & Azam M (2015) Targeting substrate-site in Jak2 kinase prevents emergence of genetic resistance. Sci Rep 5, 14538. [CrossRef]
- Meyer SC, Keller MD, Chiu S, Koppikar P, Guryanova OA, Rapaport F, Xu K, Manova K, Pankov D, O’Reilly RJ, Kleppe M, McKenney AS, Shih AH, Shank K, Ahn J, Papalexi E, Spitzer B, Socci N, Viale A, Mandon E, Ebel N, Andraos R, Rubert J, Dammassa E, Romanet V, Dolemeyer A, Zender M, Heinlein M, Rampal R, Weinberg RS, Hoffman R, Sellers WR, Hofmann F, Murakami M, Baffert F, Gaul C, Radimerski T & Levine RL (2015) CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 28, 15-28. [CrossRef]
- Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A, Christie AL, Liu H, Christodoulou A, van Bodegom D, van der Zwet J, Layer JV, Tivey T, Lane AA, Ryan JA, Ng SY, DeAngelo DJ, Stone RM, Steensma D, Wadleigh M, Harris M, Mandon E, Ebel N, Andraos R, Romanet V, Dolemeyer A, Sterker D, Zender M, Rodig SJ, Murakami M, Hofmann F, Kuo F, Eck MJ, Silverman LB, Sallan SE, Letai A, Baffert F, Vangrevelinghe E, Radimerski T, Gaul C & Weinstock DM (2015) Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 28, 29-41. [CrossRef]
- Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, Caldas-Lopes E, Ross KN, Gönen M, Gozman A, Ahn JH, Rodina A, Ouerfelli O, Yang G, Hedvat C, Bradner JE, Chiosis G & Levine RL (2010) HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest 120, 3578-3593. [CrossRef]
- Wardrop D & Steensma DP (2009) Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haematol 144, 809-817. [CrossRef]
- Apperley JF (2007) Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol 8, 1018-1029. [CrossRef]
- Downes CEJ, McClure BJ, Bruning JB, Page E, Breen J, Rehn J, Yeung DT & White DL (2021) Acquired JAK2 mutations confer resistance to JAK inhibitors in cell models of acute lymphoblastic leukemia. NPJ Precis Oncol 5, 75. [CrossRef]
- Deshpande A, Reddy MM, Schade GO, Ray A, Chowdary TK, Griffin JD & Sattler M (2012) Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia 26, 708-715. [CrossRef]
- Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y & et al. (1993) Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 92, 1736-1744. [CrossRef]
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R & Naoe T (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97, 2434-2439. [CrossRef]
- Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu SN, Knoops L & Renauld JC (2011) Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica 96, 845-853. [CrossRef]
- Marit MR, Chohan M, Matthew N, Huang K, Kuntz DA, Rose DR & Barber DL (2012) Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS One 7, e43437. [CrossRef]
- Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, Liu F, Saunders LM, Mullally A, Abdel-Wahab O, Leung L, Weinstein A, Marubayashi S, Goel A, Gönen M, Estrov Z, Ebert BL, Chiosis G, Nimer SD, Bernstein BE, Verstovsek S & Levine RL (2012) Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155-159. [CrossRef]
- Gorantla SP, Mueller TA, Albers-Leischner C, Rudelius M, von Bubnoff N & Duyster J (2023) A newly identified 45-kDa JAK2 variant with an altered kinase domain structure represents a novel mode of JAK2 kinase inhibitor resistance. Mol Oncol. [CrossRef]
- Rummelt C, Gorantla SP, Meggendorfer M, Charlet A, Endres C, Dohner K, Heidel FH, Fischer T, Haferlach T, Duyster J & von Bubnoff N (2021) Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia 35, 2017-2029. [CrossRef]
- Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR & Izraeli S (2008) Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet 372, 1484-1492. [CrossRef]
- Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, Devidas M, Atlas SR, Chen IM, Clifford RJ, Gerhard DS, Carroll WL, Reaman GH, Smith M, Downing JR, Hunger SP & Willman CL (2009) JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106, 9414-9418. [CrossRef]
- Kaplan HG, Jin R, Bifulco CB, Scanlan JM & Corwin DR (2022) PCM1-JAK2 Fusion Tyrosine Kinase Gene-Related Neoplasia: A Systematic Review of the Clinical Literature. Oncologist 27, e661-e670. [CrossRef]
- Elnaggar MM, Agersborg S, Sahoo T, Girgin A, Ma W, Rakkhit R, Zorrilla I & Leal A (2012) BCR-JAK2 fusion as a result of a translocation (9;22)(p24;q11.2) in a patient with CML-like myeloproliferative disease. Mol Cytogenet 5, 23. [CrossRef]
- Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA & Anderson KC (2011) Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol 152, 367-379. [CrossRef]
- von Bubnoff N, Gorantla SP, Engh RA, Oliveira TM, Thone S, Aberg E, Peschel C & Duyster J (2011) The low frequency of clinical resistance to PDGFR inhibitors in myeloid neoplasms with abnormalities of PDGFRA might be related to the limited repertoire of possible PDGFRA kinase domain mutations in vitro. Oncogene 30, 933-943. [CrossRef]
- Duyster J, Baskaran R & Wang JY (1995) Src homology 2 domain as a specificity determinant in the c-Abl-mediated tyrosine phosphorylation of the RNA polymerase II carboxyl-terminal repeated domain. Proceedings of the National Academy of Sciences of the United States of America 92, 1555-1559. [CrossRef]
Figure 1.
Spectrum and relative frequency of the mutations identified in the ruxolitinib screen: Single clones of Ba/F3 cells growing in 96-well plates in the presence of 4μM and 8μM ruxolitinib were picked and analyzed for the presence of JAK2 kinase domain mutations. Shown is the frequency of resistant clones per million cells (A). Resistant clones grown in the presence of 4μM ruxolitinib were expanded and analyzed for kinase domain mutations. Shown are the relative frequency of each mutation in 4μM ruxolitinib concentration (B). Similarly, relative frequency of mutations in 8μM ruxolitinib concentration is shown (C). Location of the putative JAK2 inhibitor resistant mutations is shown (D). Alignment of homologous regions in JAK2 and ABL1 (E). The blue arrows indicate the mutations identified in BCR-ABL reported to confer imatinib resistance in patients. Red arrows indicate the mutations identified by in vitro mutagenesis screening method.
Figure 1.
Spectrum and relative frequency of the mutations identified in the ruxolitinib screen: Single clones of Ba/F3 cells growing in 96-well plates in the presence of 4μM and 8μM ruxolitinib were picked and analyzed for the presence of JAK2 kinase domain mutations. Shown is the frequency of resistant clones per million cells (A). Resistant clones grown in the presence of 4μM ruxolitinib were expanded and analyzed for kinase domain mutations. Shown are the relative frequency of each mutation in 4μM ruxolitinib concentration (B). Similarly, relative frequency of mutations in 8μM ruxolitinib concentration is shown (C). Location of the putative JAK2 inhibitor resistant mutations is shown (D). Alignment of homologous regions in JAK2 and ABL1 (E). The blue arrows indicate the mutations identified in BCR-ABL reported to confer imatinib resistance in patients. Red arrows indicate the mutations identified by in vitro mutagenesis screening method.
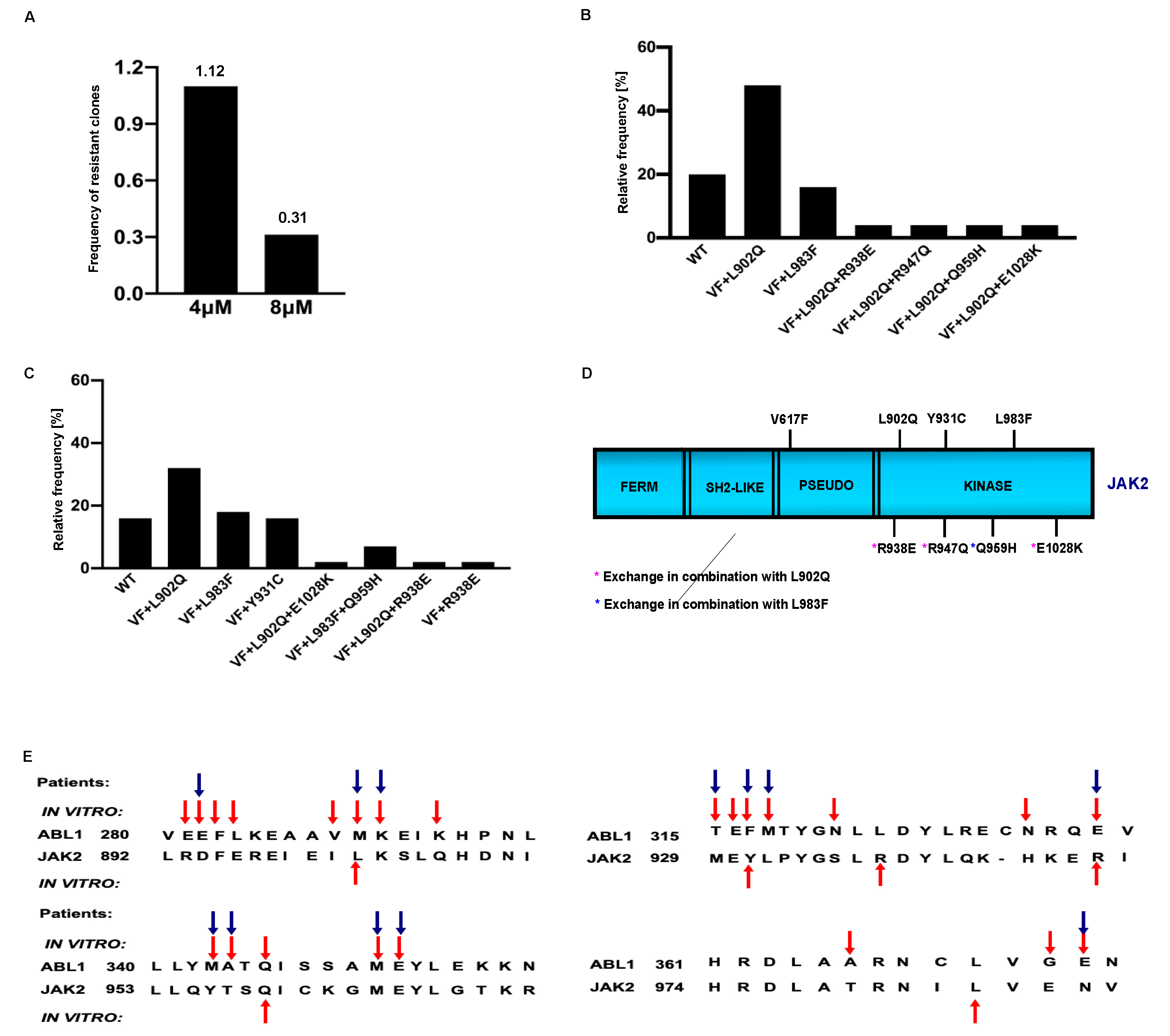
Figure 2.
Resistant mutations identified in the ruxolitinib screen transform the Ba/F3 cells and display constitutive activation of JAK2 and STAT5: Mutations identified from 4 and 8μM screen were cloned in JAK2-V617F background either as single mutation or compound mutations and stably expressed in Ba/F3 cells by retroviral gene transfer method. The transformation ability of the mutant JAK2s was measured using (MTS)- based method after 72hrs without IL-3 (n=3). JAK2-Y931C showed enhanced cell growth compared to JAK2-V617F, whereas the remaining JAK2 mutants showed equal transformation ability (A). Western blot analysis was performed using Ba/F3 cells expressing the JAK2 mutants and measured the activation of JAK2 and STAT5 (B). A representative image of n=2 two independent experiments is shown.
Figure 2.
Resistant mutations identified in the ruxolitinib screen transform the Ba/F3 cells and display constitutive activation of JAK2 and STAT5: Mutations identified from 4 and 8μM screen were cloned in JAK2-V617F background either as single mutation or compound mutations and stably expressed in Ba/F3 cells by retroviral gene transfer method. The transformation ability of the mutant JAK2s was measured using (MTS)- based method after 72hrs without IL-3 (n=3). JAK2-Y931C showed enhanced cell growth compared to JAK2-V617F, whereas the remaining JAK2 mutants showed equal transformation ability (A). Western blot analysis was performed using Ba/F3 cells expressing the JAK2 mutants and measured the activation of JAK2 and STAT5 (B). A representative image of n=2 two independent experiments is shown.
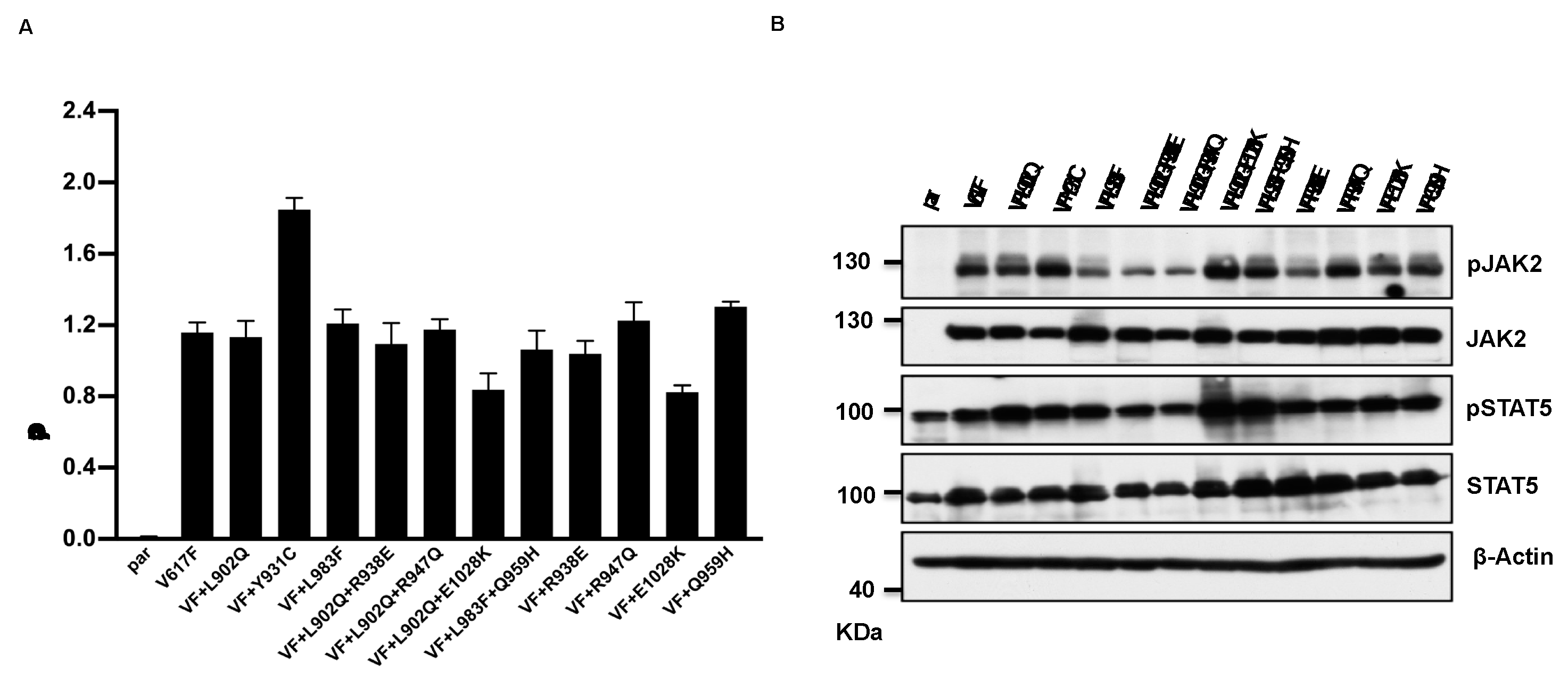
Figure 3.
JAK2 kinase domain mutations L902Q, Y931C and L983F displayed resistance phenotype towards the ruxolitinib: JAK2 mutants that were identified with the ruxolitinib including single constituents of compound mutations were recreated in JAK2-V617F using site-directed mutagenesis. Constructs were stably expressed in Ba/F3 cells. Proliferation was measured using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium (MTS)- based method after incubation for 48hrs without and in the presence of increasing concentration of the inhibitor ruxolitinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with the indicated concentration of ruxolitinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Figure 3.
JAK2 kinase domain mutations L902Q, Y931C and L983F displayed resistance phenotype towards the ruxolitinib: JAK2 mutants that were identified with the ruxolitinib including single constituents of compound mutations were recreated in JAK2-V617F using site-directed mutagenesis. Constructs were stably expressed in Ba/F3 cells. Proliferation was measured using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium (MTS)- based method after incubation for 48hrs without and in the presence of increasing concentration of the inhibitor ruxolitinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with the indicated concentration of ruxolitinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
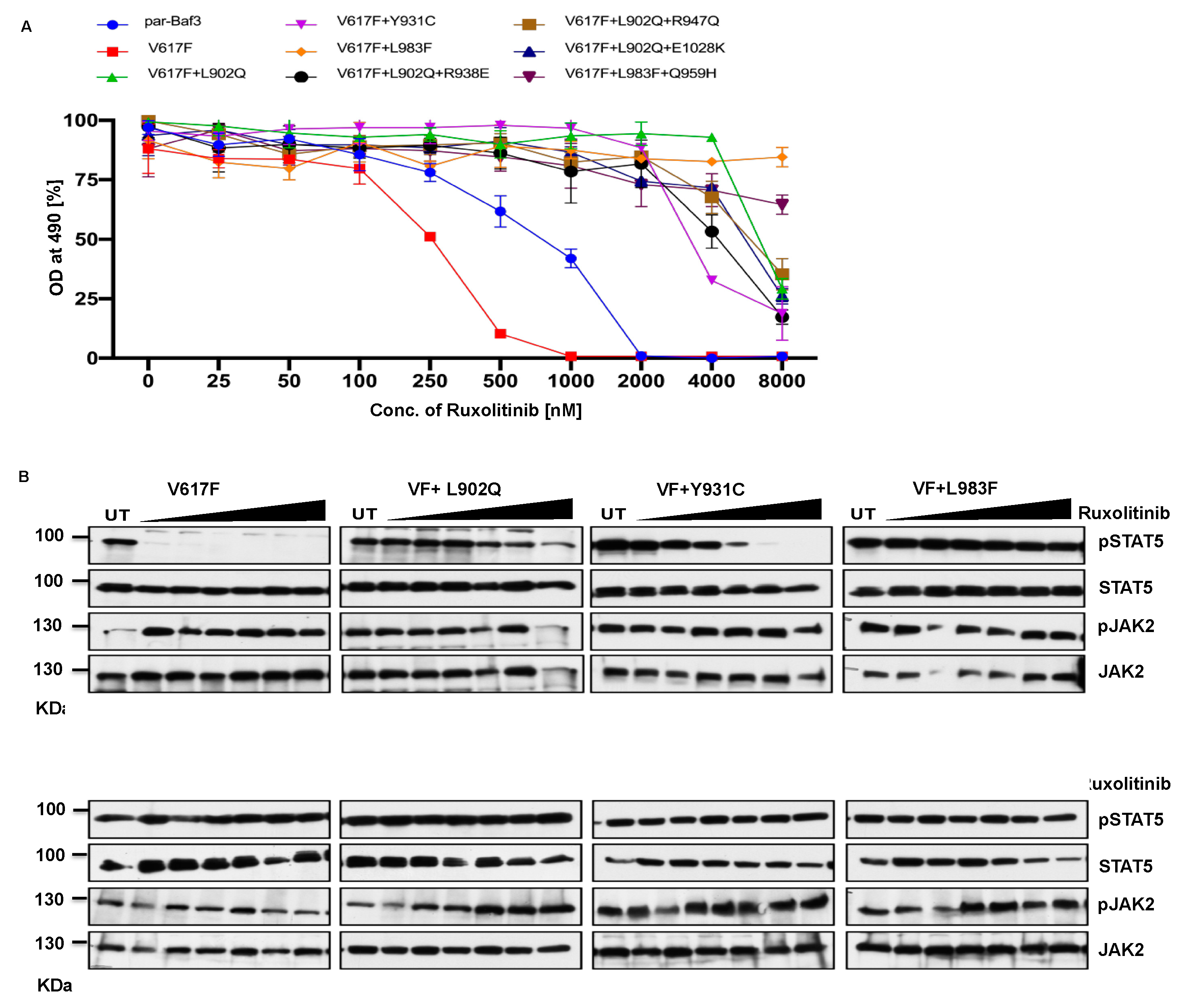
Figure 4.
L983F and Y931C display sensitivity towards fedratinib: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of fedratinib. Proliferation was measured using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium (MTS)- based method after incubation for 48hrs in the presence of increasing concentration of the inhibitor fedratinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of fedratinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Figure 4.
L983F and Y931C display sensitivity towards fedratinib: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of fedratinib. Proliferation was measured using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium (MTS)- based method after incubation for 48hrs in the presence of increasing concentration of the inhibitor fedratinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of fedratinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
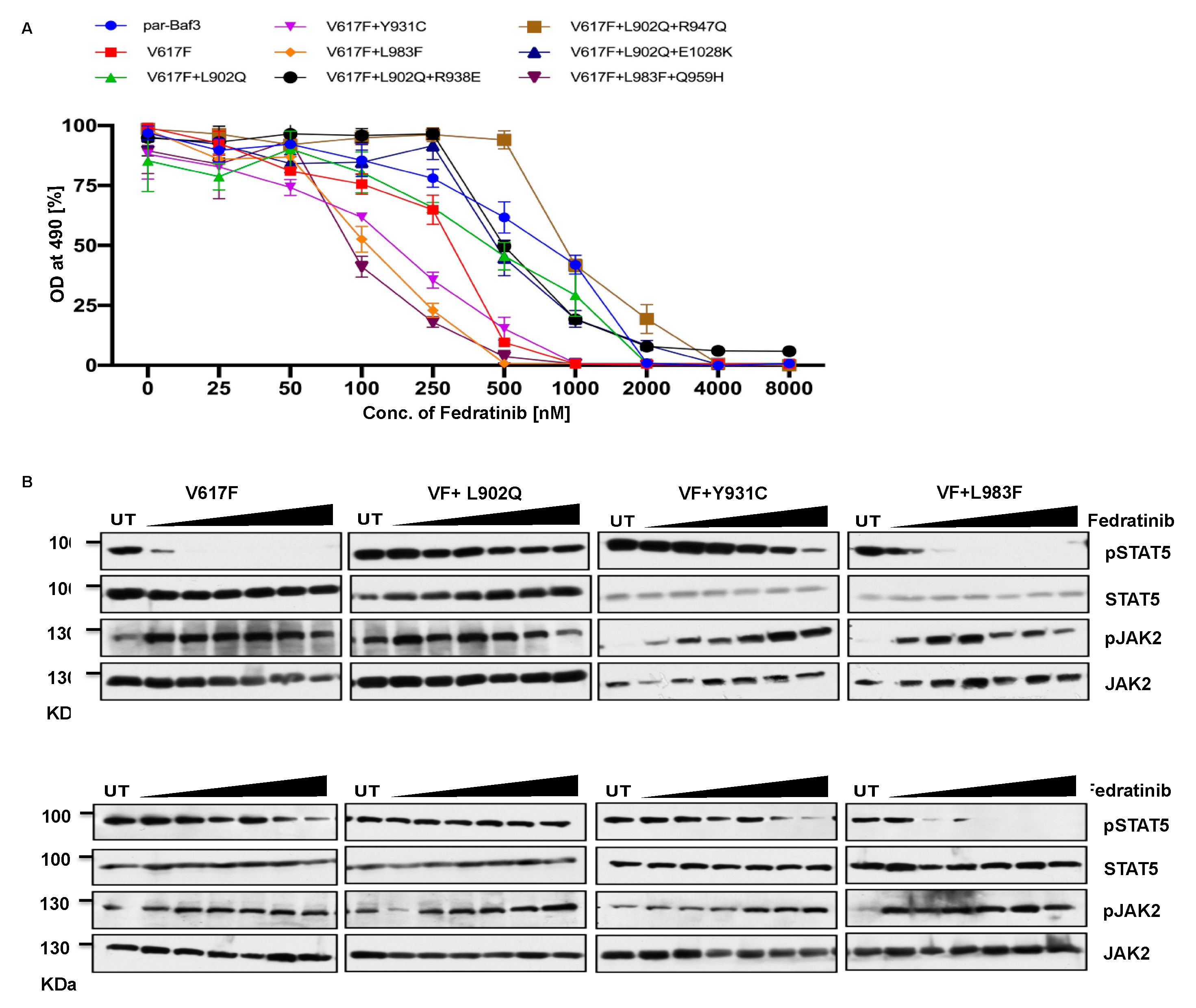
Figure 5.
L983F and Y931C display sensitivity, whereas L902Q is resistant towards lestaurtinib: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of lestaurtinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of lestaurtinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Figure 5.
L983F and Y931C display sensitivity, whereas L902Q is resistant towards lestaurtinib: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of lestaurtinib (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of lestaurtinib (0, 250, 500, 1000, 2000, 4000 and 8000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
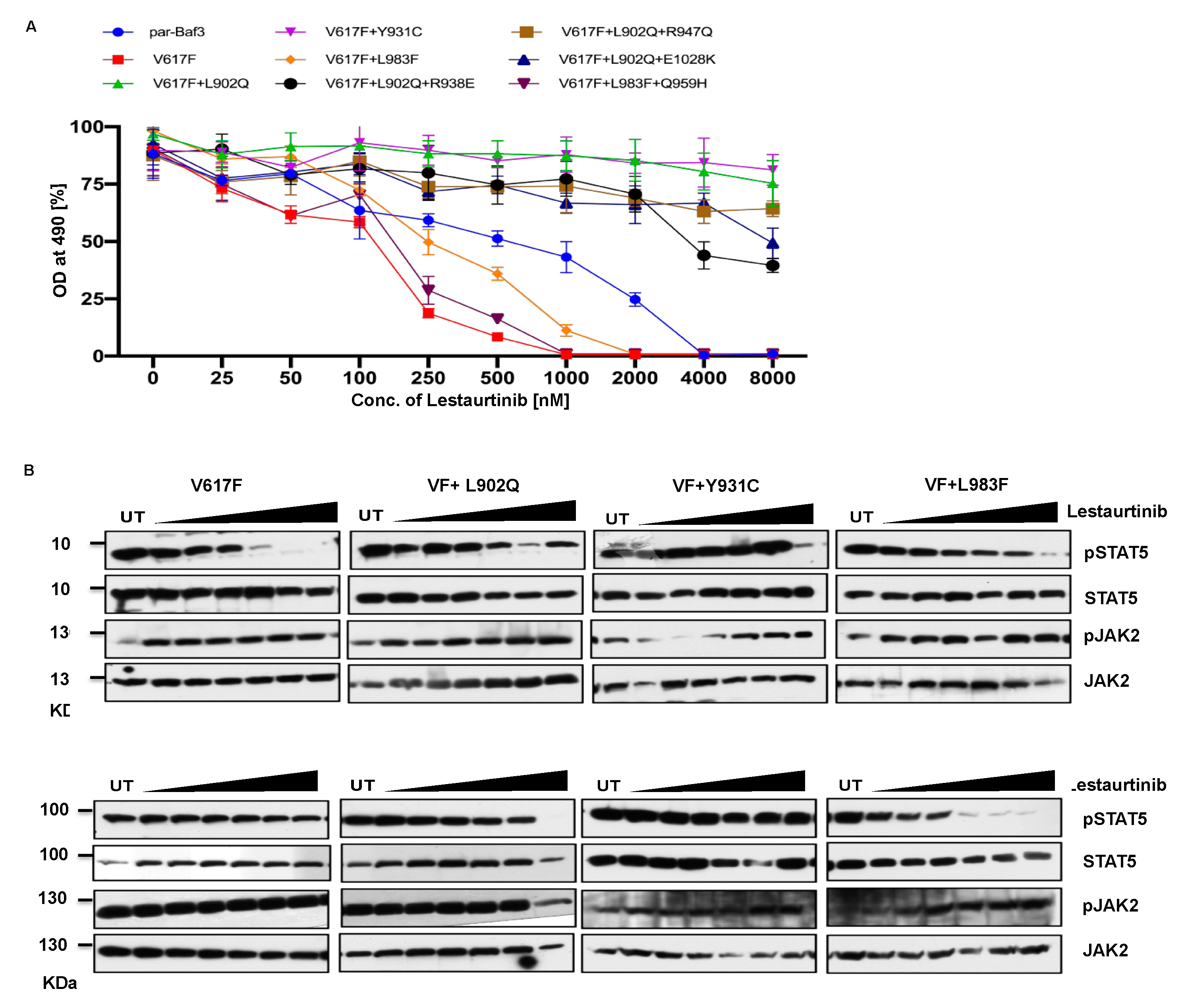
Figure 6.
Type II JAK2 inhibitor CHZ-868 is more potent towards ruxolitinib resistant variants: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of type II JAK2 inhibitor CHZ-868 with the indicated concentrations (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentration of CHZ-868 (0, 25, 50, 100, 200, 400 and 800nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Figure 6.
Type II JAK2 inhibitor CHZ-868 is more potent towards ruxolitinib resistant variants: Mutations that emerged in the ruxolitinib screen were reengineered and expressed in the Ba/F3 cells. Cells were incubated in the presence of type II JAK2 inhibitor CHZ-868 with the indicated concentrations (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentration of CHZ-868 (0, 25, 50, 100, 200, 400 and 800nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
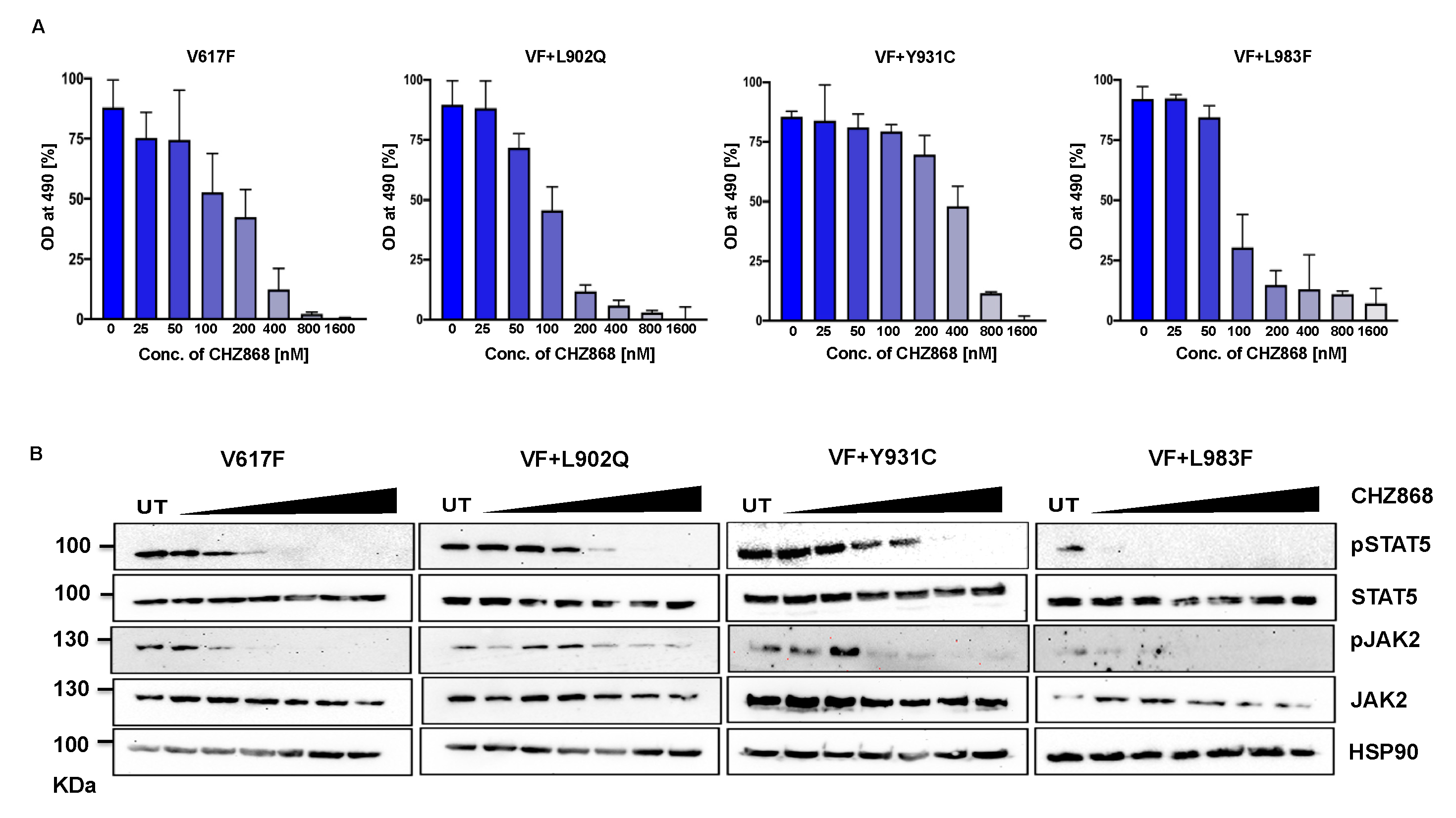
Figure 7.
HSP 90 inhibitors target JAK2-V617F and overcome resistance to ATP-competitive inhibitors: Mutations that are identified in the ruxolitinib were stably expressed in the Ba/F3 cells and proliferation was measured after incubation for 48 hrs in the presence of HSP 90 inhibitor 17-AAG (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of 17-AAG (0, 50, 100, 200, 400, 800 and 1000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown. Similarly, Ba/F3 cells expressing JAK2 mutations were treated with indicated concentrations of Geldanamycin (C). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of Geldanamycin (0, 50, 100, 200, 400, 800 and 1000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Figure 7.
HSP 90 inhibitors target JAK2-V617F and overcome resistance to ATP-competitive inhibitors: Mutations that are identified in the ruxolitinib were stably expressed in the Ba/F3 cells and proliferation was measured after incubation for 48 hrs in the presence of HSP 90 inhibitor 17-AAG (A). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of 17-AAG (0, 50, 100, 200, 400, 800 and 1000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown. Similarly, Ba/F3 cells expressing JAK2 mutations were treated with indicated concentrations of Geldanamycin (C). Data is shown as mean ± standard deviation (SD) (n=3). OD – optical density. Immunoblot analysis of Ba/F3 cells expressing JAK2 mutants cultured with increasing concentrations of Geldanamycin (0, 50, 100, 200, 400, 800 and 1000nM) for 4hrs and lysates were subjected to indicated antibodies (B). A representative image of n=2 two independent experiments is shown.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



