
Submitted:
04 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
Previous studies found high but very variable levels of tetranor-PGEM and PGD-M (urine metabolites of prostaglandin (PG) E2 and PGD2, respectively) in persons with cystic fibrosis (pwCF), which correlated with the severity of the CF transmembrane receptor (CFTR) genetic alteration. This study aims to assess the potential role of cyclooxygenase (COX)-1 and COX-2 genetic polymorphisms in the variability of PG production, and determine the potential role of PG metabolites as markers of symptoms severity and imaging findings. Thirty healthy subjects and 103 pwCF were included in the study. Clinical and radiological CF severity was evaluated using clinical scoring methods and the extent of bronchiectasis and air trapping were assessed by chest computed tomography (CT). Urine metabolites were measured using liquid chromatography/tandem mass spectrometry. The presence of variants in the COX-1 gen (PTGS1 639 C>A, PTGS1 762+14delA, and COX-2 gen: PTGS2-899G>C (-765G>C) and PTGS2 (8473T>C) were analysed. PGE-M and PGD-M urine concentrations were significantly higher in pwCF than in healthy controls. There were also statistically significant differences between clinically mild and moderate disease and severe disease. Patients with bronchiectasis and/or air trapping had higher PGE-M levels than patients without these complications. The four polymorphisms did not associate with clinical severity, air trapping, bronchiectasis, or urinary PG levels. Taken together, the present study suggests that urinary PG level testing can be used as a biomarker of CF severity. We found no relationship between COX-1 and COX-2 genetic polymorphisms in the variability of PG production.
Keywords:
cystic fibrosis
; prostaglandin E2
; prostaglandin D2
; genetic polymorphisms
; PTGS1
; PTGS2
; COX-1
; COX-2
1. Introduction
Cystic fibrosis (CF) is caused by mutations within the CF transmembrane conductance regulator (CFTR) gene leading to defective epithelial chloride transport in many organs. More than 2114 variants in the CFTR gene have been identified (1). Various variants can be grouped into different classes based on their known or predicted molecular mechanisms of dysfunction and the functional consequences for the CFTR protein [2].
Class I: Pathogenic variants in this category are associated with lack of biosynthesis or defective biosynthesis, resulting in no CFTR protein. Class II: These gene variants fail to properly process the protein to a mature form and fail to transport the protein to the apical membrane. Class III: variants of this class affect the regulation of CFTR function by preventing ATP binding at the nucleotide-binding domains required for channel activation. Class IV: variants affect the chloride conductance resulting in a normal amount of CFTR with some residual function at the apical membrane. Class V: variants may be associated with reduced biosynthesis of a fully active CFTR protein. Class VI: variants destabilise CFTR protein at the cell surface, and produce high turnover of CFTR. Class VII: large deletions
A graduated risk of developing pancreatic insufficiency and pancreatitis, according to genotype severity has been reported in various studies [3,4]. Class I, II, and III pathogenic variants are usually associated with pancreatic insufficiency and, thus, are considered severe variants, while class IV and V variants retain residual function and are usually associated with normal or slightly altered pancreatic function, being classified as mild [3,4] Numerous studies assessing the impact of CFTR variants on the severity and progression of lung disease have presented discrepant results, some showing correlations between CFTR genotypes and lung disease [5,6,7,8,9], while others did not find any consistent association [10,11]. The poor correlation between the severity of the CFTR variants and the severity of the lung disease supports the notion that other cofactors contribute to the modulation of the phenotypic expression of the primary genotype. Due to the direct contact of the lung with microbial pathogens and airborne pollutants, many modulating factors might contribute to the variable lung phenotype observed in patients with the same genotype [12].
An early, sustained, and severe inflammatory process is seen in the airways of pwCF [13], which is characterized by excessive mucus production, chronic bacterial infection, and progressive tissue damage. Airway infection occurs early in the course of the disease; however, there are observations which support that lung inflammation is, at least in part, independent of infections and being directly related to defective CFTR [14].
Various CFTR variants related with fatty acid metabolism abnormalities have been reported in CF, such as an abnormally high arachidonic acid (AA) to docosahexaenoic acid (DHA) ratio, and a linoleic acid (LA) deficiency, which is directly related to the severity of the CFTR variants [11,15,16,17].
AA release from membrane glycerophospholipids is the rate-limiting step in the enhanced production of eicosanoids such as prostaglandin E2 (PGE2) [18]. There are two cyclooxygenase (COX) enzymes, COX-1 and COX-2, involved in the conversion of AA to PGE2. COX-1 is constitutively expressed in most cells and is involved in the regulation of physiological functions, whereas COX-2 expression is rapidly induced under inflammatory conditions [18]. Increased expression of both COX-1 and COX-2 is found in CF airways [19], which accounts for the increased PGE2 production reported in these patients [20,21].
Several findings support the concept that the enhanced COX-2 expression and increased PGE2 production found in CF are directly related to CFTR dysfunction rather than to the presence of an inflammatory process associated with chronic bacterial infection [22,23]. Chen et al., [23] demonstrated that the COX-2/PGE2 positive feedback loop is negatively regulated by CFTR under normal conditions but augmented with defective CFTR. Borrot et al., showed that CFTR inhibition lead to increased eicosanoid release [24] . Moreover, the absence of CFTR can disrupt cellular signalling networks with broad functional consequences including fatty acid abnormalities [25,26].
Elevated levels of tetranor-PGEM (PGE-M), the PGE2 metabolite detected in urine, have been associated with the severity of the CFTR variants [22]. However, marked differences can be found in urine PGE-M levels among patients with similar CFTR variants, suggesting that PGE2 production is regulated by factors other than the severity of the mutated receptor [22].
Interestingly, one study found that some COX-1 and COX-2 gene polymorphisms were associated with different effects on the severity of lung disease in CF patients with the F508del pathogenic variant [27]. However, it is still unclear whether these polymorphisms play any significant role in PGE2 production and, thereby, in disease severity.
We hypothesised that the severity of the lung disease in CF correlates with the amount of PGE2 released, which in turn is related to the severity of the mutated CFTR and further regulated by the presence of some COX polymorphisms. We undertook the present study to test this hypothesis.
2. Subjects and Methods
2.1. Subjects
The study had a cross-sectional design. Thirty healthy subjects (12 male, 18 female) aged from 5 to 22 years (11.5±0.75), and 103 pwCF (56 male, 47 female) aged from 4 to 24 years (12.68±0.48) with stable CF, were included in the study. PwCF were recruited from a single paediatric CF centre (Hospital Universitari Vall d’Hebrón, Barcelona, Spain). The CF diagnosis was established based on clinical data, abnormal sweat test (sweat chloride >60 mmol/L), and genotypic characteristics consistent with CF. PwCF were in a stable clinical condition at a regular follow-up visit. Healthy control children were recruited from families of hospital workers. The demographic characteristics of patients and healthy subjects were not statistically different. Clinical and radiological characteristics of pwCF are shown in Table 1. All participants provided informed consent, with parents giving informed consent, prior to enrolment. The study was approved by the institutional Ethics Committee.
2.2. Methods
Forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) were measured by spirometry in pwCF, and the best of three manoeuvres, expressed as percentage of predicted values, was chosen.
Pancreatic sufficiency was defined as the presence of a faecal elastase value >200 μg.
All patients were genotyped using methods reported elsewhere [28]. They were classified into three groups (mild, moderate, and severe) based on the predicted functional consequences of the CFTR protein alteration, and on the accepted premise of the dominant phenotypic effect conferred by the milder of the two CFTR pathogenic variants. PwCF carrying Class I,II and Class III pathogenic variants in their alleles were considered severe, those carrying Class I,II or III mutations in one allele associated with a Class IV, V or VI in the second allele were classified as moderate, and those pwCF with any combination of Class IV,V,VI in both alleles were considered mild. PwCF were divided into three clinical groups according to severity, which was established considering the frequency of upper airway infections, pulmonary exacerbations requiring antibiotic therapy, and the number of pulmonary exacerbations requiring hospitalizations and intravenous antibiotic therapy (Table 1).
Each chest computed tomography (CT) consisted of a volumetric inspiratory and expiratory acquisition. All CTs were scored evaluating the six (lingula as a separate lobe) lung lobes for the presence of central and peripheral bronchiectasis and the extent of trapped air on expiratory CTs. Bronchiectasis was scored as 0 (no bronchiectasis), 1 (one lobe affected), 2 (two lobes affected), or 3 (three or more lobes affected). Similarly, the extension of air trapping was scored from 0 to 3. All scans were scored by a blinded observer radiologist.
Routinely, an airway sample (sputum or cough swab) was collected when pwCF attended the outpatient centre every 1-2 months. These samples were incubated in different media for the identification of bacterial and fungal organisms via standard culture protocols.
Urine samples were collected from the participants and stored at -80ºC until analysis. Measurement of PGE-M and PGD-M, final urinary metabolites of PGE2 and prostaglandin D2 (PGD2), respectively, is considered the best method to accurately assess the biosynthesis of PGE2 and PGD2 generated via the COX pathway [20]. Urinary PGE-M and PGD-M levels were measured using liquid chromatography/tandem mass spectrometry (LC/MS) with slight modifications to the method previously described (21). Briefly, 1 mL urine was converted to an O-methyloxime derivative and purified by C18 solid phase extraction before LC/MS analysis. LC was performed on a 2.0x50 mm 1.7 µm particle Acquity BEH C18 column (Water Corporation, Barcelona, Spain). Mobile phase A was 95:4.9:0.1 (v/v/v) 5 mM ammonium acetate: [22,23] acetonitrile:acetic acid, and mobile phase B was 10.0:89.9:0.1 (v/v/v) 5 mM ammonium acetate:acetonitrile:acetic acid. The samples were separated by a gradient of 85-76% of mobile phase A over 6 min at a flow rate of 900 μL/min prior to delivery to a 6500 QTRAP (Sciex) triple quadrupole mass spectrometer. Urinary creatinine (Cr) levels were measured by a Creatinine Colorimetric Assay kit from Cayman Chemical.
The DNA extracted from peripheral blood was analysed for the presence of different functional variants described in the COX-1 and COX-2 genes. Both genes were sequenced by NGS using the Generead DNA seq Targeted Panels V2 technique on MiSeq (Illumina) equipment. The following prostaglandin polymorphisms were analysed: PTGS1 639 C>A, PTGS1 762+14delA, PTGS2-899G>C (-765G>C) and PTGS2 (8473T>C, all them variants previously reported in both genes [27].
2.3. Statistical Analysis
Parametrical statistical methods were used with variables that satisfied the assumption for parametric statistical testing With variables which did not satisfy this assumption non-parametric statistical methods were applied to all data sets. For independent samples, comparisons between two groups were carried out using the Mann-Whitney U test, while the Kruskal-Wallis H test was used for multiple groups. Correlation between clinical scale severity and PG values was expressed as Spearman’s rank-correlation coefficient. Statistical significance was established at p ≤0.05.
3. Results
The distribution of patients according to CFTR gene pathogenic variants severity and clinical and radiological characteristics is shown in Table 1. CFTR variants are depicted in Table 2.
3.1. Urinary PGE-M and PGD-M Levels
Urine PGE-M and PGD-M concentrations are expressed as medians and interquartile range (25th-75th interquartile). Urine PGE-M concentrations were significantly (p<0.0001) higher in CF patients (18.10, 7.60-30.50 ng/mg Cr) versus healthy controls (5.65; 3.48-11.48 ng/mg Cr). Similarly, PGD-M levels in pwCF (5.10; 2.50-8.30 ng/mg Cr) were significantly (p<0.01) higher than in healthy controls (2.40; 1.70-5.65 ng/mg Cr).
3.2. Correlations between Urinary PGE-M and PGD-M Levels and CFTR Gene Mutation Severity
When urinary PGE-M levels were compared between healthy controls and pwCF with mild, moderate, or severe phenotypes, there were no differences between healthy controls and patients carrying either the mild or moderate phenotype (Figure 1A). In contrast, there were statistically significant differences between the severe phenotype and both healthy controls (p<0.0001) and mild pathogenic variants (p<0.0001). There were no statistically significant differences between mild and moderate phenotypes or moderate and severe phenotypes. Similar results were produced when urinary PGD-M levels were compared, with significant differences only between the severe phenotype and healthy controls (p<0.001) and patients carrying the mild phenotype (p<0.01) (Figure 1B).
3.3. Correlations between Urinary PGE-M and PGD-M Levels and CF Severity Parameters
In patients with pancreatic insufficiency (N=70), PGE-M levels (24.25;12.48-44.50 ng/mg Cr) were higher than in patients with conserved pancreatic function (N=33) (8.60; 4.60-16.80 ng/mg Cr, p<0.0001). Similarly, PGD-M levels (6.15; 3.00-9.02 ng/mg Cr) were higher than in patients with conserved pancreatic function (3.60;1.75-5.95 ng/mg Cr, p<0.01).
When urinary PGE-M levels were compared between healthy subjects and pwCF with mild, moderate, or severe clinical severity, there were differences between healthy subjects and pwCF with mild (p<0.001), moderate (p<0.0001), and severe disease (p<0.0001). Also, there were statistically significant differences between mild and moderate disease (p<0.001), and mild and severe disease (p<0.001), however, the difference between moderate and severe was not statistically significant (Figure 2A). Similar results were obtained with PGD-M levels, except that there were no statistical differences between healthy controls and pwCF with mild disease, nor between moderate and severe disease (Figure 2B).
There was no correlation between PGE-M levels with either FEV1 (r= -0.1235) or FVC (r= -0.0964). Similarly, PGD-M levels did not correlate with either FEV1 (r= -0.1901) or FVC (r= -0.1732).
Since the number of patients scoring 2 and 3 was low, the analysis of the presence of bronchiectasis and air trapping was performed binarily (with or without). Patients with bronchiectasis (n=45) had higher PGE-M (27.80;16.00-56.30 ng/mg Cr, p<0.0001) and PGD-M (7.00;3.70-9.90 ng/mg Cr,p<0.001) levels than patients without bronchiectasis (n=57) (PGE-M: 12.70;6.45-20.35 ng/mg Cr; PGD-M: 3.70;2.30-6.45 ng/mg Cr).
The presence of air trapping in the CT scan (n=64) was associated with higher PGE-M levels (22.25;11.80-41.95 ng/mg Cr) than in those without radiological findings of air trapping (n=28) (11.65;6.37-27.68 ng/mg Cr, p<0.05). In contrast, there were no differences in PGD-M levels between patients with and without air trapping (data not shown).
3.4. Correlations between Urinary PGE-M and PGD-M levels, Airway Infections, and A Docosahexaenoic (DHA) Supplemented Diet
PwCF with airways chronically colonised by fungi (n=15) had similar urinary PGE-M and PGD-M levels as non-colonised patients (n=87) (data not shown).
Patients with a DHA-supplemented diet (n=5) had lower urinary PGE-M levels (6.60;4.90-14.70 ng/mg Cr) than those without a supplemented diet (n=96) (19.10;7.80-32.95), but the difference was not statistically significant (p=0.058). On the other hand, there were no differences in urinary PGD-M levels between the two groups (data not shown).
3.5. Correlation between Prostaglandin Polymorphisms with CF Severity and Urinary Prostaglandin Levels
The four polymorphisms were analysed in 102 pwCF. The 639 C>A polymorphism was identified as homozygous in 4 patients (3.9%), heterozygous in 17 (16.7%), and negative in 81 (79.4%).
The 762+14delA polymorphism was identified as homozygous in 1 pwCF (1%), heterozygous in 17 (16.7%), and negative in 84 (82.4%).
The -765G>C polymorphism was identified as homozygous in 4 pwCF (3.9%), heterozygous in 31 (30.4%), and negative in 67 (65.7%).
The 8473T>C polymorphism was identified as homozygous in 9 pwCF (8.8%), heterozygous in 40 (39.2%), and negative in 53 (52%).
When comparing the four polymorphisms with the different variables: clinical severity, genetic variants, pancreatic function, lung function, or the presence of air trapping and/or bronchiectasis with the chest CT, no significant differences were found (data not shown). In addition, there were no significant differences between the presence of polymorphisms and chronic colonisation by Staphyloccocus aureus (SA), Pseudomonas aeruginosa (PA), and PGE-M and PGD-M urinary levels (data not shown).
4. Discussion
Forty years ago, Charlotte M. Anderson hypothesised that a disturbed PGE2 metabolism could be involved in CF [29]. Supporting this hypothesis, several studies have reported the ability of CFTR to regulate COX-2 expression and, thereby, PGE2 biosynthesis [19,20,21,22,23]. A defective CFTR protein leads to enhanced COX-2 expression resulting in an increased release of PGE2 in CF patients [19,21,23]. Moreover, it was shown that CFTR inhibition lead to membrane destabilization, favoring eicosanoid synthesis (24). In addition to its role as an ion channel, CFTR also forms complexes with a host of signaling proteins (kinases and phosphatases) involved in fatty acid metabolism [25,26]
Given the direct relationship reported between CFTR dysfunction and increased PG synthesis, we hypothesised that the amount of PGs released in CF patients would be a marker of the severity of CFTR dysfunction. According to this hypothesis, PG production should correlate with parameters of CF severity.
Our study produced several main findings: a) there is a limited relationship between the severity of CFTR genetic dysfunction and PG production; b) exocrine pancreatic insufficiency is closely associated with the severity of CFTR dysfunction and PG production; c) there is no correlation between PG levels and lung function parameters; d) PG production correlates with both clinical status and radiological findings (bronchiectasis and air trapping); e) COX-1 and COX-2 gene polymorphisms do not appear to contribute to the regulation of PG synthesis.
Previous studies found moderate or no correlation between CFTR severity and PG production. Moreover, there are notable differences between urinary PG levels in patients with the same pathogenic variants, which suggest that factors other than mutation severity are involved in the regulation of PG metabolism. These factors remain to be elucidated. A previous study demonstrated the potential role of COX-1 and COX-2 gene polymorphisms in the clinical severity of pwCF harbouring the F508del mutation [23]; however, we found no relationship between COX polymorphisms with either severity parameters or urinary levels of PGE-M and PGD-M that reflect systemic prostanoid production.
In keeping with previous reports, our study shows a relationship between the presence of normal or defective exocrine pancreatic function and the severity of CFTR pathogenic variants and urinary PG levels [20].
FEV1 measurement has been a central outcome measurement for clinical management and trials. However, various studies concluded that FEV1 has limited sensitivity in detecting disease severity and monitoring disease progression [30,31]. Our study replicates previous observations regarding the lack of correlation between lung function parameters (FEV1 and FVC) and PG biosynthesis in CF patients [21]. Based on the concept that PG biosynthesis is associated with the severity of CFTR dysfunction, our results lend further support to studies reporting that lung function assessment has a limited value in the evaluation of CF severity.
In our study, we found that urinary PGE-M levels strongly correlated with the clinical severity of the disease, detecting significant differences between healthy controls and pwCF with mild, moderate, and severe clinical severity. We also found significant differences between mild and moderate disease and mild and severe disease. Various recent studies support that bronchiectasis detected by Chest CT is a sensitive indicator of prognosis, pulmonary exacerbations, and mortality in pwCF patients [32,33,34]. Compared with bronchiectasis, the potential relevance of air trapping as a marker of disease severity remains to be clearly established. Nevertheless, a study validated the presence of trapped air in the CT scan as an independent predictor of pulmonary exacerbations [35]. We found marked differences in PG levels between pwCF with and without bronchiectasis in the CT scan. Moreover, the presence of air trapping was also associated with higher levels of PGE-M than in those without radiological findings of air trapping. Taken together, clinical, and radiological findings support the hypothesis that the level of PG production is a marker of disease severity.
Interestingly, and in accordance with our results, a very recent study found that PGE2 levels in bronchoalveolar lavage fluid collected from the lung of CF patients correlated positively and significantly with disease progression; the higher the PGE2 levels, the faster the progression of the disease [36].
Chronic infection can stimulate COX-2 expression and, therefore, induce PG release. However, in our study, we did not find any difference in urinary PG levels between chronically colonised and non-colonised patients.
A high-dose DHA supplementation diet can improve clinical outcomes, such as disease exacerbations associated with a decrease in inflammatory markers [25]. In our study, we found reduced urinary PG levels in pwCF on a supplemented DHA diet compared with those not on this diet but the difference only tended towards statistical significance. However, our study lacks statistical power as only a few patients from our cohort were on this therapy. In a very recent study, the effects of a supplemented DHA diet on urinary PGE2 levels were assessed and compared with a control placebo diet. There were no differences in the impact of the supplemented diet and placebo on PGE2 production. The study, however, was carried out in a small sample of pwCF with low baseline urinary PGE2 levels. Thus, a more extensive clinical trial will be required to assess the effects of this therapy on PG production.
A recent study investigated the effects of ivacaftor on urinary PGE-M levels in pwCF with the G551D mutation. Ivacaftor treatment significantly decreased the urine levels of PGE-M, suggesting urinary PGE-M as a marker to assess the efficacy of new CF therapies [37].
The mechanisms underlying the relationship between defective CFTR function and excessive PGE2 synthesis remain to be fully elucidated: We are tempted to speculate that the increased PGE2 release in CF is intended to stimulate the activity of the defective CFTR. When CFTR recovers its function, at least partially, with some of the new therapies, the excess PGE2 production is no longer needed.
In summary, there is a significant need to develop new, relevant biomarkers to monitor clinical care results and the effects of new therapies in CF. Taken together; the data reported in the present study suggests that measuring urinary PG levels could fulfil this role. We found no relationship between COX polymorphisms with either severity parameters or urinary levels of PGE-M and PGD-M that reflect systemic prostanoid production.
Acknowledgements
The study was carried out with the support of a Grant from the Fundació Catalana de Pneumologia (FUCAP)
References
- http://www.genet.sickkids.on.ca/.
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [CrossRef]
- Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, et al. Type of CFRT mutations determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011;140:153-161. [CrossRef]
- Kristidis, P.; Bozon, D.; Corey, M.; Markiewicz, D.; Rommens, J.; Tsui, L.C.; Durie, P. Genetic determination of exocrine pancreatic function in cystic fibrosis. 1992, 50, 1178–84.
- Cleveland, R.H.; Zurakowski, D.; Slattery, D.; Colin, A.A. Cystic Fibrosis Genotype and Assessing Rates of Decline in Pulmonary Status. Radiology 2009, 253, 813–821. [CrossRef]
- de Gracia, J.; Mata, F.; Álvarez, A.; Casals, T.; Gatner, S.; Vendrell, M.; de la Rosa, D.; Guarner, L.; Hermosilla, E. Genotype-phenotype correlation for pulmonary function in cystic fibrosis. Thorax 2005, 60, 558–563. [CrossRef]
- Cogen, J.; Emerson, J.; Sanders, D.B.; Ren, C.; Schechter, M.S.; Gibson, R.L.; Morgan, W.; Rosenfeld, M.; for the EPIC Study Group Risk factors for lung function decline in a large cohort of young cystic fibrosis patients. Pediatr. Pulmonol. 2015, 50, 763–770. [CrossRef]
- Gan, K.H.; Geus, W.P.; Bakker, W.; Lamers, C.B.; Heijerman, H.G. Genetic and clinical features of patients with cystic fibrosis diagnosed after the age of 16 years.. Thorax 1995, 50, 1301–1304. [CrossRef]
- Hubert, D.; Bienvenu, T.; Desmazes-Dufeu, N.; Fajac, I.; Lacronique, J.; Matran, R.; Kaplan, J.; Dusser, D. Genotype-phenotype relationships in a cohort of adult cystic fibrosis patients. Eur. Respir. J. 1996, 9, 2207–2214. [CrossRef]
- Borgo G, Gasparini P, Bonizzato A, Cabrini G, Mastella G, Pignatti PF. Cystic fibrosis: The delta508 mutation does not lead to an exceptional severe phenotype. A cohort study. J Pediatr 1993;152:1006-1011. [CrossRef]
- Lai, H.J.; Cheng, Y.; Cho, H.; Kosorok, M.R.; Farrell, P.M. Association between Initial Disease Presentation, Lung Disease Outcomes, and Survival in Patients with Cystic Fibrosis. Am. J. Epidemiology 2004, 159, 537–546. [CrossRef]
- Zielenski, J. Genotype and Phenotype in Cystic Fibrosis. Respiration 2000, 67, 117–133. [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [CrossRef]
- Verhaeghe, C.; Delbecque, K.; de Leval, L.; Oury, C.; Bours, V. Early inflammation in the airways of a cystic fibrosis foetus. J. Cyst. Fibros. 2007, 6, 304–308. [CrossRef]
- Shrestha, N.; McCarron, A.; Rout-Pitt, N.; Donnelley, M.; Parsons, D.W.; Hryciw, D.H. Essential Fatty Acid Deficiency in Cystic Fibrosis Disease Progression: Role of Genotype and Sex. Nutrients 2022, 14, 4666. [CrossRef]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of Cystic Fibrosis with Abnormalities in Fatty Acid Metabolism. New Engl. J. Med. 2004, 350, 560–569. [CrossRef]
- Yang, J.; Eiserich, J.P.; Cross, C.E.; Morrissey, B.M.; Hammock, B.D. Metabolomic profiling of regulatory lipid mediators in sputum from adult cystic fibrosis patients. Free. Radic. Biol. Med. 2012, 53, 160–171. [CrossRef]
- Smith WL, Dewitt DL, Garavito RM. Cyclooxygenases: Structural, cellular and molecular biology. Ann Rev Biochem 2000;69:145-182. [CrossRef]
- Roca-Ferrer, J.; Pujols, L.; Gartner, S.; Moreno, A.; Pumarola, F.; Mullol, J.; Cobos, N.; Picado, C. Upregulation of COX-1 and COX-2 in nasal polyps in cystic fibrosis. Thorax 2006, 61, 592–596. [CrossRef]
- O’connor, M.G.; Thomsen, K.; Brown, R.F.; Laposata, M.; Seegmiller, A. Elevated prostaglandin E metabolites and abnormal plasma fatty acids at baseline in pediatric cystic fibrosis patients: a pilot study. Prostaglandins, Leukot. Essent. Fat. Acids 2016, 113, 46–49. [CrossRef]
- Jabr, S.; Gartner, S.; Milne, G.L.; Roca-Ferrer, J.; Casas, J.; Moreno, A.; Gelpí, E.; Picado, C. Quantification of major urinary metabolites of PGE2 and PGD2 in cystic fibrosis: Correlation with disease severity. Prostaglandins, Leukot. Essent. Fat. Acids 2013, 89, 121–126. [CrossRef]
- Medjane S, Raymon B, Wu Y, Touqui L. Impact of CFTR delta508 mutation on prostaglandin E2 production and type IIA phospholipase A2 expression by pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 2005;289:L816-L824. [CrossRef]
- Chen J, Jiang XH, Chen H,Guo JH, Tsang LL, Yu MK, et al. CFRT negatively regulates cyclooxygenase-2-PGE2 positive feedback loop in inflammation. J Cell Physiol 2012;227:2759-2766. [CrossRef]
- Borot F, Vieu DV, Faure G, Fritsch J, Colas J, Moriceau.S Eicosanoid Release Is Increased by Membrane Destabilization and CFTR Inhibition in Calu-3 Cells. PLoS One. 2009; 4(10): e7116. [CrossRef]
- Seegmiller, A.C. Abnormal Unsaturated Fatty Acid Metabolism in Cystic Fibrosis: Biochemical Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2014, 15, 16083–16099. [CrossRef]
- Kunzelmann, K.; Mehta, A. CFTR: a hub for kinases and crosstalk of cAMP and Ca2+. FEBS J. 2013, 280, 4417–4429. [CrossRef]
- Czerska, K.; Sobczyńska-Tomaszewska, A.; Sands, D.; Nowakowska, A.; Bąk, D.; Wertheim, K.; Poznański, J.; Zielenski, J.; Norek, A.; Bal, J. Prostaglandin-endoperoxide synthase genesCOX1 andCOX2 — novel modifiers of disease severity in cystic fibrosis patients. J. Appl. Genet. 2010, 51, 323–330. [CrossRef]
- Alonso, M.J.; Heine-Suñer, D.; Calvo, M.; Rosell, J.; Giménez, J.; Ramos, M.D.; Telleria, J.J.; Palacio, A.; Estivill, X.; Casals, T. Spectrum of Mutations in the CFTR Gene in Cystic Fibrosis Patients of Spanish Ancestry. Ann. Hum. Genet. 2006, 71, 194–201. [CrossRef]
- Anderson CM. Hypothesis revisited. Cystic fibrosis: A disturbance of water and electrolyte movement of exocrine secretory tissue associated with altered prostaglandin (PGE2) metabolism. J Pediatric Gastroenterol Nutrition 1984;3:15-22.
- A de Jong, P.; Lindblad, A.; Rubin, L.; Hop, W.C.J.; de Jongste, J.C.; Brink, M.; Tiddens, H.A.W.M. Progression of lung disease on computed tomography and pulmonary function tests in children and adults with cystic fibrosis. Thorax 2005, 61, 80–85. [CrossRef]
- Bush, A.; Sly, P.D. Evolution of cystic fibrosis lung function in the early years. Curr. Opin. Pulm. Med. 2015, 21, 602–608. [CrossRef]
- McMahon, M.A.; Chotirmall, S.H.; McCullagh, B.; Branagan, P.; McElvaney, N.; Logan, P. Radiological abnormalities associated with Aspergillus colonization in a cystic fibrosis population. Eur. J. Radiol. 2012, 81, e197–e202. [CrossRef]
- Loeve M, Gerbrandts K, Hop WC, Rosenfield M, Hartman IC, Tiddens HAl. Bronchiectasis and pulmonary exacerbations in children and young adults with cystic fibrosis. Chest 2011;140:178-185. [CrossRef]
- Loeve, M.; van Hal, P.T.W.; Robinson, P.; A de Jong, P.; Lequin, M.H.; Hop, W.C.; Williams, T.J.; Nossent, G.D.; A Tiddens, H. The spectrum of structural abnormalities on CT scans from patients with CF with severe advanced lung disease. Thorax 2009, 64, 876–882. [CrossRef]
- Tepper, L.A.; Utens, E.M.; Caudri, D.; Bos, A.C.; Gonzalez-Graniel, K.; Duivenvoorden, H.J.; van der Wiel, E.C.; Quittner, A.L.; Tiddens, H.A. Impact of bronchiectasis and trapped air on quality of life and exacerbations in cystic fibrosis. Eur. Respir. J. 2013, 42, 371–379. [CrossRef]
- Horati, H.; Janssens, H.M.; Margaroli, C.; Veltman, M.; Stolarczyk, M.; Kilgore, M.B.; Chou, J.; Peng, L.; Tiddens, H.A.; Chandler, J.D.; et al. Airway profile of bioactive lipids predicts early progression of lung disease in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 902–909. [CrossRef]
- O’Connor, M.G.; Seegmiller, A. The effects of ivacaftor on CF fatty acid metabolism: An analysis from the GOAL study. J. Cyst. Fibros. 2016, 16, 132–138. [CrossRef]
Figure 1.
Urinary PGE-M and PGD-M levels and CFTR gene pathogenic variants severity. PGE-M (Panel A) and PGD-M (Panel B) concentrations were analysed by liquid chromatography/tandem mass spectrometry (LC/MS) in urine samples from healthy controls (HC, N=30) and patients with mild (MILD, N=29), moderate (MOD, N=7), and severe (SEV, N=65) CFTR gene phenotypes. The solid line indicates the median, the box indicates 25-75th percentiles, and whiskers represent minimum and maximum values. An unpaired t-test was used for statistical comparison. ****p ≤0.0001 and ***p ≤0.001 compared with HC; ####p ≤0.0001 and ##p ≤0.01 compared with MILD.
Figure 1.
Urinary PGE-M and PGD-M levels and CFTR gene pathogenic variants severity. PGE-M (Panel A) and PGD-M (Panel B) concentrations were analysed by liquid chromatography/tandem mass spectrometry (LC/MS) in urine samples from healthy controls (HC, N=30) and patients with mild (MILD, N=29), moderate (MOD, N=7), and severe (SEV, N=65) CFTR gene phenotypes. The solid line indicates the median, the box indicates 25-75th percentiles, and whiskers represent minimum and maximum values. An unpaired t-test was used for statistical comparison. ****p ≤0.0001 and ***p ≤0.001 compared with HC; ####p ≤0.0001 and ##p ≤0.01 compared with MILD.
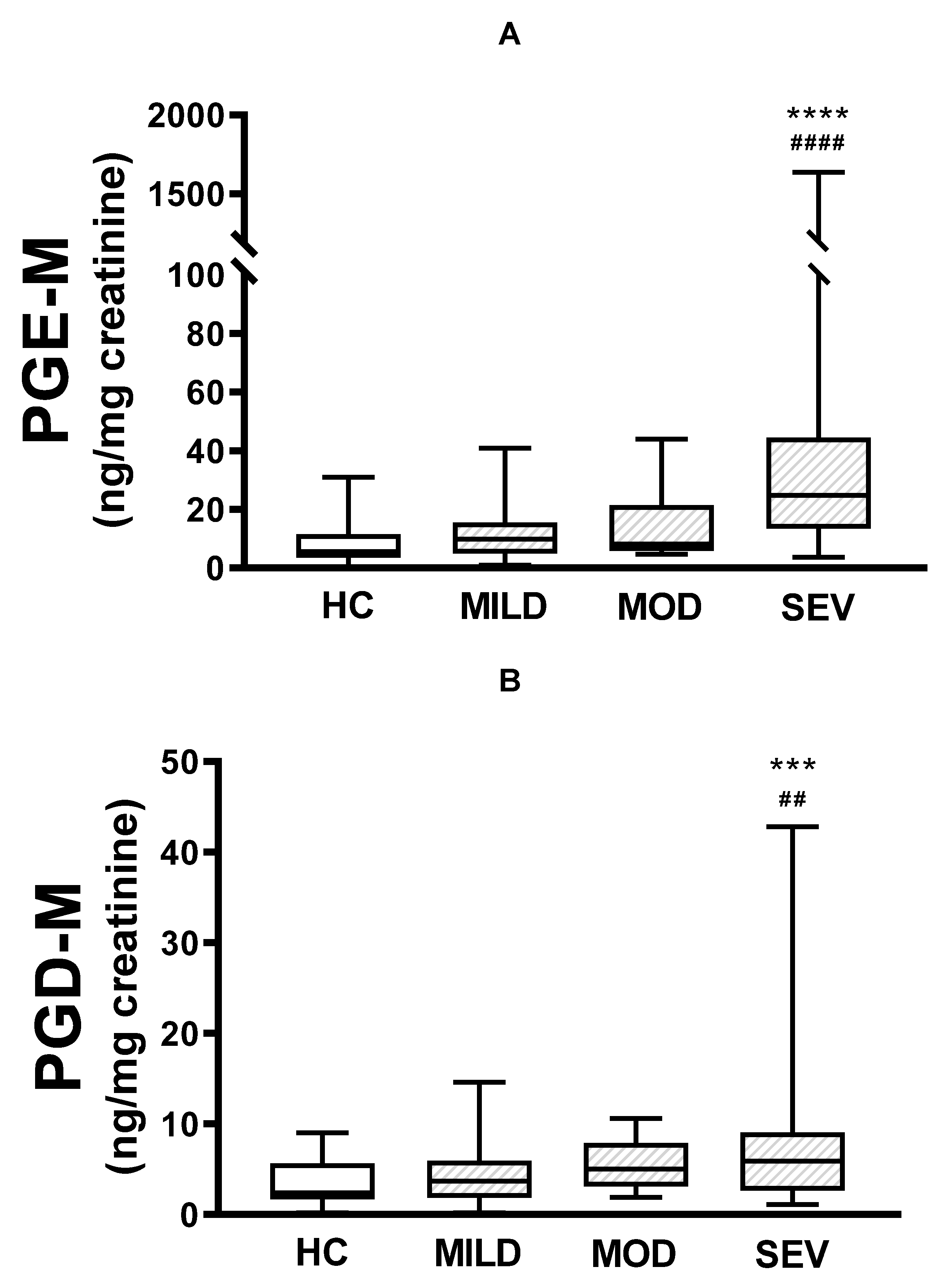
Figure 2.
Urinary PGE-M and PGD-M levels and clinical severity. PGE-M (Panel A) and PGD-M (Panel B) concentrations were analysed by liquid chromatography/tandem mass spectrometry (LC/MS) in urine samples from healthy controls (HC, N=30) and patients with mild (MILD, N=29), moderate (MOD, N=7), and severe (SEV, N=65) clinical phenotypes. The solid line indicates the median, the box indicates 25-75th percentiles, and whiskers represent minimum and maximum values. An unpaired t-test was used for statistical comparison. ****p ≤0.0001 and ***p ≤0.001 compared with HC; ###p ≤0.001 and ##p ≤0.01 compared with MILD.
Figure 2.
Urinary PGE-M and PGD-M levels and clinical severity. PGE-M (Panel A) and PGD-M (Panel B) concentrations were analysed by liquid chromatography/tandem mass spectrometry (LC/MS) in urine samples from healthy controls (HC, N=30) and patients with mild (MILD, N=29), moderate (MOD, N=7), and severe (SEV, N=65) clinical phenotypes. The solid line indicates the median, the box indicates 25-75th percentiles, and whiskers represent minimum and maximum values. An unpaired t-test was used for statistical comparison. ****p ≤0.0001 and ***p ≤0.001 compared with HC; ###p ≤0.001 and ##p ≤0.01 compared with MILD.
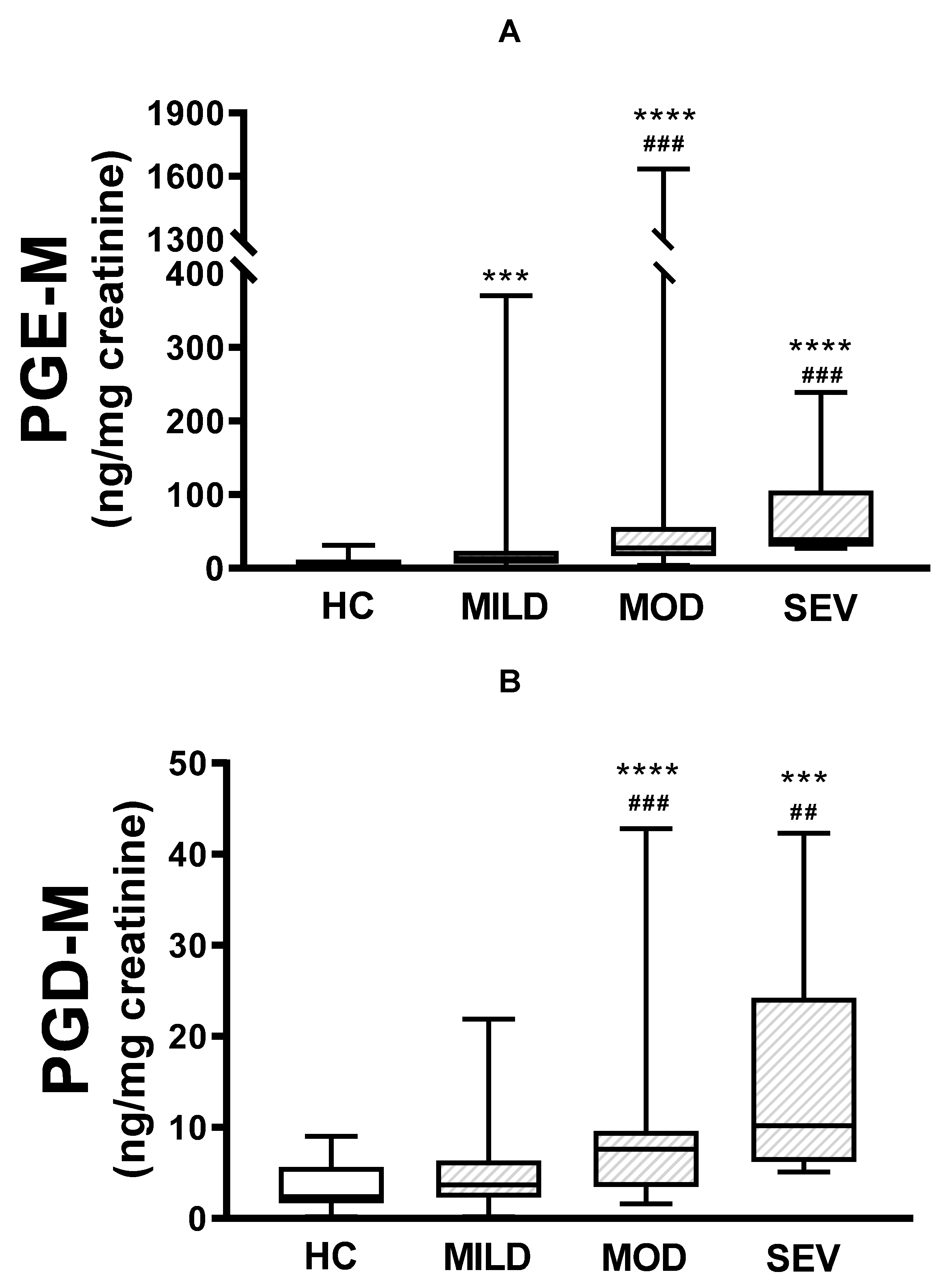

Table 1.
Clinical and radiological characteristics of the cystic fibrosis patients.
| FVC, % pred (n=93) | 104.1±1.8 |
| FEV1, % pred (n=93) | 94.3±2.3 |
| Pancreatic insufficiency, (n=103) % | 70 (68%) |
| Severity of the CFTR gene mutation (n=101): | |
| Mild, % | 28.7% |
| Moderate, % | 6.9% |
| Severe, % | 64.4% |
| Clinical severity (n=101): | |
| Mild, % | 64.4% |
| Moderate, % | 29.7% |
| Severe, % | 5.9% |
| Bronchiectasis, (n=102) % | 45(44%) |
| Air Trapping (n=92) % | 64(69.%) |
| Data presented as mean±SEM or percentage. FVC, forced vital capacity; pred, predicted; FEV1, forced expiratory volume in 1 s; CFTR, cystic fibrosis transmembrane conductance regulator | |
Table 2.
Distribution of CFTR gene variants (N=103).
| F508del homozygote, n (%) | 19 (18.4%) |
| F508del heterozygote, n (%) | 59 (57.3%) |
| Other mutations, n (%) | 25 (24.3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



