
Submitted:
04 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
Circular dichroism (CD) spectroscopy is a valuable tool in the pharmaceutical industry for determining the secondary and tertiary structure, folding, stability, and interactions of proteins in biosimilars. However, protein aggregation due to its interaction with surfactants can introduce variations in CD spectra, leading to false interpretations. To investigate this effect, we prepared aqueous solutions of α-chymotrypsin with the same amount of surfactant but at different solution preparation steps. Interestingly, the CD spectra of these solutions showed slight differences despite having the same surfactant concentration. This suggests that the order of surfactant addition can influence protein conformation. To address this issue, we propose a method for preparing aqueous protein solutions that yields reproducible CD spectra. This method is expected to benefit the pharmaceutical industry by improving the accuracy of CD spectroscopy for protein structural analysis and evaluation of protein functionality.
Keywords:
protein
; surfactant
; SDS
; protein-surfactant-interaction
; circular-diachorism
; Chirality
Introduction
Use of variety of proteins in biopharmaceutics, consumer, food processing and skin-care industry has led to significant study of proteins in terms of their structures and their correlation with application outcomes [1]. Proteins, compared to small molecule drugs, exhibits a superior therapeutical capability and has proven to be effective tools in the treatment of many complex and incurable diseases. Humira (IgG1, human monoclonal antibody) and recently Prevnar 13 have shown great success in treating rheumatoid arthritis and preventing pneumococcal infections [2]. Recent increase in the clinical approval of protein-based therapeutics has significantly progressed the development of efficient biopharmaceutical manufacturing and quality control methods [3,4]. However, there are still some problems that need attention. Primarily, the problems related to physical and chemical stabilisation of formulation during the storage is a major hinderance in commercialising many potential biomolecules. The protein molecules tend to aggregate, denature, precipitate or adsorb to the surface of vial during storage and transportation resulting into loss of therapeutic efficacy and safety. Earlier studies showed that the activity of Interleukin-1 receptor reduced to third when it undergoes physical unstable state through aggregation in solution [5]. Generally, sugar or cationic, anionic and nonionic surfactant is added for avoiding the physical destabilisation of protein solution in formulations. While surfactant or sugar addition prevents the physical instability of protein solution, it can also induce denaturisation, b-sheet elimination or other disulfide exchange and bring a same set of efficacy and safety related problems. Therefore, it is crucial to assess the structural and functional properties of biomolecules during manufacturing stage using sensitive and trustworthy analytical approaches and sample preparation methods [6].
Understanding the interaction between surfactants and proteins is crucial for ensuring the stability of protein formulations during storage and transportation. In the structural analysis of monoclonal antibodies, the Sodium Dodecyl Sulphate-Capillary Gel Electrophoresis (SDS-CGE) method is widely employed in the industry due to its ability to resolve small molecules in high molecular weight proteins, complementing size exclusion chromatography. The versatile use of SDS in various applications has led to extensive research in the field of SDS and protein interactions. This work also addresses the issue relating to SDS interactions with the protein.
Surfactant molecules, characterised by hydrophobic moieties and a hydrophilic head, can form micelles when the concentration exceeds the Critical Micelle Concentration (CMC); otherwise, they remain in a monomeric form. It is well-established that monomeric or micellar surfactants interact with free protein fragments, leading to conformational changes in protein structure or physical destabilisation in solution [7]. However, ongoing research is focused on the reversibility of SDS-induced changes in protein structure, the formation of SDS-protein complexes, and the binding between SDS molecules [8].
The presence of free proteins and protein complexes with a specific number of bound surfactant molecules has been observed to cause irreversible structural changes in proteins [9,10]. Nevertheless, recent studies have indicated the potential reversibility of surfactant-induced protein unfolding [11]. In this study, it was demonstrated that SDS interacts with denatured ferrocytochrome independently of the protein's structure, conformation, and ionization state. Moreover, the conformational transition reached a minimum at a certain concentration of SDS in the solution, suggesting reversibility [12].
The interaction or binding of SDS molecules with proteins follows a sequential pattern and is dependent on the molar ratio of protein to surfactant [13]. Previous research challenging the presence of free proteins [13] in protein-surfactant solutions suggested that observed charge discontinuity in mobility changes during capillary gel electrophoresis may be attributed to different types of protein-SDS complexes rather than the reversibility of folding. Optimal concentrations of SDS play a crucial role, with maxima and minima values determining the specific number of SDS molecules binding to proteins to form complexes or assume conformations [14].
In our study, we conducted far and near UV circular dichroism (CD) measurements on protein-SDS solutions to showcase the impact of SDS concentrations and mixing methods on CD spectra. These spectra reflect changes in the tertiary and secondary structure of the protein. Additionally, far and near UV CD spectra were obtained for protein-SDS solutions with concentrations below and near the CMC of SDS, highlighting the dependence of CD spectra on the procedure of adding SDS to achieve the final concentration in the protein solution.
Methodology
Materials
α-chymotrypsinogen A from bovine pancreas, sodium phosphate monobasic (BioReagent, for molecular biology, anhydrous, ≥98%), disodium hydrogen phosphate dihydrate (BioUltra, for molecular biology, ≥99.0% (T)), sodium chloride (BioUltra, for molecular biology, ≥99.5% (AT)), sodium dodecyl sulphate (SDS) (BioReagent, suitable for electrophoresis, for molecular biology, ≥98.5% (GC)) were purchased from Sigma-Aldrich and used as received. Ultrapure Millipore Milli-Q water (resistance 18.3 MΩ cm–1) was used available in the laboratory.
To prepare the required 10mM sodium phosphate buffer, disodium hydrogen phosphate dihydrate, sodium phosphate monobasic, and sodium chloride (NaCl) were weighed and dissolved in ultrapure Millipore water having 18 MΩ cm-1 resistivity. The pH was adjusted to 7 using Hydrochloric acid (HCl) and the solution made up to a final volume of 1L. Using an analytical balance, a-chymotrypsinogen A powder and SDS was weighed accurately. The weighed a-chymotrypsinogen A powder and SDS was transferred into a clean and dry container. To make 50 mM a-chymotrypsinogen A and 200 mM SDS stock solutions, both powders were dissolved in a small volume of buffer (10 mM, 7 pH). This was swirled gently to aid in dissolution. Once the powder was fully dissolved, both stock solutions were diluted to achieve desired stock solution concentration and transferred to a volumetric flask of a known volume.
More solvent was added to dilute and mix the stock solutions to prepare the concentrations: 40 mM, 20 mM, 40 mM+ 6 mM SDS, 20 mM+ 6 Mm SDS, 40 mM + 30 Mm SDS, 20 mM + 30 mM SDS, 6 mM SDS, 80 mM SDS, 74 mM SDS and 50 mM SDS. Each solution was mixed well to ensure uniformity. The solution was syringe filtered using 0.45 mm disposable filter to clear it of any particulate matter. All solution were made freshly and used within 30 minutes of preparation.
Circular Dichroism (CD) Spectra Collection
The CD spectra for the solutions detailed in Table 1 were acquired utilising a Circular Dichroism (CD) Spectrometer MOS-500 (Bio-Logic) spectrometer. The collection of far and near UV range CD spectra involved the use of a 0.5 s/pt, 0.25 nm step size, and a 1 nm bandwidth. A 1 mm quartz cell was employed for collecting far UV spectra, while a 10 mm quartz cell was utilized for near UV spectra. Each spectrum was an average of 5 scans. Prior to measurements, the CD spectrometer underwent setup in accordance with the manufacturer's guidelines and calibration using a standard sample. Subsequently, the cuvette was filled with the reference buffer solution and positioned in the sample holder to capture the background spectrum. The smoothed background spectrum was then subtracted from each spectrum of α-chymotrypsinogen A. All spectroscopic measurements were conducted at a controlled temperature of 20 °C.
Following the combining of each set of solutions, the spectra were promptly recorded, and subsequently, the cuvette was cleaned and dried. The subsequent pair of solutions was then mixed, followed by the recording of spectra. This iterative process, encompassing 5 replicates of the spectrum along with the time dedicated to cuvette maintenance, took approximately 30 minutes for the far-UV experiments and 40 minutes for the near-UV experiments.
Upon the completion of mixing the final pair of parent solutions, we conducted a repeat of spectrum recording for the initial pair, utilising the already prepared mixture. Three measurement cycles were executed for the far-UV range, while the near-UV range underwent two cycles. Therefore, the stability of the spectra for a given mixture implies that no discernible changes occurred for a minimum duration for all far-UV spectra or at near-UV spectra post-preparation. It is noteworthy that each recording, comprising 5 individual scans per spectrum, consumed 30 to 40 minutes, as previously mentioned.
Far UV Spectra of Each Sample
The experiment involved mixing part 1, containing a 20 μM concentration of α-chymotrypsinogen A, with part 2 in a 1:1 volumetric ratio to obtain far UV spectra. Figure 1 displays the obtained UV spectra of the mixture, which contained 10 μM α-chymotrypsinogen A in a 10 mM buffer. This qualitative match with the spectra in the data bank [16] was confirmed. Additionally, the near UV spectra obtained for all samples also exhibited qualitative matches [17]. Following the 1:1 volume ratio mixing, the final concentration of α-chymotrypsinogen A reduced to 10 μM, referred to as 20 μM CG throughout the text.
In Figure 1, a comparison of the spectra for 20 μM CG in a 10 mM buffer and 20 μM CG mixed with 80 mM SDS reveals differences due to the interaction of the surfactant with CG. When comparing the spectrum obtained from the solution of 20 μM + 30 mM SDS mixed with 50 mM SDS with the spectrum of the solution 20 μM CG mixed with 80 Mm SDS, no differences were observed. Due to same concentration of surfactant in both solutions, both spectra are found to be almost similar barring some statistical noise. However, the CD spectrum is found significantly different if it is obtained from the 20 μM CG + 6 mM SDS mixed with 74 Mm SDS solution. Though the total concentration of SDS after mixing will be same as it is in other two solutions, the spectrum is different. It can therefore be deduced that the method of obtaining final concentration of SDS in protein solution can affect the results. It was also observed with 40 mM α-chymotrypsin solution in buffer [18]. The plots Figure S1 – 3 in Supplementary Data show the effect on the UV-CD spectra due to variation in mixing protocols.
To understand the difference between secondary structure of the α-chymotrypsinogen A after interaction with surfactant molecules, the spectral CD data were processed and deconvoluted using software BeStSel [19]. BestSel determines the secondary structure by distinguishing the parallel and antiparallel and twisted b sheets, a-helix and others. A summary of the results is shown in following table.
The interaction of anionic surfactants, such as SDS in its saturated solution, with negatively charged molecules induces a transition in disordered proteins towards an α-helical structure. The data presented in Table 2 validates this effect, demonstrating an increase in the percentage of helix formation when the total SDS concentration in the solution is 40 mM. Additionally, Table 2 introduces a novel and intriguing observation, to the best of our knowledge not previously documented: all CD spectra consistently indicate that the augmentation in the α-helical population in the presence of 40 mM SDS is contingent on the manner in which the surfactant concentration is brought to this value. This particular insight stands out as the most significant outcome documented in our study.
Table 2 illustrates that the percentage of α-helix increased from 11.7 to 22.2 when a solution of 20 μM CG + 6 mM SDS was mixed with a 74 mM SDS solution to reach a final concentration of 80 mM SDS. On the other hand, if the 80 mM concentration of SDS is reached in one step or by adding a 50 mM SDS solution into a solution of 20 μM CG + 30 mM SDS, the percentage of α-helix increased from 11.7 to approximately. Notably, while the final concentration of SDS remains constant, the percentage increase in α-helix differs across few measured cases.
All measurements were repeated at least three times to ensure the reliability of the conclusions drawn from the results. Furthermore, the results from data-fitting using BestSel also align with the conclusion that the free protein in solution adopts a different secondary structure depending on whether the final SDS concentration in the solution is reached in a single or double step, as highlighted above.
Near UV-Spectra
To further validate the outcomes derived from the secondary structure analysis, the tertiary structure of the same protein is determined by obtaining the near UV spectra for all relevant samples shown in Table 2.
As seen in Figure 2, the far-UV case, the near UV CD spectra obtained for 40 mM concentration validates the data [20].
An important observation in Figure 2 is the variation observed in the spectral range of 250 to 310. This difference is evident when comparing the spectra of the 40 μM + 6 mM SDS +74 mM SDS solution with the spectra of the other two solutions, which have a final concentration of 40 mM SDS. These peaks emanates from aromatic amino acids and disulfide bonds. On other hand, the spectra between 310 and 340 nm for the solutions containing 40 mM SDS remain consistent, irrespective of the protocol used to reach this final SDS concentration. This leads us to infer that the tertiary structure of protein in the proximity of 290 nm (residue) is independent of the procedure employed to determine the final 40 mM concentration of this surfactant. The spectra for 40 mm + 30 mM + 50 mM are almost identical to 40 mm + 80 mM. It explains that the tertiary structure of α-chymotrypsinogen A in is constant and minute differences in spectra between small difference between the spectra visible in the range 260–280 nm in the Figure 3 is consequential.
The differences observed only appeared when the small amount of SDS was added to protein solution as an initial step in full protocol of mixing. Since the 6mM SDS was used as a small amount in initial step of the protocol, it is bit above the CMC for the surfactant of 4.8 mM [22] relevant to experiment parameters used in this work. Before acquiring the spectra, the solutions were mixed in equal volume with surfactant free buffer according to Table 1, which reduces the total SDS concentration below CMD to 3 mM SDS. Previous experiment similar to this one suggests that the micelle concentration of about 20 μM. Therefore, the SDS interaction are most significant for solution with 6mM concentration of SDS.
As surfactant interacts with protein in different way if the concentration is below or above the CMC. Meaning, the monomer concentration of SDS will interact differently than the micelle of SDS to induce conformal changes in secondary and tertiary structure [23]. The Figure 3 confirms the observed behaviour of SDS when the concentration is near or below the CMC concentration which is quite, distinct from the changes induced in the presence of 40 mM SDS. The spectra of 40 mm and 20 mm protein solution with final 3mM and 6 mM SDS concentration are different that suggests that the structure of the protein in both cases are different and dependent on SDS concentration higher or below CMC. It is noteworthy that these spectra were recorded approximately one and half hour after the preparation of the solutions, implying the stability of the molecular structures present in the solution over time. This suggests the irreversibility of the binding of SDS monomers by protein.
Conclusions
Proposed protocol of mixing the solutions to reach the final concentration of surfactant (SDS) is novel observation with implication of detecting the protein structure for quality control or research. Besides this work sheds a light on interaction between protein and anionic surfactant. This method is expected to benefit the pharmaceutical industry by improving the accuracy of CD spectroscopy for protein structural analysis and evaluation of protein functionality.
Patents
This section is not mandatory but may be added if there are patents resulting from the work reported in this manuscript.
Supplementary Materials
The following supporting information can be downloaded at:.
Author Contributions
Conceptualization, K.P.; methodology, K.P. and T.A.; software, K.P.; validation, T.A., K.P. and H.S.; formal analysis, T.A.; investigation, T.A.; resources, A.K.; data curation, X.X.; writing—original draft preparation, T.A.; writing—review and editing, K.P, A.K. and H.S.; visualization, H.S.; supervision, K.P.; project administration, K.P.; funding acquisition, K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding and funded by Rober Gordon University, School of Engineering, Biomedical Technology.
Data Availability Statement
Data available on request.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Song, J.G.; Lee, S.H.; Han, H.K. The stabilization of biopharmaceuticals: current understanding and future perspectives. Journal of Pharmaceutical Investigation 2017, 47, 475–496. [Google Scholar] [CrossRef]
- Zhu, M.M.; Mollet, M.; Hubert, R.S.; Kyung, Y.S.; Zhang, G.G. Industrial production of therapeutic proteins: cell lines, cell culture, and purification. Handbook of industrial chemistry and biotechnology 2017, 1639–1669. [Google Scholar]
- Agarkhed, M.; O’Dell, C.; Hsieh, M.C.; Zhang, J.; Goldstein, J.; Srivastava, A. Effect of polysorbate 80 concentration on thermal and photostability of a monoclonal antibody. AAPS PharmSciTech 2013, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Komander, D. The emerging complexity of protein ubiquitination. Biochemical Society Transactions 2009, 37(5), 937–953. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Beauvais, R.M.; Arakawa, T.; Narthi, L.O.; Dong, A.; Aparisio, D.I.; Carpenter, J.F. Formation of an active dimer during storage of interleukin-1 receptor antagonist in aqueous solution. Biophys J 1996, 71, 3399–3406. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2005, 1751(2), 119–139. [Google Scholar] [CrossRef]
- Otzen, D.E.; Pedersen, J.N.; Rasmussen H, Ø.; Pedersen, J.S. How do surfactants unfold and refold proteins? Advances in Colloid and Interface Science 2022, 308, 102754. [Google Scholar] [CrossRef]
- Takeda, K.; Moriyama, Y. Kinetic Aspects of Surfactant-Induced Structural Changes of Proteins - Unsolved Problems of Two-State Model for Protein Denaturation. J. Oleo Sci. 2015, 64, 1143–1158. [Google Scholar] [CrossRef]
- Pedersen, J.N.; Lyngsø, J.; Zinn, T.; Otzen, D.E.; Pedersen, J.S. A Complete Picture of Protein Unfolding and Refolding in Surfactants. Chem. Sci. 2020, 11, 699–712. [Google Scholar] [CrossRef]
- Moosavi-Movahedi, A.A. Thermodynamics of protein denaturation by sodium dodecyl sulfate. Journal of the Iranian Chemical Society 2005, 2, 189–196. [Google Scholar] [CrossRef]
- Anand, U.; Mukherjee, S. Reversibility in protein folding: effect of β-cyclodextrin on bovine serum albumin unfolded by sodium dodecyl sulphate. Physical Chemistry Chemical Physics 2013, 15(23), 9375–9383. [Google Scholar] [CrossRef]
- Bhuyan, A.K. On the mechanism of SDS-induced protein denaturation. Biopolymers: Original Research on Biomolecules 2010, 93(2), 186–199. [Google Scholar] [CrossRef]
- Takeda, K.A. Kinetic Study of the Interaction of Sodium Dodecyl Sulfate with Bovine Serum Albumin by Means of a Pressure-jump Method. Bull. Chem. Soc. Jpn. 1982, 55, 2547–2550. [Google Scholar] [CrossRef]
- Sasa, K.; Takeda, K. Multiple Coexisting Species of Sodium Dodecyl Sulfate-Bovine Serum Albumin Complexes as Detected by Capilary Electrophoresis. J. Colloid Interface Sci. 1993, 157, 516–517. [Google Scholar] [CrossRef]
- Anand, U.; Mukherjee, S. Binding, unfolding and refolding dynamics of serum albumins. Biochimica et Biophysica Acta (BBA)-General Subjects 2013, 1830(12), 5394–5404. [Google Scholar] [CrossRef]
- Whitmore, L.; Woollett, B.; Miles, A.J.; Klose, D.P.; Janes, R.W.; Wallace, B.A. PCDDB: the protein circular dichroism data bank, a repository for circular dichroism spectral and metadata. Nucleic acids research 2011, 39 (Suppl. S1), D480–D486. [Google Scholar] [CrossRef]
- Rinner, U.; Carreira, E.M.; Yamamoto, H. Comprehensive Chirality. 2012. [Google Scholar]
- Stachurska, K.; Marcisz, U.; Długosz, M.; Antosiewicz, J.M. Circular Dichroism Spectra of α-Chymotrypsin–SDS Solutions Depend on the Procedure of Their Preparation. ACS omega 2022, 7(27), 23782–23789. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proceedings of the National Academy of Sciences 2015, 112(24), E3095–E3103. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Völker, J.; Anafi, D.; Breslauer, K.J. The native and the heat-induced denatured states of α-chymotrypsinogen A: thermodynamic and spectroscopic studies. Journal of molecular biology 1997, 274(2), 237–252. [Google Scholar] [CrossRef]
- Li, Z.; Hirst, J.D. Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet. Chemical science 2017, 8(6), 4318–4333. [Google Scholar] [CrossRef]
- Kaur, H.; Beckman, J.; Zhang, Y.; Li, Z.J.; Szigeti, M.; Guttman, A. Capillary electrophoresis and the biopharmaceutical industry: Therapeutic protein analysis and characterization. TrAC Trends in Analytical Chemistry 2021, 144, 116407. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Andersen, K.K.; Westh, P.; Otzen, D.E. Unfolding of β-sheet proteins in SDS. Biophysical journal 2007, 92(10), 3674–3685. [Google Scholar] [CrossRef]
Figure 1.
Far-UV CD spectra of 10 μM α-chymotrypsinogen A solutions acquired following the combination of the parent solutions as specified in the legend.
Figure 1.
Far-UV CD spectra of 10 μM α-chymotrypsinogen A solutions acquired following the combination of the parent solutions as specified in the legend.
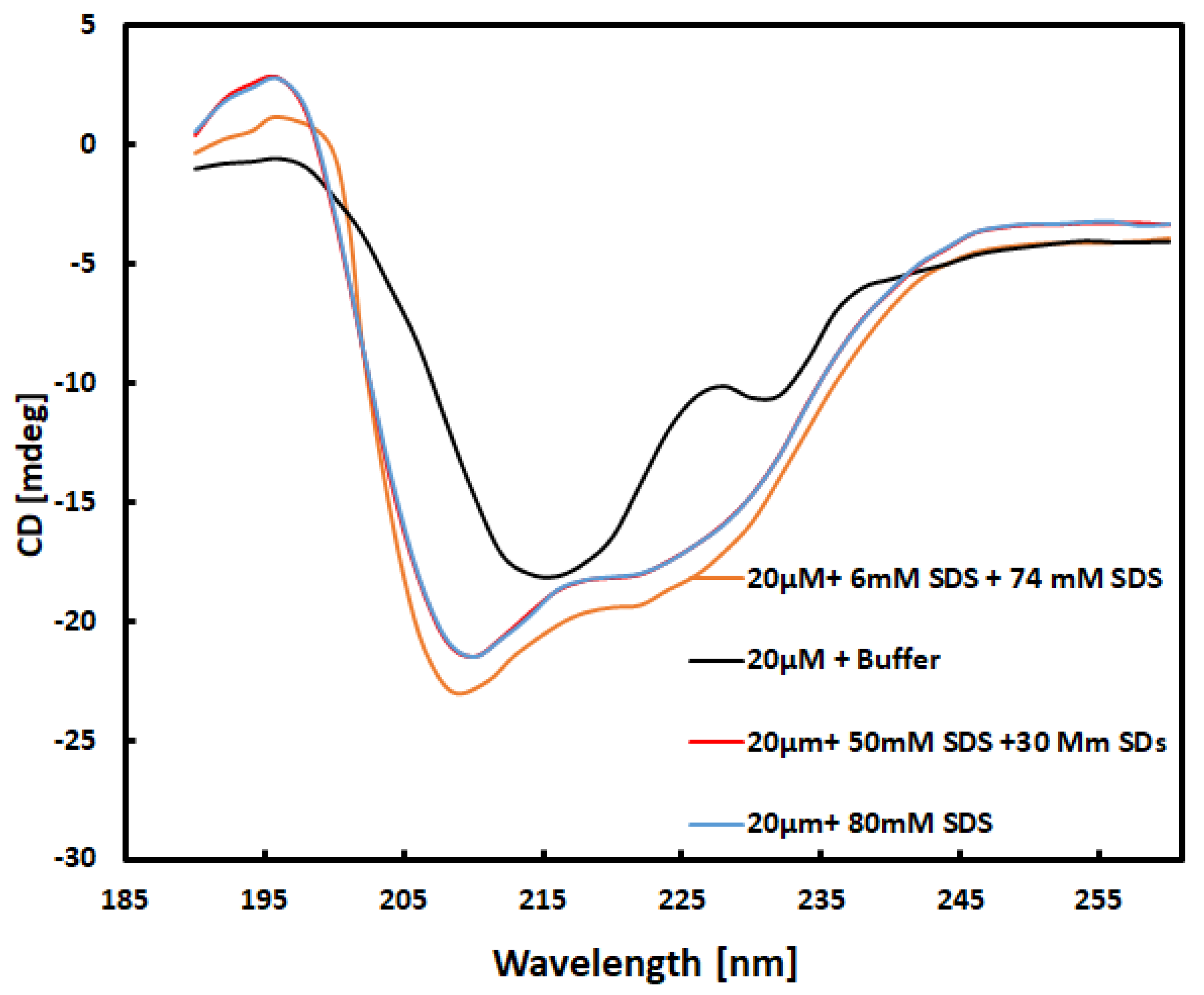
Figure 2.
Near-UV CD spectra of 40 μM α-chymotrypsinogen A solutions acquired following the combination of the solutions.
Figure 2.
Near-UV CD spectra of 40 μM α-chymotrypsinogen A solutions acquired following the combination of the solutions.
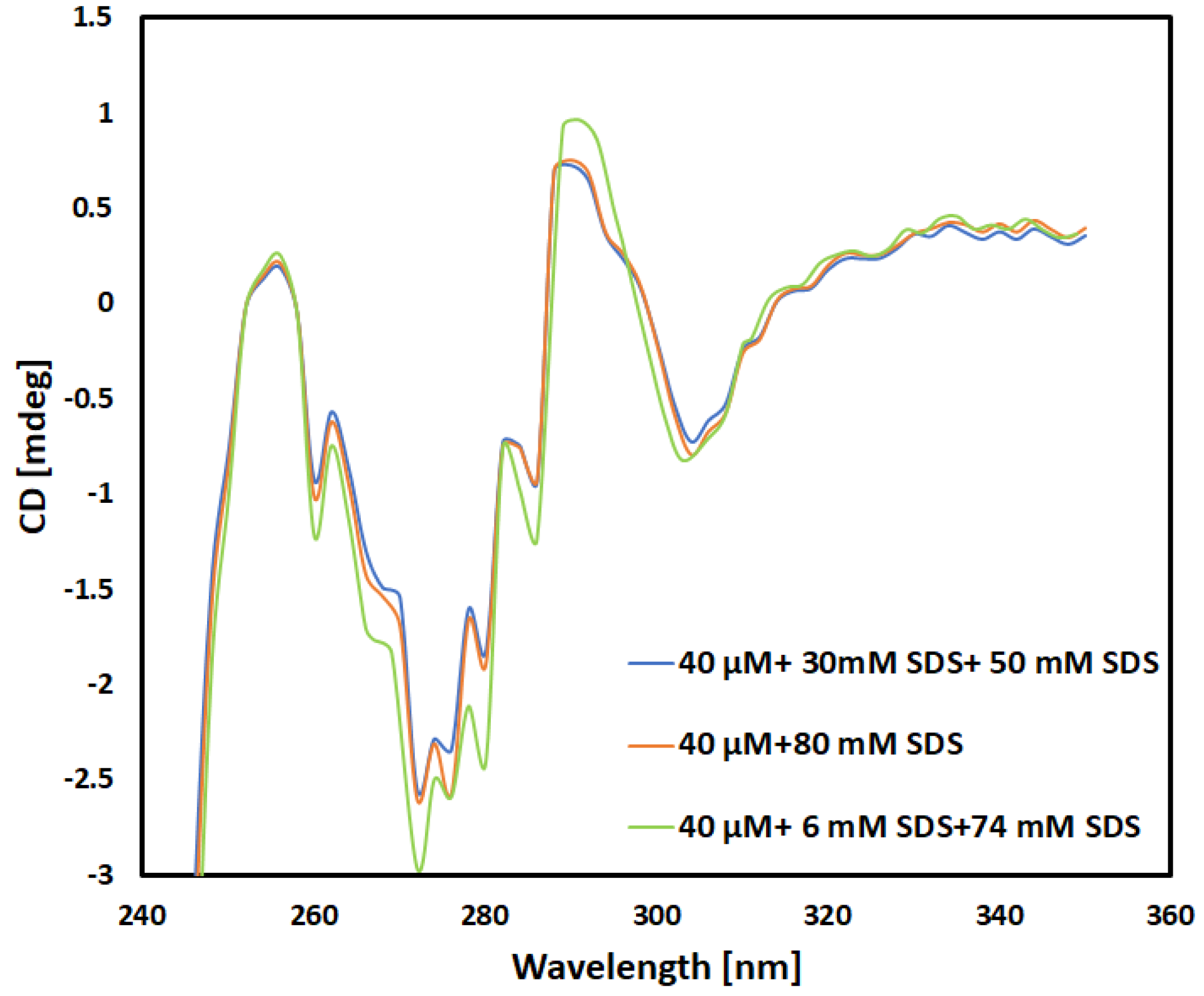
Figure 3.
Near-UV CD spectra of 20 μM (above) and 40 μM (below) α-chymotrypsinogen A solutions acquired following the combination of the solutions. These solutions either have a final concentration of protein equal 10 μM or 20 μM in the presence of the same concentration of SDS, either 40 or 3 mM.
Figure 3.
Near-UV CD spectra of 20 μM (above) and 40 μM (below) α-chymotrypsinogen A solutions acquired following the combination of the solutions. These solutions either have a final concentration of protein equal 10 μM or 20 μM in the presence of the same concentration of SDS, either 40 or 3 mM.
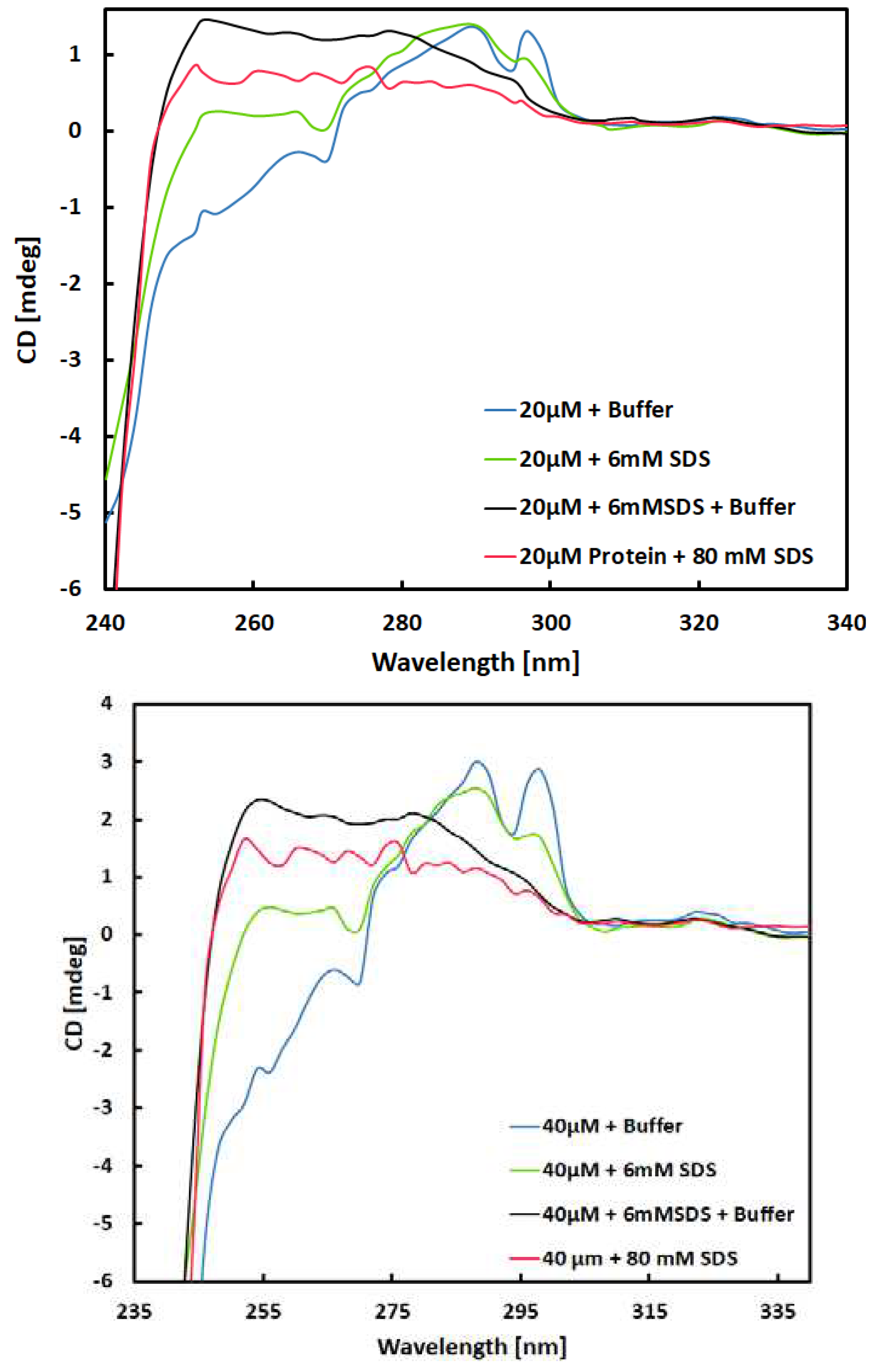

Table 1.
Compilation of Samples Prepared through the Equal Volume Mixing of Two Component Solutions (Part1 & 2).
Table 1.
Compilation of Samples Prepared through the Equal Volume Mixing of Two Component Solutions (Part1 & 2).
| Sample No. | Part 1 | Part 2 |
|---|---|---|
| 1 | 40 mM | Buffer |
| 2 | 40 mM | 80 mM SDS |
| 3 | 40 mM | 6 mM SDS |
| 4 | 40 mM+ 6 mM SDS | 74 Mm SDS |
| 5 | 40 mM + 6 mM SDS | Buffer |
| 6 | 20 mM | Buffer |
| 7 | 20 mM | 80 mM SDS |
| 8 | 20 mM | 6 mM SDS |
| 9 | 40 mM + 6 mM SDS | Buffer |
| 10 | 20 mM+ 6 mM SDS | 74 mM SDS |
| 11 | 20 mM+ 30mM SDS | 50 mM SDS |
Table 2.
Analytical results displaying the distinct secondary structure composition of α-chymotrypsinogen A, delineating the percentage contribution of each structural component.
Table 2.
Analytical results displaying the distinct secondary structure composition of α-chymotrypsinogen A, delineating the percentage contribution of each structural component.
| 20 μM CG in a 10 mM buffer (%) |
20 μM CG + 6 mM SDS mixed with 74 Mm SDS (%) |
20 μM CG mixed with 80 mM SDS (%) |
20 μM + 30 mM SDS mixed with 50 mM SDS (%) |
|
|---|---|---|---|---|
| Helix | 11.7± | 22.2 | 26.1 | 26 |
| Antiparallel b | 23.5 | 9.2 | 11.8 | 12 |
| Parallel b | 4.1 | 4.8 | 2.5 | 3 |
| Turn | 21.7 | 15.5 | 19 | 19.1 |
| Others | 39 | 48.3 | 41 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



