
Submitted:
04 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
Diabetic kidney disease (DKD) is the major cause of chronic kidney disease (CKD) and increases the risk of cardiovascular events. Hemodynamic, inflammatory and metabolic factors, which share the convergent pathway of fibrosis, are considered to involve in the pathogenesis of DKD. In spite of the emerging of angiotensin receptors blockers (ARBs)/ angiotensin converting enzyme inhibitor (ACEI), sodium-glucose cotransporter 2 (SGLT2) inhibitors, and nonsteroidal mineralocorticoid receptors antagonists (NS-MRAs), current therapies still couldn’t arrest the progression of DKD. Glucagon like peptide 1 receptor agonists (GLP-1RAs) are promising agents to play renoprotective roles in slowing the progression of DKD. Based on the treatment of heart failure, combined use of drugs is recommended for DKD rather than single use. Unearthing the mechanisms underlying DKD is urgent to investigate the management of DKD. Here, we elaborate on the potential mechanisms and the current therapies of DKD. We also discuss the additional value of the combined use of these drugs for DKD to establish novel concepts of treatment.
Keywords:
DKD
; molecular mechanisms
; ACEI/ARB
; SGLT2i
; NS-MRAs
; GLP-1RAs
1. Introduction
Diabetic kidney disease (DKD), affecting more than 700 million population, is presently the leading attributable cause of end-stage renal disease (ESRD) worldwide[1]. DKD is a serious microvascular sequela that affects approximately 30% of people with type 1 diabetes (T1D) and 40% of those with type 2 diabetes (T2D)[2]. The worldwide prevalence of diabetes is projected to reach 600 million by 2035[3] and grow to 783 million by 2045, with disproportionate growth in disadvantaged countries[4]. The climbing prevalence of DKD parallels with the dramatic rise in global prevalence of diabetes[2]. DKD confers increased risk of all-cause mortality and low quality of life. Further, the number of renal replacement treatment (RRT) recipients is extrapolated to escalate from 2.819 million to 4.35million by 2035 [5]. The expansion of RRT engenders global economic burden.
The mechanisms of kidney damage due to diabetes can be broadly classified into hemodynamic, inflammatory and metabolic factors, sharing the common pathogenesis of fibrosis[6] (Figure 1). Chronic kidney disease (CKD), attributing to diabetes or other causes, is characterized by progressive kidney fibrosis, leading to loss of function. CKD is defined as abnormities of kidney function or structure: estimated glomerular filtration rate (e GFR) <60 mL/ min/ 1.732 or markers of kidney injury, including albuminuria, present for > 3 months. The level of albuminuria is defined as albuminuria creatinine ratio (ACR) > 30mg/g or persistent albuminuria (>300mg/24h) across repeated measures of 3 or more months regardless of eGFR[7]. Albuminuria and reduced eGFR portend increased risk of cardiovascular disease (CVD) and all-cause mortality[8]. Cardiovascular mortality is the most competing risk for deaths in patients with advanced CKD (stage 4) and ESRD[9]. The foremost strategy for management of DKD is to reduce fibrosis.
RAAS: Renin angiotensin aldosterone system; AGEs: Advanced glycation end products; RAGEs: receptors for AGEs; ROS: reactive oxygen species; PKC: poyol and protein kinase C; NF- κB: nuclear factor κ light-chain enhancer of activated B cell; LPS: Lipopolysaccharide; SCFAs: short chain fatty acids; TNF-α: tumor necrosis factor α; IL-1/ IL-6/IL-18: interleukin -1/6/18; MCP-1: monocyte chemoattractant protein-1; MMP-9: matrix metalloproteinase-9; Na: sodium; MD: macula densa; ET: endothelin; TGF- β: transforming growth factor; MR: mineralocorticoid receptor; M1: M1 macrophage; M2: M2 macrophage.
Nothing in addition to blood pressure and glycemic control was available to postpone the progression of DKD until the advent of a trail of renin angiotensin system (RAS) blockade-captopril in populations with type1 diabetes in 1993[10]. RAS blockers were further consolidated in patients with diabetes during the following 8 years[11,12]. Albeit angiotensin receptors blockers (ARBs] were estimated to halt DKD by 5-7mL/min/year[10], the progression of DKD persisted. In 2014, sodium-glucose cotransporter 2 (SGLT2) inhibitors were discovered unexpectedly to further prevent the progression of DKD and multiple outcomes trials solidified the salutary effects of SGLT2 inhibitors in DKD populations[13]. Almost around the same time of the discovery of SGLT2i, trails on an innovative class of the nonsteroidal mineralocorticoid receptors antagonists (NS-MRAs), specifically finerenone, were initiated, and the agent also slowed the progression of DKD[14]. The glucagon-like peptide 1 receptor agonists (GLP-1RAs) are recommended for DKD patients to gain better glycemic treatment despite optimization with SGLT2i[15] and have been proved to curtail CV events notably by CV outcome trails (CVOT)[16]. Post-hoc-analysis of renal outcomes from CVOT show pronounced protective role of GLP-1RAs[17]. A randomized placebo-controlled trail (FLOW) that evaluates the efficiency of semaglutide in T2D and CKD is first designed with primary renal outcomes [18] and pronounced to be stopped because the results from interim analysis met the certain preassigned criteria.
In this review, we summarize current basic knowledge about pathophysiologic mechanisms of DKD, and provide advancing therapeutic interventions, including promising agents -GLP-1RAs.
2. Molecular Mechanisms of Kidney Damage in Diabetes
2.1. Glomerular Hemodynamic Perturbations
Hemodynamic effects are vital to maintain the glomerular homeostasis and surround the renin-angiotensin-aldosterone system (RAAS). SGLT2 is principal for increasing reabsorption of proximal tubular Na and glucose, which suppress the tubule-glomerular feedback due to reduced delivery of sodium chloride (NaCl) to macula densa[19] and deteriorate hyperglycemia[20]. Reduced tubule-glomerular feedback cause dilated afferent arteriole and increased angiotensin II in efferent arteriole, leading to vasoconstriction[19]. Vasodilatation of afferent arteriole and vasoconstriction of efferent arteriole could lead to hyperfiltration, which is recognized to initiate the pathogenesis of DKD[2]. Endothelin (ET) could modulate renal flow blood and glomerular filtration[21], implicating a potential vasoconstriction effect in renal blood. Dyslipidemia, hyperglycemia, endothelial dysfunction and oxidative stress elevate plasma ET[22]. Endothelin receptor (ER) blockade is demonstrated to reverse the progression of CKD[23]. Cyclo-oxygenase 2(COX-2) derived prostanoids that are expressed in endothelial cells in renal tissue have been considered to regulate renal auto-regulatory functions at the macula densa and mediate dilated function of afferent arteriole[24], resulting in hyperfiltration. Glomerular hyperfiltration results in progressive albuminuria, gradual decreased eGFR, and finally ESRD[25]. Hyperglycemia, glomerular hypertension and high levels of amino acids could exacerbate glomerular hyperfiltration[19].
Renin-Angiotensin-Aldosterone System (RAAS)
RAAS participates in the progression of DKD[26]. The renin is produced by the juxtaglomerular cells of the nephron and found contiguous to the afferent arterioles. The renin is key to trigger RAS, which generates greater vasoconstriction in efferent arteriole than that in afferent arteriole[27]. Angiotensin converting enzyme 2 (ACE2) play important roles in dilating glomerular afferent arteriole through degrading angiotensin II into angiotensin 1-7. Produced by activation of RAS, angiotensin II binds to specific receptors, namely AT1 and AT2. Activation of AT1 modulates the elevated resistance of efferent arteriole[28], contributing to hyperfiltration, and activation of AT2 exerts protective counterregulatory role in renal flow, including prostaglandin release and regulating renal vasodilation[29]. High angiotensin II accelerates renal damage through modulating calcium influx into podocyte[30], stimulating the expression of proinflammatory (tumor necrosis factor α [ TNF-α], interleukin [IL-1,IL-6, IL-18], monocyte chemoattractant protein-1[MCP-1]), matrix metalloproteinase-9 [MMP-9] and profibrotic mediators (transforming growth factor [TGF- β])[6,31], macrophage activation[32] and increased adrenal aldosterone secretion. Adrenal aldosterone could upregulate profibrotic factors such as TGF- β, which boost macrophage infiltration and fibrosis of kidney[33].
2.2. Inflammatory and Fibrotic Factors
Inflammation and fibrosis are dominant interrelated promotors of the progression of DKD. Growth factors, inflammatory cytokines and chemokines are substantiated to elevate in renal biopsy samples from DKD patients[34]. Substantial components of immune system including circulating leukocytes, chemokines and cytokines are activated in diabetes[35]. Pathological variations of DKD are characterized by nodular and diffuse mesangial expansion, thickening of the glomerular and tubular basement membranes, podocyte damage and detachment, which are attributed to sustained glomerular hypertension and hyperfiltration, subsequent to tubular atrophy and glomerular sclerosis, and eventually apparent decline in renal function[36].
TNF-α is produced by activated macrophages and resident kidney cells in glomerular and tubular, which plays vital roles in evoking chemokines, cytokines, cyto-toxic effects and apoptosis[6]. The activation of NF- κB could lead to the production of inflammatory factors such as TNF-α which prompts the progression of DKD[37]. Diabetic cohorts revealed that TNF-α receptor superfamily members were related to high risk of ESRD in diabetes[38]. The cytokines, such as IL-1, IL-6, IL-16 and IL-18 have been incriminated to involve in pathogenesis of DKD. IL-1 could cause hyperpermeability of endothelial cells and glomerular hyper-perfusion through promoting the release of phospholipase A2 and prostaglandin E[39]. Infiltrating macrophages and hyperglycemia contribute to releasing of IL-1β, which is the superfamily of IL-1, intimately involving in the pathogenesis of DKD[40]. IL-6 recruits neutrophil infiltration in the tubulointerstitium, which is correlated to podocyte hypertrophy, and GBM thickening[6], eventually resulting in albuminuria and decrease in renal function. The injection of IL-6 neutralizing antibody into diabetic mice leaded to a prominent reduction of collagen and fibrosis by ameliorating mesenchymal transition[41]. IL-18 instigates unleashing of interferon-γ, expression of adhesion molecules and apoptosis[39]. The expression of IL-18 in renal tissues is intimately associated with increased albuminuria in DKD[39].
MCP-1, also referred to CC chemokine ligand 2, have been confirmed to elevate in biopsied kidneys from patients with DKD, which may elicit inflammatory cell recruitment, migration and interplay, and finally contributing to kidney injury[42,43]. MMP-9, which is expressed in the proximal renal tubular epithelial cells, is validated to modulate the degradation of extracellular matrix during renal fibrosis[44]. Downregulation of MMP-9 leads to slowing the progression of DKD by improving creatinine and proteinuria[45].
Kidney damage in diabetes is pronouncedly featured by monocytes and macrophages. Amassment of macrophages exhibits close relationship with histological severity of kidney disease in diabetes[42,46]. Macrophages exacerbates kidney injury by modulating tissue repair and fibrosis[47,48]. Hyperglycemia, angiotensin II, endothelial cell dysfunction, oxidized low density lipoprotein (LDL) and advanced glycation end products (AGEs) promote the accumulation of macrophages[32]. M1 macrophages could switch to anti-inflammatory M2 macrophage[47]. M1 macrophages could secret substantial inflammatory factors IL-1, IL-6, MMP-9 and TNF-α after kidney injury[47]. The balance between M1 and M2 macrophage is the major challenging to develop macrophage-based therapy for DKD.
Tubulointerstitial fibrosis is inevitable outcome and the final convergent pathway of progressing kidney disease, and is correlated to extracellular matrix accumulation and tubular atrophy[49]. TGF- β, which is expressed by nearly all cell types of kidney, infiltrates macrophages and leukocytes, and plays pleiotropic effects including immunomodulation, angiogenesis and extracellular matrix formation in the progression of kidney diseases. TGF- β acts as master mediator of DKD via regulating inflammation and fibrosis[50].They summarized the pathogenetic roles of TGF- β and its downstream Smad signaling molecules in the progression of DKD. Smad3 fosters autophagy dysregulation by provoking lysosome depletion in tubular epithelial cells of DKD[51]. Recent study demonstrated leucine-rich -2-glycoprotein 1 (LRG1) could exacerbate kidney fibrosis by augmenting TGF- β/ Smad3 signal transduction[52]. Klotho, which was mainly expressed in kidney cells, was reported to be a potential therapy for DKD through regulating calcium and phosphate metabolism, downregulating apoptosis, guarding against oxidative stress, and playing anti-inflammatory and antifibrotic roles[53].
Angiotensin II – mediated reactive oxygen species (ROS) or protein kinase C (PKC) and p38 mitogen-activated protein kinase could trigger CTGF, and plasminogen activator inhibitor (PAI-1) could be activated by TGF- β[54,55]. PAI-1accelerates kidney fibrosis by restraining the production of plasmin from plasminogen, which maintains extracellular matrix accumulation.
Hyperglycemia, AGEs and glomerular hypertension could upregulate the expression of TGF- β[56]. Fibronectin is validated to result in mesangial expansion and deterioration of albuminuria, contributing to exacerbated kidney function[57]. Treatment of DKD with mesenchymal stem cell therapy could diminish fibronectin and mitigate renal function and albuminuria[58]. Metformin is reported to reduce collagen-1 together with fibronectin[59]. Studies reveal collagen-1 propels the progression of renal fibrosis and overabundant accumulation of extracellular matrix in DKD[60]. The precise mechanism of collagen-1 in the pathogenesis of DKD remains unclear and need further to be explored. The serine/threonine kinase, which is an apoptosis signal -regulating kinase 1(ASK1) induced by oxidative stress, evokes apoptosis, inflammation and fibrosis[61]. ASK1 has been incriminated to participate in the pathogenesis of DKD through phosphorylating and activating c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase[62]. Glucose dysmetabolism could activate protein kinase C (PKC) and the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathways[63]. The JAK-STAT pathway prompts the expression proinflammatory factors and multiple chemokines, enhancing inflammatory response in DKD[42]. JAK-STAT are highly expressed in glomeruli and tubulointerstitial cells in population with T2D and exhibits inverse relationship with eGFR[64].
CTGF is found to be associated with tubulointerstitial fibrosis and glomerulosclerosis in various renal disease[54]. Urinary CTGF concentrations is related to high risk of albuminuria and decreased eGFR[54]. The synthesis of fibronectin and type 1 collagen elevated when mesangial cells were exposed to CTGF[54]. Phosphatase and tensin homolog (PTEN) increases the risk of decreased eGFR of DKD patients[65]. PTEN potentiates the expression of IL-6 and CTGF[66].
2.3. Metabolic Factors
Hyperglycemia, increased adiposity and dyslipidemia could enhance the production of vasoactive mediators, including AGEs and ROS[67,68]. Upon interacting with the receptors for AGEs (RAGEs), AGEs lead to tissue damage in DKD through enhancing variation of extracellular matrix architecture and regulating cellular functions[69]. RAGE are detected throughout the kidney. The accumulation of AGEs in renal basement could upregulate the expression of RAGE on podocytes in DKD, inducing NF- κB mediated inflammation, fibrosis and oxidative stress[70]. Targeting on AGEs or AGE/RAGE induced oxidative stress could hold promising therapy for DKD. AGEs also could contribute to impaired vasodilatation in diabetes through suppressing the bioavailability of endothelium-derived nitric oxide (NO) and elevating the production of ROS[71]. ROS accelerate the progression of DKD through podocytes apoptosis and epithelial to mesenchymal transition (EMT)[68,72]. ROS are generated by nucleotide leukin rich polypeptide 3 (NLRP3) inflammasomes, and also foster the activation of NLRP3[72]. Knocking down of NLRP3 has been reported to impede podocytes injury through reducing hyperglycemia induced production of mitochondrial ROS in renal mesangial cells and preventing lipid accumulation[73]. The overexpression of pro-oxidant enzyme NADPH oxidase 5 (NOX 5) is demonstrated to promote albuminuria, inflammation and renal fibrosis in diabetes by increasing ROS formation [73]. Hyperglycemia also could modulate poyol and protein kinase C (PKC) pathways to diminish endothelial nitric oxide synthase and amplifying oxidative stress, respectively, resulting in higher vascular endothelial growth factor and endothelin levels[32]. Hyperglycemia, dyslipidemia and insulin resistance are the common features of diabetes and could potentiate vicious cycle of inflammatory and oxidative process[42,74].
2.4. Dietary AGEs and Gut Microbiome Variation
AGEs exposure could partly result from diet as well as hyperglycemia[75]. AGEs contribute to glomerular pathological alterations including glomerular hypertrophy, glomerular basement membrane widening, mesangial expansion and glomerular sclerosis[76]. Dietary AGEs could interact with gut microbiota, evoking local inflammation and the release of inflammatory factors[77]. AGEs-rich foods interrupt intestinal mucosal barrier and translocation of inflammatory mediators into systemic circulation, causing local kidney inflammation[78]. As the progression of DKD, uremic toxins result in a relocation towards Gram-negative (G-) bacteria in the gut. Gut microbiota derived phenyl sulfate has been reported to lead to podocyte injury and albuminuria. Lipopolysaccharides (LPS) from the cell wall of G- bacteria bind to Toll-like receptor (TLR)-4 to elevate local cytokine production, recruitment of inflammatory cells and the release of LPS[79]. Exposure to TLR-4 on podocytes or other kidney cells, LPS contribute to inflammation and fibrosis, ultimately resulting in podocyte damage, tubular injury, glomerular hypertrophy and hypercellularity as well as albuminuria in STZ induced diabetic mice[80]. Alteration in gut microbiota has been incriminated in the pathogenesis of DKD. Reduction in dietary associated short chain fatty acids (SCFAs) from gut microbiota could worsen podocyte damage, interstitial fibrosis and albuminuria by promoting epithelial cell dysfunction and gut inflammation[81].
3. Targeting Mechanisms and Recent Advances in the Therapy of DKD
Underlying molecular mechanisms play vital role in developing effective therapies to reduce the onset and progression of DKD. Precise medicine by connecting molecular mechanism with therapeutic strategies have been attempted to halt kidney disease progression, finally failing to be adopted for DKD (Figure 2). Long- term diabetic retinopathy study of ruboxistaurin(RBX), a protein kinase C beta (PKC- β) inhibitor, failed to prevent kidney outcomes[82]. ASCEND study of endothelial antagonist (EA) avosentan was reported to reduce albuminuria but increase fluid retention and heart failure[83]. In 2019, SONAR trail evaluates the reno-protective effect of atrasentan, a selective endothelial receptor antagonist (ERA), which draw the similar conclusion (hospitalization, anemia and fluid retention) with avosentan[84]. It is uncertain whether their benefits exceed risks of heart failure, so ERA are not recommended on DKD patients by KDIGO 2022 Guideline for Diabetes Management[85]. In 2011, a small-scale of random control trails (RCT) of pirfenidone, an oral antifibrotic and anti-inflammatory agent, was conducted to assess primary renal outcomes. The conclusion from the study is incomplete ascertainable[86]. A phase II study of pirfenidone for renal fibrosis is ongoing (NCT04258397) and will be completed by 2024. Sulodexide, a mixture of glycosaminoglycan polysaccharide components, was demonstrated to exhibit no reno-protective effect on patients with type 2 diabetes, macroalbuminuria and renal impairment[87]. The trail of Aliskiren, a renin inhibitor, was discontinued prematurely and demonstrated to be even harmful[88]. Bardoxolone methyl exerts antioxidant capacity, antiinflammation and oxidative stress through activating Keap1-nuclear 1 factor (erythroid-derived 2)-related factor 2 (Nrf2) pathway[89]. The trail of Bardoxolone methy, didn’t exhibit reduced risk of ESRD, and was terminated attributed to greater rate of cardiovascular events[89]. A following phase 2 TSUBAKI study revealed an improved eGFR and no incidence of cardiovascular events[90]. A new large multicenter phase 3 study (AYAME) for investigating the long-term efficacy and safety of bardoxolone methy has been performed in Japan[91]. The clinical trial was terminated, which indicted that bardoxolone methy failed to be adopted for DKD.
These agents failed to be adopted for DKD. PKC: poyol and protein kinase C; TGF- β: transforming growth factor; MR: mineralocorticoid receptor; Nrf2: nuclear 1 factor (erythroid-derived 2)-related factor 2; ACEI: Angiotensin converting enzyme inhibitor; ARB: angiotensin receptors blockers.
Since 2001, ARBs are demonstrated a clear benefit in preventing the progression of DKD, the following novel therapies which are mentioned above failed to show advantages until the appearance of SGLT2 inhibitors in 2014. With the approval of the NS-MRA-finerenone in 2021, we have more confidence in the management of DKD. According to interim analysis of RCT (FLOW), GLP-1RA is ushering a new era for DKD. In this section, we mainly focus on the drugs whose clinical effects have been proved as therapeutic agents for DKD (Figure 3). Pillars of therapy, primary adopted in heart failure by cardiologists, are also concerned when and how to administrate these drugs will maximally retard the progression of DKD.
Potential agents mean more clinical trials need to be conducted to validate efficiency of these drugs on DKD. RAS: Renin-Angiotensin/Aldosterone System; SGLT2: sodium-glucose cotransporter 2 ; Ns-MRAs: nonsteroidal mineralocorticoid receptors antagonists ; GLP-1RAs: Glucagon like peptide 1 receptor agonists; ASK1:apoptosis signal -regulating kinase 1; JAK: Janus kinase.
3.1. RAS Blockades
A RCT of captopril, an ACEI, was first performed to evaluate the reno-protective properties in slowing down the progression of DKD. Treatment with captopril displayed notable reduction in the risk of composite end points (death, transplantation and dialysis) independent of blood pressure management in type 1 diabetic nephropathy[10]. However, type 2 diabetic nephropathy achieved no additional benefits from captopril. Losartan, an ARB, conferred salutary effects on renal and cardiovascular outcomes on patients with type 2 diabetes and nephropathy[12]. Irbesartan exhibited similar reno-protective action for nephropathy attributed to type 2 diabetes as losartan, and the protection is independent of blood pressure control[92]. Also, standard administration of ARBs was adopted, DKD progression continues to advance. The combination therapy of ACEI and ARBs was demonstrated to be even harmful for DKD[93]. The additional novel therapies summarized above present disappointed consequences.
3.2. SGLT2 Inhibitors
The advent of SGLT2 inhibitors in 2014 generated striking excitement in strengthening DKD management. SGLT2 inhibitors have been extrapolated to involve in progression of DKD via various mechanisms, including activation of tubule-glomerular feedback to reduce glomerular hyperfiltration, reducing in circulating inflammatory and fibrotic factors of TNF receptor-1, IL-6, MMP-7 and fibronectin-1, and decreased ketone production[94]. SGLT2 inhibitors are recommended to treatment of most DKD patients with eGFR ≥ 25ml/min per 1.73m2, irrespective of the degree of glycemic management, on the basis of RCTs, associated meta-analysis and systemic review[95,96]. The Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) and the Dapagliflozin and Prevention of Adverse outcomes in Chronic Kidney Disease (DAPA-CKD) trails have exerted robust impact on clinical application of SGLT2 inhibitors[97]. Canagliflozin showed a 34% reduction in death of renal event and a 32% decrease in risk of ESRD in DKD patients[97]. Dapagliflozin showed 44% reduction in risk of ESRD or death from renal events, at least 50% decline in eGFR and a 29% relative reduction in risk of death from CV events in CKD patients irrespective of absence or presence of diabetes[98]. SGLT2 inhibitors are highly recommended in patients with severe albuminuria[99,100]. The study of Heart and Kidney Protection with Empagliflozin (EMPA-KIDNRY) trial was conducted to evaluate the impact of empagliflozin on CKD patients without diabetes. Empagliflozin revealed a lower rate of hospitalization from any cause by 14%, greater renal protective effects and lower risk of death from cardiovascular events by 28%, with their efficacy being more conspicuous in those with ACR more than 300mg/g[100]. The combined use of SGLT2 inhibitor and ACEI/ARB shows a prominent decline in renal function nearly by 30%-40%, exceeding those of ACEI/ARB alone[101]. SGLT2 inhibitors act as second validated therapy for slowing the progression of DKD.
3.3. NS-MRAs
In addition to the distal nephron, MRs are expressed on other cell types, including fibroblasts, macrophages, podocytes and vascular cells. Decreased circulating plasma volume induces RAS activation [102], further promoting aldosterone secretion, which contributes to MR activation, resulting in sodium reabsorption and potassium excretion. The activation of MRs with high sodium intake leads to hypertension, contributing to glomerular damage and fibrosis[102]. Hyperglycemia, dyslipidemia, insulin resistance and obesity upregulate the expression of MR, which elevates inflammatory (IL-1β, IL-6, TNF-α,MCP-1) and profibrotic factors (extracellular matrix proteins, PAI-1, TGF -β, CTGF), eventually resulting in progression of DKD[103,104]. The upstream accumulation of renin due to lone-term ACEI/ARB therapy could increase plasma aldosterone owing to “aldosterone escape”. Lone period use of trandolapril showed an obvious increase in aldosterone in 40% of patients at 40 weeks with mounting albuminuria[105], which indicated that the combined use of ACEI/ARBs and MRAs might present optimal treatment for DKD.
As early as 2001, animal study revealed therapeutic roles of MRAs in preventing the progression of DKD by reducing inflammation, fibrosis and albuminuria[106]. Steroid-based MRAs, including spironolactone and eplerenone are adopted for symptomatic heart failure patients[107]. In spite of reduced albuminuria and blood pressure of MRAs in DKD patients, there is scarcity of clinical trials to verify the data due to high risk of hyperkalemia and reduction in kidney function. It’s generally contraindicated to use MRAs in advanced kidney disease[108].
NS-MRAs, including finerenone, apararenone, esaxerenone and ocedurenone, which distribute between heart and kidney tissue rather than influencing the kidney alone, are conspicuously different from steroidal-based MRA[14]. Finerenone, a NS-MRA, demonstrated prominent reduction in albuminuria and blood pressure as well as risks of atherosclerotic disease and heart failure in the Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease (FIDELIO-DKD) and Finerenone in Reducing cardiovascular mortality and morbidity in Diabetic Kidney Disease (FIGCARO-DKD) clinical trials in participants with T2D and DKD treated by and ARB or ACEI as the standard care[109]. FIDELIO-DKD trail revealed that finerenone demonstrated a more than 40% reduction in risk of eGFR declining and 18% decline in death from renal disease as well as a 14% reduction in prespecified secondary endpoints of death from CV events (heart failure, hospitalization, nonfatal stroke or nonfatal myocardial infarction) [110]. Whereas, finerenone still exerted slightly higher risk of hyperkalemia than that of placebo (2.3% vs 0.9%)[110]. FIGCARO-DKD trail included patients with higher risk of CV and less advanced DKD supported the benefits of finerenone in reducing CV causes. Despite the risk of hyperkalemia, finerenone is well tolerated.
The prespecified pooled analysis of FIDELIO-DKD and FIGCARO-DKD, referred to FIDELITY, including more than 13,000 participants with type 2 diabetes and a broad range of CKD stages and albuminuria, validated a 23% reduction in doubling of creatinine, RSRD and death from renal disease, and also a 14% decrease in the risk of the composite outcomes of CV[109]. FIDELITY revealed the incidence of hyperkalemia is greater in spironolactone with resistant hypertension than that in finerenone group (64.1% vs 11.2%) [111]. A smaller phase 3 RCT clinical trial of Esaxerenone with Placebo in Japanese Type 2 Diabetic Patients with Microalbuminuria (ESAX-DN) failed to show protective effect on the progression of DKD due to short study duration in participants with early DKD or other unique characteristics of single-country study[112]. Animal study showed that the combined use of finerenone and empagliflozin in hypertensive rats contributed to obvious decrease in kidney fibrosis and albuminuria[113]. However, the retrospective analysis of DAPA-HF (Dapagliflozin in HFrEF)[114] and EMPEROR-Reduced trials (Empagliflozin Outcome Trial in Patients With Chronic Heart Failure With Reduced Ejection Fraction)[115] did not validate the efficiency of the combination of these two drugs. The therapy of SGLT2 inhibitor appears to counteract the hyperkalemia due to the adding of MRA to ACEI or ARBs[116,117]. The 2022 guidelines of the American Diabetes Association (ADA) and the Kidney Disease Improving Global Outcomes (KDIGO) recommend the use of finerenone across a wide range of DKD patients with increased albuminuria despite treatment with an ACEI/ARB and SGLT2 inhibitor[85,118]. Finerenone is the only one approved for protection for cardiorenal events, whereas other NS-MRAs are recommend only for controlling blood pressure with no outcome data in favor of use in DKD.
3.4. GLP1-RAs
GLP, which is a peptide produced by the gut epithelium, could regulate blood glucose through activating the GLP-1R in the pancreas to lower glucagon and elevate insulin secretion. Incretin drugs include GLP-1RAs and dipeptidyl peptidase 4 (DPP4) inhibitors. GLP-1RAs are approved for the management of hyperglycemia and restriction pf atherosclerotic CV disease, and/or high risk for CV events in DKD patients, in spite of optimization with metformin and SGLT2 inhibitors[119,120]. Secondary analysis from glycemic lowering and CV outcome trails confirmed reno-protective actions of the GLP-1RA in T2D through reducing albuminuria and slowing the decline in eGFR independent of glycemic control[121,122]. GLP-1RAs have been reported to play reno-protective roles through ameliorating oxidative stress, cellular apoptosis and fibrosis[119]. GLP-1RAs could deplete the generation of ROS and suppress the binding of NF- κB to its target gene, further reducing the downstream expression of cytokines (TNF-α, IL-1, IL-6) and fibrotic factors (TGF-β)[119]. A systemic review and network meta-analysis of RCTs demonstrated GLP-1RAs could reduce the risk of eGFR 15 mL/ min/ 1.732 and the need to initiate renal replacement therapy by 22% within 5 years [123]. Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) revealed liraglutide could reduce the rate of new-onset persistent albuminuria[124]. Primary evaluation of Cardiovascular and other Long-term outcomes with Semaglutide in Subjects with Type 2 Diabetes (SUSTAIN-6) showed semaglutide could result in lower rates of new or worsening nephropathy[125]. Pooled analysis of SUSTAIN-6 and LEADER revealed semaglutide and liraglutide contributed to a 24% reduction in albuminuria from baseline to 2 years[17]. Semaglutide and liraglutide reduced the risk of sustained declines in eGFR to 40% and 50%, respectively[17]. The latest published trail of Effect of Efpeglenatide on Cardiovascular Outcomes (AMPLITUDE-O) revealed efpeglenatide resulted in a 32% reduction in the risk of composite renal outcomes independent of baseline use of SGLT2 inhibitor[126]. In addition to lowering glucose, GLP-1RAs and DDP4 inhibitors can lead to decrease in blood pressure and body weight[119]. Obesity reduces the generation of adiponectin and induce the production of leptin. Leptin prompts the proinflammatory (IL-1, IL-6, TNF-α, MCP-1) and profibrotic (TGF-β, PAI-1, CTGF) factors[127]. Obesity also activates RAAS, which leads to higher intraglomerular pressure, eventually contributing to podocyte loss, progressive fibrosis and renal failure[127,128]. DDP4 inhibitors showed only modest improvement of albuminuria and failed to delay the decline in eGFR[119].
The Effect of Semaglutide Versus Placebo on the Progression of Renal Impairment in Subjects with T2D and Chronic Kidney Disease trail (FLOW) is the first performed to evaluate the primary kidney disease outcome of GLP-1RA [129], and will be stopped early for unequivocal positive efficacy based on interim analysis. It’s believable semaglutide will be recommended to slow the progression of DKD by KDIGO soon. The combination therapy of SGLT2 inhibitor and GLP-1RAs was confirmed to reduce the risk of major adverse cardiac and cerebrovascular events and heart failure in patients with type 2 diabetes[130]. The effect of combination of these two drugs on the progression will be evaluated by RCT in the near future.
3.5. Other Agents
Pentoxifylline (PTF), a nonspecific phosphodiesterase inhibitor, was demonstrated to play antiproliferative, anti-inflammatory and antifibrotic roles [131]. In 2015, PTF was demonstrated to result in a smaller decrease in eGFR and greater decline in residual albuminuria in patients with type 2 diabetes and stages 3-4 CKD under standard administration of RAS blockade[132]. A respective analysis of PTF was postulated to slow the progression through increasing the expression of Klotho, which was associated with anti-inflammatory and antialbuminuric properties[133]. More definitive trails of PTF need to be initiated to consolidate the reno-protective actions. ASK1 inhibitor has revealed protective effects on kidney injury through reducing inflammation and fibrosis in rodent models of DKD [61]. Post hoc analysis of a phase 2 clinical trial of selonsertib, a selective ASK1 inhibitor suggested selonsertib might be a potential therapeutic agent to prevent the progression of DKD despite the fact that the trail didn’t achieve the primary endpoint[62]. Multicenter Study Evaluating the Efficacy and Safety of Selonsertib in Subject With Moderate-to-Advanced Diabetic Kidney Disease (MOSAIC, NCT04026165) has been completed by 2021, and the data has not been published. A phase 2 placebo-controlled trial of baricitinib, a JAK1/2 inhibitor, predominately reduced albuminuria and inflammatory factors (intercellular adhesion molecule-1, plasma TNF receptor-1/2, and serum amyloid A) in patients with T2D and DKD[134]. Further trails need to be performed to investigate whether baricitinib could prevent the progression of DKD. JAK-STAT inhibitors are used in immune-mediated disease including psoriasis, spondyloarthritis, rheumatoid arthritis, inflammatory bowel disease [135], supporting their potential therapeutic role in slowing the progression of DKD.
3.6. Lifestyle
In addition to screening for complications and management of cardiovascular risk factors of DKD, controlling of hyperglycemia, dyslipidemia and hypertension, and weight management, life style factors including smoking, diet and physical activity play vital roles in the progression of DKD. High dietary protein intake could lead to intraglomerular hypertension, which contributes to glomerular hyperfiltration, kidney damage and proteinuria[136]. In advanced CKD, patients are advised to restrict potassium. Lower-potassium fruits, vegetables and other foods are recommended for DKD patients, and the intake of vegetables and fruits should be in accordance with normal diabetic diet recommendations. Endogenous and dietary AGEs contribute to the progression of DKD. High dietary AGEs results in inflammation, insulin resistance, diabetes and kidney injury[137]. Healthy diets, including fruits, vegetables, whole grains, legumes, fiber, unsaturated fats, plant-based proteins and nuts has been revealed to be associated with lower incidence of CKD and albuminuria[138]. Restricting sodium intake is associated with significant decrease in risk of stroke, cardiovascular disease and progression of CKD[139]. Lower levels of physical activity is related to CVD[140]. Physical activity could reduce inflammatory markers, and improve endothelial function and insulin sensitivity[141]. Physical exercise contributes to lower risk of CVD and CKD[142]. KDIGO recommends patients with DKD could undertake moderate-intensity exercise for a cumulative duration[7]. Tabacco is considered to be an explicit risk factor for the progression of DKD as well as secondhand smoke[143,144]. KDIGO recommends patients with DKD should quit smoking and avoid secondhand smoke[7].
4. The Value of Drug Combination Therapy in Clinical Application
Each drug class which was coupled with RAS blockade showed protective effects on kidney and cardiovascular events. On the basis of heart failure management, the combination of three or four drugs may be charming for reducing cardiorenal events. Practice guidelines articulate that RAS blockade should be maximally tolerated before adding other medications (SGLT2 inhibitors, NS-MRAs and GLP-1RAs)[145]. SGLT2 inhibitors and finerenone have been revealed to lead to a lesser mitigating of renal function decline[110,146,147]. The animal study also confirmed that combined treatment of empagliflozin and finerenone[113] resulted in a decrease in blood pressure, proteinuria, plasma creatinine, uric acid, vasculopathy, cardiac fibrosis and mortality regardless of the eGFR down to 25mL/ min/ 1.73m2[113]. The combination of dapagliflozin and steroidal MRA eplerenone showed additive reduction in albuminuria and risk of hyperkalemia compared with the use of eplerenone alone[148], which is in line with the lower incidence of hyperkalemia when an SGLT2 inhibitor is combined with finerenone from FIDELIO-DKD trial[116].
FIDELITY subgroup analysis further revealed that finerenone showed greater cardiorenal benefits regardless of the combined use of SGLT2 inhibitor or GLP-1RA at baseline or any time during the trail[116]. There was no trail to assess simultaneous use of all four agents in heart failure or compare combined use of different drug against each other. Each drug class that consolidated improved outcomes was combined with RAS blockade in DKD patients. The study to investigate the Combination effect of Finerenone and Empagliflozin in Participants With chronic kidney disease and Type 2 Diabetes using a UACR Endpoint study (CONFIDENCE) is an ongoing trail to evaluate whether combined treatment of finerenone and SGLT2 inhibitor outshines each drug alone[149]. A powered study of the combined therapy of four different agents together to evaluate the efficiency and safety need to be performed.
5. Conclusions
DKD is featured by a series of hemodynamic, inflammatory and metabolic process, sharing the convergent fibrotic pathway. DKD may be propagated even if glycemia is normalized due to persisted expression of proinflammatory and profibrotic mediators. Despite the emerging of RAS blockades, SGLT2 inhibitors and NS-MRAs, current therapies are ineffective for impeding kidney disease progression and abating risks of comorbidities and death among patients with DKD. RAS blockers, SGLT2 inhibitors and NS-MRAs have shown great efficacy in reducing the risk of renal disease. However, patients vary in their response to RAS blockades, the pharmacodynamic responses to SGLT2 inhibitors decrease with elevating severity of renal damage and the incidence of hyperkalemia increases in the treatment of NS-MRAs. Other agents targeting on Nrf2, fibrosis, PKC, EA, ERA, renin, glycosaminoglycan polysaccharide and phosphodiesterase achieved unmet consequences. Plentiful residual risks of progression for DKD persisted. Thus, effective therapy for DKD is yet unsatisfied. Just recently, Novo Nordisk claimed they would stop semaglutide kidney outcomes trail due to clear protective roles on the basis of interim analysis. With the advent of GLP-1RAs, the possible fourth class brings new efficacious treatment for arresting the progression of DKD. The combined therapy of these drugs for DKD is heartening.
We summarize the recent discoveries of basic and clinical research centering on novel findings and developments in regard to molecular mechanisms underlying DKD, including therapeutic interventions. Further extensive exploration is urgent to advance our understanding of the pathogenesis of DKD and to set up striking strategies for DKD.
Author Contributions
Conceptualization, Na Wang and Chun Zhang; methodology, Na Wang; validation, Na Wang and Chun Zhang; writing—original draft preparation, Na Wang; writing—review and editing, Chun Zhang; supervision, Chun Zhang; funding acquisition, Chun Zhang. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the National Natural Science Foundation of China (82370728, 81974096, 81974097, 82170773, 82100729, 82200808, 82200841, 82300843, and 82300786), National Key Research and Development Program of China (2021YFC2500200), and Key Research and Development Program of Hubei Province (2023BCB034).
Acknowledgments
Some pictures in the figures are supported by Bio-render.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol Dial Transplant 2019, 34, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Collaboration, N.C.D.R.F. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan JC, N.; Mbanya, J.C.; Pavkov, M.E.; Ramachandaran, A.; Wild, S.H.; James, S.; Herman, W.H.; Zhang, P.; Bommer, C.; Kuo, S.; Boyko, E.J.; Magliano, D.J. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 2022, 183, 109119. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; Rodgers, A.; Gallagher, M.; Kotwal, S.; Cass, A.; Perkovic, V. Worldwide access to treatment for end-stage kidney disease: a systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int 2022, 102, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes Diabetes Work, G. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Bello, A.K.; Hemmelgarn, B.; Lloyd, A.; James, M.T.; Manns, B.J.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Associations among estimated glomerular filtration rate, proteinuria, and adverse cardiovascular outcomes. Clin J Am Soc Nephrol 2011, 6, 1418–1426. [Google Scholar] [CrossRef]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Cause of Death in Patients with Reduced Kidney Function. J Am Soc Nephrol 2015, 26, 2504–2511. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Vulov, V. [Infusion treatment and parenteral feeding of the newborn]. Akush Ginekol (Sofiia) 1977, 16, 387–393. [Google Scholar]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; Investigators, R.S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Ma, Y.; Lin, C.; Cai, X.; Hu, S.; Zhu, X.; Lv, F.; Yang, W.; Ji, L. Baseline eGFR, albuminuria and renal outcomes in patients with SGLT2 inhibitor treatment: an updated meta-analysis. Acta Diabetol 2023, 60, 435–445. [Google Scholar] [CrossRef]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br J Pharmacol 2022, 179, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; Huikuri, H.V.; Johansson, I.; Juni, P.; Lettino, M.; Marx, N.; Mellbin, L.G.; Ostgren, C.J.; Rocca, B.; Roffi, M.; Sattar, N.; Seferovic, P.M.; Sousa-Uva, M.; Valensi, P.; Wheeler, D.C.; Group, E.S.C.S.D. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Shaman, A.M.; Bain, S.C.; Bakris, G.L.; Buse, J.B.; Idorn, T.; Mahaffey, K.W.; Mann JF, E.; Nauck, M.A.; Rasmussen, S.; Rossing, P.; Wolthers, B.; Zinman, B.; Perkovic, V. Effect of the Glucagon-Like Peptide-1 Receptor Agonists Semaglutide and Liraglutide on Kidney Outcomes in Patients With Type 2 Diabetes: Pooled Analysis of SUSTAIN 6 and LEADER. Circulation 2022, 145, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Naaman, S.C.; Bakris, G.L. Diabetic Nephropathy: Update on Pillars of Therapy Slowing Progression. Diabetes Care 2023, 46, 1574–1586. [Google Scholar] [CrossRef]
- Tuttle, K.R. Back to the Future: Glomerular Hyperfiltration and the Diabetic Kidney. Diabetes 2017, 66, 14–16. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; VanBeusecum, J.P.; Inscho, E.W. Endothelin and the renal microcirculation. Semin Nephrol 2015, 35, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Bjornstad, P.; van Raalte, D.H.; Heerspink, H.L.; Cherney DZ, I. The New Biology of Diabetic Kidney Disease-Mechanisms and Therapeutic Implications. Endocr Rev 2020, 41, 202–231. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 2014, 86, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Miller, J.A.; Scholey, J.W.; Nasrallah, R.; Hebert, R.L.; Dekker, M.G.; Slorach, C.; Sochett, E.B.; Bradley, T.J. Renal hyperfiltration is a determinant of endothelial function responses to cyclooxygenase 2 inhibition in type 1 diabetes. Diabetes Care 2010, 33, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Premaratne, E.; Verma, S.; Ekinci, E.I.; Theverkalam, G.; Jerums, G.; MacIsaac, R.J. () The impact of hyperfiltration on the diabetic kidney. Diabetes Metab 2015, 41, 5–17. [Google Scholar] [CrossRef]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Sochett, E.B.; Cherney, D.Z.; Curtis, J.R.; Dekker, M.G.; Scholey, J.W.; Miller, J.A. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol 2006, 17, 1703–1709. [Google Scholar] [CrossRef]
- Goodfriend, T.L.; Elliott, M.E.; Catt, K.J. Angiotensin receptors and their antagonists. N Engl J Med 1996, 334, 1649–1654. [Google Scholar] [CrossRef]
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 2000, 35, 155–163. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W., Jr.; Birnbaumer, L.; Staruschenko, A. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J Am Soc Nephrol 2018, 29, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Suzuki, Y.; Ruperez, M.; Egido, J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens 2001, 10, 321–329. [Google Scholar] [CrossRef]
- Tesch, G.H. Macrophages and diabetic nephropathy. Semin Nephrol 2010, 30, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Tomaschitz, A. Aldosterone, a vasculotoxic agent--novel functions for an old hormone. Nephrol Dial Transplant 2009, 24, 2302–2305. [Google Scholar] [CrossRef]
- Tang, S.C.; Chan, L.Y.; Leung, J.C.; Cheng, A.S.; Chan, K.W.; Lan, H.Y.; Lai, K.N. Bradykinin and high glucose promote renal tubular inflammation. Nephrol Dial Transplant 2010, 25, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat Rev Nephrol 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Han, Q.; Zhu, H.; Chen, X.; Liu, Z. Non-genetic mechanisms of diabetic nephropathy. Front Med 2017, 11, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev 2002, 15, 414–429. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; Satake, E.; Simeone, C.A.; Shah, H.; Qiu, C.; Looker, H.C.; Fiorina, P.; Ware, C.F.; Sun, J.K.; Doria, A.; Kretzler, M.; Susztak, K.; Duffin, K.L.; Nelson, R.G.; Krolewski, A.S. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Johnson, E.J.; Tuttle, K.R. Inflammatory Mechanisms as New Biomarkers and Therapeutic Targets for Diabetic Kidney Disease. Adv Chronic Kidney Dis 2018, 25, 181–191. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J Cell Physiol 2019, 234, 5674–5682. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat Commun 2021, 12, 2368. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol 2017, 312, F716–F731. [Google Scholar] [CrossRef] [PubMed]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; Ix, J.H.; Lash, J.P.; Parikh, C.R.; Rebholz, C.M.; Sabbisetti, V.; Sarnak, M.J.; Shlipak, M.G.; Waikar, S.S.; Kimmel, P.L.; Vasan, R.S.; Feldman, H.I.; Schelling, J.R.; Consortium, C.K.D.B.; the Chronic Renal Insufficiency Cohort Study, I. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J Am Soc Nephrol 2021, 32, 115–126. [Google Scholar] [CrossRef]
- Yang, H.; Chen, H.; Liu, F.; Ma, Q. Up-regulation of matrix metalloproteinases-9 in the kidneys of diabetic rats and the association with neutrophil gelatinase-associated lipocalin. BMC Nephrol 2021, 22, 211. [Google Scholar] [CrossRef]
- Yue, Y.; Yeh, J.N.; Chiang, J.Y.; Sung, P.H.; Chen, Y.L.; Liu, F.; Yip, H.K. Intrarenal arterial administration of human umbilical cord-derived mesenchymal stem cells effectively preserved the residual renal function of diabetic kidney disease in rat. Stem Cell Res Ther 2022, 13, 186. [Google Scholar] [CrossRef]
- Perez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammation in Diabetic Kidney Disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef]
- Guiteras, R.; Flaquer, M.; Cruzado, J.M. Macrophage in chronic kidney disease. Clin Kidney J 2016, 9, 765–771. [Google Scholar] [CrossRef]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J Histochem Cytochem 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Yang, C. Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, X.C.; Li, Z.H.; Wu, H.L.; Jing, K.P.; Huang, X.R.; Ye, L.; Wei, B.; Lan, H.Y.; Liu, H.F. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy 2021, 17, 2325–2344. [Google Scholar] [CrossRef]
- Hong, Q.; Cai, H.; Zhang, L.; Li, Z.; Zhong, F.; Ni, Z.; Cai, G.; Chen, X.M.; He, J.C.; Lee, K. Modulation of transforming growth factor-beta-induced kidney fibrosis by leucine-rich ⍺-2 glycoprotein-1. Kidney Int 2022, 101, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Typiak, M.; Piwkowska, A. Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm Regen 2018, 38, 14. [Google Scholar] [CrossRef]
- Brown, N.J.; Vaughan, D.E.; Fogo, A.B. The renin-angiotensin-aldosterone system and fibrinolysis in progressive renal disease. Semin Nephrol 2002, 22, 399–406. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front Cell Dev Biol 2020, 8, 187. [Google Scholar] [CrossRef]
- Klemis, V.; Ghura, H.; Federico, G.; Wurfel, C.; Bentmann, A.; Gretz, N.; Miyazaki, T.; Grone, H.J.; Nakchbandi, I.A. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int 2017, 91, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Li, H.Y.; Yang, Q.; Chen, G.; Lin, S.; Liao, C.; Zhou, T. Administration of mesenchymal stem cells in diabetic kidney disease: a systematic review and meta-analysis. Stem Cell Res Ther 2021, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; Li, A.M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.Q.; Peng, C.H.; Zhang, H.; Yang, S. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis 2021, 12, 925. [Google Scholar] [CrossRef]
- Yang, Y.L.; Hu, F.; Xue, M.; Jia, Y.J.; Zheng, Z.J.; Li, Y.; Xue, Y.M. Early growth response protein-1 upregulates long noncoding RNA Arid2-IR to promote extracellular matrix production in diabetic kidney disease. Am J Physiol Cell Physiol 2019, 316, C340–C352. [Google Scholar] [CrossRef]
- Liles, J.T.; Corkey, B.K.; Notte, G.T.; Budas, G.R.; Lansdon, E.B.; Hinojosa-Kirschenbaum, F.; Badal, S.S.; Lee, M.; Schultz, B.E.; Wise, S.; Pendem, S.; Graupe, M.; Castonguay, L.; Koch, K.A.; Wong, M.H.; Papalia, G.A.; French, D.M.; Sullivan, T.; Huntzicker, E.G.; Ma, F.Y.; Nikolic-Paterson, D.J.; Altuhaifi, T.; Yang, H.; Fogo, A.B.; Breckenridge, D.G. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest 2018, 128, 4485–4500. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Pergola, P.E.; Chen, F.; Kirby, B.J.; Sundy, J.S.; Patel, U.D.; Investigators, G.-U.-. . Effects of Selonsertib in Patients with Diabetic Kidney Disease. J Am Soc Nephrol 2019, 30, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J Clin Invest 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; Argetsinger, L.S.; Rastaldi, M.P.; Brosius, F.C.; Kretzler, M. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Looker, H.C.; Lin, C.; Nair, V.; Kretzler, M.; Mauer, M.; Najafian, B.; Nelson, R.G. Serum Level of Polyubiquitinated PTEN and Loss of Kidney Function in American Indians With Type 2 Diabetes. Am J Kidney Dis 2022, 79, 497–506. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Q.; Li, C.; Liang, K.; Xiang, Y.; Hsiao, H.; Nguyen, T.K.; Park, P.K.; Egranov, S.D.; Ambati, C.R.; Putluri, N.; Hawke, D.H.; Han, L.; Hung, M.C.; Danesh, F.R.; Yang, L.; Lin, C. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J Clin Invest 2019, 129, 1129–1151. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J Cell Physiol 2017, 232, 3209–3217. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D'Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol 2000, 11, 1656–1666. [Google Scholar] [CrossRef]
- Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Tan, Y.Q.; Yu, X.Y.; Zhao, Y.Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic Biol Med 2021, 171, 260–271. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N. Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease. Toxins (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc 2010, 110, 911–916. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci U S A 1994, 91, 11704–11708. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Beeri, M.S.; de la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The potential role of dietary advanced glycation endproducts in the development of chronic non-infectious diseases: a narrative review. Nutr Res Rev 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; Ziemann, M.; Steer, D.; El-Osta, A.; Davies, M.J.; Donnellan, L.; Deo, P.; Kellow, N.J.; Cooper, M.E.; Woodruff, T.M.; Mackay, C.R.; Forbes, J.M.; Coughlan, M.T. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS One 2014, 9, e97985. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; Chadban, S.J.; Wu, H. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J Am Soc Nephrol 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Tuttle, K.R.; McGill, J.B.; Haney, D.J.; Lin, T.E.; Anderson, P.W.; Pkc-Drs, P.-D.; Groups, P.-D. S. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clin J Am Soc Nephrol 2007, 2, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; Group, A.S. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; Melnick, J.Z.; Miller, M.G.; Pergola, P.E.; Perkovic, V.; Tobe, S.; Yi, T.; Wigderson, M.; de Zeeuw, D.; Committees, S.; Investigators. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; Sadusky, T.; Tandon, N.; Tuttle, K.R.; Wanner, C.; Wilkens, K.G.; Zoungas, S.; Craig, J.C.; Tunnicliffe, D.J.; Tonelli, M.A.; Cheung, M.; Earley, A.; de Boer, I.H. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: an update based on rapidly emerging new evidence. Kidney Int 2022, 102, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; Donohue, M.; Ramachandrarao, S.; Xu, R.; Fervenza, F.C.; Kopp, J.B. Pirfenidone for diabetic nephropathy. J Am Soc Nephrol 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Packham, D.K.; Wolfe, R.; Reutens, A.T.; Berl, T.; Heerspink, H.L.; Rohde, R.; Ivory, S.; Lewis, J.; Raz, I.; Wiegmann, T.B.; Chan, J.C.; de Zeeuw, D.; Lewis, E.J.; Atkins, R.C.; Collaborative Study, G. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J Am Soc Nephrol 2012, 23, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; Richard, A.; Xiang, Z.; Brunel, P.; Pfeffer, M.A.; Investigators, A. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012, 367, 2204–2213. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; McMurray, J.J.; Meyer, C.J.; Parving, H.H.; Remuzzi, G.; Toto, R.D.; Vaziri, N.D.; Wanner, C.; Wittes, J.; Wrolstad, D.; Chertow, G.M.; Investigators, B.T. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int Rep 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Nangaku, M.; Takama, H.; Ichikawa, T.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; Kashihara, N.; Kadowaki, T.; Hase, H.; Akizawa, T. Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: design and baseline characteristics of the AYAME study. Nephrol Dial Transplant 2023, 38, 1204–1216. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; Collaborative Study, G. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O'Connor, T.; Palevsky, P.M.; Reilly, R.F.; Seliger, S.L.; Warren, S.R.; Watnick, S.; Peduzzi, P.; Guarino, P.; Investigators, V.N.-D. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013, 369, 1892–1903. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- American Diabetes, A. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S135–S151. [Google Scholar] [CrossRef]
- Wanner, C.; Marx, N. SGLT2 inhibitors: the future for treatment of type 2 diabetes mellitus and other chronic diseases. Diabetologia 2018, 61, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; Cannon, C.P.; Capuano, G.; Chu, P.L.; de Zeeuw, D.; Greene, T.; Levin, A.; Pollock, C.; Wheeler, D.C.; Yavin, Y.; Zhang, H.; Zinman, B.; Meininger, G.; Brenner, B.M.; Mahaffey, K.W.; Investigators, C.T. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; Sjostrom, C.D.; Toto, R.D.; Langkilde, A.M.; Wheeler, D.C.; Committees, D.-C. T.; Investigators. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Salah, H.M.; Al'Aref, S.J.; Khan, M.S.; Al-Hawwas, M.; Vallurupalli, S.; Mehta, J.L.; Mounsey, J.P.; Greene, S.J.; McGuire, D.K.; Lopes, R.D.; Fudim, M. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am Heart J 2021, 232, 10–22. [Google Scholar] [CrossRef]
- The, E.-K. C. G.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; Ng, S.Y.A.; Sammons, E.; Zhu, D.; Hill, M.; Stevens, W.; Wallendszus, K.; Brenner, S.; Cheung, A.K.; Liu, Z.H.; Li, J.; Hooi, L.S.; Liu, W.; Kadowaki, T.; Nangaku, M.; Levin, A.; Cherney, D.; Maggioni, A.P.; Pontremoli, R.; Deo, R.; Goto, S.; Rossello, X.; Tuttle, K.R.; Steubl, D.; Petrini, M.; Massey, D.; Eilbracht, J.; Brueckmann, M.; Landray, M.J.; Baigent, C.; Haynes, R. Empagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2023, 388, 117–127. [Google Scholar]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Erratum. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257. Diabetes 2019, 68, 1094. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Girerd, S.; Jaisser, F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int 2019, 96, 302–319. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am J Hypertens 2021, 34, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Young, M.J. Mineralocorticoid Receptor Signaling as a Therapeutic Target for Renal and Cardiac Fibrosis. Front Pharmacol 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hayashi, K.; Naruse, M.; Saruta, T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003, 41, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Miric, G.; Dallemagne, C.; Endre, Z.; Margolin, S.; Taylor, S.M.; Brown, L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol 2001, 133, 687–694. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; Cleland, J.G.F.; Coats, A.J.S.; Crespo-Leiro, M.G.; Farmakis, D.; Gilard, M.; Heymans, S.; Hoes, A.W.; Jaarsma, T.; Jankowska, E.A.; Lainscak, M.; Lam, C.S.P.; Lyon, A.R.; McMurray, J.J.V.; Mebazaa, A.; Mindham, R.; Muneretto, C.; Francesco Piepoli, M.; Price, S.; Rosano, G.M.C.; Ruschitzka, F.; Kathrine Skibelund, A. Corrigendum to: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2021, 42, 4901. [Google Scholar]
- Charytan, D.M.; Himmelfarb, J.; Ikizler, T.A.; Raj, D.S.; Hsu, J.Y.; Landis, J.R.; Anderson, A.H.; Hung, A.M.; Mehrotra, R.; Sharma, S.; Weiner, D.E.; Williams, M.; DiCarli, M.; Skali, H.; Kimmel, P.L.; Kliger, A.S.; Dember, L.M.; Hemodialysis Novel Therapies, C. Safety and cardiovascular efficacy of spironolactone in dialysis-dependent ESRD (SPin-D): a randomized, placebo-controlled, multiple dosage trial. Kidney Int 2019, 95, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; Bakris, G.L.; Fidelio, D.K.D.; investigators, F.-D. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: the FIDELITY pooled analysis. Eur Heart J 2022, 43, 474–484. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; Filippatos, G.; Investigators, F.-D. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl J Med 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Agarwal, R.; Pitt, B.; Palmer, B.F.; Kovesdy, C.P.; Burgess, E.; Filippatos, G.; Malyszko, J.; Ruilope, L.M.; Rossignol, P.; Rossing, P.; Pecoits-Filho, R.; Anker, S.D.; Joseph, A.; Lawatscheck, R.; Wilson, D.; Gebel, M.; Bakris, G.L. A comparative post hoc analysis of finerenone and spironolactone in resistant hypertension in moderate-to-advanced chronic kidney disease. Clin Kidney J 2023, 16, 293–302. [Google Scholar] [CrossRef]
- Ito, S.; Kashihara, N.; Shikata, K.; Nangaku, M.; Wada, T.; Okuda, Y.; Sawanobori, T. Esaxerenone (CS-3150) in Patients with Type 2 Diabetes and Microalbuminuria (ESAX-DN): Phase 3 Randomized Controlled Clinical Trial. Clin J Am Soc Nephrol 2020, 15, 1715–1727. [Google Scholar] [CrossRef]
- Kolkhof, P.; Hartmann, E.; Freyberger, A.; Pavkovic, M.; Mathar, I.; Sandner, P.; Droebner, K.; Joseph, A.; Huser, J.; Eitner, F. Effects of Finerenone Combined with Empagliflozin in a Model of Hypertension-Induced End-Organ Damage. Am J Nephrol 2021, 52, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kristensen, S.L.; Bengtsson, O.; Bohm, M.; de Boer, R.A.; Docherty, K.F.; Inzucchi, S.E.; Katova, T.; Kober, L.; Kosiborod, M.N.; Langkilde, A.M.; Lindholm, D.; Martinez, M.F.A.; O'Meara, E.; Nicolau, J.C.; Petrie, M.C.; Ponikowski, P.; Sabatine, M.S.; Schou, M.; Sjostrand, M.; Solomon, S.D.; Jhund, P.S.; McMurray, J.J.V. Dapagliflozin in HFrEF Patients Treated With Mineralocorticoid Receptor Antagonists: An Analysis of DAPA-HF. JACC Heart Fail 2021, 9, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Zannad, F.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Jamal, W.; Steubl, D.; Schueler, E.; Packer, M. Interplay of Mineralocorticoid Receptor Antagonists and Empagliflozin in Heart Failure: EMPEROR-Reduced. J Am Coll Cardiol 2021, 77, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; Wilson, D.J.; Bakris, G.L.; Investigators, F.-D. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J Am Soc Nephrol 2022, 33, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Chan, J.C.N.; Kooy, A.; McCafferty, K.; Schernthaner, G.; Wanner, C.; Joseph, A.; Scheerer, M.F.; Scott, C.; Bakris, G.L.; Investigators, F.-D. Finerenone in Predominantly Advanced CKD and Type 2 Diabetes With or Without Sodium-Glucose Cotransporter-2 Inhibitor Therapy. Kidney Int Rep 2022, 7, 36–45. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: a consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2022, 102, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: biological mechanisms and clinical evidence. Nat Rev Nephrol 2021, 17, 227–244. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes Diabetes Work, G. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int 2020, 98, S1–S115. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Lakshmanan, M.C.; Rayner, B.; Busch, R.S.; Zimmermann, A.G.; Woodward, D.B.; Botros, F.T. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol 2018, 6, 605–617. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; Rosenstock, J.; Gerstein, H.C. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; Nicolucci, A.; Johnson, D.W.; Tonelli, M.; Rossi, M.C.; Badve, S.V.; Cho, Y.; Nadeau-Fredette, A.C.; Burke, M.; Faruque, L.I.; Lloyd, A.; Ahmad, N.; Liu, Y.; Tiv, S.; Millard, T.; Gagliardi, L.; Kolanu, N.; Barmanray, R.D.; McMorrow, R.; Raygoza Cortez, A.K.; White, H.; Chen, X.; Zhou, X.; Liu, J.; Rodriguez, A.F.; Gonzalez-Colmenero, A.D.; Wang, Y.; Li, L.; Sutanto, S.; Solis, R.C.; Diaz Gonzalez-Colmenero, F.; Rodriguez-Gutierrez, R.; Walsh, M.; Guyatt, G.; Strippoli, G.F.M. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; Steinberg, W.M.; Stockner, M.; Zinman, B.; Bergenstal, R.M.; Buse, J.B.; Committee, L.S.; Investigators, L.T. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; Woo, V.; Hansen, O.; Holst, A.G.; Pettersson, J.; Vilsboll, T.; Investigators, S.-. . Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; Dyal, L.; Branch, K.; Investigators, A.-O. T. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N Engl J Med 2021, 385, 896–907. [Google Scholar] [CrossRef]
- Sarathy, H.; Henriquez, G.; Abramowitz, M.K.; Kramer, H.; Rosas, S.E.; Johns, T.; Kumar, J.; Skversky, A.; Kaskel, F.; Melamed, M.L. Abdominal Obesity, Race and Chronic Kidney Disease in Young Adults: Results from NHANES 1999-2010. PLoS One 2016, 11, e0153588. [Google Scholar] [CrossRef]
- Ejerblad, E.; Fored, C.M.; Lindblad, P.; Fryzek, J.; McLaughlin, J.K.; Nyren, O. Obesity and risk for chronic renal failure. J Am Soc Nephrol 2006, 17, 1695–1702. [Google Scholar] [CrossRef]
- Rossing, P.; Baeres, F.M.M.; Bakris, G.; Bosch-Traberg, H.; Gislum, M.; Gough, S.C.L.; Idorn, T.; Lawson, J.; Mahaffey, K.W.; Mann, J.F.E.; Mersebach, H.; Perkovic, V.; Tuttle, K.; Pratley, R. The rationale, design and baseline data of FLOW, a kidney outcomes trial with once-weekly semaglutide in people with type 2 diabetes and chronic kidney disease. Nephrol Dial Transplant 2023, 38, 2041–2051. [Google Scholar] [CrossRef]
- Wright, A.K.; Carr, M.J.; Kontopantelis, E.; Leelarathna, L.; Thabit, H.; Emsley, R.; Buchan, I.; Mamas, M.A.; van Staa, T.P.; Sattar, N.; Ashcroft, D.M.; Rutter, M.K. Primary Prevention of Cardiovascular and Heart Failure Events With SGLT2 Inhibitors, GLP-1 Receptor Agonists, and Their Combination in Type 2 Diabetes. Diabetes Care 2022, 45, 909–918. [Google Scholar] [CrossRef]
- Davila-Esqueda, M.E.; Martinez-Morales, F. Pentoxifylline diminishes the oxidative damage to renal tissue induced by streptozotocin in the rat. Exp Diabesity Res 2004, 5, 245–251. [Google Scholar] [CrossRef]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Chahin, J.; Mendez, M.L.; Gallego, E.; Macia, M.; del Castillo, N.; Rivero, A.; Getino, M.A.; Garcia, P.; Jarque, A.; Garcia, J. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J Am Soc Nephrol 2015, 26, 220–229. [Google Scholar] [CrossRef]
- Navarro-Gonzalez, J.F.; Sanchez-Nino, M.D.; Donate-Correa, J.; Martin-Nunez, E.; Ferri, C.; Perez-Delgado, N.; Gorriz, J.L.; Martinez-Castelao, A.; Ortiz, A.; Mora-Fernandez, C. Effects of Pentoxifylline on Soluble Klotho Concentrations and Renal Tubular Cell Expression in Diabetic Kidney Disease. Diabetes Care 2018, 41, 1817–1820. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C.; 3rd, Adler, S. G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; Cardillo, T.E.; Duffin, K.L.; Haas, J.V.; Macias, W.L.; Nunes, F.P.; Janes, J.M. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant 2018, 33, 1950–1959. [Google Scholar]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford) 2019, 58, i43–i54. [Google Scholar] [CrossRef]
- Ko, G.J.; Rhee, C.M.; Kalantar-Zadeh, K.; Joshi, S. The Effects of High-Protein Diets on Kidney Health and Longevity. J Am Soc Nephrol 2020, 31, 1667–1679. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep 2014, 14, 453. [Google Scholar] [CrossRef]
- Bach, K.E.; Kelly, J.T.; Palmer, S.C.; Khalesi, S.; Strippoli, G.F.M.; Campbell, K.L. Healthy Dietary Patterns and Incidence of CKD: A Meta-Analysis of Cohort Studies. Clin J Am Soc Nephrol 2019, 14, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D. Health effects of dietary risks in 195 countries, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [Google Scholar]
- Pandey, A.; Garg, S.; Khunger, M.; Darden, D.; Ayers, C.; Kumbhani, D.J.; Mayo, H.G.; de Lemos, J.A.; Berry, J.D. Dose-Response Relationship Between Physical Activity and Risk of Heart Failure: A Meta-Analysis. Circulation 2015, 132, 1786–1794. [Google Scholar] [CrossRef]
- Bowlby, W.; Zelnick, L.R.; Henry, C.; Himmelfarb, J.; Kahn, S.E.; Kestenbaum, B.; Robinson-Cohen, C.; Utzschneider, K.M.; de Boer, I.H. Physical activity and metabolic health in chronic kidney disease: a cross-sectional study. BMC Nephrol 2016, 17, 187. [Google Scholar] [CrossRef]
- Kosmadakis, G.C.; John, S.G.; Clapp, E.L.; Viana, J.L.; Smith, A.C.; Bishop, N.C.; Bevington, A.; Owen, P.J.; McIntyre, C.W.; Feehally, J. Benefits of regular walking exercise in advanced pre-dialysis chronic kidney disease. Nephrol Dial Transplant 2012, 27, 997–1004. [Google Scholar] [CrossRef]
- Xia, J.; Wang, L.; Ma, Z.; Zhong, L.; Wang, Y.; Gao, Y.; He, L.; Su, X. Cigarette smoking and chronic kidney disease in the general population: a systematic review and meta-analysis of prospective cohort studies. Nephrol Dial Transplant 2017, 32, 475–487. [Google Scholar] [CrossRef]
- Jhee, J.H.; Joo, Y.S.; Kee, Y.K.; Jung, S.Y.; Park, S.; Yoon, C.Y.; Han, S.H.; Yoo, T.H.; Kang, S.W.; Park, J.T. Secondhand Smoke and CKD. Clin J Am Soc Nephrol 2019, 14, 515–522. [Google Scholar] [CrossRef]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes Management in Chronic Kidney Disease: A Consensus Report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Diabetes Care 2022, 45, 3075–3090. [Google Scholar] [CrossRef]
- Oshima, M.; Jardine, M.J.; Agarwal, R.; Bakris, G.; Cannon, C.P.; Charytan, D.M.; de Zeeuw, D.; Edwards, R.; Greene, T.; Levin, A.; Lim, S.K.; Mahaffey, K.W.; Neal, B.; Pollock, C.; Rosenthal, N.; Wheeler, D.C.; Zhang, H.; Zinman, B.; Perkovic, V.; Heerspink, H.J.L. Insights from CREDENCE trial indicate an acute drop in estimated glomerular filtration rate during treatment with canagliflozin with implications for clinical practice. Kidney Int 2021, 99, 999–1009. [Google Scholar] [CrossRef]
- Kraus, B.J.; Weir, M.R.; Bakris, G.L.; Mattheus, M.; Cherney, D.Z.I.; Sattar, N.; Heerspink, H.J.L.; Ritter, I.; von Eynatten, M.; Zinman, B.; Inzucchi, S.E.; Wanner, C.; Koitka-Weber, A. Characterization and implications of the initial estimated glomerular filtration rate 'dip' upon sodium-glucose cotransporter-2 inhibition with empagliflozin in the EMPA-REG OUTCOME trial. Kidney Int 2021, 99, 750–762. [Google Scholar] [CrossRef]
- Provenzano, M.; Puchades, M.J.; Garofalo, C.; Jongs, N.; D'Marco, L.; Andreucci, M.; De Nicola, L.; Gorriz, J.L.; Heerspink, H.J.L.; group, R.-s. , and members, R.-s. g. Albuminuria-Lowering Effect of Dapagliflozin, Eplerenone, and Their Combination in Patients with Chronic Kidney Disease: A Randomized Crossover Clinical Trial. J Am Soc Nephrol 2022, 33, 1569–1580. [Google Scholar] [CrossRef]
- Green, J.B.; Mottl, A.K.; Bakris, G.; Heerspink, H.J.L.; Mann, J.F.E.; McGill, J.B.; Nangaku, M.; Rossing, P.; Scott, C.; Gay, A.; Agarwal, R. Design of the COmbinatioN effect of FInerenone anD EmpaglifloziN in participants with chronic kidney disease and type 2 diabetes using a UACR Endpoint study (CONFIDENCE). Nephrol Dial Transplant 2023, 38, 894–903. [Google Scholar] [CrossRef]
Figure 1.
Metabolic, inflammatory and hemodynamic perturbations pathways in the pathogenesis of DKD.
Figure 1.
Metabolic, inflammatory and hemodynamic perturbations pathways in the pathogenesis of DKD.

Figure 2.
The timeline of major therapy targeting on precise molecular of DKD.
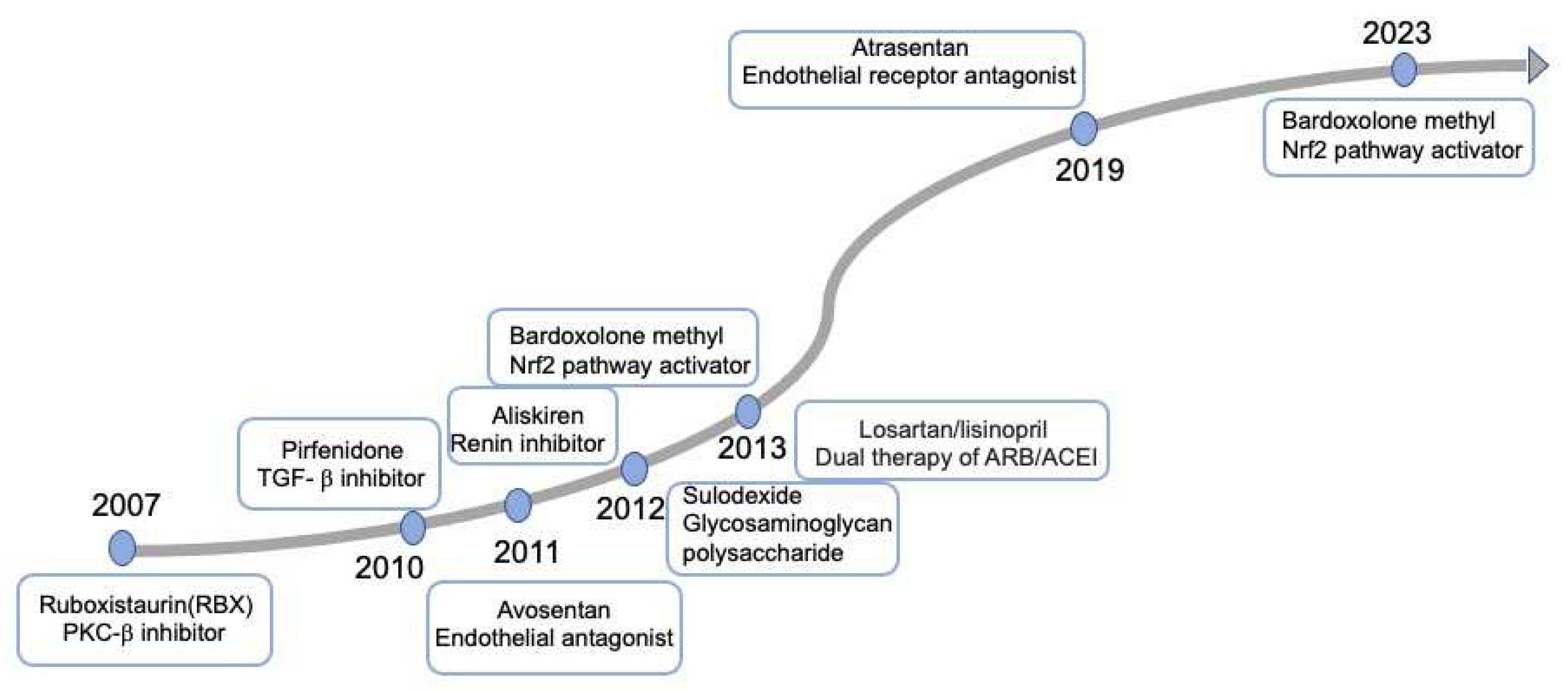
Figure 3.
Recent advances in the therapy and potential mechanisms in slowing the progression of DKD .
Figure 3.
Recent advances in the therapy and potential mechanisms in slowing the progression of DKD .
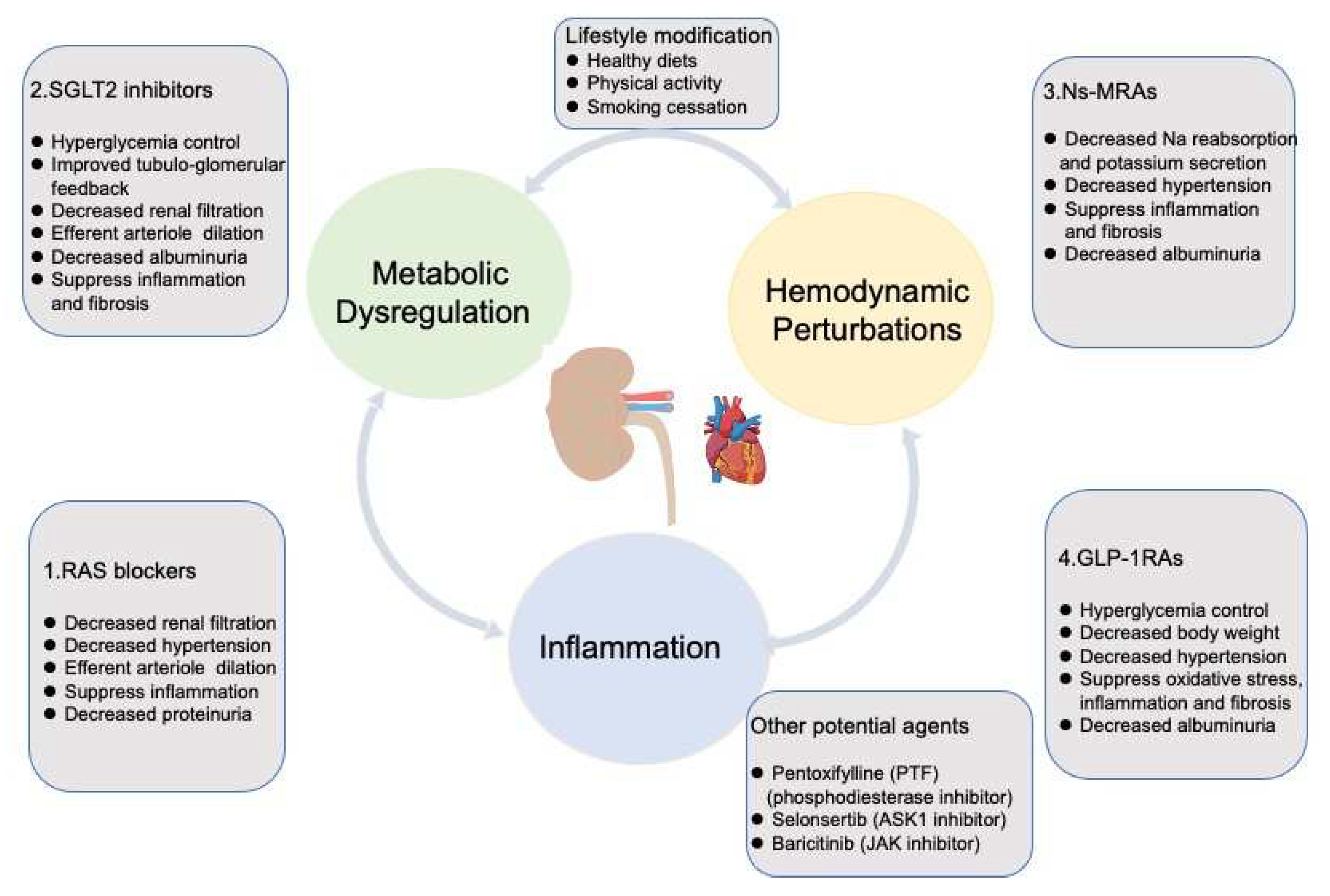
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



