
Submitted:
06 January 2024
Posted:
08 January 2024
You are already at the latest version
Abstract
The current review comprehensively explores the multifaceted landscape of anaplastic lymphoma kinase (ALK) and its pivotal role, specifically within non-small cell lung cancer (NSCLC). It traces the trajectory of ALK's discovery, notably its fusion with nucleolar phosphoprotein (NPM)-1 in anaplastic large cell non-Hodgkin's lymphoma (ALCL) in 1994. It unfolds the subsequent impact of ALK gene alterations across various malignancies, including inflammatory myofibroblastoma (IMT) and NSCLC. The prevalence of intricate ALK rearrangements affecting roughly 3–5% of NSCLC patients prompted the approval of six ALK-tyrosine kinase inhibitors (TKIs) by 2022, revolutionizing the treatment landscape for advanced metastatic ALK+ NSCLC. Second-generation TKIs like alectinib, ceritinib, and brigatinib have notably emerged to combat resistance issues initially associated with crizotinib, the pioneer ALK-TKI approval. Moreover, this review delves into the intersection of ALK and immunotherapy, exploring the potential of immune checkpoint inhibitors in ALK-altered NSCLC tumors. Despite promising preclinical data, challenges encountered in trials evaluating combinations like nivolumab-crizotinib, primarily due to severe hepatic toxicity, underscore the need for cautious exploration of these novel approaches. Additionally, the review delves into innovative avenues such as ALK vaccines and biosensors, shedding light on their promising potential within ALK-driven cancers. This comprehensive analysis encompasses molecular mechanisms, therapeutic strategies, and immune interactions associated with ALK-rearranged NSCLC. Ultimately, the review is a pivotal resource guiding future research and therapeutic interventions in ALK-targeted therapy for NSCLC.
Keywords:
NSCLC
; ALK inhibitors
; lung cancer
; targeted therapy
; precision medicine
1. Introduction
Anaplastic lymphoma kinase (ALK), belonging to the insulin receptor superfamily, plays a significant role in various cancers. The ALK gene is in the 2p23.2-p23.1 chromosomal region, encoding a protein of 26 exons and 1620 amino acid residues. In 1994, a groundbreaking discovery revealed the fusion between nucleolar phosphoprotein (NPM)-1 and ALK in anaplastic large cell non-Hodgkin’s lymphoma (ALCL), emphasizing ALK’s importance. Understanding the t(2;5)(p23;q35) chromosomal translocation in ALCL led to identifying ALK and the resulting NPM-ALK oncogenic protein [1,2]. On the other hand, different changes in the ALK gene—like alternative splicing, amplification, and mutations—are linked to various tumors, such as inflammatory myofibroblastoma (IMT) and non-small cell lung cancer (NSCLC) [2,3,4,5]. In addition, ALK gene rearrangements impact immune systems, affecting T-cell activation, cytokine secretion, and immune evasion within tumors [6]. This diversity makes ALK gene variations promising targets for cancer therapies [7,8].
Lung cancer comprises NSCLC (81% of cases) and small cell lung cancer (SCLC) (14% of cases). In the U.S., NSCLC dominates and is projected to affect around 238,340 adults by 2023, resulting in 127,070 deaths. Globally, 2,206,771 people were diagnosed with lung cancer in 2020, encompassing both NSCLC and SCLC cases [9,10]. In 2007, the ALK gene rearrangement in NSCLC patients revealed the initial fusion between echinoderm microtubule-associated protein-like 4 (EML4) and ALK in lung cancer [11,12]. Around 3–5% of NSCLC patients exhibit ALK rearrangement, often associated with a non-smoking history, younger age, and adenocarcinoma histology [13,14]. Six ALK-tyrosine kinase inhibitors (TKIs) (crizotinib, ceritinib, alectinib, brigatinib, ensartinib, and lorlatinib) received approval by 2022 for advanced metastatic ALK+ NSCLC treatment. Among these, alectinib, brigatinib, and lorlatinib are recommended for advanced ALK+ NSCLC in the United States [15]. Crizotinib was the first approved ALK-TKI inhibitor for treating ALK-rearranged NSCLCs. Studies indicated its significant efficacy compared to standard chemotherapy [16]. Second-generation ALK–TKIs (alectinib, ceritinib, brigatinib) were developed to combat resistance emerging with crizotinib. Initially, these TKIs showed promising effectiveness, validated in several phase III trials as the primary treatment for newly diagnosed ALK+ NSCLC [17,18,19,20,21].
Moreover, immunotherapies targeting programmed cell death 1 (PD-1), programmed cell death 1 ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) show potential for NSCLC treatment. Still, their efficacy in oncogenic mutated proteins like epidermal growth factor (EGF) receptor (EGFR) or ALK remains uncertain. ALK alterations have been linked to increased immune checkpoint expression, raising questions about the effectiveness of immunotherapy alone or combined with targeted therapies in this subset of patients. However, trials evaluating immunotherapy in NSCLC often need more representation of ALK-rearranged patients, limiting robust conclusions about their clinical benefit in this population [22,23]. Besides, the combination of ALK-targeted therapy, immune checkpoint blockade, and immune checkpoint blockade monotherapy for ALK-modified NSCLC is being clinically investigated. Preclinical data initially supported the nivolumab-crizotinib combination, demonstrating increased PD-L1 expression in the presence of ALK-EML4 fusion protein and how both checkpoint and ALK inhibitors reduced T cell apoptosis and crizotinib-resistant cell survival [24]. Despite this promise, adverse events in the trial obstructed the evaluation of the combo’s efficacy, underscoring the necessity for robust phase 1 studies despite encouraging preclinical evidence. In a phase 1/2 study, the combination of nivolumab and crizotinib was examined as a first-line treatment for advanced NSCLC with ALK translocation. This study aimed to assess the safety and tolerability of nivolumab in various NSCLC treatment settings. Patients with confirmed ALK-translocation positive NSCLC were administered nivolumab intravenously every two weeks alongside crizotinib orally twice daily. However, initial findings from the first 13 patients revealed severe hepatotoxicity in 38% of cases, leading to the discontinuation of treatment for some due to grade 3 or higher hepatotoxicity [25]. Further enrollment was stopped due to this severe hepatic toxicity, and the trial was ceased. Although the combination showed partial responses in 38% of patients, the trial did not meet its primary safety endpoint, prompting the authors to advise against further exploration of this combination for ALK-translocation NSCLC [26].
The current review comprehensively analyzes ALK-rearranged NSCLC, delving into mechanisms and updated data concerning ALK-targeted therapy and immunotherapy. Its scope includes ALK’s structural biology, tissue-specific functions, and diverse roles in ALK-targeted therapy. Additionally, it thoroughly explores the signaling pathways activated by ALK fusion proteins and mutations, addressing challenges in ALK-targeted therapy resistance and proposing innovative strategies, notably combination therapies. Ultimately, this review is a valuable resource, offering insights for future research and guiding therapeutic interventions in the domain of ALK-targeted therapy for NSCLC.
2. The ALK structural biology
2.1. ALK extracellular side
The ALK extracellular domain (ECD) consists of distinct segments believed to serve specific roles like binding ligands, interacting with potential co-receptors and secreted regulatory proteins, and facilitating dimerization. These functions could trigger structural changes initiating activation within the intracellular protein tyrosine kinase (PTK) domain. The ECD of ALK stands out among receptor tyrosine kinases (RTKs) due to its distinct glycine-rich section. At the same time, ALK includes an additional low-density lipoprotein receptor class A (LDLa) and two meprin, A5 protein, and receptor protein tyrosine phosphatase mu (MAM) domains (Figure 1). Pleiotrophin (PTN) and midkine (MK) are recognized as triggers for mammalian ALK, playing pivotal roles in neural development, survival, and tumorigenesis [27]. These growth factors, binding to heparin, can activate various receptors, including receptor protein tyrosine phosphatase-β (RPTPβ), RPTPζ, N-syndecan, low-density lipoprotein receptor-related protein (LRP), and integrins. PTN can specifically engage RPTPβ and RPTPζ phosphatases to initiate ALK signaling. However, the actual activation of ALK by PTN and MK remains contentious, with conflicting reports among studies. This debate contrasts with findings in non-vertebrate models like Drosophila melanogaster and Caenorhabditis elegans, underscoring the ongoing uncertainty about the natural ligand for mammalian ALK [1].
The structure of ALK deviates from the typical architecture due to its unique domain composition. One distinctive element is the glycine-rich domain (GRD) near the membrane. This GRD, which contains a cysteine-rich area resembling the fold of EGF, is an unconventional and less defined region. Interestingly, this peculiar GRD is a shared feature between ALK and LTK. Despite its high glycine content, often associated with structural disorders, the GRD alone can drive receptor activity regulated by its ligands. Vertebrate ALKs’ ligands, known as ALKALs (ALK and LTK Activating Ligands), include Fam150A (AUGβ) and Fam150B (AUGα). These ligands, consisting of approximately 100 amino acids, contain a highly conserved domain termed the ALKAL domain, which stimulates ALK activity [28,29,30]. A recent crystallographic study of the GRD reveals a ß-sandwich structure with N- and C-termini positioned close to each other, resembling a portion of the tumor necrosis factor-α (TNF-α) domain. Despite this similarity, the ß-sandwich structure of ALK differs from TNF-α. The loop in the C-terminal contributes to a fold, creating a binding epitope [28]. The crystal structure of ALK demonstrates that two ALKALs bind to the dimeric complex of two ALKs, stabilizing the complex. The ALK GRD comprises short α-helices, ß-sheets, and glycine helices, while the ALKAL domain forms a disulfide helical hairpin [28,31,32,33]. The interaction between ALK and ALKAL initially occurs through the ALKAL domain and the TNF-α-like region within the GRD, attributed to the high positive charge on the ALKAL domain surface, which supports ALK protein activation (Figure 1) [28].
2.2. ALK intracellular side
The activation loop (A-loop), a pivotal segment governing access to the active site, commences with a conserved Asp-Phe-Gly (DFG) sequence, significantly regulating ALK’s active and inactive states. ALK boasts two distinct hydrophobic spines named the ‘regulatory’ and ‘catalytic,’ contributing to vital allosteric control within and between the lobes. These spines, housing conserved hydrophobic amino acids, facilitate the transition between active and inactive states. The ALK regulatory spine, encompassing I1171, C1182, H1247, F1271, and D1311, assembles during kinase activation and disengages during inactivation [34]. Researchers also explored the structure of the unphosphorylated human ALK kinase domain in tandem with ATP-competitive ligands like PHA-E429 and NVP-TAE684. This analysis provided invaluable insights into the distinct attributes of the ALK active site, aiding in the quest for selective ALK inhibitors. Specifically, the ALK-PTK-PHA-E429 structure uncovered a potential regulatory mechanism, linking a brief helical segment after the DFG motif to a two-stranded beta-sheet at the N-terminal. The ALK structure begins with an initial 13-residue segment featuring two β-strands (β1′ and β2′) before the bilobal protein kinase fold. This configuration encompasses an N-terminal lobe housing a core five-stranded β-sheet and an α-helix.
In contrast, the C-terminal lobe is primarily α-helical and accommodates the critical activation loop pivotal for enzyme activation. The gaps in the ALK structures, particularly in the complexes with PHA-E429 and TAE684, indicate regions of structural disorder within the protein. Comparative analysis with other kinase structures highlighted discrepancies in lobe closure and αC helix positioning, suggesting potential inactivity in the ALK-PTK structure due to these structural deviations (Figure 1).
A crucial hydrogen bond between specific residues implies an active kinase state. The study also underscored variations in hydrogen bond formations and structural conflicts within the ALK-PTK structures, influencing the initiation of a specific structural element termed β9 and affecting the formation of the substrate binding site. However, confirming these observations remains challenging due to the absence of an apoenzyme ALK structure, which would depict the natural state devoid of inhibitors. Nonetheless, consistent features observed in most ALK structures, like the DFG helix and interactions involving specific residues, hint at a potential regulatory role in enzyme function [31].
Furthermore, ALK exhibits a distinctive autophosphorylation motif, Y’XXX’YY (Y’RAS’YY), within the A-loop. In instances of ALK fusions, the tyrosine at position Y1278 is the primary site for phosphorylation within this sequence. Notably, an inhibitory structural feature within the ALK kinase domain involves a short α-helix in the A-loop closely associated with the αC-helix. At the same time, a β-turn motif containing C1097 obstructs the region for substrate binding. This arrangement prevents Y1278 from being accessible for phosphorylation as it forms a bond with C1097 within the amino-terminal β-sheet [35,36,37]. These insights suggest that the initial activation of ALK could potentially involve the regulation of Y1278 phosphorylation, thereby freeing ALK from inactive structural constraints (Figure 2) [38].
The ALK protein’s structural intricacies and regulatory mechanisms are governed by specific amino acid residues within its sequence. Residues within the range of 1095-1401 display intermittent gaps due to structural disorder, encapsulating crucial segments such as the glycine-rich loop (1123-1128) and the activation loop (1271-1288), essential for ALK’s functional modulation. Among these residues, 1150 (K1150) and 1167 (E1167) stand out for their involvement in pivotal hydrogen bond formations, contributing to ALK’s enzymatic activity. Residue 1245 (F1245) notably interacts, potentially impeding the initiation of a critical structural element termed β9. Additionally, residues 1274 (A1274) and 1278 (Y1278) are significant: A1274 engages in clashes with F1245, impacting structural conformation, while Y1278 marks the conclusion of the DFG helix, influencing the formation of the substrate binding site. These residues within the ALK protein sequence intricately contribute to its structural stability, functional regulation, and inhibitor interactions, which are crucial for understanding its biological significance and potential therapeutic targeting [31].
2.3. Crizotinib and ALK vs. c-MET
Crizotinib, a drug with diverse effects on various kinases, demonstrates different inhibitory patterns in enzyme and cellular assays. While it affects multiple kinases in enzymatic tests, its cellular actions show potent inhibition, specifically on mesenchymal-epithelial transition factor (c-MET) and ALK (Figure 2). This selectivity is linked to distinct binding sites shaped by unphosphorylated c-MET’s unique conformation. However, in the ALK-PTK, crizotinib shows similarities in binding to c-MET but lacks a crucial interaction in ALK, possibly explaining its weaker potency against ALK. Critical interactions with specific amino acids (M1211 in c-MET) play a vital role in maintaining crizotinib’s inhibitory effect and are found similarly in RON (a c-MET-related receptor) and ALK [32]. This drug demonstrates promising potential in inhibiting c-MET and ALK phosphorylation, curbing tumor cell growth, exhibiting antiangiogenic properties, and inducing apoptosis in specific cancer cells. Eventually, preclinical and animal studies support its efficacy against cancers harboring ALK mutations, indicating its potential as a therapeutic agent. Clinical trials have shown remarkable effectiveness in several cancers, particularly in NSCLC and other tumors carrying fusion ALK genes or amplified c-MET genes [39].
Overall, crizotinib presents itself as a promising targeted therapy across diverse cancer types by selectively inhibiting c-MET and ALK, leading to crizotinib accelerated approval by the U.S. Food and Drug Administration (FDA) on August 26, 2011, for treating ALK+ locally advanced or metastatic NSCLC. The approval relied on two single-arm trials, showcasing objective response rates (ORRs) of 50% and 61%, along with median response durations lasting 42 and 48 weeks [41].
Furthermore, the co-crystal structure of the ALK kinase domain complexed with crizotinib shares a binding configuration like c-MET. However, unlike c-MET, the interaction involving tyrosine π–π stacking is absent in the ALK co-crystal structure, potentially contributing to a slight decrease in potency against ALK compared to c-MET. Examining wild-type and L1196M ALK co-crystals aimed to delineate binding interactions and protein conformations [33]. The binding mode of crizotinib in both proteins exhibited remarkable similarity. Specific hydrogen bonds were formed between crizotinib and hinge residues M1199 and E1197 in wild-type and L1196M structures.
Additionally, interactions with the gatekeeper residue and other critical elements of the inhibitor were consistent across both structures, maintaining similar distances and positions. Although the overall protein conformations between the two crystal structures were broadly comparable, a notable difference was observed in the gatekeeper residue (L1196 to M1196). Further optimization was needed for the pyrazolopiperidine tail of crizotinib to enhance its potency against ALK and its ADME (absorption, distribution, metabolism, and excretion) properties. The pyrazole portion of the tail group occupied a lipophilic pocket near the solvent-exposed area. In contrast, the piperidinyl group extended toward the solvent, resulting in low permeability and high efflux. Strategies aimed at improving both the 2,6-dichloro-3-fluorophenyl head and the pyrazolopiperidine tail of crizotinib were explored to enhance its effectiveness against ALK [33].
In addition, ALK possesses multiple LC3-interacting region (LIR) motifs across different domains, hinting at a direct connection between ALK and autophagy. This suggests a complex interplay between inhibiting ALK kinase activity and activating autophagy, potentially complicating targeted therapies for NSCLC and other conditions. Understanding the dual roles of autophagy in cancer—serving as both an immune response facilitator and a tumor growth promoter—underscores the need to categorize ALK+ NSCLC based on hepatocyte growth factor (HGF)/c-MET signaling or autophagy-related subtypes to guide treatment decisions for optimal patient outcomes (Figure 3) [42].
3. ALK cleavage and modifications
ALK protein manifests diverse sizes and variations, encompassing an 8.0 kb message detected in rhabdomyosarcoma, small intestine, and brain, alongside transcripts of around 6.5 kb, presumed as typical cDNA. Additionally, various ALK messages—approximately 6.0 kb in the human testis, placenta, and fetal liver, and a distinct 4.4 kb transcript found solely in the testis—highlight tissue-specific isoforms likely arising from alternative transcriptional start sites or polyadenylation signals. These distinct sizes potentially signify tissue-specific functions generated via alternative splicing, leading to diverse receptor forms with varying ligand-binding capabilities and biological activities. Investigations have striven to unravel the precise roles of these diverse ALK isoforms in specific tissues, shedding light on their significance in mammalian development and growth, particularly in neural signaling pathways and development [43].
A study delved into the expression of ALK in a specific subset of neurons associated with nociception and explored factors influencing its cleavage, shedding light on the potential roles of ALK in sensory neuron development and pain perception [44]. The study identified that 73% of these sensory neurons expressed ALK, with a significant portion also expressing markers for nociception. This suggests that ALK might be a marker for neurons sensing pain. Additionally, the study explored the impact of glial cells on ALK metabolism. It observed that Schwann cells release a factor that inhibits the cleavage of the ALK receptor into its two forms. The study proposes two hypotheses: one suggests direct binding of a factor to ALK, and another suggests inhibition of proteases involved in ALK cleavage. However, the experiments did not definitively identify the factor or clarify its mechanism [44].
The ALK gene has emerged as a significant player in neuroblastoma development, making it an attractive target for therapeutic interventions. Studies identified specific mutations at F1174 and R1275 in neuroblastoma tumor cells that activate ALK, establishing its role in the disease. Researchers clarified distinct behaviors between the standard and mutated forms of the ALK receptor. They identified that the altered ALK receptors are primarily inside the cell, notably in the reticulum/Golgi structures. This internal retention was particularly noticeable in the F1174L mutation compared to the R1275Q variation.
Treatments inhibiting ALK kinase activity resulted in the translocation of mutated receptors to the cell membrane. This sheds light on potential therapeutic avenues, suggesting that targeting ALK with kinase inhibitors or specific antibodies could hold promise in neuroblastoma treatment, especially considering the possibility of these antibodies inducing receptor internalization and downregulation. These findings open avenues for therapeutic approaches targeting both the wild-type and mutated ALK receptors in neuroblastoma treatment, offering potential complementary strategies to kinase inhibitors [45]. Furthermore, another study highlighted that different mutations in ALK could result in varying oncogenic potentials, with the ALK F1174L mutation exhibiting heightened activity. Importantly, it was found that the mutated receptor, especially the ALK F1174L variant, had altered trafficking patterns, predominantly retained inside the cell. Remarkably, treatment with specific inhibitors restored normal trafficking of the mutated receptor, suggesting a potential therapeutic approach. Additionally, the study unveiled complex mechanisms of ALK degradation, contingent upon its cellular location, offering insights into potential strategies to inhibit neuroblastoma proliferation by targeting these degradation pathways. Ultimately, this research underscores the complexity of ALK behavior and its implications for targeted therapies in neuroblastoma treatment [46].
Besides, a recent study introduced a potentially groundbreaking therapeutic approach targeting ALK using a peptide derived from neuronal growth regulator 1 (Negr1) [47]. Negr1 has been linked to regulating various RTKs, and the researchers observed that acute treatment with soluble Negr1 reduced ALK protein levels, suggesting it prompts ALK protein degradation [48]. The study proposed that the Negr1-derived peptide might influence ALK levels and downstream signaling pathways impacting cell proliferation. The peptides derived from Negr1 may interact differently with ALK compared to the full-length protein, potentially leading to ALK degradation. This could be a promising strategy as ALK activation is known to drive neuroblastoma growth, and therapies targeting ALK have shown efficacy but are prone to resistance and adverse effects. The Negr1-derived peptide demonstrated the ability to degrade ALK and slow tumor growth both in vitro and in vivo, presenting a promising avenue for treating aggressive neuroblastoma resistant to current ALK inhibitors [47].
On the flip side, although cleaving ALK’s intracellular domain may help ALK-targeted therapy, cleavage of its extracellular side fosters ALK-related tumor formation and the movement of cells in neuroblastoma. Another potential regulatory process involves the proteolytic breakdown of the full-length ALK receptor ECD [49], releasing an ECD fragment approximately 80 kDa in size alongside a significantly tyrosine-phosphorylated 140 kDa truncated receptor. The precise physiological significance and the molecular mechanisms driving this cleavage event remain ambiguous; it is uncertain whether cleaved ALK might be more stable or active than intact ALK and whether this cleavage plays a role in ligand-mediated activation [38].
Researchers noted that blocking this cleavage in neuroblastoma cells reduced migration and invasion. Intriguingly, introducing the cleavable form of ALK in cells with minimal ALK expression significantly boosted their migration, whereas mutations preventing cleavage did not have the same impact [50]. This indicates the critical role of this cleavage process in driving cell movement, supported by changes in gene activity linked to cell motion. Grasping this process’s developmental role is vital, as abnormal expression in neuroblastoma cells might heighten tumor spread. The study suggests this cleavage could affect a protein called β-catenin, regulating cell motion. When ALK undergoes cleavage at the N654-L655 site, it might release β-catenin, enabling its movement into the cell nucleus to activate genes related to cell motion. Conversely, obstructing matrix metallopeptidase 9 (MMP-9) could impede ALK cleavage, reducing migration and invasion of neuroblastoma cells, hinting at a promising therapeutic approach [50]. The study also explores the impact of ALK cleavage in other cancers where ALK is present and whether blocking this process could aid in devising new treatment approaches (Figure 4).
On the other hand, N-linked glycosylation impacts ALK function in neuroblastoma cells. However, it is essential to note that the N654 cleavage site identified by Huang et al. (2021) targeted by MMP-9 activates β-catenin signaling. A substantial decrease in the binding of β-catenin to the truncated membrane-bound ALK 655–1604 receptor indicated that the cleavage of the ECD releases β-catenin from ALK, allowing its transportation to the nucleus [50]. Nonetheless, previous findings showed that N-linked glycosylations impact ALK [51] and other RTKs [52]. Researchers detected decreased ALK phosphorylation, specifically in neuroblastoma cells that depend on ALK for survival, when employing tunicamycin, a substance renowned for broadly disrupting glycosylation. Interestingly, this inhibition only affected cell proliferation and survival in ALK+ neuroblastoma cells, suggesting a potential therapeutic strategy. While tunicamycin broadly affects glycoproteins, its impact seemed selective for ALK-dependent cells. However, as tunicamycin might not be suitable for clinical use, the study’s findings emphasize a proof-of-concept, prompting further exploration into alternative approaches targeting N-linked glycosylation as a potential strategy for ALK-dependent neuroblastoma treatment [51].
Comprehensive research has delineated intricate molecular pathways involving diverse ALK isoforms, mutations, and regulatory mechanisms, particularly emphasizing their implications in neuroblastoma development and potential therapeutic strategies. Nevertheless, exploring ALK’s function, glycosylation impact, and cleavage mechanisms in NSCLC remains an uncharted territory. Therefore, future research should concentrate on elucidating the role of ALK in NSCLC, investigating how its glycosylation and cleavage intricately contribute to treatment resistance, which is pivotal for advancing effective therapeutic interventions in NSCLC patients relying on ALK-targeted therapy.
4. ALK signaling and TKI resistance
While several ALK inhibitors, like crizotinib, alectinib, and ceritinib, have been utilized clinically for ALK+ NSCLC treatment, resistance commonly develops against these inhibitors. The mutated forms of ALK, along with ALK fusion proteins like NPM–ALK, can activate various signaling pathways that contribute to cell transformation and the maintenance of a cancerous state. This persistent activation triggers the recruitment of several adaptors, initiating multiple signaling pathways. Mutated ALK and ALK chimeras induce mitogenic signaling, predominantly through the RAS/mitogen-activated protein (MAP) kinase pathway, facilitated by the direct binding of IRS1, SHC, and SRC to specific tyrosine residues within ALK’s intracytoplasmic segment. The SHP2/growth factor receptor-bound protein 2 (GRB2) complex interaction with p130Cas alters cytoskeletal organization. Activation of the phosphatidylinositol 3 kinase (PI3K) pathway by ALK results in a significant anti-apoptotic signal, mainly mediated by pAKT1/2 and its downstream molecules that inhibit BAD and FOXO3a-mediated transcription, while regulating cell cycle progression. Additionally, phospholipase C (PLC)-γ, directly binding to activated ALK, generates diacylglycerol and IP3, activating PKC and mobilizing calcium stores from the endoplasmic reticulum [53,54,55,56]. The Janus kinase (JAK)/Signal transducer and activator of transcription (STAT)-3 pathway activated by ALK provides crucial survival signals and regulates cellular metabolism via the mitochondrial oxidation chain. STAT3 activation, directly or through JAK, leads to a unique gene expression profile distinguishing ALCL from other T-cell neoplasms. Its downstream effectors include BCL2 family members (BCL2, BCL-XL, MCL-1), anti-apoptotic proteins like survivin, and multiple transcription factors such as C/EBPβ. ALK fusion proteins also stimulate the upregulation of CD30 via RAS and AP-1 transcription factors, providing anti-apoptotic signals through TNF receptor-associated factor 2 (TRAF2) (Figure 5) [57].
Further, in neuroendocrine prostate cancer (NEPC), a severe form of prostate cancer, a mutation (ALK F1174C) in the ALK gene responded well to alectinib. An experimental model combining ALK F1174C and N-Myc led to aggressive NEPC, mirroring poor outcomes seen in human datasets. This combination also activated the wnt/β-catenin pathway [58]; however, as mentioned earlier, ALK cleavage by MMP-9 in neuroblastoma results in β-catenin release from ALK [50]. However, inhibiting ALK suppressed this pathway, hindering NEPC and neuroblastoma growth in lab experiments and live models. Combining ALK and Wnt inhibitors showed potential against NEPC and neuroblastoma, underscoring ALK’s significance and proposing a therapeutic strategy targeting both ALK and Wnt pathways in ALK-related tumors, linking insights between NEPC and neuroblastoma [58]. However, a subgroup of lung cancers relies on the ALK for survival, and treatment with the ALK inhibitor crizotinib initially yields remarkable tumor responses. Long-term effectiveness is limited due to emerging drug resistance.
Further investigation revealed that ALK controls MYC’s transcriptional expression and activates c-MYC’s regulation of target genes in NSCLC. Silencing MYC, either through RNAi or small molecules, sensitizes ALK+ cells to crizotinib. These findings illuminate a dual oncogenic mechanism whereby ALK stimulates the MYC signaling axis, suggesting that targeting MYC could potentially prevent or overcome crizotinib resistance [59].
In addition, findings suggested that targeting Src signaling could be a promising therapeutic strategy for ALK+ NSCLC cases that have developed resistance to ALK-TKIs. Researchers discovered that Src signaling is a key resistance mechanism to alectinib, and combining ALK and Src inhibitors effectively halted the growth of ALK-TKI-resistant cells. Further, blocking Src in alectinib-resistant cells effectively countered the activation of phospho-receptor tyrosine kinases and downstream PI3K/AKT signaling. This combined inhibition of ALK and Src also displayed effectiveness against other ALK+ NSCLC cell lines resistant to ceritinib or lorlatinib [60]. In addition, another group of researchers established ALK+ lung cancer cell lines resistant to ceritinib. Hence, treatment with ceritinib significantly increased Src activity. The silencing of Src alone using siRNA effectively restored sensitivity to ceritinib in ALK+ cells. Moreover, inhibiting Src with saracatinib was effective in ALK-resistant cancer cells. Therefore, ceritinib’s inhibition of ALK may trigger an upsurge in Src signaling, and saracatinib could potentially serve as a therapeutic agent for treating lung cancer patients resistant to ALK inhibitors [61].
On the other hand, in NSCLC, where ALK genes are rearranged, resistance to ALK-TKIs remains a challenge despite their success. Research into resistance mechanisms uncovered a new adaptive resistance mechanism linked to JNK/c-Jun signaling. This pathway contributes to the survival of cells tolerant to alectinib and brigatinib. Blocking JNK/c-Jun improved the effectiveness of ALK-TKI treatment in curbing cell growth and promoting cell death. Combining the inhibition of JNK with ALK-TKIs increased cell death by suppressing Bcl-xL proteins, surpassing the effects observed with ALK-TKI treatment alone. Targeting both JNK signaling and ALK might be a promising method to improve outcomes for ALK-rearranged NSCLC [62].
Additionally, treatment advancements for NSCLC with the EML4-ALK fusion gene have been made with ALK-TKIs. In ALK-TKI-resistant cells, the expression of EML4-ALK decreased at the transcriptional level, while the phosphorylation of EGFR, HER2, and HER3 increased compared to parental-sensitive cells. This increase in the activation of HER family proteins coincided with a higher secretion of EGF. Treatment with an EGFR-TKI induced apoptosis in ALK-TKI resistant cells but not in sensitive cells. In the parental cells, the inhibition of extracellular signal-regulated kinase (ERK) and STAT3 phosphorylation by the selective ALK-TKI TAE684 was disrupted when these cells were exposed to exogenous EGF, leading to reduced sensitivity in cell growth to TAE684 [63]. However, resistance, notably the G1202R mutation in ALK, limits their effectiveness. A recent study demonstrated that the EML4-ALK G1202R mutation prompts an epithelial-mesenchymal transition (EMT), likely boosting cell migration and invasion through increased STAT3 and Slug expression. Combining ALK and STAT3 inhibitors restores sensitivity to ceritinib, offering a potential approach to counter ALK mutation-driven resistance in NSCLC therapy [64]. Nonetheless, the ALK-TKI TAE684 suppressed cell growth, triggered cell death, and blocked the activation of STAT3 and ERK in H3122 cells carrying the EML4-ALK fusion gene, but not in H2228 cells with the same fusion gene demonstrated that TAE684 predominantly inhibited STAT3 activation without significantly impacting cell growth or apoptosis. However, the combined use of TAE684 and an MEK inhibitor induced cell death by concurrently targeting the STAT3 and ERK pathways in H2228 cells. This combined inhibition reduced levels of the antiapoptotic protein survivin and increased levels of the proapoptotic protein BIM [65].
In addition, research exploring protein methylation, notably SET and MYND domain containing 2 (SMYD2) methyltransferases, discovered their role in methylating specific lysine residues (1451, 1455, and 1610) in the ALK protein. Lowering SMYD2 levels or using an SMYD2 inhibitor reduced EML4-ALK protein phosphorylation in NSCLC cell lines. Modification of these lysine residues hindered ALK methylation and inhibited downstream AKT phosphorylation, impeding cell growth. Combining SMYD2 and ALK inhibitors demonstrated enhanced efficacy in restraining NSCLC cell growth. Hence, this SMYD2-mediated ALK methylation is suggested as a novel treatment avenue for ALK fusion gene-related tumors [66].
A patient with EGFR mutation and EML4-ALK rearrangement, post-EGFR-TKI resistance, showed promising responses to combined EGFR and ALK inhibitors, suggesting a viable therapeutic approach for managing NSCLC with concurrent mutations. A patient with NSCLC developed both an EGFR mutation and an EML4-ALK rearrangement after resistance to EGFR-TKI treatment. Researchers engineered EGFR mutant cells with ALK variants to understand how these molecular combinations function and tested various inhibitors in laboratory and animal settings. The findings revealed that cells expressing these variants resisted individual treatments but responded positively to a combination of ALK and EGFR inhibitors, showing elevated effectiveness in killing cancer cells. In animal experiments, this combination therapy significantly reduced tumor growth compared to individual treatments. Particularly noteworthy was a patient with liver metastases experiencing a decrease in liver tumor size after receiving a combination of osimertinib (an EGFR-TKI) and ceritinib (an ALK-TKI). The study suggests that employing both EGFR and ALK inhibitors might be a promising therapeutic approach for managing NSCLC marked by simultaneous EGFR mutation and EML4-ALK rearrangement [67].
Moreover, scientists aimed to develop more potent ALK inhibitors to combat drug resistance in ALK rearrangement-related NSCLC [68,69]. They identified ZX-29 as a potent inhibitor that caused G1 phase cell cycle arrest and subsequent cell death via endoplasmic reticulum stress. Notably, ZX-29 induced protective autophagy, and blocking this process enhanced its effectiveness against tumors. Additionally, ZX-29 effectively hindered mouse tumor growth and showcased its ability to overcome drug resistance stemming from the ALK G1202R mutation [68]. Moreover, scientists developed XMU-MP-5, a new ALK inhibitor to combat crizotinib resistance in NSCLC. In laboratory and mouse model studies, XMU-MP-5 successfully targeted ALK pathways, inhibiting cell proliferation in both wild-type and mutant EML4-ALK cells. These preclinical outcomes underscore XMU-MP-5 as a promising, highly selective ALK inhibitor capable of addressing clinically relevant secondary ALK mutations [69]. An extensive analysis was also carried out on 31 cancer tissues and 90 cfDNA samples. Among cancers resistant to crizotinib, 16% displayed ALK mutations (like L1196M, I1171T, D1203N, G1269A/F1174L) and three potential bypass mutations. Ceritinib-resistant cancers exhibited 22% ALK mutations (including G1128A, G1202R, G1269A, I1171T/E1210K) and similar bypass mutations. Alectinib-resistant cancers showed 17% ALK mutations (including G1202R, W1295C, G1202R/L1196M) and one potential bypass mutation. Lorlatinib-resistant cancers had 11% ALK mutations (including G1202R/G1269A) and two potential bypass mutations. Cases with both tissue and cfDNA samples revealed mutations in 45% and 30%, respectively, with a matching rate of 45% [70].
Eventually, researchers have discovered a direct relationship between ALK and cyclin-dependent kinase 9 (CDK9) in breast cancer, where ALK phosphorylates CDK9, leading to resistance against Poly(ADP-Ribose) Polymerase (PARP) inhibitors and encouraging homologous recombination repair. This phosphorylation boosts CDK9’s activity, promoting gene transcription linked to HR-repair within the nucleus. When ALK is inhibited, CDK9 is degraded by Skp2, an E3 ligase. These discoveries propose a treatment avenue based on specific biomarkers, combining ALK and PARP inhibitors to induce synthetic lethality in PARP inhibitor-/platinum-resistant tumors expressing elevated p-ALK-p-Tyr19-CDK9 [71]. Moreover, considering the promising suppression of NSCLC with CDK9 inhibitors seen in EGFR-TKI resistant NSCLC [72], further investigation into their potential in ALK+ NSCLC is warranted.
In conclusion, the diverse mechanisms of resistance and intricate signaling pathways associated with ALK+ cancers, particularly in NSCLC and other malignancies, reveal the complexity of targeting ALK alterations. Various studies have highlighted the signaling cascades activated by mutated forms of ALK and ALK fusion proteins, such as NPM-ALK, elucidating their role in cell transformation, survival, and resistance to therapy. Strategies involving dual inhibition of ALK and other pathways, including MYC, Src, JNK/c-Jun, and Wnt/β-catenin, have demonstrated potential in overcoming resistance and hindering tumor growth in ALK+ cancers. The discovery of ALK mutations and their association with resistance to different ALK inhibitors has spurred the development of newer, more potent inhibitors like ZX-29 and XMU-MP-5, showcasing promising preclinical efficacy against various ALK mutations, including the challenging G1202R mutation. These findings underscore the need for a multifaceted approach to combat ALK-related resistance and advance therapeutic strategies, emphasizing the need for further investigation and clinical trials to optimize treatment outcomes in ALK+ cancers.
5. FDA-approved ALK inhibitors
Studies comparing various ALK inhibitors for advanced NSCLC have highlighted distinct efficacy profiles and safety concerns associated with each medication. Examining the efficacy of crizotinib, alectinib, brigatinib, ceritinib, and lorlatinib in treating ALK+ NSCLC has provided valuable insights into their performance, safety, and unique adverse event profiles.
Clinical studies [73] highlighted crizotinib’s superior performance over chemotherapy, emphasizing extended progression-free survival (PFS) and higher ORR. Similarly, another research [74] reinforced these findings in PROFILE 1029 (NCT01639001), underlining the significant advantages of crizotinib in terms of PFS, ORR, and prompt response time among East Asian patients despite negligible differences in overall survival (OS). Conversely, several studies have consistently drawn attention to alectinib’s advantages compared to crizotinib. Peters et al. [75] and Gadgeel et al. [76] independently demonstrated alectinib’s superior PFS and CNS activity in untreated ALK+ NSCLC patients, irrespective of prior CNS disease. Further studies examining circulating Cell-free DNA (cfDNA) as a prognostic biomarker [77] and ALK+ tumor responses [78] consistently favored alectinib over crizotinib. Additionally, patient-reported outcomes from the ALEX trial [79] showcased alectinib’s prolonged benefits in lung cancer symptom management and superior CNS progression control.
On the other hand, in the ALTA-1L trial (NCT02737501), brigatinib’s effectiveness was evaluated against crizotinib among patients with locally advanced or metastatic ALK+ NSCLC who had not previously received ALK inhibitors [80]. Brigatinib demonstrated significant superiority over crizotinib in terms of PFS and ORR. The trial highlighted brigatinib’s substantial increase in response duration compared to crizotinib, with an OS rate of 86% for crizotinib and 85% for brigatinib. Notably, the adverse effect profiles differed between the two treatments, with distinct patterns of side effects observed in each group. Further analysis from the second interim assessment reinforced brigatinib’s superior PFS over crizotinib, supporting its efficacy in delaying disease progression [81]. Despite similar OS probabilities at two years, brigatinib consistently outperformed crizotinib in PFS. However, adverse effects, especially those in grades 3-5, were higher in the brigatinib group. Besides, researchers [82] conducted a comparative analysis of the ALTA-1L trial outcomes in Asian and non-Asian patients, affirming brigatinib’s notable advantages in PFS across both subgroups, while overall safety remained comparable.
Additionally, Ng et al. [83] focused on the unique pulmonary-related adverse events associated with brigatinib among ALK inhibitors, highlighting their rarity but significance. Their assessment across phase I to III trials revealed early onset pulmonary events, albeit at a low percentage, emphasizing the importance of monitoring for these specific adverse events during brigatinib treatment. In the future, the ALTA-3 trial (NCT03596866) aims to assess how well brigatinib performs compared to alectinib in individuals with advanced ALK+ NSCLC resistant to crizotinib, offering additional information about the effectiveness and relative results of these therapies [84].
In the multicenter, randomized ASCEND-4 trial (NCT01828099) comparing ceritinib to platinum-based chemotherapy, the efficacy and safety of ceritinib in ALK-rearranged nonsquamous NSCLC were assessed [85]. Ceritinib demonstrated significantly longer PFS than chemotherapy, with substantial, rapid, and durable responses observed in the ceritinib group. However, adverse events, particularly diarrhea, nausea, and vomiting, were more familiar with ceritinib, including higher-grade events like elevated liver enzymes. The effectiveness and tolerability of ceritinib were further analyzed in a Japanese subgroup of patients from the ASCEND-5 (NCT01828112) study [17,86]. The ceritinib group exhibited prolonged PFS compared to chemotherapy, though it came with a higher incidence of suspected drug-related adverse events. Another comparison, presented by Li et al. [73], evaluated the outcomes of PROFILE 1014 (NCT01154140) and ASCEND-4 (NCT01828099) phase III trials. Ceritinib significantly improved PFS compared to crizotinib, showcasing a notable reduction in disease progression or mortality risk. Despite a comparable OS rate at 12 months, ceritinib exhibited a clinically meaningful advantage by maintaining a higher PFS rate. This analysis indicated a substantial improvement in the treatment of first-line metastatic NSCLC. In the ALUR trial (NCT02604342), a randomized, multicenter, open-label, phase III study, researchers compared the efficacy of alectinib to chemotherapy in patients with advanced or metastatic ALK+ NSCLC. This study specifically enrolled 107 patients from Europe and Asia who had previously undergone platinum-based doublet chemotherapy and crizotinib. Novello et al. [20] summarized the findings of the ALUR trial, revealing substantial improvements in PFS with alectinib compared to chemotherapy. Patients treated with alectinib experienced significantly longer PFS durations than those receiving chemotherapy (9.6-7.1 months versus 1.4-1.6 months, p < 0.001). Additionally, adverse events of grade 3 or higher and those leading to discontinuation of the study drug were less prevalent in the alectinib group. Furthermore, the duration of alectinib treatment was notably prolonged compared to chemotherapy (20.1 weeks versus 6.0 weeks).
In the CROWN clinical trial (NCT03052608), a phase III, open-label, multicenter, randomized study, researchers investigated the efficacy of lorlatinib as a first-line treatment for advanced ALK+ NSCLC in comparison to crizotinib [87]. The trial enrolled 296 patients without previously received systemic therapy for their metastatic disease. Patients were randomized to receive either lorlatinib (100 mg daily) or crizotinib (250 mg twice daily) for 28 days. The study revealed significant advantages in favor of lorlatinib over crizotinib. The proportion of patients alive at 12 months without disease progression was notably higher in the lorlatinib arm compared to the crizotinib arm (78% versus 39%, p < 0.001). Additionally, lorlatinib demonstrated superior PFS within one year compared to crizotinib (80% versus 35%). Objective responses were observed in 76% of patients in the lorlatinib group and 58% in the crizotinib group. The most common adverse effects associated with lorlatinib were hyperlipidemia and edema, while crizotinib was linked to different adverse effects. Assessing the quality of life (QoL), both groups experienced overall improvements and delayed declines [88]. Crizotinib showed better results in cognitive functioning, while lorlatinib demonstrated advantages in physical, role, emotional, and social functioning. The lorlatinib group showed more noticeable enhancements in symptoms like fatigue, nausea, vomiting, insomnia, appetite loss, constipation, diarrhea, and congestion, whereas crizotinib was more beneficial for improving peripheral neuropathy.
In conclusion, examining various ALK inhibitors for advanced NSCLC has unveiled their distinct efficacy profiles and safety considerations. While crizotinib showcased notable advantages regarding PFS and ORRs, alectinib consistently emerged as a superior performer across multiple studies. Alectinib’s prolonged PFS heightened CNS activity, and excellent patient-reported outcomes stood out prominently. Despite its effectiveness in extending PFS, brigatinib exhibited more adverse effects than crizotinib. Ceritinib’s ability to improve PFS and reduce the risk of disease progression or mortality underscored its clinical significance. As a first-line treatment, lorlatinib demonstrated promising outcomes, displaying superior PFS and a higher proportion of patients without disease progression at the 12-month than crizotinib. However, it is essential to note that lorlatinib showed varying adverse effect profiles and impacts on different facets of patients’ quality of life. These findings provide clinicians with invaluable insights to tailor therapies according to individual patient needs and tolerability, ultimately advancing the development of personalized treatment strategies for ALK+ NSCLC.
6. ALK-targeted therapy and future directions
6.1. ALK and immunotherapy
Changes in the structure of the ALK gene significantly contribute to the onset of different human cancers, and therapies aimed at this gene have revolutionized how we treat these tumors driven by this specific oncogene. However, overcoming inherent or acquired resistance remains a significant hurdle. Variations in the ALK gene, like gene rearrangements or mutations, also influence the immune environment within tumors. Harnessing immunotherapy to target the ALK gene holds promise in clinical settings [8]. In addition, interleukin (IL)-6, IL-8, and IL-10 have been associated with disease progression in NSCLC, specifically in ALK+ patients. The interactions between TLRs and various interleukins underscore their involvement in lung cancer pathogenesis, progression, and potential prognostic value [89].
Serum-soluble IL-2R levels are a reliable marker for disease activity in hairy cell leukemia and adult T-cell leukemia/lymphoma patients. In addition, ALCL patients often display CD30 and CD25 expression in malignant cells. A study measured serum soluble IL-2R and CD30 levels in ALCL patients treated with etoposide, prednisone, Oncovin, Cytoxan, and hydroxydaunorubicin (EPOCH) chemotherapy. Soluble CD30 levels were initially high and decreased with treatment. This study also demonstrated that patients with the ALK gene had higher soluble IL-2R levels than those without ALK, whose soluble IL-2R levels were normal and whose tumors lacked CD25 expression. Elevated soluble IL-2R levels were observed in patients with recurrent disease, regardless of ALK status [90].
Further, the research explored lymphoma immune profiles, which are crucial for accurate diagnosis and new treatment avenues. In ALCL, microRNA-135b (miR-135b), influenced by the NPM-ALK oncogene, promoted cancer growth and instigated an immune profile producing IL-17. NPM-ALK activated miR-135b via STAT3, targeting FOXO1 and impacting ALCL cells’ chemotherapy response. Additionally, miR-135b hindered Th2 regulators STAT6 and GATA3, altering IL-17 production and resembling the immune profile of Th17 cells in ALCL. Blocking miR-135b reduced tumor growth and blood vessel formation in experiments, indicating its potential as a therapeutic target. This highlights how cancer-causing pathways affect tumor immune profiles and the surrounding environment [91].
Furthermore, the PD-1/PD-L1 axis in ALK+ tumors favors regulatory T cells (Tregs), limiting T cell activity. Additionally, PD-L1 upregulation promotes an immunosuppressive environment, hindering effective antitumor responses. Meanwhile, ALK release into the tumor microenvironment stimulates tumor-associated macrophages (TAM) and B-cells, shaping a milieu conducive to tumor growth and immune evasion. Understanding these dynamics is pivotal in developing targeted therapies against ALK-driven tumors, aiming to recalibrate the immune landscape and disrupt tumor-promoting interactions.
Alternatively, in ALCL patients, specific levels of serum cytokines could indicate the tumor’s size and variations in the body’s response against the lymphoma. Levels of IL-9, IL-10, IL-17a, HGF, soluble IL-2R, and soluble CD30 collectively create a distinct cytokine profile specific to ALK+ ALCL, distinguishing them from both remission samples and samples from children of similar age with low-stage B-cell non-Hodgkin lymphoma, serving as special control groups. Furthermore, cytokine levels like IL-6 and interferon (IFN)-γ correlated with the disease stage, patient condition, and the likelihood of relapse among ALCL patients, with IL-6 displaying individual predictive value. These initial cytokine profiles in ALCL patients might reflect the tumor characteristics and the strength of their immune responses [92]. Furthermore, the researchers explored how circulating cytokines might be markers for monitoring disease progression in ALK+ NSCLC when treated with TKIs. They examined eight cytokines in serum samples from 38 patients. Higher levels of IL-6, IL-8, and IL-10 correlated with disease advancement, particularly IL-8, which displayed the most substantial potential as a biomarker. Although the combination of alterations in IL-8 alongside circulating tumor DNA parameters enhanced the ability to detect disease progression, it did not exceed the accuracy achieved by using circulating tumor DNA alone. This study suggested that serum cytokine levels might indicate disease progression in ALK+ NSCLC, potentially enhancing current monitoring methods pending further confirmation in more extensive studies [93].
The occurrence of any mutation in ALK results in the promotion of PD-L1 expression. Increasing the expression of immunosuppressive molecules like PD-L1 may lead to tolerance and immune evasion in patients with tumors and cancers. Tian et al. have shown that upregulation of PD-L1 can be identified as a biomarker for ALK-rearrange NSCLC. In addition, it has been recognized that the TME in the presence of upregulated expression of PD-L1 encompasses an immunosuppressive condition [24,94]. Immune checkpoint inhibitors (ICIs) have shown significant promise in various cancers [95,96]. In ALK+ NSCLC, these inhibitors have been explored, mainly due to the upregulation of PD-L1 expression in ALK+ tumors [97,98]. However, studies on the prognosis of ALK+ patients using ICIs have yielded conflicting results, necessitating further investigation [97]. Initial data from randomized studies suggested lower effectiveness of immunotherapies in ALK+ tumors compared to wild-type tumors. For instance, a global retrospective study found no objective response in ALK+ NSCLC patients treated with ICI monotherapy, with a higher incidence of rapid progression in this group. Subsequent investigations in this area mainly focused on understanding ALK inhibitor resistance [99]. In addition, another study studied how ALK fusion proteins regulate PD-L1 expression and immune function in ALK+ NSCLC. Researchers observed a correlation between PD-L1 expression, EGFR mutations, and ALK fusion genes in NSCLC cell lines. Elevating ALK fusion protein levels boosted PD-L1 expression, leading to T-cell apoptosis in co-culture systems. Blocking ALK with TKIs amplified IFN-γ production. Anti-PD-1 antibodies were effective in both crizotinib-sensitive and --resistant NSCLC cells. However, combining ALK-TKIs with anti-PD-1 antibodies did not benefit co-culture systems. ALK-TKIs suppressed tumor growth and indirectly bolstered antitumor immunity by reducing PD-L1 expression. While anti-PD-1/PD-L1 antibodies could be an option for ALK+ NSCLC patients, especially crizotinib-resistant ones, combining ALK-TKIs with anti-PD-1/PD-L1 antibodies requires further study before clinical use [24].
On the other hand, attempts to combine ICIs with ALK-TKIs showed some promise. Preclinical research demonstrated that combining ceritinib (an ALK inhibitor) with a PD-L1 inhibitor suppressed PD-L1 expression and enhanced lymphocyte activity in ALK-rearranged NSCLC [100]. However, several phase 1/2 clinical trials combining nivolumab or pembrolizumab (PD-1 inhibitors) with crizotinib or lorlatinib (ALK inhibitors) reported dose-limiting toxicities, impacting their efficacy. A few studies indicated potential benefits with ceritinib plus nivolumab, suggesting activity, especially in patients with high PD-L1 expression [100,101,102]. Furthermore, combining avelumab or atezolizumab (PD-L1 inhibitors) with lorlatinib or alectinib (ALK inhibitors) demonstrated promising efficacy in ALK+ NSCLC. The use of alectinib combined with atezolizumab showed promising results, but heightened toxicity was observed compared to using each agent separately. Due to limited sample sizes and relatively short observation periods, definitive conclusions about the treatment’s effectiveness against tumors could not be drawn. Nonetheless, more extensive studies are required to verify these outcomes further [103]. However, the level of PD-L1 expression may not reliably indicate the expected response to initial treatment with alectinib in patients with ALK+ NSCLC [104].
Another path being explored is the fusion of ICIs with anti-angiogenesis therapy. While atezolizumab combined with chemotherapy did not exhibit enhanced survival compared to chemotherapy alone in ALK+ patients, the combination of atezolizumab with bevacizumab, an anti-vascular endothelial growth factor (VEGF) agent, along with chemotherapy demonstrated marked improvements in PFS and hinted at a potential benefit for OS [105]. This combined approach might be favored for ALK+ NSCLC patients’ post-resistance to ALK inhibitors. Studies scrutinizing the synergy between ICIs and anti-VEGF agents have underscored their combined mechanisms, indicating the potential to counteract resistance to ICIs by reversing VEGF-mediated immunosuppression and bolstering the response to ICIs in ALK+ tumors. Overall, the confluence of ICIs with anti-angiogenesis therapy presents a promising treatment strategy for ALK+ NSCLC [106].
Angiogenic factors create vascular abnormalities and hinder antigen presentation, suppress immune cells, and boost cell activity that inhibits the immune system. ALK signaling in the PI3K/AKT/mTOR pathway stimulates VEGF expression, potentially heightening sensitivity to bevacizumab in ALK+ patients. Following treatment with ALK inhibitors, individuals with ALK+ tumors encounter a decline in the infiltration of CD8+ T cells and a rise in regulatory T cells, resulting in a diminished response to immune checkpoint inhibitors [107]. Besides, clinical studies indicate that combining bevacizumab and atezolizumab overcomes ICI resistance by reversing VEGF-induced immunosuppression and enhancing CD8+ TIL in tumors. Evidence suggests that bevacizumab can overcome ALK inhibitor resistance when combined with targeted therapy. Recent research highlights VEGFR2 inhibition as a promising strategy for inhibiting tumor angiogenesis and directly impeding cancer cell growth in oncogene-driven NSCLC [108]. However, two patients with ALK-rearranged lung cancer, previously treated with multiple ALK-TKIs and other therapies, showed promising outcomes when treated with a combination of bevacizumab and lorlatinib. They experienced disease regression and control for 5-9 months, surpassing the duration of single-agent erlotinib therapy. This combination was well tolerated and could benefit patients after lorlatinib failure, especially against both on-target (such as ALK kinase domain mutations) and off-target resistance mechanisms. Additionally, it might hold promise for patients before lorlatinib treatment or those with ROS1-rearranged lung cancers. The study suggests exploring this combination further in clinical settings for ALK+ lung cancer patients [109].
6.2. ALK-innovative approaches
In ALK+ cancer models, DNA vaccines directed against the ALK gene exhibited notable effectiveness [110]. These vaccines prompted specific immune reactions against ALK, fostering CD8+ T cell-mediated cytotoxicity and provoking IFN-γ responses. When combined with chemotherapy, ALK DNA vaccination significantly extended the survival of mice afflicted with ALK+ lymphomas. In the context of ALK+ NSCLC models, the ALK DNA vaccine triggered robust immune responses, curtailing tumor growth and elongating survival. However, lung tumors with ALK rearrangements establish an immunosuppressive setting, diminishing the efficacy of the ALK vaccine by upregulating PD-L1 expression. However, administering anti-PD-1 immunotherapy reinstates the vaccine’s effectiveness. This implies that combining ALK vaccines with TKIs and immune checkpoint inhibitors could present a robust treatment strategy for ALK-driven NSCLC [111]. Its pairing with ALK-TKIs notably delayed tumor relapse post-TKI treatment.
Additionally, alternative ALK vaccines utilizing peptides or lipid vesicles encapsulating ALK antigens showcased potential in restraining tumor advancement in preclinical models. Further, the application of anti-EGF-VacAbs targeting EGF in ALK+ NSCLC cell lines amplified the effectiveness of ALK-TKIs, impeding the emergence of resistance and intercepting downstream oncogenic pathways [112]. These experimental findings propose an encouraging strategy for managing ALK-driven tumors, hinting at the possibility of ALK vaccines entering clinical trials.
On the other hand, scientists developed a highly sensitive NanoBiT LATS bioluminescent biosensor (BS) to track LATS kinase activity in the Hippo signaling pathway in lab settings and living organisms. This new biosensor showed greater sensitivity and stability than previous versions, even when expressed at significantly lower levels. Using this advanced biosensor, they could monitor LATS activity in live cells at physiologically relevant levels and simplify kinase activity analysis in vitro. Moreover, their study revealed an unprecedented interaction between ALK and the Hippo pathway, identifying a new mechanism involving the YAP/TAZ-PD-L1 axis. Targeting YAP/TAZ alone or in combination with ALK could offer a promising strategy for more effective treatment of cancers involving ALK or facing resistance to ALK inhibitors [113].
Alternatively, researchers recently investigated the role of the Nuclear Interaction Partner of ALK (NIPA) in a specific type of lymphoma induced by the NPM-ALK gene. Previous studies highlighted NIPA’s significance in cell division control and bone marrow failure but had yet to explore its involvement in NPM-ALK-driven lymphomas [114]. Researchers demonstrated that NIPA interacts with NPM-ALK, and its absence or downregulation led to significant impairment in the growth and transformation of cells associated with this lymphoma in lab tests. Further experiments in mice confirmed that removing or reducing NIPA in cells related to NPM-ALK-driven tumors prolonged survival without altering the tumors’ characteristics. Interestingly, the absence of NIPA affected a specific subpopulation of cells within the lymphoma, possibly impacting the disease’s onset and progression. These findings suggest that NIPA plays a crucial role in initiating this type of lymphoma, highlighting its potential as a target for future therapeutic interventions in this disease. Further investigations into the specific mechanisms of NIPA’s interaction with NPM-ALK and its role in tumor development could offer valuable insights for potential treatment strategies [115].
Furthermore, T-LAK cell-oriented protein kinase (TOPK), recognized as a potential therapeutic target in cancer, has been scrutinized in ALK+ NSCLC. The study identified ALK as an upstream kinase of TOPK, phosphorylating it specifically at Y74. This phosphorylation notably promotes tumor growth in ALK+ lung cancer cells, a finding supported by a phosphoproteomic analysis delineating downstream pathway involvement [116]. Comparatively, TOPK emerges as a superior target for cancer therapy compared to other direct downstream molecules of ALK, including Smad4, STAT3, PI3K, and PLC-γ. Clinical studies have consistently associated TOPK with a marker of poor prognosis in various cancers and an independent predictor for OS [117,118,119,120]. Encouragingly, inhibitors such as HI-032 and SKLB-C05, which target TOPK, have demonstrated promising potential.
Moreover, combining TOPK inhibition with alectinib, an ALK inhibitor, has shown remarkable synergy in impeding cell proliferation and promoting apoptosis. This combined approach proposes a promising strategy to counter drug resistance in ALK+ NSCLC [116]. These research findings advance our understanding of ALK’s oncogenic signaling network and suggest the potential efficacy of co-inhibition of ALK and TOPK as a novel therapeutic strategy to treat ALK+ NSCLC and potentially delay the onset of drug resistance.
7. Concluding remarks
Evaluating FDA-approved ALK inhibitors in advanced NSCLC highlights their unique effectiveness and safety profiles. Crizotinib exhibits notable benefits regarding PFS and ORR; however, multiple studies consistently position alectinib as the superior option. Alectinib distinguishes itself with extended PFS, increased CNS activity, and excellent patient-reported outcomes. In contrast, while brigatinib effectively extends PFS, it has a higher incidence of adverse effects than crizotinib. Besides, ceritinib’s ability to enhance PFS and reduce disease progression or mortality emphasizes its clinical significance. As a primary treatment, lorlatinib displays promising outcomes by demonstrating superior PFS and more patients without disease progression at 12 months than crizotinib. However, it is essential to note that these treatments have varying impacts on patients’ quality of life due to their distinct adverse effect profiles. These findings provide essential insights for tailoring treatments based on individual patient’s needs and tolerability, thereby shaping personalized strategies for managing ALK+ NSCLC. Additionally, exploring interactions between ALK and immunotherapy and innovative methods such as ALK vaccines, biosensors, and targeted pathway approaches offers potential avenues for future interventions in ALK-driven cancers.
Funding
NA.
CRediT Author Statement
Conceptualization, P.J.K.; Writing – Original Draft, H.P, F.G., and P.B.; Writing – Review & Editing, P.J.K. and P.B.; Visualization, G.R.; Supervision, P.J.K.
Data Availability
Not applicable.
Conflict of Interests
The authors have no conflict of interest.
Acknowledgments
The authors would like to acknowledge that the Figures presented in this work were created using BioRender, utilizing a licensed account purchased by Parham Jabbarzadeh Kaboli.
Abbreviations
| Akt | Protein Kinase B |
| ALCL | Anaplastic large cell lymphoma |
| ALK | Anaplastic lymphoma kinase |
| CDK | Cyclin-dependent kinase |
| c-MET | Mesenchymal-epithelial transition factor |
| ECD | Extracellular domain |
| EGFR | Epidermal growth factor receptor |
| EML4 | Echinoderm microtubule-associated protein-like 4 |
| ERK | Extracellular Signal-Regulated Kinase |
| FDA | Food and Drug Administration |
| GRB2 | Growth factor receptor-bound protein 2 |
| GRD | Glycine-rich domain |
| ICI | Immune checkpoint inhibitor |
| LDLa | Low-density lipoprotein receptor class A |
| MAM | Meprin, A5 protein and receptor protein tyrosine phosphatase mu |
| MMP-9 | Matrix metallopeptidase 9 |
| NPM | Nucleolar phosphoprotein |
| NSCLC | Non-small cell lung cancer |
| ORR | Objective response rate |
| OS | Overall survival |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3 kinase |
| PTK | Protein tyrosine kinase |
| RAS | Rat sarcoma viral oncogene |
| STAT | Signal transducer and activator of transcription |
| TKI | Tyrosine kinase inhibitor |
| VEGF | Vascular endothelial growth factor |
References
- Hallberg, B.; Palmer, R.H. Mechanistic Insight into ALK Receptor Tyrosine Kinase in Human Cancer Biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Pearson, J.D.; Lee, J.K.H.; Bacani, J.T.C.; Lai, R.; Ingham, R.J. NPM-ALK: The Prototypic Member of a Family of Oncogenic Fusion Tyrosine Kinases. J. Signal Transduct. 2012, 2012, 123253. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Rossi, P.; Myasnikov, A.G.; Sowaileh, M.; Mohanty, J.; Nourse, A.; Miller, D.J.; Lax, I.; Schlessinger, J.; Kalodimos, C.G. Mechanism for the Activation of the Anaplastic Lymphoma Kinase Receptor. Nature 2021, 600, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Andraos, E.; Dignac, J.; Meggetto, F. NPM-ALK: A Driver of Lymphoma Pathogenesis and a Therapeutic Target. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Sankar, K.; Nagrath, S.; Ramnath, N. Immunotherapy for ALK-Rearranged Non-Small Cell Lung Cancer: Challenges Inform Promising Approaches. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Cameron, L.B.; Hitchen, N.; Chandran, E.; Morris, T.; Manser, R.; Solomon, B.J.; Jordan, V. Targeted Therapy for Advanced Anaplastic Lymphoma Kinase (<I>ALK</I>)-Rearranged Non-Small Cell Lung Cancer. Cochrane database Syst. Rev. 2022, 1, CD013453. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, H.; Zhang, Y.; Cui, J. Anaplastic Lymphoma Kinase-Special Immunity and Immunotherapy. Front. Immunol. 2022, 13, 908894. [Google Scholar] [CrossRef]
- Panagiotidis, E. The Role of Positron Computed Tomography (PET/CT) in Lung Cancer Staging. Hell. J. Nucl. Med. 2023, 26 Suppl, 22–29. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Liu, S.; Huang, T.; Liu, M.; He, W.; Zhao, Y.; Yang, L.; Long, Y.; Zong, D.; Zeng, H.; Liu, Y.; et al. The Genomic Characteristics of ALK Fusion Positive Tumors in Chinese NSCLC Patients. Front. Oncol. 2020, 10, 726. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical Features and Outcome of Patients with Non-Small-Cell Lung Cancer Who Harbor EML4-ALK. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef]
- Chia, P.L.; Mitchell, P.; Dobrovic, A.; John, T. Prevalence and Natural History of ALK Positive Non-Small-Cell Lung Cancer and the Clinical Impact of Targeted Therapy with ALK Inhibitors. Clin. Epidemiol. 2014, 6, 423–432. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Lee, A.T.M.; Nagasaka, M. From Preclinical Efficacy to 2022 (36.7 Months Median Follow -up) Updated CROWN Trial, Lorlatinib Is the Preferred 1st-Line Treatment of Advanced ALK+ NSCLC. Crit. Rev. Oncol. Hematol. 2023, 187, 104019. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus Chemotherapy in Patients with ALK-Rearranged Non-Small-Cell Lung Cancer Previously given Chemotherapy and Crizotinib (ASCEND-5): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet. Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.-M.; Garcia, L.; Yu, L.; et al. Safety and Activity of Alectinib against Systemic Disease and Brain Metastases in Patients with Crizotinib-Resistant ALK-Rearranged Non-Small-Cell Lung Cancer (AF-002JG): Results from the Dose-Finding Portion of a Phase 1/2 Study. Lancet. Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Ahn, J.S.; DePetris, L.; Govindan, R.; Yang, J.C.-H.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in Crizotinib-Refractory ALK-Rearranged Non-Small-Cell Lung Cancer: A Phase II Global Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef]
- Novello, S.; Mazières, J.; Oh, I.-J.; deCastro, J.; Migliorino, M.R.; Helland, Å.; Dziadziuszko, R.; Griesinger, F.; Kotb, A.; Zeaiter, A.; et al. Alectinib versus Chemotherapy in Crizotinib-Pretreated Anaplastic Lymphoma Kinase (ALK)-Positive Non-Small-Cell Lung Cancer: Results from the Phase III ALUR Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1409–1416. [Google Scholar] [CrossRef]
- Kim, D.-W.; Tiseo, M.; Ahn, M.-J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.-W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef]
- Jahanzeb, M.; Lin, H.M.; Pan, X.; Yin, Y.; Baumann, P.; Langer, C.J. Immunotherapy Treatment Patterns and Outcomes Among ALK-Positive Patients With Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 49–57. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Zhou, T.; Huang, J.; et al. Upregulation of PD-L1 by EML4-ALK Fusion Protein Mediates the Immune Escape in ALK Positive NSCLC: Implication for Optional Anti-PD-1/PD-L1 Immune Therapy for ALK-TKIs Sensitive and Resistant NSCLC Patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation - Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 682–688. [Google Scholar] [CrossRef]
- Patel, M.; Jabbour, S.K.; Malhotra, J. ALK Inhibitors and Checkpoint Blockade: A Cautionary Tale of Mixing Oil with Water? J. Thorac. Dis. 2018, 10, S2198–S2201. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic Lymphoma Kinase: Signalling in Development and Disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Stayrook, S.E.; Tsutsui, Y.; Zhang, J.; Wang, Y.; Li, H.; Proffitt, A.; Krimmer, S.G.; Ahmed, M.; Belliveau, O.; et al. Structural Basis for Ligand Reception by Anaplastic Lymphoma Kinase. Nature 2021, 600, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B Are Activating Ligands for Anaplastic Lymphoma Kinase. Elife 2015, 4, e09811. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor α and β (FAM150) Are Ligands of the Receptor Tyrosine Kinases ALK and LTK: Hierarchy and Specificity of Ligand-Receptor Interactions. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal Structures of Anaplastic Lymphoma Kinase in Complex with ATP Competitive Inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Huang, Q.; Johnson, T.W.; Bailey, S.; Brooun, A.; Bunker, K.D.; Burke, B.J.; Collins, M.R.; Cook, A.S.; Cui, J.J.; Dack, K.N.; et al. Design of Potent and Selective Inhibitors to Overcome Clinical Anaplastic Lymphoma Kinase Mutations Resistant to Crizotinib. J. Med. Chem. 2014, 57, 1170–1187. [Google Scholar] [CrossRef]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.L.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique Substrate Specificity of Anaplastic Lymphoma Kinase (ALK): Development of Phosphoacceptor Peptides for the Assay of ALK Activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.L.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of Some Molecular Mechanisms Governing Autoactivation of the Catalytic Domain of the Anaplastic Lymphoma Kinase. J. Biol. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef]
- Wang, P.; Wu, F.; Zhang, J.; McMullen, T.; Young, L.C.; Ingham, R.J.; Li, L.; Lai, R. Serine Phosphorylation of NPM-ALK, Which Is Dependent on the Auto-Activation of the Kinase Activation Loop, Contributes to Its Oncogenic Potential. Carcinogenesis 2011, 32, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The Role of the ALK Receptor in Cancer Biology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 Suppl 3, iii4–iii15. [Google Scholar] [CrossRef]
- Loong, H.H.; Mok, K.; Leung, L.K.S.; Mok, T.S.K. Crizotinib in the Management of Advanced-Stage Non-Small-Cell Lung Cancer. Future Oncol. 2015, 11, 735–745. [Google Scholar] [CrossRef]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA Approval Summary: Crizotinib for the Treatment of Metastatic Non-Small Cell Lung Cancer with Anaplastic Lymphoma Kinase Rearrangements. Oncologist 2014, 19, e5–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. The Potential Role of HGF-MET Signaling and Autophagy in the War of Alectinib versus Crizotinib against ALK-Positive NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 33. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the Chromosome 2 Gene Locus Altered by the t(2;5) in Non-Hodgkin’s Lymphoma, Encodes a Novel Neural Receptor Tyrosine Kinase That Is Highly Related to Leukocyte Tyrosine Kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) Expression in DRG Neurons and Its Involvement in Neuron-Schwann Cells Interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Mazot, P.; Cazes, A.; Boutterin, M.C.; Figueiredo, A.; Raynal, V.; Combaret, V.; Hallberg, B.; Palmer, R.H.; Delattre, O.; Janoueix-Lerosey, I.; et al. The Constitutive Activity of the ALK Mutated at Positions F1174 or R1275 Impairs Receptor Trafficking. Oncogene 2011, 30, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Mazot, P.; Cazes, A.; Dingli, F.; Degoutin, J.; Irinopoulou, T.; Boutterin, M.-C.; Lombard, B.; Loew, D.; Hallberg, B.; Palmer, R.H.; et al. Internalization and Down-Regulation of the ALK Receptor in Neuroblastoma Cell Lines upon Monoclonal Antibodies Treatment. PLoS One 2012, 7, e33581. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Ghirelli, A.; Tripathi, V.; Piccoli, G. Negr1-Derived Peptides Trigger ALK Degradation and Halt Neuroblastoma Progression In Vitro and In Vivo. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Venkannagari, H.; Kasper, J.M.; Misra, A.; Rush, S.A.; Fan, S.; Lee, H.; Sun, H.; Seshadrinathan, S.; Machius, M.; Hommel, J.D.; et al. Highly Conserved Molecular Features in IgLONs Contrast Their Distinct Structural and Biological Outcomes. J. Mol. Biol. 2020, 432, 5287–5303. [Google Scholar] [CrossRef]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Créminon, C.; Vigny, M. Activation and Inhibition of Anaplastic Lymphoma Kinase Receptor Tyrosine Kinase by Monoclonal Antibodies and Absence of Agonist Activity of Pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef]
- Huang, H.; Gont, A.; Kee, L.; Dries, R.; Pfeifer, K.; Sharma, B.; Debruyne, D.N.; Harlow, M.; Sengupta, S.; Guan, J.; et al. Extracellular Domain Shedding of the ALK Receptor Mediates Neuroblastoma Cell Migration. Cell Rep. 2021, 36, 109363. [Google Scholar] [CrossRef]
- DelGrosso, F.; DeMariano, M.; Passoni, L.; Luksch, R.; Tonini, G.P.; Longo, L. Inhibition of N-Linked Glycosylation Impairs ALK Phosphorylation and Disrupts pro-Survival Signaling in Neuroblastoma Cell Lines. BMC Cancer 2011, 11, 525. [Google Scholar] [CrossRef]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Rehemtulla, A.; Lawrence, T.S. Inhibition of N-Linked Glycosylation Disrupts Receptor Tyrosine Kinase Signaling in Tumor Cells. Cancer Res. 2008, 68, 3803–3809. [Google Scholar] [CrossRef]
- Dedoni, S.; Scherma, M.; Camoglio, C.; Siddi, C.; Fratta, W.; Fadda, P. Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa. Nutrients 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef]
- Xin, S.; Fang, W.; Li, J.; Li, D.; Wang, C.; Huang, Q.; Huang, M.; Zhuang, W.; Wang, X.; Chen, L. Impact of STAT1 Polymorphisms on Crizotinib-Induced Hepatotoxicity in ALK-Positive Non-Small Cell Lung Cancer Patients. J. Cancer Res. Clin. Oncol. 2021, 147, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Barreca, A.; Lasorsa, E.; Riera, L.; Machiorlatti, R.; Piva, R.; Ponzoni, M.; Kwee, I.; Bertoni, F.; Piccaluga, P.P.; Pileri, S.A.; et al. Anaplastic Lymphoma Kinase in Human Cancer. J. Mol. Endocrinol. 2011, 47, R11–23. [Google Scholar] [CrossRef]
- Unno, K.; Chalmers, Z.R.; Pamarthy, S.; Vatapalli, R.; Rodriguez, Y.; Lysy, B.; Mok, H.; Sagar, V.; Han, H.; Yoo, Y.A.; et al. Activated ALK Cooperates with N-Myc via Wnt/β-Catenin Signaling to Induce Neuroendocrine Prostate Cancer. Cancer Res. 2021, 81, 2157–2170. [Google Scholar] [CrossRef]
- Pilling, A.B.; Kim, J.; Estrada-Bernal, A.; Zhou, Q.; Le, A.T.; Singleton, K.R.; Heasley, L.E.; Tan, A.C.; DeGregori, J.; Doebele, R.C. ALK Is a Critical Regulator of the MYC-Signaling Axis in ALK Positive Lung Cancer. Oncotarget 2018, 9, 8823–8835. [Google Scholar] [CrossRef]
- Yoshida, R.; Sasaki, T.; Minami, Y.; Hibino, Y.; Okumura, S.; Sado, M.; Miyokawa, N.; Hayashi, S.; Kitada, M.; Ohsaki, Y. Activation of Src Signaling Mediates Acquired Resistance to ALK Inhibition in Lung Cancer. Int. J. Oncol. 2017, 51, 1533–1540. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Xu, Y.; Lu, S.; Jian, H. AZD0530 Sensitizes Drug-Resistant ALK-Positive Lung Cancer Cells by Inhibiting SRC Signaling. FEBS Open Bio 2017, 7, 472–476. [Google Scholar] [CrossRef]
- Tanimura, K.; Yamada, T.; Horinaka, M.; Katayama, Y.; Fukui, S.; Morimoto, K.; Nakano, T.; Tokuda, S.; Morimoto, Y.; Iwasaku, M.; et al. Inhibition of C-Jun N-Terminal Kinase Signaling Increased Apoptosis and Prevented the Emergence of ALK-TKI-Tolerant Cells in ALK-Rearranged Non-Small Cell Lung Cancer. Cancer Lett. 2021, 522, 119–128. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of HER Family Signaling as a Mechanism of Acquired Resistance to ALK Inhibitors in EML4-ALK-Positive Non-Small Cell Lung Cancer. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2012, 18, 6219–6226. [Google Scholar] [CrossRef]
- Shen, J.; Meng, Y.; Wang, K.; Gao, M.; Du, J.; Wang, J.; Li, Z.; Zuo, D.; Wu, Y. EML4-ALK G1202R Mutation Induces EMT and Confers Resistance to Ceritinib in NSCLC Cells via Activation of STAT3/Slug Signaling. Cell. Signal. 2022, 92, 110264. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Takezawa, K.; Sakai, K.; Azuma, K.; Kuwata, K.; Yamaguchi, H.; Hatashita, E.; Nishio, K.; Janne, P.A.; et al. Combined Effect of ALK and MEK Inhibitors in EML4-ALK-Positive Non-Small-Cell Lung Cancer Cells. Br. J. Cancer 2012, 106, 763–767. [Google Scholar] [CrossRef]
- Wang, R.; Deng, X.; Yoshioka, Y.; Vougiouklakis, T.; Park, J.-H.; Suzuki, T.; Dohmae, N.; Ueda, K.; Hamamoto, R.; Nakamura, Y. Effects of SMYD2-Mediated EML4-ALK Methylation on the Signaling Pathway and Growth in Non-Small-Cell Lung Cancer Cells. Cancer Sci. 2017, 108, 1203–1209. [Google Scholar] [CrossRef]
- Huang, M.-H.; Lee, J.-H.; Hung, P.-S.; Chih-Hsin Yang, J. Potential Therapeutic Strategy for EGFR-Mutant Lung Cancer With Concomitant EML4-ALK Rearrangement-Combination of EGFR Tyrosine Kinase Inhibitors and ALK Inhibitors. JTO Clin. Res. reports 2022, 3, 100405. [Google Scholar] [CrossRef] [PubMed]
- Gou, W.; Li, Z.; Xu, X.; Shen, J.; Guo, M.; Zhou, X.; Zhang, X.; Wu, Y.; Zhai, X.; Zuo, D. ZX-29, a Novel ALK Inhibitor, Induces Apoptosis via ER Stress in ALK Rearrangement NSCLC Cells and Overcomes Cell Resistance Caused by an ALK Mutation. Biochim. Biophys. acta. Mol. cell Res. 2020, 1867, 118712. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Fan, Z.; Zhu, S.-J.; Huang, X.; Zhuang, Z.; Li, Y.; Deng, Z.; Gao, L.; Hong, X.; Zhang, T.; et al. A New ALK Inhibitor Overcomes Resistance to First- and Second-Generation Inhibitors in NSCLC. EMBO Mol. Med. 2022, 14, e14296. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Chiang, C.-L.; Hung, J.-Y.; Lee, M.-H.; Su, W.-C.; Wu, S.-Y.; Wei, Y.-F.; Lee, K.-Y.; Tseng, Y.-H.; Su, J.; et al. Resistance Profiles of Anaplastic Lymphoma Kinase Tyrosine Kinase Inhibitors in Advanced Non-Small-Cell Lung Cancer: A Multicenter Study Using Targeted next-Generation Sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- Chu, Y.-Y.; Chen, M.-K.; Wei, Y.; Lee, H.-H.; Xia, W.; Wang, Y.-N.; Yam, C.; Hsu, J.L.; Wang, H.-L.; Chang, W.-C.; et al. Targeting the ALK-CDK9-Tyr19 Kinase Cascade Sensitizes Ovarian and Breast Tumors to PARP Inhibition via Destabilization of the P-TEFb Complex. Nat. cancer 2022, 3, 1211–1227. [Google Scholar] [CrossRef]
- Wu, T.; Yu, B.; Xu, Y.; Du, Z.; Zhang, Z.; Wang, Y.; Chen, H.; Zhang, L.A.; Chen, R.; Ma, F.; et al. Discovery of Selective and Potent Macrocyclic CDK9 Inhibitors for the Treatment of Osimertinib-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2023, 66, 15340–15361. [Google Scholar] [CrossRef]
- Li, J.; Knoll, S.; Bocharova, I.; Tang, W.; Signorovitch, J. Comparative Efficacy of First-Line Ceritinib and Crizotinib in Advanced or Metastatic Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer: An Adjusted Indirect Comparison with External Controls. Curr. Med. Res. Opin. 2019, 35, 105–111. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lu, S.; Lu, Y.; Zhou, J.; Shi, Y.-K.; Sriuranpong, V.; Ho, J.C.M.; Ong, C.K.; Tsai, C.-M.; Chung, C.-H.; et al. Results of PROFILE 1029, a Phase III Comparison of First-Line Crizotinib versus Chemotherapy in East Asian Patients with ALK-Positive Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 1539–1548. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Gadgeel, S.; Peters, S.; Mok, T.; Shaw, A.T.; Kim, D.W.; Ou, S.I.; Pérol, M.; Wrona, A.; Novello, S.; Rosell, R.; et al. Alectinib versus Crizotinib in Treatment-Naive Anaplastic Lymphoma Kinase-Positive (ALK+) Non-Small-Cell Lung Cancer: CNS Efficacy Results from the ALEX Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2214–2222. [Google Scholar] [CrossRef]
- Dziadziuszko, R.; Peters, S.; Mok, T.; Camidge, D.R.; Gadgeel, S.M.; Ou, S.-H.I.; Konopa, K.; Noé, J.; Nowicka, M.; Bordogna, W.; et al. Circulating Cell-Free DNA as a Prognostic Biomarker in Patients with Advanced ALK+ Non-Small Cell Lung Cancer in the Global Phase III ALEX Trial. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2022, 28, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Peters, S.; Camidge, D.R.; Noé, J.; Gadgeel, S.; Ou, S.-H.I.; Kim, D.-W.; Konopa, K.; Pozzi, E.; Liu, T.; et al. Outcomes According to ALK Status Determined by Central Immunohistochemistry or Fluorescence In Situ Hybridization in Patients With ALK-Positive NSCLC Enrolled in the Phase 3 ALEX Study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Pérol, M.; Pavlakis, N.; Levchenko, E.; Platania, M.; Oliveira, J.; Novello, S.; Chiari, R.; Moran, T.; Mitry, E.; Nüesch, E.; et al. Patient-Reported Outcomes from the Randomized Phase III ALEX Study of Alectinib versus Crizotinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer. Lung Cancer 2019, 138, 79–87. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor-Naive ALK-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3592–3603. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; Garcia Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in ALK Inhibitor-Naive Advanced ALK-Positive NSCLC: Final Results of Phase 3 ALTA-1L Trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 2091–2108. [Google Scholar] [CrossRef]
- Ng, T.L.; Narasimhan, N.; Gupta, N.; Venkatakrishnan, K.; Kerstein, D.; Camidge, D.R. Early-Onset Pulmonary Events Associated With Brigatinib Use in Advanced NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1190–1199. [Google Scholar] [CrossRef]
- Popat, S.; Liu, G.; Lu, S.; Song, G.; Ma, X.; Yang, J.C.-H. Brigatinib vs Alectinib in Crizotinib-Resistant Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer (ALTA-3). Future Oncol. 2021, 17, 4237–4247. [Google Scholar] [CrossRef]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-Line Ceritinib versus Platinum-Based Chemotherapy in Advanced ALK-Rearranged Non-Small-Cell Lung Cancer (ASCEND-4): A Randomised, Open-Label, Phase 3 Study. Lancet (London, England) 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Kiura, K.; Imamura, F.; Kagamu, H.; Matsumoto, S.; Hida, T.; Nakagawa, K.; Satouchi, M.; Okamoto, I.; Takenoyama, M.; Fujisaka, Y.; et al. Phase 3 Study of Ceritinib vs Chemotherapy in ALK-Rearranged NSCLC Patients Previously Treated with Chemotherapy and Crizotinib (ASCEND-5): Japanese Subset. Jpn. J. Clin. Oncol. 2018, 48, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; deMarinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Iadeluca, L.; Shaw, A.T.; Solomon, B.J.; Bauer, T.M.; deMarinis, F.; Felip, E.; Goto, Y.; Kim, D.-W.; Mok, T.; et al. Patient-Reported Outcomes from the Randomized Phase 3 CROWN Study of First-Line Lorlatinib versus Crizotinib in Advanced ALK-Positive Non-Small Cell Lung Cancer. Lung Cancer 2022, 174, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Patra, R.; Behzadi, P.; Masotti, A.; Paolini, A.; Sarshar, M. Toll-like Receptor-Guided Therapeutic Intervention of Human Cancers: Molecular and Immunological Perspectives. Front. Immunol. 2023, 14, 1244345. [Google Scholar] [CrossRef] [PubMed]
- Janik, J.E.; Morris, J.C.; Pittaluga, S.; McDonald, K.; Raffeld, M.; Jaffe, E.S.; Grant, N.; Gutierrez, M.; Waldmann, T.A.; Wilson, W.H. Elevated Serum-Soluble Interleukin-2 Receptor Levels in Patients with Anaplastic Large Cell Lymphoma. Blood 2004, 104, 3355–3357. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Suzuki, H.I.; Nishimori, H.; Noguchi, M.; Yao, T.; Komatsu, N.; Mano, H.; Sugimoto, K.; Miyazono, K. MiR-135b Mediates NPM-ALK-Driven Oncogenicity and Renders IL-17-Producing Immunophenotype to Anaplastic Large Cell Lymphoma. Blood 2011, 118, 6881–6892. [Google Scholar] [CrossRef]
- Knörr, F.; Damm-Welk, C.; Ruf, S.; Singh, V.K.; Zimmermann, M.; Reiter, A.; Woessmann, W. Blood Cytokine Concentrations in Pediatric Patients with Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma. Haematologica 2018, 103, 477–485. [Google Scholar] [CrossRef]
- Angeles, A.K.; Janke, F.; Daum, A.-K.; Reck, M.; Schneider, M.A.; Thomas, M.; Christopoulos, P.; Sültmann, H. Integrated Circulating Tumour DNA and Cytokine Analysis for Therapy Monitoring of ALK-Rearranged Lung Adenocarcinoma. Br. J. Cancer 2023, 129, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, Y.; Huang, Q.; Zeng, H.; Wei, Q.; Tian, P. High PD-L1 Expression Correlates with an Immunosuppressive Tumour Immune Microenvironment and Worse Prognosis in ALK-Rearranged Non-Small Cell Lung Cancer. Biomolecules 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Jabbarzadeh Kaboli, P.; Shabani, S.; Sharma, S.; Partovi Nasr, M.; Yamaguchi, H.; Hung, M.-C. Shedding Light on Triple-Negative Breast Cancer with Trop2-Targeted Antibody-Drug Conjugates. Am. J. Cancer Res. 2022, 12, 1671–1685. [Google Scholar] [PubMed]
- Zhao, Q.; Guo, J.; Zhao, Y.; Shen, J.; Kaboli, P.J.; Xiang, S.; Du, F.; Wu, X.; Li, M.; Wan, L.; et al. Comprehensive Assessment of PD-L1 and PD-L2 Dysregulation in Gastrointestinal Cancers. Epigenomics 2020, 12, 2155–2171. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Auclin, E.; Mosteiro, M.; Dempsey, N.; Majem, M.; Lobefaro, R.; López-Castro, R.; Bosch-Barrera, J.; Pilotto, S.; Escalera, E.; et al. Durvalumab Consolidation in Patients with Unresectable Stage III Non-Small Cell Lung Cancer with Driver Genomic Alterations. Eur. J. Cancer 2022, 167, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Baldacci, S.; Grégoire, V.; Patrucco, E.; Chiarle, R.; Jamme, P.; Wasielewski, E.; Descarpentries, C.; Copin, M.-C.; Awad, M.M.; Cortot, A.B. Complete and Prolonged Response to Anti-PD1 Therapy in an ALK Rearranged Lung Adenocarcinoma. Lung Cancer 2020, 146, 366–369. [Google Scholar] [CrossRef]
- Hebart, H.; Lang, P.; Woessmann, W. Nivolumab for Refractory Anaplastic Large Cell Lymphoma: A Case Report. Ann. Intern. Med. 2016, 165, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Abbou, S.; Minard-Colin, V.; Geoerger, B.; Scoazec, J.Y.; Vassal, G.; Jaff, N.; Heuberger, L.; Valteau-Couanet, D.; Brugieres, L. Efficacy of Nivolumab in a Patient with Systemic Refractory ALK+ Anaplastic Large Cell Lymphoma. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef]
- Kim, D.-W.; Gadgeel, S.; Gettinger, S.N.; Riely, G.J.; Oxnard, G.R.; Mekhail, T.; Schmid, P.; Dowlati, A.; Heist, R.S.; Wozniak, A.J.; et al. Brief Report: Safety and Antitumor Activity of Alectinib Plus Atezolizumab From a Phase 1b Study in Advanced ALK-Positive NSCLC. JTO Clin. Res. reports 2022, 3, 100367. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, X.; Zhang, W.; Wang, W.; Wang, H.; Luo, L.; Jia, K.; Shao, C.; Mao, S.; Qiu, T.; et al. Association of PD-L1 Expression with Efficacy of Alectinib in Advanced NSCLC Patients with ALK Fusion. Lung Cancer 2023, 181, 107233. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11, 598877. [Google Scholar] [CrossRef]
- Pyo, K.-H.; Lim, S.M.; Park, C.-W.; Jo, H.-N.; Kim, J.H.; Yun, M.-R.; Kim, D.; Xin, C.-F.; Lee, W.; Gheorghiu, B.; et al. Comprehensive Analyses of Immunodynamics and Immunoreactivity in Response to Treatment in ALK-Positive Non-Small-Cell Lung Cancer. J. Immunother. cancer 2020, 8. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination with Bevacizumab versus Sunitinib in Renal Cell Carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Young, R.J.; Sellitti, M.; Miller, A.; Drilon, A. Lorlatinib and Bevacizumab Activity in ALK-Rearranged Lung Cancers After Lorlatinib Progression. JCO Precis. Oncol. 2020, 4. [Google Scholar]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The Anaplastic Lymphoma Kinase Is an Effective Oncoantigen for Lymphoma Vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef]
- Voena, C.; Menotti, M.; Mastini, C.; DiGiacomo, F.; Longo, D.L.; Castella, B.; Merlo, M.E.B.; Ambrogio, C.; Wang, Q.; Minero, V.G.; et al. Efficacy of a Cancer Vaccine against ALK-Rearranged Lung Tumors. Cancer Immunol. Res. 2015, 3, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; García-Roman, S.; Molina-Vila, M.Á.; Bertran-Alamillo, J.; Viteri, S.; d’Hondt, E.; Rosell, R. Anti-Epidermal Growth Factor Vaccine Antibodies Increase the Antitumor Activity of Kinase Inhibitors in ALK and RET Rearranged Lung Cancer Cells. Transl. Oncol. 2021, 14, 100887. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A Kinome-Wide Screen Using a NanoLuc LATS Luminescent Biosensor Identifies ALK as a Novel Regulator of the Hippo Pathway in Tumorigenesis and Immune Evasion. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 12487–12499. [Google Scholar] [CrossRef]
- Kreutmair, S.; Erlacher, M.; Andrieux, G.; Istvanffy, R.; Mueller-Rudorf, A.; Zwick, M.; Rückert, T.; Pantic, M.; Poggio, T.; Shoumariyeh, K.; et al. Loss of the Fanconi Anemia-Associated Protein NIPA Causes Bone Marrow Failure. J. Clin. Invest. 2020, 130, 2827–2844. [Google Scholar] [CrossRef]
- Kreutmair, S.; Lippert, L.J.; Klingeberg, C.; Albers-Leischner, C.; Yacob, S.; Shlyakhto, V.; Mueller, T.; Mueller-Rudorf, A.; Yu, C.; Gorantla, S.P.; et al. NIPA (Nuclear Interaction Partner of ALK) Is Crucial for Effective NPM-ALK Mediated Lymphomagenesis. Front. Oncol. 2022, 12, 875117. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, L.; Yi, H.; Zou, L.; Mo, J.; Xue, F.; Zheng, J.; Huang, Y.; Lu, H.; Wu, H.; et al. Inhibiting ALK-TOPK Signaling Pathway Promotes Cell Apoptosis of ALK-Positive NSCLC. Cell Death Dis. 2022, 13, 828. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Wang, R.; Zhang, X. Prognostic Value of PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase (PBK/TOPK) in Patients with Cancer. J. Cancer 2019, 10, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Park, J.-H.; Miyamoto, T.; Takamatsu, N.; Kato, T.; Iwasa, A.; Okabe, S.; Imai, Y.; Fujiwara, K.; Nakamura, Y.; et al. T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2016, 22, 6110–6117. [Google Scholar] [CrossRef]
- Zlobec, I.; Molinari, F.; Kovac, M.; Bihl, M.P.; Altermatt, H.J.; Diebold, J.; Frick, H.; Germer, M.; Horcic, M.; Montani, M.; et al. Prognostic and Predictive Value of TOPK Stratified by KRAS and BRAF Gene Alterations in Sporadic, Hereditary and Metastatic Colorectal Cancer Patients. Br. J. Cancer 2010, 102, 151–161. [Google Scholar] [CrossRef]
- Ohashi, T.; Komatsu, S.; Ichikawa, D.; Miyamae, M.; Okajima, W.; Imamura, T.; Kiuchi, J.; Kosuga, T.; Konishi, H.; Shiozaki, A.; et al. Overexpression of PBK/TOPK Relates to Tumour Malignant Potential and Poor Outcome of Gastric Carcinoma. Br. J. Cancer 2017, 116, 218–226. [Google Scholar] [CrossRef]
Figure 1.
ALK Domain Structure and Regulatory Elements. This comprehensive depiction illustrates ALK’s extracellular and intracellular aspects, emphasizing key structural domains and functional motifs. The extracellular domain comprises sections crucial for ligand binding and potential activation, including MAMs and LDLa domains. Within the intracellular protein kinase domain (PTK), essential regulatory segments governing ALK’s active and inactive states are highlighted, shedding light on its allosteric control and potential therapeutic targeting. Notably, the A-loop, housing pivotal amino acid residues Y1278, Y1282, and Y1283, drives ALK activation and downstream signaling. In contrast, specific C-terminal lysine residues serve as targets for methylation, contributing to regulatory functions. Furthermore, this figure delineates the duality of ALK functionality: ligand-dependent dimerization (Left) and ligand-independent monomeric activity (Right).
Figure 1.
ALK Domain Structure and Regulatory Elements. This comprehensive depiction illustrates ALK’s extracellular and intracellular aspects, emphasizing key structural domains and functional motifs. The extracellular domain comprises sections crucial for ligand binding and potential activation, including MAMs and LDLa domains. Within the intracellular protein kinase domain (PTK), essential regulatory segments governing ALK’s active and inactive states are highlighted, shedding light on its allosteric control and potential therapeutic targeting. Notably, the A-loop, housing pivotal amino acid residues Y1278, Y1282, and Y1283, drives ALK activation and downstream signaling. In contrast, specific C-terminal lysine residues serve as targets for methylation, contributing to regulatory functions. Furthermore, this figure delineates the duality of ALK functionality: ligand-dependent dimerization (Left) and ligand-independent monomeric activity (Right).
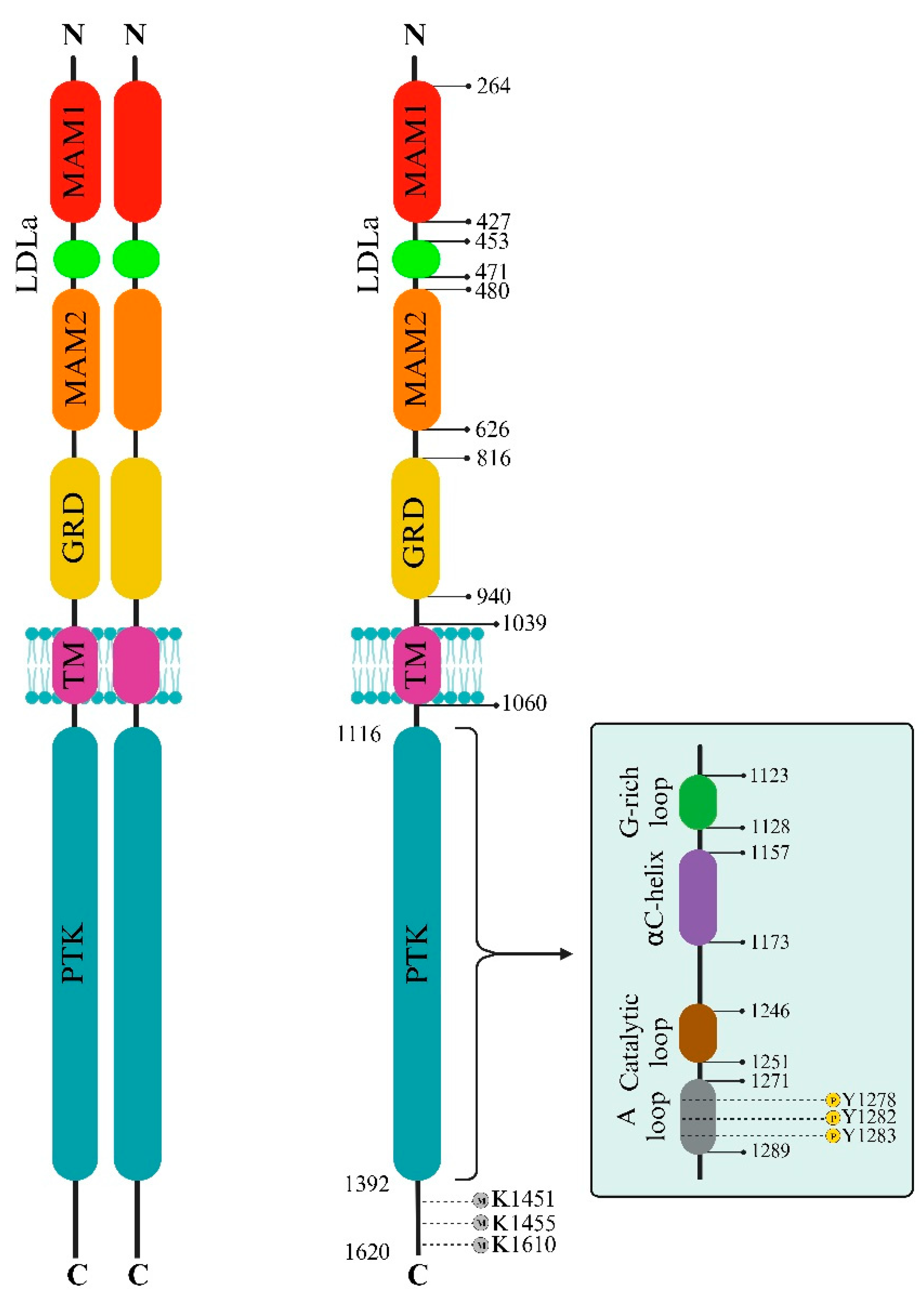
Figure 2.
Crizotinib Interactions with ALK and c-MET. (a-b) Comparison of Crizotinib’s Interactions with (a) ALK (PDB id: 5aaa [40]) and (b) c-MET (PDB id: 2wgj [32]). The figure illustrates distinct binding sites of Crizotinib within unphosphorylated c-MET and ALK-PTK, highlighting crucial π interactions and conventional hydrogen bonds. Fundamental interactions, such as those with specific amino acids (e.g., M1211 in c-MET), contribute significantly to Crizotinib’s inhibitory effect. (c) ALK-PTK (PDB id: 5aaa). The figure details the binding interactions of Crizotinib within the ALK-PTK domain, emphasizing critical regions like the G-loop, A-loop, and catalytic loop. ALK’s activation loop (A-loop) with the conserved Asp-Phe-Gly (DFG) sequence plays a pivotal role in regulating ALK’s active and inactive states. Hydrophobic spines named the ‘regulatory’ and ‘catalytic’ contribute to vital allosteric control between lobes, impacting the transition between active and inactive states. This figure was designed using the BIOVIA Discovery Studio Visualizer (v.21.1).
Figure 2.
Crizotinib Interactions with ALK and c-MET. (a-b) Comparison of Crizotinib’s Interactions with (a) ALK (PDB id: 5aaa [40]) and (b) c-MET (PDB id: 2wgj [32]). The figure illustrates distinct binding sites of Crizotinib within unphosphorylated c-MET and ALK-PTK, highlighting crucial π interactions and conventional hydrogen bonds. Fundamental interactions, such as those with specific amino acids (e.g., M1211 in c-MET), contribute significantly to Crizotinib’s inhibitory effect. (c) ALK-PTK (PDB id: 5aaa). The figure details the binding interactions of Crizotinib within the ALK-PTK domain, emphasizing critical regions like the G-loop, A-loop, and catalytic loop. ALK’s activation loop (A-loop) with the conserved Asp-Phe-Gly (DFG) sequence plays a pivotal role in regulating ALK’s active and inactive states. Hydrophobic spines named the ‘regulatory’ and ‘catalytic’ contribute to vital allosteric control between lobes, impacting the transition between active and inactive states. This figure was designed using the BIOVIA Discovery Studio Visualizer (v.21.1).
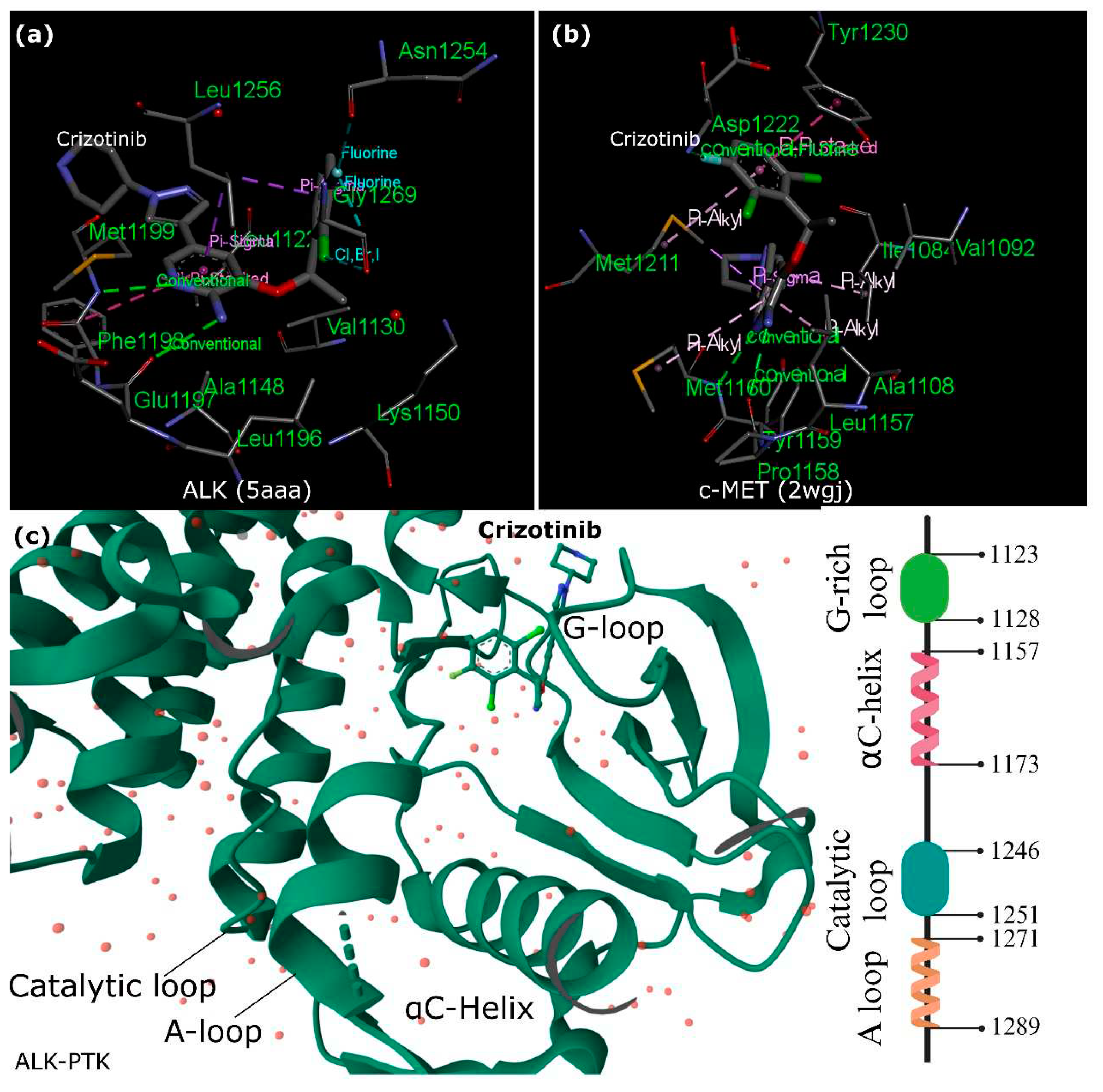
Figure 3.
ALK Sequences as LC3 Interaction Targets. LC3, a mammalian counterpart to yeast Atg8, is a precise marker for autophagy monitoring. The diagram highlights various LC3-interacting region (LIR) motifs across ALK’s diverse domains, indicating a direct link between ALK and autophagy. This connection suggests a complex interplay between suppressing ALK kinase activity and activating autophagy, potentially complicating therapeutic approaches for NSCLC and similar conditions. Understanding autophagy’s dual role in cancer, both as an immune response facilitator and a promoter of tumor growth, underscores the need to classify ALK+ NSCLC based on HGF/c-MET signaling or autophagy-related subtypes. This stratification enables customized treatment strategies for optimizing patient outcomes.
Figure 3.
ALK Sequences as LC3 Interaction Targets. LC3, a mammalian counterpart to yeast Atg8, is a precise marker for autophagy monitoring. The diagram highlights various LC3-interacting region (LIR) motifs across ALK’s diverse domains, indicating a direct link between ALK and autophagy. This connection suggests a complex interplay between suppressing ALK kinase activity and activating autophagy, potentially complicating therapeutic approaches for NSCLC and similar conditions. Understanding autophagy’s dual role in cancer, both as an immune response facilitator and a promoter of tumor growth, underscores the need to classify ALK+ NSCLC based on HGF/c-MET signaling or autophagy-related subtypes. This stratification enables customized treatment strategies for optimizing patient outcomes.
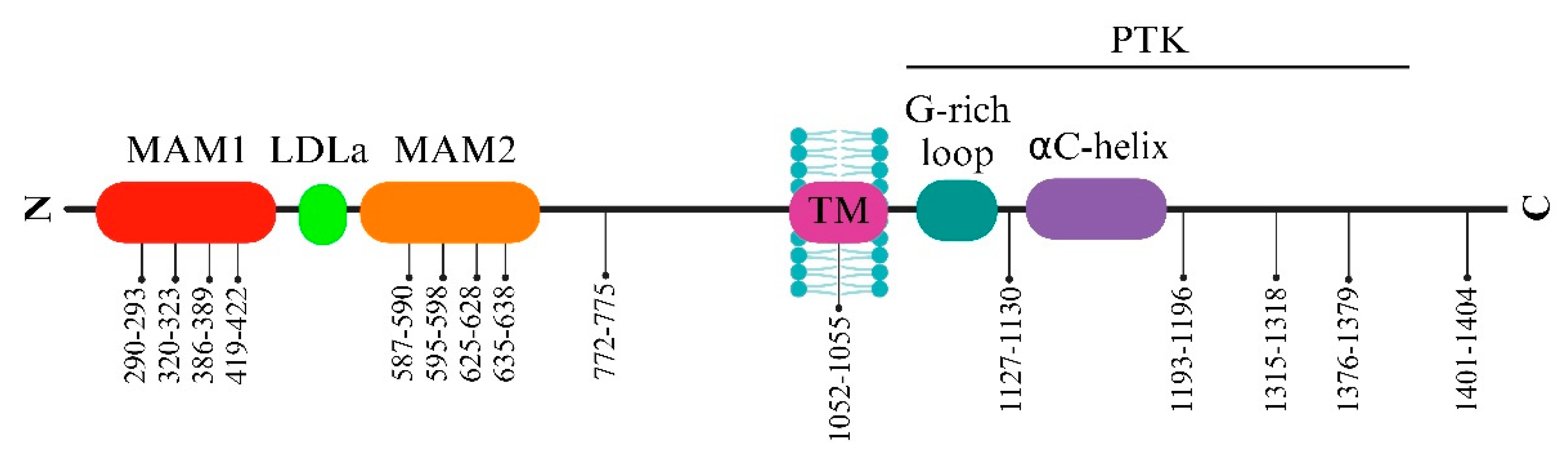
Figure 4.
ALK Cleavage and its Impact on Tumor Cell Behavior. This illustration demonstrates the complex process of ALK cleavage in the tumor microenvironment by MMP9, resulting in the release of an 80 kDa extracellular fragment and a 140 kDa truncated ALK at the membrane. The cleavage of ALK by MMP9 in the tumor microenvironment has diverse effects on immune cell activity and tumor cell migration. While intracellular domain cleavage may aid ALK-targeted therapy, extracellular cleavage fosters ALK-related tumor formation and cellular movement, particularly in neuroblastoma.
Figure 4.
ALK Cleavage and its Impact on Tumor Cell Behavior. This illustration demonstrates the complex process of ALK cleavage in the tumor microenvironment by MMP9, resulting in the release of an 80 kDa extracellular fragment and a 140 kDa truncated ALK at the membrane. The cleavage of ALK by MMP9 in the tumor microenvironment has diverse effects on immune cell activity and tumor cell migration. While intracellular domain cleavage may aid ALK-targeted therapy, extracellular cleavage fosters ALK-related tumor formation and cellular movement, particularly in neuroblastoma.
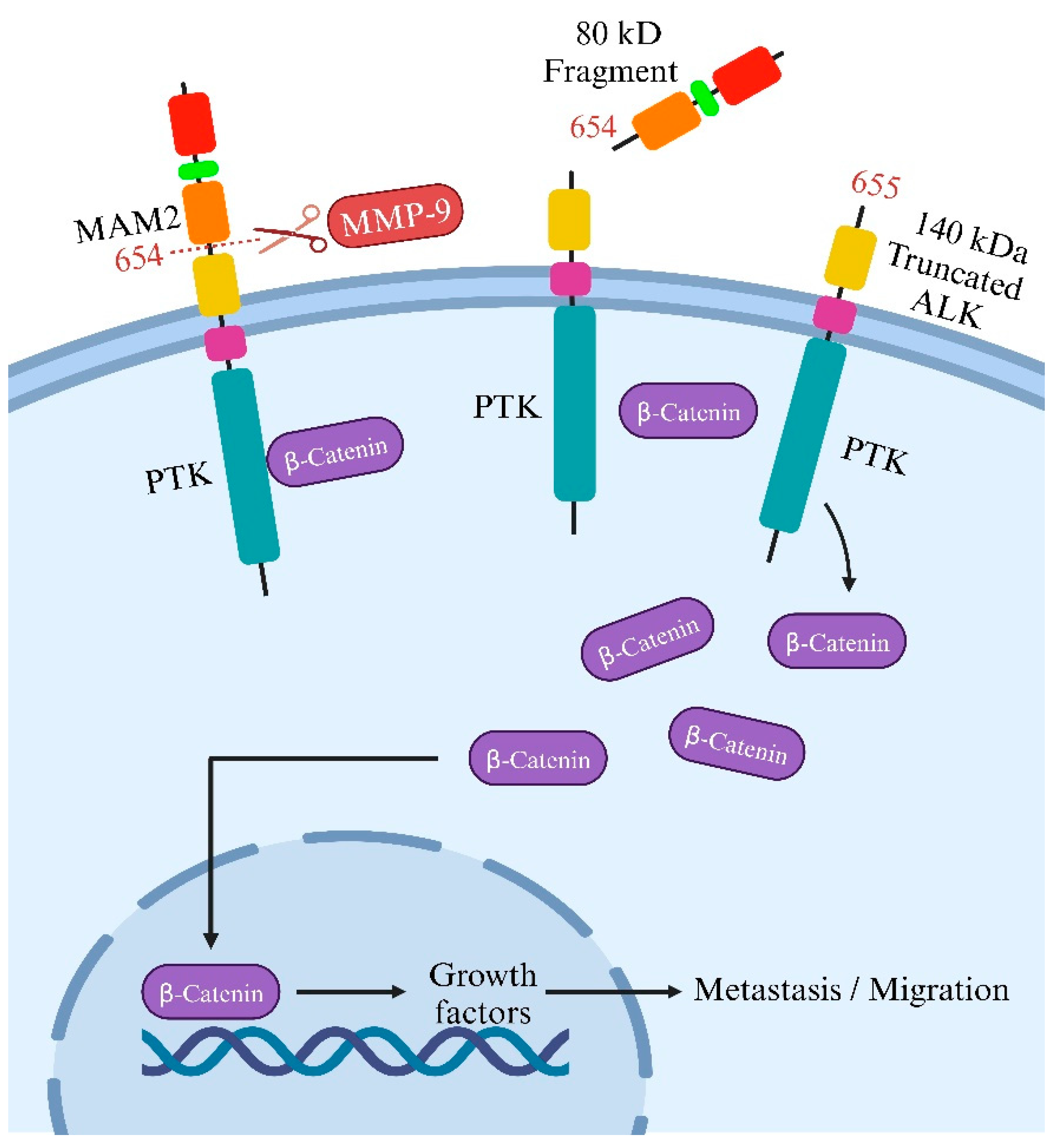
Figure 5.
ALK Signaling Pathways and Therapeutic Implications. This comprehensive visualization outlines the intricate network of ALK signaling pathways, depicting the influence of both membrane wild-type ALK and cytoplasmic NPM–ALK on Akt, MAPK, and STAT3 cascades. ALK mutations and fusion proteins drive varied signaling pathways linked to cell transformation, survival, and therapeutic resistance in ALK+ cancers, notably NSCLC. Despite clinical use of ALK inhibitors (crizotinib, alectinib, ceritinib), resistance emerges due to diverse ALK mutations and fusion proteins, activating multiple adaptors and signaling cascades. These alterations activate mitogenic signaling (RAS/MAP kinase), PI3K, and PLC-γ pathways, impacting anti-apoptotic signaling, cell cycle regulation, and cellular metabolism. Strategies targeting ALK, MYC, Src, JNK/c-Jun, and Wnt/β-catenin pathways show potential against ALK resistance. Novel inhibitors (ZX-29, XMU-MP-5) demonstrate efficacy against diverse ALK mutations. Moreover, the co-expression of CD30, a TNFR superfamily member, alongside ALK in certain cancers—especially ALCL—establishes a distinct ALK+, CD30+ immunophenotype. This co-expression aids ALCL diagnosis and guides targeted therapies, underscoring the diagnostic and therapeutic significance of ALK-CD30 co-expression patterns.
Figure 5.
ALK Signaling Pathways and Therapeutic Implications. This comprehensive visualization outlines the intricate network of ALK signaling pathways, depicting the influence of both membrane wild-type ALK and cytoplasmic NPM–ALK on Akt, MAPK, and STAT3 cascades. ALK mutations and fusion proteins drive varied signaling pathways linked to cell transformation, survival, and therapeutic resistance in ALK+ cancers, notably NSCLC. Despite clinical use of ALK inhibitors (crizotinib, alectinib, ceritinib), resistance emerges due to diverse ALK mutations and fusion proteins, activating multiple adaptors and signaling cascades. These alterations activate mitogenic signaling (RAS/MAP kinase), PI3K, and PLC-γ pathways, impacting anti-apoptotic signaling, cell cycle regulation, and cellular metabolism. Strategies targeting ALK, MYC, Src, JNK/c-Jun, and Wnt/β-catenin pathways show potential against ALK resistance. Novel inhibitors (ZX-29, XMU-MP-5) demonstrate efficacy against diverse ALK mutations. Moreover, the co-expression of CD30, a TNFR superfamily member, alongside ALK in certain cancers—especially ALCL—establishes a distinct ALK+, CD30+ immunophenotype. This co-expression aids ALCL diagnosis and guides targeted therapies, underscoring the diagnostic and therapeutic significance of ALK-CD30 co-expression patterns.
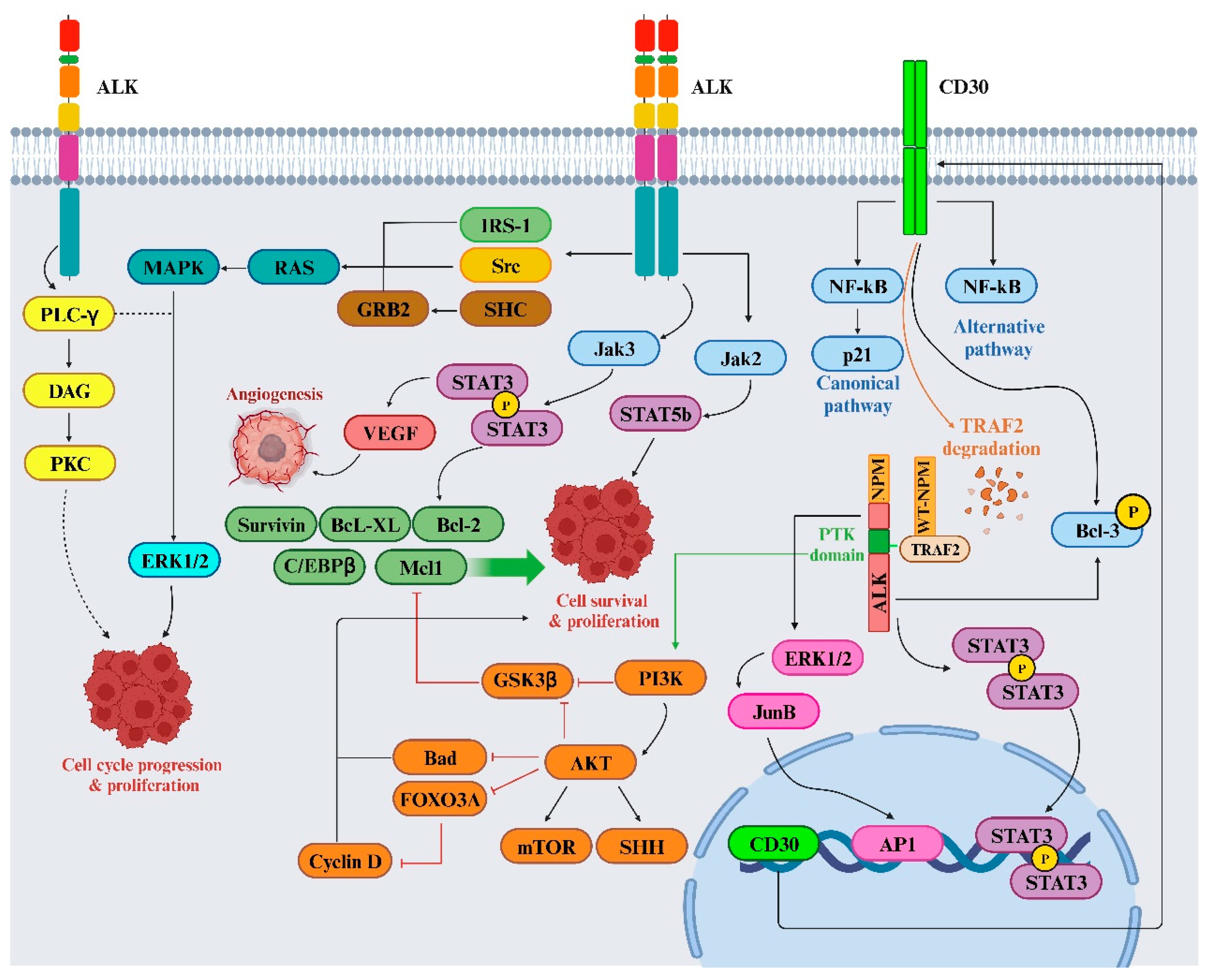
Figure 6.
Interaction between ALK+ Tumors and Immune Cells. The complex landscape of ALK gene alterations significantly contributes to the diverse spectrum of human cancers, revolutionizing therapeutic approaches. These alterations influence the immune milieu within tumors, prompting the exploration of immunotherapy as a promising clinical avenue. Interleukins (IL-6, IL-8, IL-10) are implicated in NSCLC progression, notably in ALK+ patients, affecting the immune landscape via Toll-like receptors (TLRs) and interleukin interactions. Serum markers such as soluble IL-2R (sIL-2R) and CD30 provide insights into disease activity and immune response in ALCL, particularly ALK+ patients, impacting prognosis and treatment assessment. Cytokine profiles in ALCL patients reveal distinct patterns correlating with disease stages, reflecting tumor characteristics and immune responses. Moreover, cytokine levels like IL-6, IL-8, and IL-10 serve as potential indicators for disease progression in ALK+ NSCLC, offering additional monitoring options.
Figure 6.
Interaction between ALK+ Tumors and Immune Cells. The complex landscape of ALK gene alterations significantly contributes to the diverse spectrum of human cancers, revolutionizing therapeutic approaches. These alterations influence the immune milieu within tumors, prompting the exploration of immunotherapy as a promising clinical avenue. Interleukins (IL-6, IL-8, IL-10) are implicated in NSCLC progression, notably in ALK+ patients, affecting the immune landscape via Toll-like receptors (TLRs) and interleukin interactions. Serum markers such as soluble IL-2R (sIL-2R) and CD30 provide insights into disease activity and immune response in ALCL, particularly ALK+ patients, impacting prognosis and treatment assessment. Cytokine profiles in ALCL patients reveal distinct patterns correlating with disease stages, reflecting tumor characteristics and immune responses. Moreover, cytokine levels like IL-6, IL-8, and IL-10 serve as potential indicators for disease progression in ALK+ NSCLC, offering additional monitoring options.
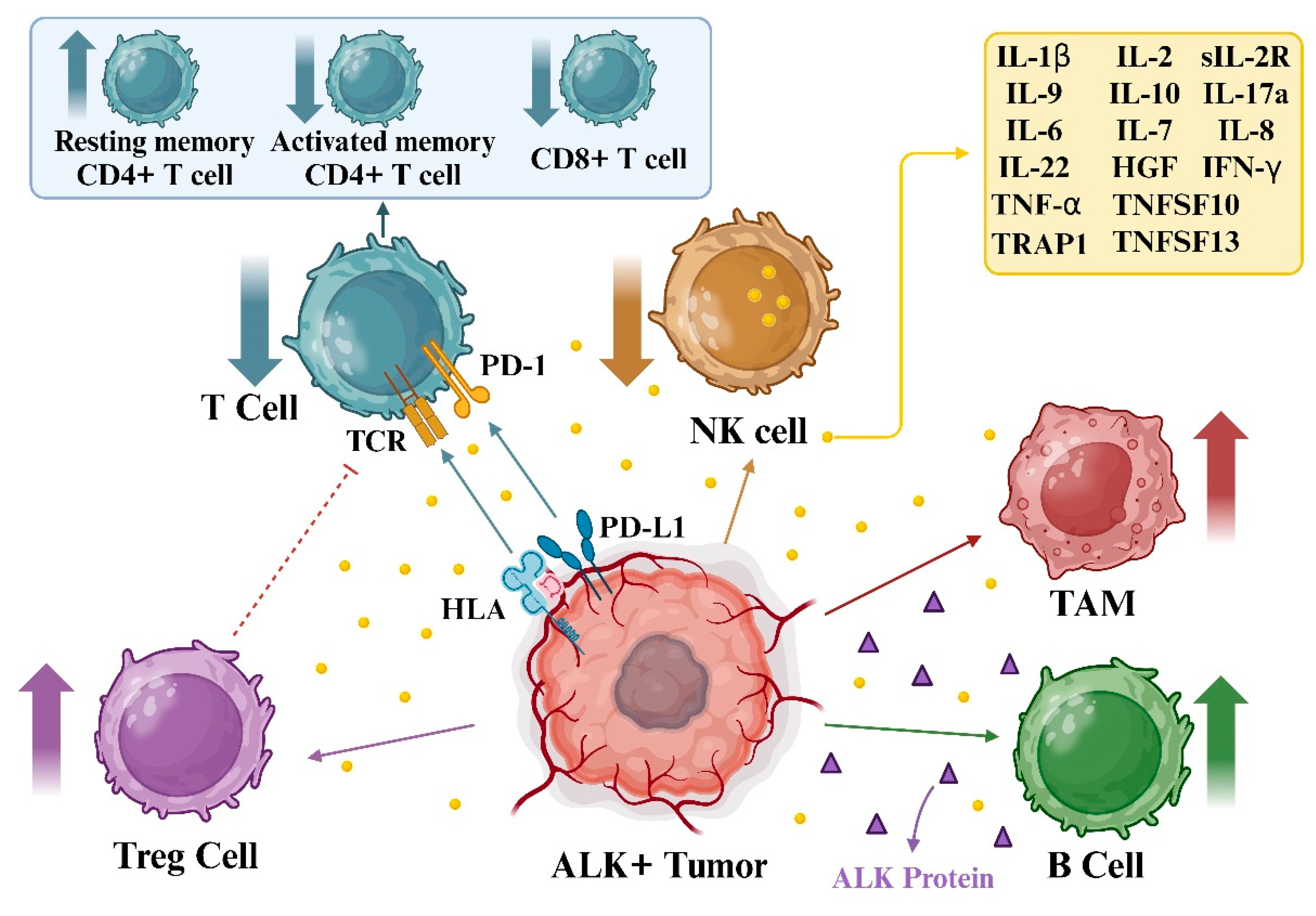
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



