
Submitted:
07 January 2024
Posted:
08 January 2024
Read the latest preprint version here
Abstract
Microfluidic systems offer precise control over physiological, biochemical, and mechanical stimuli in cell culture, allowing in vitro emulation of tissue or tumor microenvironments. This study aims to advance our understanding of tumor biology and contribute to personalized therapy development. Overcoming challenges in cell culture, including cell density and microfluidic device properties, is essential for this purpose. We designed a microfluidic system to facilitate the interaction of diverse cell lineages, incorporating human brain microvascular endothelial cells (HBEC5i), glioblastoma multiforme cells (U87MG), and astrocytes (ScienceCells 1800). Post-fabrication, we assessed the system's functionality and systematically evaluated cell inoculation conditions. Photographic acquisitions comprehensively captured cell culture and their interactions. The device successfully enabled assessment of cellular proliferation, invasion, and migration, supporting the growth of monolayers and tumor organoids. Importantly, each channel demonstrated independence in study processes while allowing cellular intercommunication, both physically and biochemically. In conclusion, the functional microfluidic device facilitated the study of cell migration, tumor invasion, and organoid growth, faithfully replicating biological features observed in a tumor microenvironment. This research contributes to advancing our understanding of tumor biology for the development of personalized therapies.
Keywords:
microfluidic system
; glioblastoma
; tumoral biology
; organoid culture
1. Introduction
Brain tumors (BTs) constitute approximately 2 to 4% of all malignant neoplasms [1]. They have been classified as an extremely heterogeneous disease with a wide range of various somatic genomic alterations, ranging from single nucleotide somatic alterations to structural chromosomal rearrangements, dictated and modulated by the biological environment [2]. Tumor cells coexist with non-mutated glial cells, immune cells, endothelial cells (ECs), as well as the extracellular matrix deriving in a highly complex 3D structure, cell heterogeneity and dynamic cell interaction [3,4,5].
A common characteristic in many neurological diseases such as Alzheimer's, depressive disorders, cognitive impairment, and central infectious diseases is the loss of the integrity of the blood-brain barrier, a structure formed by tight junctions between endothelial cells and communication with glial cells. When its integrity is compromised, it allows the entry of immune cells into the central nervous system (CNS) [6,7,8,9]. Conversely, the function of the blood-brain barrier hinders the entry of medications into the CNS and their effective functioning [9]. This will depend closely on the location and development of the tumor. It is considered that primary tumors of different cellular origins can exist in the glial differentiation axis [10], which acquire phenotypic and functional characteristics that resemble "normal" stem cells [11]. In the cancer stem cell (CSC) paradigm, various surface proteins have been identified providing an useful method for their identification, such as CD133+, CD15, CD49f+, CD36+, A2B5+, CD44+, L1CAM+, and the epidermal growth factor receptor (EGFR+). In addition to these surface markers, several transcription factors have been identified that play an important role in the CSC subpopulation, including: BMI1+, Olig2+, Oct3/4, SOX2+, NESTIN, MYC, and IDH1 (an inhibitory differentiation protein) [12,13,14].
The development of new tools for prognosis and effective therapies is based on a deep understanding of the molecular mechanisms involved in health and disease, including essential aspects such as the microenvironment [15,16]. There are various cell culture methods that can provide answers to some of the current disease-related questions. One of the important steps in the history of tissue cultures has been the transition from 2D cultures to three-dimensional (3D) culture techniques in behalf of 3D cultures have allowed for greater physiological relevance (Figure 1). However, 3D cell culture is method that does not allows the control or modulation of cell interaction, hence have not yet managed to fully replicate the complexity of multicellularity in tumor tissues, as well as the lack of interaction with vascularization process which is essential for cell tumor proliferation and migration, in fact the “index of vascularity” has major clinical implications [17]. Cancer is an example of complex pathophysiological mechanisms that are difficult to mimic in vitro, and it is even challenging to extrapolate the results from animal models to humans [18]. Faced with these limitations, the development of new technologies for culturing human cells, such as microfluidics cell culture systems, aims to mimic human physiological systems with respect to tissue complexity, biophysical and biochemical characteristics [18,19,20,21], with the potential to study formation, progression, and therapy response [20].
Furthermore, it should be noted that certain types of nervous system cancer exhibit specific regulation due to the unique characteristics of the blood-brain barrier, which represents a microenvironment where fluid-cell permeability and interaction are highly regulated. This concept broadly to glial cell tumors, for which there is currently no effective therapeutic strategy, and the chemotherapeutic drugs used show poor efficacy and a high recurrence of tumor invasion [22]. Therefore, there is a need for an experimental blood-brain barrier model that accurately represents the complexity of the tumor environment and its cellular interactions, allowing for the testing of new, more effective, and safer treatments as part of the path toward personalized medicine.
Despite cell culture in microfluidic systems provides a wide control of cell – cell interaction, up to now still faces various factors and challenges that can influence its development. In terms of physics, the design of channels in microfluidic devices, including their shape and size, affects cell behavior. Cells can experience different flow patterns and shear stress in straight or curved channels, and the properties of the device surfaces, such as roughness, chemistry, and surface charge, also have an impact.
In addition to device design, there are other hurdles to consider for optimizing successful cell culture. These include cell density, media viscosity, cell size and shape, speed of inoculation, and prevention of bacterial and fungal contamination. All these factors converge to obtain a cell culture that allows for proper cell-cell interaction, migration, and proliferation.
The cell inoculation process is one of the most important challenges. During this process, cells can be damaged due to the generated shear forces, affecting their viability and functionality. Additionally, cells could clump together and block the channels. Therefore, an efficient cell inoculation process must consider cell density to avoid clustering and channel blockages, as well as ensure proper adhesion for cell proliferation and migration in the microchannels of the device.
The vehicle used for cell inoculation is also relevant. A highly viscous liquid can hinder cell loading, while one that is too fluid may not keep the cells suspended during the loading process. The viscosity of the inoculation vehicle affects flow speed, where high speed can damage cells and reduce their ability to adhere, meanwhile low speed may not stimulate cell migration. Furthermore, microfluidic devices can be prone to bacterial or fungal contamination, hence sterile handling methods should be excerted. The manipulation and loading of cells into the devices can introduce cellular or chemical contaminants, requiring precautions to maintain proper culture conditions.
In summary, cell culture in microfluidic systems faces challenges related to devise design, cell inoculation, media viscosity, flow speed, and contamination, all of which must be carefully addressed to achieve successful cell culture.
2. Materials and Methods
The project was approved by the ethics, research, and biosafety committees of the Centro Medico Nacional 20 de Noviembre, belonging to the Institute of Social Security and Services for State Workers (ISSSTE), with approval identification number: 468.2020. Human U87mg cells (glioblastoma multiforme) and HBEC5i endothelial cells from the American Type Culture Collection (ATCC) were used. It was a prospective design for proof of concept, with each experiment and variable being performed in triplicate unless otherwise stated.
2.1. Design and Fabrication of Microfluidic System
The microfluidic system was designed in AutoCAD 2021 and consists of 3 interconnected main channels with microchannels measuring 6 µm wide x 30 µm high; as shown in Figure 2. It was fabricated using polydimethylsiloxane (PDMS) material, and the microchannel inlets and outlets have a diameter of 2 mm. Each microfluidic system was attached to a coverslip using corona discharge. The microfluidic systems were designed at CMN 20 de Noviembre – ISSSTE and manufactured at Laboratorio de Micro y Nanotecnología del Laboratorio Nacional de Soluciones Biomimeticas Diagnostico y Tratamiento (LaNSBioDyT), UNAM.
2.2. Characterization of the Microfluidic System
It was evaluated that within the microfluidic system, there was a proper communication through the micro connections between each of the channels. For this purpose, 70% ethanol with vegetable dye and with Texas red dextran (10WD-Thermofisher) was placed in one each of the channels of the microfluidic system in order to evaluate microfluidic communication (Figure 3). Subsequently, in another microfluidic system (at room temperature) supplementary Figure 1, a total of 1 x 104 cells (U87MG) • µl were inoculated, and photographs were acquired every 15 minutes over a total period of 5.25 hours to visualize the effect of microflow on the inoculated cells.
2.3. Immunophenotype Cells
The cell lines underwent expansion in cell culture plates according to the indication of each data sheet and were subsequently seeded in 24-well plates at a density of 1 x 104 cells •cm2. Following a 72-hour incubation period, they were fixed using 4% paraformaldehyde. In the epifluorescence setting, the process for immunostaining is outlined for acquiring (Figure 4).
2.4. Cell Proliferation Kinetics
For the cell proliferation assay, 1 × 104 cells•cm-2 were inoculated in culture plates in triplicate for 264 hours for HBEC5i, U87MG, and Astrocytes cells, without media replacement (Figure 5). The cell viability (%) was evaluated every 24 hours for a maximum of 11 days (264 hours) of culture using trypan blue exclusion test. The quantification of the specific growth rate (µ) and doubling time (Td) was calculated using the following equations:
Doubling Time (Td):
2.5. Protein Matrix Test for HBEC5i Endothelial Cells
Commercially pre-treated 24-well plates and Falcon 8-well culture slides were treated with different adherent cell culture proteins either for one hour at 37oC or overnight at 4oC. The proteins used to evaluate HBEC5i endothelial cell optimal cell culture and expansion were gelatin (0.1%), Matrigel (1:50), laminin (20 μg•ml-1), vitronectin (3.75 μg•ml-1), and absence of matrix. Representative cell culture with extracellular matrix is shown in Figure 6A.
2.6. Endothelial Vascularization Test
The feasibility of HBEC5i endothelial cells to form microvasculature was evaluated. Previous studies in our laboratory implemented human circulating vascular precursor cells in peripheral blood and found that by replacing the culture medium every 96 hours over a period of 30 days, allowed to obtain in vitro microvasculature processes (data not shown), hence We implemented this method for HBEC5i microvasculature induction. In addition, the inoculation of cells on a fibrin matrix obtained from blood plasma, either immersively or in a three-dimensional manner, has also been evaluated, resulting in vascularization processes as early as 96 hours of cell culture. In this context, we performed the same functionality tests on HBEC5i cells in different conditions (without matrix, gelatin-0.1%, Matrigel-1:50, Laminina-20μg • ml-1, vitronectina-3.75μg • ml-1), at different cells density (1 x 104, 2 x 104, 3 x 104, 4 x 104, 5 x 104, and 6 x 104 cells • cm-2). Representative cell culture with extracellular matrix is shown in Figure 6B.
2.7. Epifluorescence Microscopy
The cells were fixed using 4% paraformaldehyde. After fixation, three washes with PBS were performed. To detect intercellular proteins, 0.1% tween20 was used. Subsequently, the primary antibody was applied and incubated for 60 minutes at room temperature or overnight at 4°C. Next, 10% fetal bovine serum in PBS was used as a blocking agent, followed by three washes with PBS. DAPI staining was used for nuclear staining. Finally, immunopositive fluorescent cells were identified using an inverted epifluorescence microscope, Olympus IX71.
The cellular immunophenotype was determined using the antibodies listed in Table 1:
2.8. Cell Line Cultures in the Microfluidic System
The microfluidic culture experiments were conducted at passages 3-6 of each cell line. The cell lines were cultured and expanded in accordance with the technical specifications provided by ATCC and Science Cell respectively. For each experimental case, cultures were utilized at 80% confluency in T25 or T75 tissue culture flasks. For preliminary Optimization Test - The microfluidic system was initially evaluated by coating the glass surface (cover slips) with a protein matrix. Subsequently, modifications were made to the microfluidic system inputs to serve as reservoirs for culture medium, and finally, different cell densities were tested (see Supplementary Table 1 and Supplementary Figure 2). First, the microfluidic systems were placed individually in Petri dishes to prevent exposure to the environment. In this closed system, they were irradiated with ultraviolet light for 20 minutes in a vertical laminar flow hood. Subsequently, 1000 microliter micropipette tips were cut to a height of 0.5 cm to create cones at each inlet and outlet of the microfluidic system. This was done to create reservoirs of culture medium larger than the capacity of the main 1- or 2-mm inlet.
Different concentrations of HBEC5i and U87MG cells were tested both individually and in combination across multiple devices. The seeding included cell concentrations of 3 x 102, 5 x 102, 1 x 103, 2.5 x 103, 5 x 103, 7.5 x 103, 1 x 104, and 2 x 104 cells•µl-1, with medium changes every 48 hours (Figure 7, Figure 8, Figure 9 and Figure 10 and Supplementary Figure 3).
Similarly, the cellular detachment capability was assessed using trypsin or a mechanical air injection process (200 microliters) with 1000-microliter pipettes. This process involved cell densities of 2.5 x 103, 5 x 103, 7.5 x 103, and 1 x 104 cells•µl-1 (Supplementary Figure 4).
The influence on the maintenance and proliferation of HBEC5i cells within the microfluidic system was evaluated. Initially, a starting density of 1 x 104 cells•µl-1 with 50 µl of medium daily renewed, was maintained for 24-120 hours. In a subsequent experiment, the medium volume was increased to 100 µl with daily changes during the cultivation period of 144-172 hours (Supplementary Figure 5).
The behavior of U87MG cell culture was assessed at a density of 2 x 104 cells•µl-1 with daily medium changes (100 microliters) in each channel. After 260 hours of cultivation, the expression of GFAP was evaluated (Supplementary Figure 6).
The use of a matrix for cell encapsulation in a single channel, such as human fibrin from blood plasma (obtained with vacutainer tubes-citrate), was implemented as shown in Supplementary Figure 7. In this process, a cellular density of 2.5 x 104 cells•µl-1 in 6 microliters of fibrin was utilized. The coagulation cascade was activated with reactive calcium chloride (Supplementary Figure 7). Upon inoculating the same quantity of HBEC5i and U87MG cells into their respective channels in another system, a parallel alignment of endothelial cells migrating to channel 2 was observed, while U87MG cells exhibited spheroid growth (Supplementary Figure 8)
2.9. Microenvironment of Hematoencefalic Barrier
Based on a blood-brain barrier structure, it is constituted in part by the interaction of endothelial cells interacting with astrocytes, and in other sections by the tight junctions of endothelial cells. In studies as the reported by Deosarcar et al., (2018) [23], the importance of cellular interactions for the maturation of microvasculature in the microfluidic device has been demonstrated. Likewise, we agree with the relevance of this cellular interaction process, accordingly We carried out a systematic scaling of the functionalization of the microfluidic system for the interaction with a preformed blood-brain barrier and subsequently the cellular interaction with glioblastoma multiforme (U87MG) cells.
There were two different methodological approaches. In the first approach, the extreme cell lines (channel 1 - U87MG and channel 3 - HBEC5i) were cultured at a cell density of 1 x 104 cells•µl-1. HBEC5i cells were cultured with Texas Red Dextran for the first 96 hours, followed by the inoculation of U87MG cells with maintenance during the period of 96-432 hours. Finally, the inoculation of Science-cell 1800 astrocytes was performed in the last 48 hours (432-480 hours) (Supplementary Figure 9).
In contrast, the second approach involved first scaling the astrocyte culture in channel 2, followed by endothelial cells in channel 3, and finally, U87MG cells in channel 1. In this experiment, we initially inoculated astrocytes in the middle channel (channel 2), followed by endothelial cells (labeled with Texas Red Dextran), and finally, U87MG cells labeled with MitoTracker Green Green to ease cell identification during cell culture. The culture medium collected every 24 hours in this experiment was stored in freezing conditions (-80 degrees Celsius) until cytokine measurements were conducted.
2.10. Confocal Microscopy
Cells in the microfluidic systems were fixed with 4% paraformaldehyde at room temperature and washed with PBS. Antibodies and DAPI were then overnight incubated. Subsequently, the images were acquired at the Laboratorio Nacional de Microscopía Avanzada, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, using a Nikon A1 inverted confocal microscope; and at the Centro de Desarrollo de Productos Bióticos (CEPROBI) at Instituto Politécnico Nacional. The specimens were observed using a CLSM (Carl Zeiss, LSM800, Germany) with a motorized stage (X, Y, Z) and coupled to an AxioCam HD color camera (Carl Zeiss, Model 305, Germany). The ZEN (Zeiss efficient navigation) software version 2.6 Blue edition was employed.
2.11. Cytokine Expression Assay
The supernatants from the media were collected from the three replicate experiments every 24 hours. Cytokine expression was assessed using a multiplex ELISA assay (arigoplex) kit for IL2, IL6, IL10, and gamma interferon, following the kit's technical specifications.
3. Results
3.1. The Microfluidic System: Device Design
In order to resemble the dynamic haemato-encephalic barrier, a parallel microfluidic system was chosen as the basic structure supporting cells from different lineages, including endothelial, astrocytes, and tumor glioblastoma cells (Figure 2). Three main chambers were designed with the following characteristics; channel 1 (for U87MG culture): 600 µm wide x 6780 µm length x 100 µm depth; Central channel or channel 2 ( for Astrocyte culture): 200 µm wide x 16900 µm length x 100 µm depth, and finally channel 3 (for Vascular cell formation): 400 µm wide x 6750 µm length x 100 µm depth; meanwhile chamber interconnecting channels were width 6 µm, height 30 µm and length 50 µm (Figure 2A and 2B).
3.2. The Microfluidic System Permeability Test
The microfluidic system allowed for a proper diffusion of vegetable dye within each of the microchannels and their connections, as illustrated by the diffusion process from the outer channel (Figure 3) in a period of 2 minutes for all channels.
3.3. Characterization of Cellular Population
Cell populations considered to integrate into the Microfluidic System Device were those resembling the microenvironment interactions of tumor cells and the hemato-encephalic barrier. Therefore, tumor cells were constituted by glioblastoma tumoral cells (U87MG) while hemato-encephalic barrier was constituted by the interaction of endothelial cells (HBEC5i) and astrocytes (ScienceCell1800) cell lines.
In order to characterize the morphology and cell function lines, in vitro assays of immunophenotype (Figure 4) and proliferation were performed. We observed that endothelial cells have an average growth rate of 0.20 ± 0.0035 cells•h-1and a doubling time of 37 ± 6.034 h. U87 MG cells exhibited an average growth rate of 0.018 ± 0.00070 cells•h-1 and a doubling time of 35.2 ± 1.98 h. As for astrocytes, the growth rate was 0.010 ± 0.00040, with a doubling time of 67.84 ± 2.3 h (Figure 5). Maintenance and vasculogenic (Figure 6) ability were performed, out from the microfluidic device. In our experience, once human derived endothelial cells reach passage 9th they lose the ability to duplicate and to form microvessels (data do not show). In comparison, HBEC5i cells, which are immortalized, and u87MG cells, which are tumoral, exhibit distinct characteristics, surpassing more than 20 passages without any difficulty for cell expansion or for microvessel formation in the case of endothelial cells (HBEC5i). It is important to note that during optimization assays, matrix-covered surface resulted in a higher rate of proliferation and vasculogenic ability, while there was no difference related to the type of the matrix-cover use.
3.4. Optimization of Inoculation Cell Density in the Microfluidic System
This model allowed studying different cellular processes such as growth, adaptability, migration, and cell interactions, while cells are arranged in layers within the device (Figure 7). To achieve these results, a process of standardization through trial and errors was necessary in order to indicate the best path forward for the cell culture system. These approaches are outlined in Supplementary Table 1, with illustrative images provided in Supplementary Figure 2. The optimal working options include placing a culture medium reservoir with a capacity equal to or greater than 100 microliters (Supplementary Image 5). For studies involving cell migration, it is advisable to use a matrix such as gelatin with a minimal coating time of one hour in all channels, without allowing the device to dry. Additionally, cell detachment tests were conducted (Supplementary Figure 4), revealing that mechanical cell detachment is the most viable option.
The U87MG culture process was optimized at a density of 1 x 104 cells•µl-1 for uniform distribution and population growth within the microfluidic system (Figure 7) and 2 x 104 cells•µl-1 for three-dimensional growth resembling tumor organoids (Figure 8 and Supplementary 6). Meanwhile, the co-culture cell interaction between HBEC5i and U87MG was optimized at a density of 1 x 104 cells•µl-1 respectively (Figure 9). It is worth noting that the approach of culturing endothelial cells on a matrix of human plasma fibrin was evaluated at lower cell densities (Supplementary Figure 7). It was found that it is possible to encapsulate the cells within the channel immediately after the activation of the coagulation cascade. Within 48-72 hours, they undergo a migration process to other channels. However, it was possible to identify that cells cultured with a fibrin matrix tend to exhibit a cellular alignment (polarization) process from the first 24 hours (Supplementary Figure 8). To recreate a brain tumor microenvironment in our microfluidic device, we focused on ensuring that different cell strains did not mix but proliferated individually in their respective channels, as illustrated in Supplementary Figure 9. We initiated the cultivation with HBEC5i cells, followed by U87MG cells, and eventually introduced astrocyte cells to facilitate communication between the two aforementioned cell lines. Finally, the optimal strategy involved the staggered and organized seeding of cells, starting with astrocytes as the initial instance, followed by HBEC5i cells, and subsequently, U87MG cells at density of 1 x 10 4 cells•µl-1 (Figure 10), with this arrangement, we were able to mimic a blood brain barrier in the channel where HBEC5i cells were seeded, meanwhile we also observed cell – cell interaction between glioblastoma cells (U87MG) and brain endothelial cells (HEBC5i) as shown in Figure 9 and Figure 10 confocal images. The interaction of glioblastoma and endothelial cells within this microfluidic device mimics tumor microenviroment as shown by the 3-fold increase of IL6 due to organized and controlled cell interaction after 360 to 384 h of culture (Figure 10). All these studies were performed in the absence of a coating matrix.
4. Discussion
The present study aimed to characterize and optimize conditions of cell culture and physical- chemical properties of a microfluidic device, suitable to carry out experiments that increase the understanding of tumor biology in the central nervous system and potential development of personalized therapies. In the process, we found unexpected cellular behaviors that increase similarity of the device with processes occurring during tumor invasion in the hemato-encephalic barrier.
The cells implemented in this study have been adequate, maintaining the functional capacities of each corresponding lineage. It was evidenced that U87MG cells show a higher cellular duplication rate compared to the other cells, followed by endothelial cells, and finally, astrocytes. Therefore, these cellular lineages exhibit behavior corresponding to their nature. It is worth mentioning that astrocytes constitute a primary cell culture line, whereas HBEC5i has been immortalized. However, despite this distinction, HBEC5i displays functional characteristics remarkably like physiological endothelial cells, as observed in the expression of endothelial cell proteins and capacity for microvascular formation. This renders them a valuable cellular tool for in vitro studies (Figure 5B). Additionally, other research groups have used these cells as a cellular model for brain interactions proving to be more suitable than the implementation of HUVEC cells or any other cell line cellular endothelial. On the other hand, U87MG cells showed the expression of Oct3/4 and Sox2, characteristic of stem cells, as well as the expression of glial fibrillary acidic protein (GFAP).
The microfluidic device design allowed the culture and interaction of different cell lines, as seen in the previous results, demonstrating proper diffusion of the dye through the channels and their micro connection, with a diffusion time of 10 seconds between the first and second channel and 60 seconds to permeate to the 3rd channel of the microfluidic device.
Several reports have shown that cellular adhesion to microfluidic channels can be greatly enhanced by implementing different proteins, such as fibrin, collagen, gelatin, and even poly-L-lysine [21,24,25,26]. This was indeed reflected in cell culture plates (Figure 6A). However, we observed that when implementing this protein handling within the microfluidic system, it does not have a significant direct impact on cell adhesion. In our experiments, when using ECM the channels were at risk of blockage or retaining material residues, rendering the channel or system useless (supplementary 2). On the other hand, when the surface tension of the culture medium inoculated into each of the channels is disrupted, it promotes communication between them, if there are no air bubbles within the system. We observed that by inoculating only one cell line into the device and filling all the channels with a culture medium, the cells started to migrate from one channel to another. Regarding experiments focused on the success rate of cellular adhesion, manipulating the variables of cell concentration versus time, it was identified that by integrating only one cell line at a time, whether U87MG or HBEC5i, into a single channel and filling all other channels with culture medium allows the observation of a cell migration processes. This cell migration process was primarily observed with the U87MG cell line, as well as for cell HBEC5i. Once again, the nature of each cell line was highlighted by demonstrating the migration capacity of the U87MG cell line to colonize all three channels of the microfluidic system, with percentages varying based on cell density. Interestingly, at higher cell densities, a higher migration rate was observed for all cell lines. In contrast, in the HBEC5i culture, cell proliferation was only observed in the vascular channel (channel 3), and a small percentage, positively correlated with cell density, migrated to the intermediate channel (channel 2). One of the remarkable benefits of working with these types of devices is the ability to monitor the study 24/7, depending largely on the conditions under investigation. In terms of cell culture, we have observed a positively correlated impact between the inoculated density and development within the device, whether through migration or proliferation.
The cell detachment experiment conclusively demonstrates that each channel can be treated differently while maintaining biochemical communication and cellular contact. When trypsin was introduced into a single channel, it only had an effect on the channel inoculated with very low cell densities. In cases where more complex cellular conglomerates were present, the enzyme did not have an evident effect. Therefore, a mechanical approach is recommended for detaching and rescuing cells, whether from a single channel or the entire device (see Supplementary Figure 4).
We increased the cell density of U87MG, as it is particularly resistant to pH changes and cellular stress. We observed that the cells began to grow in three-dimensional clusters, known in other reports as organoids, and due to their nature, they do not exhibit contact inhibition, resembling a tumor microenvironment; to more detailed, the study was conducted with U87MG cells at a density of 2 x 10 4 cells•µl-1, reflecting that at higher cell densities, cellular conglomerates can be obtained within the inoculation channel. Over time, these conglomerates undergo three-dimensional development, leading to the formation of organoids mimicking microtumor shape. The migration process of U87MG in the microfluidic system was successfully visualized through confocal microscopy. It is crucial to highlight this process since, considering the measurement of cellular nuclei, they can range from 12 to 16 micrometers in diameter. However, as observed, cells have modified their cellular structure to migrate from one channel to another, despite the initial restriction that the connecting microchannels have a diameter of 6 micrometers. This was visualized in experiments of cell densities in the microfluidic system and through immunofluorescence.
Devising a scenario to hinder cellular migration from the outset, we implemented the encapsulation of HBEC5i cells within a hydrogel containing sufficient nutrients to enhance endothelial cell capabilities. This hydrogel, previously demonstrated to stimulate vascular formation by endothelial cells, was based on human fibrin. In this study, it was observed that endothelial cells exhibited proper maintenance and even demonstrated cellular alignment or polarization (Supplementary Figure 7 and Figure 8). However, migration processes to other channels were still evident.
To prevent this large-scale cellular migration, two approaches were employed to mimic a cerebral tumor microenvironment. The first involved the initial structuring of the vascular channel, followed by the U87MG channel, and finally, the introduction of astrocyte cells. It is noteworthy that the culture medium was only added at the time of cell inoculation, thus allowing isolation of cell growth in a single channel. Intercommunication was achieved by completing the cell culture with astrocytes. This approach resulted in a shorter time for cellular migration or invasion compared to the total cell culture process, proving to be successful with a 480-hour culture duration (see Supplementary Figure 9).
The second cellular cultivation approach aimed to replicate a non-pathological blood-brain barrier initially, leading to a different cellular scaling process, revealed that the majority of cell lineages saturated their respective channels within 72 hours of cultivation. Astrocytes were the first to be present in the system, followed by the inoculation of HBEC5i cells (with Texas Red-dextran), indicating that these endothelial cells began to interact in the astrocyte channel. After 240 hours of cultivation, U87MG cells were inoculated with MitoTracker, and the two initial channels showed red fluorescence (Texas Red-dextran). After a total of 384 hours of cultivation, with the assistance of confocal microscopy, a cellular invasion process from the U87MG channel (green) to the Astrocyte and HBEC5i channel was observed.
Tumor cell proliferation and invasion as well as the interaction with endothelial cells correlates with the 3-fold increase expression of IL6 and IL10, showing a slight fluctuation for interferon-gamma. No significant change was observed for IL2, interestingly the increase in IL6 expression occurred with the interaction with U87MG from 216 hours. Fluctuations in IL10 and interferon-gamma are processes described in tumor microenvironments, where interleukin 6 facilitates the disruption of tight junctions in endothelial cells, allowing mobilization in the vasculature. This has been described as biochemical signaling for cells of the immune system. Hence, the achieved tumor-brain blood barrier microenvironment was generated by the interaction of the three cell lines. In light of the above, the system exhibited stimulation through cellular interaction of the implemented cell lines, despite we still can enhance cell interaction in order to more effectively represent a blood-brain barrier. One potential improvement is connecting the vascular system to a constant pressure medium culture system, as Arvanitis et al. 2020 [27] described the importance of the blood-brain barrier and its influence on the central nervous system homeostasis. It is known that the application of shear stress stimulates mechanoreceptors on endothelial cells, influencing various cellular functions, including cytoskeletal remodeling, gene expression, cell viability, and calcium homeostasis, thereby controlling myogenic tone [28]. This also affects the formation of tight junctions, leading to selective permeability and, consequently, improving barrier function artificially [16,29,30].
5. Conclusions
The microfluidic proposed deviced fulfills and is suitable for providing interactions between 2-3 cell lines, achieving tumor-brain blood barrier microenvironment generated by the interaction of the three cell lines. It demonstrated the independence of each channel, and the maintenance of cell culture for a maximum of 480 hours (figure supplementary 9). Culturing in a microfluidic system presents many challenges, even greater than those required in traditional cultures. To address these issues, it is important to use appropriate techniques for cell loading, avoid high flow velocities, use suitable surfaces for the microfluidic device, and implement adequate measures to prevent contamination. Additionally, optimization of experimental parameters and the use of proper controls can reduce experimental variability. In summary, despite the potential challenges, microfluidic systems continue to be a valuable tool for cell manipulation and analysis in many fields of research.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Cell inoculation in the microfluidic system. Over a period of 5.25 hours, cells are gradually transported to the ends of the microfluidic system. Figure S2. Strategies employed for proper cellular adhesion within the microfluidic system. A) Surface view of the microfluidic system with and without steps (left and right sides, respectively), B) Side view of the devices on the right (with steps) and left (without steps), C) Adaptation of medical-grade silicone tubing at each entry and exit of the device, D) Longer adapted tubes using a clamping system, E) Adaptation of the microfluidic system with thousand-microliter pipette tips as a reservoir for culture medium, F) Adaptation of the microfluidic system with two-hundred-microliter pipette tips as a reservoir for culture medium, and G) Representative results showing poor cell adhesion, bubble generation, or, as shown in the bottom right, extracellular matrix blockage. Figure S3. Cell culture maintenance within the microfluid system, the upper image shows the different densities of U87MG inoculated in the microfluid system and its maintenance over a period of 144h, the lower image shows the different densities of HBEC5i inoculated in the micro fluid system and its maintenance in a period of 144 hours. Figure S4. Cell detachment. Cell detachment was evaluated at various cell densities using the trypsinization process conducted over a 5-20 minute standard incubation period. At the bottom, mechanical detachment via air injection is demonstrated. Figure S5. Influence of cell culture medium on maintenance and proliferation in the devices. Two quantities of culture medium were evaluated with constant changes every day. From 24-120 hours, 50 microliters of culture medium were used, while from 144-172 hours, the volume was increased to 100 microliters." Figure S6. Glial Fibrillary Acidic Protein (GFAP) expression in U87MG microfluidic systems. Following the seeding of U87MG cells in the microfluidic system, the expression of GFAP was assessed within the device (green), with cell nuclei stained in blue (DAPI). Figure S7. Cellular encapsulation of HBEC5i with human fibrin. The cellular behavior was assessed within microfluidic devices utilizing a fibrin matrix at cellular density of2.5x103 cells•µl-1. Figure S8. Influence of human fibrin on the cultivation of HBEC5i in a microfluidic device. The growth of endothelial cells is observed to be influenced by the fibrin matrix. In the cellular growth within the U87 MG channel, spherical cell growth is evident, whereas in channel 2 originally populated by astrocytes, the migrated HBEC5i cells exhibit cellular polarization (graph on the right). Figure S9. Brain Tumor Microenvironment. The temporal scaling of cell culture is observed, starting with the inoculation of HBEC5i cells (labeled with Texas Red Dextran). Subsequently, cells from the opposite end (U87MG) were introduced, and finally, astrocyte cells were inoculated. F-actin expression is shown in green (Phalloidin), and cell nuclei are stained in blue (DAPI).
Author Contributions
DS-C conducted the experiments, analyzed, and interpreted the data. • AEC-R, MCAH, and AG-A, along with DA-B, provided administrative support. • JAS-C guided the manuscript construction. • VP-K carried out confocal microscope acquisition. • CGT-L, SG, TGM-A and ME-T collaborated with the graphs, tables and design of the paper • PM-T. Designed and supervised the experiments, managed funding acquisition, and critically reviewed the manuscript. • All authors read and approved the final manuscript.
Funding
This study was funded through the "Investigación Científica y Tecnológica" (E015-2022) of the Institute of Security and Social Services for State Workers (ISSSTE). This funding enabled the acquisition of all necessary supplies for the project, particularly the manufacturing of the microfluidic system, cells, and antibodies. Additionally, a scholarship was granted to student Daniel Santillán-Cortez under grant number #761521 by the Consejo Nacional de Ciencia y Tecnología (CONACYT).
Institutional Review Board Statement
The project was approved by the ethics, research, and biosafety committees of the National Medical Center “20 de Noviembre”, belonging to the Institute of Social Security and Services for State Workers (ISSSTE), with approval identification number: 468.2020. Consent to participate do not apply.
Informed Consent Statement
“Not applicable.”.
Acknowledgments
Daniel Santillán-Cortez is a doctoral student in the Programa de Doctorado en Ciencias Biomédicas at the Universidad Nacional Autónoma de México (UNAM) and has been awarded fellowship #761521 from the Consejo Nacional de Ciencia y Tecnología (CONACYT). We acknowledge the support from the Laboratorio Nacional de Microscopía Avanzada at Centro Médico Nacional Siglo XXI, Centro de Desarrollo de Productos Bióticos (CEPROBI) at Instituto Politécnico Nacional for confocal image acqusition. The authors express gratitude to the Laboratorio de Micro y Nanotecnología del LaNSBioDyT, Técnico Benito Juárez García for the manufacturing services provided, and finally to the Laboratorio de Medicina Regenerativa e Ingeniería de Tejidos at the Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado (ISSSTE) is equally acknowledged. PMT acknowledges CONACYT financial support for the establishment of the Regenerative Medicine Laboratory through the National Scientific Infrastructure Call with approval No. 188458.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2017, 21, 2683–710. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Andersen, M.H.; Schrama, D.; thor Straten, P. Immune-suppressive properties of the tumor microenvironment. Cancer immunology, immunotherapy 2013, 62, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Hacking, R.; Hunt, K. Cerebral abscess: A review of its pathophysiology, diagnosis and management. British Journal of Neuroscience Nursing 2007, 3, 400–403. [Google Scholar] [CrossRef]
- Lecuyer, M.A.; Kebir, H.; Prat, A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2016, 1862, 472–482. [Google Scholar] [CrossRef]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Svendsen, C.N. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell stem cell 2019, 24, 995–1005. [Google Scholar] [CrossRef]
- Wang, X.; Hou, Y.; Ai, X.; Sun, J.; Xu, B.; Meng, X.; Zhang, S. Potential applications of microfluidics-based blood-brain barrier (BBB)-on-chips for in vitro drug development. Biomedicine & Pharmacotherapy 2020, 132, 110822. [Google Scholar]
- Gronseth, E.; Wang, L.; Harder, D.R.; Ramchandran, R. The Role of Astrocytes in Tumor Growth and Progression. Astrocyte-Physiology and Pathology 2018. [Google Scholar]
- Lee, G.; Hall, I.I.I.; RR; Ahmed, A.U. Cancer stem cells: cellular plasticity, niche, and its clinical relevance. Journal of stem cell research & therapy 2016, 6. Ahmed, A.U. Cancer stem cells: cellular plasticity, niche.
- Abou-Antoun, T.J.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain cancer stem cells in adults and children: cell biology and therapeutic implications. Neurotherapeutics 2017, 14, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.H.V.; Middleton, K.; You, L.; Sun, Y. A review of microfluidic approaches for investigating cancer extravasation during metastasis. Microsystems & Nanoengineering 2018, 4, 17104. [Google Scholar]
- Ruiz-Garcia, H.; Alvarado-Estrada, K.; Schiapparelli, P.; Quinones-Hinojosa, A.; Trifiletti, D.M. Engineering three- dimensional tumor models to study glioma cancer stem cells and tumor microenvironment. Frontiers in Cellular Neuroscience 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Yissachar, N.; Zhou, Y.; Ung, L.; Lai, N.Y.; Mohan, J.F.; Ehrlicher, A.; Benoist, C. An intestinal organ culture system uncovers a role for the nervous system in microbe-immune crosstalk. Cell 2017, 168, 1135–1148. [Google Scholar] [CrossRef]
- Basma, E.; KS, B.R. A dynamic perfusion based blood-brain barrier model for cytotoxicity testing and drug permeation. Scientific Reports (Nature Publisher Group) 2020, 10. [Google Scholar]
- van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: from tools to tissue models. Current opinion in biotechnology 2015, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Plummer, S.; Wallace, S.; Ball, G.; Lloyd, R.; Schiapparelli, P.; Quiñones-Hinojosa, A.; Pamies, D. A Human iPSC-derived 3D platform using primary brain cancer cells to study drug development and personalized medicine. Scientific reports 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.F.; Trubelja, A.; Shen, A.Q.; Bao, G. Tumour-on-a-chip: microfluidic models of tumour morphology, growth and microenvironment. Journal of the Royal Society Interface 2017, 14, 20170137. [Google Scholar] [CrossRef]
- Meng, F.; Meyer, C.M.; Joung, D.; Vallera, D.A.; McAlpine, M.C.; Panoskaltsis-Mortari, A. 3D Bioprinted In Vitro Metastatic Models via Reconstruction of Tumor Microenvironments. Advanced Materials 2019, 1806899. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Y.; Shen, Y. Microfluidics for cancer nanomedicine: from fabrication to evaluation. Small 2018, 14, 1800360. [Google Scholar] [CrossRef]
- Logun, M.; Zhao, W.; Mao, L.; Karumbaiah, L. Microfluidics in malignant glioma research and precision medicine. Advanced biosystems 2018, 2, 170022. [Google Scholar] [CrossRef]
- Deosarkar, S.P.; Prabhakarpandian, B.; Wang, B.; Sheffield, J.B.; Krynska, B.; Kiani, M.F. A novel dynamic neonatal blood-brain barrier on a chip. PloS one 2015, 10, e0142725. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nature Reviews Cancer. 2019. [CrossRef]
- Kamei, K.I.; Ohashi, M.; Gschweng, E.; Ho, Q.; Suh, J.; Tang, J.; Tseng, H.R. Microfluidic image cytometry for quantitative single-cell profiling of human pluripotent stem cells in chemically defined conditions. Lab on a Chip 2010, 10, 1113–1119. [Google Scholar] [CrossRef]
- Lee, H.; Kim, N.; Rheem, H.B.; Kim, B.J.; Park, J.H.; Choi, I.S. A Decade of Advances in Single-Cell Nanocoating for Mammalian Cells. Advanced Healthcare Materials 2021, 10, 2100347. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nature Reviews Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Tovar-Lopez, F.; Thurgood, P.; Gilliam, C.; Nguyen, N.; Pirogova, E.; Khoshmanesh, K.; Baratchi, S. A microfluidic system for studying the effects of disturbed flow on endothelial cells. Frontiers in bioengineering and biotechnology 2019, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood–brain barrier. Lab on a Chip 2017, 17, 448–459. [Google Scholar] [CrossRef] [PubMed]
- May, S.; Evans, S.; Parry, L. Organoids, organs-on-chips and other systems, and microbiota. Emerging Topics in Life Sciences 2017, 1. [Google Scholar]
Figure 1.
Complexity of cell culture models to study brain tumor. The figure represents increasing complexity of cellular models used to study brain tumors. The complexity is related to the ability to mimic real biological microenviroment as long as 3D architecture and cell – cell and micro vascularity interaction.
Figure 1.
Complexity of cell culture models to study brain tumor. The figure represents increasing complexity of cellular models used to study brain tumors. The complexity is related to the ability to mimic real biological microenviroment as long as 3D architecture and cell – cell and micro vascularity interaction.
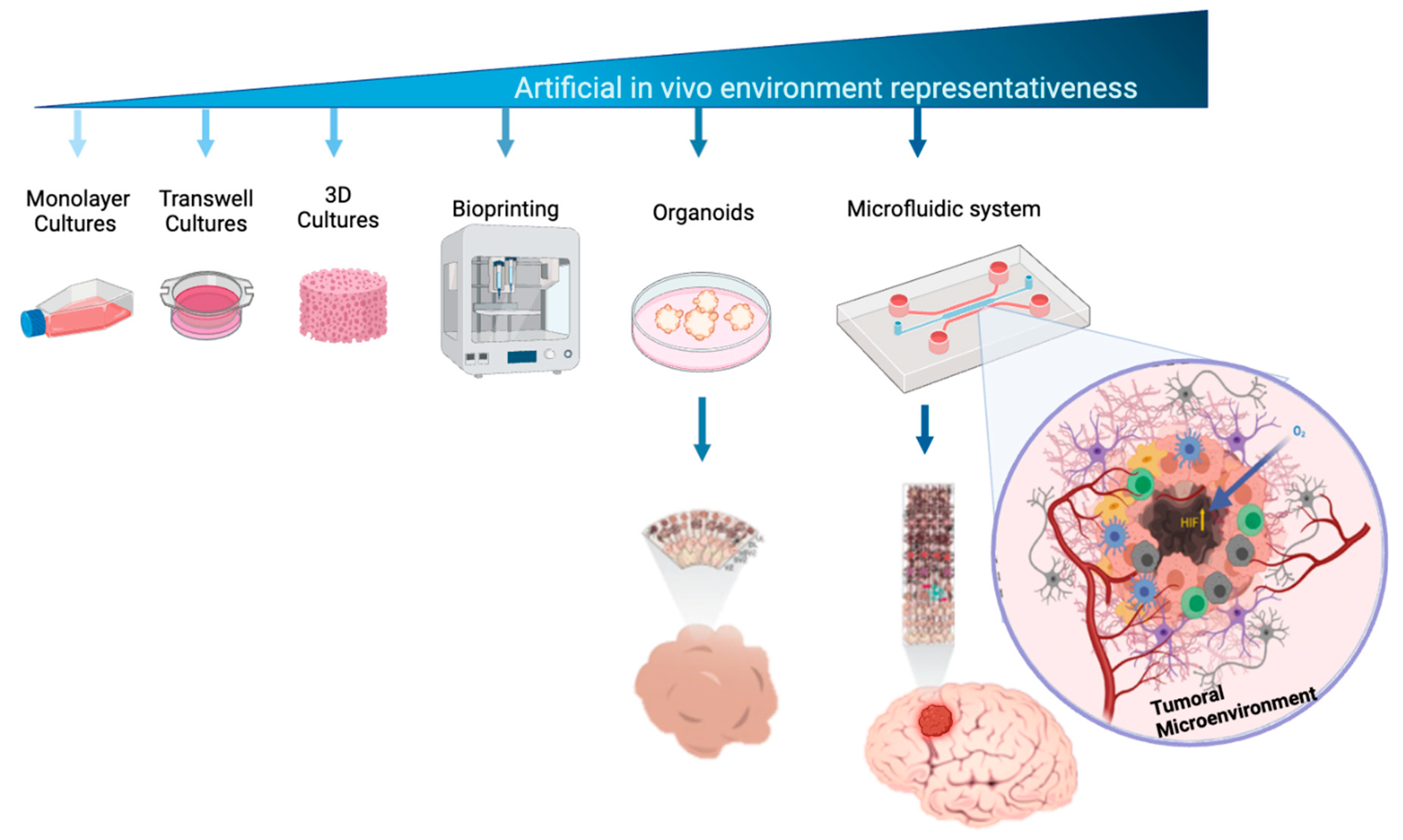
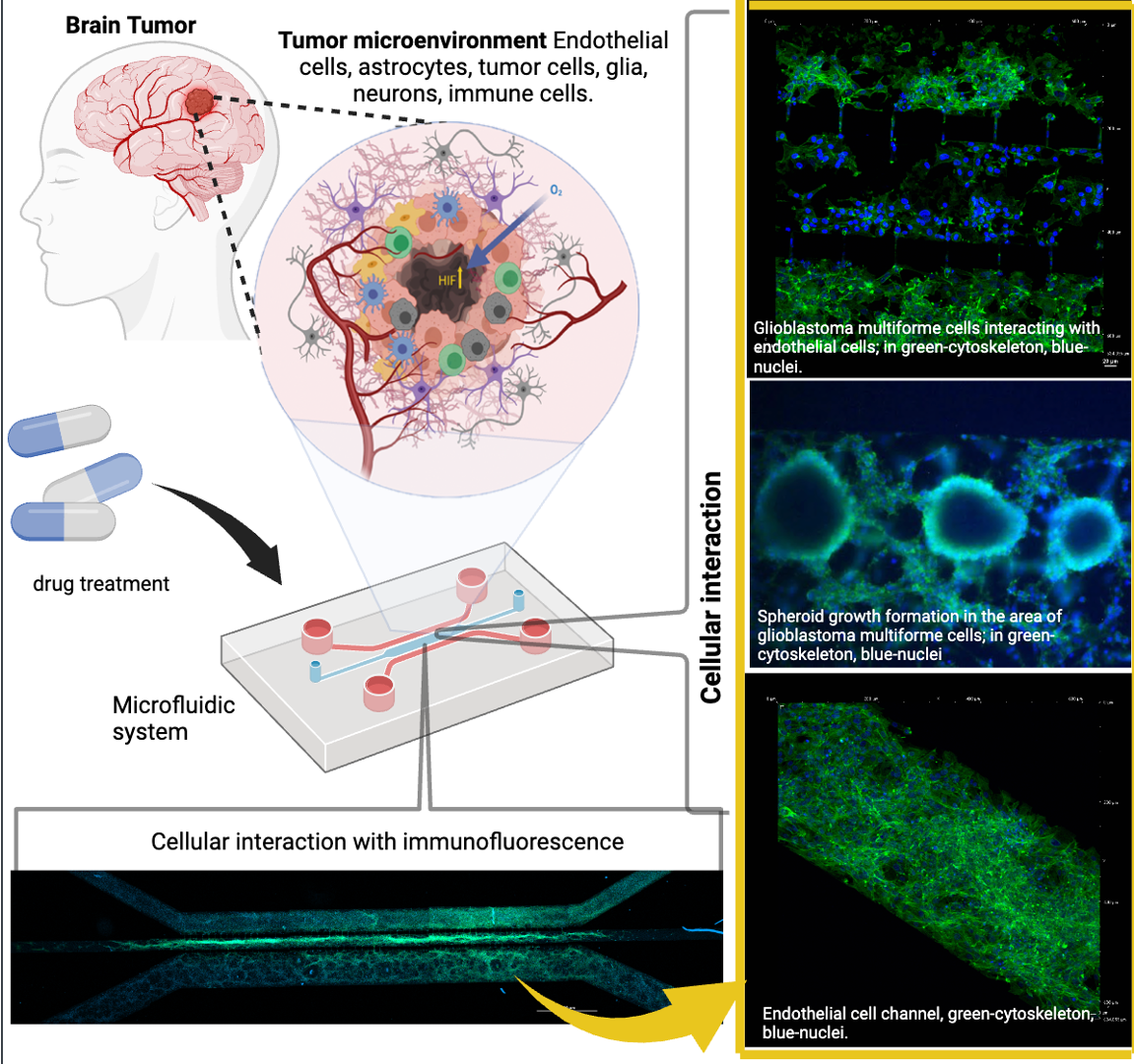
Figure 2.
Device Design of Microfluidic System. The figure shows different characteristics of the microfluidic system: A) Top view of the microfluidic system design; B) Tilted top-side view; C) Side view of the microfluidic system; D) comparative size of device and E) lateral view of device.
Figure 2.
Device Design of Microfluidic System. The figure shows different characteristics of the microfluidic system: A) Top view of the microfluidic system design; B) Tilted top-side view; C) Side view of the microfluidic system; D) comparative size of device and E) lateral view of device.

Figure 3.
Micro connection function test. Progressive permeability in the device was demonstrated by permeation of either: blue vegetal stain (upper panel) and Texas red stain (10WD) (lower panel). Time is also indicated for comparison.
Figure 3.
Micro connection function test. Progressive permeability in the device was demonstrated by permeation of either: blue vegetal stain (upper panel) and Texas red stain (10WD) (lower panel). Time is also indicated for comparison.
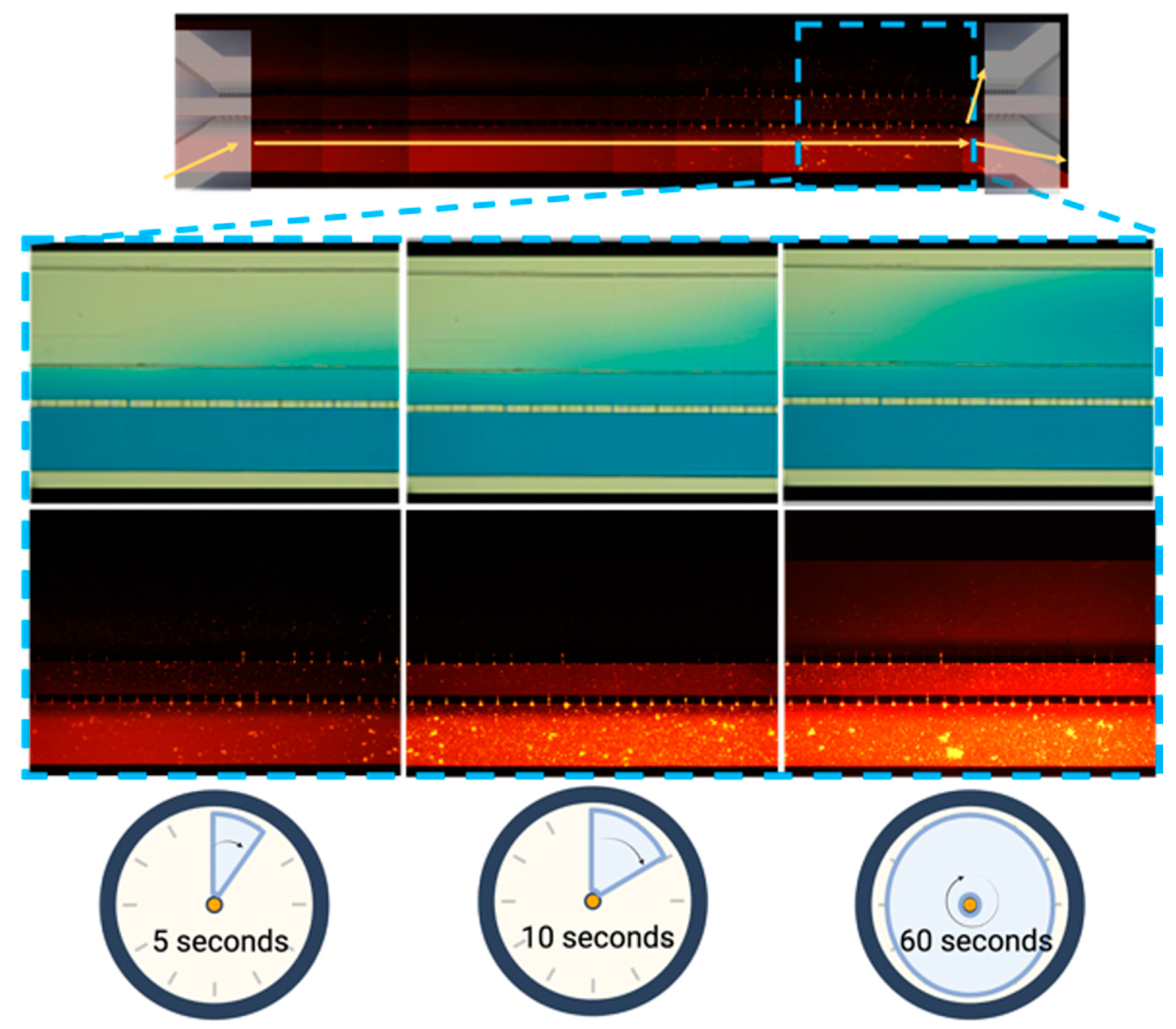
Figure 4.
Immunophenotype characteristics. The figure shows immunophenotype characterization of HBEC5i (endothelial cells) and U87MG (glioblastoma tumoral cells) cell lines in culture plate; as indicated by images of bright field, immunofluorescence, DAPI and merge. Percentage of microscope field positive for immunostaining is also showed. .
Figure 4.
Immunophenotype characteristics. The figure shows immunophenotype characterization of HBEC5i (endothelial cells) and U87MG (glioblastoma tumoral cells) cell lines in culture plate; as indicated by images of bright field, immunofluorescence, DAPI and merge. Percentage of microscope field positive for immunostaining is also showed. .
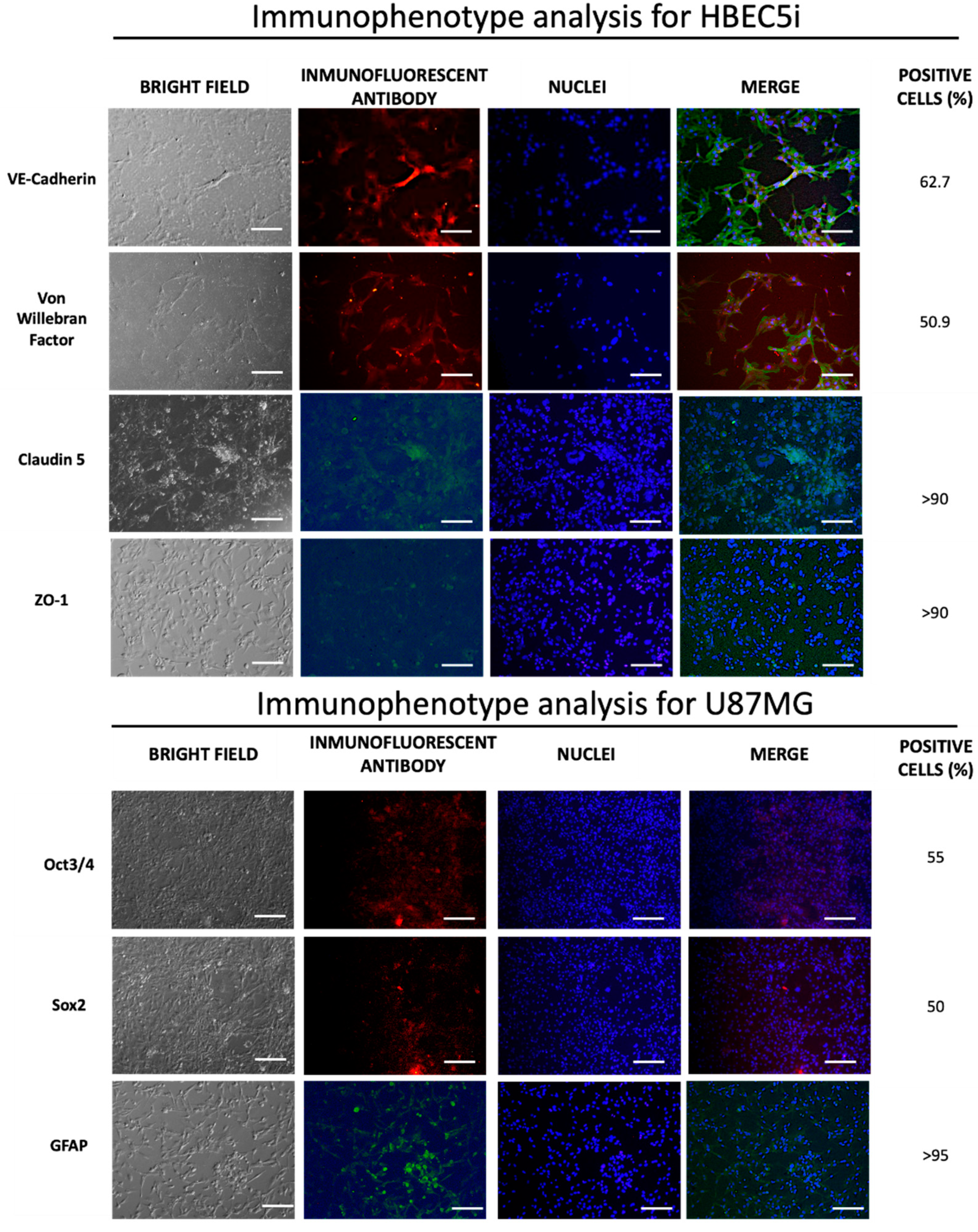
Figure 5.
Proliferation kinetics. The figure depicts growth kinetics of HBEC5i (endothelial), U87MG (glioblastoma tumoral cells) and astrocyte cell lines. Representative micrographs of time curse of proliferation in culture plate are showed at both sides and the central graph shows quantification of growth rate, the withe scale represents 100 µm.
Figure 5.
Proliferation kinetics. The figure depicts growth kinetics of HBEC5i (endothelial), U87MG (glioblastoma tumoral cells) and astrocyte cell lines. Representative micrographs of time curse of proliferation in culture plate are showed at both sides and the central graph shows quantification of growth rate, the withe scale represents 100 µm.
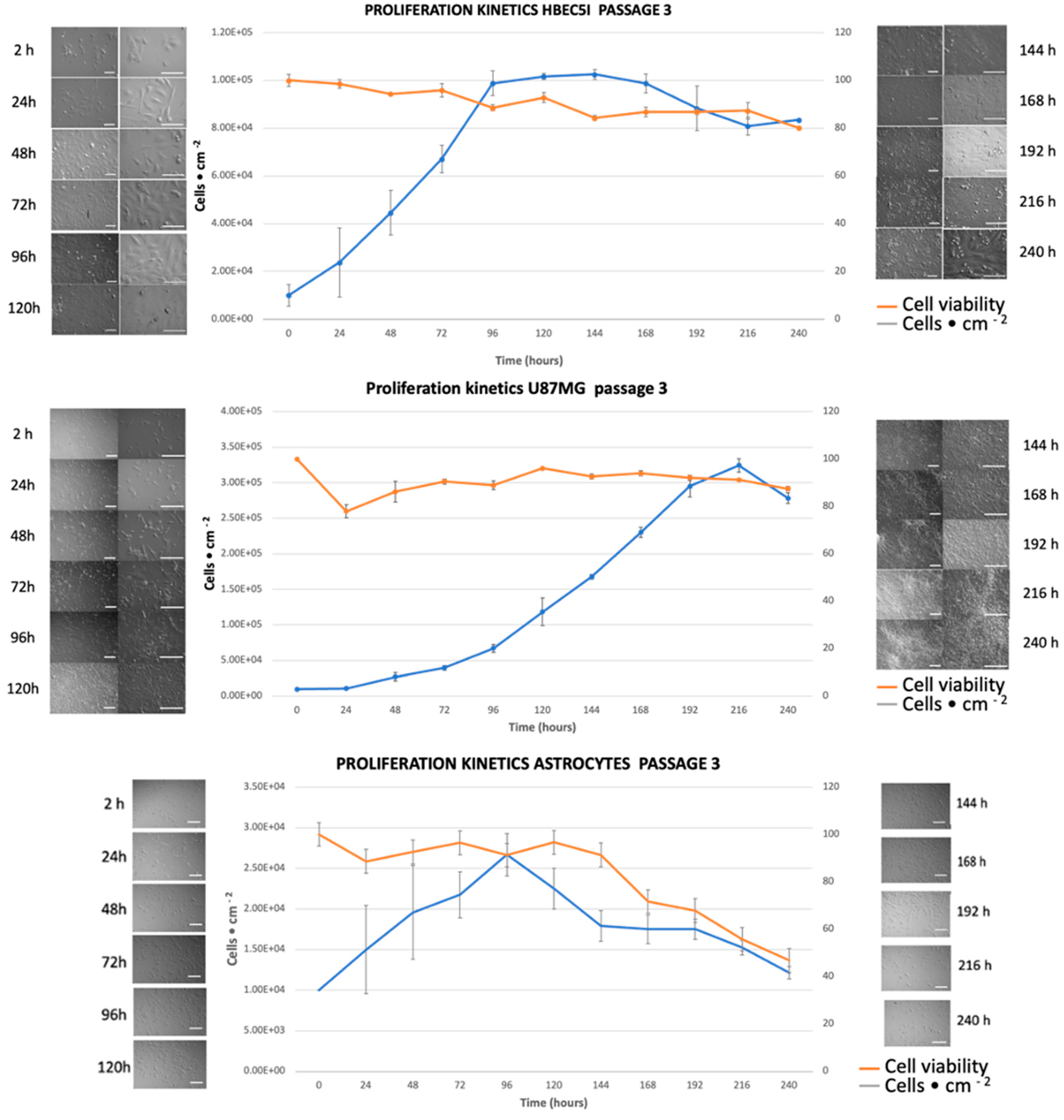
Figure 6.
Vasculogenesis ability of HBEC5i. A. Optimization of cell maintainance for HBEC5i cells. B. Vasculogenic assay. C. Vasculogenic structure on gelatin matrix at 96 hours culture. The figure shows the ability of endothelial cells HBEC5i to generate vascular shapes, regardless the matrix used. Initial cell culture 6 x 104 cells•cm -2.
Figure 6.
Vasculogenesis ability of HBEC5i. A. Optimization of cell maintainance for HBEC5i cells. B. Vasculogenic assay. C. Vasculogenic structure on gelatin matrix at 96 hours culture. The figure shows the ability of endothelial cells HBEC5i to generate vascular shapes, regardless the matrix used. Initial cell culture 6 x 104 cells•cm -2.
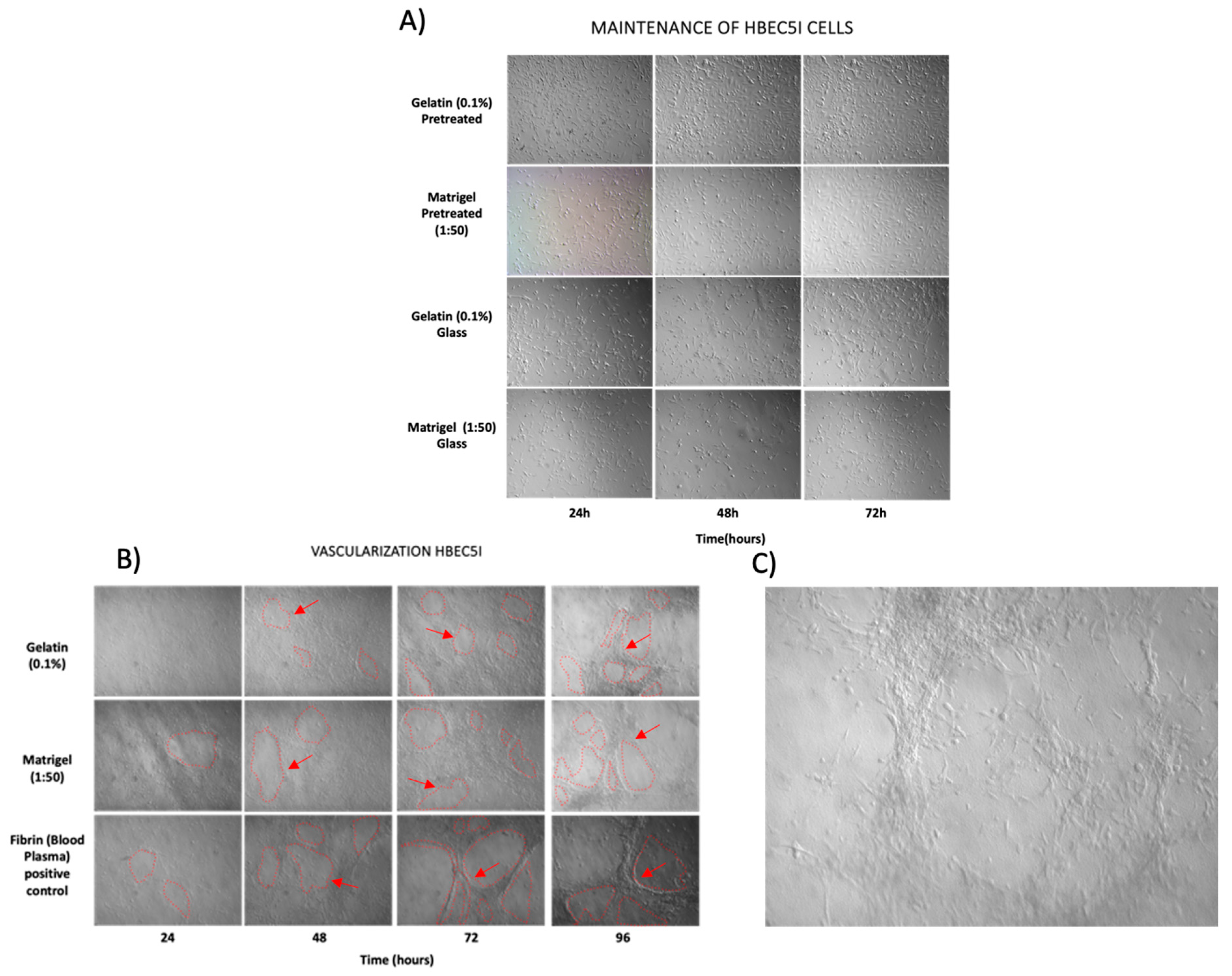
Figure 7.
Cell proliferation and migration tests. The figure shows cell migration ability of U87MG endothelial cells and HBEC51, at different starting cell densities. Migrating cells were indicated using digital yellow highlight. Density bars graphics are also provided below each micrograph.
Figure 7.
Cell proliferation and migration tests. The figure shows cell migration ability of U87MG endothelial cells and HBEC51, at different starting cell densities. Migrating cells were indicated using digital yellow highlight. Density bars graphics are also provided below each micrograph.
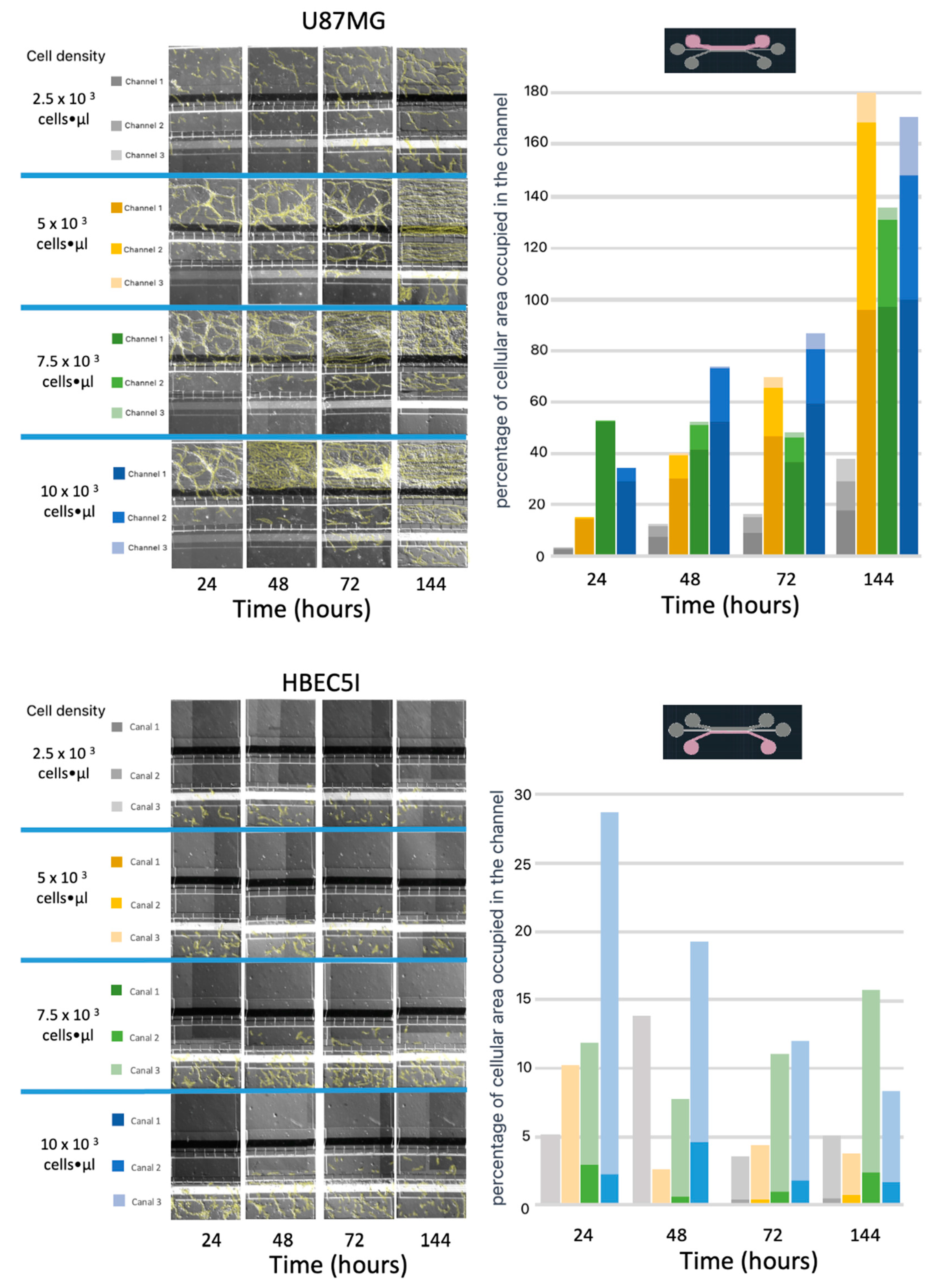
Figure 8.
Culture U87MG in microfluidic system culture. The cell culture process is demonstrated at a density of 2 x 104 cells•µl-1. At 0 hours, a heatmap of the cell distribution is shown for cell loading, and as time progresses (48-150h), cell clusters that were initially formed exhibit 3D projections, which have been described as organoids, And acquisition by confocal microscopy at 150 hours. The cellular cytoskeleton is shown through the expression of phalloidin (green), and the nuclei were assessed with DAPI (blue), the withe scale represents 100 µm.
Figure 8.
Culture U87MG in microfluidic system culture. The cell culture process is demonstrated at a density of 2 x 104 cells•µl-1. At 0 hours, a heatmap of the cell distribution is shown for cell loading, and as time progresses (48-150h), cell clusters that were initially formed exhibit 3D projections, which have been described as organoids, And acquisition by confocal microscopy at 150 hours. The cellular cytoskeleton is shown through the expression of phalloidin (green), and the nuclei were assessed with DAPI (blue), the withe scale represents 100 µm.
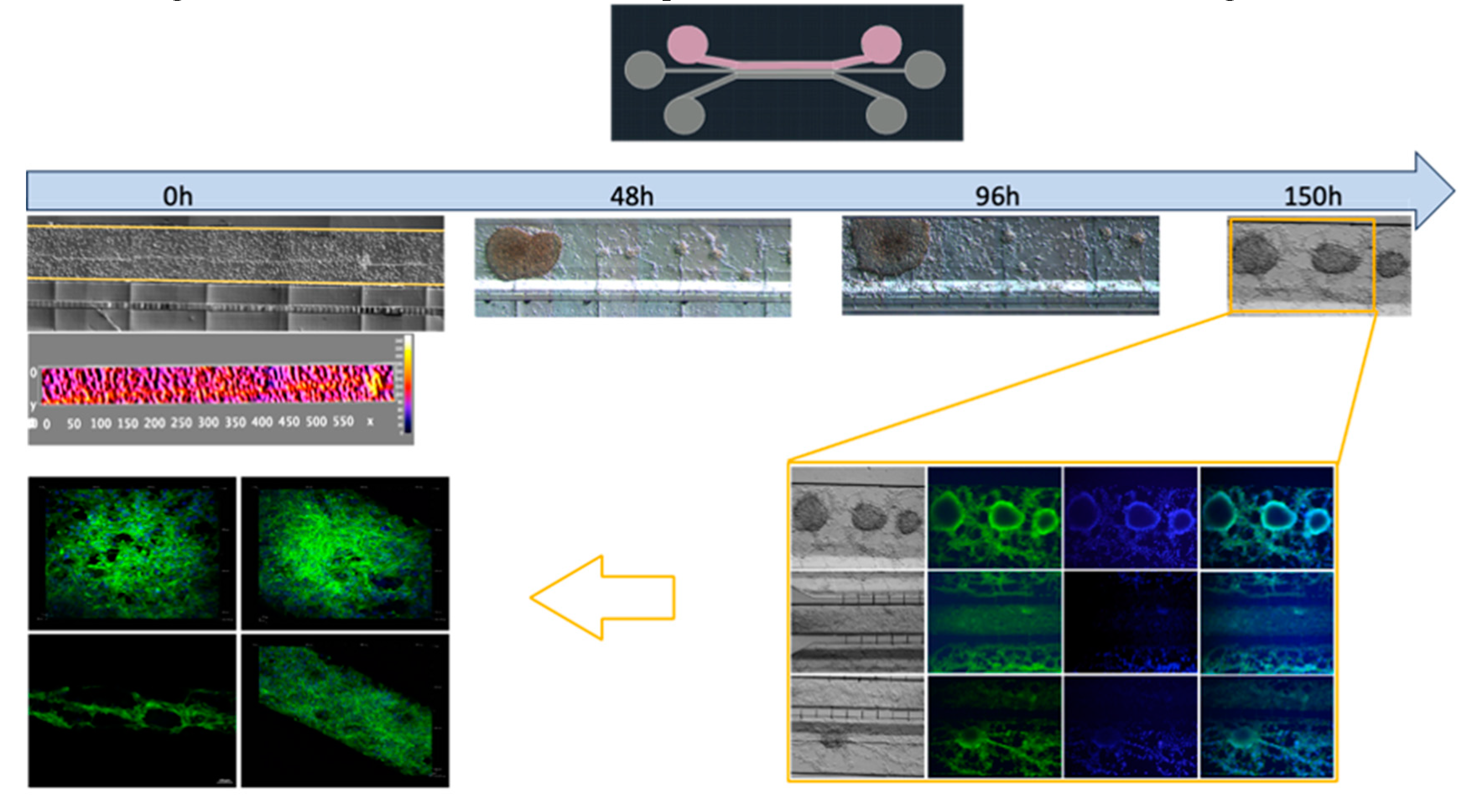
Figure 9.
Co- Culture U87MG-HBEC5i microfluidic system culture. acquisition by confocal microscopy the cell migration process. The cellular cytoskeleton is shown through the expression of phalloidin (green), and the nuclei were assessed with DAPI (blue). Cell – cell interaction is shown with yellow arrows. Such interactions are performed trough interconnecting channels, the withe scale represents 100 µm.
Figure 9.
Co- Culture U87MG-HBEC5i microfluidic system culture. acquisition by confocal microscopy the cell migration process. The cellular cytoskeleton is shown through the expression of phalloidin (green), and the nuclei were assessed with DAPI (blue). Cell – cell interaction is shown with yellow arrows. Such interactions are performed trough interconnecting channels, the withe scale represents 100 µm.
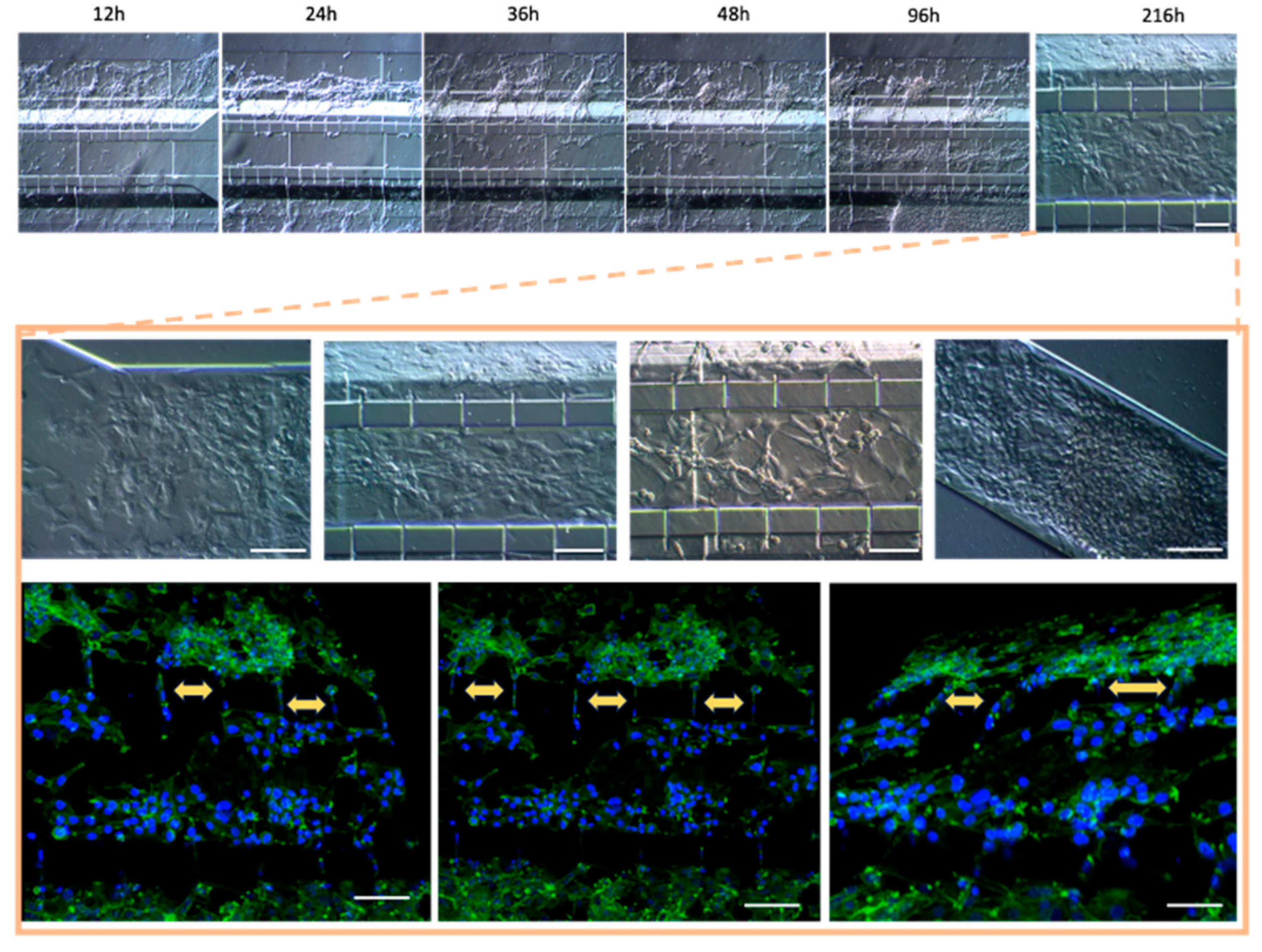
Figure 10.
Ordered scaling cell culture. The culture evolution at different time points is shown, as well as a process of cell invasion. Independent cell cultures were achieved for each channel as shown with glioblastoma (U87MG) cells labeled with Mitotracker (green), and endothelial (HBEC5i) cells in red, and cell nuclei in blue (DAPI); as well as their interaction through interconnecting channels (yellow arrows). In addition, in the lower graph, cytokines analyzed are shown in relation to the cell culture time.
Figure 10.
Ordered scaling cell culture. The culture evolution at different time points is shown, as well as a process of cell invasion. Independent cell cultures were achieved for each channel as shown with glioblastoma (U87MG) cells labeled with Mitotracker (green), and endothelial (HBEC5i) cells in red, and cell nuclei in blue (DAPI); as well as their interaction through interconnecting channels (yellow arrows). In addition, in the lower graph, cytokines analyzed are shown in relation to the cell culture time.
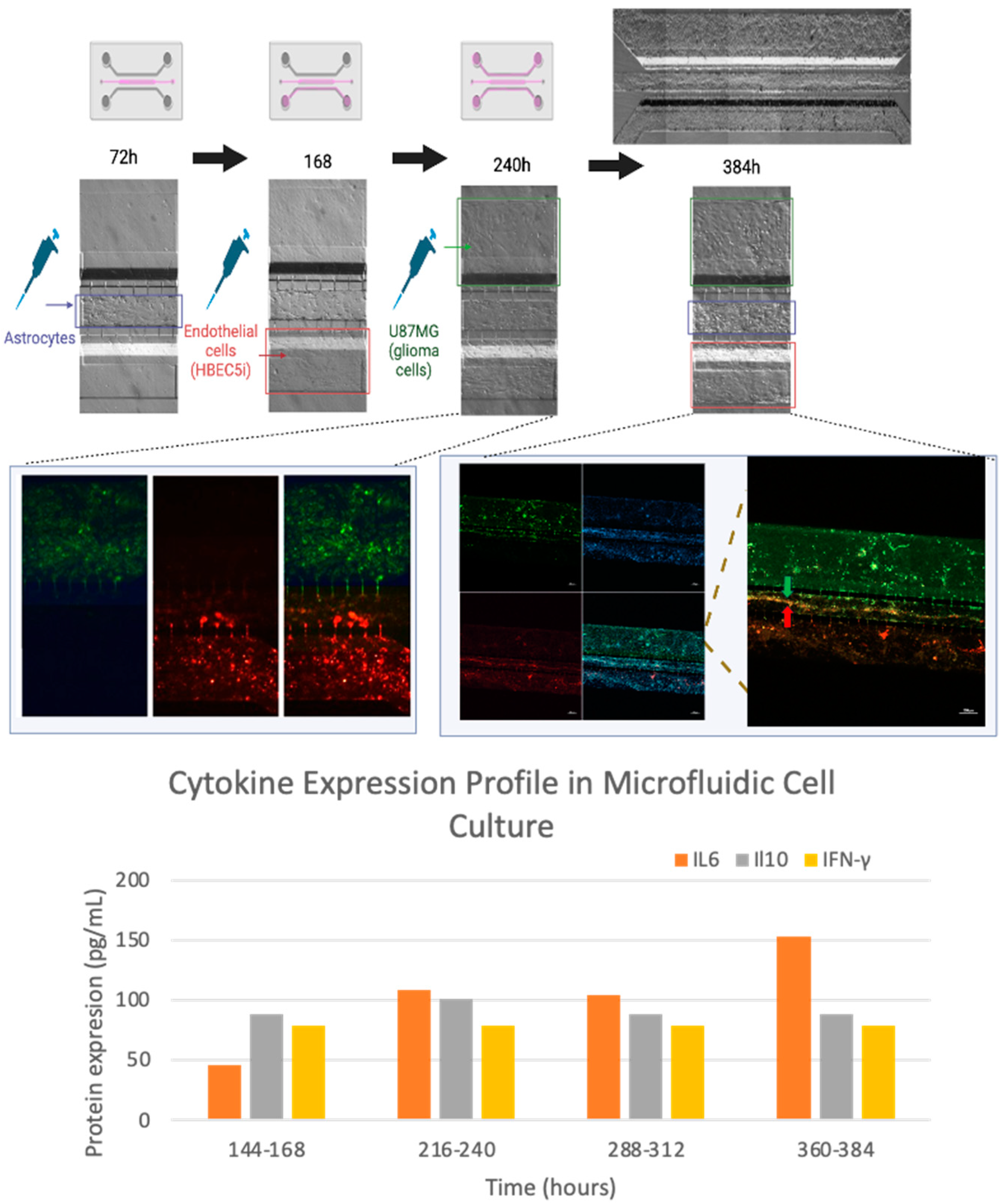

Table 1.
Antibodies used for phenotypic characterization.
|
Antibody |
Primary antibody | Secondary antibody |
Target description |
| Anti Ve-cadherin | 1:200 (Ab33168) | 1:1000 (Ab6718) | The Ve-cadherin is an adhesion protein present in endothelial cells that form the inner lining of blood vessels |
| Anti Von Willebrand | 1:200 (Ab6994) | 1:1000 (Ab6718) | The Von Willebrand protein is mainly found in endothelial cells that line the blood vessels. |
| Anti VEGFR | 1:200 (ab9530) | 1:1000 (ab6786) | The anti-VEGFR antibody is a specific antibody that targets the vascular endothelial growth factor receptor (VEGFR), a protein that plays a crucial role in angiogenesis and blood vessel growth. |
| Anti Oct3/4 | 1:200(C10-Z210Ms) | 1:1000 (ab6786) | The expression of Oct-3/4 in glioblastoma cells, such as U87MG, is an indication of these tumor cells' ability to acquire more undifferentiated and aggressive characteristics. |
| Anti Sox2 | 1:200 (ZM57-Z236MS) | 1:1000 (ab6786) | Sox-2 expression in glioblastoma cells, including U87MG, signals the existence of stem cells capable of self-renewal and generating differentiated tumor cell subpopulations. This phenomenon contributes to therapy resistance and tumor recurrence |
| Anti GFAP | 1:200 (G3893) | 1:1000 (ab6785) | GFAP (glial fibrillary acidic protein) is a valuable tool for identifying and locating astrocytes in cell cultures, brain tissues, and histological sections. |
| Anti Falloidin-Alexa Fluor 488 | 1:100 (A12379) | --------------------- | Cells and tissues. By using the anti-phalloidin antibody labeled with a fluorescent substance or an enzyme, it is possible to visualize the distribution, organization, and dynamics of actin filaments in living cells or fixed samples |
| Claudin5 | 1:50 (Ab15106) | 1:1000 (ab6785) | Claudin 5 is a protein expressed in the tight junctions of endothelial cells, playing a crucial role in the formation and maintenance of the blood-brain barrier. This membrane protein has a vital function in contributing to the regulation of capillary permeability in the brain. |
| ZO1 | 1:50 (Ab221547) | 1:1000 (Ab6717) | Zonula occludens-1, commonly referred to as ZO-1, is a tight junction protein that plays a key role in the formation and maintenance of cellular tight junctions. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



