
Submitted:
09 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Apple red flesh coloration is a result of the performance of biochemical pathway involved in the biosynthesis of anthocyanins and anthocyanidins. Based on apple genome analysis, high number of regulatory genes, mainly transcription factors such as MYB, which are components of regulatory complex MYB-bHLH-WD40, and several structural genes (PAL, 4CL, CHS, CHI, F3H, DFR, ANS, UFGT) involved in anthocyanin biosynthesis have been identified so far. In this study, we investigated previously unrecognized genes involved in the constitution of red-flesh apple phenotype. These genes could be concerned as molecular markers for early selection of new apple cultivars. Based on comparative transcriptome analysis of apples with different fruit flesh coloration, we successfully identified and characterized ten genes from plant hormone transduction pathway of: auxin (GH3); cytokinins (B-ARR); gibberellins (DELLA); abscisic acid (SnRK2 and ABF); brassinosteroids (BRI1, BZR1 and TCH4); jasmonic acid (MYC2); and salicylic acid (NPR1). In presented studies an analysis of their expression profiles was performed in immature and ripe fruits of red-fleshed cultivars. Generally, we have uncovered, the set of the novel genes, mediating regulation of the abscisic acid, salicylic acid, cytokinins and jasmonic acid signaling and preliminarily described their role in anthocyanin biosynthesis, accumulation and degradation. Their expression was not characterized in apple fruits so far. Presented study, allowed to underline the relationship of genes from hormone signal transduction pathway with UFGT gene, directly responsible for anthocyanin color transformation as well as confirmed their crucial role of plant hormone regulation and anthocyanin accumulation during apple fruit ripening.
Keywords:
apple
; expression profiling
; KEGG
; red-flesh fruits
; RNA-seq
; RT-qPCR
; transcriptome
1. Introduction
Apples are one of the most widely cultivated crops in the world temperate regions worldwide. Their cultivation is very popular, especially due to their high ecological adaptability and nutritional value of the fruits [1,2]. The global production of apples is about 78.4 million tons. Poland is the largest producer of apple fruits in the European Union (4.3 million tons), third in the world after China (35-40 million tons) and USA (over 4.5 million tons) [3]. The management of such massive fruit production raises challenges in terms of cultivation and storage. However, an opportunity to address this situation lies in increasing of consumption of fresh apples and their products. This can be achieved by introduction into apple industry red-fleshed varieties, rich in anthocyanins, having health-promoting properties [4]. In addition apples with red flesh are an interesting raw material for the production of cloudy apple juices [5].
Apple wildly spread breeding efforts allowed to confirm that Malus sieviersii is the most closely related to the commonly cultivated ‘Golden Delicious’ cultivar [6,7]. Moreover, the primary donors of genes for the red skin and red flesh of apple fruits are Malus sieversii f. niedzwetzkyana and Malus pumila var. niedzwetzkyana [7]. As red fruits are highly attractive to consumers and often associated with apple ripeness and good flavor, their pigmentation becomes an important trait in horticultural research [2,8]. However, there are only few breeding programs focused on developing new red-fleshed cultivars with high quality of the fruits [9]. An analysis of apple segregating F1 populations combined with QTL mapping as well as transcriptomic studies, have highlighted the complexity of this trait [10,11,12,13].
The anthocyanins (glycosides of anthocyanidins), which are synthetized by flavonoid pathway, are the most important fruit component contributing in red-skinned and red-fleshed apple fruits, as well as red apple plant tissues (e.g. stems, leaves and seeds). They have been extensively studied especially in regard to human health, playing the huge role in reduction of risks diseases such as: cardiovascular, asthma, gastrointestinal, weigh regulation etc. [12,14,15]. The flavonoid pathway is well-recognized and consist the number of different enzymes catalyzes biosynthesis of anthocyanidins and anthocyanins [16]. In apple, anthocyanidins are divided into several subclasses: pelargonidin, cyanidin, delphinidin, peonidin, petunidin, malvidin, and almost 80% of them are cyanidins, thus the reconstruction of galactose into form cyanidin galactoside is mainly based on transformation of galactose to cyanidin-3-galactoside [17]. Determination of anthocyanidins coloration in specific tissues is revealed by different types and numbers of substituents in the ring of the anthocyanin [9,18,19,20,21,22].
Initial precursor in anthocyanins and flavonoids biosynthesis pathway is a phenylalanine. It is subsequently catalyzed by phenylalanine ammonialyase (PAL), 4-coumarate:coenzyme A ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), flavonoid 3’5′-hydroxylase (F3’5’H), and dihydroflavonol 4-reductase (DFR) leading to colorless metabolites production. Then, the colored anthocyanins are synthesized by anthocyanidin synthase (ANS). Finally, they are transformed into brick-red, magenta or even blue–violet glycosides through addition of UDP-glucose by flavonoid 3-O-glucosyltransferase (UFGT) [23,24,25,26,27].
Based on apple genome analysis, a number of genes encoding MYB transcription factors involved in anthocyanin biosynthesis regulation were identified [9,28,29,30]. Overexpression of MYB10 strongly correlates with apple type 1 flesh color regulation by anthocyanin increment in skin, shoots and fruit flesh (expressing the pigmentation of fruit flesh, cortex and white core). This can be a result of autoregulation of MYB10 leading to red flesh apple phenotype [19,31,32,33,34]. Meanwhile in the type 2, apple red-fleshed fruit phenotype did not co-segregate with MYB10 [9]. An analysis performed by Chagné et al. [11] and Mahmoudi et al. [26] confirmed the relationship between the MYB allele combinations and fruit flesh color. Characterization of 16 apple cultivars with red and white flesh as well as segregating population derived from the cross of ‘Geneva’ and ‘Braeburn’, confirmed that the seedlings possessing MYB1/MYB1 allele combination had red skin and white fruit flesh phenotype, while the presence of MYB10 alleles (MYB1/MYB10 or MYB10/MYB10) shaped the genotypes with red skin, red fruit flesh and red seeds [11,26].
Recent studies, established by the analysis of different set of apple genotypes as well as transgenic ‘Royal Gala’, allowed to investigate new genes such as MdJa2, MdNAC, WD40, bHLH or SEPALLATA and MADS-box transcription factors, involved in brassinosteroids accumulation and affecting anthocyanin and proantocyanidins (PA) biosynthesis. MYB-bHLH-WD40 (MBW; a composition of R2R3-MYB and WD transcription factors) was described as a part of the transcriptional complex of the anthocyanin structural genes regulation [2,13,19,35,36,37]. Simultaneously, current studies also confirmed that transcription factors such as MYB17, MYB111, MYBL2 and MYB16, interacting with HLH33 and HB1 may negatively regulate the anthocyanin biosynthesis [38,39].
Since the mechanism of regulation of red-fleshed apple fruits is complex and still unclear, the objective of this study was to investigate new, so far unrecognized, genes involved in apple fruit flesh coloration. They can become candidates to develop reliable molecular markers to accelerate an early selection of apple cultivars with red-fleshed fruits, applicable in apple breeding programs.
2. Results
2.1. Transcriptome Profiling
The major goal of this analysis was to determine the difference between gene transcription in fruits of red- and white-fleshed apple cultivars. RNA-seq experiment with fruits of Red Love® ‘General’ and ’Early Fuji’, characterized by red and white fruit flesh respectively, has been performed. For each cultivar 3 independent fruit samples (3 replications) were used, six RNA-seq libraries were constructed and over 40G of clean reads were obtained. The effective data volume for each sample was in the range from 6.06 to 7.23G. The quality of reads was high, with Q30 ratio from 92.33 to 92.78% of inferred base call accuracy. The GC content was similar in all samples with an average of 47.46% (Supplementary Table S1). Clean reads were mapped on the apple reference genome at a similar ratio, on average 93.63%, with 89.7% of uniquely mapping reads and 85.91% of reads mapped in proper pairing.
The expression of protein-coding genes was calculated using FPKM method, which indicates the number of fragments per kilobase length of a protein-coding gene per million of sequenced fragments. For 35 890 genes annotated in apple genome, similar number in each analyzed sample, on average 25 666 genes, were found to be expressed (Supplementary Table S2). Sample-to-sample cluster analysis and principal component analysis (PCA) of expression profiles indicated that the results were consistent and investigated cultivars samples clearly are grouped together (Figure 1).
2.2. Differentially Expressed Genes
Based on gene expression profiling, differentially expressed genes (DEGs) in Red Love® ‘General’ as compared to ‘Early Fuji’ control were identified (log2 fold-change >1 false discovery rate FDR < 0.05) (Supplementary Table S3). In total 4 839 genes were found to be differentially expressed in Red Love® ‘General’ with 2 259 genes up-regulated and 2 580 down-regulated (Figure 2).
2.3. Functional Categories of DEGs
2.3.1. GO Enrichment Analysis of DEGs
After finding the differentially expressed genes in red-fleshed Red Love® ‘General’, we performed Gene Ontology (GO) enrichment analysis to describe their functions (Figure 3).
The GO enrichment analysis allowed classifying the general role of differentially expressed genes in: biological processes (BP), cellular component integration (CC) and molecular function (MF). The total GO enrichment categorization of 3 500 DEGs (Supplementary Table S4 and Supplementary Figure S1) led to assigned 1 635 genes as up-regulated and 1 865 as down-regulated in red-fleshed fruits of Red Love® ‘General’ as compared to white-fleshed ’Early Fuji’ (Figure 3 and Supplementary table S5). In total most of DEGs were clustered into 65 different GO terms: 23 for biological process, 20 for cell component and 21 for molecular function (Figure 3). In terms of biological process the highest number of DEGs was assigned to categories: biological regulation, cellular process, metabolic process, response to stimulus and single-organism process. In terms of cellular component, the highest number of DEGs was assigned to the categories: cell and cell part, membrane and membrane part integration and organelle and organelle part. In terms of molecular function, most of genes were assigned into two categories: binding and catalytic activity (Figure 3).
Next, DEGs from top 30 GO enriched categories were analyzed (Supplementary Figure S2). In the main category of biological process, top up-regulated DEGs (Supplementary Figure S3) were classified in terms to: GO:0042445 – hormone metabolism process, GO:0008202 – steroid metabolic process, GO:0009850 – auxin metabolic process and GO:0009684 – indoleacetic acid biosynthesis. There were other terms additionally enriched: GO:0023052 – signaling, GO:0050896 – response to stimulus, GO:0044700 – single organism signaling, GO:0006952 – defense response, GO:0007165 – signal transduction, GO:0007154 – cell communication, GO:0034050 – host programmed cell death, and GO:0009626 – plant hypo-sensitivity. In the category of cell component genes, two top GO enrichment terms were extracted: GO:0071944 - cell periphery and GO:0005886 – plasma membrane (cell membrane integration) (Supplementary Figure S4). In the category of molecular function, top GO enrichment terms were assigned to: GO:0043531 – ADP binding and GO:0016491 - oxidoreductase activity (Supplementary Figure S5).
2.3.2. KEGG Enrichment Analysis of DEGs
Analysis of significantly enriched pathways according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) revealed that the highest numbers of DEGs were classified in six main groups: cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, organismal systems (Figure 4a). The biggest group included genes involved in metabolic pathway regulation (in total 812 DEGs; blue blocks) and second - in genetic information processing (in total 205 DEGs; green blocks). In addition, the third group of genes was classified in pathways of plant metabolism in diverse environments (in total 99 DEGs; brown blocks). The subgroups of DEGs, classified in metabolism were assigned to: carbohydrate metabolism (170), amino acid metabolism (110), energy metabolism (105), and lipid metabolism (99). From the other groups the highest number of DEGs (90), was classified to the signal transduction (Figure 4b). Extracted up and down regulated genes recovered from Red Love® ‘General’ vs. ‘Early Fuji’ compartments are presented in Supplementary Figure S6. From this grouping, 30 top GO terms uncovered, three most abundant one from biological process were assigned to the galacturonate biosynthetic process and signal transduction; from cellular components – plasma membrane and extracellular space and form the group of molecular functions – genes involved in ADP binding and representing chalcone isomerase activity (Figure 4b)
Detailed analysis of the KEGG pathways enrichment classification showed that the group of DEGs most significantly up-regulated in red-fleshed Red Love® ‘General’ was assigned to plant hormone signal transduction pathway (mdm04075) (Figure 5). In the next, ten up-regulated DEGs from this group, involved in auxin, cytokinine, gibberellin, abscisic acid, brassinosteroids, jasmonic acid and salicylic acid hormonal signaling and regulation, were selected for further study (Figure 6).
The functions and cell localization of selected genes are presented in Table 3 and the selected genes were addressed for detailed expression profiling in the apple cultivars characterized by red-fleshed fruits.
2.4. Validation of Activity of Genes from Plant Hormone and Signal Transduction Pathway by RT-qPCR
The expression profiles of selected genes were analyzed in immature and ripe fruits of seven red-fleshed apple cultivars in comparsion to the immature and ripe white-fleshed fruits of ‘Free Redstar’ (Figure 7).
Overall, in comparison to ‘Free Redstar’, significant down-regulation of five, out of ten selected genes: DELLA (involved in gibberelin pathway), ABF (involved in abscisic acid pathway), BZR1 and TCH4 (brassinosteroids pathway), and MYC2 (responsible fo jasmonic acid transduction) was observed in immature fruits of: ‘Trinity’, ‘Alex Red’, Red Love® ‘Era’, Red Love® ‘Circe’, ‘Roxana’ and ‘Red Love® ‘Sirena’ (Figure 8). Interestly the most significant up regulation of those genes was evaluated for M. sieviersii f. niedzwetzkyana (characterized for red coloration of leaves, flower petals, core, stem, seeds, skin and flesh), representing wild apple variety, and concidered as the donor of target genes. Simultinously relevant overexpression of the same set of genes was noted for control immature white fruit samples of ‘Free Redstar’,
In case of ripe fruits of red fleshed cultivars all genes applied in this study showed significant variation in expression profiles. The activities of GH3, SnRK2, BRI1 and TCH4 genes were significanlty higher in fruit samples of ‘Alex Red’, Red Love® ‘Circe’ and Red Love® ‘Sirena’. In case of ‘Trinity’, Red Love® ‘Era’ and ‘Roxana’ cultivars four genes: B-ARR (cytokinine pathway), DELLA (gibberellin pathway), BZR1 (brassinosteroids pathway), and MYC2 (jasmonic acid pathway) showed significant down regulation. High expression level of ABF gene (abscisic acid pathway) (ranged between 10x to 50x number of transcript fold change) in comparsion to ‘Free Redsar’ white ripe fruit control, was noted for all red fleshed apple cultivars evaluated. Overall, the activity of analyzed genes mantained overexpressed in immature and ripe fruits of M. sieviersii f. niedzwetzkyana, meanwhile inactivation of all selected genes was observed in ripe fruits of white-fleshed ‘Free Redstar’ (Figure 9).
2.5. The activity of the Structural Genes in Red-Fleshed Apple Cultivars
Six genes, from polyphenol and anthocyanin biosynthesis pathway were analyzed to confirm their expression in red fleshed apple cultivars. Significantly high level of expression of all structural genes was noted in immature fruits of M. sieviersii f. niedzwetzkyana (i.e. showing about 18x fold changes for F3H) and ‘Free Redstar’ white-fleshed apple fruits (Figure 10). In case of DFR and UFGT negligible activity was noted for analyzed immature fruit flesh samples (data not shown).
Additionally, CHS, F3H and UFGT genes were significantly up-regulated in ripe fruits of ‘Trinity’, Red Love® ‘Circe’ and Red Love® ‘Sirena’. In general, significant up-regulation (fold change ranged from 10 to 60x) of PAL, CHS, ANS and F3H (encoding enzymes for anthocyanin precursors synthesis) was observed for all ripe fruits of red-fleshed apples. As we could expect, the activity of tested genes was significantly lower in ripe fruits of white-fleshed ‘Free Redstar’ (Figure 11). Simultaneously, the light red fruit flesh color of ’Roxana’ cv. may be the result of significantly higher expression of the PAL (60 fold change) precursor gene and relative inhibition of other structural genes, probably causing the breakdown of intensity of anthocyanin biosynthesis in the genome of this cultivar. Interestly, probably similar mechanism of this regulation was noted for white-fleshed ’Free Redstar’ (Figure 11). Generally, in red-fleshed apple cultivars four of structural genes (PAL, CHS, F3H, ANS), were significantly up-regulated, while high activity of DFR and UFGT genes was noted only for ‘Alex Red’, Red love® ‘Circe’ and ‘Red Love® ‘Sirena’.
Additionally, the lower activity of all tested genes was observed for ripe, in comparison to immature, fruits of M. sieversii f. niedzwetzkyana.
2.6. Genes from Signal Transduction Pathway Shows Inconsistencies in Regard Expression Profiles in Comparison to RNA-seq Experiment but Significant Intergenic Correlation
Genes from the plant signal transduction pathway selected on the basis of the RNA-seq experiment were expressed at high level in the fruits of the Red Love® ’General’ cultivated in China. They were chosen for further expression profiling with regard to transcriptome analysis. However, in the validation studies performed on the red-fleshed apple collection, grown in Poland, variable and inconsistent expression profiles of those genes were observed.
Gene-to-gene correlation coefficient between structural and uncovered genes was evaluated individually for a set of immature and ripe fruits of analyzed apple cultivars. The assessment of interaction between structural genes and those selected from the hormone signal transduction pathway in immature fruits of all tested cultivars, showed significant correlation between, i.e.: DFR F3H, UFGT and B-ARR, NPR1, GH3, SnRK2, BRI1 (Table 4A). In ripe fruits, the correlation was noted between the all identified genes and the structural UFGT gene (encoding flavonoid 3-O-glucosyl transferase, the gene responsible for final glycosides transformation and their pigmentation). This may explain that the most of the structural genes thus from anthocyanin pathway as well as plant hormone signal transduction pathway are potentially activated at early fruit developmental stage and shows significant intergenic correlation.
Generally, in ripe fruits significant correlation was observed between chalcone synthase gene (CHS) and B-AAR, GH3, SnRK2 and TCH4 (respectively involved in pathways of: cytokinins, auxin, salicylic acid and brassinosteroids), between gene encoding phenylalanine ammonialyase (PAL) and the same set of genes, as well as between UFGT and all genes selected form hormone transduction pathway (Table 4B.). The B-ARR, GH3, SnRK2 and TCH4 genes (from cytokinine, auxin, abscisic acid and brassinosteroids pathways respectively) showed correlation with CHS, PAL and UFGT genes and could be considered as possible indicators of regulation of glucoside color transformation in apple red fleshed fruits ready for harvesting.
3. Discussion
In the presented study, utilizing transcriptome comparisons and gene expression analysis of two apple cultivars producing red- and white-fleshed fruits, we have revealed new genes putatively related with the regulation of anthocyanin biosynthesis. Among of over 40 Gbase of filtered sequence data, two major groups of genes mapped on the reference apple genome were recovered. One group of sequence reads (43%) were assigned to be involved in metabolic pathways and second (31%) - in secondary metabolite biosynthesis. Based on differentially expressed genes (DEGs) form top 20 KEGG Gene Ontology (GO) term classifications (Figure 5; Results), we selected a set of 10 genes from plant hormone signal transduction pathway, involved in auxin (AUX), cytokinine (CK), gibberellin (GA), abscisic acid (ABA), brassinosteroids (BR), jasmonic acid (JA) and salicylic acid (SA) biosynthesis. Following RT-qPCR analysis of the genes of interests we have observed their variable activity with up-regulation noted for GH3, SnRK2, ABF and TCH4, and down-regulation for B-ARR, DELLA, BZR1, BRI1, MYC2 and NPR1. However, although all the selected genes showed up-regulation in red-fleshed Red Love® ‘General’ cultivar used in RNA-seq experiment, the results obtained for the RT-qPCR, performed for seven cultivars studied, were not consistently uniform. A similar observation of inconsistencies between RNA-seq and RT-qPCR comprehensive analysis was reported by Everaert et al. [40]. The authors concluded that, depending on the analysis workflow, 15–20% of genes usualy are considered as ‘non-concordant’ in regard to the results obtained with RNA-seq and RT-qPCR. Those ‘non-concordant’ genes are defined when both approaches yield differential expression in opposing directions or when one method shows differential expression while the other does not [40].
The analysis carried out for representative set of seven red-fleshed apple cultivars collected at two fruit developmental stages, and compared to white-fleshed cv. ’Free Redstar’, allowed to show, that the changes in gene expression are closely related to the fruit ripening process [34,41,42]. All structural genes and the newly revealed GH3 (auxin response), SnRK2 and ABF (both involved in ABA signaling) as well as TCH4 (BR pathway) were activated in the ripe red-fleshed fruits. In accordance to fold change calculation, DELLA (GA response), B-ARR (CK response), BZR1 and BRI1 (BR receptors), as well as MYC2 and NPR1 (genes from JA and SA signaling pathways respectively), were defined as to be significantly inhibited in the ripe red-fleshed fruits. The putative mechanism of gene regulation of the plant hormone and transduction pathway is presented on the scheme below (Figure 12).
In accordance to presented scheme, similar observation was described by Su and coworkers [37]. They postulated that the structural genes from anthocyanin biosynthesis pathway were up-regulated in apple calli, suppressing expression of brassinosteroids resistant gene (BZR1) (BR accumulation = anthocyanin pathway activation). They also have reported that interaction of BZR1 with MdJa2 modulates the target genes transcription, and generally regulate plant response to brassinosteroids [37]. Based on observation of apple calli development, Zheng and coworkers also underlined the anthocyanin biosynthesis regulation by plant hormones, including brassinosteroids, which strictly influenced their accumulation [43]. Simultaneously, as it was reported by Su et al., application of external BR also promoted fruit maturation and pro-anthocyanin synthesis in apple tissue [37,43]. Our observations led to the comparable conclusion that the inactivation of BZR1 is positively correlated with promotion of anthocyanin accumulation in red-fleshed ripe apple fruits.
Brassinosteroids signaling is perceived by receptors BRI1 and BZR1 in cell membrane. As the recent research explains it is one of the major factors (similarly to bHLH) playing key role in regulation of brassinosteroids gene expression [43,44. Both, brassinosteroid receptor genes, analyzed in presented study, showed significantly low expression in the series of red-fleshed fruit cultivars, probably leading to brassinosteroids accumulation, thus promoting an anthocyanin synthesis. This selecting mechanism was described in previous studies of tomato, cucumber, strawberry and grape, indicates the key role of brassinosteroids in fleshy fruits development and ripening [42,45,46,47,48].
In parallel, based on our observations, we have found the significant inactivation of genes NPR1 and MYC2, respectively responsible for salicylic acid (SA) and jasmonic acid (JA) signal transduction, influencing in negative correlation in anthocyanin regulation in apple fruits. Therefore, our observation gives new insight in anthocyanin biosynthesis dependence on salicylic and jasmonic acids signal inhibition, not reported so far.
In this concern, previous research of Sasaki et al. and summarized by Wang and Chang explained the role of JA in regulation of secondary metabolites, plant defense response and organ development [49,2]. Then, later studies of Qi, An, and coworkers showed, that the activation of MYB and bHLH, significantly correlated with anthocyanin accumulation in A. thaliana and it is promoted by complex mechanism, including degradation of repressor protein (so called JA ZIM DOMAIN) [50,51]. Recent reports of Wang et al. also underline the role of jasmonic acid as signaling factor influencing the MYB24L gene and affecting the anthocyanin biosynthesis [30].
In conducted research, the newly studied DEGs such as DELLA (gibberellin pathway) and B-ARR (cytokinin pathway), showed significant down-regulation in the red-fleshed ripe fruits and up-regulation in white fruit flesh of ‘Free Redstar’. The observations are similar to the results obtained by Nawaz and coworkers, which confirmed up regulation of histidine kinase and DELLA genes in ripe fruits of white-fleshed apple ’Hanfu’ and its mutant. The authors explained, the level of gene transcript number is genotype-depended, resulting in apple flesh fruit pigmentation intensity; from flush flesh, pinky flesh, to dark red flesh [42,52].
Another founding of our work underlines negative correlation between gene activity and auxin and cytokinin production (gene activation = less auxins or gene inactivation = more cytokinin production) expressed by up-regulation of auxin response gene (GH3) and down regulation of (B-AAR) in red-fleshed cultivars (Figure 10, Results). This gene activity and metabolite production relationship was highlighted by Ji, Dikeman and coworkers who observed that increment of auxins together with decreasing of cytokinin in the cells can inhibit anthocyanin synthesis [53,54]. This phenomenon was observed in callus tissue of both A. thaliana and apple and underlined by Nguyen, Ji and coworkers as they have elucidated that the increased concentration of cytokinin and decreasing of auxin can significantly promote the expression of MYB transcription factors, leading to anthocyanin accumulation [55,56].
Additionally, we have noted up-regulation of ABF and SnRK2 genes, (involved in abscisic acid pathway) in red-fleshed ripe fruits. Notably, this was observed in all cultivars except ‘Roxana’ (pink fruit flesh) and M. sieversii f. niedzwetzkyana (a wild apple variety). This observation suggests a distinct regulation of fruit flesh coloration in these two cultivars. The gene activity in this context is genotype-dependent, highlighting the complexity of regulation of apple fruit flesh coloration. As it was described by Jiang and Joyce ABA plays the kay role in strawberry fruit color development (generally through PAL activity stimulation), as well as in grapes and litchi promotes the fruit coloration stage [57,58,59].
Through the effect of regulating the number of gene transcripts, we have noted the changes in targeted hormonal pathways, which, by regulating the content of hormonal proteins, influence: the mechanism of anthocyanins synthesis and accumulation in apple fruits. For the DELLA, B-ARR, BZR1, BRI1, MYC and NPR1 genes, we have observed a negative regulatory mechanism, by blocking the gene activity and thus resulted in hampering recognition or degradation of hormonal proteins.
The uncovered genes were correlated with the functional gene UFGT (responsible for final anthocyanin color transformation). To date, only limited studies have examined the negative regulatory mechanisms underlying anthocyanin synthesis in apple. The novel technologies applied in our research allowed understanding the mechanism of apple fruit color development. Uncovered genes being the candidates for molecular markers can accelerate the breeding of high quality red-fleshed apple cultivars. Application of such research provides the knowledge of regulation of fruit color development and anthocyanin biosynthesis, accumulation and degradation.
4. Materials and Methods
4.1. Plant Material
In RNA-seq experiment red fleshed Red Love® ‘General’ and white fleshed ‘Early Fuji’ apple cultivars were used. Apple fruits were collected from trees grown in experimental orchard of the Yantai Academy of Apple Research in China. Fruits (min. 3) of both cultivars were picked up at the harvest time (middle of September). From each fruit peeled flesh discs with the diameter of 2 cm and 1 cm of depth, cut with sterile blade, were collected. The samples were placed directly into liquid nitrogen and stored in -80℃ until RNA extraction.
In validation of gene expression experiments a set of apple cultivars characterized by different patterns of fruit pigmentation was used. This set included: ‘Alex Red’, ‘Roxana’, ‘Trinity’, Red Love® ‘Circe’ (‘Lureone’), Red Love® ‘Era’, Red love® ‘Sirena’ and Malus sieviersii f. niedzwetzkyana and a control white fruit flesh ‘Free Redstar’ (flesh pigmentation control). Tissue samples were collected, from the apple trees grown in the experimental orchard maintained at the National Institute of Horticultural Research in Skierniewice, Poland. For each cultivar three fruits were collected at two developmental stages: immature fruits sampled 100 days after full bloom (DAFB) and ripe fruits collected at harvest time. Then, the apple fruit flesh discs (as describe above) were prepared and immediately placed in liquid nitrogen for the RNA isolation. The samples were used for Reverse Transcriptase quantitative PCR (RT-qPCR) gene expression analysis of target structural genes as well as for differentially expressed genes revealed by RNA-seq.
4.2. RNA Extraction
For RNA-seq experiment, total RNA was isolated using MINIBEST Plant RNA Extraction Kit (TaKaRa, China) according to manufacture protocol. The concentration and quality of RNA was evaluated after electrophoresis in 1% agarose gel stained with GelRed Dye (Biotiun, China). High quality RNA preparations were sent to the commercial company for transcriptome sequencing.
For RT-qPCR total RNA was isolated using method described by Zeng and Yang [60] with minor modifications. The procedure was started with sample incubation for 20 minutes in CTAB buffer heated to 65℃. After centrifugation in a mixture of chloroform and isoamyl alcohol (24:1, v/v) RNA was resolved from the upper solution fraction and precipitated with 10 M lithium chloride overnight at 4°C. Then, the precipitate was dissolved in RNase-free water. The quality, degree of integration and concentration of RNA samples were assessed by using the Agilent 2100 Bioanalyzer and Expert 2100 software (Agilent Technologies, CA, USA).
4.3. RNA-seq Analysis
The library construction and sequencing were completed by Shanghai Ouyi Biomedical Technology Co., Ltd. The transcriptome sequencing by the Illumina technology was conducted by OE biotech Co., Ltd. (Shanghai, China). To perform quality check of the raw data and summarize the number of reads Trimmomatic software [61] was used. To map sequence reads on apple reference genome Htseq-count software [62] was used. As a reference ‘Golden Delicious’ apple genome assembly ASM211411v1 was applied: https://www.ncbi.nlm.nih.gov/data-hub/genome/GCF_002114115.1/. The DESeq [63] allowed standardize the number of counts of each single gene, calculate the multiple of difference and perform significant difference tests. To obtain the number of reads compared to protein-coding genes in each sample cufflinks [64] was applied. Finally, the gene expression level was calculated as FPKM (Fragments Per kb Per Million Reads), giving the number of fragments per kilobase length of a protein-coding gene per million fragments sequenced. The raw sequence data were stored in FASTQ file format. Evaluation of differentially expressed genes (DEGs) between Red Love® ‘General’ and ‘Early Fuji’ was performed with use of DEseq2 R package software. For this purpose RNA-seq data were compared and analyzed weather the same gene is differentially expressed within two samples. For this purpose, LogFC >1 (logarithm of fold change in the expression level of the same gene in two analyzed samples) and filtering data condition p-value<0.05 criteria were applied.
4.4. GO Enrichment Analysis
The gene function enrichment was performed based on compilation of the list of all protein coding genes and differential protein-coding genes (counted based on FPKM), using hypergeometric distribution test to calculate the representative GO function set. Than p-value of significant enrichment in the list of differential protein-coding genes was corrected by Benjamini & Hochberg’s multiple test to obtain fails discovery rate (FDR). The procedure translates experimental results into a series of regulated gene lists at multiple false discovery rate FDR cutoffs, and computes the p-value of the overrepresentation of the genes.
The top30 GO enrichments analysis was performed based on screened GO entries corresponding to the number of differential genes greater than 2 in the three categories (Biological Process (BP), Cell Components (CC), and Molecular Function (MF)), and sorted 10 entries according to the -log10Pvalue corresponding to each entry from large to small layout.
The fisher algorithm was used to conduct Cellular Components (CC), Biological Process (BP), and Molecular Function (MF) enrichment analysis on the differential genes between samples [65], and topGO was used to draw a directed acyclic graph for the enriched term to visually display the GO term of differentially expressed gene enrichment and their hierarchical (more specific) relationship.
4.5. KEGG Enrichment Analysis of Differential Genes
KEGG (Kyoto Encyclopedia of Genes and Genomes) database was screened for pathway analysis on differential protein coding genes. The hypergeometric distribution test was used to calculate the significance p-value of differential gene enrichment in each pathway entry. KEGG enrichment analysis of top20 pathway entries corresponding to the number of differential genes greater than 2, were sorted by the -log10Pvalue and presented by bubble chart (Figure 5; Results).
4.6. cDNA Synthesis and RT-qPCR
Total RNA (1µg) was reverse-transcribed into cDNA using the AffinityScript QPCR cDNASynthesis Kit (Agilent, CA, USA). The reverse transcription reaction was carried out with universal oligo-dT primer and reverse transcriptase (RT) in optimized thermal conditions: 25°C for 5 min., 42°C for 5 min. (oligo-dT annealing), 55°C for 15 min. (reverse transcription), 95°C for 5 min. (enzyme inactivation) in Biometra Basic thermocycler (Biometra, Germany). For differentially expressed genes uncovered in this study oligonucleotides were designed using Primer3plus software (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). For structural genes, encoding key enzymes of anthocyanin biosynthesis pathway, oligonucleotides published by Kondo et al. [24] were used. Gene encoding ACTIN [39] was used as a RT-qPCR data normalizer (Table 5).
RT-qPCR was performed with Kapa SYBR qPCR kit in the presence of SYBR Green fluorescent dye (KapaBiosystems, USA) using RotorGen 6000 thermal cycler (Corbett Research, Australia). In each experimental setup, two pairs of specific primers, complementary to the evaluated structural gene and the selected target DEGs, were used in analogous reactions. The cDNA template was prepared in dilutions of known concentrations, enabling the preparation of a standard amplification reaction curve. The thermal profile of the RT-qPCR reaction was as follow: 95°C for 5 min. (polymerase activation), than 40 cycles including steps: 95°C for 15 s (denaturation), 60°C for 20 s (oligonucleotide annealing), 72°C for 20 s (fluorescence level detection). Relative expression (fold change) normalized in regard to ACTIN was determined on the basis of single data points derived from real-time PCR amplification curve threshold cycles (Ct values) (2-∆∆Ct) described by Livak and Schmittgen [66]. For this purpose Rotor-Gene 6000 Series Software 1.7 was used (Corbett Research, Australia). The average of relative expression was normalized to the white flesh fruit control ‘Free Redstar’. Standard error of the mean ± SEM and t-significance at the levels of p-value> 0.05 (*), 0.01 (**), 0.001 (***) between ‘Free Redstar’ and red fleshed cvs were calculated separately (GraphPad Prism 10.0.3). Relative fold changes diagrams for each gene were drawn using GraphPad Prism 10.0.3. The gene-to-gen correlation matrix between structural and newly uncovered gens was calculated using Pearson correlation coefficient (r=1, -1) with p-value<0,05 (GraphPad Prism 10.0.3 software).
5. Conclusions
In this research we have carried out the evaluation of the expression profiles of genes related to the plant hormonal pathway in apple fruits. We have defined the set of genes showing diverse activity in immature and ripe fruits of red fleshed apple cultivars. The group of selected genes, ABF, SnRK2, as well as MYC2, NPR1 and DELLA seem to be involved in positive or negative regulation of plant signaling pathways and may play crucial role, mediating the abscisic acid, jasmonic acid, salicylic acid and gibberellin pathways respectively, thus influencing in anthocyanin transformation, accumulation and biosynthesis. All investigated genes showed significant correlation with the UFGT gene in ripe red-fleshed fruits. So far, there are many reports devoted to research on the involvement of auxins, cytokinine and brassinosteroids in apple fruit ripening and fruit flesh pigmentation. The molecular basis for the regulation of other hormonal transduction factors and their impact on apple red fruit flesh phenotype constitution have not been deeply investigated so far. Despite the preliminary research conducted, further analyzes, including important fruit quality parameters, such as fruits vitamin C, polyphenols, and anthocyanins value, are necessary to determine the suitability of uncovered genes to be used for early selection of red-fleshed apple varieties by molecular marker assisted selection (MAS) procedure.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, and methodology, Sylwia Keller-Przybylkowicz and Xiaoyun Du; expression analysis, writing and original draft preparation, Sylwia Keller-Przybylkowicz; software, Michal Oskiera; validation, Xueqing Liu, Laiqing Song and Lingling Zhao; visualization, Agnieszka Walencik and Norbert Kowara; supervision of the data analysis and manuscript verification, Grzegorz Bartoszewski; project administration, Dorota Kruczyńska. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the external project “Double-Hundred experts exchange” with the agreement between The National Institute of Horticultural Science, Poland and Yantai Academy of Agricultural Science, Yantai, China.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
“The authors declare no conflict of interest.”.
References
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Wang, N.; Chen, X. Genetics and genomics of fruit color development in apple. In The Apple Genome; Korban, S.S., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 271–295. [Google Scholar]
- Available online: https://www.fao.org/faostat/en.
- Van Nocker, S.; Berry, G.; Najdowski, J.; Michelutti, R.; Luffman, M.; Forsline, P.; Alsmairat, N.; Beaudry, R.; Nair, M.G.; Ordidge, M. Genetic diversity of red-fleshed apples (Malus). Euphytica 2012, 185, 281–293. [Google Scholar] [CrossRef]
- Mieszczakowska-Frąc, M.; Buczek, M.; Kruczyńska, D.; Markowski, J. Cloudy red-fleshed apple juice production and quality. Pol. J. Natur. Sc. 2015, 30, 59–72. [Google Scholar]
- Zhou, Z.Q.; Li, Y.N. The RAPD evidence for the phylogenetic relationship of the closely related species of cultivated apple. Genet. Resour. Crop Evol. 2000, 47, 353–357. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, S.; Zhang, Z.; Fang, H.; Xu, H.; Wang, Y.; Chen, X. Malus sieversii: the origin, flavonoid synthesis mechanism, and breeding of red-skinned and red-fleshed apples. Hortic. Res. 2018, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, L.; Liu, W.; Zhang, J.; Wang, N.; Chen, X. Research progress of fruit color development in apple (Malus domestica Borkh.). Plant Physiol. Biochem. 2021, 162, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, H.; Jiang, S.; Zhang, Z.; Lu, N.; Qiu, H.; Qu, C.; Wang, Y.; Wu, S.; Chen, X. MYB12 and MYB22 play essential roles in proanthocyanidin and flavonol synthesis in red-fleshed apple (Malus sieversii f. niedzwetzkyana). Plant J. 2017, 90, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, C.; Alston, F.H.; Van Arkel, G.; Brown, L.M.; Chevreau, E.; Dunemann, F.; Evans, K.M.; Gardiner, S.; Guilford, P.; Van Heusden, A.W.; Janse, J.; Laurens, F.; Lynn, J.R.; Manganaris, A.G.; den Nijs, A.P.M.; Periam, N.; Rikkerink, E.; Roche, P.; Ryder, C.; Sansavini, S.; Schmidt, H.; Tartarini, S.; Verhaegh, J.J.; Vrielink-van Ginkel, M.; King, G.J. Aligning male and female linkage maps of apple (Malus pumila Mill.) using multi-allelic markers. Theor. Appl. Genet. 1998, 97, 60–73. [Google Scholar] [CrossRef]
- Chagné, D.; Carlisle, C.M.; Blond, C.; Volz, R.K.; Whitworth, C.J.; Oraguzie, N.C.; Crowhurst, R.N.; Allan, A.C.; Espley, R.V.; Hellens, R.P.; Gardiner, S.E. Mapping a candidate gene (MdMYB10) for red flesh and foliage colour in apple. BMC Genom. 2007, 8, 212. [Google Scholar] [CrossRef]
- Chagné, D.; Krieger, C.; Rassam, M.; Sullivan, M.; Fraser, J.; André, C.; Pindo, M.; Troggio, M.; Gardiner, S.E.; Henry, R.A.; Allan, A.C.; McGhie, T.K.; Laing, W.A. QTL and candidate gene mapping for polyphenolic composition in apple fruit. BMC Plant Biol. 2012, 12, 12. [Google Scholar] [CrossRef]
- Yang, W.; Feng, H.; Zhang, X.; Zhang, J.; Doonan, J.H.; Batchelor, W.D.; Xiong, L.; Yan, J. Crop Phenomics and High-Throughput Phenotyping: Past Decades, Current Challenges, and Future Perspectives. Mol. Plant 2020, 13, 187–214. [Google Scholar] [CrossRef]
- Hertog, M.G.; Feskens, E.J.; Hollman, P.C.; Katan, M.B.; Kromhout, D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet 1993, 342, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Knekt, P.; Kumpulainen, J.; Järvinen, R.; Rissanen, H.; Heliövaara, M.; Reunanen, A.; Hakulinen, T.; Aromaa, A. Flavonoid intake and risk of chronic diseases. Am. J. Clin. Nutr. 2002, 76, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Takos, A.M.; Jaffé, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Treutter, D. Biosynthesis of phenolic compounds and its regulation in apple. Plant Growth Regul. 2001, 34, 71–89. [Google Scholar] [CrossRef]
- Pelletier, M.K.; Shirley, B.W. Analysis of flavanone 3-hydroxylase in Arabidopsis seedlings. Coordinate regulation with chalcone synthase and chalcone isomerase. Plant Physiol. 1996, 111, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red coloration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Dick-Pérez, M.; Zhang, Y.; Hayes, J.; Salazar, A.; Zabotina, O.A.; Hong, M. Structure and interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR. Biochemistry 2011, 50, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef]
- Honda, C.; Kotoda, N.; Wada, M.; Kondo, S.; Kobayashi, S.; Soejima, J.; Zhang, Z.; Tsuba, T.; Moriguchi, T. Anthocyanin biosynthetic genes are coordinately expressed during red coloration in apple skin. Plant Physiol. Biochem. 2002, 40, 955–962. [Google Scholar] [CrossRef]
- Kondo, S.; Hiraoka, K.; Kobayashi, S.; Honda, C.; Terahara, N. Changes in the expression of anthocyanin biosynthetic genes during apple development. J. Am. Soc. Hortic. Sci. 2002, 127, 971–997. [Google Scholar] [CrossRef]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, E.; Soltani, B.M.; Yadollahi, A.; Hosseini, E. Independence of color intensity variation in red flesh apples from the number of repeat units in promoter region of the MdMYB10 gene as an allele to MdMYB1 and MdMYBA. Iran. J. Biotechnol. 2012, 10, 153–160. [Google Scholar]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, Z.; Jiang, S.; Xu, H.; Wang, Y.; Feng, S.; Chen, X. Synergistic effects of light and temperature on anthocyanin biosynthesis in callus cultures of red-fleshed apple (Malus sieversii f. niedzwetzkyana). Plant Cell, Tissue Organ Cult. 2016, 127, 217–227. [Google Scholar] [CrossRef]
- Wang, N.; Liu, W.; Zhang, T.; Jiang, S.; Xu, H.; Wang, Y.; Zhang, Z.; Wang, C.; Chen, X. Transcriptomic Analysis of Red-Fleshed Apples Reveals the Novel Role of MdWRKY11 in Flavonoid and Anthocyanin Biosynthesis. J. Agric. Food Chem. 2018, 66, 7076–7086. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Jiang, H.; Mao, Z.; Wang, N.; Jiang, S.; Xu, H.; Yang, G.; Zhang, Z.; Chen, X. The R2R3-MYB transcription factor MdMYB24-like is involved in methyl jasmonate-induced anthocyanin biosynthesis in apple. Plant Physiol. Biochem. 2019, 139, 273–282. [Google Scholar] [CrossRef]
- Espley, R.V.; Brendolise, C.; Chagné, D.; Kutty-Amma, S.; Green, S.; Volz, R.; Putterill, J.; Schouten, H.J.; Gardiner, S.E.; Hellens, R.P.; Allan, A.C. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 2009, 21, 168–183. [Google Scholar] [CrossRef]
- Espley, R.V.; Bovy, A.; Bava, C.; Jaeger, S.R.; Tomes, S.; Norling, C.; Crawford, J.; Rowan, D.; McGhie, T.K.; Brendolise, C.; Putterill, J.; Schouten, H.J.; Hellens, R.P.; Allan, A.C. Analysis of genetically modified red-fleshed apples reveals effects on growth and consumer attributes. Plant Biotechnol. J. 2013, 11, 408–419. [Google Scholar] [CrossRef]
- Chagné, D.; Crowhurst, R.N.; Troggio, M.; Davey, M.W.; Gilmore, B.; Lawley, C.; Vanderzande, S.; Hellens, R.P.; Kumar, S.; Cestaro, A.; Velasco, R.; Main, D.; Rees, J.D.; Iezzoni, A.; Mockler, T.; Wilhelm, L.; Van de Weg, E.; Gardiner, S.E.; Bassil, N.; Peace, C. Genome-Wide SNP Detection, Validation, and Development of an 8K SNP Array for Apple. PLoS ONE 2012, 7, e31745. [Google Scholar] [CrossRef]
- Honda, C.; Moriya, S. Anthocyanin Biosynthesis in Apple Fruit. Hort. J. 2018, 87, 305–314. [Google Scholar] [CrossRef]
- Ireland, H.S.; Guillen, F.; Bowen, J.H.; Tacken, E.; Putterill, J.; Schaffer, R.J.; Johnston, J.W. Mining the apple genome reveals a family of nine ethylene receptor genes. Postharvest Biol. Technol. 2012, 72, 42–46. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Y.; Zhao, L.; Li, C.; Yu, J.; Li, T.; Yang, W.; Zhang, S.; Su, H.; Wang, L. A novel NAC transcription factor, MdNAC42, regulates anthocyanin accumulation in red-fleshed apple by interacting with MdMYB10. Tree Physiol. 2020, 40, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, S.; Liu, W.; Yang, M.; Zhang, Z.; Wang, N.; Chen, X. MdJa2 Participates in the Brassinosteroid Signaling Pathway to Regulate the Synthesis of Anthocyanin and Proanthocyanidin in Red-Fleshed Apple. Front. Plant Sci. 2022, 13, 830349. [Google Scholar] [CrossRef]
- Wang, N.; Qu, C.; Jiang, S.; Chen, Z.; Xu, H.; Fang, H.; Su, M.; Zhang, J.; Wang, Y.; Liu, W.; Zhang, Z.; Lu, N.; Chen, X. The proanthocyanidin-specific transcription factor MdMYBPA1 initiates anthocyanin synthesis under low-temperature conditions in red-fleshed apples. Plant J. 2018, 96, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, N.; Liu, J.; Qu, C.; Wang, Y.; Jiang, S.; Lu, N.; Wang, D.; Zhang, Z.; Chen, X. The molecular mechanism underlying anthocyanin metabolism in apple using the MdMYB16 and MdbHLH33 genes. Plant Mol. Biol. 2017, 94, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, I.; Tariq, R.; Nazir, T.; Khan, I.; Basit, A.; Gul, H.; Anwar, T.; Awan, S.A.; Bacha, S.A.S.; Zhang, L.; Zhang, C.; Cong, P. RNA-Seq profiling reveals the plant hormones and molecular mechanisms stimulating the early ripening in apple. Genomics 2021, 113, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; An, Y.Y.; Wang, L.J. 24-Epibrassinolide enhances 5-ALA-induced anthocyanin and flavonol accumulation in calli of ‘Fuji’ apple flesh. Plant Cell, Tissue Organ Cult. 2018, 134, 319–330. [Google Scholar] [CrossRef]
- Ye, H.; Li, L.; Yin, Y. Recent advances in the regulation of brassinosteroid signaling and biosynthesis pathways. J. Integr. Plant Biol. 2011, 53, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Lisso, J.; Altmann, T.; Müssig, C. Metabolic changes in fruits of the tomato dx mutant. Phytochemistry 2006, 67, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Symons, G.M.; Davies, C.; Shavrukov, Y.; Dry, I.B.; Reid, J.B.; Thomas, M.R. Grapes on steroids. Brassinosteroids are involved in grape berry ripening. Plant Physiol. 2006, 140, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.Q.; Mao, W.H.; Shi, K.; Zhou, Y.H.; Asami, T.; Yu, J.Q. A role of brassinosteroids in early fruit development in cucumber. J. Exp. Bot. 2008, 59, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.M.; Zhang, Q.; Tian, L.; Li, C.L.; Xing, Y.; Qin, L.; Shen, Y.Y. Brassinosteroid is involved in strawberry fruit ripening. Plant Growth Regul. 2013, 69, 63–69. [Google Scholar] [CrossRef]
- Sasaki, Y.; Asamizu, E.; Shibata, D.; Nakamura, Y.; Kaneko, T.; Awai, K.; Amagai, M.; Kuwata, C.; Tsugane, T.; Masuda, T.; Shimada, H.; Takamiya, K.; Ohta, H.; Tabata, S. Monitoring of methyl jasmonate-responsive genes in Arabidopsis by cDNA macroarray: Self-activation of jasmonic acid biosynthesis and crosstalk with other phytohormone signaling pathways. DNA Res. 2001, 8, 153–161. [Google Scholar] [CrossRef]
- Qi, T.; Song, S.; Ren, Q.; Wu, D.; Huang, H.; Chen, Y.; Fan, M.; Peng, W.; Ren, C.; Xie, D. The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 2011, 23, 1795–1814. [Google Scholar] [CrossRef]
- An, X.H.; Tian, Y.; Chen, K.Q.; Liu, X.J.; Liu, D.D.; Xie, X.B.; Cheng, C.G.; Cong, P.H.; Hao, Y.J. MdMYB9 and MdMYB11 are involved in the regulation of the JA-induced biosynthesis of anthocyanin and proanthocyanidin in apples. Plant Cell Physiol. 2015, 56, 650–662. [Google Scholar] [CrossRef]
- Chen, L.; Xiang, S.; Chen, Y.; Li, D.; Yu, D. Arabidopsis WRKY45 Interacts with the DELLA Protein RGL1 to Positively Regulate Age-Triggered Leaf Senescence. Mol. Plant 2017, 10, 1174–1189. [Google Scholar] [CrossRef]
- Ji, X.H.; Wang, Y.T.; Zhang, R.; Wu, S.J.; An, M.M.; Li, M.; Wang, C.Z.; Chen, X.L.; Zhang, Y.M.; Chen, X.S. Effect of auxin, cytokinin and nitrogen on anthocyanin biosynthesis in callus cultures of red-fleshed apple (Malus sieversii f. niedzwetzkyana). Plant Cell, Tissue Organ Cult. 2015, 120, 325–337. [Google Scholar] [CrossRef]
- Deikman, J.; Hammer, P.E. Induction of Anthocyanin Accumulation by Cytokinins in Arabidopsis thaliana. Plant Physiol. 1995, 108, 47–57. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Jeong, C.Y.; Kang, G.; Yoo, S.; Hong, S.; Lee, H. MYBD employed by HY5 increases anthocyanin accumulation via repression of MYBL2 in Arabidopsis. Plant J. 2015, 84, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.H.; Zhang, R.; Wang, N.; Yang, L.; Chen, X.S. Transcriptome profiling reveals auxin suppressed anthocyanin biosynthesis in red-fleshed apple callus (Malus sieversii f. niedzwetzkyana). Plant Cell Tissue Organ Cult. 2015, 123, 389–404. [Google Scholar] [CrossRef]
- Jing, Y.; Joyce, D.C. ABA effects n ethylene production, PAL activity, anthocyanin and polyphenol contents of strawberry fruit. Plant Grow Regul. 2023, 39, 171–174. [Google Scholar] [CrossRef]
- Ban, T.; Shiozaki, S.; Ogata, T.; Horiuchi, S. Effects of abscisic acid and shading treatments on the level of anthocyanin and resveratrol in skin of kyoho grape berry. Acta Hortic. 2000, 514, 83–90. [Google Scholar] [CrossRef]
- Singh, S.P.; Saini, M.K.; Sinhg, J.; Pongener, A.; Singh, G.S. Postharvest application of abscisic acid promotes anthocyanin accumulation in pericarp of litchi fruit without adversely affecting postharvest quality. Postharv. Biol. Technol. 2014, 96, 92–100. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, T. RNA Isolation From Highly Viscous Samples Rich in Polyphenols and Polysaccharides. Plant. Mol. Biol. Rep. 2002, 20, 417–417. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level–the DESeq Package. Eur. Mol. Biol. Lab. 2012, 10, f1000research. [Google Scholar]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. R Package Version 2010, 2, 2010. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta DeltaC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Heatmap of Pearson’s correlation (a), and hierarchical clustering (b) between Red Love® ‘General’ and ‘Early Fuji’ samples calculated based on gene expression FPKM values.
Figure 1.
Heatmap of Pearson’s correlation (a), and hierarchical clustering (b) between Red Love® ‘General’ and ‘Early Fuji’ samples calculated based on gene expression FPKM values.
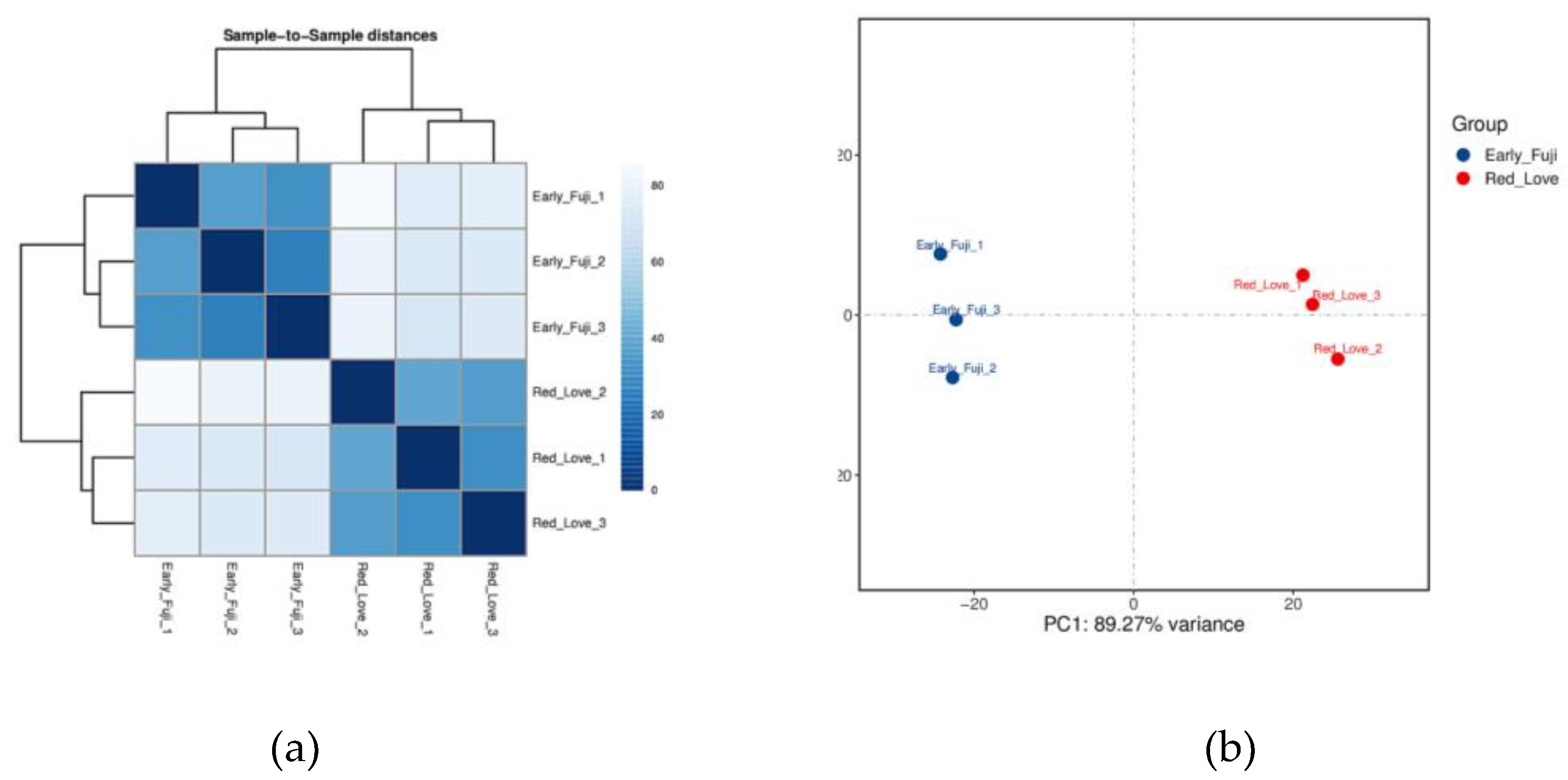
Figure 2.
Summary of the differentially expressed genes (DEGs) identified in fruits of red-fleshed apple cultivar Red Love® ‘General’, as compared to white-fleshed ‘Early Fuji’. (a) The volcano plot shows the filtered data extraction of genes up vs. down regulated in accordance to p-value. (b) Numbers of DEGs detected in comparison.
Figure 2.
Summary of the differentially expressed genes (DEGs) identified in fruits of red-fleshed apple cultivar Red Love® ‘General’, as compared to white-fleshed ‘Early Fuji’. (a) The volcano plot shows the filtered data extraction of genes up vs. down regulated in accordance to p-value. (b) Numbers of DEGs detected in comparison.
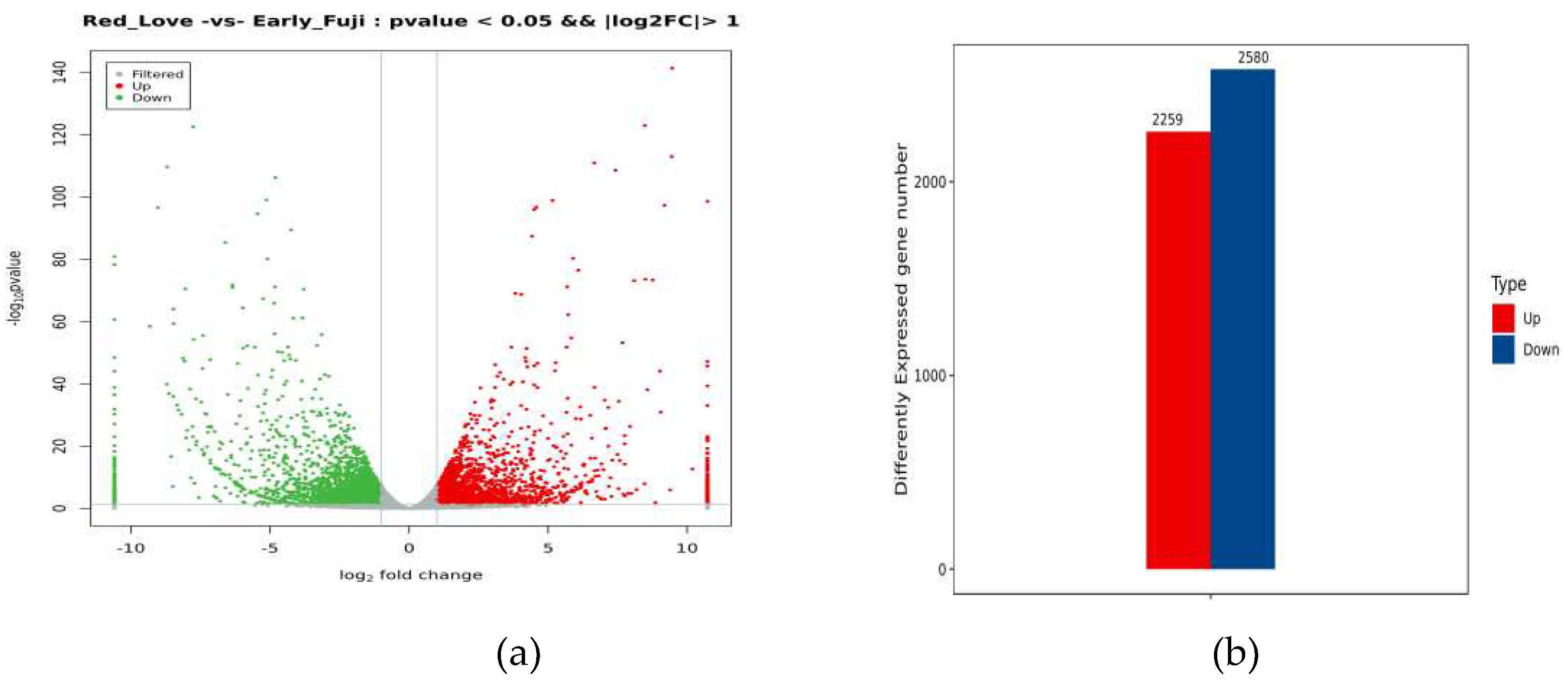
Figure 3.
Gene ontology (GO) classification.
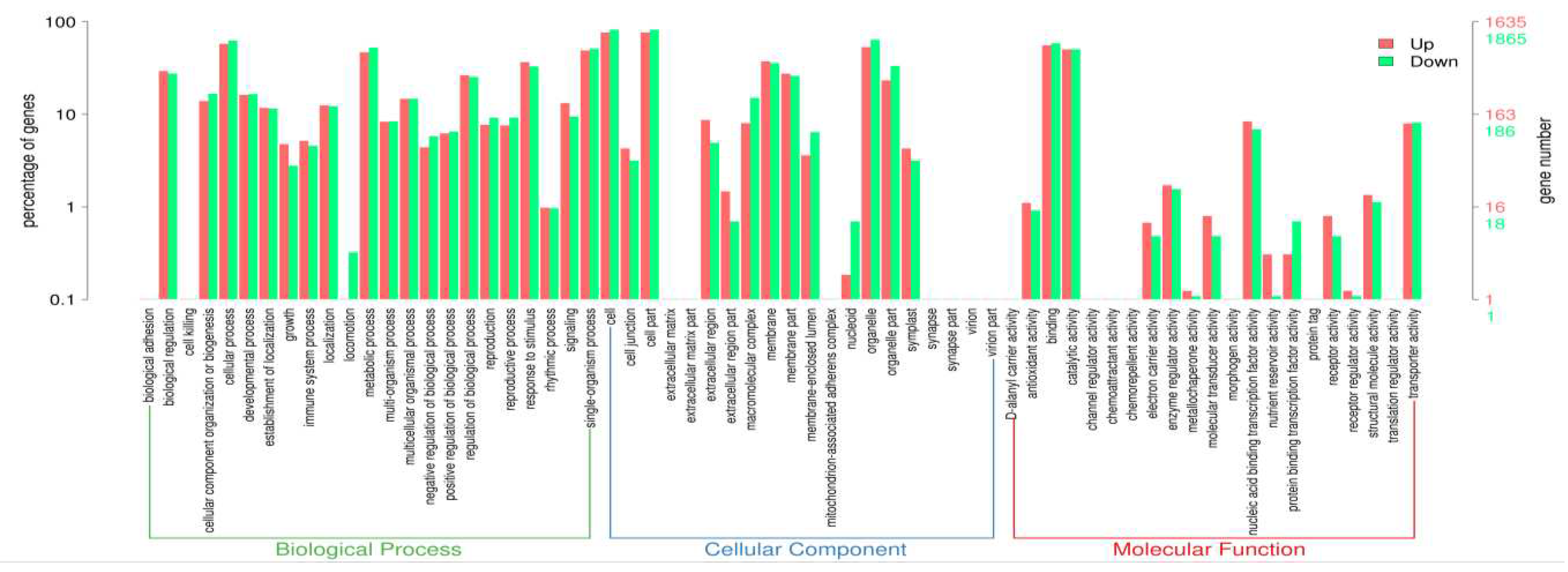
Figure 4.
Summary of the DEGs functional enrichment analysis. (a) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway classification of DEGs identified in red-fleshed fruits of Red Love® ‘General’ in comparison with white-fleshed ‘Early Fuji’. (b) GO term classification of top genes up regulated in red flesh fruits of Red Love® ‘General’.
Figure 4.
Summary of the DEGs functional enrichment analysis. (a) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway classification of DEGs identified in red-fleshed fruits of Red Love® ‘General’ in comparison with white-fleshed ‘Early Fuji’. (b) GO term classification of top genes up regulated in red flesh fruits of Red Love® ‘General’.
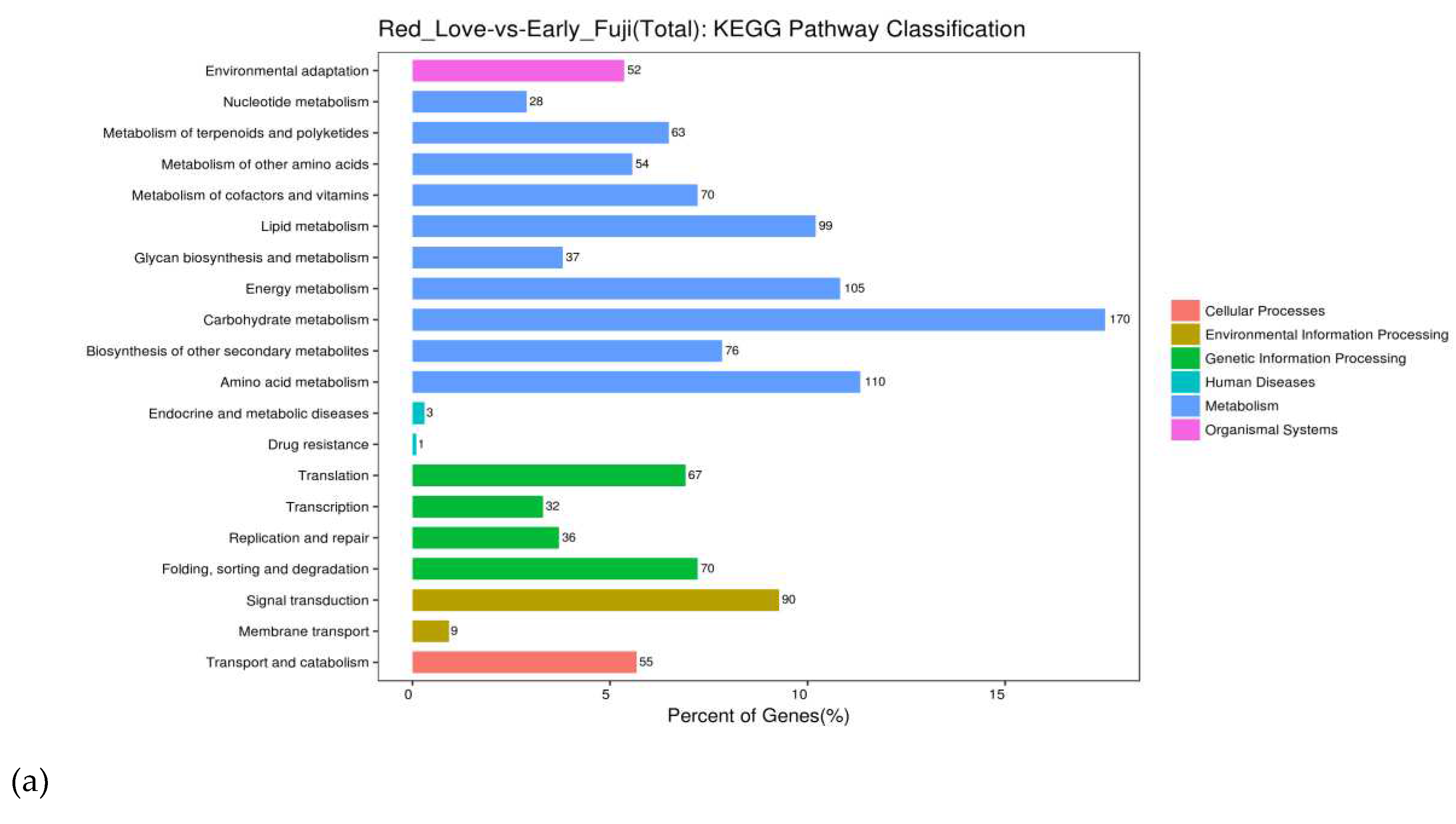
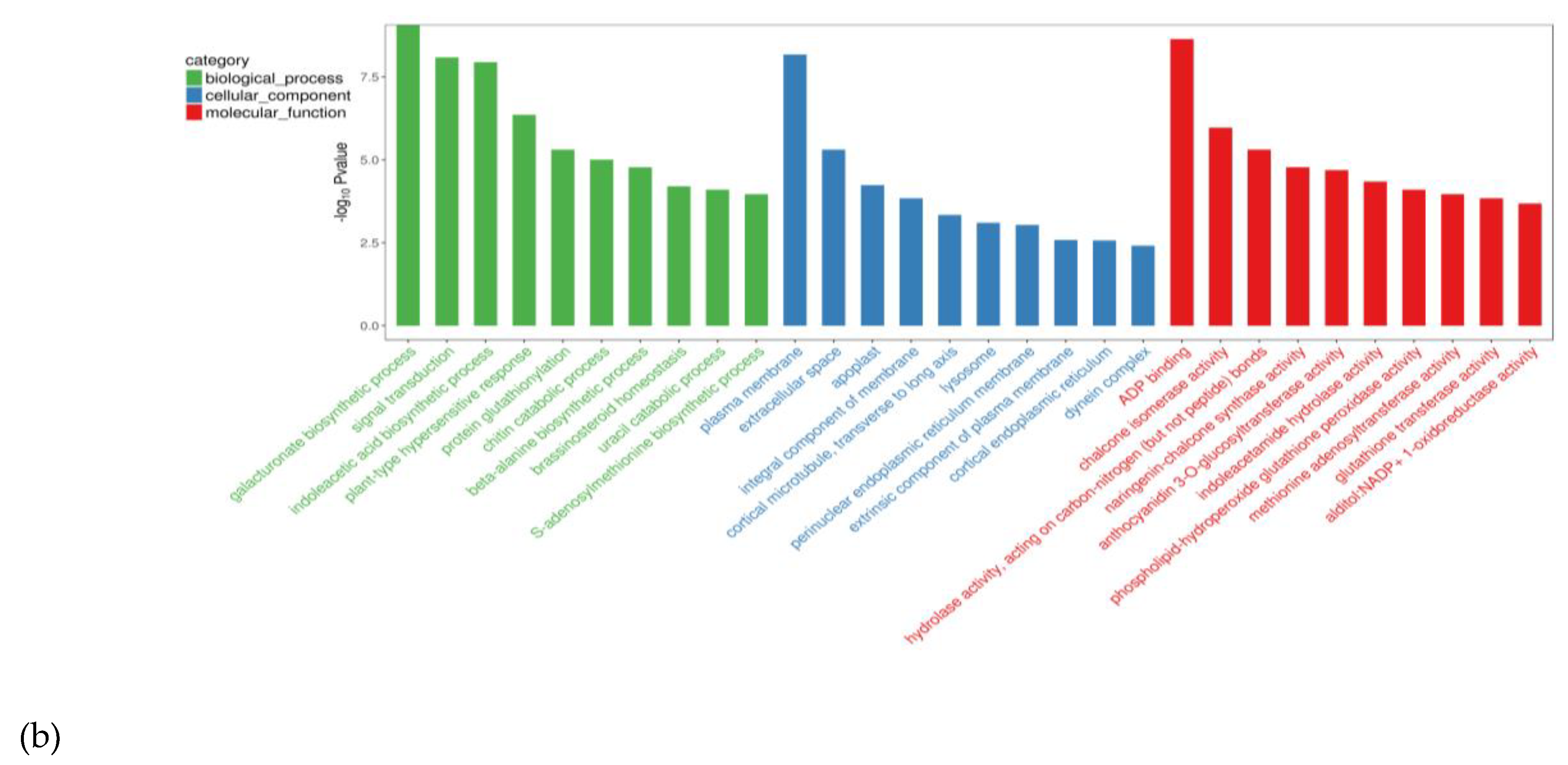
Figure 5.
Classification of genes from top 20 selected KEGG enrichments based on fold change significance value.
Figure 5.
Classification of genes from top 20 selected KEGG enrichments based on fold change significance value.
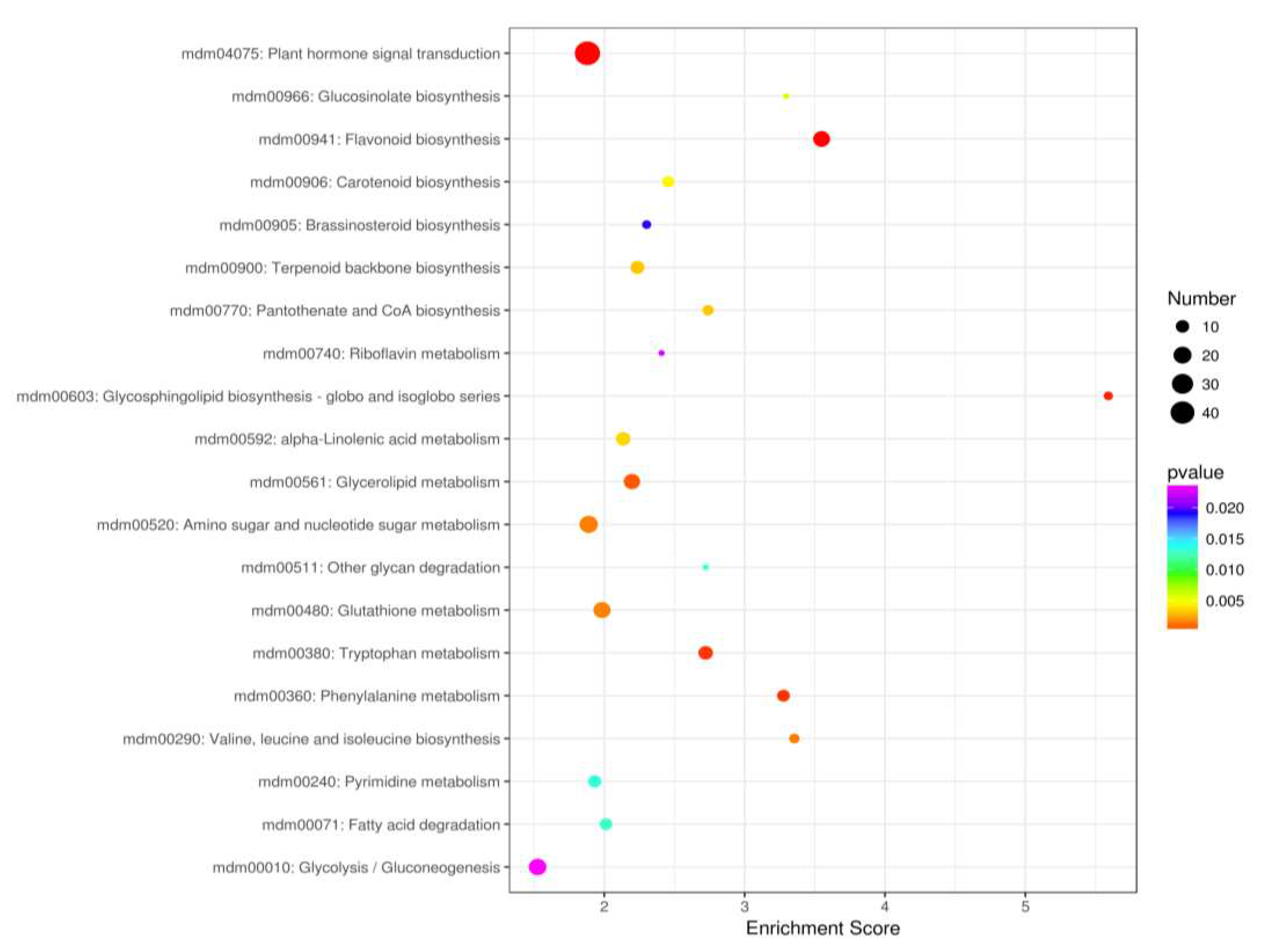
Figure 6.
KEGG plant hormone signal transduction pathway with differentially expressed genes in fruits of red-fleshed Red Love® ‘General’. Red marked genes are up-regulated, green down-regulated, and yellow up- or down- regulated.
Figure 6.
KEGG plant hormone signal transduction pathway with differentially expressed genes in fruits of red-fleshed Red Love® ‘General’. Red marked genes are up-regulated, green down-regulated, and yellow up- or down- regulated.
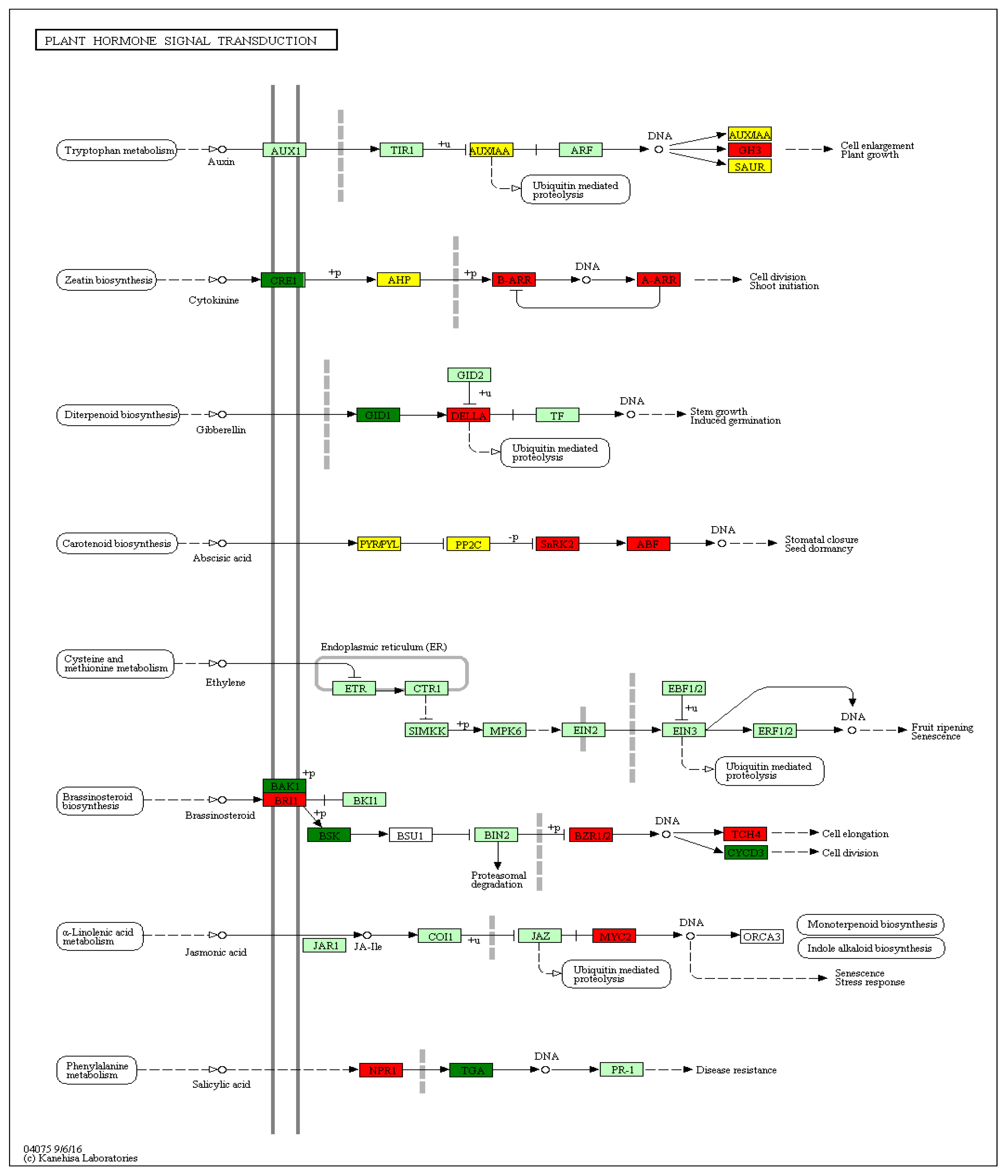
Figure 7.
Visualization of apple fruit flesh coloration; ‘Free Redstar’ – fruit flesh control; 1-‘Trinity’, 2-‘Alex Red’, 3-Red Loce® ‘Era’, 4-Red Love® ‘Circe’, 5-‘Roxana’, 6- M. sieviersii f. niedzwetzkyana, 6-Red Love® ‘Sirena’; white and black ruler (with default 1 x 1cm dimension squares) illustrates fruit size.
Figure 7.
Visualization of apple fruit flesh coloration; ‘Free Redstar’ – fruit flesh control; 1-‘Trinity’, 2-‘Alex Red’, 3-Red Loce® ‘Era’, 4-Red Love® ‘Circe’, 5-‘Roxana’, 6- M. sieviersii f. niedzwetzkyana, 6-Red Love® ‘Sirena’; white and black ruler (with default 1 x 1cm dimension squares) illustrates fruit size.
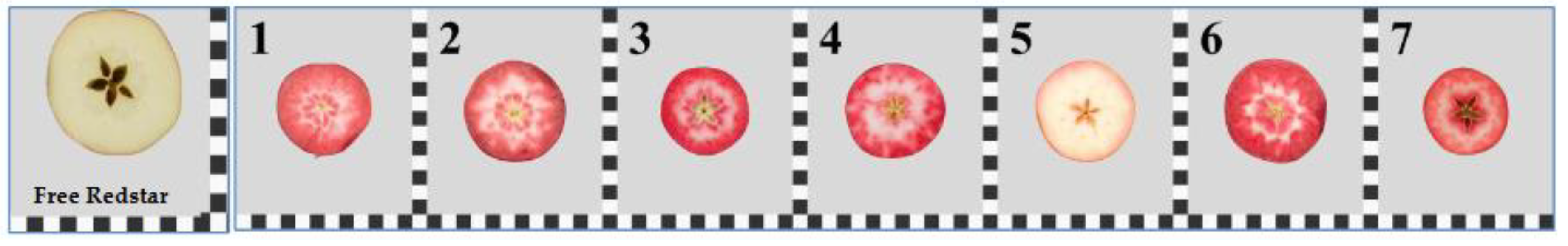
Figure 8.
Validation of differentially expressed genes for immature fruits of apple cultivars with different flesh color (only significant changes in gene expression was showed). Diagrams present an average relative gene expression data with standard error of the mean (±SEM) compared to white flesh ‘Free Redstar’ and t-test significance calculation level p<0,05*, 0,01 **, 0,001***, normalized to ACTIN gene (showing stable expression in the experiment layout). The relative expression of gene of interests was calculated using mathematical equation 2-ΔΔCT (RotorGene 6000 Series software 1.7) and visualized with GraphPad Prism10.0.3 software.
Figure 8.
Validation of differentially expressed genes for immature fruits of apple cultivars with different flesh color (only significant changes in gene expression was showed). Diagrams present an average relative gene expression data with standard error of the mean (±SEM) compared to white flesh ‘Free Redstar’ and t-test significance calculation level p<0,05*, 0,01 **, 0,001***, normalized to ACTIN gene (showing stable expression in the experiment layout). The relative expression of gene of interests was calculated using mathematical equation 2-ΔΔCT (RotorGene 6000 Series software 1.7) and visualized with GraphPad Prism10.0.3 software.
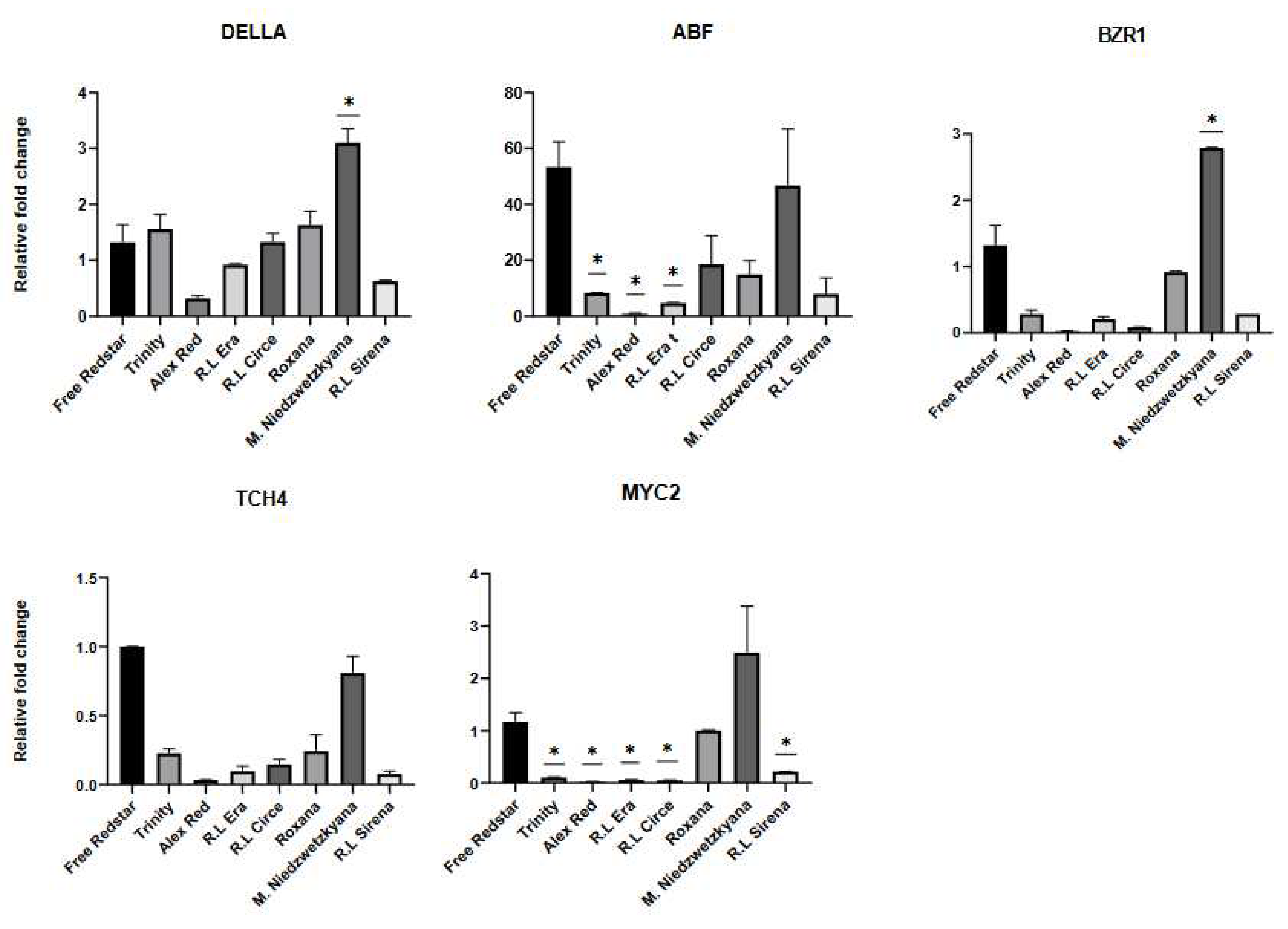
Figure 9.
Validation of differentially expressed genes in ripe fruits of apple cultivars analyzed.
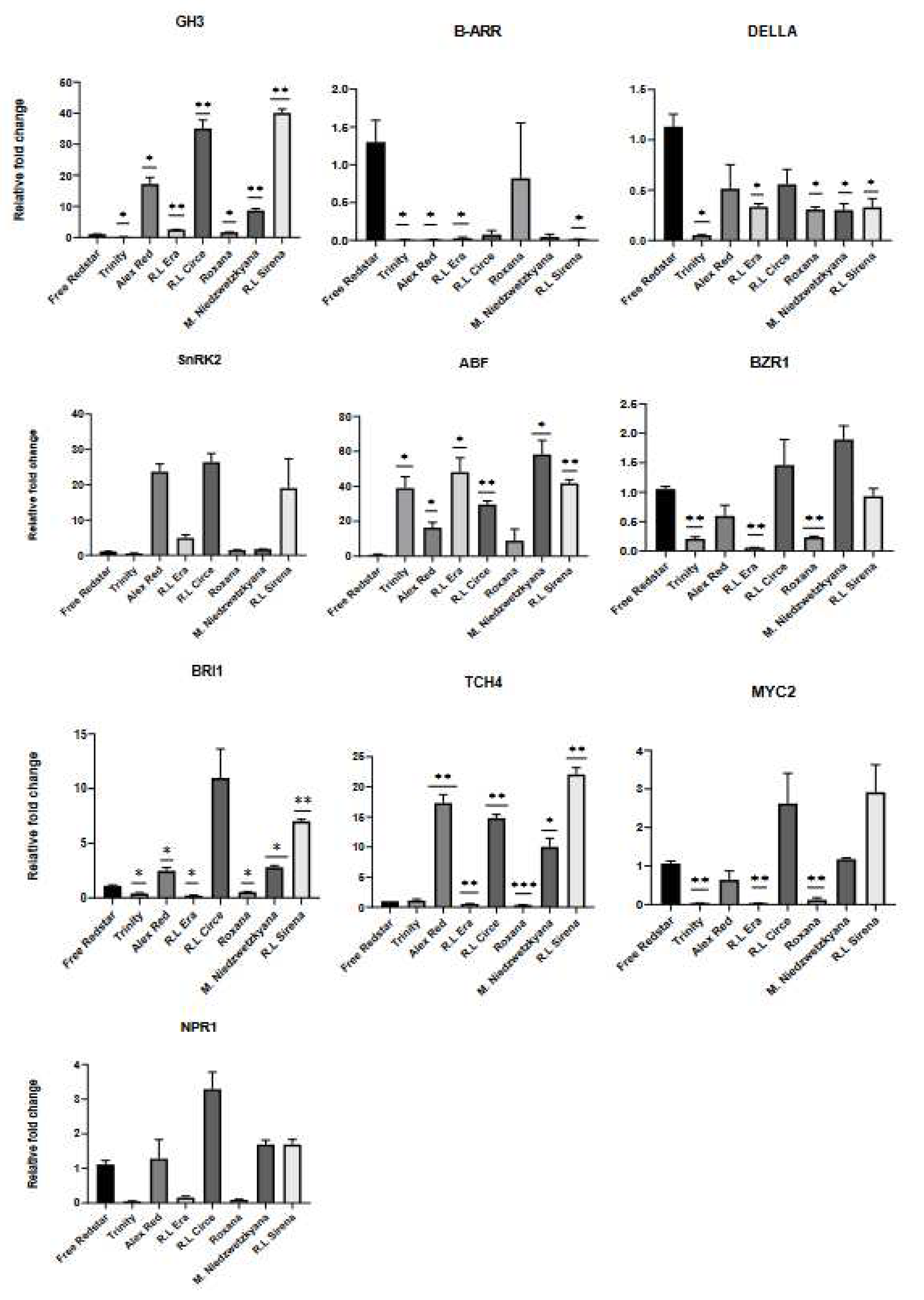
Figure 10.
Expression profiles of structural genes of anthocyanins biosynthesis pathway, evaluated in immature fruits of apple cultivars.
Figure 10.
Expression profiles of structural genes of anthocyanins biosynthesis pathway, evaluated in immature fruits of apple cultivars.
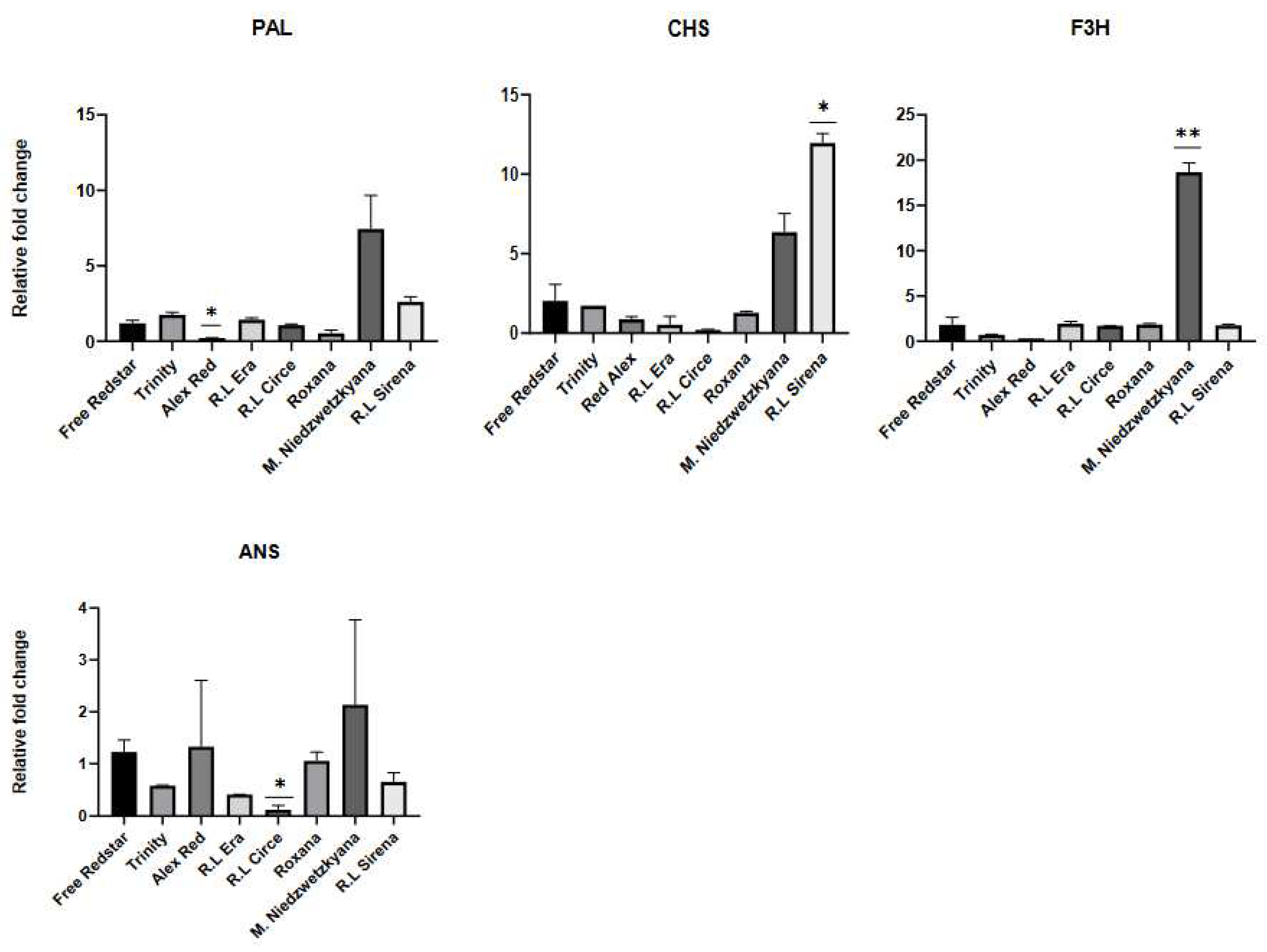
Figure 11.
Expression profiles of structural genes of anthocyanins biosynthesis pathway, evaluated in ripe fruits of apple cultivars.
Figure 11.
Expression profiles of structural genes of anthocyanins biosynthesis pathway, evaluated in ripe fruits of apple cultivars.
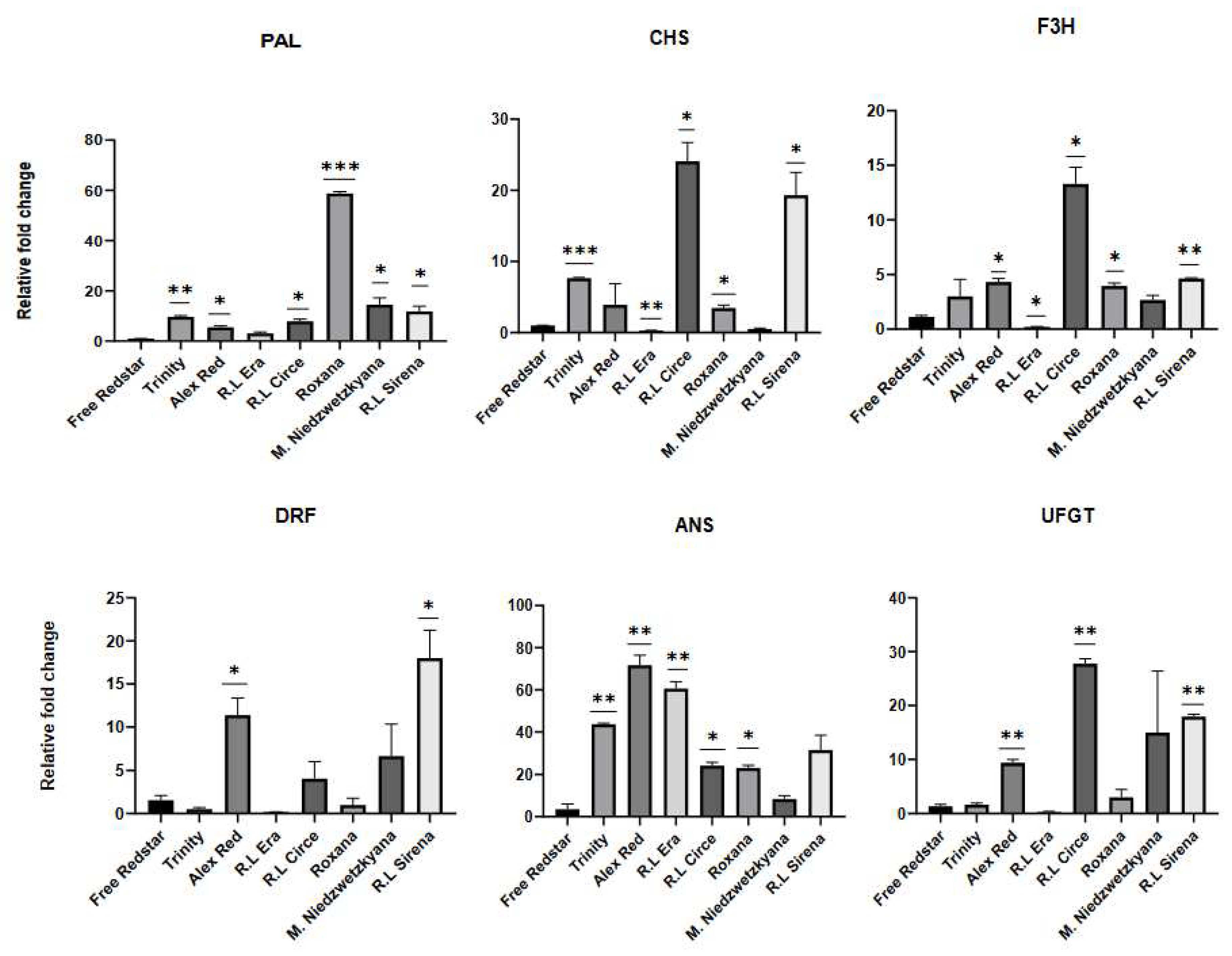
Figure 12.
Simulated scheme of the mechanism for anthocyanin synthesis regulation predicted by the activity modulation of genes from plant hormone and transduction pathway. Green arrows and red prohibition signs show gene regulation (positive and negative regulatory mechanism respectively) in response to the hormone signaling in red/white flesh apple phenotype. The right column shows the description of the putative mechanisms for anthocyanin accumulation in red fleshed apples.
Figure 12.
Simulated scheme of the mechanism for anthocyanin synthesis regulation predicted by the activity modulation of genes from plant hormone and transduction pathway. Green arrows and red prohibition signs show gene regulation (positive and negative regulatory mechanism respectively) in response to the hormone signaling in red/white flesh apple phenotype. The right column shows the description of the putative mechanisms for anthocyanin accumulation in red fleshed apples.
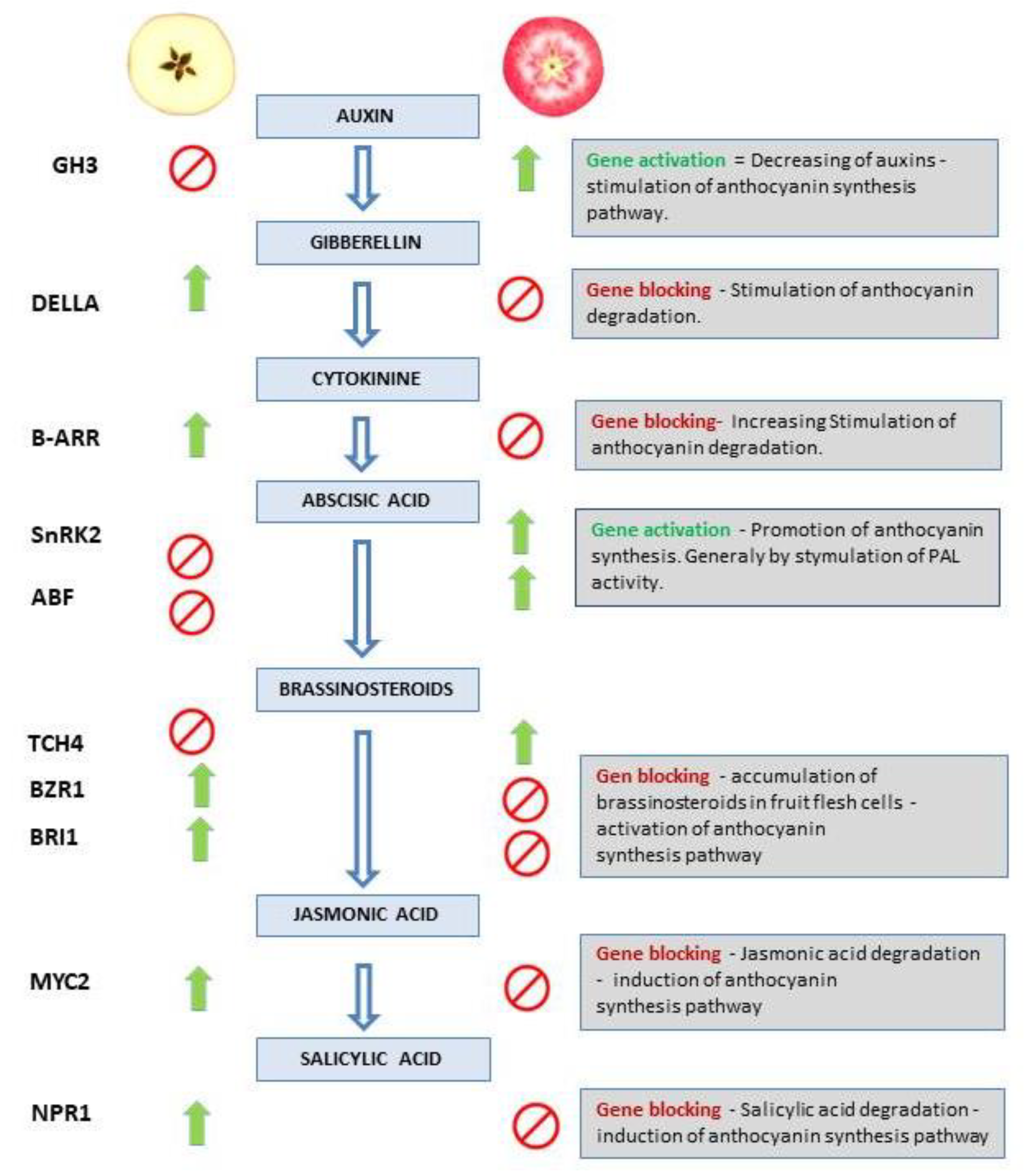

Table 3.
Characterization of the DEGs involved in hormonal signallig, up-regulated in red-fleshed fruits of RedLove® ‘General’.
Table 3.
Characterization of the DEGs involved in hormonal signallig, up-regulated in red-fleshed fruits of RedLove® ‘General’.
| Gene name | Locus | Hormone signaling pathway | Cellular localization and gene function | FoldChange (FC) |
| GH3 | LOC103436425 | Auxin | probable indole-3-acetic acid-amido synthetase GH3.1; auxin responsive GH3 gene family | 24,2 |
| B-ARR | LOC103400015 | Cytokinin | two-component response regulator ARR1-like isoform X1; regulator ARR-B family; Response_reg Myb_DNA-binding | 4,1 |
| DELLA | LOC103406747 | Gibberellin | DELLA protein GAI-like; DELLA protein | 2,6 |
| SnRK2 | LOC103429475 | ABA | serine/threonine-protein kinase SRK2; kinase PK_Tyr_Ser-Thr Choline_kinase PPP1R21_C; Cytoplasm, signaling pathway kinase family. | 2,7 |
| ABF | LOC103446587 | ABA | ABSCISIC ACID-INSENSITIVE 5-like protein 2; ABA responsive element binding factor. | 20,6 |
| BRI1 | LOC103410973 | Brassinosteroids | Plasma membrane, receptor S160/ brassinosteroid insensitive protein 1 | 2,2 |
| BZR1/2 | LOC103440434 | Brassinosteroids | BES1/BZR1 homolog protein 2-like; brassinosteroid resistant 1/2 | 5,2 |
| TCH4 | LOC103409272 | Brassinosteroids | probable xyloglucan endotransglucosylase/hydrolase protein 23 precursor; cell wall xyloglucan:xyloglucosyl transferase TCH4; | 5,7 |
| MYC2 | LOC103404780 | Jasmonic acid | Transcription factor; bHLH-MYC | 2,1 |
| NPR1 | LOC103454562 | Salicylic acid | Protein ubiqitination, BTB/POZ domain and ankyrin repeat-containing regulatory protein NPR1 | 2,9 |
Table 4.
Correlation matrix calculated between activity of genes from plant signal and hormone transduction and anthocyanin biosynthesis pathways, calculated by Pearson rank evaluation with the significance of p>0,05 (*), 0,01 (**) , 0,001 (***) and 0,0001 (****) evaluated for flesh of immature (a) and ripe (b) apple fruits.
Table 4.
Correlation matrix calculated between activity of genes from plant signal and hormone transduction and anthocyanin biosynthesis pathways, calculated by Pearson rank evaluation with the significance of p>0,05 (*), 0,01 (**) , 0,001 (***) and 0,0001 (****) evaluated for flesh of immature (a) and ripe (b) apple fruits.
| (a) | ||||||||||||||||||
| ABF | B-ARR | BZR1 | NPR1 | DELLA | GH3 | MYC2 | SnRK2 | BRI1 | TCH4 | |||||||||
| ANS | ** | |||||||||||||||||
| CHS | ||||||||||||||||||
| DRF | **** | *** | ** | ** | **** | |||||||||||||
| F3H | **** | * | **** | * | ** | **** | ||||||||||||
| PAL | * | * | ||||||||||||||||
| UFGT | ** | * | **** | *** | *** | * | ||||||||||||
| (b) | ||||||||||||||||||
| ABF | B-ARR | BZR1 | NPR1 | DELLA | GH3 | MYC2 | SnRK2 | BRI1 | TCH4 | |||||||||
| ANS | ||||||||||||||||||
| CHS | * | * | * | * | ||||||||||||||
| DFR | ||||||||||||||||||
| F3H | ||||||||||||||||||
| PAL | ** | ** | ** | |||||||||||||||
| UFGT1 | *** | **** | ** | * | ** | **** | * | *** | **** | *** | ||||||||
1 The significant correlation between genes from plant hormone transduction pathway and UFGT (anthocyanin biosynthesis gene) is underlined in ripe red-fleshed fruits.
Table 5.
Names and sequences of primers used to study expression profiles of genes involved in anthocyanin biosynthesis in apple fruit.
Table 5.
Names and sequences of primers used to study expression profiles of genes involved in anthocyanin biosynthesis in apple fruit.
| Gene abbreviation | Oligo 5’ | Oligo 3’ | Reference | |
| Differentially expressed genes | TCH4 | ctcaactggggaaccctaca | ggcattccaaaaagttgcat | This study - revealed in RNA-seq |
| BZR1 | tagtccgtcgtcttcgtcct | gagacggcgtaaaatgggta | ||
| BRI1 | gctttggaccaccttgacat | cacaagctctgcacacgaat | ||
| MYC2 | tgtttgggctgcagactatg | tccttcatttccatggtgg | ||
| NPR1 | gccttgagctcgtacagtcc | agaccccatttgatgagctg | ||
| GH3 | acagatccttcccgcctagt | aattggtgcgccataggtag | ||
| B-ARR | acttgcttcgccaaaagaaa | tgccatatattgcgcagttc | ||
| DELLA | tagtgacggttgtggagcag | ctccacttgcttagcggttc | ||
| SnRK2 | ggcgaatccttactgtacgc | gtctatgctctgggctggag | ||
| ABF | acaacggtcaccatcaacaa | ctgacgtcctcttccctcac | ||
| Structural genes | ANS | caatttggcctcaaacacct | tgagcttcaacaccaagtgc | Kondo et al. [24] |
| PAL | cggaaacttggactcggtaa | gatggagcctcttgcttgtc | ||
| DFR | gagtccgaatccgtttgtgtca | atgtttgtgggggctgtcgatg | ||
| UFGT | tccctttcactagccatgcaag | gtggaggatggagtttttacc | ||
| F3H | ggtgaactcaaacagcagca | ccactttggctttctccaag | ||
| CHS | acccacttggtcttttgcac | actaggccctcggaaggtaa | ||
| Reference | ACTIN | gactgtgaaactgcgaatggctca | catgaatcatcagagcaacgggca | Xu et al. [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



