
Submitted:
08 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Current research suggests that polycystic ovary syndrome (PCOS) might originate in utero and implicates the placenta in its pathogenesis. Kisspeptin (KISS1) and neurokinin B (NKB) are produced by the placenta in high amounts, and they have been implicated in several pregnancy complications associated with placental dysfunction. However, their placental expression has not been studied in PCOS. We isolated mRNA after delivery from the placentae of 31 PCOS and 37 control women with term, uncomplicated, singleton pregnancies. The expression of KISS1, NKB, and neurokinin receptors 1, 2, and 3 was analyzed with real-time polymerase chain reaction, using β-actin as the reference gene. Maternal serum and umbilical cord levels of total testosterone, sex hormone binding globulin (SHBG), free androgen index (FAI), androstenedione, dehydroepiandrosterone sulfate (DHEAS), Anti-Mullerian hormone (AMH), and estradiol were also assessed. NKB placental mRNA expression was higher in PCOS versus controls in pregnancies with female offspring. NKB expression depended on fetal gender, being higher in pregnancies with male fetuses. NKB was positively correlated with umbilical cord FAI and AMH, and KISS1 with cord testosterone and FAI; there was also a strong positive correlation between NKB and KISS1 expression. Women with PCOS had higher serum AMH and FAI and lower SHBG than controls. Our findings indicate that NKB might be involved in PCOS-related placental dysfunction, and warrant further investigation. Studies assessing the placental expression of NKB should take fetal gender into consideration.
Keywords:
polycystic ovary syndrome (PCOS)
; placenta
; pregnancy
; Neurokinin B (NKB)
; Kisspeptin (KISS1)
; placental expression
1. Introduction
Polycystic ovary syndrome (PCOS) is the commonest endocrine abnormality in reproductive-aged women, affecting 5-18% of them, depending on the diagnostic criteria used [1,2]. The syndrome is characterized by considerable phenotypic heterogeneity: menstrual disturbances (mostly in the form of oligomenorrhea), clinical manifestations of hyperandrogenemia (acne, hirsutism, androgenic alopecia), obesity and/or insulin resistance (IR) may affect the patients in different degrees [3]. Besides, women with PCOS frequently exhibit traditional risk factors for cardiovascular disease, including dyslipidemia, hypertension, metabolic syndrome, and type 2 diabetes [4]. PCOS is a major source of fertility problems, and it is the main cause of infertility related to anovulation [3]. Women with PCOS often need to use assisted reproduction technologies (ART) to achieve pregnancy and, even when this is accomplished, pregnancy is associated with increased risk of adverse outcomes such as gestational diabetes, hypertension and preeclampsia (PE), miscarriage, and preterm delivery [5]. Anxiety, depression, and eating disorders are among the psychological comorbidities of the syndrome [6].
It is evident that PCOS is a highly prevalent disease causing a significant burden to both patients and health care systems; hence, research aiming to understand its pathogenesis is intense. Even though PCOS etiology is a topic attracting considerable attention, the cause of this disorder remains elusive. Several hypotheses pertaining to PCOS origins have been formulated, and most of them assume an interplay between genetic, epigenetic, and environmental factors [7]. Genome Wide Association Studies (GWAS) have identified several genetic loci with genetic susceptibility for PCOS, but the evidence supporting the relationship between these loci and the clinical manifestations of the syndrome are considered inconclusive [7]. Furthermore, it is estimated that these loci can explain less than 10% of PCOS heritability [8]. Obesity and exposure to endocrine disrupting chemicals are also considered significant environmental contributors favoring PCOS development in genetically susceptible individuals [9]. Epigenetic mechanisms could also explain why only a subset of genetically predisposed individuals will manifest the syndrome, as it is known that factors external to the DNA can affect gene expression. A plausible theory is that hyper-exposure of the female fetus to androgens in utero influences the expression of several genes, particularly those regulating ovarian steroid production, insulin action and GnRH pulsatility, leading to a “re-programming” of the reproductive axis which later in life manifests as PCOS [7].
Indeed, data from animal studies have shown that prenatal exposure to excess androgen results in reproductive as well as metabolic disturbances that are typical of PCOS [10]. In humans, data are very limited but suggest that fetuses of PCOS women are exposed to increased androgen levels in utero and this has been correlated with PCOS traits in later life [10].
The placenta, being the main steroidogenic organ in pregnancy, as well as a theoretical barrier to the overexposure of the fetus to maternally-derived androgens (via androgen aromatization), could be implicated in the aforementioned theory of PCOS pathogenesis. It has been shown that women with PCOS demonstrate alterations in placental structure and function, and there is increasing evidence that androgen excess and insulin resistance, two characteristics of PCOS, may negatively affect the placenta [11]. As the placenta is a mediator of both pregnancy complications and developmental programming, mechanisms known to be involved in pregnancy complications such as PE and gestational diabetes might also be implicated in PCOS pregnancies.
Kisspeptin (KISS1) and neurokinin-B (NKB) are neuropeptides acting synergistically at the hypothalamus and stimulating pulsatile GnRH release. Their role is fundamental in puberty initiation, and they are also involved in the disturbed hypothalamic-pituitary function characterizing PCOS [12,13]. KISS1 and NKB levels increase in pregnancy due to placental production, and these peptides along with neurokinin receptors have been implicated in the pathogenesis of several pregnancy complications (PE, gestational diabetes, intrauterine growth restriction-IUGR, preterm delivery) [14,15]. However, studies regarding the expression of these peptides in the human placenta in PCOS are lacking.
The aim of our study was to compare the placental expression of KISS1, NKB, and neurokinin receptors in women with PCOS and healthy pregnant women, and to correlate data from gene expression with the maternal and cord blood sex steroid levels.
2. Materials and Methods
Participants
This was a prospective, single-center case-control study. Participants were recruited from the Department of Obstetrics and Gynecology of the University Hospital of Patras from January 2020 to December 2022. Only women with term, uncomplicated, singleton pregnancies, who gave birth to healthy babies were included in the study. The presence of PCOS prior to pregnancy was ascertained according to the Rotterdam criteria, when at least two of the following were present: oligo-anovulation, clinical and/or biochemical hyperandrogenemia, polycystic ovarian morphology, after the exclusion of other pathologies with similar presentation [16]. The control group consisted of healthy women without a prior history of menstrual irregularity, clinical or biochemical hyperandrogenemia, or polycystic ovarian morphology. Exclusion criteria were: any major medical condition, drug or alcohol use during pregnancy, and use of hormonal or anti-diabetic medication up to 3 months prior to conception. Sixty-eight women participated in the study, 31 PCOS and 37 controls. The study was approved by the University Hospital of Patras Ethics Committee, and all participants provided written informed consent.
Placental Tissue Sampling and Gene Expression Analysis
Placental tissue was collected within 15 minutes of placental delivery. Full-depth samples 1x1cm were excised from three areas at the middle point of the placental radius, away from obvious infarcts or damage. The maternal decidua and chorionic plate tissues were removed, and each sample was further divided into 2-3 pieces of 0.5-1cm3. Sampled tissues were thoroughly rinsed with saline to remove blood and were then submerged in RNAlater solution and stored at 4oC for 24 hours and then at -20oC until analysis.
We studied the mRNA expression of kisspeptin (KISS1), neurokinin B (NKB), and neurokinin receptors 1, 2, and 3 (NK1R, NK2R, NK3R). Isolation of total RNA from placental specimens was carried out using the commercially available RNeasy Lipid Tissue Mini kit (QIAGEN, Hilden, Germany), according to the manufacturer’s protocol (including a 15-minute DNAse I treatment). RNA concentration and purity were estimated by measuring optical absorption at 260nm and calculating the ratio 260/280nm, respectively. Complementary DNA (cDNA) synthesis was performed using the Transcriptor First Strand cDNA Synthesis Kit (04379012001; Roche, Basel, Switzerland) with a mixture of anchored-oligo(dT)18 primer and 1μg of total RNA, according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in the LightCycler 2.0 Instrument (Roche, Basel, Switzerland), using 50ng of template cDNA and FastStart Universal SYBR Green 100 Master (Roche Hellas). PCR primers can be provided upon request for the genes ACTB (NM_001101), KISS1 (NM_002256), NKB (NM_001006667), NK1R (NM_001058), NK2R (NM_001057), and NK3R (NM_001059). Reactions were run in triplicates. The quality of the PCR reactions and specificity of the primers were confirmed by melting curve analysis. Relative gene expression was assessed using the ΔΔCt method, using β-actin (ACTB) as a reference gene because of its suitability for this experimental setting [17].
Hormone Measurements
At delivery, blood samples were collected from the mother (from a peripheral vein) and from the umbilical cord for hormonal determinations. Samples were immediately centrifuged, and the serum was collected and stored at -80oC until analysis. Total testosterone, sex hormone binding globulin (SHBG), androstenedione, dehydroepiandrosterone sulfate (DHEAS), Anti-Mullerian hormone (AMH) and estradiol were measured by electrochemiluminescence quantization in the Cobas e601 analyzer (Roche Diagnostics®, Mannheim, Germany). The samples were assayed in a single large batch. The intra- and inter-assay precision CV (%) values were 2.1%-14.8% and 2.5%–18.1% for testosterone, 1.1%–1.7% and 1.8%–4.0% for SHBG, 1.8%-3% and 3.7%-4.6% for androstenedione, 1.5%-3.2% and 2.2%-2.7% for DHEAS, 0.9%-1.7% and 2.7%-3.5% for AMH, and 1.1%–6.7% and 1.9%–10.6% for estradiol. The free androgen index (FAI) was calculated according to the formula: FAI = testosterone (nmol/l) × 100/SHBG(nmol/l).
Statistical Analysis
Data were analyzed using IBM SPSS Statistics for Windows, version 27.0 (IBM Corp., Armonk, N.Y., USA). Variables were tested for normality with the Kolmogorov-Smirnov test. Categorical data are presented as number (percentage) and continuous data as mean ± standard deviation (SD) (normally distributed variables) or as median (interquartile range-IQR) (non-normally distributed variables). Comparisons between the two study groups were conducted using the independent samples t-test for normally distributed continuous data and the Mann-Whitney U test for non-normally distributed continuous data, while the chi-squared test was used for comparisons concerning categorical variables. Correlations were estimated by Pearson or Spearman correlation tests, as appropriate. All tests were 2-tailed and a p-value of less than 0.05 was considered significant.
3. Results
The study included 68 women (PCOS-n=31, Controls-n=37) with a mean age of 31.81±5.49 years. The demographic data and pregnancy characteristics of the two groups are shown in Table 1. There was no significant difference between PCOS and controls regarding age, BMI, presence of gestational diabetes, pregnancy duration, mode of delivery, and offspring gender, weight, or length.
Women with PCOS had higher serum FAI and AMH and lower SHBG levels than controls, while no significant difference was observed between the two groups regarding serum total testosterone, androstenedione, DHEAS, and estradiol (Table 2). The umbilical cord blood hormone levels were studied separately in male and female offspring, as there are considerable differences between genders. In both the male and female subgroups, the cord blood hormone levels did not differ significantly between PCOS and control women’s offspring (Table 2).
Maternal serum total testosterone levels were strongly positively correlated with androstenedione (Spearman’s r=0.875, p<0.001), DHEAS (r=0.385, p<0.001), and AMH (r=0.407, p<0.001). AMH was positively correlated with testosterone, as well as with estradiol (r=0.287, p=0.018) and androstenedione (r=0.454, p=0.002). Maternal SHBG was negatively correlated with the 1st visit BMI (r=-0.305, p=0.013), and positively with estradiol (r=0.371, p=0.002).
Umbilical cord total testosterone was positively correlated with maternal serum testosterone (r=0.249, p=0.043), DHEAS (r=0.428, p<0.001), and androstenedione (r=0.329, p=0.029). Cord blood DHEAS and androstenedione were also positively correlated with maternal serum androgens, while cord blood estradiol was positively correlated with maternal estradiol, testosterone, DHEAS, and androstenedione (data not shown).
We did not detect a statistically significant difference in the placental mRNA expression of the studied genes between PCOS and controls, although NKB and KISS1 showed a trend for increased expression in PCOS (p=0.16 for NKB and p=0.12 for KISS1) (Table 3). Similarly, there was no difference regarding placental gene expression between PCOS and controls in the male offspring subgroup. However, in the female offspring group, NKB expression was significantly increased in PCOS versus controls (Median 2-ΔCt for relative expression to ACTB was 0.0008 for PCOS and 0.0001 for Controls, p=0.021) (Table 3, Figure 1). Besides, placental NKB expression was higher in women with male offspring versus those with female offspring, regardless of PCOS status (p=0.034).
Regarding correlations of placental gene expression with demographic and pregnancy characteristics, NK3R expression was positively correlated with the maternal BMI at delivery (Spearman’s r=0.409, p=0.005). There was no correlation between placental gene expression and maternal serum hormone levels. Placental NKB expression showed a positive correlation with umbilical cord FAI (r=0.356, p=0.021) and AMH (r=0.336, p=0.028) levels, while KISS1 expression was positively correlated with cord estradiol (r=0.324, p=0.032), testosterone (r=0.432, p=0.003), and FAI (r=0.415, p=0.006). Last, NKB and KISS1 expression were strongly positively correlated (r=0.496, p<0.001), while the expression of each neurokinin receptor was positively correlated with the expression of the other two (NK1R with NK2R and NK3R, and so forth).
4. Discussion
The present study showed that the placental mRNA expression of NKB is increased in women with PCOS versus controls, in pregnancies with female offspring. It also confirmed that pregnant women with PCOS have higher serum AMH and higher amounts of circulating free, bioactive androgens (higher FAI and lower SHBG) compared to healthy pregnant women. Umbilical cord blood hormones did not differ between the groups in neither male nor female offspring. Notably, our study included only women with term, uncomplicated, singleton pregnancies.
The presence of a genetic component in PCOS has been anticipated since the 1960’s, with studies showing familial segregation of the syndrome [18]. Since then, several reports have confirmed these findings, demonstrating high prevalence of PCOS in first degree relatives of the probands [19], as well as high concordance in twins [8]. However, family and twin studies have failed to provide a consistent pattern of inheritance, while genetic loci identified by GWAS are estimated to explain only a small part of PCOS heritability [7,19].
The Barker or thrifty phenotype hypothesis was introduced in the 1990’s, after a study showed that IUGR and low birth weight were correlated with cardiovascular disease in middle age [20]. This hypothesis supports that the intrauterine environment can “program” the future health of the fetus and determine the risk of chronic diseases, by inducing epigenetic changes in the fetal DNA. In the context of PCOS, this theory proposes that a hyperandrogenic intrauterine environment has epigenetic consequences affecting the expression of genes that regulate the future endocrine and metabolic functions of the fetus (i.e. GnRH pulsatility, folliculogenesis, ovarian production of sex steroids, insulin resistance). This theory for PCOS inheritance has been supported by animal studies showing that the administration of androgens during pregnancy leads to manifestation of PCOS-like phenotypes in adult life in the female offspring; results have been consistent across a variety of species, such as rodents, sheep, and monkeys [21,22,23]. These studies have proven that prenatal androgen excess produces reproductive defects, such as morphological changes / increased kisspeptin expression and reduced sex steroid feedback in the hypothalamus and increased LH secretion, leading to increased ovarian androgen production, anovulation, and infertility [10,23,24,25,26,27]. It has also been shown that androgen excess in utero is associated with metabolic perturbations in adult life: beta cell dysfunction, insulin resistance, hyperinsulinemia, increased adiposity, and obesity have been reported [28,29,30]. In humans, obviously, there are and will be no studies exploring the effect of exogenous androgen administration in pregnancy. Some studies have longitudinally followed up the offspring of hyperandrogenic mothers with PCOS and showed that daughters of PCOS women exhibit PCOS features, like elevated testosterone, increased LH, and increased ovarian volume [31]. Besides, daughters of mothers with congenital adrenal hyperplasia (CAH), another disease state of intrauterine hyperandrogenemia, frequently manifest a PCOS phenotype [32]. Although data regarding the longitudinal metabolic phenotyping of PCOS or CAH offspring is sparse, insulin resistance, dyslipidemia, and increased hospitalization for metabolic disorders have been reported in this population [33,34,35].
The placenta is the source of oxygen and nutrients for the fetus, and also the major producer of the steroid hormones required for the maintenance of pregnancy. As the main determinant of the intrauterine hormonal and metabolic/nutritional milieu, it is consequently implicated in the hypothesis linking intrauterine hyperandrogenemia with PCOS inheritance. In support of this view, placental insufficiency leads to infants born small for gestational age (SGA), which has been shown to be a predisposing factor for PCOS [36]. There is a great body of evidence exposing the central role of the placenta in a variety of pregnancy complications (spontaneous abortion, PE, preterm labor, IUGR, SGA) [11]. PCOS pregnancies are characterized by increased rates of such adverse outcomes which are directly related to placental dysfunction [5]. It is, therefore, possible that there are common pathogenic mechanisms / placental alterations underlying both these complications and PCOS. Khan et al showed, for example, that there is an overlap of five proteomic biomarkers between PCOS and PE [37]; however, there are several mediators of placental pathophysiology that are still not fully investigated in PCOS [11].
There is no doubt that the process of placentation can be problematic in PCOS, as evidenced by studies demonstrating increased occurrence of structural abnormalities in the placenta of women suffering from the syndrome. Palomba et al found that PCOS placentae had significantly lower weight, thickness, density, and volume, and increased frequency of lesions such as fibrosis compared to those of healthy pregnant women [38]. Another study showed that PCOS was characterized by higher incidence of placental anomalies associated with increased hypoxic state, such as chorioamnionitis, funisitis, villitis, vascular thrombosis, infarction, and villous immaturity [39]. Notably, in both these studies the increased rate of placental anomalies in PCOS was not accounted for by other pregnancy complications, as the former study included only uncomplicated pregnancies, while in the latter adjustment for pregnancy complications was performed. Finally, a recent study examining placentas of women with PCOS who underwent in-vitro-fertilization revealed that PCOS was associated with important anatomic as well as vascular placental abnormalities [40].
Apart from anomalies in placental structure, it seems that placental steroidogenic function is also impaired in PCOS. Androgen levels rise during physiologic pregnancy, serving as estrogen precursors but also in initiation of parturition. Normally, maternal and fetal over-exposure to androgen is hampered by a significant increase in SHBG production by the maternal liver and a high level of placental aromatase activity [10]. However, it is possible that in pregnant PCOS women supra-normal ovarian androgen production exceeds the capacity of these mechanisms, facilitating the hyperexposure of the fetus to maternal androgens. Indeed, increased 3b-hydroxysteroid dehydrogenase and decreased aromatase activities have been observed in placental tissue from PCOS women, compatible with increased androgen production [41]. Besides, PCOS pregnancies are characterized by elevated androgen levels in the maternal serum [42,43,44], as well as in the amniotic fluid [45] compared to controls; studies investigating umbilical cord blood androgen levels and their correlation with maternal androgens have yielded conflicting results [11,44]. In our study, pregnant women with PCOS had significantly higher serum FAI, hence increased concentration of free androgens relative to controls; the levels of total testosterone, androstenedione and DHEAS did not differ between the groups. With regards to umbilical cord hormone levels, there was no significant difference between the groups, regardless of the offspring gender. However, cord blood androgens were positively correlated with maternal serum androgens. Aside from altered steroidogenesis, PCOS status affects placental function in a more generalized way, as proteomic analysis showed differential expression between PCOS and controls in 258 placental proteins [44].
Little is known concerning the mechanisms involved in the placental dysfunction complicating PCOS, and most data come from animal models. Both animal and human placentae express the androgen receptor and are therefore susceptible to androgen effects [46]. Exogenous administration of androgens during gestation in animal models of PCOS leads to decreased placental weight and fetal growth restriction [47,48,49,50,51,52], implying a negative effect of hyperandrogenemia on placental function and efficiency. Altered regulation of placental nutrient transport (decreased free fatty acid and amino acid transport, increased signal transducer and activator of transcription 3 (STAT3) expression) has been shown in prenatally androgenized rodents and monkeys [47,52,53]. Increased placental STAT3 signaling has also been found in humans with PCOS [54]. Despite increased free serum androgens, our PCOS women did not differ from controls regarding the offspring weight and length; however, our study included only uncomplicated pregnancies and therefore cases of placental insufficiency, IUGR or SGA were excluded.
Besides, androgen administration results in increased placental expression of estrogen and androgen receptors in rodents [49,55], further enhancing steroid hormone action. Gestational hyperandrogenism has also been linked with placental vasculopathy; treatment with testosterone led to decreased uterine artery blood flow, elevated vascular resistance, and increased expression of hypoxia-responsive genes in rats [50,51], while in sheep it led to increased placental mRNA and protein expression of vascular endothelial growth factor (VEGF) [48]. Insulin resistance is another cardinal characteristic of PCOS, which might adversely affect placentation. Insulin resistance impairs human trophoblast invasion in vitro, while insulin sensitizers promote appropriate trophoblast migration and invasion [56]. Hyperandrogenic and insulin resistant rats show disrupted trophoblast invasion/differentiation, associated with placental mitochondrial dysfunction and over-production of reactive oxygen species [57]. Insulin resistance predisposes to gestational diabetes mellitus (GDM), which commonly complicates PCOS pregnancies [5]. GDM is in turn associated with placental dysfunction, via several mechanisms such as reduced placental apoptosis, impaired vasculogenesis, increased ischemia, and villous immaturity [11]. A small number of women with GDM were included in our study (4 PCOS and 4 Controls). These women were diagnosed by an oral glucose tolerance test conducted at the 24th-28th week of pregnancy; none received insulin treatment, and all achieved excellent glycemic control with diet only. This mild abnormality in glucose metabolism would be expected to have minor, if any, effects on the placenta. The placental expression of the studied genes did not differ between women with and without GDM.
Another potential factor implicated in PCOS heritability and acting, at least in part, at the level of the placenta is AMH. The administration of AMH to pregnant mice led to increased maternal GnRH activity, increased LH and testosterone, and decreased estradiol and progesterone; at the placenta, AMH treatment induced decreased aromatase and 3b-hydroxysteroid dehydrogenase, and increased LH receptor expression; the female offspring of AMH-treated mice demonstrated altered GnRH activity and phenotypic features of PCOS [58]. In humans, AMH levels are normally low in pregnancy, reflecting ovarian suppression. However, in PCOS, high AMH levels persist and even increase in pregnancy [58]. It is therefore possible that increased AMH contributes to maternal hyperandrogenemia via GnRH/LH activation. Moreover, since the human placenta expresses the AMH receptor type 2, increased maternal AMH might block placental aromatase and contribute to fetal androgen overexposure. Our study confirmed that PCOS is characterized by non-suppression of AMH during pregnancy, since we found that serum AMH levels measured at delivery are significantly higher in PCOS versus controls.
KISS1 and NKB are increased in pregnancy as a result of placental production. Although the role of kisspeptin in regulating trophoblast invasion and embryo implantation is established [15], little is known regarding the function of NKB in normal pregnancy [14]. Kisspeptins may have a role in the pathogenesis of GDM, as lower KISS1 plasma levels and higher placental protein expression of KISS1 and the kisspeptin-1 receptor have been found in women with GDM [59,60]. KISS1 levels are elevated in preeclamptic compared to healthy pregnant women, and its concentrations correlate with the severity of PE [61,62], while KISS1 mRNA expression is increased in the preeclamptic placenta [63,64]. Besides, decreased maternal KISS1 has been correlated with fetal growth restriction [65]. KISS1 might also exert antiapoptotic effects on the placenta: in hypothyroid pregnant rats, the administration of kisspeptin suppresses the apoptotic effect of hypothyroidism, by blocking the activation of the inflammasome NLRP3 pathway [66].
It is well-established that PE is characterized by significantly increased maternal NKB levels, and by increased placental NKB mRNA and protein expression; the placental expression of neurokinin receptors is also increased in PE [14,67]. Chronic infusion of high-dose NKB in rats induces hypertension [68], implying a possible etiologic role of NKB in PE development. The concentration of NKB is increased in the maternal plasma of IUGR pregnancies [69], while increased placental mRNA expression of NKB has been shown in preterm labor [70].
Although KISS1 and NKB are implicated in PCOS pathogenesis (both at the level of the hypothalamus [12,13] and at the level of the ovary [71]) and in several pregnancy complications characterized by placental dysfunction, data concerning their placental expression in PCOS are lacking. To our knowledge, our study was the first to assess the expression of these peptides in PCOS placentae. We found that the mRNA expression of NKB was increased in the placenta of women with PCOS and female offspring compared to controls. When all women were examined together regardless of the offspring gender, there was a trend for increased placental expression of NKB and KISS1, but this did not reach statistical significance. NKB expression was higher in women with male versus those with female offspring, regardless of PCOS status. Furthermore, NKB demonstrated a positive correlation with umbilical cord FAI and AMH levels, while KISS1 was positively correlated with cord testosterone and FAI. Maybe the fact that the percentage of male offspring was higher in our control group (57% vs 45% in PCOS) attenuated the difference in NKB and KISS1 expression between PCOS and controls in the whole group analysis. The notion that fetal sex might affect the placental adaptation to external insults is not new; testosterone administration in pregnant in sheep leads to fetal growth restriction more frequently in female fetuses, underscoring the possibility of sex-specific placental alterations in PCOS pregnancies [72]. Finally, we observed a strong positive correlation in the placental expression of NKB and KISS1, compatible with data from studies in rat placental cells showing that NKB upregulates KISS1 mRNA expression [73].
Our study does not lack limitations. We used the Rotterdam criteria to diagnose PCOS, which led to the inclusion of all PCOS phenotypes in our cohort, increasing the heterogeneity of this subgroup and possibly affecting the results; for example, since increased maternal androgens are a potential mediator of placental dysfunction, placental alterations might differ in hyperandrogenic versus normoandrogenic PCOS women. PCOS was retrospectively diagnosed in this study, increasing the risk of recall bias. Besides, our study cannot answer whether the observed increase in placental NKB expression contributes to PCOS-related placental dysfunction, or it is a compensatory mechanism. In support of the former, NKB suppresses several proteins involved in antioxidant defense and in inhibition of intravascular coagulation in human cytotrophoblast cells [74], suggesting that NKB might be a crucial mediator in placental dysfunction.
In conclusion, our study showed that NKB placental mRNA expression is increased in women with PCOS versus controls in pregnancies with female offspring. NKB expression depends on fetal gender, and it is positively correlated with placental KISS1 expression as well as with umbilical cord blood FAI and AMH levels. More studies are needed to clarify the potential role of NKB in the placental abnormalities characterizing PCOS.
Author Contributions
Conceptualization, G.K.M.; methodology, G.K.M., V.K., A.K., G.A. and N.A.G.; software, G.K.M.; formal analysis, G.K.M.; investigation, E.P., V.K., I.M., A.K. and G.A.; resources, A.K., G.A. and N.A.G.; data curation, G.K.M. and E.P..; writing—original draft preparation, G.K.M.; writing—review and editing, E.P., V.K., I.M., A.K., G.A. and N.A.G.; supervision, G.K.M. and N.A.G.; project administration, G.K.M. and N.A.G.; funding acquisition, G.K.M. and N.A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research is co-financed by Greece and the European Union (European Social Fund - ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning 2014-2020” in the context of the project “Exploring the developmental theory for polycystic ovary syndrome: the role of alterations in placental gene expression and in fetal DNA methylation (MIS: 5047128).” The APC (publication fees) of this manuscript have been financed by the Research Council of the University of Patras.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the University Hospital of Patras, Greece (protocol code 679-15/10/2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are available upon request.
Acknowledgements
N/A.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Azziz, R.; Woods, K.S.; Reyna, R.; Key, T.J.; Knochenhauer, E.S.; Yildiz, B.O. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004, 89, 2745-2749. [CrossRef]
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.W.; Norman, R.J.; Davies, M.J. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod. 2010, 25, 544–551. [CrossRef]
- The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS). Hum Reprod. 2012, 27, 14–24.
- Wild, R.A.; Carmina, E.; Diamanti-Kandrarakis, E., et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010, 95, 2038–2049. [CrossRef]
- Yu, H.F.; Chen, H.S.; Rao, D.P.; Gong, J. Association between polycystic ovary syndrome and the risk of pregnancy complications: A PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore). 2016, 95, e4863.
- Rassi, A.; Veras, A.B.; dos Reis, M., et al. Prevalence of psychiatric disorders in patients with polycystic ovary syndrome. Compr Psychiatry. 2010, 51, 599-602. [CrossRef]
- Raperport, C.; Homburg, R. The Source of Polycystic Ovarian Syndrome. Clin Med Insights Reprod Health. 2019, 13, 1179558119871467. [CrossRef]
- Crespo, R.P.; Bachega, T.A.; Mendonça, B.B.; Gomes, L.G. An Update of Genetic Basis of PCOS Pathogenesis. Arch Endocrinol Metab. 2018, 62, 352–361. [CrossRef]
- Palioura, E.; Diamanti-Kandarakis, E. Polycystic ovary syndrome (PCOS) and endocrine disrupting chemicals (EDCs). Rev Endocr Metab Disord. 2015, 16, 365-371. [CrossRef]
- Hakim, C.; Padmanabhan, V.; Vyas, A.K. Gestational Hyperandrogenism in Developmental Programming. Endocrinology. 2017, 158, 199-212. [CrossRef]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A Narrative Review of Placental Contribution to Adverse Pregnancy Outcomes in Women With Polycystic Ovary Syndrome. Clin Endocrinol Metab. 2019, 104, 5299–5315. [CrossRef]
- Gorkem, U.; Togrul, C.; Arslan, E.; Sargin Oruc, A.; Buyukkayaci Duman, N. Is there a role for kisspeptin in pathogenesis of polycystic ovary syndrome? Gynecol Endocrinol. 2018, 34, 157-160.
- George, J.T.; Kakkar, R.; Marshall, J., et al. Neurokinin B Receptor Antagonism in Women With Polycystic Ovary Syndrome: A Randomized, Placebo-Controlled Trial. J Clin Endocrinol Metab. 2016, 101, 4313-4321. [CrossRef]
- Page, N.M. Neurokinin B and pre-eclampsia: a decade of discovery. Reprod Biol Endocrinol. 2010, 8, 4. [CrossRef]
- Szydełko-Gorzkowicz, M.; Poniedziałek-Czajkowska, E.; Mierzyński, R.; Sotowski, M.; Leszczyńska-Gorzelak, B. The Role of Kisspeptin in the Pathogenesis of Pregnancy Complications: A Narrative Review. Int J Mol Sci. 2022, 23, 6611. [CrossRef]
- The Rotterdam ESHRE/ASRM – Sponsored PCOS Consensus Workshop Group 2004. Revised 2003 consensus on the diagnostic criteria and long term health risks related to polycystic ovary syndrome. Fertil Steril. 2003, 81, 19-25.
- Panagodimou, E.; Koika, V.; Markatos, F.; Kaponis, A.; Adonakis, G.; Georgopoulos, N.A.; Markantes, G.K. Expression stability of ACTB, 18S, and GAPDH in human placental tissues from subjects with PCOS and controls: GAPDH expression is increased in PCOS. Hormones (Athens). 2022, 21, 329-333. [CrossRef]
- Cooper, H.E.; Spellacy, W.E.; Prem, K.A.; Cohen, W.D. Hereditary factors in Stein-Leventhal syndrome. Am J Obstet Gynecol. 1968, 100, 371–382. [CrossRef]
- Jones, M.R.; Goodarzi, M.O. Genetic Determinants of Polycystic Ovary Syndrome: Progress and Future Directions. Fertil Steril. 2016, 106, 25–32. [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. Br Med J. 1990, 301, 1111. [CrossRef]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E.; Dunaif, A.; Padmanabhan, V. Animal models and fetal programming of PCOS. In Contemporary Endocrinology: Androgen Excess Disorders in Women: Polycystic Ovary Syndrome and Other Disorders; Azziz, J.E., Nestler, J.E., Dewailly, D., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 259-272.
- Franks, S. Animal models and the developmental origins of polycystic ovary syndrome: increasing evidence for the role of androgens in programming reproductive and metabolic dysfunction. Endocrinology. 2012, 153, 2536-2538. [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013, 373, 8-20. [CrossRef]
- Cernea, M.; Padmanabhan, V.; Goodman, R.L.; Coolen, L.M.; Lehman, M.N. Prenatal testosterone treatment leads to changes in the morphology of KNDy neurons, their inputs, and projections to GnRH cells in female sheep. Endocrinology. 2015, 156, 3277–3291. [CrossRef]
- Kondo, M.; Osuka, S.; Iwase, A.; Nakahara, T.; Saito, A.; Bayasula; Nakamura, T.; Goto, M.; Kotani, T.; Kikkawa, F. Increase of kisspeptin-positive cells in the hypothalamus of a rat model of polycystic ovary syndrome. Metab Brain Dis. 2016, 31, 673–681. [CrossRef]
- Dumesic, D.A.; Abbott, D.H.; Eisner, J.R.; Goy, R.W. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil Steril. 1997, 67, 155–163. [CrossRef]
- Foecking, E.M.; Szabo, M.; Schwartz, N.B.; Levine, J.E. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005, 72, 1475–1483. [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A.; Abbott, D.H.; Recabarren, S.E.; Herkimer, C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010, 151, 595–605. [CrossRef]
- Lu, C.; Cardoso, R.C.; Puttabyatappa, M.; Padmanabhan, V. Developmental programming: prenatal testosterone excess and insulin signaling disruptions in female sheep. Biol Reprod. 2016, 94, 113. [CrossRef]
- Eisner, J.R.; Dumesic, D.A.; Kemnitz, J.W.; Colman, R.J.; Abbott, D.H. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003, 11, 279–286. [CrossRef]
- Sir-Petermann, T.; Codner, E.; Pérez, V.; Echiburú, B.; Maliqueo, M.; Ladrón de Guevara, A.; Preisler, J.; Crisosto, N.; Sánchez, F.; Cassorla, F.; Bhasin, S. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009, 94, 1923–1930. [CrossRef]
- Hague, W.M.; Adams, J.; Rodda, C.; Brook, C.G.; de Bruyn, R.; Grant, D.B.; Jacobs, H.S. The prevalence of polycystic ovaries in patients with congenital adrenal hyperplasia and their close relatives. Clin Endocrinol (Oxf). 1990, 33, 501–510. [CrossRef]
- Boomsma, C.M.; Eijkemans, M.J.; Hughes, E.G.; Visser, G.H.; Fauser, B.C.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum Reprod Update. 2006, 12, 673–683. [CrossRef]
- Recabarren, S.E.; Smith, R.; Rios, R.; Maliqueo, M.; Echiburú, B.; Codner, E.; Cassorla, F.; Rojas, P.; Sir-Petermann, T. Metabolic profile in sons of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008, 93, 1820–1826. [CrossRef]
- Speiser, P.W.; Serrat, J.; New, M.I.; Gertner, J.M. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1992, 75, 1421–1424. [CrossRef]
- de Zegher, F.; Reinehr, T.; Malpique, R., et al. Reduced Prenatal Weight Gain and/or Augmented Postnatal Weight Gain Precedes Polycystic Ovary Syndrome in Adolescent Girls. Obesity (Silver Spring). 2017, 25, 1486-1489. [CrossRef]
- Khan, G.H.; Galazis, N.; Docheva, N.; Layfield, R.; Atiomo, W. Overlap of proteomics biomarkers between women with pre-eclampsia and PCOS: a systematic review and biomarker database integration. Hum Reprod. 2015, 30, 133-148. [CrossRef]
- Palomba, S.; Russo, T.; Falbo, A., et al. Macroscopic and microscopic findings of the placenta in women with polycystic ovary syndrome. Hum Reprod. 2013, 28, 2838-2847. [CrossRef]
- Koster, M.P.; de Wilde, M.A.; Veltman-Verhulst, S.M.; Houben, M.L.; Nikkels, P.G.; van Rijn, B.B.; Fauser, B.C. Placental characteristics in women with polycystic ovary syndrome. Hum Reprod. 2015, 30, 2829-2837. [CrossRef]
- Hochberg, A.; Mills, G.; Volodarsky-Perel, A.; Nu, T.N.T.; Machado-Gedeon, A.; Cui, Y.; Shaul, J.; Dahan, M.H. The impact of polycystic ovary syndrome on placental histopathology patterns in in-vitro fertilization singleton live births. Placenta. 2023, 139, 12-18. [CrossRef]
- Maliqueo, M.; Lara, H.E.; Sánchez, F.; Echiburú, B.; Crisosto, N.; Sir-Petermann, T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013, 166, 151-155. [CrossRef]
- Sir-Petermann, T.; Maliqueo, M.; Angel, B.; Lara, H.E.; Pérez-Bravo, F.; Recabarren, S.E. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002, 17, 2573–2579. [CrossRef]
- Glintborg, D.; Jensen, R.C.; Bentsen, K.; Schmedes, A.V.; Brandslund, I.; Kyhl, H.B.; Bilenberg, N.; Andersen, M.S. Testosterone levels in third trimester in polycystic ovary syndrome. Odense Child Cohort. J Clin Endocrinol Metab. 2018, 103, 3819–3827. [CrossRef]
- Sun, M.; Sun, B.; Qiao, S.; Feng, X.; Li, Y.; Zhang, S.; Lin, Y.; Hou, L. Elevated maternal androgen is associated with dysfunctional placenta and lipid disorder in newborns of mothers with polycystic ovary syndrome. Fertil Steril. 2020, 113, 1275-1285.e2. [CrossRef]
- Palomba, S.; Marotta, R.; Di Cello, A., et al. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study. Clin Endocrinol. 2012, 77, 898-904. [CrossRef]
- Hsu, T.Y.; Lan, K.C.; Tsai, C.C.; Ou, C.Y.; Cheng, B.H.; Tsai, M.Y.; Kang, H.Y.; Tung, Y.H.; Wong, Y.H.; Huang, K.E. Expression of androgen receptor in human placentas from normal and preeclamptic pregnancies. Taiwan J Obstet Gynecol. 2009, 48, 262–267. [CrossRef]
- Sathishkumar, K.; Elkins, R.; Chinnathambi, V.; Gao, H.; Hankins, G.D.; Yallampalli, C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011, 9, 110. [CrossRef]
- Cleys, E.R.; Halleran, J.L.; Enriquez, V.A.; da Silveira, J.C.; West, R.C.; Winger, Q.A.; Anthony, R.V.; Bruemmer, J.E.; Clay, C.M.; Bouma, G.J. Androgen receptor and histone lysine demethylases in ovine placenta. PLoS One. 2015, 10, e0117472. [CrossRef]
- Sun, M.; Maliqueo, M.; Benrick, A.; Johansson, J.; Shao, R.; Hou, L.; Jansson, T.; Wu, X.; Stener-Victorin, E. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am J Physiol Endocrinol Metab. 2012, 303, E1373–E1385. [CrossRef]
- Gopalakrishnan, K.; Mishra, J.S.; Chinnathambi, V.; Vincent, K.L.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Hankins, G.D.; Sathishkumar, K. Elevated testosterone reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant rats. Hypertension. 2016, 67, 630–639. [CrossRef]
- Chinnathambi, V.; Blesson, C.S.; Vincent, K.L.; Saade, G.R.; Hankins, G.D.; Yallampalli, C.; Sathishkumar, K. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension. 2014, 64, 405–414.
- Hu, M.; Richard, J.E.; Maliqueo, M.; Kokosar, M.; Fornes, R.; Benrick, A.; Jansson, T.; Ohlsson, C.; Wu, X.; Skibicka, K.P.; Stener-Victorin, E. Maternal testosterone exposure increases anxiety-like behavior and impacts the limbic system in the offspring. Proc Natl Acad Sci USA. 2015, 112, 14348–14353. [CrossRef]
- Abbott, D.H.; Cristin, R.; Bruns, C.R.; Barnett, D.K.; Dunaif, A.; Theodore, L.; Goodfriend, T.L.; Daniel, A.; Tarantal, D.; Tarantal, A. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab. 2010, 299, E741–E751. [CrossRef]
- Maliqueo, M.; Sundström Poromaa, I.; Vanky, E.; Fornes, R.; Benrick, A.; Åkerud, H.; Stridsklev, S.; Labrie, F.; Jansson, T.; Stener-Victorin, E. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum Reprod. 2015, 30, 692-700. [CrossRef]
- Fornes, R.; Maliqueo, M.; Hu, M.; Hadi, L.; Jimenez-Andrade, J.M.; Ebefors, K.; Nyström, J.; Labrie, F.; Jansson, T.; Benrick, A.; Stener-Victorin, E. The effect of androgen excess on maternal metabolism, placental function and fetal growth in obese dams. Sci Rep. 2017, 7, 8066. [CrossRef]
- Mayama, R.; Izawa, T.; Sakai, K.; Suciu, N.; Iwashita, M. Improvement of insulin sensitivity promotes extravillous trophoblast cell migration stimulated by insulin-like growth factor-I. Endocr J. 2013, 60, 359–368. [CrossRef]
- Zhang, Y.; Zhao, W.; Xu, H.; Hu, M.; Guo, X.; Jia, W.; Liu, G.; Li, J.; Cui, P.; Lager, S.; Sferruzzi-Perri, A.N.; Li, W.; Wu, X.K.; Han, Y.; Brännström, M.; Shao, L.R.; Billig, H. Hyperandrogenism and insulin resistance-induced fetal loss: evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. J Physiol. 2019, 597, 3927-3950. [CrossRef]
- Tata, B.; Mimouni, N.E.H.; Barbotin, A.L.; Malone, S.A.; Loyens, A.; Pigny, P.; Dewailly, D.; Catteau-Jonard, S.; Sundström-Poromaa, I.; Piltonen, T.T.; Dal Bello, F.; Medana, C.; Prevot, V.; Clasadonte, J.; Giacobini, P. Elevated prenatal anti-Müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med. 2018, 24, 834-846. [CrossRef]
- Kapustin, R.V.; Drobintseva, A.O.; Alekseenkova, E.N.; Onopriychuk, A.R.; Arzhanova, O.N.; Polyakova, V.O.; Kvetnoy, I.M. Placental protein expression of kisspeptin-1 (KISS1) and the kisspeptin-1 receptor (KISS1R) in pregnancy complicated by diabetes mellitus or preeclampsia. Arch Gynecol Obstet. 2020, 301, 437–445. [CrossRef]
- Bowe, J.E.; Hill, T.G.; Hunt, K.F.; Smith, L.I.; Simpson, S.J.; Amiel, S.A.; Jones, P.M. A role for placental kisspeptin in β cell adaptation to pregnancy. JCI Insight 2019, 4, e124540. [CrossRef]
- Fang, L.; Gao, Y.; Wang, Z.; Li, Y.; Yan, Y.; Wu, Z.; Cheng, J.-C.; Sun, Y.-P. EGF stimulates human trophoblast cell invasion by downregulating ID3-mediated KISS1 expression. Cell Commun Signal. 2021, 19, 101. [CrossRef]
- Adali, E.; Kurdoglu, Z.; Kurdoglu, M.; Kamaci, M.; Kolusari, A.; Yildizhan, R. Metastin levels in pregnancies complicated by pre-eclampsia and their relation with disease severity. J Matern Fetal Neonatal Med. 2012, 25, 2671–2675. [CrossRef]
- Qiao, C.; Wang, C.; Zhao, J.; Liu, C.; Shang, T. Elevated expression of KiSS-1 in placenta of Chinese women with early-onset preeclampsia. PLoS ONE 2012, 7, e48937. [CrossRef]
- Vazquez-Alaniz, F.; Galaviz-Hernandez, C.; Marchat, L.A.; Salas-Pacheco, J.M.; Chairez-Hernandez, I.; Guijarro-Bustillos, J.J.;Mireles-Ordaz, A. Comparative expression profiles for KiSS-1 and REN genes in preeclamptic and healthy placental tissues. Eur J Obstet Gynecol Reprod Biol. 2011, 159, 67–71. [CrossRef]
- Smets, E.M.L.; Deurloo, K.L.; Go, A.T.J.I.; van Vugt, J.M.G.; Blankenstein, M.A.; Oudejans, C.B.M. Decreased plasma levels of metastin in early pregnancy are associated with small for gestational age neonates. Prenat Diagn. 2008, 28, 299–303. [CrossRef]
- Santos, B.R.; Cordeiro, J.M.D.A.; Santos, L.C.; Santana, L.D.S.; Nascimento, A.E.J.; Silva, J.F. Kisspeptin Suppresses Inflammasome-NLRP3 Activation and Pyroptosis Caused by Hypothyroidism at the Maternal-Fetal Interface of Rats. Int J Mol Sci. 2023, 24, 6820. [CrossRef]
- Liu, Y.; Chen, X.; Chen, H. Placental and umbilical cord levels of neurokinin B and neurokinin B receptor in pre-eclampsia. Int J Gynaecol Obstet. 2009, 107, 58-59. [CrossRef]
- Yang, J.; Dhawan, V.; Morrish, D.W.; Kaufman, S. Bimodal effects of chronically administered neurokinin B (NKB) on in vivo and in vitro cardiovascular responses in female rats. Regul Pept. 2007, 143, 136-142. [CrossRef]
- D'Anna, R.; Baviera, G.; Corrado, F.; Crisafulli, A.; Ientile, R.; Buemi, M.; Squadrito, F. Neurokinin B and nitric oxide plasma levels in pre-eclampsia and isolated intrauterine growth restriction. BJOG. 2004, 111, 1046-1050. [CrossRef]
- Torricelli, M.; Giovannelli, A.; Leucci, E.; Florio, P.; De Falco, G.; Torres, P.B.; Reis, F.M.; Leoncini, L.; Petraglia, F. Placental neurokinin B mRNA expression increases at preterm labor. Placenta. 2007, 28, 1020-1023. [CrossRef]
- Blasco, V.; Pinto, F.M.; Fernández-Atucha, A.; Prados, N.; Tena-Sempere, M.; Fernández-Sánchez, M.; Candenas, L. Altered expression of the kisspeptin/KISS1R and neurokinin B/NK3R systems in mural granulosa and cumulus cells of patients with polycystic ovarian syndrome. J Assist Reprod Genet. 2019, 36, 113-120. [CrossRef]
- Beckett, E.M.; Astapova, O.; Steckler, T.L.; Veiga-Lopez, A.; Padmanabhan, V. Developmental programing: impact of testosterone on placental differentiation. Reproduction. 2014, 148, 199–209. [CrossRef]
- Oride, A.; Kanasaki, H.; Mijiddorj, T.; Sukhbaatar, U.; Ishihara, T.; Kyo, S. Regulation of kisspeptin and gonadotropin-releasing hormone expression in rat placenta: study using primary cultures of rat placental cells. Reprod Biol Endocrinol. 2015, 13, 90. [CrossRef]
- Sawicki, G.; Dakour, J.; Morrish, D.W. Functional proteomics of neurokinin B in the placenta indicates a novel role in regulating cytotrophoblast antioxidant defences. Proteomics. 2003, 3, 2044-2051. [CrossRef]
Figure 1.
Relative placental mRNA expression of NKB in PCOS and controls. The expression level in controls is considered as reference. *p<0.05 (Mann-Whitney U test).
Figure 1.
Relative placental mRNA expression of NKB in PCOS and controls. The expression level in controls is considered as reference. *p<0.05 (Mann-Whitney U test).
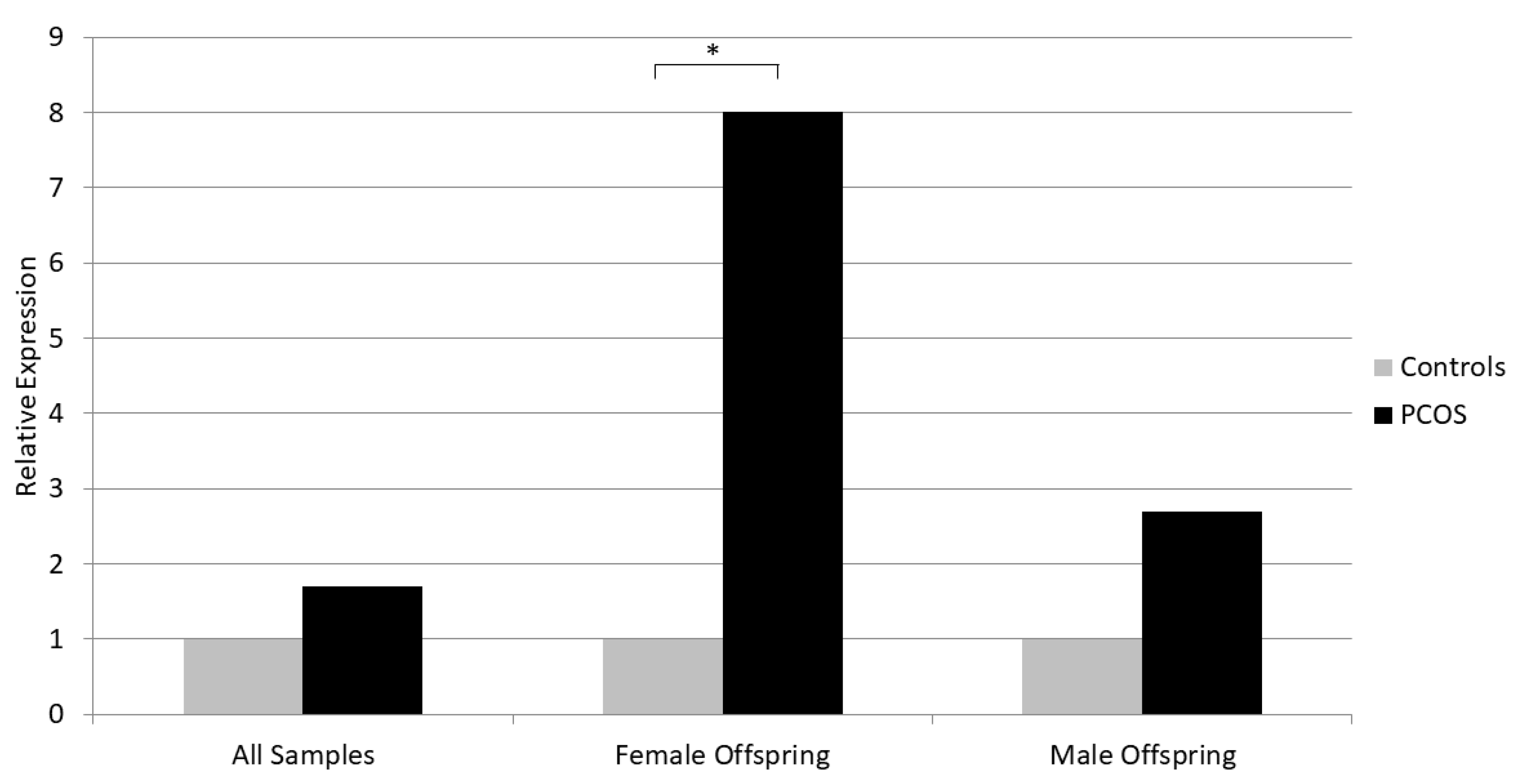

Table 1.
Demographic data and pregnancy characteristics of the PCOS and Control women included in the study. VD: vaginal delivery, CS: cesarean section, M: male, F: female.
Table 1.
Demographic data and pregnancy characteristics of the PCOS and Control women included in the study. VD: vaginal delivery, CS: cesarean section, M: male, F: female.
| PCOS (n=31) | Controls (n=37) | p Value | |
|---|---|---|---|
| Age (years) | 31.52±5.32 | 32.06±5.78 | 0.677 |
| BMI at 1st visit (kg/m2) | 26.81±5.06 | 25.27±4.44 | 0.191 |
| BMI at delivery (kg/m2) | 31.98±5.49 | 29.84±4.44 | 0.105 |
| Gestational diabetes | 4 (12.9%) | 4 (10.8%) | 0.790 |
| Delivery week | 39 (2) | 39 (2) | 0.785 |
| Mode of delivery (VD/CS) | 15 (48.4%) / 16 (51.6%) | 19 (51.4%) / 18 (48.6%) | 0.808 |
| Offspring gender (M/F) | 14 (45.2%) / 17 (54.8%) | 21 (56.8%) / 16 (43.2%) | 0.274 |
| Offspring weight (g) | 3161.33±555.14 | 3330.27±462.90 | 0.179 |
| Offspring length (cm) | 49.75±2.44 | 50.88±2.27 | 0.139 |
Table 2.
Maternal serum and umbilical cord blood hormone levels in PCOS and controls. SHBG: sex hormone binding globulin, FAI: free androgen index, DHEAS: dehydroepiandrosterone sulfate, AMH: Anti-Mullerian hormone.
Table 2.
Maternal serum and umbilical cord blood hormone levels in PCOS and controls. SHBG: sex hormone binding globulin, FAI: free androgen index, DHEAS: dehydroepiandrosterone sulfate, AMH: Anti-Mullerian hormone.
| Maternal Serum | PCOS (n=31) | Controls (n=37) | p Value |
|---|---|---|---|
| Total testosterone (ng/dL) | 88.29 (98.77) | 91.27 (70.99) | 0.538 |
| SHBG (nmol/L) | 415.99±135.61 | 478.20±120.47 | 0.049 |
| FAI | 0.68 (0.40) | 0.56 (0.67) | 0.048 |
| Androstenedione (ng/mL) | 2.19 (2.10) | 1.64 (2.12) | 0.387 |
| DHEAS (μg/dL) | 105.97±54.46 | 120.99±72.82 | 0.347 |
| AMH (pmol/L) | 7.23 (5.41) | 3.84 (6.07) | 0.012 |
| Estradiol (pg/mL) | 8860 (16301) | 6698 (15510) | 0.310 |
| Umbilical cord blood | |||
| Female Offspring | PCOS (n=17) | Controls (n=16) | |
| Total testosterone (ng/dL) | 142.54±56.71 | 130.97±37.27 | 0.501 |
| SHBG (nmol/L) | 32.16 (18.15) | 32.50 (23.31) | 0.624 |
| FAI | 14.57 (9.37) | 12.69 (11.52) | 0.468 |
| Androstenedione (ng/mL) | 0.52±0.12 | 0.44±0.11 | 0.185 |
| DHEAS (μg/dL) | 425.58±194.81 | 353.89±151.84 | 0.255 |
| AMH (pmol/L) | 1.50 (2.03) | 1.19 (1.56) | 0.624 |
| Estradiol (pg/mL) | 2577.19±929.94 | 2959.82±1095.36 | 0.295 |
| Male Offspring | PCOS (n=14) | Controls (n=21) | |
| Total testosterone (ng/dL) | 168.76±69.34 | 166.62±56.62 | 0.923 |
| SHBG (nmol/L) | 33.54 (9.02) | 37.06 (12.43) | 0.255 |
| FAI | 17.14 (9.13) | 16.12 (6.73) | 0.893 |
| Androstenedione (ng/mL) | 0.56±0.24 | 0.48±0.17 | 0.341 |
| DHEAS (μg/dL) | 394.01±166.72 | 349.44±161.11 | 0.449 |
| AMH (pmol/L) | 165.84 (80.60) | 180.10 (90.90) | 0.439 |
| Estradiol (pg/mL) | 3266.23±1375.43 | 2730.19±1769.12 | 0.363 |
Table 3.
Placental mRNA expression of NKB, NK1R, NK2R, NK3R, and KISS1 genes in PCOS and controls. Results are expressed as the median (IQR) relative expression to ACTB according to the ΔΔCt method.
Table 3.
Placental mRNA expression of NKB, NK1R, NK2R, NK3R, and KISS1 genes in PCOS and controls. Results are expressed as the median (IQR) relative expression to ACTB according to the ΔΔCt method.
| All Samples | PCOS (n=31) | Controls (n=37) | p value |
|---|---|---|---|
| NKB | 0.0017 (0.04) | 0.0010 (0.02) | 0.160 |
| NK1R | 2.58x10-5 (5.3x10-5) | 2.44x10-5 (9.0x10-5) | 0.514 |
| NK2R | 3.66x10-6 (9x10-6) | 4.14 x10-6 (14x10-6) | 0.298 |
| NK3R | 5.41x10-5 (18.8x10-5) | 2.80x10-5 (7.4x10-5) | 0.394 |
| KISS1 | 0.0160 (0.07) | 0.0079 (0.07) | 0.120 |
| Female Offspring | PCOS (n=17) | Controls (n=16) | |
| NKB | 0.0008 (0.03) | 0.0001 (0.0001) | 0.021 |
| NK1R | 1.51x10-5 (2.9x10-5) | 2.43x10-5 (7.0x10-5) | 0.762 |
| NK2R | 0.77x10-6 (24x10-6) | 1.57 x10-6 (4x10-6) | 0.579 |
| NK3R | 1.66x10-5 (37.0x10-5) | 1.42 x10-5 (6.3x10-5) | 0.631 |
| KISS1 | 0.0136 (0.03) | 0.0022 (0.03) | 0.315 |
| Male Offspring | PCOS (n=14) | Controls (n=21) | |
| NKB | 0.0135 (0.07) | 0.0050 (0.02) | 0.586 |
| NK1R | 5.62x10-5 (9.4x10-5) | 3.56x10-5 (31.4x10-5) | 0.867 |
| NK2R | 4.63x10-6 (12x10-6) | 2.97x10-6 (5x10-6) | 0.660 |
| NK3R | 8.40x10-5 (18.7x10-5) | 2.20x10-5 (6.8x10-5) | 0.363 |
| KISS1 | 0.0435 (0.26) | 0.0089 (0.08) | 0.135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



