
Submitted:
10 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Extensive genomic analyses of Enterococcus cecorum isolates from sepsis outbreaks in broilers suggests a polyphyletic origin likely arising from core genome mutations rather than gene acquisition. This species is normal intestinal flora in avian species with particular isolates associated with osteomyelitis. More recently, the species has been associated with sepsis outbreaks affecting broilers during the first 3 weeks post hatch. Understanding the genetic and management basis of this new phenotype is critical for developing strategies to mitigate this emerging problem. Phylogenomic analyses of 227 genomes suggest that sepsis isolates are polyphyletic and closely related to both commensal and osteomyelitis isolate genomes. Pangenome analyses detect no gene acquisitions that distinguish all sepsis isolates. Core genome SNP analyses identify a number of mutations, affecting protein coding sequence, that are enriched in sepsis isolates. Analysis of the protein substitutions supports mutational origins of sepsis isolates.
Keywords:
broiler
; Enterococcus
; sepsis
; osteomyelitis
; phylogenomics
; core genome mutations
; pathogenesis
1. Introduction
Enterococci are Gram positive firmicutes comprising commensals and pathogens in a wide range of vertebrates, and plants [1,2], that are prevalent in animal gastrointestinal tracts [1,2,3,4], nature [1,5], and even food processing systems [6]. Some species are found in diverse vertebrates, such as Enterococcus faecalis, Enterococcus faecium, and Enterococcus avium, which have been isolated from both mammals and aves [1,2]. In humans these species have been associated with infections of the urinary tract, blood stream, heart, and central nervous system [7,8]. Some Enterococcal species appear to be restricted to particular host species or classes [2]. Enterococcus cecorum is primarily associated with avian species and has been isolated from the intestines or cloaca of galliforms and neoornithes [1,2,9]. Comparative genomics based on the Enterococus core genome places E. cecorum most closely related to Enterococcus columbae (host birds) and within a clade associated with plant, aquatic and bird hosts [2]. E. cecorum typically colonizes the intestines of chickens around 3 weeks of age [10,11,12]. Pathogenic strains have been associated with osteomyelitis affecting the flexible thoracic vertebrae (kinky back) and the proximal femoral (femoral head necrosis) or tibial (tibial head necrosis) growth plates. Borst et al., [13] identified a cluster of twelve genes that appear to distinguish osteomyelitis isolates from commensals, likely to be involved in virulence through capsular modifications. Evolution of pathogenesis and drug resistance in E. faecalis and E. faecium, have been associated with mobile elements and acquisition of capsular gene clusters [14,15]. Phylogenomic analysis of more than 100 genomes of E. cecorum isolates from French broilers suggested that a specific lineage of pathogenic isolates is infecting broilers in Europe and the United States [16].
The recent occurrence of sepsis outbreaks caused by E. cecorum is characterized by onset during the first few weeks post hatch, prior to the normal colonization of the intestinal tract [17]. However, there are older reports of sepsis outbreaks with losses of 5-8%, characterized by pericarditis, bacteremia, and osteomyelitis [18]. Mechanisms of E. cecorum horizontal or vertical spread in flocks are not known, although particular facilities or flocks may experience repetitive outbreaks [17,18]. Recent outbreaks may be more problematic with the removal of early administration of antibiotic growth promoters in flocks [17,19,20]. Questions we wished to address included: whether specific mutations, or gene acquisition are driving recent sepsis outbreaks, whether sepsis and osteomyelitis isolates are distinct lineages, and whether high resolution bacterial genomics can inform responses to sepsis outbreaks. Answers to these questions will be critical for developing treatments, vaccines, or management strategies for mitigating sepsis outbreaks.
2. Materials and Methods
E. cecorum isolates were provided by the University of Arkansas Poultry Diagnostic Laboratory. Cultures were grown in tryptic soy broth (Difco) +5% chicken serum (Gibco ThermoFisher, Waltham, MA USA) in 5% CO2, and archived in 40% glycerol at -80 oC. DNA isolation was from 20 ml overnight stationary-phase, broth cultures by treatment with lysozyme, followed by standard SDS-lysis, organic extraction, RNAse/protease digestion, and ethanol precipitation [21,22]. Final purification used NanoSep 100k Omega spin cups according to manufacturer’s instructions (Pall Corporation, Port Washington, NY). Purified bacterial total DNAs were submitted to SeqCenter (Pittsburgh, PA) for 2x151 paired-end sequencing. Illumina paired-end sequencing. Sequencing data for 22 additional isolates was obtained from the laboratory of Luke Borst (North Carolina State University). Sequence data was uploaded to BV-BRC [23] for processing through the Trim Galore pipeline and Unicycler genome assembly. Assemblies were analyzed for comparative genomics using Proksee [24], and for location of mobile element genes [25]. Genomic islands were identified using IslandViewer 4 [26]. Prophage were identified using Phaster [27]. NCBI genome assemblies were downloaded with genome_updater v0.6.3 (https://github.com/pirovc/genome_updater). Phylogenomic trees of genome assemblies were produced based on kmer comparisons using PopPUNK v2.6.0 [28] and based on core genome SNPs using ParSNP v1.7.4 [29]. Newick trees were midpoint rooted with Archeopteryx v 0.9928 beta [30] and rendered in MicroReact [31]. Assemblies were annotated using Prokka v1.14.5 [32]. Pan and core gene partitioning was with Roary v3.13.0 [33]. Scoary v1.6.16 [34] was used for pan-genome wide association. ParSNP and Maast v1.0.8 [35] were used to generate vcf for core genome SNPs which were then processed further in Microsoft Excel to identify possible trait-associated SNPs. Specific polypeptide sequences were extracted from Prokka annotation files based on description text using the linux grep command. Fasta files were converted to and from .tab format using a Biopython script (https://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_tab.php). Excel was used to curate .tab formatted sequences to filter for evident orthologs based on amino and terminal sequences, and for full-length orthologs.. For polypeptide variant identification the sequences were aligned using Clustal W in MegAlignPro (Lasergene v17 (DNAStar, Inc.; Madison, WI, USA) and variation tables exported to Microsoft Excel. Signal sequences and cleavage sites were predicted using SignalP-6.0 [36].
Whole genome sequence reads, and genome assemblies were submitted to NCBI under BioProject PRJNA1050746, BioSamples SAMN38750234 through SAMN38750267, and accessions JAXOGD000000000 through JAXOHK000000000.
3. Results and Discussion
Enterococcus cecorum Isolates from commercial operation samplings were obtained from poultry diagnostic laboratories representing samplings from 2020 through 2021 (Table 1). These included environmental samples as well as necropsy samples, including internal organs. We also included two bacterial chondronecrosis with osteomyelitis (BCO) isolates from our own sampling of commercial broiler farms [22], and our poultry research farm (UAPRF). Total DNA was purified from each isolate and submitted for Illumina 2x151 sequencing. Reads were trimmed and assembled as described in Materials and Methods. The assemblies (Table 1) ranged from 32 to 105 contigs with an average of 62 contigs. Total assembled base pairs ranged from 2.15 to 2.49 Mbp with an average of 2.27. The N50 values ranged from 49.7 to 209.1 kbp with an average of 108.9. These 34 genomes along with 193 E. cecorum genomes deposited in NCBI were used to generate phylogenomic trees to discern the relationships and possible origins of the BCO and sepsis isolates. A major issue in any of these analyses is knowing the phenotypic trait of each isolate regarding virulence/pathogenicity. BioSample data from NCBI may not clearly identify host disease state or the actual anatomical source of the culture. For example, if an isolate was obtained from bone marrow should that be classified as a case of sepsis or BCO? If an isolate was obtained from hatchery egg waste from an infected breeder flock, the virulence potential of that isolate is unknown. We therefore made every effort to be strict in our interpretation (see legend to Figure 1) as to whether an isolate was from BCO (osteomyelitis or bone marrow) or from sepsis (peritoneum, heart, liver, blood). For the purposes of our analyses, we reserved sepsis isolates to be those isolated from an internal organ or compartment while BCO isolates were from bone marrow or infected joint. We also defined a Chicken Disease group (145 genomes) as BCO isolates plus the strictly defined sepsis isolates which excludes isolates from air sac, egg waste, environmental swabs, intestines, cecum, and unspecified clinical samples. All genomes not in Chicken Disease were assigned to the None phenotypic group. Strain assignments to phenotypic groups are listed in Table S1.
Phylogenomic trees were generated for all 227 genomes using either kmer genome comparisons using PopPUNK (Figure 1) or shared core genome SNPs using ParSNP (Figure S1). Although branch lengths were different for the two methods the overall topology was similar, and clustering very similar. Regardless of using a strict or relaxed definition of BCO or sepsis the phylogenomic analyses all support a polyphyletic origin for both the BCO and sepsis isolates. Red circles for BCO appear in all branches and the green triangles/circles for chicken sepsis appear in multiple locations. There is one large cluster (A) of US sepsis isolates that is related to a BCO isolate we obtained from tibial pus from a broiler in 2016 (1415). Within this cluster are two vertebral osteomyelitis isolates (LB5924 & LB5925). Cluster B contains LB5800, a sepsis isolate from heart, four closely related isolates from egg transfer residues (LB5857, LB 5916, LB5876), an isolate from a case of air-saacculitis (LB5880), and two distantly related isolates, a BCO femoral head necrosis isolate (1675) and an intestinal isolate (LB5923). Cluster C contains one sepsis isolate (1750) and two vertebral osteomyelitis isolates (LB5836 & LB5843). Cluster D contains no evidence of BCO or sepsis isolates as it consists of isolates from cull eggs, or egg transfer residue, along with one isolate (LB5922) with incomplete documentation of isolation source. The tree contains 100 genomes from isolates collected from 16 poultry farms in France from 2007 to 2017. Some of these genomes are from probable sepsis cases (red circles) with many representing pericarditis isolates [16]. These isolates appear throughout the tree with clusters including BCO and probable sepsis virulence phenotypes. There are also six clinical isolates from humans (green plus nodes, Figure 1 and Figure S1) that are distributed amongst the chicken isolates. Of note, the phylogenomic analyses also included cloacal isolates from Harris Hawk (ESM13C1 & ESM13AIC1), and black vulture (E1062, ESM877AIC1), pig feces (WCA-380), and internal organs from duck (D-104), and goose (G-29). The Harris Hawk isolates appear to be the most distant from the other 225 isolates. While the goose G-29 isolate is within the branches leading to cluster D. The pig and one of the black vulture isolates bracket a solitary USA sepsis isolate, 1752, The duck internal organ/sepsis isolate D-104, isolated in Poland, places in a cluster with French sepsis and BCO isolates. We interpret these data as most consistent with pathogenic isolates arising from non-pathogenic isolates and that sepsis isolates are closely related to BCO isolates. Our conclusions contrast with those from previous analyses of smaller collections of genomes that suggested distinct lineages for commensal and pathogenic isolates of E. cecorum [13,16,37].
If osteomyelitis and/or sepsis are polyphyletic then this could be driven by gene acquisition and/or by mutation. Based on comparison of genomes of three BCO isolates with three cecal isolates, Borst et al. [13], proposed that a cluster of 12 genes possibly related to capsular modification distinguished the BCO isolates from the cecal isolates. Using the SA1 assembly of Borst as our reference genome, the Prokka annotation places the 12 gene cluster as protein encoded genes (PEG) 312 through 323. We used BLASTp to search the Prokka predicted polypeptides from all 227 E. cecorum genomes for these 12 polypeptides. BLASTp results were tabulated combining values for percent identity and query coverage (pid x qcov/100) and averaged across all genomes according to phenotypic group: None, Chicken Disease, BCO, Strict Sepsis (summarized in Table 2 with complete results in Table S2). The results show that the cluster of capsular genes are conserved in approximately 70-80% of the isolates classified into Chicken Disease and for those classified as BCO, the conservation may be higher in strict sepsis group, but the number of those isolates is significantly lower (34 vs 113). Additionally, presence of these capsular genes in the isolates within the None group (no disease or disease association not known) is around 30 to 40% of the isolates.
Unicycler assemblies for our 34 assemblies were surveyed for circular elements. This identified 3 potential plasmids in isolate 1754. We used BLASTn searches of all 227 genomes to determine how widespread these contigs are, as a signal for horizontal genetic exchange. Contig 25 (6237 bp) from 1745 appears to be more than 75% conserved in 24 assemblies: 1415, 1754, An144, ARS48, ARS60, ARS65, CHD182EN, CHD270EN, CHD278EN, CHD341EN, CHD347EN, CHD83EN, CHD96EN, CIRMBP-1218, CIRMBP-1233, CIRMBP-1259, CIRMBP-1276, CIRMBP-1282, CIRMBP-1284, CIRMBP-1285, CIRMBP-1294, PS10, PS2, and SA2. Contig 27 (4525 bp) is more than 75% conserved in 10 assemblies: 1415, 1754, An144, ARS60, CHD83EN, CHD96EN, CIRMBP-1234, CIRMBP-1238, CIRMBP-1273, and PS2. Contig 31 (2418 bp) is more than 75% conserved in only the 1754 genome; the closest related contig is in An144 and only 71% conserved. The isolates containing homologous contigs are scattered around the phylogenomic tree (Figure 1), but sometimes found in highly similar genomes, indicative of both horizontal transfer or shared origins. Only some genomes containing contig 25 homologs contain contig 27 homologs (and vice versa). Analysis of contig 25 using Prokka annotation, and BLASTp of NCBI NonRedundant indicates six predicted CDS that annotate as four hypothetical proteins, a replication RepB initiation protein, and a plasmid recombination protein. Similarly, for contig 27 there are four annotated CDS: a hypothetical protein, a plasmid recombination protein, a CopG transcriptional regulator, and a replication RepB protein. Contig 31 annotates as three CDS: a hypothetical protein, a transcriptional regulator, and a replication RebB protein. Therefore, mobile plasmids appear to exist in E. cecorum but do not seem to be widespread or the drivers of the emergence of sepsis. However, this is only based on identification of circular elements from the Unicycler assemblies in the 34 genomes we have generated.
Our Prokka-Roary analysis of 227 genomes suggests that the core+ pan genome for this species contains 14728 genes with 878 genes in the strict-core genome (99%≤strains≤100%), 335 genes in the soft-core genome (95%≤strains<99%), 1689 genes in the shell genome (15%≤strains<95%), and 11826 cloud genes (strains<15%). Prior analysis of 140 genomes had suggested a pangenome of 8523 gene clusters with 1207 genes in the strict-core genome, 4664 genes in the accessory genome, and a unique genome of 2,652 [16]. Thus, our results appear to expand the pangenome by over 6000 genes with similar gene content for the sum of the strict-core and soft-core to the pervious numbers for a strict-core genome. Scoary analyses of the pan genome with respect to phenotypic traits (Table S2) were employed to determine whether any particular loci, or gene clusters had high specificity for either Chicken Disease isolates or for Strict Sepsis. We also analyzed for Chicken Disease which compared BCO isolates, along with the sepsis isolates, to the isolates for which there is no disease phenotype known. Since many of the isolates for which there is no disease phenotype known may actually include pathogenic isolates, we were skeptical of any meaningful results for that particular comparison. The data for loci positively or negatively associated with Chicken Disease identified 195 genes with corrected P-values <0.05 (Table S3). Of the 159 loci positively associated (Odds ratio≥5) with Chicken Disease trait, 63 annotated as something other than hypothetical protein. Of the 20 loci negatively associated (Odds ratio≤0.2) only 6 annotated as something other than hypothetical protein. For these 159 loci we identified 12 clusters of two or more loci based on position in the genomes for either of three BCO isolates. Seven of the locus clusters were present and clustered in all three isolate genomes examined (Table S3). Functions associated with these clusters include glycosylation loci including the capsular genes identified by Borst [13], carbohydrate/sugar transferase systems, probable transposable elements, and vitamin/biotin transferase systems. When we analyzed all the genomes for loci associated with the Strict Sepsis group, the data identified 101 loci with 97 positively associated and 4 negatively associated (Table S4) However, of these loci 94 were apparently identified because they are found in primarily in the genomes present in cluster A in Figure 1. To reduce the bias of Cluster A we reduced representation from that cluster by excluding some of the Cluster A isolates to generate the trait group Sepsis Strict Reduced (Table S1). The Scoary output using Sepsis Strict Reduced indicated that there were no loci with Bonferonni corrected P-values <0.05 (Table S5). Inspection of this data identifies 24 loci with sensitivity ≥50 (measure of frequency of presence in isolates with the trait), but only 10 of those loci had a specificity score ≥50 (measure of how specificity the gene is isolates with the trait). Of those 10 loci with higher specificity, five annotate as hypothetical protein and another a transposase. The other four loci encode functions in vitamin B12 import, ascorbate uptake, hexulose conversion, and a transcriptional regulator for glucosamine utilization. We interpret these Roary-Scoary results to indicate that there is no evidence of specific gene acquisitions that are shared by the majority of sepsis isolates. Therefore, either there are no specific gene acquisitions that can convert a BCO pathogen into a sepsis pathogen, or individual clades have specific independent gene sets that must be acquired for transition to a sepsis pathogen based on their particular pan genome composition.
To assess clade specific gene acquisition we used Scoary to analyze gene presence/absence for sepsis and BCO isolates in cluster 1 comprised of French isolates (trait CIRMBP Cluster 1 in Table S1). This compares four sepsis isolates to 22 BCO isolates within this clade. The Scoary results (Table S6) identified only seven genes, with six annotated as hypothetical proteins and one a transposase. None of these loci had significant corrected P-values for association but did show high specificity scores. Five of the loci are clustered in at least two of the sepsis isolate genomes and Phaster results show this region is likely a prophage (contig 1 from 109451-157474). Thus, our analyses failed to reveal compelling evidence for gene acquisition driving the emergence of sepsis.
For the transition from BCO pathogen to sepsis pathogen, an alternative to gene acquisition through horizontal transfer, is that the transition involves mutation of resident genes. Evidence from sequence analysis of ancient Marek’s Disease Virus strongly support that this virus has become more pathogenic not by gene acquisition or gene rearrangement, but through point mutations [38]. Emergence of sepsis through point mutations would also be consistent with the polyphyletic origins of sepsis isolates (Figure 1). Existing pathogens or commensals could experience a few key mutations which would allow them to survive in blood and colonize organs.
We first used ParSNP to identify and determine core genome SNPs that are differentially represented in BCO vs sepsis isolates. The ParSNP analysis was limited to those genomes known to be from cases of chicken disease (scored as 1 in Chicken Disease; Table S1). For this analysis we also employed the same reduced representation from Cluster A (exclude NA isolates as in Sepsis Strict Reduced; Table S1). This compared 17 sepsis isolates to 113 BCO isolates using the finished/complete genome BCO isolate SA1 as the reference. Since the reference genome was a BCO isolate, only SNPs over-represented in sepsis should represent mutations favoring sepsis. The core genome SNPs (n=78150) were filtered for those present in >94% of Sepsis Strict Reduced isolates (n=967) and then for frequencies ≥ 0.30 in the sepsis isolates relative to the BCO isolates. This identified 34 SNPs; 32 bi-allelic and 2 tri-allelic (Table S7). These 34 potentially diagnostic SNPs occur in only six genes. Eight SNPs are in first or second codon base positions and most likely to be missense while 26 are in the wobble position and more likely to be silent. Interestingly, four of the genes (Smc, YaaA, hypothetical protein, and AldC) appear to be clustered, as they represent annotated PEG 229, 230, 231 and 234.
We analyzed the same reduced representation Chicken Disease genomes using Maast as an alternative to ParSNP because the two programs use different alignment algorithms to genotype SNPs and are known to produce different results [35]. The reference genome was again BCO isolate SA1. The maximum SNP genotype frequencies for Sepsis genomes from Maast were 0.54 (Table S8); considerably lower than the 1.00 SNP frequencies identified by ParSNP (Table S7). The Maast genotype data were filtered for SNPs >50% in Sepsis (n=252) and then frequency difference (sepsis - BCO) ≥ 0.20; resulting in a total of 77 SNPs (74 biallelic and 3 triallelic); 7 intergenic, and 70 in coding sequences for 51 protein encoding genes (Table S8). Fifteen of the genes that flank or span the SNPs annotated as hypothetical proteins, but none of these hypothetical genes is PEG 231 identified in the ParSNP analyses. Of the four clustered genes from the ParSNP data, only the Smc gene was identified also in the Maast analyses. Both ParSNP and Maast identified SNPs in PTS system fructose-specific EIIABC component. Potential clusters of SNPs identified in the Maast analyses include those affecting PEG 346 to 373, 570 to 577, 629 to 637, 744 to 753. There is also the potential for clustering of genes affected for seven SNPs affecting four genes from PEG 594 to 603.
We extracted and aligned the encoded polypeptides for the six genes from ParSNP and 12 additional genes from Maast to identify all variant polypeptide positions (Table S9). The analyses included predicted polypeptides from all chicken disease isolates and included those from Cluster A that had been excluded from the initial ParSNP and Maast analyses. Each variant position was scored for frequency in the Sepsis Strict group of isolates relative to the BCO isolates. This identified 114 polypeptide variants where Sepsis percentage was greater than 20 points higher than the percentage in BCO (green highlight in Table S9). The highest frequency protein variations were the R555Q and A590V substitutions in PTS system fructose-specific EIIABC which were found in 75% of sepsis isolates but only in 33 and 29%, respectively, of BCO isolates (Table S9). All 114 polypeptide variants were also scored for frequency in the Sepsis Strict Reduced (to reduce influence from the higher number of sepsis isolates from Cluster A). Of the 114 protein variants there were 15 that were ≥ 50% in Sepsis Strict Reduced in 7 genes (yellow highlight in Table S9). Those 15 SNPs are summarized in Table 3 and are candidates to be key mutations driving the adaptation from BCO to sepsis.
Annotation of these seven genes and the literature support possible roles in virulence of E. cecorum. Hypothetical protein PEG231 was identified in the ParSNP analysis (Table S7). BLASTp analysis at uniport.org identifies this as a serine aminopeptidase S33 domain-containing protein. Hip1 in Mycobacterium tuberculosis is a S33 serine aminopeptidase that inhibits macrophage and dendritic cell functions [39]. PTS (phosphotransferase transport system) fructose-specific EIIABC has been found to regulate virulence expresssion and stress response in Lysteria monocytogenes [40], and biofilm and endocarditis in Enterococcus faecium [41]. CBCL1 orthologs in Pseudomonas aeruginosa are involved in production of quorum sensing signals [42]. BLASTp at uniprot.org identifies ElaA as a GNAT family N-acetyltransferase, a family of proteins involved in bacterial adaptation to diverse habitats [43]. EttA regulates protein synthesis in energy-depleted cells and is critical for bacterial survival in long-term stationary phase [44]. RpoN encodes a sigma factor, σ54, for directing RNA polymerase to alternative promoters. Orthologs have been shown to regulate virulence determinants including pili, flagella, and type III secretion systems (reviewed in [45]). YheH is known to function in the signaling pathway for sporulation in Bacillus subtilis [46]. Therefore, the genes identified have to do with environmental response and gene regulation. Further, there are at least an additional 39 genes from the Maast analysis that we have yet to assess whether they have coding variations differentially present in sepsis vs BCO isolate genomes (Table S8). There are also intergenic region SNPs that could affect regulation of flanking genes.
Future work should be focused on obtaining additional isolates from sepsis outbreaks to determine how much genomic diversity is present in each outbreak, and to assess whether the same genotypes are present in successive outbreaks in a facility. Previous work has failed to identify vertical transmission of E. cecorum using standard culture methods [10; 18]. More sensitive DNA-based methods need to be applied to breeder hens, hatcheries, embryos, and broiler houses [47]. Virulence testing of isolates is also problematic. Others have relied on chicken embryo lethality assays for evaluation of E.cecorum isolates [17,48,49], but the direct relevance to pathogenicity post hatch has been a concern [50]. Experimental infections of young chicks with isolates of E. cecorum have shown some promise [10] but have not been used to evaluate relative virulence of different isolates. Some insects have been used for virulence assays of human isolates of other Enterococcus species (reviewed in [51]). Alternative models could be used to discern whether sepsis isolates and BCO isolates have measurable differences in pathogenesis. Further, many of the genomes in the national databases are poorly documented for specific clinical source and host disease state, thus the need for expanding the available genomes from well documented cases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: ParSNP phylogenomic tree of 227 Enterococcus cecorum isolates midpoint-rooted with E. cecorum SA1 as the reference genome; Table S1: Phenotypic group assignments for the 227 isolate genomes used in this work; Table S2: BLASTp results for the 227 genomes queried with the 12 encoded polypeptides (peg 312 to 323) from the capsular cluster identified by Borst; Table S3: Scoary output for gene presence-absence for phenotypic trait Chicken Disease.; Table S4: Scoary output for gene presence-absence for phenotypic trait Sepsis_Strict; Table S5: Scoary output for gene presence-absence for phenotypic trait Sepsis_Strict_Reducced (SSR); Table S6. Scoary output for gene presence-absence for phenotypic trait CIRMBP Cluster 1; Table S7: Most differentially represented core genome SNPs distinguishing BCO and sepsis isolate genomes as determined using ParSNP; Table S8: Most differentially represented core genome SNPs distinguishing BCO and sepsis isolate genomes as determined using mast; Table S9: Protein variations of selected annotated polypeptides from sepsis and BCO isolate genomes.
Author Contributions
DR performed most of the genomic characterizations and wrote the initial draft of the manuscript, JP provided advice, guidance, and expertise regarding the bioinformatics. AA, JP and DR secured funding and oversaw the research. DR was responsible for the final draft of the manuscript with contributions from AA and JP.
Funding
This research was funded by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
Data Availability Statement
All genomes analyzed are available in the databases at National Cener for Biotechnology Information (https://www.ncbi.nlm.nih.gov).
Conflicts of Interest
The authors declare no conflicts of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- F. Lebreton, R.J. Willems, and M.S. Gilmore, Enterococcus diversity, origins in nature, and gut colonization. in: M.S. Gilmore, D.B. Clewell, Y. Ike, and N. Shankar, (Eds.), Enterococci: from commensals to leading causes of drug resistant infection [Internet], Massachusetts Eye and Ear Infirmary, Boston, MA, 2014, pp. 1-46.
- Z. Zhong, W. Zhang, Y. Song, W. Liu, H. Xu, X. Xi, B. Menghe, H. Zhang, and Z. Sun, Comparative genomic analysis of the genus Enterococcus. Microbiol. Res. 196 (2017) 95-105. [CrossRef]
- S.M. Naser, M. Vancanneyt, E. De Graef, L.A. Devriese, C. Snauwaert, K. Lefebvre, B. Hoste, P. Švec, A. Decostere, and F. Haesebrouck, Enterococcus canintestini sp. nov., from faecal samples of healthy dogs. Int. J. Syst. Evol. Microbiol. 55 (2005) 2177-2182. [CrossRef]
- G.K. Walker, M.M. Suyemoto, S. Gall, L. Chen, S. Thakur, and L.B. Borst, The role of Enterococcus faecalis during co-infection with avian pathogenic Escherichia coli in avian colibacillosis. Avian Pathol. 49 (2020) 589-599. [CrossRef]
- V. Sistek, A.F. Maheux, M. Boissinot, K.A. Bernard, P. Cantin, I. Cleenwerck, P. De Vos, and M.G. Bergeron, Enterococcus ureasiticus sp. nov. and Enterococcus quebecensis sp. nov., isolated from water. Int. J. Syst. Evol. Microbiol. 62 (2012) 1314-1320. [CrossRef]
- M.F. Moreno, P. Sarantinopoulos, E. Tsakalidou, and L. De Vuyst, The role and application of enterococci in food and health. Int. J. Food. Microbiol. 106 (2006) 1-24. [CrossRef]
- T. O’Driscoll, and C.W. Crank, Vancomycin-resistant enterococcal infections: epidemiology, clinical manifestations, and optimal management. Infection and drug resistance (2015) 217-230. [CrossRef]
- R.C. Moellering Jr, Emergence of Enterococcus as a significant pathogen. Clin. Infect. Dis. (1992) 1173-1176. [CrossRef]
- C.R. Jackson, P.J. Fedorka-Cray, and J.B. Barrett, Use of a Genus- and Species-Specific Multiplex PCR for Identification of Enterococci. J. Clin. Microbiol. 42 (2004) 3558-3565. [CrossRef]
- L.B. Borst, M.M. Suyemoto, A.H. Sarsour, M.C. Harris, M.P. Martin, J.D. Strickland, E.O. Oviedo, and H.J. Barnes, Pathogenesis of Enterococcal Spondylitis Caused by Enterococcus cecorum in Broiler Chickens. Vet. Pathol. 54 (2017) 61-73. [CrossRef]
- A. Jung, L.R. Chen, M.M. Suyemoto, H.J. Barnes, and L.B. Borst, A Review of Enterococcus cecorum Infection in Poultry. Avian Dis. 62 (2018) 261-271. [CrossRef]
- C.M. Logue, C.B. Andreasen, L.B. Borst, H. Eriksson, D.J. Hampson, S. Sanchez, and R.M. Fulton, Other Bacterial Diseases. in: D.E. Swayne, M. Boulianne, C.M. Logue, L.R. McDougald, V. Nair, D.L. Suarez, S. Wit, T. Grimes, D. Johnson, M. Kromm, T.Y. Prajitno, I. Rubinoff, and G. Zavala, (Eds.), Diseases of Poultry, John Wiley & Sons, 2020, pp. 995-1085.
- L.B. Borst, M.M. Suyemoto, E.H. Scholl, F.J. Fuller, and H.J. Barnes, Comparative Genomic Analysis Identifies Divergent Genomic Features of Pathogenic Enterococcus cecorum Including a Type IC CRISPR-Cas System, a Capsule Locus, an epa-Like Locus, and Putative Host Tissue Binding Proteins. PLoS One 10 (2015) e0121294. [CrossRef]
- I.T. Paulsen, L. Banerjei, G.S. Myers, K.E. Nelson, R. Seshadri, T.D. Read, D.E. Fouts, J.A. Eisen, S.R. Gill, J.F. Heidelberg, H. Tettelin, R.J. Dodson, L. Umayam, L. Brinkac, M. Beanan, S. Daugherty, R.T. DeBoy, S. Durkin, J. Kolonay, R. Madupu, W. Nelson, J. Vamathevan, B. Tran, J. Upton, T. Hansen, J. Shetty, H. Khouri, T. Utterback, D. Radune, K.A. Ketchum, B.A. Dougherty, and C.M. Fraser, Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299 (2003) 2071-4. [CrossRef]
- K.L. Palmer, P. Godfrey, A. Griggs, V.N. Kos, J. Zucker, C. Desjardins, G. Cerqueira, D. Gevers, S. Walker, J. Wortman, M. Feldgarden, B. Haas, B. Birren, and M.S. Gilmore, Comparative Genomics of Enterococci: Variation in Enterococcus faecalis, Clade Structure in E. faecium, and Defining Characteristics of E. gallinarum and E. casseliflavus. mBio 3 (2012) 10.1128/mbio.00318-11. [CrossRef]
- J. Laurentie, V. Loux, C. Hennequet-Antier, E. Chambellon, J. Deschamps, A. Trotereau, S. Furlan, C. Darrigo, F. Kempf, J. Lao, M. Milhes, C. Roques, B. Quinquis, C. Vandecasteele, R. Boyer, O. Bouchez, F. Repoila, J.L. Guennec, H. Chiapello, R. Briandet, E. Helloin, C. Schouler, I. Kempf, and P. Serror, Comparative Genome Analysis of Enterococcus cecorum Reveals Intercontinental Spread of a Lineage of Clinical Poultry Isolates. mSphere 8 (2023) e00495-22. [CrossRef]
- M. Arango, A. Forga, J. Liu, G. Zhang, L. Gray, R. Moore, M. Coles, A. Atencio, C. Trujillo, J.D. Latorre, G. Tellez-Isaias, B. Hargis, and D. Graham, Characterizing the impact of Enterococcus cecorum infection during late embryogenesis on disease progression, cecal microbiome composition, and early performance in broiler chickens. Poult. Sci. 102 (2023) 103059. [CrossRef]
- M.J. Kense, and W.J.M. Landman, Enterococcus cecorum infections in broiler breeders and their offspring: molecular epidemiology. Avian Pathol. 40 (2011) 603-612.
- G. Dunnam, J. Thornton, and M. Pulido-Landinez, An Emerging Enterococcus cecorum Outbreak in a Broiler Integrator in the Southern US: Analysis of Antimicrobial resistance Trends. Proceeedings of the 71st Western Poultry Disease Conference 2022 (2022) 55-61.
- C.R. Hodak, D.M. Bescucci, K. Shamash, L.C. Kelly, T. Montina, P.B. Savage, and G.D. Inglis, Antimicrobial Growth Promoters Altered the Function but Not the Structure of Enteric Bacterial Communities in Broiler Chicks ± Microbiota Transplantation. Animals 13 (2023) 997. [CrossRef]
- J. Sambrook, E.F. Fritsch, and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 1989.
- N.S. Ekesi, A. Hasan, A. Alrubaye, and D. Rhoads, Analysis of Genomes of Bacterial Isolates from Lameness Outbreaks in Broilers. Poult. Sci. 100 (2021) 101148. [CrossRef]
- R.D. Olson, R. Assaf, T. Brettin, N. Conrad, C. Cucinell, James J. Davis, Donald M. Dempsey, A. Dickerman, Emily M. Dietrich, Ronald W. Kenyon, M. Kuscuoglu, Elliot J. Lefkowitz, J. Lu, D. Machi, C. Macken, C. Mao, A. Niewiadomska, M. Nguyen, Gary J. Olsen, Jamie C. Overbeek, B. Parrello, V. Parrello, Jacob S. Porter, Gordon D. Pusch, M. Shukla, I. Singh, L. Stewart, G. Tan, C. Thomas, M. VanOeffelen, V. Vonstein, Zachary S. Wallace, Andrew S. Warren, Alice R. Wattam, F. Xia, H. Yoo, Y. Zhang, Christian M. Zmasek, Richard H. Scheuermann, and Rick L. Stevens, Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acid Res. 51 (2022) D678-D689. [CrossRef]
- J.R. Grant, and P. Stothard, The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acid Res. 36 (2008) W181-W184. [CrossRef]
- C.L. Brown, J. Mullet, F. Hindi, J.E. Stoll, S. Gupta, M. Choi, I. Keenum, P. Vikesland, A. Pruden, and L. Zhang, mobileOG-db: a Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl. Environ. Microbiol. 88 (2022) e0099122.
- C. Bertelli, M.R. Laird, K.P. Williams, Simon Fraser University Research Computing Group, B.Y. Lau, G. Hoad, G.L. Winsor, and F.S. Brinkman, IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acid Res. 45 (2017) W30-W35. [CrossRef]
- A. Pon, A. Marcu, D. Arndt, J.R. Grant, T. Sajed, Y. Liang, and D.S. Wishart, PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acid Res. 44 (2016) W16-W21. [CrossRef]
- J.A. Lees, S.R. Harris, G. Tonkin-Hill, R.A. Gladstone, S.W. Lo, J.N. Weiser, J. Corander, S.D. Bentley, and N.J. Croucher, Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 29 (2019) 304-316. [CrossRef]
- T.J. Treangen, B.D. Ondov, S. Koren, and A.M. Phillippy, The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15 (2014) 524. [CrossRef]
- M.V. Han, and C.M. Zmasek, phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10 (2009) 356. [CrossRef]
- S. Argimón, K. Abudahab, R.J.E. Goater, A. Fedosejev, J. Bhai, C. Glasner, E.J. Feil, M.T.G. Holden, C.A. Yeats, H. Grundmann, B.G. Spratt, and D.M. Aanensen, Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2 (2016). [CrossRef]
- T. Seemann, Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (2014) 2068-2069. [CrossRef]
- A.J. Page, C.A. Cummins, M. Hunt, V.K. Wong, S. Reuter, M.T.G. Holden, M. Fookes, D. Falush, J.A. Keane, and J. Parkhill, Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31 (2015) 3691-3693. [CrossRef]
- O. Brynildsrud, J. Bohlin, L. Scheffer, and V. Eldholm, Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17 (2016) 238. [CrossRef]
- Z.J. Shi, S. Nayfach, and K.S. Pollard, Maast: genotyping thousands of microbial strains efficiently. Genome Biol. 24 (2023) 186. [CrossRef]
- F. Teufel, J.J. Almagro Armenteros, A.R. Johansen, M.H. Gíslason, S.I. Pihl, K.D. Tsirigos, O. Winther, S. Brunak, G. von Heijne, and H. Nielsen, SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotech. (2022). [CrossRef]
- Y. Huang, V. Eeckhaut, E. Goossens, G. Rasschaert, J. Van Erum, G. Roovers, R. Ducatelle, G. Antonissen, and F. Van Immerseel, Bacterial chondronecrosis with osteomyelitis related Enterococcus cecorum isolates are genetically distinct from the commensal population and are more virulent in an embryo mortality model. Vet. Res. 54 (2023) 13. [CrossRef]
- S.R. Fiddaman, E.A. Dimopoulos, O. Lebrasseur, L. du Plessis, B. Vrancken, S. Charlton, A.F. Haruda, K. Tabbada, P.G. Flammer, S. Dascalu, N. Marković, H. Li, G. Franklin, R. Symmons, H. Baron, L. Daróczi-Szabó, D.N. Shaymuratova, I.V. Askeyev, O. Putelat, M. Sana, H. Davoudi, H. Fathi, A.S. Mucheshi, A.A. Vahdati, L. Zhang, A. Foster, N. Sykes, G.C. Baumberg, J. Bulatović, A.O. Askeyev, O.V. Askeyev, M. Mashkour, O.G. Pybus, V. Nair, G. Larson, A.L. Smith, and L.A.F. Frantz, Ancient chicken remains reveal the origins of virulence in Marek’s disease virus. Science 382 (2023) 1276-1281. [CrossRef]
- R. Madan-Lala, J.K. Sia, R. King, T. Adekambi, L. Monin, S.A. Khader, B. Pulendran, and J. Rengarajan, Mycobacterium tuberculosis Impairs Dendritic Cell Functions through the Serine Hydrolase Hip1. J Immunol 192 (2014) 4263-4272. [CrossRef]
- Y. Liu, B.B. Yoo, C.-A. Hwang, Y. Suo, S. Sheen, P. Khosravi, and L. Huang, LMOf2365_0442 Encoding for a Fructose Specific PTS Permease IIA May Be Required for Virulence in L. monocytogenes Strain F2365. Front. Microbiol. 8 (2017). [CrossRef]
- F.L. Paganelli, J. Huebner, K.V. Singh, X. Zhang, W. van Schaik, D. Wobser, J.C. Braat, B.E. Murray, M.J.M. Bonten, R.J.L. Willems, and H.L. Leavis, Genome-wide Screening Identifies Phosphotransferase System Permease BepA to Be Involved in Enterococcus faecium Endocarditis and Biofilm Formation. J. Infect. Dis. 214 (2016) 189-195. [CrossRef]
- J.P. Coleman, L.L. Hudson, S.L. McKnight, J.M. Farrow, M.W. Calfee, C.A. Lindsey, and E.C. Pesci, Pseudomonas aeruginosa PqsA Is an Anthranilate-Coenzyme A Ligase. J. Bact. 190 (2008) 1247-1255.
- A. Dash, and R. Modak, Protein Acetyltransferases Mediate Bacterial Adaptation to a Diverse Environment. J. Bact. 203 (2021) 10.1128/jb.00231-21. [CrossRef]
- G. Boël, P.C. Smith, W. Ning, M.T. Englander, B. Chen, Y. Hashem, A.J. Testa, J.J. Fischer, H.-J. Wieden, J. Frank, R.L. Gonzalez, and J.F. Hunt, The ABC-F protein EttA gates ribosome entry into the translation elongation cycle. Na.t Struct. Mol. Biol. 21 (2014) 143-151. [CrossRef]
- M.J. Kazmierczak, M. Wiedmann, and K.J. Boor, Alternative Sigma Factors and Their Roles in Bacterial Virulence. Microbiology and Molecular Biology Reviews 69 (2005) 527-543. [CrossRef]
- S. Fukushima, M. Yoshimura, T. Chibazakura, T. Sato, and H. Yoshikawa, The putative ABC transporter YheH/YheI is involved in the signalling pathway that activates KinA during sporulation initiation. FEMS Microbiol. Lett. 256 (2006) 90-97. [CrossRef]
- A. Shwani, B. Zuo, A. Alrubaye, J. Zhao, and D.D. Rhoads, A Simple, Inexpensive Alkaline Method for Bacterial DNA Extraction from Environmental Samples for PCR Surveillance and Microbiome Analyses. Appl. Sci. 14 (2024) 141. [CrossRef]
- L.B. Borst, M.M. Suyemoto, S. Keelara, S.E. Dunningan, J.S. Guy, and H.J. Barnes, A Chicken Embryo Lethality Assay for Pathogenic Enterococcus cecorum. Avian Dis. 58 (2014) 244-248. [CrossRef]
- B. Dolka, D. Chrobak-Chmiel, M. Czopowicz, and P. Szeleszczuk, Characterization of pathogenic Enterococcus cecorum from different poultry groups: Broiler chickens, layers, turkeys, and waterfowl. PLoS One 12 (2017) e0185199. [CrossRef]
- N. Ekesi, A. Hasan, A. Parveen, A. Shwani, and D. Rhoads, Embryo Lethality Assay for Evaluating Virulence of Isolates from Bacterial Chondronecrosis with Osteomyelitis in Broilers. Poult. Sci. (in review) (2021).
- D.A. Garsin, K.L. Frank, J. Silanpää, F.M. Ausubel, A. Hartke, N. Shankar, and B.E. Murray, Pathogenesis and Models of Enterococcal Infection. in: M.S. Gilmore, D.B. Clewell, Y. Ike, and N. Shankar, (Eds.), Enterococci: from commensals to leading causes of drug resistant infection [Internet], Massachusetts Eye and Ear Infirmary, Boston, MA, 2014, pp. 139-192.
Figure 1.
PopPUNK phylogenomic tree of 227 Enterococcus cecorum isolates. Strain names are the leaves. Clusters of USA isolates from the sepsis survey are bracketed with letters denoting cluster (Table 1), and orange arrows indicating solo isolates. Node colors indicate isolation anatomical location: Pink- air sac, intestine, cloaca, feces, egg residue, feces, rinsate, swab; Green- blood, heart, liver, spleen, peritoneum; Red- air sacculitis, bone marrow, leg joint, spine, osteomyelitis; Gray- unknown; Yellow- meat; Black- reactor. Node shape indicates the isolate source: Circle- UA poultry research farm, osteomyelitis, research clinic; Triangle- corporate survey; Plus- human; Square- not available/unknown.
Figure 1.
PopPUNK phylogenomic tree of 227 Enterococcus cecorum isolates. Strain names are the leaves. Clusters of USA isolates from the sepsis survey are bracketed with letters denoting cluster (Table 1), and orange arrows indicating solo isolates. Node colors indicate isolation anatomical location: Pink- air sac, intestine, cloaca, feces, egg residue, feces, rinsate, swab; Green- blood, heart, liver, spleen, peritoneum; Red- air sacculitis, bone marrow, leg joint, spine, osteomyelitis; Gray- unknown; Yellow- meat; Black- reactor. Node shape indicates the isolate source: Circle- UA poultry research farm, osteomyelitis, research clinic; Triangle- corporate survey; Plus- human; Square- not available/unknown.
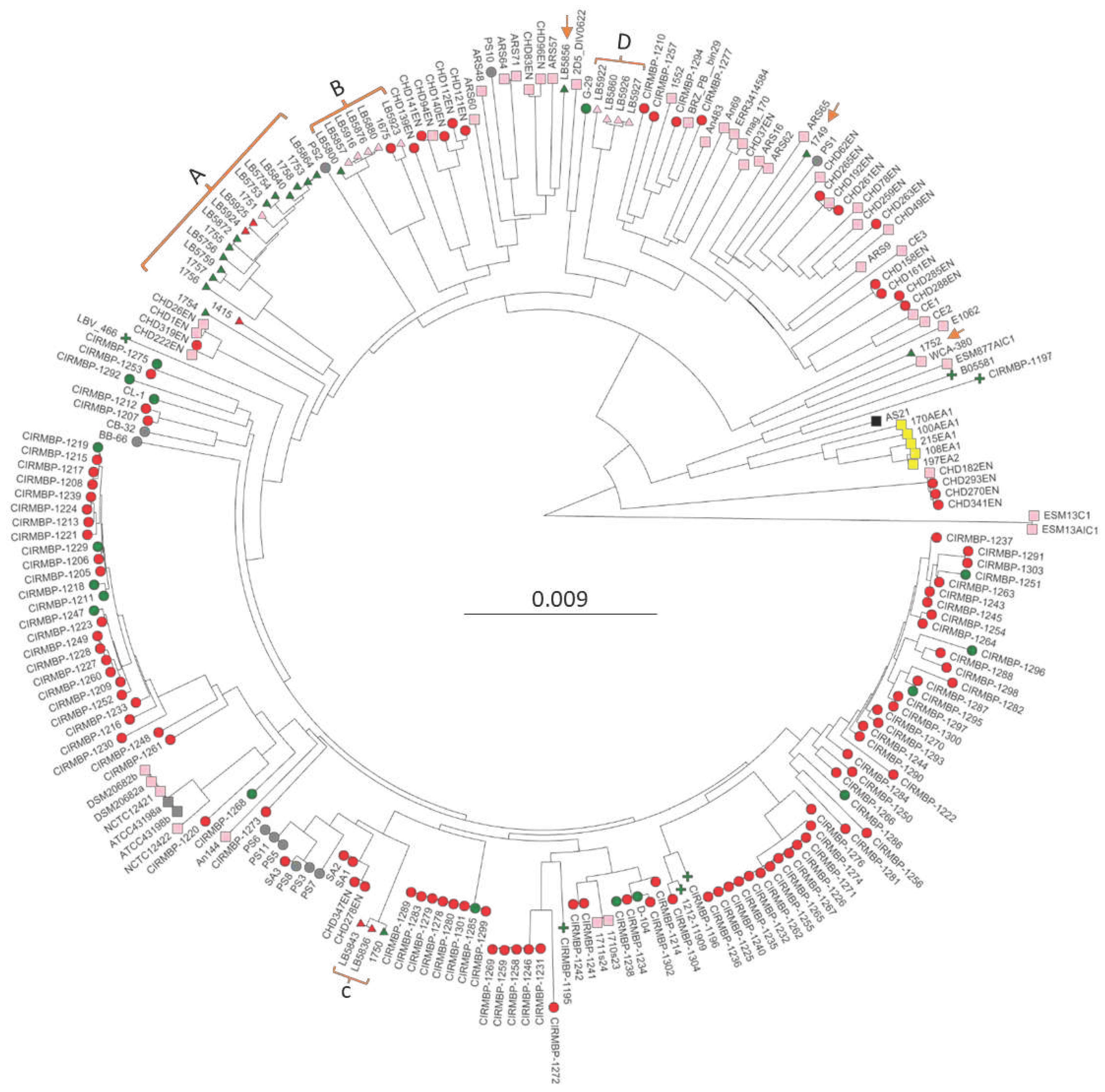

Table 1.
Details for Enterococcus cecorum strain source, date, location and bird age. For each strain the genome assembly in number of assembled contigs, total assembly in base pairs and NCBI BioSample are presented. For each DNA, the BLASTp results (average for percent identity * query coverage for protein gene products gene 312 to 323) for the Borst virulence gene cluster. PopPUNK cluster is for the groupings based on phylogenomic comparisons. Not available: n/a.
Table 1.
Details for Enterococcus cecorum strain source, date, location and bird age. For each strain the genome assembly in number of assembled contigs, total assembly in base pairs and NCBI BioSample are presented. For each DNA, the BLASTp results (average for percent identity * query coverage for protein gene products gene 312 to 323) for the Borst virulence gene cluster. PopPUNK cluster is for the groupings based on phylogenomic comparisons. Not available: n/a.
| Strain | Source | Date Collected | Collect Location | Bird Age | Assembly contigs | Total bp | BioSample | BLASTp | PopPUNK cluster |
| 1415 | Tibial pus | 6/23/2016 | farm16 | 4.3 | 65 | 2411963 | SAMN38750234 | 100 | A |
| 1675 | Femoral head necrosis | 11/26/2020 | UAPRF | 8 | 56 | 2188186 | SAMN38750235 | 100 | B |
| 1749 | Liver | n/a | farm15 | 0.5 | 93 | 2494030 | SAMN38750236 | 15 | solo |
| 1750 | Heart | n/a | farm22 | 3 | 49 | 2281603 | SAMN38750237 | 100 | C |
| 1751 | n/a | n/a | farm19 | 2.5 | 49 | 2300397 | SAMN38750238 | 100 | A |
| 1752 | Liver | n/a | farm8 | 3.6 | 32 | 2279238 | SAMN38750239 | 17 | solo |
| 1753 | Heart | n/a | farm15 | 3.1 | 51 | 2240969 | SAMN38750240 | 100 | A |
| 1754 | n/a | n/a | farm2 | n/a | 47 | 2180738 | SAMN38750241 | 100 | A |
| 1755 | Heart | n/a | farm2 | n/a | 58 | 2366287 | SAMN38750242 | 100 | A |
| 1756 | Heart | n/a | farm8 | 3.2 | 62 | 2288864 | SAMN38750243 | 100 | A |
| 1757 | Heart | n/a | farm4 | 3.3 | 53 | 2297190 | SAMN38750244 | 100 | A |
| 1758 | Heart | n/a | farm22 | 3 | 53 | 2249959 | SAMN38750245 | 100 | A |
| LB5753 | Heart | 2/5/2021 | farm17 | 3 | 56 | 2226311 | SAMN38750246 | 100 | A |
| LB5754 | Heart | 2/12/2021 | farm9 | 5 | 62 | 2234879 | SAMN38750247 | 100 | A |
| LB5756 | Heart | 3/8/2021 | farm13 | 2.3 | 62 | 2239264 | SAMN38750248 | 100 | A |
| LB5759 | Heart | 3/8/2021 | farm11 | 2.6 | 75 | 2392627 | SAMN38750249 | 100 | A |
| LB5800 | Heart | 3/11/2021 | farm3 | 3 | 42 | 2212442 | SAMN38750250 | 100 | B |
| LB5836 | Vertebral osteomyelitis | 9/29/2020 | farm6 | 12 | 69 | 2320142 | SAMN38750251 | 100 | C |
| LB5840 | Heart | 10/13/2020 | farm5 | n/a | 57 | 2258228 | SAMN38750252 | 100 | A |
| LB5843 | Vertebral osteomyelitis | 10/20/2020 | farm7 | 8 | 70 | 2319059 | SAMN38750253 | 100 | C |
| LB5856 | Heart | 12/21/2020 | farm14 | n/a | 81 | 2378014 | SAMN38750254 | 40 | solo |
| LB5857 | Egg transfer residue | 3/26/2021 | hatchery 1 | Eggs | 47 | 2180187 | SAMN38750255 | 100 | B |
| LB5860 | Cull eggs | 3/24/2021 | hatchery 1 | Eggs | 102 | 2230151 | SAMN38750256 | 100 | D |
| LB5864 | Heart | 12/9/2020 | farm20 | 2.3 | 61 | 2239173 | SAMN38750257 | 100 | A |
| LB5872 | Heart | 4/21/2021 | farm18 | n/a | 65 | 2365322 | SAMN38750258 | 100 | A |
| LB5876 | Egg transfer residue | 5/10/2021 | hatchery 1 | Eggs | 37 | 2183317 | SAMN38750259 | 100 | B |
| LB5880 | Air sacculitis | 4/29/2021 | farm21 | 2 | 38 | 2146928 | SAMN38750260 | 100 | B |
| LB5916 | Egg transfer residue | 6/4/2021 | hatchery 1 | Eggs | 40 | 2193104 | SAMN38750261 | 100 | B |
| LB5922 | n/a | 6/16/2021 | farm12 | 1 | 88 | 2169308 | SAMN38750262 | 99 | D |
| LB5923 | intestinal tract | 6/21/2021 | farm 1 | 1.3 | 58 | 2239406 | SAMN38750263 | 100 | B |
| LB5924 | Air sacculitis | 4/2/2021 | farm10 | 2.4 | 61 | 2296440 | SAMN38750264 | 100 | A |
| LB5925 | Air sacculitis | 4/2/2021 | farm10 | 2.4 | 61 | 2297136 | SAMN38750265 | 94 | A |
| LB5926 | Egg transfer residue | 4/16/2021 | hatchery 2 | Eggs | 103 | 2277367 | SAMN38750266 | 100 | D |
| LB5927 | Egg transfer residue | 4/16/2021 | hatchery 2 | Eggs | 105 | 2276597 | SAMN38750267 | 97 | D |
Table 2.
BLASTp scores for protein encoded gene products from Borst gene cluster, based on strain categorization by phenotype for not chicken disease (none), chicken disease (CD), bacterial chondronecrosis with osteomyelitis (BCO) or strict sepsis (SS). See results for definitions of phenotype categorization. For each protein encoded gene the value is the average for that phenotype of the BLASTp scores for (percent identity*query coverage)/100. For each phenotype the count is the number of strains in that phenotypic category. the average±SEM were computed for all 12 polypeptides. Individual gene scores for each isolate are provided in Table S2.
Table 2.
BLASTp scores for protein encoded gene products from Borst gene cluster, based on strain categorization by phenotype for not chicken disease (none), chicken disease (CD), bacterial chondronecrosis with osteomyelitis (BCO) or strict sepsis (SS). See results for definitions of phenotype categorization. For each protein encoded gene the value is the average for that phenotype of the BLASTp scores for (percent identity*query coverage)/100. For each phenotype the count is the number of strains in that phenotypic category. the average±SEM were computed for all 12 polypeptides. Individual gene scores for each isolate are provided in Table S2.
| phentotype | count | Borst gene cluster protein encoded gene | average±SEM | |||||||||||
| 312 | 313 | 314 | 315 | 316 | 317 | 318 | 319 | 320 | 321 | 322 | 323 | |||
| None | 82 | 53 | 35 | 37 | 49 | 30 | 43 | 31 | 31 | 45 | 43 | 39 | 91 | 44±5 |
| CD | 145 | 85 | 77 | 79 | 83 | 75 | 79 | 74 | 75 | 84 | 85 | 82 | 98 | 81±2 |
| BCO | 113 | 84 | 76 | 77 | 82 | 73 | 77 | 72 | 73 | 81 | 83 | 79 | 97 | 80±2 |
| SS | 34 | 88 | 80 | 83 | 86 | 81 | 83 | 79 | 79 | 89 | 88 | 88 | 98 | 85±2 |
Table 3.
Summary of fifteen polypeptide variants most prevalent in Enterococcus cecorum sepsis isolates. Entries for each variant are the Gene Name, predicted Function, amino acid positions (AA Pos) affected, the reference residues (Ref), substitution (Alt), and percentage of isolates in each of three Phenotypic Trait Groups.
Table 3.
Summary of fifteen polypeptide variants most prevalent in Enterococcus cecorum sepsis isolates. Entries for each variant are the Gene Name, predicted Function, amino acid positions (AA Pos) affected, the reference residues (Ref), substitution (Alt), and percentage of isolates in each of three Phenotypic Trait Groups.
| Gene | Phenotypic Trait Group | ||||||
| Name | Function | AA Pos | Ref | Alt | BCO | Sepsis Strict | Sepsis Strict Reduced |
| PEG231 | Serine aminopeptidase S33 domain-containing protein | 261 | T | A | 35 | 66 | 50 |
| EIIABC | PTS fructose-specific EIIABC component | 555 | R | Q | 33 | 75 | 64 |
| 590 | A | V | 29 | 75 | 64 | ||
| CBCL1 | 4-chlorobenzoate--CoA ligase | 244 | G | H | 43 | 69 | 55 |
| 246 | I | L | 43 | 69 | 55 | ||
| 249 | H | Y | 43 | 69 | 55 | ||
| ElaA | Gcn5-related N-acetyltransferase (GNAT family) | 128 | N | K | 34 | 69 | 55 |
| EttA | Energy-dependent translational throttle protein | 106 | S | A | 40 | 69 | 55 |
| 534 | T | A | 39 | 66 | 50 | ||
| RpoN | RNA polymerase σ54 factor | 12-13 | TQ | -- | 25 | 38 | 50 |
| 22 | T | S | 26 | 38 | 50 | ||
| YheH | putative multidrug resistance ABC transporter ATP-binding/permease protein | 13 | I | L | 43 | 72 | 59 |
| 578 | D | N | 28 | 66 | 50 | ||
| 590 | S | I | 28 | 66 | 50 | ||
| 592-593 | EEI | GAD | 28 | 69 | 55 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



