
Submitted:
11 January 2024
Posted:
11 January 2024
You are already at the latest version
Abstract
Global warming and the global energy crisis are two major challenges humanity is currently confronting that are pressuring the scientific community to find efficient, low cost and environmentally sustainable solutions. Within this context, hydrogen has emerged as a clean and efficient energy carrier promising to replace the environmentally hazardous fossil fuels. The present study, of relevance to the water splitting domain, concerns the synthesis of two novel hybrid structures, based on perovskite materials functionalized with polyvinylpyrrolidone (PVP). The water electrolysis catalytic activity of these new materials was evaluated in a strongly alkaline medium. Perovskite-based modified electrodes were manufactured through four different procedures. The samples displayed electrocatalytic activity for the O2 evolution reaction and the most active electrode was the one obtained by drop-casting a mixture of LaMnO3:Ag/PVP, Carbon Black and Nafion on graphite support. The study is aimed at and succeeds in increasing the scientific database concerning the role of perovskite oxides in the water splitting field.
Keywords:
perovskite oxide
; electrocatalyst
; water splitting
; water electrolysis
; oxygen evolution reaction
1. Introduction
Perovskites represent interesting and promising materials due to advantages such as chemical stability, photostability, low production cost, modifiable energy in the band gap, high absorption properties, as well as long carrier lifetime and diffusion length, which can be used in green and sustainable environmental applications [1]. Despite the high dielectric constant and multifunctionality of perovskite materials, they possess high density, brittleness and low dielectric strength, as well as poor processability [2].
Polymer-perovskite nanocomposites have attracted the attention of researchers as multifunctional materials for the development of flexible components/devices with many significant technological uses, in which the favorable characteristics of inorganic perovskite nanofiller and organic polymer are effectively integrated [2].
Nanosized filler polymer composites exhibit outstanding properties due to the unique characteristics of nanoparticles, such as the high surface-to-volume ratio and large interfacial area formed between the matrix and nanoparticles, with enhanced mechanical, electrical, and thermal properties. Nanocomposites that combine the advantages of polymer and filler (ceramics) can be processed more easily and are viable alternatives to plain/doped ceramic materials [3].
PVP is a water-soluble, non-toxic amorphous nonionic polymer, with high solubility in polar solvents, widely used in the synthesis of nanoparticles [4] acting as a nanoparticle dispersant, growth modifier, surface stabilizer, preventing agglomeration of nanoparticles [5]. Due to its amphiphilic nature, PVP can affect the morphology and growth of nanoparticles by providing solubility in various solvents, discriminative surface stabilization, controlled crystal growth, playing the role of a shape control agent and facilitating the growth of specific crystal faces, while preventing the growth others [5].
Climate change, the worldwide increase in the demand for electricity and the depletion of the natural resources from which non-renewable energy is obtained are among the major problems facing humanity. In an attempt to resolve these issues, researchers have come to advance environmentally friendly technologies that exploit renewable energy sources and provide energy carriers [6]. Hydrogen is one such energy carrier that has attracted the attention of the scientific community due to its properties, such as its carbon-free composition and very high energy density [6,7]. This gas is currently regarded as an efficient source of environmentally friendly energy that can replace the fossil fuels considered to play a significant role in global warming [8].
There is more than one available technology for generating hydrogen and not all of them are greenhouse gas-free. For example, H2 can be obtained via biomass and fossil fuel burning [9], but in order to avoid damaging the environment the go-to processes are electrolysis and photoelectrolysis [10]. Today, electrochemical water splitting performed by exploiting solar energy either directly or indirectly constitutes a very promising approach for the large-scale generation of hydrogen in an environmentally friendly manner [11,12].
Water electrolysis is a well-known, simple, quick and non-polluting way of decomposing water molecules into O2 and H2 via two main half-reactions: the oxygen and hydrogen evolution reactions (OER and HER) unfolding at the anode and cathode, respectively. If the electricity required by this process is supplied from renewable energy sources - such as solar, wind and tidal energy – it becomes even more environmentally friendly [13]. One of the issues facing water electrolysis that stands out is constituted by the sluggish reaction kinetics of the OER and HER. To address this problem researchers are aiming to synthesize materials with high electrocatalytic activity for at least one of the two half-reactions. In order to make water splitting viable for large-scale H2 production the electrocatalysts must also be stable and low cost [14]. Currently, Pt-based catalysts for the HER and Ru- and Ir-based catalysts for the OER are benchmarks when it comes to the performance evaluation of the other catalysts reported in the literature [15,16,17]. However, their scarcity and high cost are significant obstacles for large-scale applications, making the identification of non-noble materials that are both highly efficient and stable a main concern. The scientific literature contains a large number of functional materials that have been studied regarding their electrocatalytic water splitting properties, ranging from noble metal-based to non-noble metal-based and metal-free ones [18,19,20].
Perovskite oxides are a category of functional materials that have been revealed to be very promising for the water splitting domain [21,22]. These compounds share a crystal structure characterized by the chemical formula ABO3, where the A-site is occupied by a bigger metal cation than the one occupying the B-site, and they possess properties such as piezoelectricity, superconductivity, ferroelectricity and enormous magnetoresistivity which make them useful for various industrial and commercial domains [23]. The full potential of perovskite oxides in the water splitting field has yet to be revealed, but the evaluation of their relevant catalytic properties is of current interest to researchers, as reflected in the recent publications of Sfirloaga et al. [24,25]. In one of the studies, hybrid materials based on montmorillonite functionalized with LaMnO3 perovskite were obtained for the first time and were tested regarding their water splitting electrocatalytic activity in alkaline media, which led to the identification of the modified electrode possessing the best properties for the HER [24]. A subsequently published investigation outlines the HER electrocatalytic properties in alkaline media of unsubstituted, Ca-substituted and Pd-substituted LaMnO3. The most significant results were obtained for the electrodes modified with compositions containing the Pd-substituted perovskite oxide [25].
The current paper continues the work of Sfirloaga et al. [25] concerning the water electrolysis catalytic properties of perovskite oxide-based modified electrodes via the study of two novel materials (LaMnO3:Ag/PVP (P11) and LaMnO3:Pd/PVP (P14)) in a strong alkaline medium. The modified samples whose OER and HER activity were evaluated experimentally were obtained using four different procedures. The results show that while both perovskite compounds display negligible electrocatalytic activity for the HER, the electrode modified with a composition containing P11 and manufactured with one of the procedures (designated as Procedure 4) is more active toward the OER than the rest of the investigated samples. The study complements the scientific literature of relevance to the water splitting field.
2. Materials and Methods
2.1. Materials and reagents
Perovskite materials (1% Ag-doped LaMnO3 and 1% Pd-doped LaMnO3) were obtained by a previously reported sol-gel technique [26,27,28]. Nafion solution of 5 % concentration was purchased from Sigma Aldrich (Saint Louis, MO, USA) as Nafion® 117 and Carbon Black - Vulcan XC 72 was acquired from Fuell Cell Store (Bryan, TX, USA). The glassy carbon (GC) pellets were from Andreescu Labor & Soft SRL (Bucharest, Romania) while the spectroscopic graphite bars (type SW.114) were manufactured at the National Corporation "Kablo Bratislava", the "ElectrocarbonTopolcany" factory (Bratislava, Slovakia). Polyvinylpyrrolidone (Sigma-Aldrich), potassium hydroxide (Merck, Darmstadt, Germany), potassium nitrate (Merck), potassium hexacyanoferrate(III) (Sigma-Aldrich), ethanol (Chimreactiv, Bucharest, Romania) and acetone (Chimreactiv) were also used in the study. All aqueous solutions were obtained with double-distilled water produced in the laboratory.
2.2. Synthesis of hybrid materials
The procedure for obtaining the new hybrid materials based on polyvinylpyrrolidone (PVP) polymer functionalized with LaMnO3 type perovskite structures doped with Ag (P11) or Pd (P14) consists in mixing the precursors in a mass ratio of 20:1 (PVP:perovskite) and dispersing them in distilled water. The resulting suspensions were stirred for 2 hours maintaining the temperature at 80 °C and 400 rpm using a magnetic stirrer SMHS-3 (WitegLabortechnik GmbH, Germany). The resulting viscous mixture was cast into thin film of 2-5 mm thickness on a flat surface (polypropylene film) and dried at room temperature for 12 hours. For further analyzes, the polymer-perovskite film was triturated until a homogeneous mixture with small grain size (up to 1 mm) was obtained and then dried in a forced convection oven, at 60 °C for 12 hours.
2.3. Characterization of hybrid materials
X-ray diffraction (XRD) data were collected using an X’Pert PRO MPD diffractometer (PANalytical, Netherlands) with Cu-Kα radiation in the 2θ range of 10-80 °. The ATR-FT-IR spectra were recorded at room temperature in the 4000–600 cm-1 range using a Bruker Vertex 70 spectrometer (Bruker Optik GmbH, Rosenheim, Germany) equipped with a Platinium ATR unit, Bruker Diamond A225/Q.1. The morphology of the samples and the atomic content were registered using a scanning electron microscope equipped with energy dispersive X-ray detector (Inspect S + EDAX, FEI, Holland) in low vacuum mode.
2.4. Procedures for manufacturing the modified electrodes
Four different procedures were employed to obtain the studied modified electrodes. Procedures 1 and 2 involved the use of the GC supports, while graphite was utilized as substrate for the samples obtained with Procedures 3 and 4. Prior to modification, the GC pellets were washed with an aqueous detergent solution and subsequently washed with water and rinsed with double-distilled water, acetone and ethanol. The modification process was initiated after a drying stage at 23 ± 2 °C. The preparation of the graphite supports involved several steps. Each bar-shaped piece of graphite was inserted into a polyethylene tube and subjected to a heat treatment at 180 °C that ensured the sealing of the two materials. The two ends of each bar were left uncovered. One of them was required to connect the graphite substrate to the potentiostat while the other was modified with catalytic material after a preliminary treatment. The treatment consisted of the polishing of the graphite surface with silicon carbide paper (grit size = 800 and 1200) and felt. This was followed by a washing stage involving water, double-distilled water, acetone and ethanol. After drying at 23 ± 2 °C the surface (φ = 6 mm) was modified by either Procedure 3 or Procedure 4.
Regarding Procedure 1, five suspensions in ethanol were obtained having the composition presented in Table 1. The role of Nafion was that of binder, enhancing the adhesion of the catalysts and Carbon Black to the substrate, while the use of Carbon Black was aimed at increasing the electron transfer among electrode surface and electroactive species [29]. Each suspension was utilized to manufacture a type of modified electrode. The construction of an electrode consisted of taking a volume of 10 µL from one of the suspensions and applying it via the drop-casting method to one of the surfaces of a glassy carbon pellet prepared according to the previously mentioned specifications. The modified sample resulted after a drying step at 23 ± 2 °C. To perform the electrochemical experiments the electrodes obtained using Procedure 1 were inserted into a polyamide support that ensured a constant geometric surface of 0.28 cm2 exposed to the electrolyte solution.
Five suspensions were also prepared for Procedure 2, but in accordance with the specifications in Table 2. Each modified electrode was manufactured by following the same steps described in the case of Procedure 1 and was inserted into the same polyamide support before being evaluated in terms of its water splitting electrocatalytic activity.
Procedure 3 is the same as Procedure 1 and Procedure 4 is the same as Procedure 2 except for the carbon substrate – the graphite support was used instead of the GC.
2.5. Electrochemical experiments
The electrochemical assembly used during the study of the OER and HER electrocatalytic activity of the electrodes constructed with the four procedures consisted of a Voltalab PGZ 402 potentiostat from Radiometer Analytical (Lyon, France), a glass electrolysis cell, an auxiliary plate-type Pt electrode with the geometric surface area of 0.8 cm2, the Ag/AgCl (sat. KCl) reference electrode and the working electrode. The role of the working electrode was fulfilled by each of the samples obtained with Procedures 1 to 4, but also by the unmodified glassy carbon and graphite electrodes. The electrolyte employed during the water splitting experiments was the strongly alkaline 1M KOH solution. The OER activity of the samples was evaluated by recording anodic linear sweep voltammograms (LSVs) while their HER activity was investigated by tracing cathodic LSVs. The iR-corrected polarization curves were obtained in unstirred solution at the scan rate (v) of 5 mV/s. Before each HER experiment the electrolyte solution was deaerated by high-purity nitrogen bubbling.
Except if stated differently, the electrochemical potential values (E) are expressed in terms of the Reversible Hydrogen Electrode (RHE) via Equation (1) and the current density values (i) refer to the geometric current density.
The water electrolysis electrocatalytic properties of the electrodes identified as having the highest activity for this process were subjected to additional electrochemical evaluation. Their electrochemically active surface area (EASA), the diffusion coefficient of hexacyanoferrate (III) ions (D), the values of their Tafel slopes and their stability were studied as well. The OER overpotential was calculated with Equation (2) and the HER overpotential with Equation (3). The Tafel slope was determined with Equation (4) and the EASA and D values were estimated using Equation (5) – the Randles-Sevcik equation – together with experimental data obtained from cyclic voltammetry experiments. The voltammetry curves were recorded in 1 M KNO3 solution, in the absence and presence of 4 mM potassium hexacyanoferrate (III), at increasing scan rate values (v = 50, 100, 150, 200, 250, 300 and 350 mV/s) and in the 0 – 0.8 V potential range vs. Ag/AgCl (sat. KCl).
where: ERHE = Reversible Hydrogen Electrode potential (V), EAg/AgCl(sat. KCl) = potential expressed in terms of the Ag/AgCl (sat. KCl) reference electrode (V), ηOER and ηHER = O2 and H2 evolution overpotentials (V); η = overpotential (V); i = current density (mA/cm2); b = Tafel slope (V/dec); Ip = peak current (A); n = number of electrons involved in the redox process at T = 298 K; A = surface of the working electrode (cm2); D = diffusion coefficient of the electroactive species (cm2/s); C = concentration of the electroactive species (M) and v = scan rate (V/s).
3. Results and discussion
3.1. XRD analysis
Figure 1 shows the XRD patterns of the hybrid materials based on polyvinylpyrrolidone (PVP) functionalized with Ag or Pd doped LaMnO3 type perovskite structures. The structural parameters were refined by fitting the XRD patterns using the Rietveld refinement technique with High Score software was used to investigate the crystalline structure of the new hybrid materials obtained. Thus, the XRD pattern of pure PVP showed two broad characteristic peaks at 2θ of 11.25 ° and 21.21 °, which is in good agreement with results reported by Li et al. [34] and El Hotaby et al. [35]. The obtained hybrid materials present well defined peaks, mainly corresponding to the perovskite phase, which leads to the conclusion that the polymer did not mask the perovskite material.
3.2. FT-IR analysis
The FT-IR spectra for the precursors (PVP, LaMnO3:Ag, and LaMnO3:Pd) and the two hybrid materials (LaMnO3:Ag/PVP (P11), LaMnO3:Pd/PVP (P14)) are shown in Figure 2.
The FT-IR spectrum of pure PVA exhibits the following significant absorption bands: 3453 cm-1 related to O-H stretching, 2950 cm-1 corresponding to asymmetric C−H2 stretching [4,36], 1493−1421 cm−1 assigned to C−H2 deformations [37], 1286 cm−1 attributed to C–N bending vibration from the pyrrolidone structure [38].
A strong absorption peak located at 1652 cm-1 can be assigned to the C=O stretching vibration in the pyrrolidone group [4,36,38,39]. However, Mireles et al. [37] supported the assignment of the peak at ∼1660 cm−1 as amide I rather than as a carbonyl stretch.
All the major functional groups of host polymer can be found in the spectra of the hybrid materials. The FT-IR spectra of the hybrid materials show certain differences regarding the position of the absorption bands corresponding to PVP and increased intensities.
The C-H stretching vibration peaks blue-shift to 2953 cm-1 for sample P11, and 2952 cm-1 for sample P14, respectively. The broad band around 3600–3200 cm-1 corresponding to O-H stretching vibration is centered at ~3453 cm−1 in PVP spectra, while in both hybrid materials it is centered at 3418 cm−1.
The C=O stretching band is shifted from 1652 cm-1 to lower wavenumbers in both hybrid materials (1646 cm-1 for sample P11, and 1647 for sample P14). This red shift could be explained by the coordination between metallic species and carbonyl oxygen from PVP [39,40,41].
The band shifting and their variation in intensities in the FT-IR spectrum indicate the formation of new hybrid materials. The bands of both perovskites are not visible in the spectra of the hybrid materials in the 4000–600 cm-1 range, probably due to the large amount of PVP used compared to the amount of perovskite materials.
3.3. SEM micrographs
Figure 3 shows the surface morphologies revealed by SEM. Figure 3a presents the qualitative analysis of PVP. As can be seen, the particles have a spherical shape of several tens of micrometers. In the case of perovskite materials doped with Ag or Pd, functionalized with PVP (Figure 3c and e), it can be observed that the hybrid materials obtained show agglomerations in asymmetric formations. In order to make a comparative study, the SEM images of perovskite materials doped with Ag (Figure 3b) and Pd (Figure 3d) are presented. Thus, it can be seen that perovskite materials have a spherical shape and nanometric sizes.
3.4. Electrochemical experiments
The OER studies performed on the electrodes modified with Procedure 1 revealed GC1P11+CB and GC1P14+CB as the only samples having higher electrocatalytic activity than the sample modified only with Carbon Black (GC1CB). The polarization curves recorded on these electrodes are shown in Figure 4. At i = 10 mA/cm2 – the current density value at which ηOER is usually specified [42,43,44] – ηOER = 1.145 V for GC1P11+CB and 1.14 V for GC1P14+CB.
The samples obtained using Procedure 1 did not exhibit electrocatalytic activity for the HER, and those obtained by applying Procedure 2 exhibited negligible water splitting activity. In light of these results subsequent investigations focused only on the OER and the electrodes manufactured with Procedures 3 and 4 were evaluated in terms of their electrocatalytic activity for this half-reaction.
Figure 5 presents the LSVs recorded on the most catalytically active samples modified using Procedure 3. It can be observed that at current densities > 25 mA/cm2 the polarization curves traced on Gr3P11 and Gr3P14+CB overlap, but at lower values of this parameter ηOER for the former electrode is <ηOER for the latter. However, at i = 10 mA/cm2 the ηOER value determined for the sample obtained using only Carbon Black is higher than for Gr3P14+CB and very close to that of Gr3P11 (0.91 V vs. 0.9 V). Considering this, the OER catalytic activity of Gr3P11 is too low to be considered significant.
The linear voltammograms shown in Figure 6 were traced on the unmodified graphite electrode and on the samples modified according to Procedure 4. Perovskite-containing electrodes exhibited higher OER electrocatalytic activity compared with Gr0 and the Carbon Black-only modified graphite sample (Gr3CB). At i = 10 mA/cm2ηOER = 0.875 V for Gr4P11, 0.84 V for Gr4P14 and Gr4P14+CB, and 0.83 for Gr4P11+CB. At higher i values it is observed that the polarization curves are very close to each other and in some cases even overlap.
By comparing the LSVs traced on the perovskite-containing electrodes manufactured via the four procedures it can be concluded that the best results were obtained for the samples resulted by using Procedure 4. Because of the closeness of the ηOER values Gr4P11, Gr4P14, Gr4P11+CB and Gr4P14+CB were all considered for the further evaluation of their electrochemical properties, including their OER electrocatalytic ones. Firstly, their EASA and the diffusion coefficient of the hexacyanoferrate (III) ions (D) were estimated. To do so, the Randles-Sevcik equation was employed together with data from experiments performed as described in section 2.5. The obtained values are presented in Table 3.
High values for both parameters are to be desired since a higher D indicates a faster diffusion process while a higher EASA implies a larger number of catalytic active centers involved in an electrochemical process [45]. As can be seen in Table 3, the highest values were found for the Gr4P11+CB electrode.
The data acquired during the cyclic voltammetry experiments were used to plot the dependence between the peak current densities of the anodic and cathodic peaks corresponding to the ferrocyanide/ferricyanide redox couple and the square root of the scan rate (Figure 7). In all cases, the absolute values of the peak current densities increase proportionally with the scan rate. This increase denotes the specific behavior of diffusion-controlled electron transfer processes [46].
The OER kinetics at the interfaces between the four electrodes and the electrolyte solution were also studied. The obtained Tafel plots are shown in Figure 8, and the determined values of the Tafel slopes can be seen in Table 4. To represent the Tafel curve belonging to each sample the current density was normalized to their estimated EASA value (iEASA).
The smallest Tafel slope value is the one calculated for the Gr4P11+CB sample. A smaller value indicates more favorable kinetics and a higher reaction rate [47].
The electrochemical stability of the four electrodes was evaluated during 7 h chronoamperometric tests at the constant potential value corresponding to i = 5 mA/cm2. Gr4P11+CB was revealed to be more stable than the other investigated samples. The ivs. time curves recorded for this electrode and for Gr4P14+CB are presented in Figure 9. In the case of Gr4P11+CB, the i value stabilized at 5.8 mA/cm2. LSVs were obtained on this sample before and after the experiment and the results can be seen in Inset a. At higher i values the two polarization curves overlap almost perfectly, but this is not the case at values < 24 mA/cm2 where the stability test led to a decrease in the OER activity of the electrode. For example, at i = 10 mA/cm2ηOER = 0.88 V. This 50 mV increase is probably due to the high electrochemical potential applied throughout the chronoamperometric study. Still, the Gr4P11+CB electrode was more electrochemically stable than the other samples. To exemplify this, the ivs. time curve recorded under the same conditions but for Gr4P14+CB is also included in Figure 9. Apparently, the behavior of the two samples is not very different, but a closer look at the amperogram (Insets b and c) reveals the presence of features attributable to intense alternate processes of O2 bubble accumulation and release that are not observed on the curve traced on Gr4P11+CB [29]. More importantly, once the test ended and the surface of the sample was investigated visually it was observed that the perovskite-containing composition was partially detached from the graphite surface it was deposited on during the manufacturing process.
3.5. Further considerations
The results of the electrochemical experiments evidence the superior properties of the Gr4P11+CB electrode. Compared with the Gr4P11, Gr4P14 and Gr4P14+CB samples, Gr4P11+CB displays higher OER electrocatalytic activity, higher EASA and D values, a lower Tafel slope value and is more electrochemically stable. The higher OER catalytic activity of the electrode modified with the P11 perovskite material, Carbon Black and Nafion is probably the outcome of structural and transport effects [48]. However, it should also be pointed out that the higher EASA value estimated for this sample indicates its surface is more discontinuous, probably due to features including edges and defects that generate more catalytically active centers [49]. Furthermore, it is sometimes the case that electrodes manufactured using compositions containing at least two materials benefit from improved charge transfer and adjusted electron density at the catalytic centers as a result of their interplay [50]. The acquired experimental data implies this is not the case with Gr4P14+CB and Gr4P11+CB which were manufactured using the perovskite oxides mixed with Carbon Black. At i = −10 mA/cm2 the ηOER values for these samples are very close to those determined for the electrodes obtained without utilizing Carbon Black. This observation is rather unexpected since the material in question was added for electron transfer enhancement purposes. A potential explanation has to do with the way in which the particles constituting the solid phase of the suspension applied on the surface of the carbon substrate were arranged. Arrangements in the shape of thick layers prevent the electrolyte solution from reaching the pores situated deep into the deposited material. The result is a decrease in the number of catalytically active centers that would otherwise participate in the electrochemical reaction [24,51]. According to this explanation, the EASA values of Gr4P14+CB and Gr4P11+CB could have been higher and the same is true of their OER electrocatalytic activity.
While Gr4P11+CB possesses superior OER electrocatalytic properties compared with the electrodes investigated during the present study the same cannot be said regarding other electrodes reported in the scientific literature, including other perovskite-based ones [52,53,54,55]. The OER catalytic activity of Gr4P11+CB is not as high and its electrochemical stability is not as good as those of the electrodes from recently published studies. In light of this observation, the future water electrolysis studies that will be performed on P11, P14 and other novel perovskite oxide structures will probably involve a more complex electrode manufacturing method than drop-casting (e.g., pulsed laser deposition and spray pyrolysis).
4. Conclusions
The water electrolysis electrocatalytic properties of two novel perovskite oxides were investigated in a strong alkaline medium. The perovskite-based electrodes were manufactured using four different procedures. The samples displayed OER activity but negligible HER activity. The electrode obtained by drop-casting a composition containing LaMnO3:Ag/PVP (P11), Carbon Black and Nafion on graphite substrate was identified as being the most electrocatalytically active for the OER. Its OER overpotential at i = 10 mA/cm2 is 0.83 V and its Tafel slope is 0.197 V/dec. It was also found to be more electrochemically stable than the other samples.
The present study evaluates for the first time the electrocatalytic properties of new perovskite materials and the obtained results extend the available knowledge of relevance to the water splitting domain.
Author Contributions
Conceptualization, A.C. and P.S.; methodology, A.C. and P.S.; validation, A.C., B.-O. T., I.M.C.I., P.S.; formal analysis, A.C., B.-O. T., I.M.C.I., P.S.; investigation, A.C., B.-O. T., I.M.C.I., P.S.; writing—original draft preparation, A.C., B.-O. T., P.S..; supervision, P.S.; project administration, P.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
This work was supported by the Experimental Demonstrative Project 683 PED/2022- Executive Unit for Financing Higher Education, Research, Development and Innovation (UEFISCDI). The authors thank D. Ursu, N. S. Tolea and C. Moșoarcă from National Institute of Research and Development for Electrochemistry and Condensed Matter Timisoara for materials characterization.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brahmi, C.; Benltifa, M.; Vaulot, C.; Michelin, L.; Dumur, F.; Airoudj, A.; Morlet-Savary, F.; Raveau, B.; Bousselmi, L.; Lalevée, J. New hybrid perovskites/polymer composites for the photodegradation of organic dyes. Eur. Polym. J. 2021, 157, 110641. [Google Scholar] [CrossRef]
- Al-Muntaser, A.A.; Pashameah, R.A.; Sharma, K.; Alzahrani, E.; Hameed, S.T.; Morsi, M.A. Boosting of structural, optical, and dielectric properties of PVA/CMC polymer blend using SrTiO3 perovskite nanoparticles for advanced optoelectronic applications. Opt. Mater. 2022, 132, 112799. [Google Scholar] [CrossRef]
- Basturk, S.B.; Dancer, C.E.J.; McNally, T. Fabrication and Characterization of Composites of a Perovskite and Polymers with High Dielectric Permittivity. Mater. Res. Bull. 2021, 135, 111126. [Google Scholar] [CrossRef]
- Koczkur, K.M.; Mourdikoudis, S.; Polavarapu, L.; Skrabalak, S.E. Polyvinylpyrrolidone (PVP) in nanoparticle synthesis. Dalton Trans. 2015, 44, 17883. [Google Scholar] [CrossRef]
- Voronova, M.I.; Surov, O.V.; Rubleva, N.V.; Kochkina, N.E.; Afineevskii, A.V.; Zakharov, A.G. Nanocrystalline cellulose composites with polyvinylpyrrolidone: Selfassembly and dispersibility in water. Compos. Commun. 2019, 15, 108–112. [Google Scholar] [CrossRef]
- Ahmad, F.; Rafiq, K.; Najam, T.; Hussain, E.; Sohail, M.; Abid, M.Z.; Mahmood, A.; Javed, M.S.; Shah, S.S.A. Metal-organic frameworks for electrocatalytic water-splitting: Beyond the pyrolysis. Int. J. Hydrog. Energy 2023, 48, 35075–35111. [Google Scholar] [CrossRef]
- Roy, S.; Huang, Z.; Bhunia, A.; Castner, A.; Gupta, A.K.; Zou, X.; Ott, S. Electrocatalytic hydrogen evolution from a cobaloxime-based metal−organic framework thin film. J. Am. Chem. Soc. 2019, 141, 15942–15950. [Google Scholar] [CrossRef]
- Hosseini, S.E. Fossil fuel crisis and global warming. In Fundamentals of low emission flameless combustion and its applications; Hosseini, S.E., Ed.; Academic Press: 125 London Wall, London, United Kingdom, 2022; pp. 1–11. [Google Scholar]
- Lanjekar, P.R.; Panwar, N.L.; Agrawa, C. A comprehensive review on hydrogen production through thermochemical conversion of biomass for energy security. Bioresour. Technol. Rep. 2023, 21, 101293. [Google Scholar] [CrossRef]
- Bulfin, B.; Carmo, M.; Van de Krol, R.; Mougin, J.; Ayers, K.; Gross, K.J.; Marina, O.A.; Roberts, G.M.; Stechel, E.B.; Xiang, C. Editorial: Advanced water splitting technologies development: Best practices and protocols. Front. Energy Res. 2023, 11, 1–4. [Google Scholar] [CrossRef]
- Peng, Y.; Jiang, K.; Hill, W.; Lu, Z.; Yao, H.; Wang, H. Large-scale, low-cost, and high-efficiency water-splitting system for clean H2 generation. ACS Appl. Mater. Interfaces 2019, 11, 3971–3977. [Google Scholar] [CrossRef]
- Son, M.-K. Design and demonstration of large scale Cu2O photocathodes with metal grid structure for photoelectrochemical water splitting. Energies, 2021, 14, 7422. [Google Scholar] [CrossRef]
- Xu, X.; Sun, H.; Jiang, S.P.; Shao, Z. Modulating metal–organic frameworks for catalyzing acidic oxygen evolution for proton exchange membrane water electrolysis. Sus. Mat. 2021, 1, 460–481. [Google Scholar] [CrossRef]
- Wang, S.; Lu, A.; Zhong, C.-J. Hydrogen production from water electrolysis: role of catalysts. Nano Converg. 2021, 8, 4. [Google Scholar] [CrossRef]
- Guo, F.; Macdonald, T.J.; Sobrido, A.J.; Liu, L.; Feng, J.; He, G. Recent advances in ultralow-Pt-loading electrocatalysts for the efficient hydrogen evolution. Adv. Sci. 2023, 10, 2301098. [Google Scholar] [CrossRef]
- Sun, H.; Jung, W.C. Recent advances in doped ruthenium oxides as high-efficiency electrocatalysts for the oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 15506–15521. [Google Scholar] [CrossRef]
- Galyamin, D.; Tolosana-Moranchel, A.; Retuerto, M.; Rojas, S. Unraveling the most relevant features for the design of iridium mixed oxides with high activity and durability for the oxygen evolution reaction in acidic media. JACS Au 2023, 3, 2336–2355. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Wang, Q.; Huang, C.; Zhang, M.; Yan, Y.; Liu, T.; Zhou, W. Noble metal-based heterogeneous catalysts for electrochemical hydrogen evolution reaction. Appl. Sci. 2023, 13, 2177. [Google Scholar] [CrossRef]
- Li, X.-P.; Huang, C.; Han, W.-K.; Ouyang, T.; Liu, Z.-Q. Transition metal-based electrocatalysts for overall water splitting. Chin. Chem. Lett. 2021, 32, 2597–2616. [Google Scholar] [CrossRef]
- Taranu, B.-O.; Fagadar-Cosma, E. Catalytic properties of free-base porphyrin modified graphite electrodes for electrochemical water splitting in alkaline medium. Processes 2022, 10, 611. [Google Scholar] [CrossRef]
- Alom, S.; Kananke-Gamage, C.C.W.; Ramezanipour, F. Perovskite oxides as electrocatalysts for hydrogen evolution reaction. ACS Omega 2022, 7, 7444–7451. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Zhang, K.; Xu, J.; Wu, Q.; Xie, Z.; An, W.; Liang, X.; Zou, X. Electrocatalytic water splitting over perovskite oxide catalysts. Chinese J. Catal. 2023, 50, 109–125. [Google Scholar] [CrossRef]
- Rahman, S.; Hussain, A.; Noreen, S.; Bibi, N.; Arshad, S.; Rehman, J.U.; Tahir, M.B. Structural, electronic, optical and mechanical properties of oxide-based perovskite ABO3 (A = Cu, Nd and B = Sn, Sc): A DFT study. J. Solid State Chem. 2023, 317, 123650. [Google Scholar] [CrossRef]
- Taranu, B.-O.; Vlazan, P.; Svera, P.; Poienar, M.; Sfirloaga, P. New functional hybrid materials based on clay minerals for enhanced electrocatalytic activity. J. Alloys Compd. 2022, 892, 162239. [Google Scholar] [CrossRef]
- Sfirloaga, P.; Taranu, B.-O.; Poienar, M.; Vlazan, P. Addressing electrocatalytic activity of metal-substituted lanthanum manganite for the hydrogen evolution reaction. Surf. Interfaces 2023, 39, 102881. [Google Scholar] [CrossRef]
- Sfirloaga, P.; Malaescu, I.; Nicolae Marin, C.; Vlazan, P. The effect of partial substitution of Pd in LaMnO3 polycrystalline materials synthesized by sol–gel technique on the electrical performance. J. Sol-Gel Sci. Technol. 2019, 92, 537–545. [Google Scholar] [CrossRef]
- Sfirloaga, P.; Vlase, T.; Malaescu, I.; Nicolae Marin, C.; Vlazan, P. Silver doping in lanthanum manganite materials: structural and electrical properties. J. Therm. Anal. Calorim. 2020, 142, 1817–1823. [Google Scholar] [CrossRef]
- Sfirloaga, P.; Ivanovici, M.-G.; Poienar, M.; Ianasi, C.; Vlazan, P. Investigation of Catalytic and Photocatalytic Degradation of Methyl Orange Using Doped LaMnO3 Compounds. Processes 2022, 10, 2688. [Google Scholar] [CrossRef]
- Poienar, M.; Taranu, B.-O.; Svera, P.; Sfirloaga, P.; Vlazan, P. Disclosing the thermal behaviour, electrochemical and optical properties of synthetic Fe3(PO4)2(OH)2 materials. J. Therm. Anal. Calorim. 2022, 147, 11839–11855. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, H.; He, H.; Xu, X.; Jin, Y. Self-standing non-noble metal (Ni–Fe) oxide nanotube array anode catalysts with synergistic reactivity for high-performance water oxidation. J. Mater. Chem. A 2015, 3, 7179–7186. [Google Scholar] [CrossRef]
- Baciu, A.; Remes, A.; Ilinoiu, E.; Manea, F.; Picken, S.J.; Schoonman, J. Carbon nanotubes composite for environmentally friendly sensing. Environm. Eng. Manag. J. 2012, 11, 239–246. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical methods: Fundamentals and applications, 2nd ed.; John Wiley & Sons: New York, USA, 2001; pp. 186–191. [Google Scholar]
- Hrapovic, S.; Liu, Y.; Male, K.B.; Luong, J.H.T. Electrochemical biosensing platforms using platinum nanoparticles and carbon nanotubes. Anal. Chem. 2004, 76, 1083–1088. [Google Scholar] [CrossRef]
- Li, X.G.; Kresse, I.; Springer, J.; Nissen, J.; Yang, Y.L. Morphology and gas permselectivity of blend membranes of polyvinylpyridine with ethylcellulose. Polymer 2001, 42, 6859–6869. [Google Scholar] [CrossRef]
- El Hotaby, W.; Sherif, H.H.A.; Hemdan, B.A.; Khalil, W.A.; Khalil, S.K.H. Assessment of in situ-Prepared Polyvinylpyrrolidone-Silver Nanocomposite for Antimicrobial Applications. Acta Phys. Pol. A 2017, 131, 1554–1560. [Google Scholar] [CrossRef]
- Baganizi, D.R.; Nyairo, E.; Duncan, S.A.; Singh, S.R.; Dennis, V.A. Interleukin-10 Conjugation to Carboxylated PVP-Coated Silver Nanoparticles for Improved Stability and Therapeutic Efficacy. Nanomaterials 2017, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- Mireles, L.K.; Wu, M.-R.; Saadeh, N.; Yahia, L’H.; Sacher, E. Physicochemical Characterization of Polyvinyl Pyrrolidone: A Tale of Two Polyvinyl Pyrrolidones. ACS Omega 2020, 5, 30461–30467. [Google Scholar] [CrossRef] [PubMed]
- Safo, I.A.; Werheid, M.; Dosche, C.; Oezaslan, M. The role of polyvinylpyrrolidone (PVP) as a capping and structure-directing agent in the formation of Pt nanocubes. Nanoscale Adv., 2019, 1, 3095. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-J.; Wang, M.; Zhang, Y.-Y.; Wu, J.-Y.; Zhang, T. Investigation of the role of the molecular weight of polyvinyl pyrrolidone in the shape control of high-yield silver nanospheres and nanowires. Nanoscale Res. Lett. 2014, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Min, B.R.; Kim, C.K.; Won, J.; Kang, Y.S. Spectroscopic Interpretation of Silver Ion Complexation with Propylene in Silver Polymer Electrolytes. J. Phys. Chem. B 2002, 106, 2786–2790. [Google Scholar] [CrossRef]
- Bryaskova, R.; Daniela Pencheva, D.; Nikolov, S.; Kantardjiev, T. Synthesis and comparative study on the antimicrobial activity of hybrid materials based on silver nanoparticles (AgNps) stabilized by polyvinylpyrrolidone (PVP). J. Chem. Biol. 2011, 4, 185–191. [Google Scholar] [CrossRef]
- Menezes, P.W.; Panda, C.; Loos, S.; Bunschei-Bruns, F.; Walter, C.; Schwarze, M.; Deng, X.; Dau, H.; Driess, M. A structurally versatile nickel phosphite acting as a robust bifunctional electrocatalyst for overall water splitting. Energy Environ. Sci. 2018, 11, 1287–1298. [Google Scholar] [CrossRef]
- Mani, V.; Anantharaj, S.; Mishra, S.; Kalaiselvi, N.; Kundu, S. Iron hydroxyphosphate and Sn-incorporated iron hydroxyphosphate: Efficient and stable electrocatalysts for oxygen evolution reaction. Catal. Sci. Technol. 2017, 7, 5092–5104. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Zhou, X.; Xia, Z.; Gao, W.; Ma, Y.; Qu, Y. Highly efficient and robust nickel phosphides as bifunctional electrocatalysts for overall water-splitting. ACS Appl. Mater. Interfaces 2016, 8, 10826–10834. [Google Scholar] [CrossRef]
- Zhou, Z.; Zaman, W.Q.; Sun, W.; Cao, L.M.; Tariq, M.; Yang, J. Cultivating crystal lattice distortion in IrO2: Via coupling with MnO2 to boost the oxygen evolution reaction with high intrinsic activity. Chem. Commun. 2018, 54, 4959–4962. [Google Scholar] [CrossRef]
- Fratilescu, I.; Lascu, A.; Taranu, B.O.; Epuran, C.; Birdeanu, M.; Macsim, A.-M.; Tanasa, E.; Vasile, E.; Fagadar-Cosma, E. One A3B porphyrin structure—Three successful applications. Nanomaterials 2022, 12, 1930. [Google Scholar] [CrossRef]
- Taranu, B.O.; Ivanovici, M.G.; Svera, P.; Vlazan, P.; Sfirloaga, P.; Poienar, M. Ni11□(HPO3)8(OH)6 multifunctional materials: Electrodes for oxygen evolution reaction and potential visible-light active photocatalysts. J. Alloys Compd. 2020, 848, 156595. [Google Scholar] [CrossRef]
- Taranu, B.O.; Vlazan, P.; Racu, A. Water splitting studies in alkaline medium using graphite electrodes modified with transition metal oxides and compositions containing them. Stud. Univ. Babes-Bolyai Chem. 2022, 67, 79–95. [Google Scholar] [CrossRef]
- Taranu, B.O.; Fagadar-Cosma, E. The pH influence on the water-splitting electrocatalytic activity of graphite electrodes modified with symmetrically substituted metalloporphyrins. Nanomaterials 2022, 12, 3788. [Google Scholar] [CrossRef]
- Taranu, B.O.; Fagadar-Cosma, E.; Sfirloaga, P.; Poienar, M. Free-base porphyrin aggregates combined with nickel phosphite for enhanced alkaline hydrogen evolution. Energies 2023, 16, 1212. [Google Scholar] [CrossRef]
- Lahiri, A.; Li, G.; Endres, F. Highly efficient electrocatalytic hydrogen evolution reaction on carbonized porous conducting polymers. J. Solid State Electrochem. 2020, 24, 2763–2771. [Google Scholar] [CrossRef]
- Tang, J.; Xu, X.; Tang, T.; Zhong, Y.; Shao, Z. Perovskite-Based Electrocatalysts for Cost-Effective Ultrahigh-Current-Density Water Splitting in Anion Exchange Membrane Electrolyzer Cell. Small Methods 2022, 6, 2201099. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, Y.; Zhao, Y.; Ge, X.; Lu, Q.; Zhang, T.; Xie, D.; Li, M.; Bu, Y. Design strategies of perovskite nanofibers electrocatalysts for water splitting: A mini review. Chem. Eng. J. 2023, 451, 138710. [Google Scholar] [CrossRef]
- Kim, D.; Oh, L.S.; Park, J.H.; Kim, H.J.; Lee, S.; Lim, E. Perovskite-based electrocatalysts for oxygen evolution reaction in alkaline media: A mini review. Front Chem. 2022, 10, 1024865. [Google Scholar] [CrossRef] [PubMed]
- Si, C.; Zhang, W.; Lu, Q.; Guo, E.; Yang, Z.; Chen, J.; He, X.; Luo, J. Recent Advances in Perovskite Catalysts for Efficient Overall Water Splitting. Catalysts 2022, 12, 601. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns for the precursors and hybrid materials: (a) LaMnO3:Ag, PVP, and LaMnO3:Ag/PVP (P11); (b) LaMnO3:Pd, PVP, and LaMnO3:Pd/PVP (P14).
Figure 1.
XRD patterns for the precursors and hybrid materials: (a) LaMnO3:Ag, PVP, and LaMnO3:Ag/PVP (P11); (b) LaMnO3:Pd, PVP, and LaMnO3:Pd/PVP (P14).
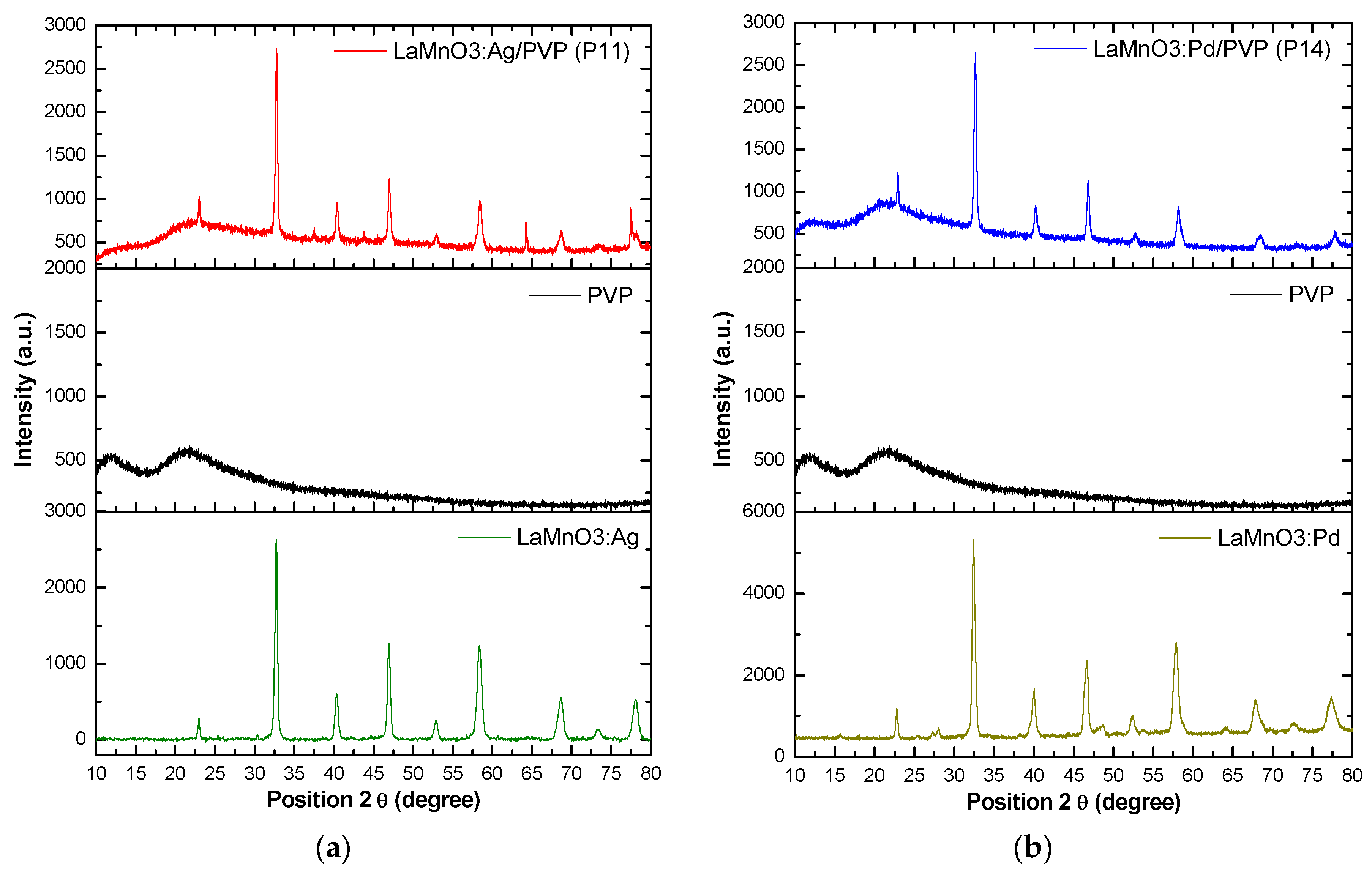
Figure 2.
The FT-IR spectra of the precursors and hybrid materials: (a) LaMnO3:Ag, PVP, and LaMnO3:Ag/PVP (P11); (b) LaMnO3:Pd, PVP, and LaMnO3:Pd/PVP (P14).
Figure 2.
The FT-IR spectra of the precursors and hybrid materials: (a) LaMnO3:Ag, PVP, and LaMnO3:Ag/PVP (P11); (b) LaMnO3:Pd, PVP, and LaMnO3:Pd/PVP (P14).
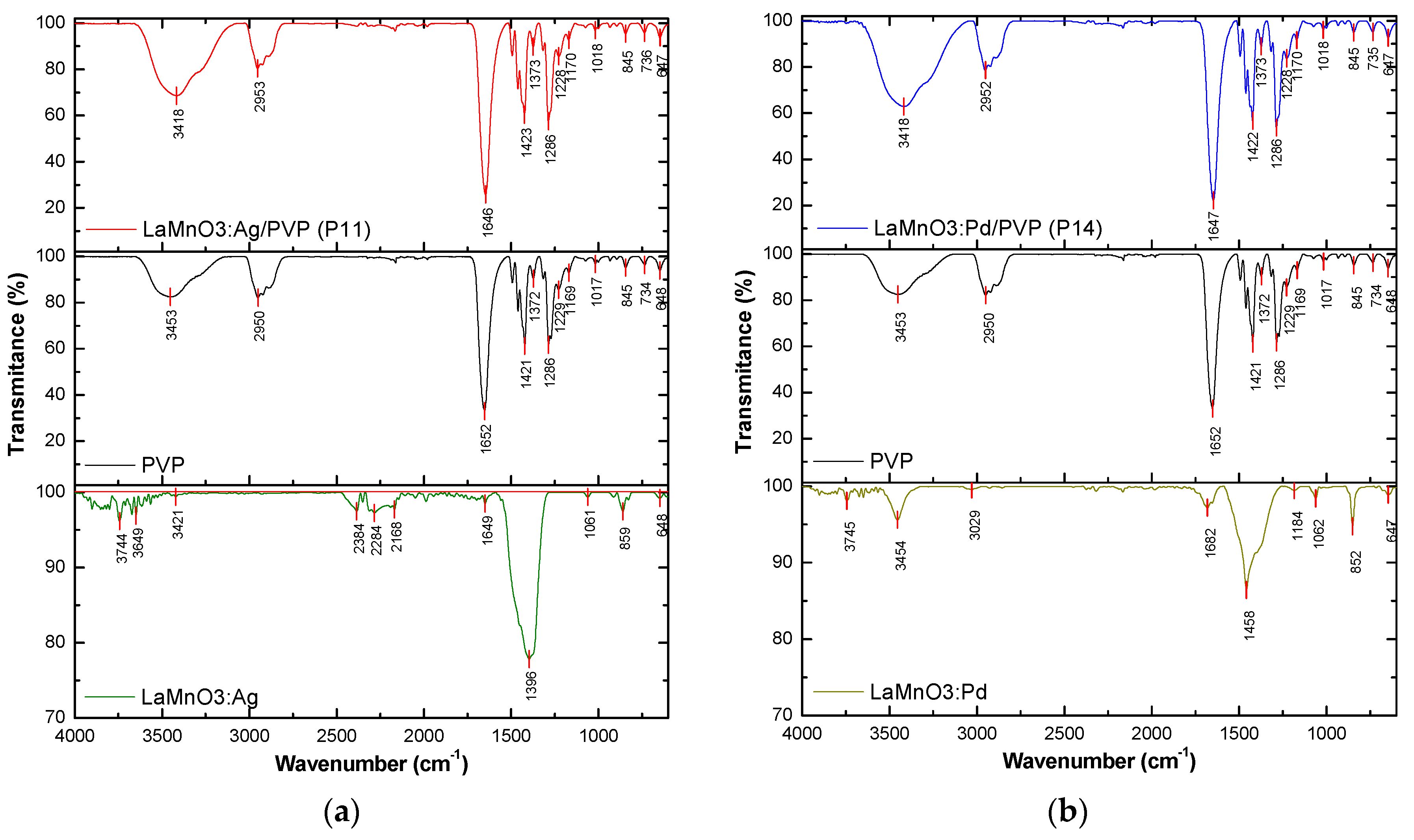
Figure 3.
SEM images for (a) PVP); (b) LaMnO3:Ag; (c) LaMnO3:Ag/PVP (P11), (d) LaMnO3:Pd; (e) LaMnO3:Pd/PVP (P14).
Figure 3.
SEM images for (a) PVP); (b) LaMnO3:Ag; (c) LaMnO3:Ag/PVP (P11), (d) LaMnO3:Pd; (e) LaMnO3:Pd/PVP (P14).

Figure 4.
Anodic LSVs recorded on the unmodified glassy carbon electrode (GC0) and on the modified electrodes GC1CB, GC1P11+CB and GC1P14+CB, in 1 M KOH solution, at v = 5 mV/s.
Figure 4.
Anodic LSVs recorded on the unmodified glassy carbon electrode (GC0) and on the modified electrodes GC1CB, GC1P11+CB and GC1P14+CB, in 1 M KOH solution, at v = 5 mV/s.
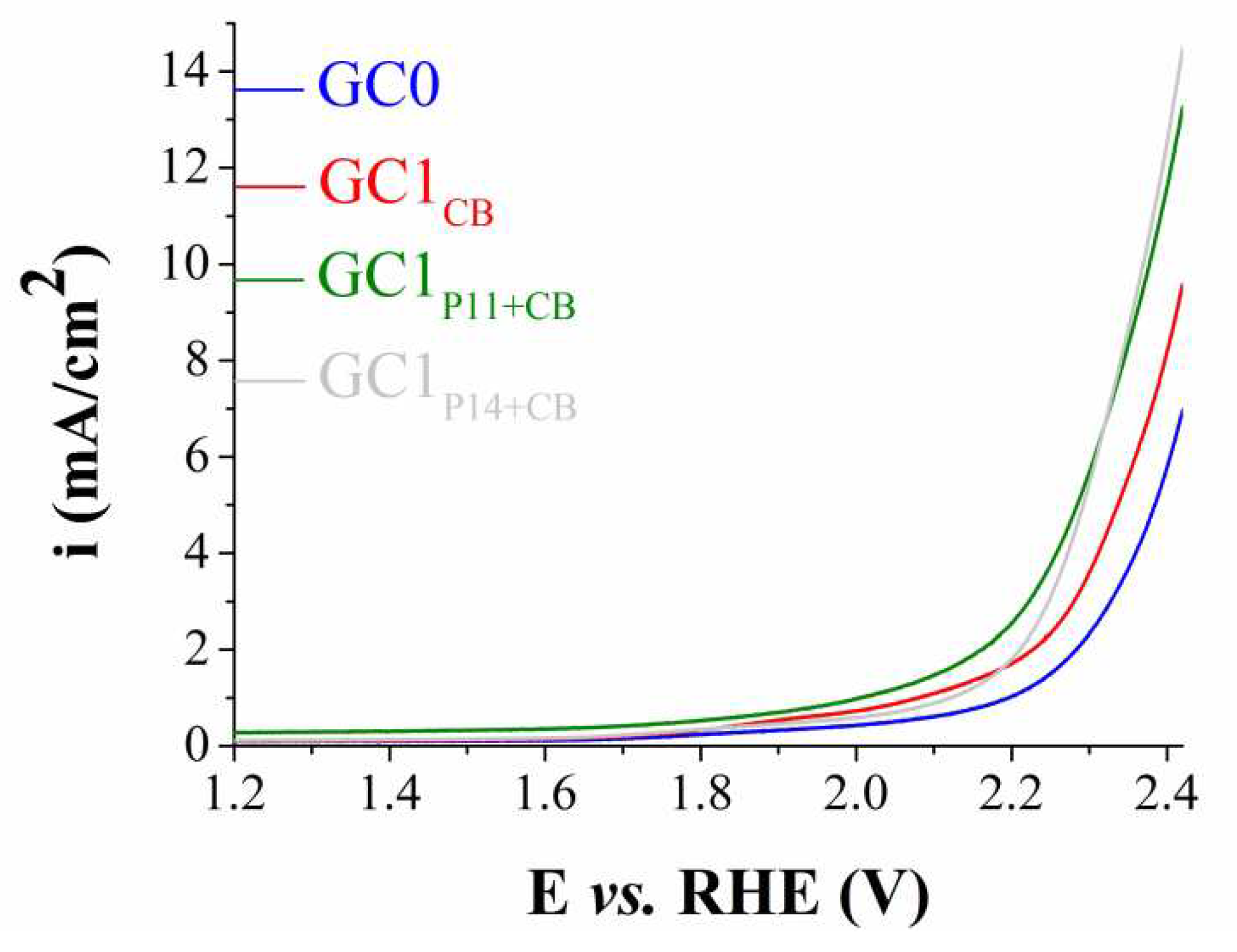
Figure 5.
Anodic polarization curves recorded on the unmodified graphite electrode (Gr0) and on the modified electrodes Gr3CB, Gr3P11 and Gr3P14+CB, in 1 M KOH solution, at v = 5 mV/s.
Figure 5.
Anodic polarization curves recorded on the unmodified graphite electrode (Gr0) and on the modified electrodes Gr3CB, Gr3P11 and Gr3P14+CB, in 1 M KOH solution, at v = 5 mV/s.
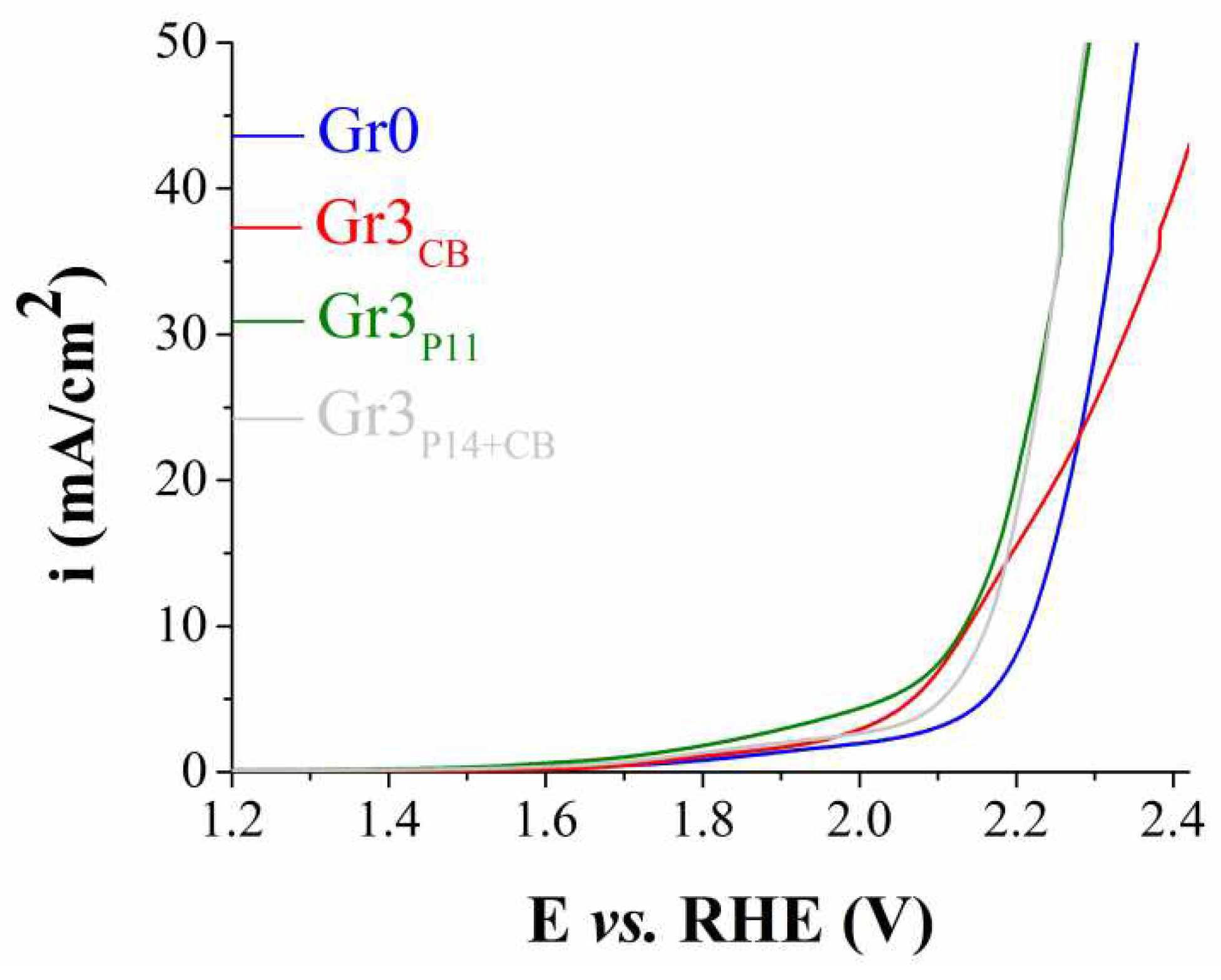
Figure 6.
Anodic polarization curves recorded on Gr0, Gr4CB, Gr4P11, Gr4P14, Gr4P11+CB and Gr4P14+CB electrodes, in 1 M KOH solution, at v = 5 mV/s.
Figure 6.
Anodic polarization curves recorded on Gr0, Gr4CB, Gr4P11, Gr4P14, Gr4P11+CB and Gr4P14+CB electrodes, in 1 M KOH solution, at v = 5 mV/s.
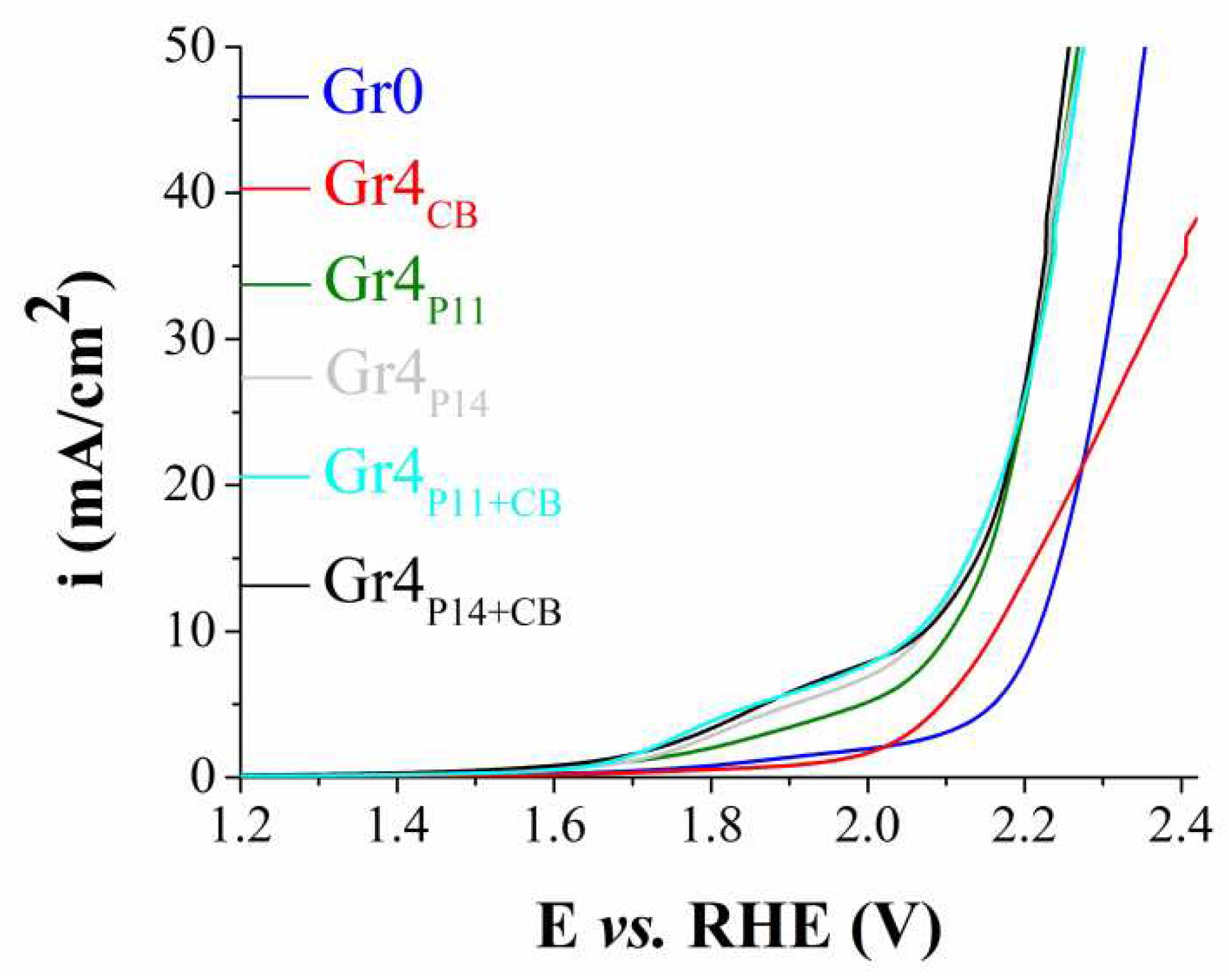
Figure 7.
The graphical representation of the dependence between the peak current densities of the anodic and cathodic peaks corresponding to the ferrocyanide/ferricyanide redox couple and the square root of the scan rate for the Gr4P11 (a), Gr4P14 (b), Gr4P11+CB (c) and Gr4P14+CB (d) electrodes.
Figure 7.
The graphical representation of the dependence between the peak current densities of the anodic and cathodic peaks corresponding to the ferrocyanide/ferricyanide redox couple and the square root of the scan rate for the Gr4P11 (a), Gr4P14 (b), Gr4P11+CB (c) and Gr4P14+CB (d) electrodes.
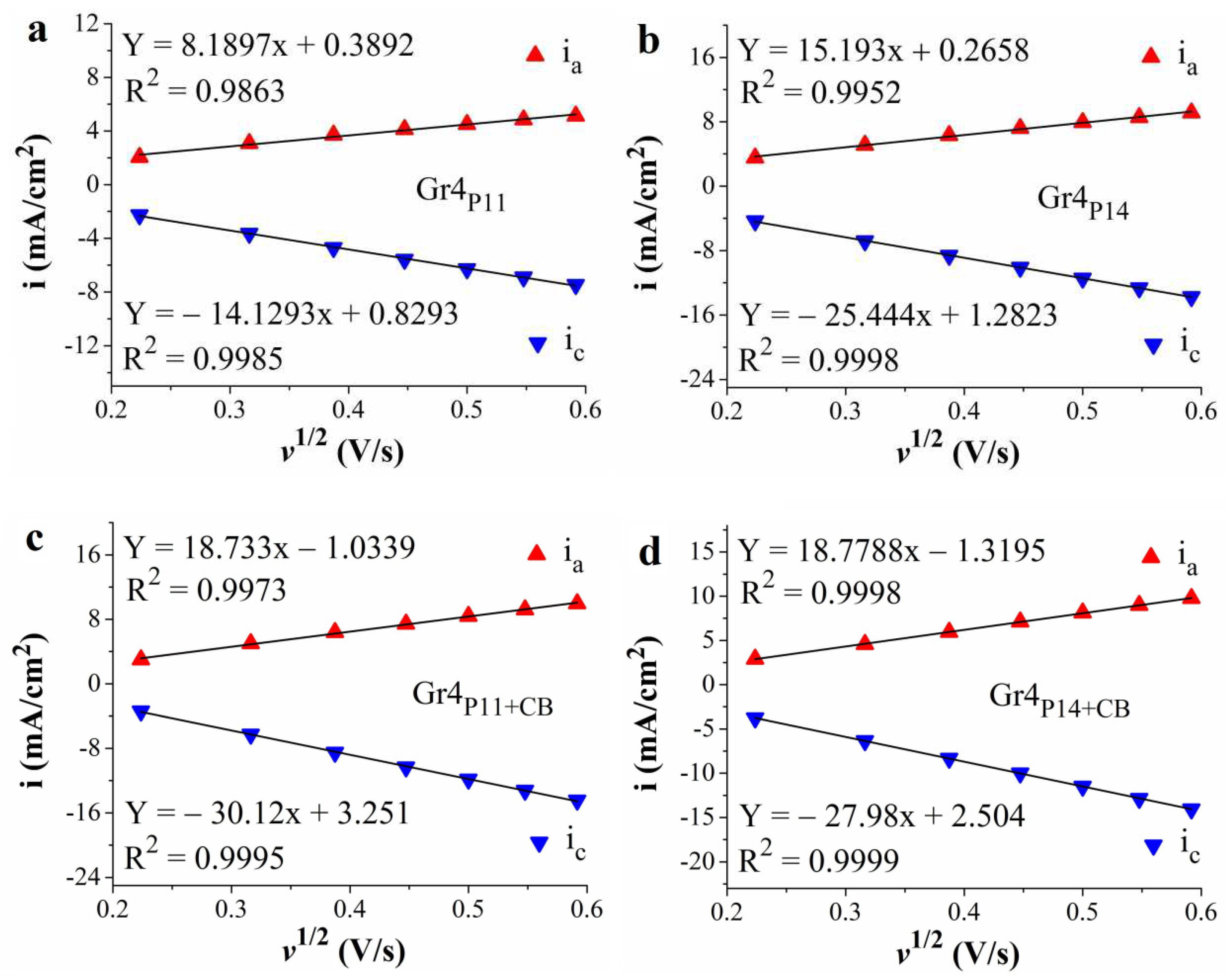
Figure 8.
The Tafel plots obtained for the O2 evolution reaction on the Gr4P11 (a), Gr4P14 (b), Gr4P11+CB (c) and Gr4P14+CB (d) electrodes, in 1 M KOH solution.
Figure 8.
The Tafel plots obtained for the O2 evolution reaction on the Gr4P11 (a), Gr4P14 (b), Gr4P11+CB (c) and Gr4P14+CB (d) electrodes, in 1 M KOH solution.
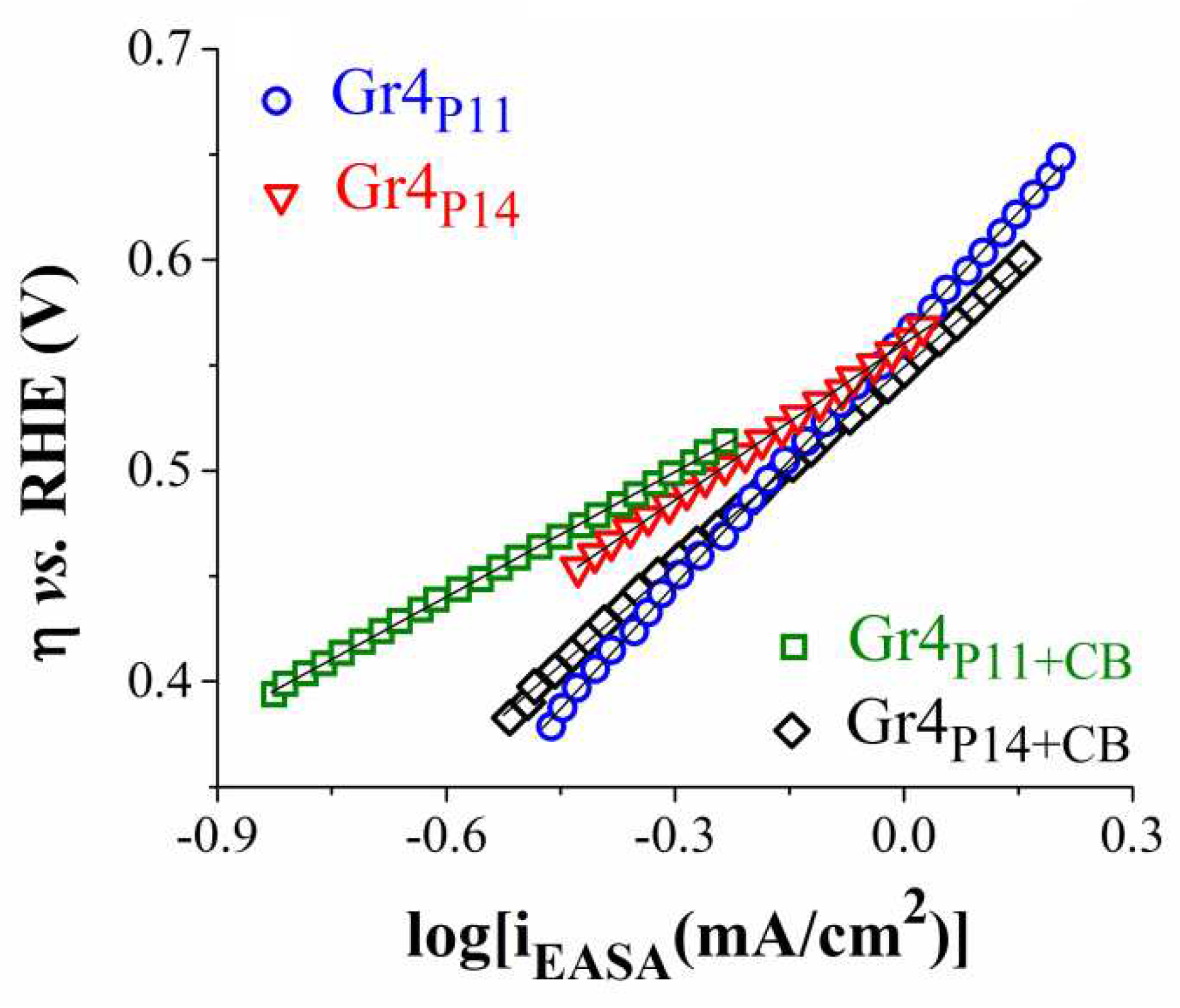
Figure 9.
The graphical representation of the dependence between current density and time for the Gr4P11+CBand Gr4P14+CB electrodes immersed in 1 M KOH solution. Insets: (a) The overlap between the polarization curves obtained on the Gr4P11+CB electrode before and after the chronoamperometric experiment, denoted as Gr4P11+CB and Gr4P11+CB’, respectively; (b) and (c) enlarged areas from the overlapped ivs. time curves.
Figure 9.
The graphical representation of the dependence between current density and time for the Gr4P11+CBand Gr4P14+CB electrodes immersed in 1 M KOH solution. Insets: (a) The overlap between the polarization curves obtained on the Gr4P11+CB electrode before and after the chronoamperometric experiment, denoted as Gr4P11+CB and Gr4P11+CB’, respectively; (b) and (c) enlarged areas from the overlapped ivs. time curves.
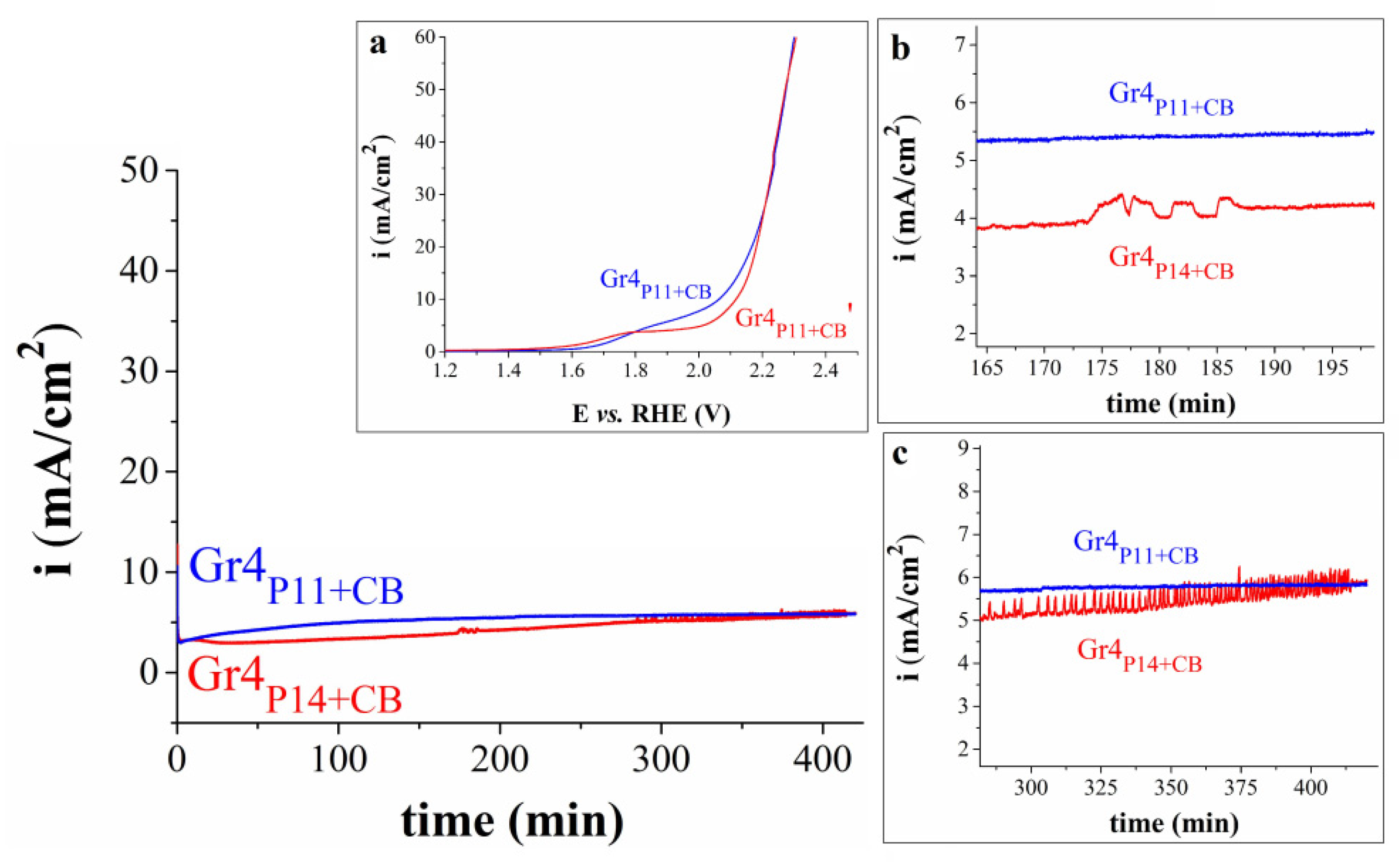

Table 1.
Electrodes obtained with Procedures 1 and 3 and the suspensions used to construct them.
| Electrode | Suspension composition | |||||
|---|---|---|---|---|---|---|
| Procedure 1 | Procedure 3 | P11 (mg) |
P14 (mg) |
Nafion solution (µL) | Carbon Black (mg) | Ethanol (µL) |
| GC1CB | Gr3CB | - | - | 50 | 5 | 450 |
| GC1P11 | Gr3P11 | 5 | - | 50 | - | 450 |
| GC1P11+CB | Gr3P11+CB | 5 | - | 100 | 5 | 900 |
| GC1P14 | Gr3P14 | - | 5 | 50 | - | 450 |
| GC1P14+CB | Gr3P14+CB | - | 5 | 100 | 5 | 900 |
Table 2.
Electrodes obtained with Procedures 2 and 4 and the suspensions used to construct them.
| Electrode | Suspension composition | |||||
|---|---|---|---|---|---|---|
| Procedure 2 | Procedure 4 | P11 (mg) |
P14 (mg) |
Nafion solution (µL) | Carbon Black (mg) | Ethanol (µL) |
| GC2CB | Gr4CB | - | - | 50 | 5 | 150 |
| GC2P11 | Gr4P11 | 5 | - | 50 | - | 150 |
| GC2P11+CB | Gr4P11+CB | 5 | - | 100 | 5 | 300 |
| GC2P14 | Gr4P14 | - | 5 | 50 | - | 150 |
| GC2P14+CB | Gr4P14+CB | - | 5 | 100 | 5 | 300 |
Table 3.
The EASA and D values determined in the case of Gr4P11, Gr4P14, Gr4P11+CB and Gr4P14+CB.
| Electrode | EASA (cm2) | D (cm2/s) |
|---|---|---|
| Gr4P11 | 0.54 | 2.55 x 10-5 |
| Gr4P14 | 0.74 | 4.85 x 10-5 |
| Gr4P11+CB | 1.19 | 1.21 x 10-4 |
| Gr4P14+CB | 0.80 | 5.51 x 10-5 |
Table 4.
Tafel slope values determined for the Gr4P11, Gr4P14, Gr4P11+CB and Gr4P14+CB electrodes.
| Electrode | Tafel slope (V/dec) | R2 |
|---|---|---|
| Gr4P11 | 0.392 | 0.9996 |
| Gr4P14 | 0.247 | 0.9993 |
| Gr4P11+CB | 0.197 | 0.9994 |
| Gr4P14+CB | 0.314 | 0.9995 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



