Submitted:
11 January 2024
Posted:
12 January 2024
You are already at the latest version
Abstract
Abstract: There are a large number of cross-linked chemical bonds such as tight disulfide bonds, hydrogen bonds,and salt bonds in keratin, which makes it difficult to break the structural stability of the protein using conventional methods. The degradation of feather waste using keratinases produced by microorganisms has the advantage of being efficient and environmentally friendly. In this study, 23 strains capable of producing transparent circles were obtained via preliminary screening with skim milk as the sole carbon and nitrogen source, and 9 strains capable of degrading feathers were obtained via rescreening with feathers as the sole carbon and nitrogen source. Finally, through the determination of enzyme activity, the strain CY-A with a high ability to degrade feathers was obtained. The strain was identified as Bacillus tequilensis via morphological observation, physiological and biochemical identification, and 16 S rRNA molecular biology. The keratinase gene (bta) of Bacillus tequila was cloned, and its full-length gene sequence was 1110 bp, containing an open reading frame of 1089, encoding 369 amino acids (GeneBank accession number OR733336). The physicochemical properties, secondary structure, and tertiary structure of the recombinant protein were analyzed using bioinformatics tools such as ProtParm, SOPMA, MEGA, and SWISS-MODELS. It was found that the relative molecular mass was 37.953 kD and that the isoelectric point was 8.48. The protein was a hydrophilic protein. The keratinase bta gene was ligated to the expression vector pET28a(+) using T4 ligase, and the recombination expression plasmid pET28-bta was constructed and transformed into E. coli BL21(DE3) to obtain recombinant engineered bacteria. The expressed keratinase was purified using a Ni2+ affinity chromatography column to obtain recombinant bta keratinase bta with a molecular weight of 37.953 kD. The identification and characterization of this keratinase gene provides some theoretical basis for the further enhancement of keratinase activity by means of genetic engineering.
Keywords:
feather degradation
; keratinase
; Bacillus
; gene expression
; E. coli
1. Introduction
The increase in chicken farming has led to increased chicken feather waste generated from high-speed abattoir industries and other chicken slaughterhouses. This feather waste requires special treatment as it has a significant impact on environmental pollution [1]. Every year, more than one million tonnes of chicken feathers are casually discarded, burned, and landfilled globally, causing unimaginable environmental problems [2,3]. Currently, only a few waterfowl feathers, such as duck down, are processed into higher-value products such as down jackets, while most feathers are discarded [3]. Feathers themselves contain 75–80% keratin. Nowadays, how to deal with the feathers left by poultry slaughtering with high efficiency, no pollution and a low cost has become an important issue [4]. Keratin is a fibrous and insoluble structural protein, which is widely cross-linked with disulfide, hydrogen and hydrophobic bonds. It has mechanical stability and resistance to common proteolytic enzymes such as pepsin, trypsin and papain [5,6].
Keratin is widely found in feathers, wool, hooves and nails, and it can be degraded by keratinases, which are a class of protein hydrolases with a high specificity for keratin[7,8,9]. Keratinases are mainly metalloproteases and serine proteases, and are active over a wide range of temperatures and pHs [7]. At present, some bacteria, actinomycetes and fungi are known to produce keratinases. In bacteria, keratinase has been widely recorded in Gram-positive bacteria such as Bacillus (B. subtilis, B. licheniformis, B. halodurans). Among the Gram-negative bacteria, keratinase is produced by Xanthomonas maltophilia, Vibrio, Stenotrophomonas, Crystalline Bacillus and Iron Bacillus [10]. Using keratinases, we can save energy, avoid environmental pollution and utilize keratin waste by generating value-added products such as soluble proteins and amino acids. Therefore, microbial degradation of horny waste is currently being explored and converted into valuable products, such as animal feed, nitrogen-rich organic fertilizers, amino acid supplements, peptides and ammonium ions [11,12]. In addition, keratinases produced by these microorganisms have a wide range of applications in the food, detergent, leather and cosmetic industries [9].
With the development of molecular biology, people began to study keratinases from the aspects of molecular structure and genetically engineered expression. At present, there are many reports on keratinase genes. Lin et al. (1995)[13] cloned the kerA gene from Bacillus licheniformis PWD-1 and analyzed its expression. Liang Bin et al. (2003) [14]amplified a kerB gene fragment from a keratinase-producing strain of Bacillus licheniformis L-25, which was introduced into sensitive cells of the enzyme-deficient Bacillus subtilis strain DB104 for expression, and found that the successfully expressed strain could grow on feather medium and hydrolyze the feathers completely. Radha et al. (2007)[15] cloned Bacillus licheniformis keratinase using the T7 promoter and successfully expressed it in Bacillus megaterium using a xylose-inducible expression system. The optimization of process parameters using response surface methodology resulted in a 3-fold increase in recombinant keratinase production. Fakhfakh et al. (2009)[16] conducted an in-depth study of keratinases produced by Bacillus licheniformis and sequenced and cloned its keratinase (kerA) gene. Lin Wang et al. (2019)[17] cloned the keratinase (kerT1) gene (1170 bp) of the genus Actinobacillus thermophilus and expressed it in Escherichia coli BL21 (DE3). Revathi et al. (2021) [18]cloned, expressed and purified the protease gene of Bacillus cereus. The protein sequence of the 38 kDa protease SLSP-k was determined using mass spectrometry analyses and identified as a B. subtilisin serine protease.
In the present study, feather meal was used as the sole source of carbon and nitrogen to screen for high keratinase-producing strains from the soil environment, and the keratinase gene bta was successfully cloned using the designed primers. In order to further improve the extracellular expression of keratinase, the recombinant strain pET-28a(+)-bta was constructed by choosing pET-28a(+) as the vector and Escherichia coli BL21 as the host in this study, which provided molecular materials for the improvement of keratinase activity, and provided a specific theoretical basis for the industrial production of keratinases.
2. Materials and Methods
2.1. Materials
2.1.1. Soil Samples, Strains and Plasmids
Soil samples were collected from a chicken pen of a resident in Changji City, Xinjiang Uygur Autonomous Region. The feathers were procured from a farmer’s market in Anning Canal, Urumqi City. The reagents used in this study included the following: E. coli DH5α and E. coli BL21 (DE3) (Bao Bioengineering (Dalian) Co. Ltd., Dalian, China); Pichia pastoris X-33, plasmid pET-28a(+) and cloning vector pPICZαA (Invitrogen, Waltham, MA, USA); DNA restriction endonuclease, DNA polymerase and DNA ligase purchased from TaKaRa; and a plasmid extraction kit, DNA fragment purification and recovery kit, and DNA fragment cutting and recovery kit purchased from TIANGEN.
2.1.2. Culture Media
LB medium: 1.0% peptone, 0.5% yeast powder, 1.0% NaCl, 2.0% agar powder, pH 7.0.
Skimmed milk medium: 1.5% skimmed milk powder, 2.0% agar powder, pH 7.0.
Feather fermentation medium: feather 0.5 g, 0.05% KH2PO4, 0.12% K2HPO4, 0.05% NaCl, 0.01% MgSO4, pH 7.0.
2.1.3. Buffer Solutions
Buffer A (bacteriophage buffer): 50 mmol/L Tris-HCl, 300 mmol/L NaCl, pH 8.0)
Buffer B (Inclusion Body Lysis Solution/Inclusion Body Purification Equilibrium Solution): 8 mol/L urea, 50 mmol/L Tris-HCl, 300 mmol/L NaCl, pH 8.0
Buffer C (inclusion body purification eluent): 8 mol/L urea, 500 mmol/L imidazole, 50 mmol/L Tris-HCl, 300 mmol/L NaCl, pH 8.0
Buffer D (inclusion body dialysis rehydration solution): 4 mol/L urea, 50 mmol/L Tris-HCl, pH 8.0
Buffer E (inclusion body dialysis rehydration solution): 2 mol/L urea, 50 mmol/L Tris-HCl, pH 8.0
Buffer F (inclusion body dialysis rehydration solution): 50 mmol/L Tris-HCl, pH 8.0
2.2. Methods
2.2.1. Screening of Keratinase-Producing Strains
Approximately 1 g of soil was added to enrichment medium and incubate at 37 °C and 220 r/min for 12 h. A 1 mL volume of the above bacterial suspension was diluted to 10−6, and 100 μL of each dilution was use to coat a skimmed milk plate, which was then cultured in an incubator at 37 °C for 1 d. Single colonies producing hyaline rings were singled out and cultured in multiple passages on LB plates. After several rounds of purification, a single colony was picked and used to inoculate 50 mL of LB; the culture was incubate at 37 °C and 180 r/min for 12 h, and then used to inoculate 50 mL of feather meal fermentation medium (inoculum volume of 2%), which was then incubated at 37 °C and 180 r/min for 3 d. The degradation of the feather was observed and the culture’s keratinase enzyme activity was measured.
2.2.2. Morphological Observations of Colonies and Bacteria
The isolated and purified strains were streaked in LB and milk plates, respectively, and cultured at a constant temperature of 37 °C for 18–24 h. The characteristics of colony morphology, size, color, colony edge and surface texture were observed. A single colony was picked and the individual morphology of the bacteria was observed under an optical microscope after Gram staining.
2.2.3. Determination of Keratinase Activity
In this study, according to the determination method of Yamamura et al.)[19], the fermentation broth was centrifuged at 4 °C and 12,000 r/min for 10 min. A 250 μL volume of the crude enzyme solution was taken, and 250 μL of 0.05 mol/L Gly-NaOH buffer (pH 10.0) was added to dissolve 1% of the substrate (keratin). The reaction was carried out at 60 °C for 10 min, and 500 μL of a 0.4 mol/L trichloroacetic acid solution was added to terminate the reaction (trichloroacetic acid was added to the control group first). The reaction was centrifuged at 12,000 r/min for 5 min; 500 μL of the supernatant was taken, mixed with 2.5 mL of a 0.4 mol/L Na2CO3 solution and 500 μL of Folin–Ciocalteu reagent (1:2 V/V), and the chromogenic reaction was carried out at 60 °C for 20 min. The absorbance was detected at a wavelength of 680 nm. The enzyme activity was expressed in U/g (U/mL) where 1 g of solid enzyme powder (or 1 mL of liquid enzyme) at 60 °C and pH 10.0, a unit of enzyme activity is equivalent to 1 μg of tyrosine produced after 1 min of hydrolysis of casein. The regression equation of the standard curve of the enzyme activity was obtained by calculating the enzyme activity:
where X is the enzyme activity of the sample, U/g (U/mL); A is the average absorbance of the sample in parallel tests; K is the absorbance constant; 1 is the total volume of the reaction reagent (mL); 10 is the reaction time of 10 min; and n is the number of dilutions.
y = 0.0082x + 0.0485, R2 = 0.9989
Enzyme activity calculation formula: X = A × K × 1/10 × n × 4
2.2.4. Determination of Strain Growth Curves
To six 150 mL triangular flasks, 50 mL of autoclaved LB medium was added, which was then inoculated with the CY-A bacterial solution cultured overnight in LB seed medium (inoculum volume of 2%). The flasks were incubated at 37 °C and 180 r/min; the absorbance at 600 nm of the culture solution was measured every 4 h.
2.2.5. Physiological and Biochemical Experimental Identification
The effects of the strains on lactose, glucose, litmus milk, VP assay, L-arabinose, citrate, sucrose, and ammonia production were determined separately, and a preliminary identification was carried out according to the Handbook of Identification of Common Bacteria[20].
2.2.6. 16S rRNA Sequence Determination and Phylogenetic Tree Construction
The sequencing results of the CY-A strain were submitted to NCBI’s Blast software for homology comparison to obtain information on gene sequences with high homology to it. The BLAST comparison results showed that the strains with 99% homology to the CY-A strain were Bacillus tequilensis and Bacillus subtilis.
2.2.7. Extraction of Total DNA from Bacillus Tequilensis CY-A
The total DNA of Bacillus tequilensis CY-A was extracted from the bacterial solution according to the instructions of the Bacterial DNA Extraction Kit.
2.2.8. Primer Design and bta Gene Amplification
Based on the CDS sequence of the keratinase gene (ker, GenBank: S78160. 1)from GenBank, suitable upstream and downstream primers were designed using the biological software SnapGene:
BT-F: 5′-CGCGGATCCCGCGTTAACGTTAATCTT-3′ (BamH I cleavage site is underlined).
BT-R: 5′-CGCGGATCCCGCGTTAACGTTAATCTT-3′ (Xho I cleavage site is underlined).
The primers were synthesized by Shanghai Bio-Bio Industry Co. The composition of the PCR amplification reaction was as follows (25 µL): DNA template, 1 μL; upstream and downstream primers, 1.0 μL each; 10× Buffer, 2.0 μL; dNTP mixture, 2.5 μL; and Taq enzyme, 0.25 μL. The PCR procedure was as follows: pre-denaturation at 95 °C for 3 min; denaturation at 95 °C for 30 s, annealing at 69 °C for 30 s and extension at 72 °C for 70 s for 35 cycles; and finally, extension at 72 °C for 5 min. The PCR products were separated by 1.5% agarose gel electrophoresis, and then the target bands were recovered using the Gel Recovery Purification Kit from TianGen according to the manufacturer’s instructions. The recovered PCR products were stored at −20 °C
The recovered target fragment was ligated into the pMD19-T cloning vector, transformed into E. coli DH5α receptor cells and screened using ampicillin; the positive plasmid was identified and sent to Shanghai Sangong Biologicals Co. (Shanghai, China) for sequencing.
2.2.9. Sequence Analysis
Referring to the article by Xiao Jiping et al. (2015 )[21], the resulting DNA sequences and their corresponding amino acid sequences were analyzed using the following bioinformatics tools:
- (1)
- ProtParm website (https://web.expasy.org/protparam/, accessed on) to analyze their physicochemical properties including relative molecular mass, amino acid composition, isoelectric point (pI), extinction coefficient, half-life, instability coefficient, and total average hydrophilicity;
- (2)
- SignalP (http://www.cbs.dtu.dk/services/SignalP/index.php, accessed on) to predict signal peptides;
- (3)
- The TMHMM program (http://www.cbs.dtu.dk/services/TMHMM, accessed on) to analyze protein transmembrane regions;
- (4)
- SOPMA(http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html, accessed on) to analyze secondary structures;
- (5)
- SWISS-MODELS (https://www.swissmodel.expasy.org/, accessed on) to predict tertiary structures.
2.2.10. Expression of the bta Gene in Escherichia coli
Plasmid pMD19-T-bta and vector pET28a(+) were double digested with the restriction enzymes BamH I and Xho I and then recovered and purified, after which, bta obtained by double digestion was ligated with double-cleaved pET28a(+) using T4 ligase (Takara, Dalian, China) and transformed (pET28a(+)-bta) into E. coli BL21 (DE3). LB plates containing 50 µg/mL kanamycin were used to select positive clones. Positive clones were identified using bacteriophage PCR, double digestion and sequencing. Recombinant keratinase was expressed by culturing the recombinant strain in 50 mL of kanamycin-containing LB medium at 37 °C and 180 r/m. When OD600 reached 0.4–0.6, 0.1 mM IPTG (Takara, China) was added to continue the induction for 4 h. At the end of induction, the bacteria were centrifuged at 8000 r/min for 10 min in a 50 mL centrifuge tube, the supernatant was discarded, and the bacteria were resuspended using 10 mL of PBS buffer (pH 7.4). They were dispersed completely using a spiralizer, the centrifugation described above was repeated, and the supernatant was discarded. The bacteria were then resuspended using 10 mL of 50 mmol/L Tris-HCl (pH 10.0) and fragmented by ultrasound under ice-bath conditions, which was programmed to work for 2 s with 5 s intervals at 200 w for a total run time of 1.5 h. After sonication, the samples were centrifuged at 9000 r/min for 20 min, the supernatant was collected, and the precipitate was resuspended with the appropriate amount of PBS (pH 10.0) and collected and then subjected to SDS-PAGE.
2.2.11. Ni2+ Chelation Chromatographic Purification of Recombinant Keratinase Bta
- (1)
- Inclusion of body fragmentation and purification
Buffer A containing 0.5% and 1% TritonX-100 was used to wash the inclusion bodies three times. The washed inclusion bodies were broken using 5–10 mL Buffer B. The column was loaded with 2 mL of Ni2+-affinity chromatography medium (6-His Fast Flow, GE), equilibrated with Buffer B, and the inclusion body protein solution was filtered through a filter membrane (pore size of 0.45 μm), then 3 mL of it was taken for sampling, and the flow-through solution was repeatedly sampled twice. Firstly, the sample was washed with 4 mL of Buffer B, and then it was eluted with eluent containing 20 mmol/L, 50 mmol/L, 100 mmol/L, 150 mmol/L, 200 mmol/L, 300 mmol/L and 500 mmol/L imidazole, and then, the purified sample was collected in separate tubes. The distribution of proteins in the eluent was detected by SDS-PAGE.
- (2)
- Dialysis of inclusion bodies for dialysis reversibility and enzyme activity measurement
Separate stepwise dialysis of these purified protein samples was performed at 4 °C. Dialysis was first performed with Buffer D for 12 h, the dialysate was renewed, and then the dialysis was continued for another 12 h. Then, the dialysate was replaced with Buffer E and dialyzed in the same manner as for Buffer D for 24 h. The dialysate was then replaced with Buffer F, and the stepwise dialysis was similarly completed. Finally, the enzyme activity of the complexed proteins was determined in the same way as described in Section 2.2.3, using three replicates.
3. Results
3.1. Screening of Strains
A total of 23 strains that degraded casein and produced hyaline rings were obtained by the primary screening in skimmed milk medium (Figure 1).
3.2. Re-Screening of Strains and Determination of Enzyme Viability
Using the feather liquid fermentation medium, the 23 strains obtained from the initial screening that could produce hyaline rings were subjected to shake bottle rescreening, and their feather degradation ability was observed after 48 h of fermentation. As shown in Figure 2, only nine strains could degrade the feathers. The keratinase activity results are shown in Table 1. The strain numbered CY-A was found to be the most effective in degrading feathers, with an enzyme activity of 28.64 U/mL. Strain CY-A was further investigated in the subsequent experiments.
3.3. Growth Curve of Strain CY-A
The strain was cultured in LB medium for 72 h, and sampled every 4 h. The OD600 was measured using an ultraviolet spectrophotometer. The results are shown in Figure 3. By analyzing the stable period of the growth curve, the strain was found to be in the growth delay phase at 0–4 h, in the logarithmic phase at 4–16 h, in the stable phase at 16–48 h, and in the decline phase at 48–72 h.
3.4. Strain Identification
3.4.1. Morphological Characteristics
The morphological characteristics of the colonies and cells of strain CY-A are shown in Figure 4. The colonies were round, shiny and raised, with neat edges, a smooth surface, and were easy to pick. The Gram stain was positive and the cells of the bacteriophage were in the form of short rods.
3.4.2. Physiological and Biochemical Characteristics
The physiological and biochemical characteristics of strain CY-A are shown in Table 2. From Table 2, it can be seen that strain CY-A cannot use citrate, arabinose, malonate, lactose or sucrose. However, it can use glucose fermentation but it does not produce gas, litmus milk became red, and then the CY-A strain produced acid, and the bacterial liquid became red in the methyl red test, proving that it is a positive bacterium. If the solution turns red in the vol-poo reaction, the result is positive. In the ammonia production test, a yellow-brown precipitate appeared in the solution indicating that it is an ammonia-producing bacterium. According to the morphological characteristics of the colonies and the results of the physiological and biochemical experiments, and consulting Berger’s Handbook of Bacterial Identification, the strain CY-A was preliminarily identified as a Gram-positive bacillus.
3.4.3. 16S rRNA Sequence Determination and Phylogenetic Tree Construction
The obtained strains were purified several times and sent for sequencing, and 16S rRNA primers were designed and synthesized.
The sequences obtained from the sequencing of the CY-A strain were spliced using the Multiple Sequence Comparison software SeqMan to remove promiscuous bases. Using the NCBI Blast program (blast.ncbi.nlm.nih.gov/Blast.cgi), it was revealed that strain CY-A had the highest 16S rRNA gene sequence similarity with Bacillus tequilensis, with 99.05% similarity. The constructed phylogenetic tree showed that the closest relative to strain CY-A was Bacillus tequilensis (GenBank accession number: MW009674) (Figure 4). Combining the morphological characteristics of the colonies and bacteria, the physiological and biochemical characteristics and the results of the 16S rRNA analysis, CY-A was finally identified as Bacillus tequilensis, and the strain was named Bacillus tequilensis CY-A.
3.5. Extraction of Total DNA
The total DNA of Bacillus tequilensis was extracted and subjected to agarose gel electrophoresis, producing a visible genomic band (Figure 5).
3.6. Cloning of the bta Gene
The extracted DNA of Bacillus tequilensis CY-A was used as a template for PCR amplification with the synthesized primers, and the amplified fragments were detected by agarose electrophoresis (Figure 6) The size of the amplified DNA fragments was about 1100 bp, which is consistent with the theoretical size. The bta gene fragment was ligated into the pMD19-T vector and verified by double enzyme digestion. The results are shown in Figure 7b.
3.7. Sequence Analysis
According to the nucleotide sequence obtained from the sequencing analysis, the corresponding amino acid sequence was deduced using DNAMAN software (Figure 7). The keratin gene was found to be 1110 bp in length, encoding 369 amino acids. The gene sequence was submitted to GenBank under the accession number OR733336. A homology comparison of the amino acid sequences of Bacillus tequilensis with Subtilisin E using MEGA7 and DNAMAN software revealed 98.92% similarity to the amino acids of Subtilisin E and that four amino acids were mutated: V189A, T256S, T224S and L327F (Figure 8).
Figure 8.
Sequence of the keratinase gene and putative amino acid sequence. Red triangles indicate the active sites of serine proteases, and the red boxes indicate the conserved sequences of serine proteases.
Figure 8.
Sequence of the keratinase gene and putative amino acid sequence. Red triangles indicate the active sites of serine proteases, and the red boxes indicate the conserved sequences of serine proteases.
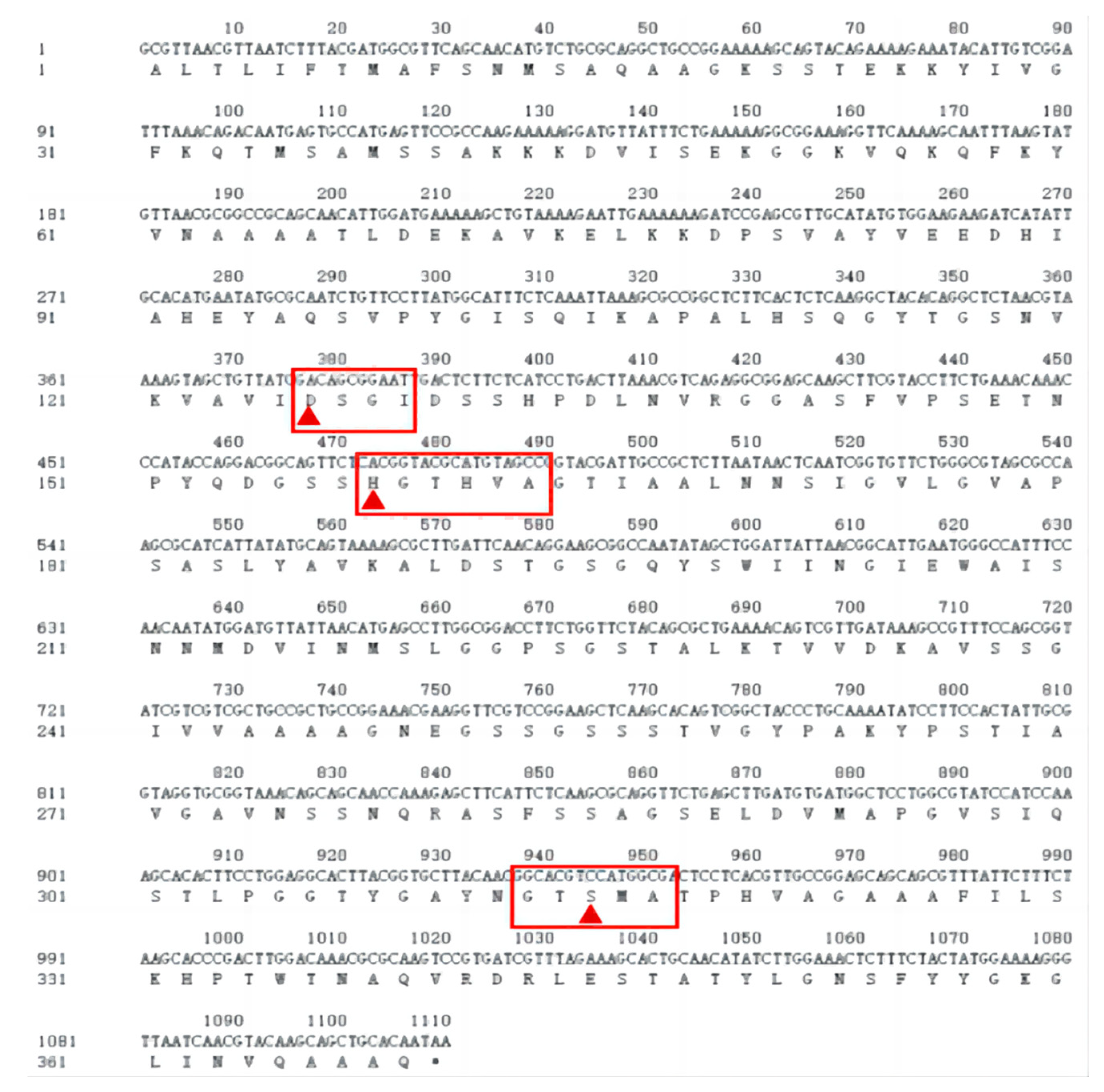
Figure 9.
Amino acid sequence comparison of Bta and arpE.

3.8. Predicting Basic Protein Physicochemical Properties
Theoretically, the encoded protein has a molecular weight of 37.953 kD and an isoelectric point of 8.48, according to predictions on the ProtParm website. The atomic composition is 1668 C atoms, 2629 H atoms, 455 N atoms, 540 O atoms and 8 S atoms; the extinction coefficient is 1.126; the instability coefficient is 30.31, which is less than 40, indicating that the protein is stable; and the average hydrophilicity coefficient is −0.089, meaning that the protein is hydrophilic.
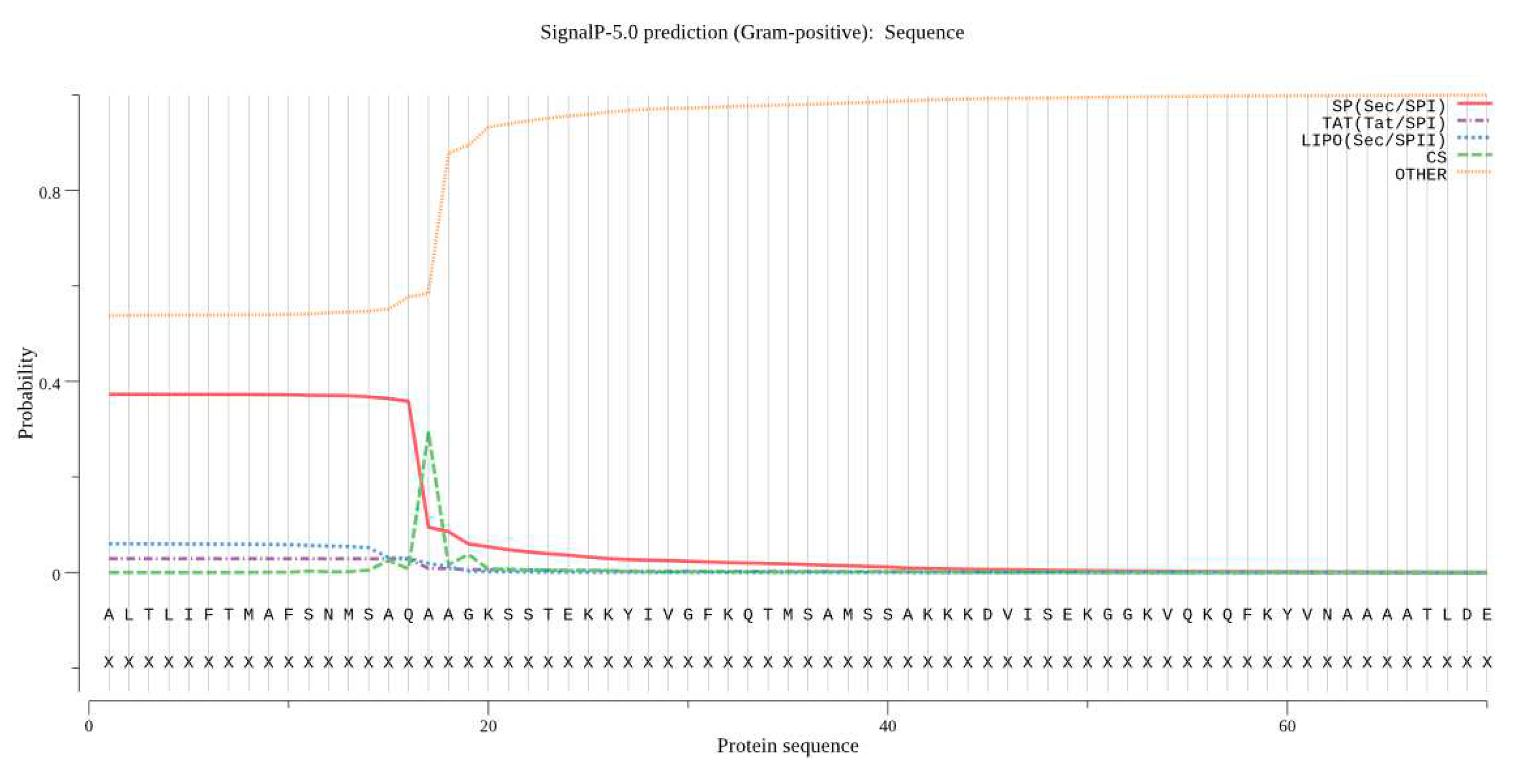
Signal peptide prediction using SignalP identified amino acids 16 and 17 as the signal peptide shear site. The TMHMM analysis showed that the protein does not have a transmembrane domain.
Figure 10.
Signal peptide prediction for Bta proteins.
3.9. Structural Analysis
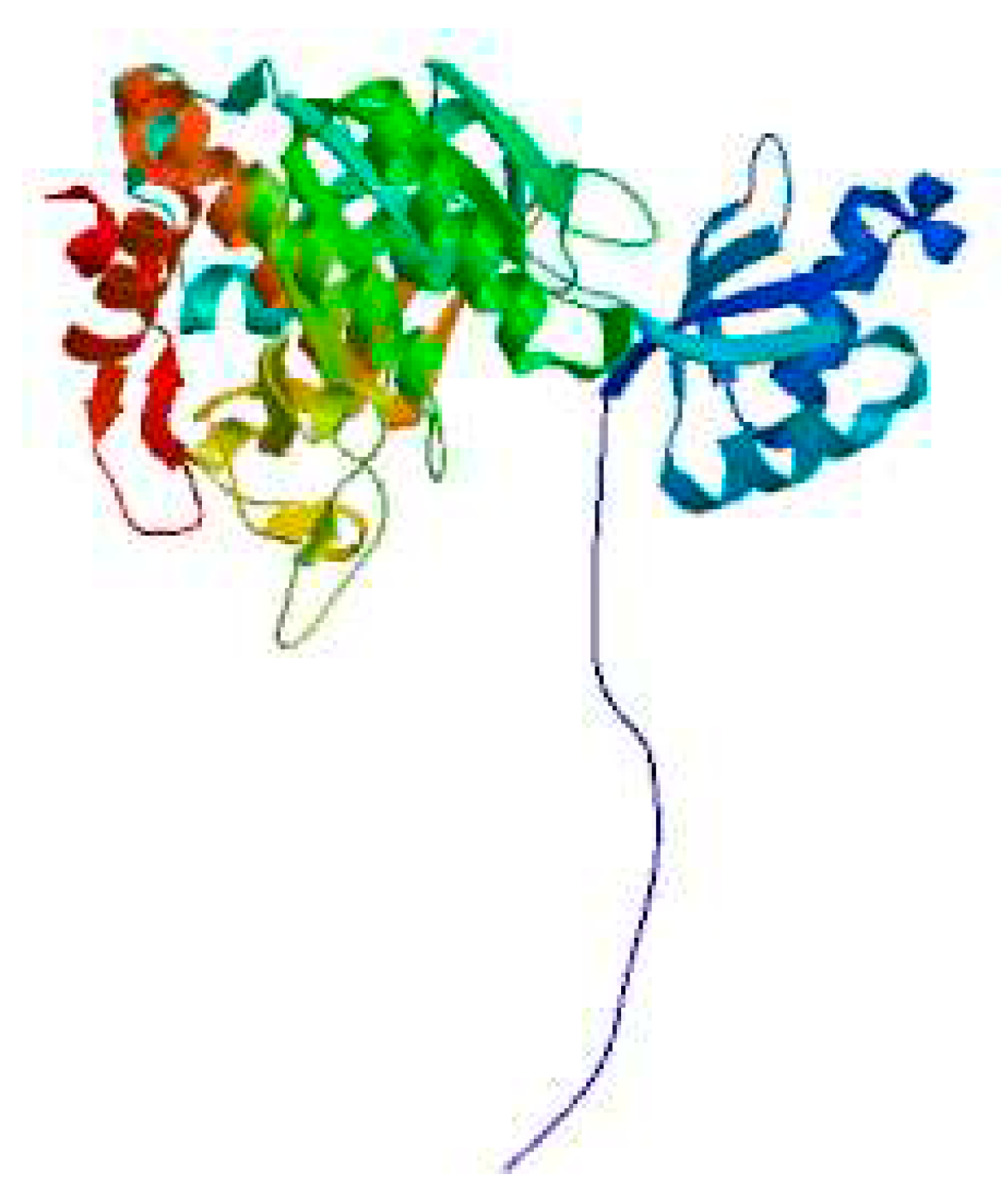
The secondary structure of the protein was predicted using the SOPMA method. It was found that it contained 39.30% irregular coils followed by 30.08% α-helices, 22.22% extended strands and 8.40% β-turns (Figure 11). The tertiary structure of the Bta protein was predicted using SWISS-MODELS, an automated modeling approach, and the tertiary structure similarity between the Bta used for modeling and the homologous sequence arpE was 98.92% (Figure 12), which further suggests that Bta keratinase belongs to the Bacillus subtilis protease family.
Figure 11.
Analysis of Bta protein for transmembrane domains.
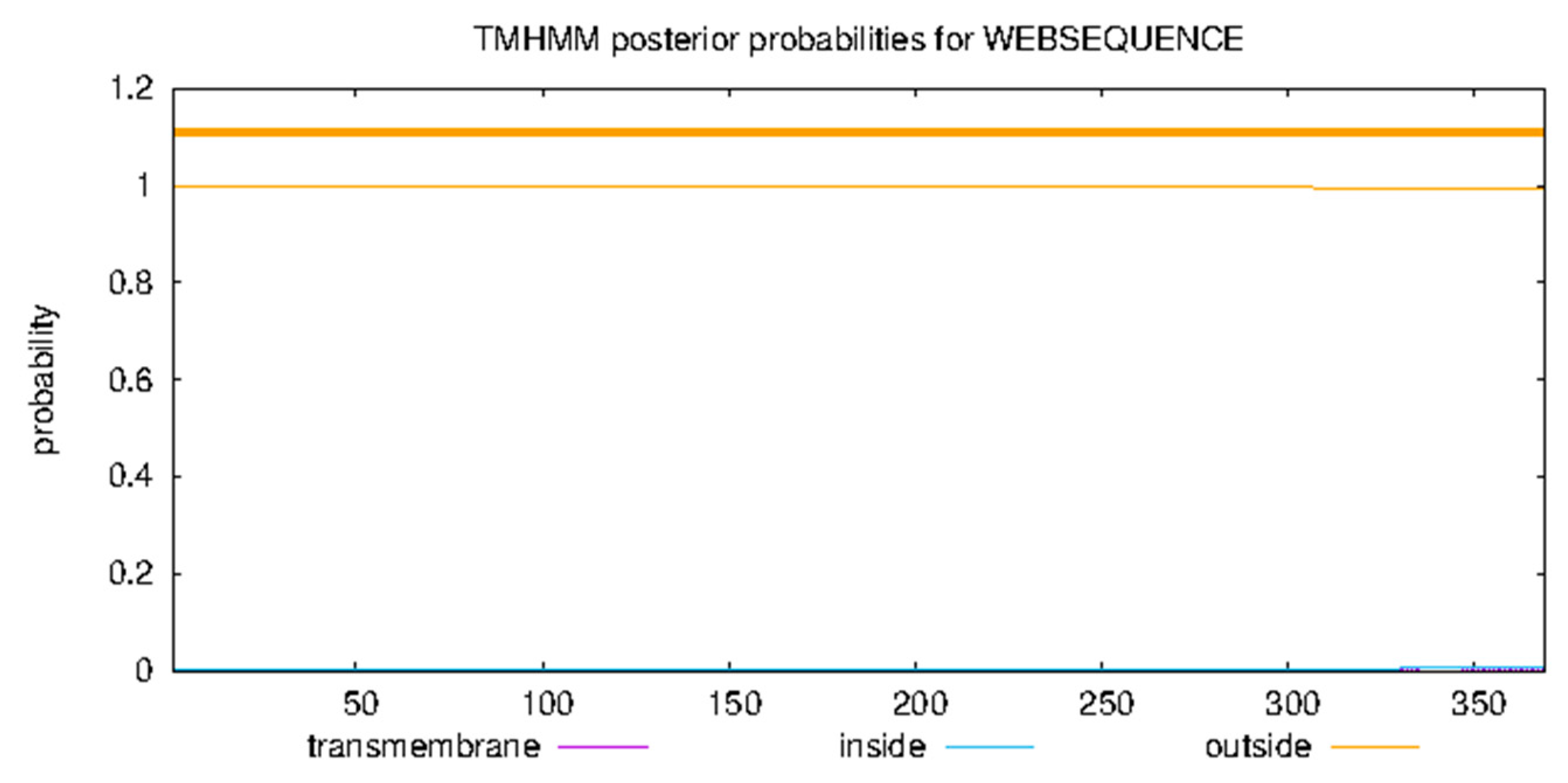
Figure 12.
Prediction of the secondary structure of kerC protein: alpha helices (blue); beta turns (green); extended strands (red); random coils (purple).
Figure 12.
Prediction of the secondary structure of kerC protein: alpha helices (blue); beta turns (green); extended strands (red); random coils (purple).
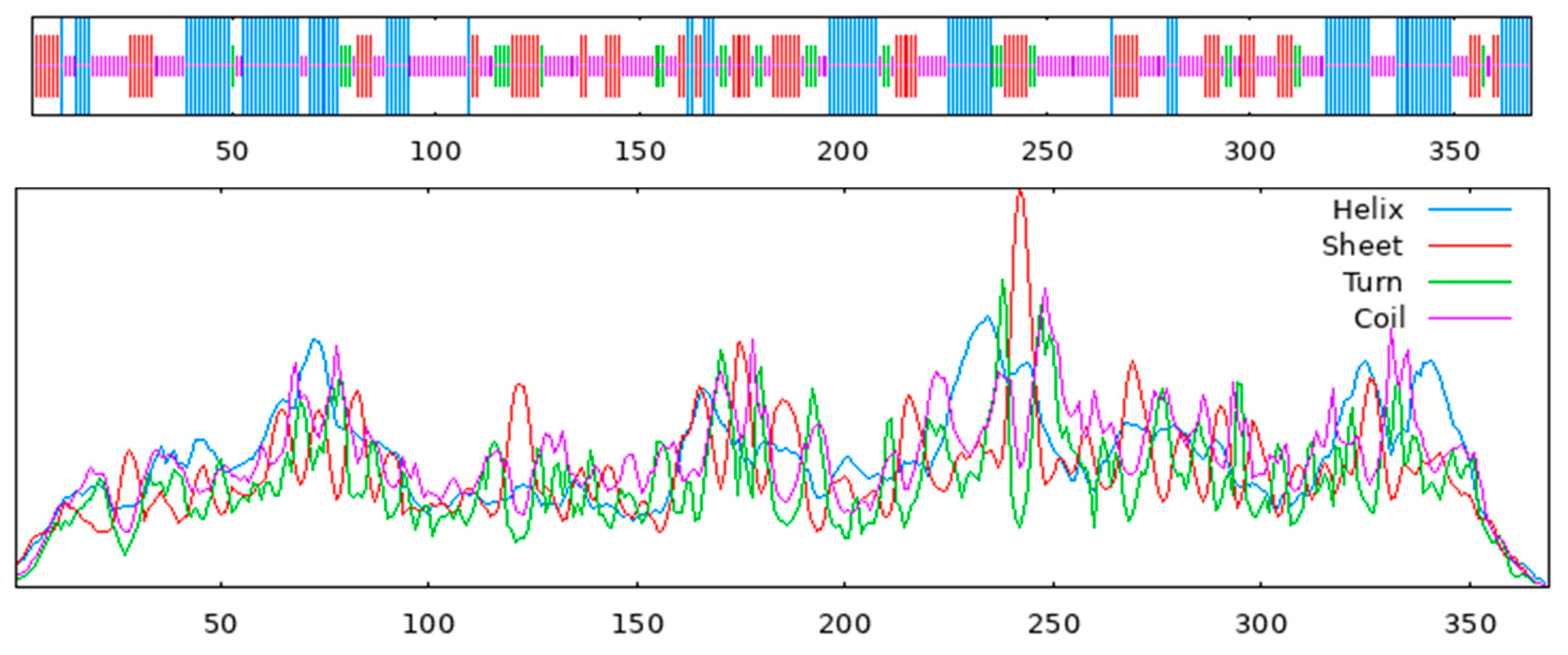
Figure 13.
Prediction of the tertiary structure of Bta protein.
3.10. Construction of Prokaryotic Expression Vectors
The recovered digest products were ligated into the recombinant plasmid pMD19-T-bta and vector pET-28a and used to transform E. coli BL21(DE3), and the positive clones were verified using bacteriophage PCR identification as well as double enzyme digestion identification.
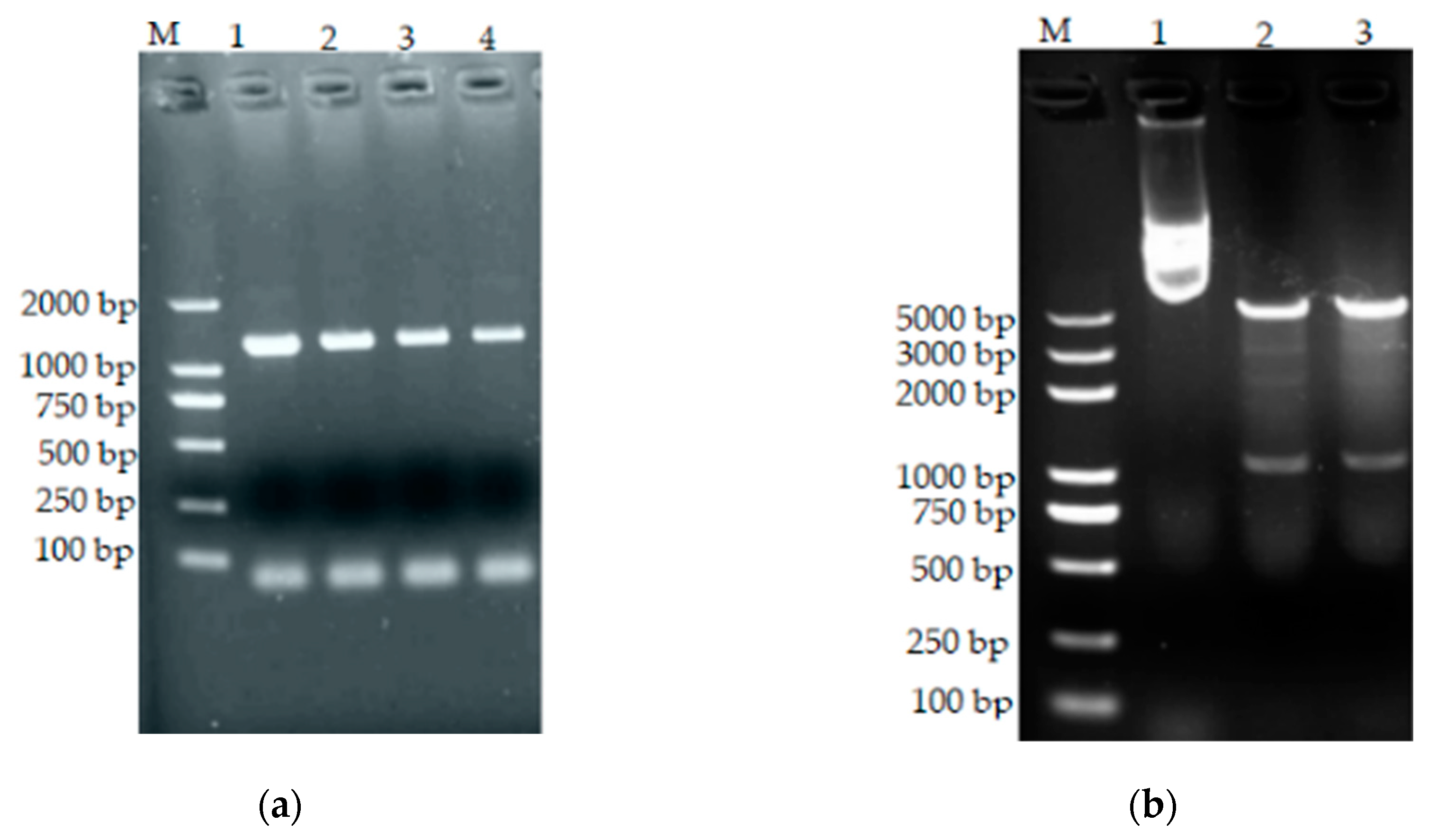
Figure 14.
(a) PCR of transformed bacteria. M: standard molecular weight DNA (DL2000); 1–4: positive clones. (b) pET28a(+)-bta double enzyme digestion results. M: DNA standard molecular weight (DL5000); 1: pET28a(+)-bta positive control; 2 and 3: pET28a(+)-bta double digestion.
Figure 14.
(a) PCR of transformed bacteria. M: standard molecular weight DNA (DL2000); 1–4: positive clones. (b) pET28a(+)-bta double enzyme digestion results. M: DNA standard molecular weight (DL5000); 1: pET28a(+)-bta positive control; 2 and 3: pET28a(+)-bta double digestion.
3.11. Induced Expression of Recombinant Proteins
The correctly sequenced recombinant plasmid pET28a-bta was transferred into E. coli (DE3) receptor cells to obtain a recombinant strain. The recombinant strain was inoculated into 50 mL of LB medium containing 100 mg/mL kanamycin and cultured for 12 h to prepare the seed solution, which was transferred to 50 mL of LB medium containing the same antibiotic at an inoculum concentration of 2% (v/v), and incubated at 37 °C and 180 r/min for 4–5 h. When the OD600 was about 0.4–0.6, IPTG was added at a final concentration of 0.5 mmol/L, and the induction was continued for four hours. After centrifugation, the bacteria were taken for SDS-PAGE.
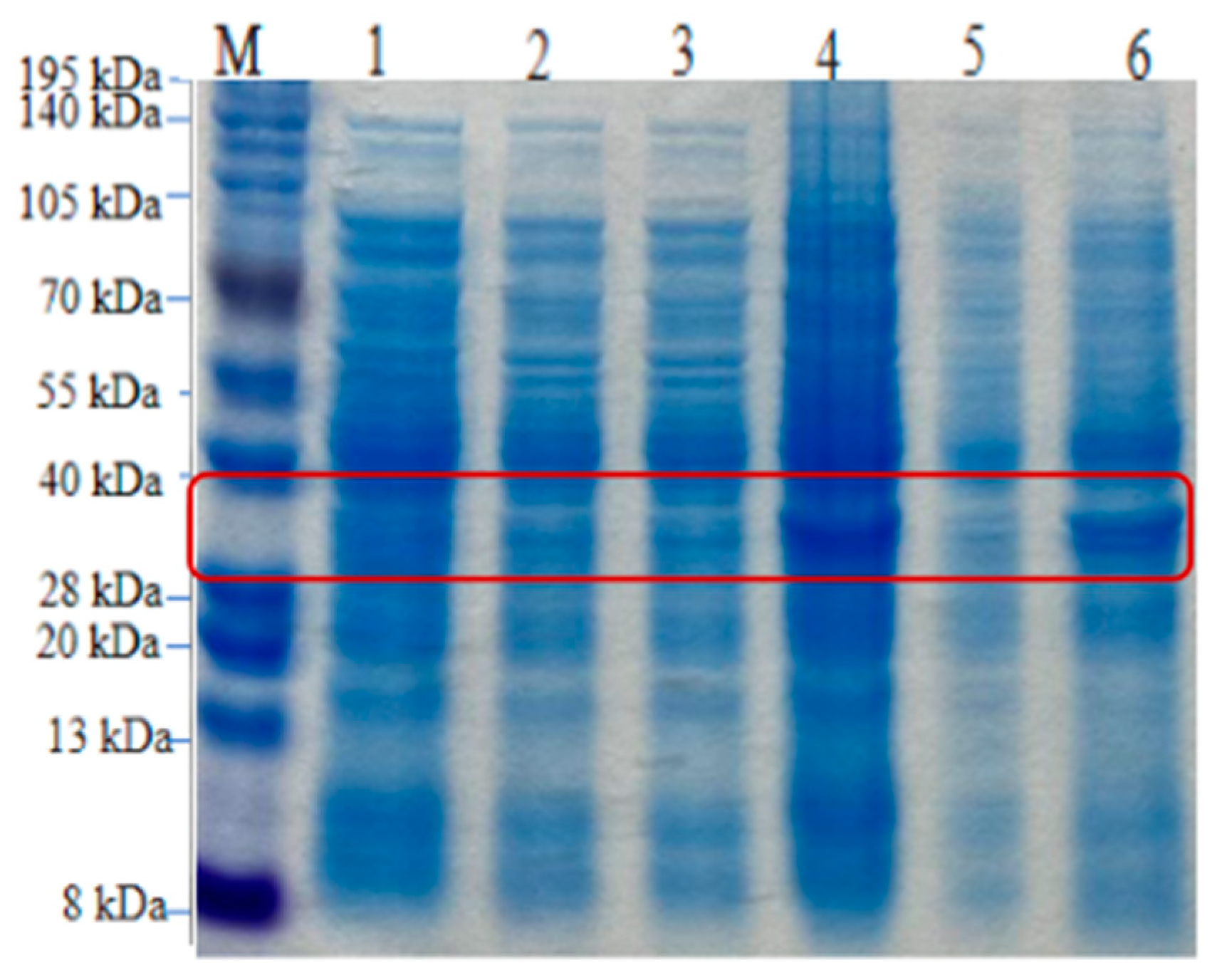
Figure 15.
SDS-PAGE analysis of the expression products of recombinant strains. M, protein standard molecular weight; lane 1, pre-induction whole protein; lane 2, pre-induction supernatant; lane 3, pre-induction precipitate; lane 4, post-induction whole protein lysate; lane 5, post-induction supernatant; lane 6, post-induction precipitate.
Figure 15.
SDS-PAGE analysis of the expression products of recombinant strains. M, protein standard molecular weight; lane 1, pre-induction whole protein; lane 2, pre-induction supernatant; lane 3, pre-induction precipitate; lane 4, post-induction whole protein lysate; lane 5, post-induction supernatant; lane 6, post-induction precipitate.
3.12. Purification of Recombinant Keratin
The column was loaded with 2 mL of Ni2+-affinity chromatography medium (6-His Fast Flow, GE), equilibrated with Buffer B, and the inclusion body protein solution was filtered through a filter membrane (pore size of 0.45 μm); 3 mL of the solution was sampled, and the flow-through solution was sampled twice. First, 5 mL of Buffer B was used to rinse the column, followed by elution with 20 mmol/L, 50 mmol/L, 100 mmol/L, 150 mmol/L, 200 mmol/L, 300 mmol/L and 500 mmol/L imidazole (in that order). The purified samples were collected in separate tubes, and the protein distribution in the eluate was examined by SDS-PAGE.
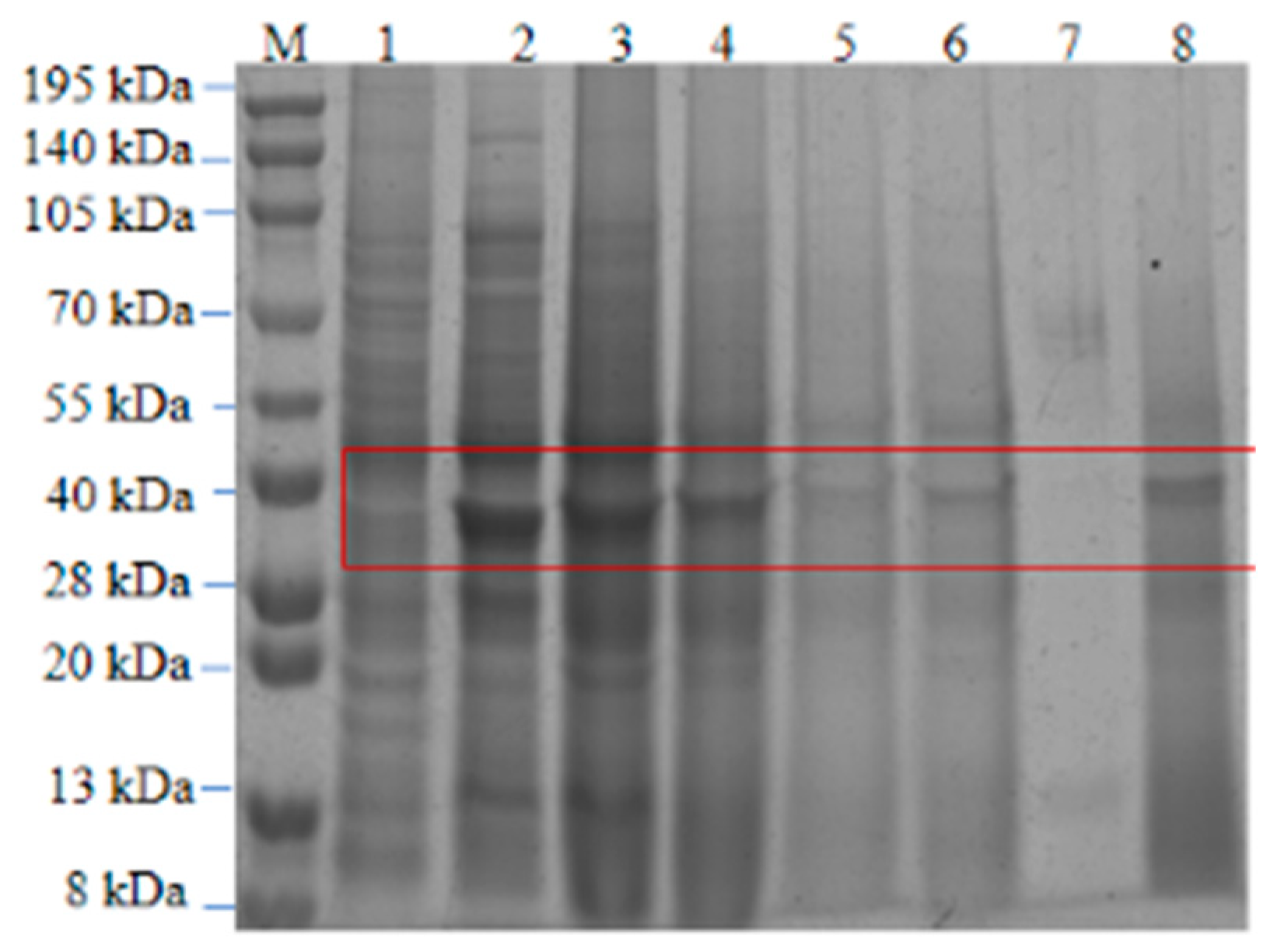
Figure 16.
SDS-PAGE of purified recombinant keratinase. M, protein standard molecular weight; lane 1, post-induction supernatant; lane 2, post-induction precipitate; lane 3, supernatant after inclusion body fragmentation; lane 4, effluent after Ni2+ binding; lanes 5 and 6, different imidazole eluents; lanes 7 and 8, keratinase after dialysis and reconstitution.
Figure 16.
SDS-PAGE of purified recombinant keratinase. M, protein standard molecular weight; lane 1, post-induction supernatant; lane 2, post-induction precipitate; lane 3, supernatant after inclusion body fragmentation; lane 4, effluent after Ni2+ binding; lanes 5 and 6, different imidazole eluents; lanes 7 and 8, keratinase after dialysis and reconstitution.
4. Discussion
Feather waste is the most abundant keratin substance in nature, containing 91% β-keratin, and is produced in large quantities in poultry slaughterhouses [22]. It is recorded that 5 million tons of feather waste are accumulated globally every day. These wastes are generally discarded into garbage dumps and landfills or are incinerated, causing large amounts of environmental pollution [9,23,24]. Therefore, there is a demand for methods to process feathers and produce value-added products. Currently, biodegradation is the most logical processing approach.
The research on keratin-degrading bacteria mainly focuses on bacteria, fungi and actinomycetes. Compared with fungi and actinomycetes, bacteria have the advantages of fast growth, high enzyme activity and safe industrial application, and have become the main source for keratinases. Bacterial protease research for industrial applications has high application prospects [25]. Sun Rong et al. [6] screened the Bacillus subtilis strain BS10 for the efficient degradation of feathers, and the enzyme activity was determined as 1.88 ± 0.10 U/mL using feather meal as the substrate. Zhang et al. [26] obtained a strain of Bacillus paramycoides Gxun-30 that can efficiently degrade feather keratin from the soil of feather waste from a sea duck breeding area in Beibu Gulf, Guangxi. The optimum temperature for the enzyme production of this strain was 35 °C. After fermentation for 48 h, the whole feather was almost completely degraded, and the enzyme activity reached 434.54 U/mL. Dong Mengmeng et al. [27] isolated a strain of feather-degrading Bacillus cereus (DWH-06) from high-temperature-treated soil samples. They determined its optimal enzyme-producing conditions and enzymatic properties, resulting in a maximum enzyme activity of 129.47 U/mL. In an earlier study, Hossain et al. [28] achieved a maximum level of keratinolytic activity (51 U/mL) from B. licheniformis MZK-3 at 37 °C and pH 7.5 using the feather meal broth medium used in this study. A strain isolated from the soil of a residential chicken pen efficiently degraded feathers with an enzyme activity of 28.64 U/mL. Regarding the actual degradation effect, the strain could degrade a complete feather (with stalk) in 3 d, and its degradation ability was comparable to that of Bacillus subtilis-derived keratinases. After morphological observation, physiological and biochemical characterization, analysis of 16S rRNA sequences and construction of a phylogenetic tree, CY-A was identified as Bacillus tequilensis.
Keratinases have received increasing attention from researchers with reports of their multiple promising applications, and more keratinase-producing microorganisms have been identified [29]. In recent years, several keratinase genes have been cloned and the heterologous expression of some of these genes has been successfully achieved [30,31]. For example, some keratinases encoded by the kerA gene extracted from bacteria of the genus Bacillus (most commonly, Bacillus licheniformis) have been cloned and expressed [32,33,34,35]. In this study, a strain of feather-degrading Bacillus sp. was successfully screened to have a functional keratinase gene, and a keratinase-producing genetically engineered bacterium, BL21 (pET-28a(+)-bta), was successfully constructed using molecular cloning. The obtained keratinase gene sequence was subjected to homologous sequence comparison using BLAST and was found to be 99% similar to Bacillus subtilis aprE as well as to Bacillus tequilensis strain BK324. Thus, the complete keratinase gene was found to be 1110 bp long, containing a 1089 bp open reading frame encoding 369 amino acids. The molecular weight size of the recombinant keratinase was determined to be approximately 37.953 kD according to the ProtParm website and SDS-PAGE analysis, which is similar to the molecular weights of keratinases from Bacillus licheniformis PWD-1 (33 kDa), Pseudomonas aeruginosa MS21 (30 kDa) and Bacillus subtilis KD-N2 (30.5 kDa) [36,37]. The molecular weight of keratinases produced by the Bacillus subtilis species generally ranges from 30 to 50 kD (the molecular weight of keratinases reported by Rao et al. was 37.5 kD [38], and the molecular weight of keratinases reported by Zhang Q. et al. was 30.5 kD [39]). The molecular weight of Bta in the present study was 37.95 kD. In addition, the amino acid sequence of B. tequila was homologous to Subtilisin E. Four amino acid mutations, V189A, T256S, T224S, and L327F, were found, and the amino acid similarity with Subtilisin E was 98.92%. The physicochemical properties, secondary structure, and tertiary structure of the recombinant protein were analyzed using bioinformatics tools such as BLAST, ProtParm, SOPMA, and SWISS-MODELS. The tertiary structure of Bta keratinase had 98.92% similarity with the tertiary structure of the keratinase from Bacillus subtilis, aprE. Thus, it was shown that Bta keratinase belongs to the peptidase S8 family of serine proteases. Most of the reported keratinases are present in the lysate, but in this study, the recombinant protein detected after the induction of the recombinant plasmid was present in the inclusion bodies, so 8M urea denaturation, as well as dialysis reconstitution, was used to break and reconstitute the inclusion bodies, and then the target proteins were collected via purification using Ni2+ columns, and, ultimately, the active keratinases were obtained. Although this method will cause some loss of cutinase activity in the process of renaturation and purification, it also provides a reference method to study the renaturation conditions of prokaryotic keratinase expression in inclusion bodies.
Author Contributions
D.M. and Y.F. participated in the thesis topic selection and design, conceived the study and provided feedback on the analytic approach. C.M. designed the analytic protocol, conducted the analyses, interpreted the results, and drafted the manuscript. C.M. and Y.F. generated all the data, supervised the analysis, and interpreted the results.Y.Z., B.C. and Z.M. contributed feedback to the study protocol and provided data. All authors revised and approved the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Special Fund for Key Science & Technology Program in Xinjiang Province of China, grant no. 2022B02053-2.
Data Availability Statement
All data necessary to assess the conclusions of the study are present in the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gozali, C.; Suryanto, D.; Hartanto, A. Partial Purification and Characterization of Keratinase Produced by Keratinolytic Fungi, Earliella Scabrosa A2 and Aspergillus Flavus A11; IOP Publishing Ltd.: Bristol, UK, 2023. [Google Scholar]
- Bhari, R.; Kaur, M.; Singh, R.S.; Pandey, A.; Larroche, C. Bioconversion of chicken feathers by Bacillus aerius NSMk2: A potential approach in poultry waste management. Bioresour. Technol. Rep. 2018, 3, 224–230. [Google Scholar] [CrossRef]
- Khumalo, M.; Sithole, B.; Tesfaye, T. Valorisation of waste chicken feathers: Optimisation of keratin extraction from waste chicken feathers by sodium bisulphite, sodium dodecyl sulphate and urea. J. Environ. Manag. 2020, 262, 110329.1–110329.7. [Google Scholar] [CrossRef]
- Yahaya, R.S.R.; Normi, Y.M.; Lai Yee, P.; Ahmad, S.A.; Abdullah, J.O.; Sabri, S. Molecular strategies to increase keratinase production in heterologous expression systems for industrial applications. Appl. Microbiol. Biotechnol. 2021, 5, 3955–3969. [Google Scholar] [CrossRef]
- Fang, Z.; Yong, Y.C.; Zhang, J.; Du, G.; Chen, J. Keratinolytic protease: A green biocatalyst for the leather industry. Appl. Microbiol. Biotechnol. 2017, 101, 7771–7779. [Google Scholar] [CrossRef]
- Mamangkey, J.; Suryanto, D.; Munir, E.; Mustopa, A. Isolation, Molecular Identification and Verification of Gene Encoding Bacterial Keratinase from Crocodile (Crocodylus porosus) Feces. In Proceedings of the 4th International Conference on Biological Sciences and Biotechnology, Kuala Lumpur, Malaysia, 21–22 February 2019. [Google Scholar]
- Gupta, R.; Ramnani, P. Microbial keratinases and their prospective applications: An overview. Appl. Microbiol. Biotechnol. 2006, 70, 21–33. [Google Scholar] [CrossRef]
- Lange, L.; Huang, Y.; Busk, P.K. Microbial decomposition of keratin in nature—A new hypothesis of industrial relevance. Appl. Microbiol. Biotechnol. 2016, 100, 2083–2096. [Google Scholar] [CrossRef]
- Sharma, I.; Kango, N. Production and characterization of keratinase by Ochrobactrum intermedium for feather keratin utilization. Int. J. Biol. Macromol. 2021, 166, 1046–1056. [Google Scholar] [CrossRef]
- Gopinath, S.C.B.; Periasamy, A.; Thangavel, L.; Tang, T.-H.; Chen, Y.; Hashim, U.; Ruslinda, R.; Arshad, M.K.M. Biotechnological Aspects and Perspective of Microbial Keratinase Production. J. Biomed. Biotechnol. 2015, 2015, 140726. [Google Scholar] [CrossRef]
- Vidmar, B.; Vodovnik, M. Microbial Keratinases: Enzymes with Promising Biotechnological Applications. Food Technol. Biotechnol. 2018, 56, 312–328. [Google Scholar] [CrossRef]
- Imania, G.; Aqsa, I.; Ali, H.; Arshad, J.; Faiza, J.; Muhammad, A.; Javed, I.Q. Microbial production and industrial applications of keratinases: An overview. Int. Microbiol. 2018, 21, 163–174. [Google Scholar]
- Cheng, S.W.; Hu, H.M.; Shen, S.W.; Takagi, H.; Asano, M.; Tsai, Y.-C. Production and characterization of keratinase of a feather-degrading Bacillus licheniformis PWD-1. Biosci. Biotechnol. Biochem. 1995, 59, 2239–2243. [Google Scholar] [CrossRef]
- Liang, B. Extraction, Cloning and Expression of Keratinase Gene kerB; Jilin University: Changchun, China, 2003. [Google Scholar]
- Radha, S.; Gunasekaran, P. Sustained expression of keratinase gene under PxylA and PamyL promoters in the recombinant Bacillus megaterium MS941. Bioresour. Technol. 2008, 99, 5528–5537. [Google Scholar] [CrossRef]
- Fakhfakh, N.; Kanoun, S.; Manni, L.; Nasri, M. Production and biochemical and molecular characterization of a keratinolytic serine protease from chicken feather-degrading Bacillus licheniformis RPk. Can. J. Microbiol. 2009, 55, 427. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.; Huang, Y.; Wei, Q.; Huang, H.; Guo, C. Cloning and expression of a thermostable keratinase gene from Thermoactinomyces sp. YT06 in Escherichia coli and characterization of purified recombinant enzymes. World J. Microbiol. Biotechnol. 2019, 35, 135. [Google Scholar] [CrossRef]
- Gurunathan, R.; Huang, B.; Ponnusamy, V.K.; Hwang, J.-S.; Dahms, H.-U. Novel recombinant keratin degrading subtilisin like serine alkaline protease from Bacillus cereus isolated from marine hydrothermal vent crabs. Sci. Rep. 2021, 11, 12007. [Google Scholar] [CrossRef]
- Yamamura, S.; Morita, Y.; Hasan, Q.; Rao, S.R.; Murakami, Y.; Yokoyama, K.; Tamiya, E. Characterization of a new keratin-degrading bacterium isolated from deer fur. J. Biosci. Bioeng. 2002, 93, 595–600. [Google Scholar] [CrossRef]
- Zhu, J.G. Manual of Identification of Common Clinical Bacteria; Beijing Medical University and Peking Union Medical College Joint Publishing House: Beijing, China, 1993. [Google Scholar]
- Xiao, J.; Yang, X.; Guo, H. Bioinformatics and expression analysis of the flavonoid-3′5′-hydroxylase (F3′5′H) gene of the colourful potato variety ‘Turning Heart U’. 2015(7):9.
- Mabrouk, M.E.M. Feather degradation by a new keratinolytic Streptomyces sp. MS-2. World J. Microbiol. Biotechnol. 2008, 24, 2331–2338. [Google Scholar] [CrossRef]
- Arokiyaraj, S.; Varghese, R.; Ahmed, B.A.; Duraipandiyan, V.; Al-Dhabi, N. Optimizing the fermentation conditions and enhanced production of keratinase from Bacillus cereus isolated from halophilic environment. Saudi J. Biol. Sci. 2019, 26, 378–381. [Google Scholar] [CrossRef]
- Sharma, I.; Kango, N. Production and characterization of keratinase by Ochrobactrum intermedium for feather keratin utilization. Int. J. Biol. Macromol. 2021, 166, 1046–1056. [Google Scholar] [CrossRef]
- Mamangkey, J.; Suryanto, D.; Munir, E.; Mustopa, A. Isolation, Molecular Identification and Verification of Gene Encoding Bacterial Keratinase from Crocodile (Crocodylus porosus) Feces. In Proceedings of the 4th International Conference on Biological Sciences and Biotechnology, Kuala Lumpur, Malaysia, 21–22 February 2019. [Google Scholar]
- ZHANG Ni, ZHANG Hongyan, YANG Mengying, et al. Screening, characterisation and enzymatic properties of a marine-derived strain of highly efficient keratinase-producing bacteria[J]. Food and Fermentation Industry 2020, 46, 7.
- DONG Mengmeng, HAO Pengze, WANG Jialiu, et al. Isolation and identification of feather-degrading bacterium DHW-06 and study on enzyme-producing characteristics[J]. Journal of Northwest Agriculture and Forestry University: Natural Science Edition 2021, 49, 9.
- Hossain, M.S.; Azad, A.K.; Abu Sayem, S.M.; Mostafa, G.; Hoq, M. Production and partial characterization of feather-degrading keratinolytic serine protease from Bacillus licheniformis MZK-3. J. Biological. Sci. 2007, 7, 599–606. [Google Scholar]
- Manivasagan, P.; Sivakumar, K.; Gnanam, S.; Venkatesan, J.; Kim, S.-K. Production, Biochemical Characterization and Detergents Application of Keratinase from the Marine Actinobacterium Actinoalloteichus sp. MA-32. J. Surfactants Deterg. 2014, 17, 669–682. [Google Scholar] [CrossRef]
- Mwanza, E.P.; van der Westhuizen, W.A.; Boucher, C.E.; Charimba, G.; Hugo, C. Heterologous expression and characterisation of a keratinase produced by Chryseobacterium carnipullorum. Protein Expr. Purif. 2021, 186, 105926. [Google Scholar] [CrossRef]
- Hu, H.; Gao, J.; He, J.; Yu, B.; Zheng, P.; Huang, Z.; Mao, X.; Yu, J.; Han, G.; Chen, D. Codon Optimization Significantly Improves the Expression Level of a Keratinase Gene in Pichia pastoris. PLoS ONE 2013, 8, e58393. [Google Scholar] [CrossRef]
- Hu, H.; He, J.; Yu, B.; Zheng, P.; Huang, Z.; Mao, X.; Yu, J.; Han, G.; Chen, D. Expression of a keratinase (kerA) gene from Bacillus licheniformis in Escherichia coli and characterization of the recombinant enzymes. Biotechnol. Lett. 2013, 35, 239–244. [Google Scholar] [CrossRef]
- Porres, J.M.; Benito, M.J.; Lei, X.G. Functional expression of keratinase (kerA) gene from Bacillus licheniformis in Pichia pastoris. Biotechnol. Lett. 2002, 24, 631–636. [Google Scholar] [CrossRef]
- Wang, J.-J.; Rojanatavorn, K.; Shih, J.C.H. Increased production of Bacillus keratinase by chromosomal integration of multiple copies of the kerA gene. Biotechnol. Bioeng. 2004, 87, 459–464. [Google Scholar] [CrossRef]
- Lin, X.; Lee, C.G.; Casale, E.S.; Shih, J.C.H. Purification and Characterization of a Keratinase from a Feather-Degrading Bacillus licheniformis Strain. Appl. Environ. Microbiol. 1992, 58, 3271–3275. [Google Scholar] [CrossRef]
- Tork, S.; Aly, M.M.; Nawar, L. Biochemical and Molecular Characterization of a New Local Keratinase Producing Pseudomomanas sp. MS21. Asian J. Biotechnol. 2010, 2, 1–13. [Google Scholar] [CrossRef]
- Cai, C.; Chen, J.S.; Qi, J.J.; Yin, Y.; Zheng, X.-D. Purification and characterization of keratinase from a new Bacillus subtilis strain. J. Zhejiang Univ. Sci. B 2008, 9, 713–720. [Google Scholar] [CrossRef]
- Rao, C.S.; Sathish, T.; Ravichandra, P.; Prakasham, R. Characterization of thermo- and detergent stable serine protease from isolated Bacillus circulans and evaluation of eco-friendly applications. Process Biochem. 2009, 44, 262–268. [Google Scholar]
- ZHANG Qi, SUN Dan, YANG Wenbo. Purification process, physicochemical properties and application of keratinase produced by Bacillus sphaericus L_4[J]. Ion exchange and adsorption 2009, 25, 448–455.
Figure 1.
Degradation activity of strain CY-A on milk screening plate.
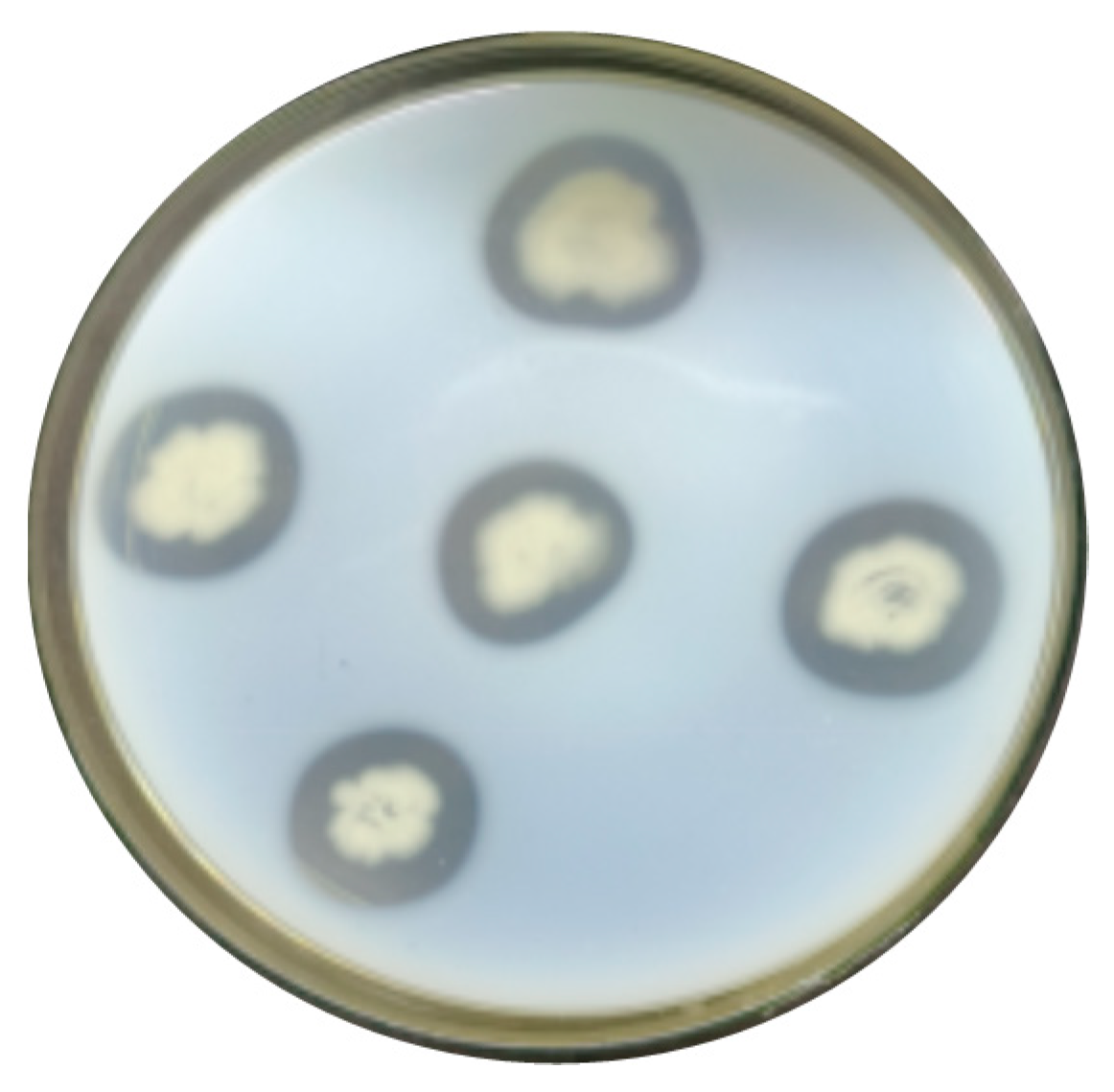
Figure 2.
Feather degradation capacity of rescreened strains of bacteria.

Figure 3.
Growth curve of strain CY-A.
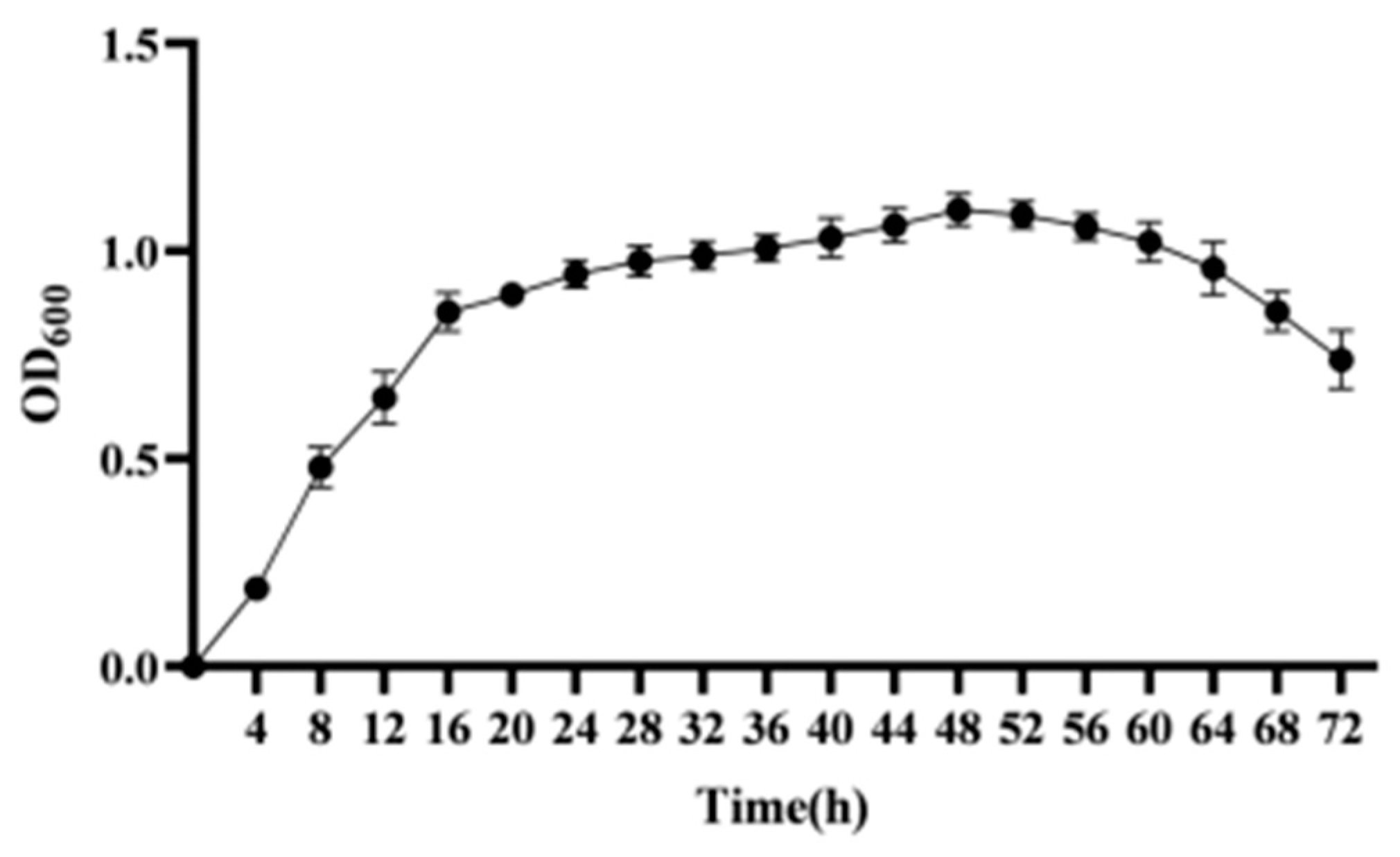
Figure 4.
Morphological characteristics of strain CY-A.
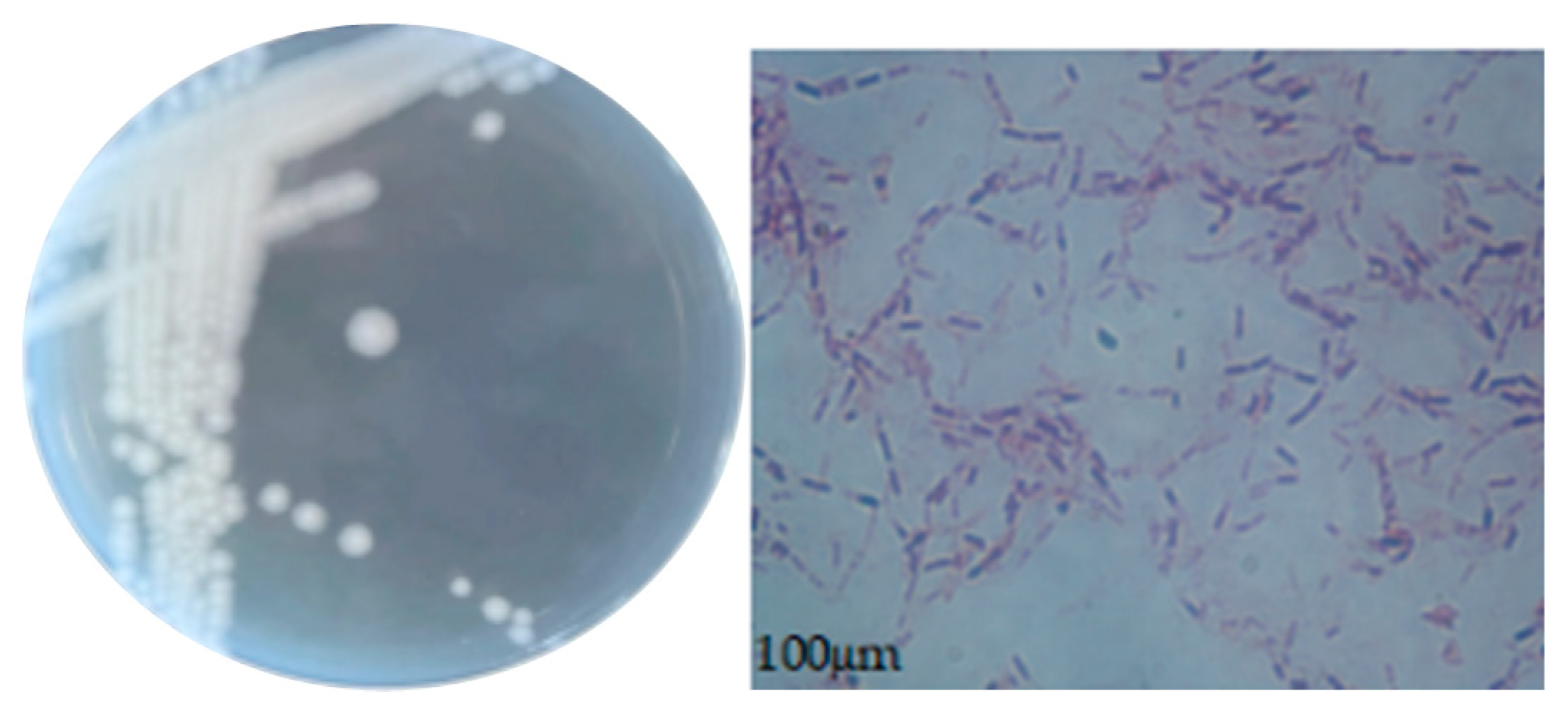
Figure 5.
Phylogenetic evolutionary tree of strain CY-A.
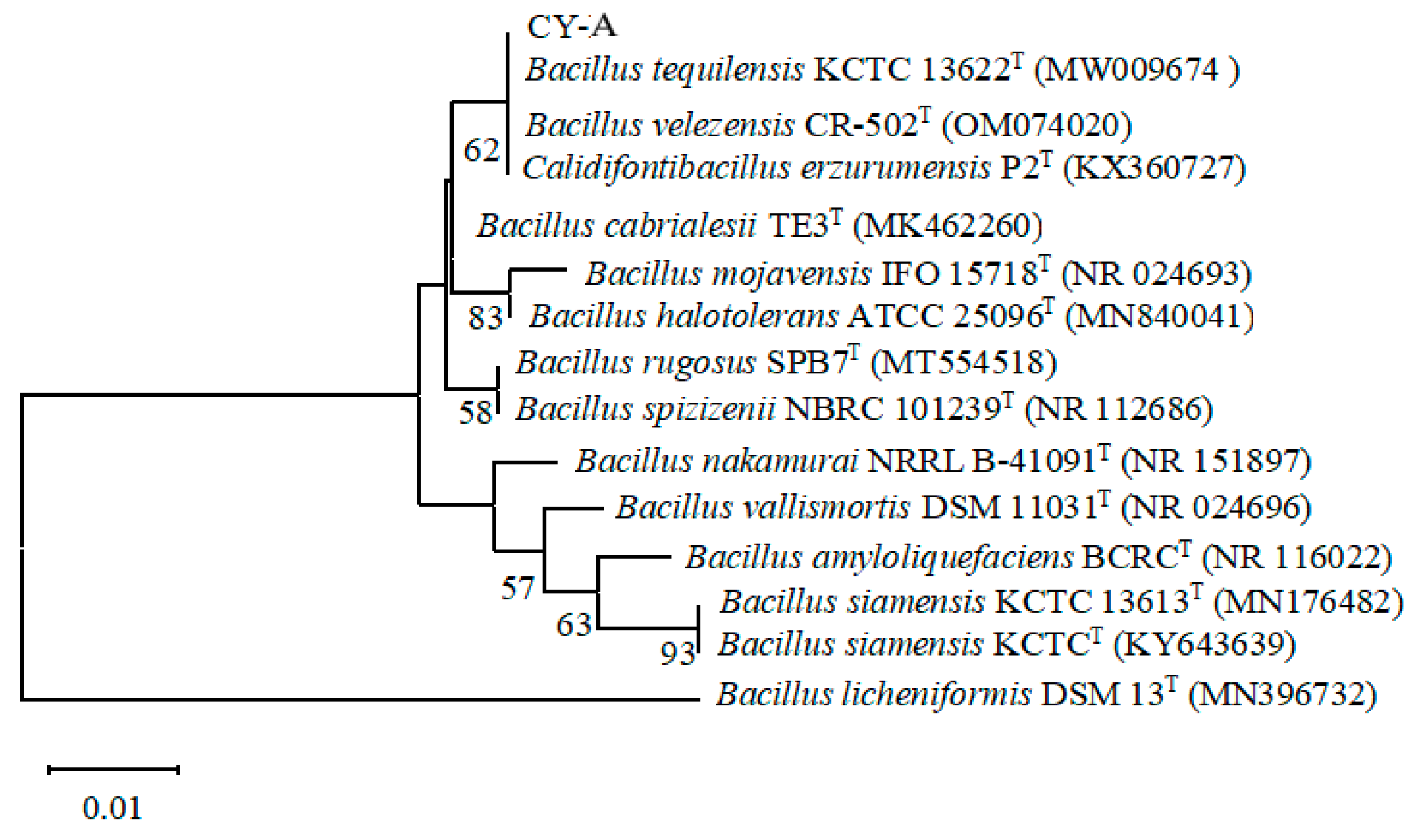
Figure 6.
Results of total DNA extraction Bacillus tequilensis CY-A. M: DNA marker (DL15,000); 1: total DNA of Bacillus tequilensis CY-A.
Figure 6.
Results of total DNA extraction Bacillus tequilensis CY-A. M: DNA marker (DL15,000); 1: total DNA of Bacillus tequilensis CY-A.
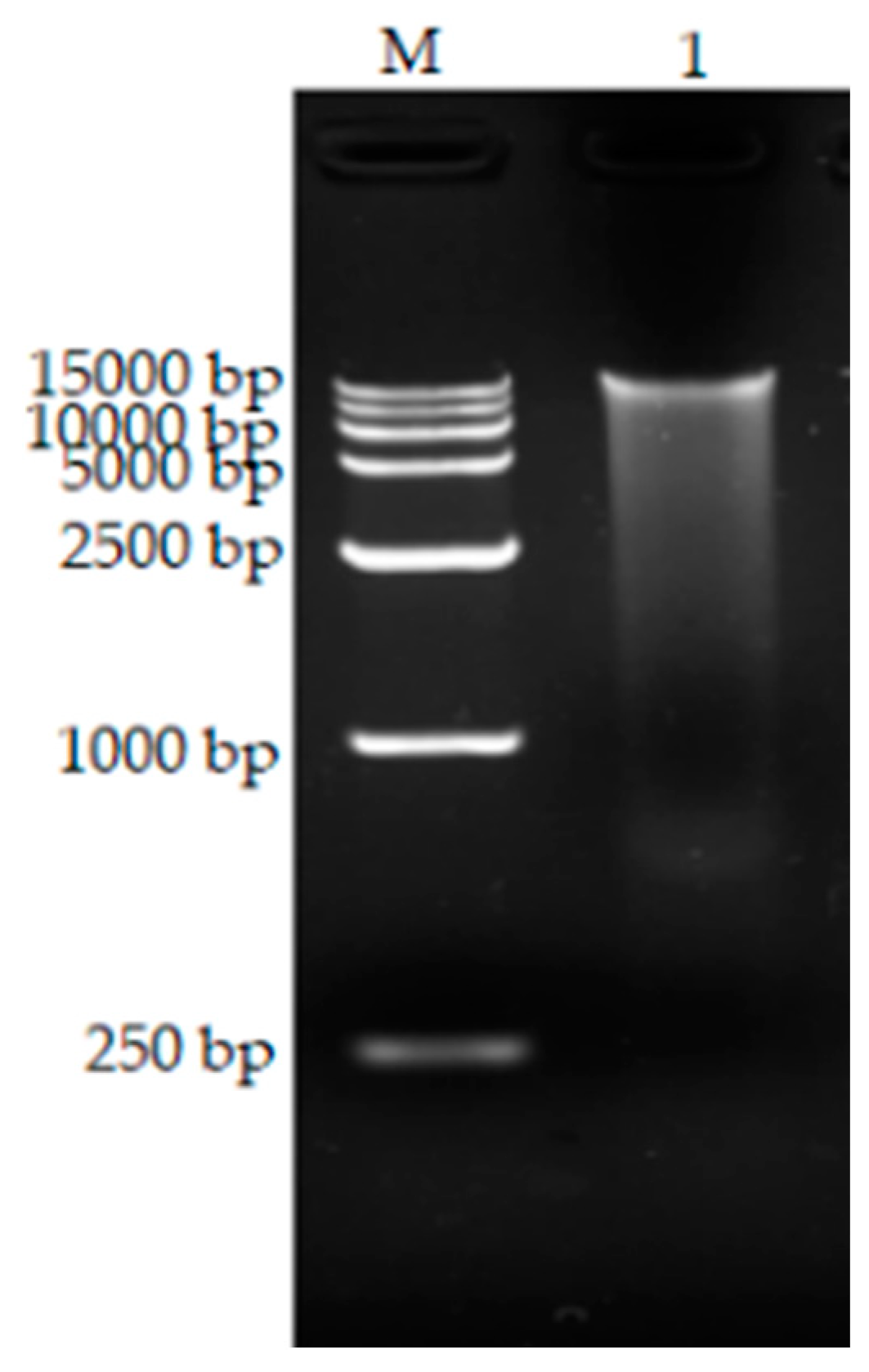
Figure 7.
(a) Results of gene amplification in Bacillus tequilensis CY-A. M: DNA standard molecular weight (DL2000); 1–2: gene amplification results. (b) pMD19-T-bta double digestion results. M: DNA standard molecular weight (DL5000); 1: pMD19-T-bta positive control; 2 and 3: pMD19-T-bta double digestion results.
Figure 7.
(a) Results of gene amplification in Bacillus tequilensis CY-A. M: DNA standard molecular weight (DL2000); 1–2: gene amplification results. (b) pMD19-T-bta double digestion results. M: DNA standard molecular weight (DL5000); 1: pMD19-T-bta positive control; 2 and 3: pMD19-T-bta double digestion results.
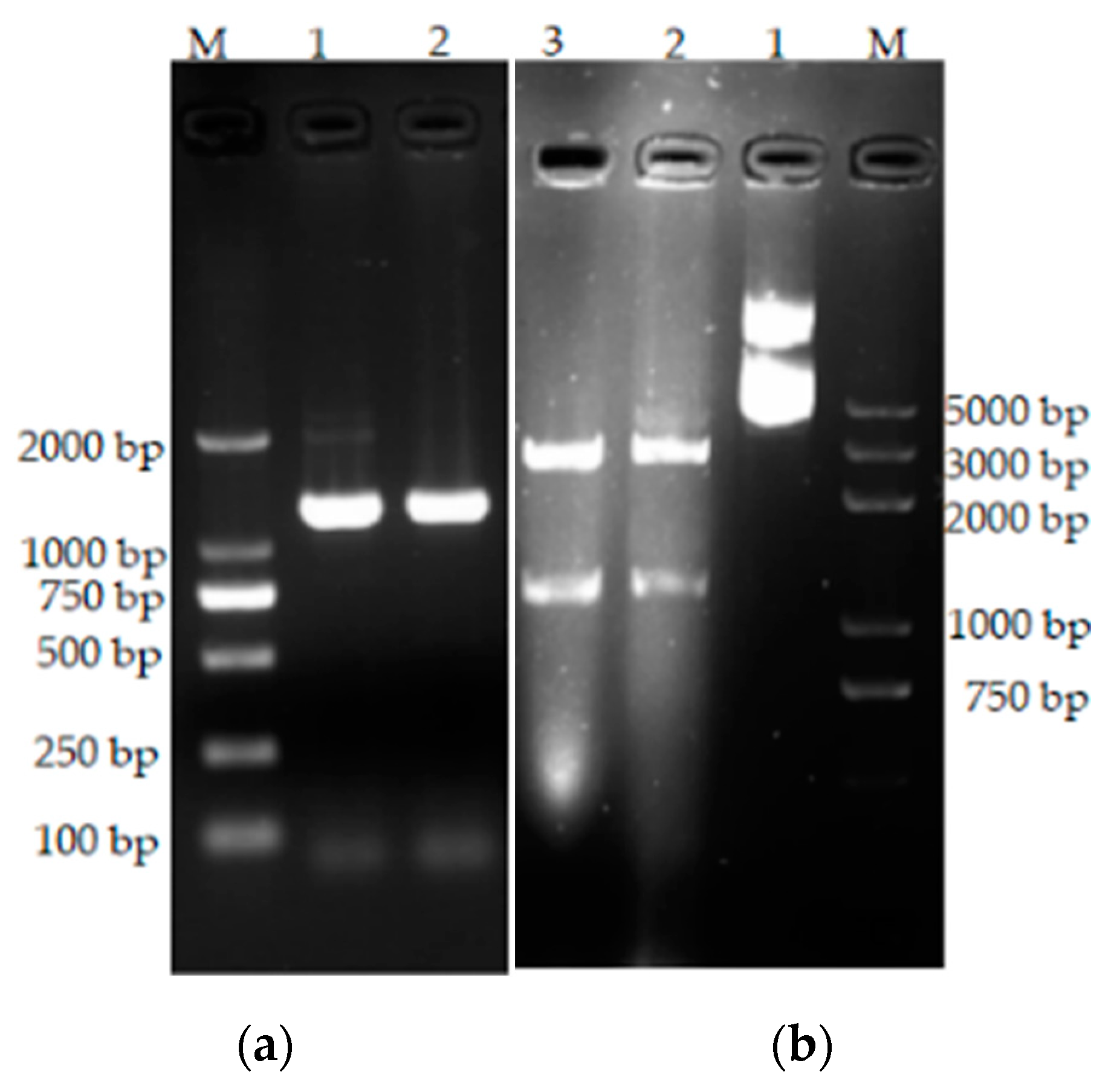

Table 1.
Keratinase activity of 9 strains of bacteria.
| Strain | Keratinase Activity (U/mL) |
|---|---|
| CY-A | 28.64 |
| CY-C | 19.99 |
| CY-E | 25.04 |
| CY-F | 28.14 |
| CY-G | 25.42 |
| WQ4-3 | 16.70 |
| WO5-3 | 20.83 |
| QT4-1 | 27.23 |
| QT4-3 | 25.42 |
Table 2.
Physiological and biochemical characteristics of strain CY-A.
| Assay | Reaction Result |
|---|---|
| CY-A | |
| Glucose fermentation | + |
| Lactose fermentation | - |
| Sucrose fermentation | - |
| Ammonia production experiment | + |
| Malonic acid utilization | - |
| Decomposition of milk with litmus | + |
| Methyl red | + |
| Citrate decomposition | - |
| L-Arabinose fermentation | - |
| V-P experiment | + |
Note: + indicates a positive reaction; - indicates a negative reaction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



