Submitted:
08 January 2024
Posted:
12 January 2024
Read the latest preprint version here
Abstract
Two ferrocene derivatives, namely, 1,2-(tetramethylene)-ferrocene and 1,2,1’,2’-bis(tetramethylene)-ferrocene, were synthesized in a four-step reaction sequence starting from ferrocene. Friedel-Craft acylation of ferrocene with succinic anhydride gave mono- or bis(3-carboxypropinoyl)ferrocene depending on the stoichiometry of succinic anhydride. The reduction of the keto groups to methylene, followed by ring-closing using trifluoroacetic anhydride gave 1,2-(α-ketotetramethylene)-ferrocene or 1,2,1’,2’-bis(α-ketotetramethylene)ferrocene. The diastereomeric mixture of the later diketones was separated by column chromatography, characterized by single-crystal X-ray analysis, and assigned their stereochemistry. Reduction of the keto groups to methylene under Clemmensen conditions gave homoannular mono- or bis(tetramethylene) ferrocene derivatives. The molecular structure of 1,2-(tetramethylene)-ferrocene reveals that ipso carbons of the cyclopentadienyl group are 0.023(3) Å farther away from Fe(II) in comparison to its remaining three carbons. Both complexes exhibit lower half-wave oxidation potentials than ferrocene possibly due to the electron-releasing effects of the tetramethylene bridges.
Keywords:
1
; 2-(α-ketotetramethylene)-ferrocene
; 2-(tetramethylene)-ferrocene
; ferrocene
; Friedel-Crafts acylation
; Clemmensen reduction
; X-ray crystallography
; planar chirality
; puckering parameters
; electrochemistry
1. Introduction
Ferrocene and its derivatives play important roles in modern-day materials chemistry owing to their air stability, aromatic reactivity, reversible electrochemistry, and low toxicity [1]. After the discovery of ferrocene in 1951 [2], research was focused on the conjugation of carbocyclic rings to one or both cyclopentadienyl rings. The most straightforward method of such conjugation was the cyclization of ω-ferrocenylaliphatic acids. The ferrocenylaliphatic acids containing four or five carbon atoms on their side chain undergo electrophilic ring-closing to give homoannular ferrocene derivatives under electrophilic conditions [3]. Such ferrocene derivatives with unsymmetrically substituted cyclopentadienyl rings represented prototypes of planar chirality in metallocene chemistry due to the “sandwich” structure of the ferrocenyl moiety [4].
With our continuous interest in extending the π-conjugation of metallocenes [5], we synthesized 1’,2’,3’,4’,5’-pentamethylruthenocene-fused quinones from the double Friedel-Crafts acylation between metallocene-1,2-diacylchloride and organic aromatics [6]. Later, we realized that switching the functionality of two reaction partners can give such complexes in a much simpler method. Since ferrocene is an excellent nucleophile for Friedel-Crafts acylation, its reaction with succinic anhydride can eventually lead us to synthesize ferrocene-fused ring(s). These compounds can serve as the simplest possible model for extending the π-conjugation of metallocene. Further, ferrocene and its derivatives undergo reversible electrochemistry. Numerous studies have been concerned with the effects of substituents on the redox potential of the ferrocenyl moiety [7,8,9,10]. In general, electron-releasing substituents lower the oxidation potentials and electron-withdrawing substituents increase the potential in comparison to unsubstituted ferrocene [11]. In this contribution, we report the synthesis of 1,2-(tetramethylene)-ferrocene and 1,2,1’,2’-bis(tetramethylene)-ferrocene starting from ferrocene, their crystal structures, and their half-wave oxidation potentials.
2. Materials and Methods
2.1. General Procedures
Reactions were carried out using standard Schlenk line techniques under nitrogen unless otherwise mentioned. Solvents were used as received without further drying or purification. Ferrocene, succinic anhydride, aluminum chloride, zinc powder, mercuric chloride (Alfa Aesar), and trifluoroacetic anhydride (Oakwood Chemicals) were used as purchased. The organic phases were dried using anhydrous magnesium sulfate (Mallinckrodt). Flash chromatography was performed using 60-Å pore size, 230 x 400 mesh silica gel (Sorbent Technologies).
1H and 13C NMR spectra were recorded on a JEOL – 400 ESZ spectrometer at ca. 22 °C and were referenced to residual solvent peaks. Infrared spectra were recorded on Bruker Alpha-E FT-IR spectrometer with a diamond crystal ATR accessory. Melting points were taken on a Thomas-Hoover capillary melting point apparatus and were uncorrected. The oxidation potentials of the compounds were measured with BASi Epsilon – Electrochemical Workstation.
2.2. X-ray Crystallography
X-ray diffraction data were measured at T = 90 K on a Bruker Kappa Apex-II diffractometer equipped with a sealed-tube MoKα source (λ=0.71073 Å), a Triumph focusing monochromator and a CCD detector. Structures were solved using SHELXT [12] and refined using SHELXL [13]. Hydrogen atoms were visible in difference maps and were placed in idealized positions during refinement and treated as riding. The structure of 3b’ had a disorder involving partially occupied carbonyl groups at CH2 sites. The crystal structures and refinement data are presented in Table 1.
2.3. Experimental Procedures
Synthesis of C14H14FeO3 (1a). In a 500 mL flask, a mixture of succinic anhydride (1.83 g, 18.1 mmol) and aluminum chloride (4.20 g, 32.1 mmol) suspended in dichloroethane (80 mL) was added to the solution of ferrocene (3.02 g, 16.3 mmol) in dichloroethane (40 mL) over 45 minutes. The reaction mixture was allowed to stir for an additional 2 hours at room temperature and then poured into water (50 mL). The organic phase was collected. The product was extracted in 1.0 M NaOH (3 x 20 mL) and acidified with concentrated HCl until the pH of the solution dropped below 7. The precipitate was collected by filtration, washed with water, and dried to give 1a (1.78 g, 38%) as an orange powder. Melting point: 165–166 °C (Lit. [14] 164 – 165). 1H NMR (400 mHz, CDCl3 ): δ 2.72 – 2.76 (t, 2 H, 3J = 6.8 Hz), 3.06 – 3.09 (t, 2 H, 3J = 6.4 Hz), 4.23 (s, 5 H, Cp), 4.51 – 4.52 (t, 2 H, 3J = 2.4 Hz), 4.80 – 4.81 (t, 2 H, 3J = 2.0 Hz). IR (ATR, cm – 1): 1705 (C=O), 1657 (C=O).
Synthesis of C18H18FeO6 (1b). In a 500 mL flask, succinic anhydride (10.8 g, 108 mmol) and anhydrous aluminum chloride (28.8 g, 216 mmol) were mixed in dichloroethane (80 mL). Ferrocene (10.0 g, 53.8 mmol) dissolved in dichloroethane (80 mL) was added dropwise into the flask using a dropping funnel. The reaction was stirred for 48 hours at room temperature and poured into ice water (100 mL). The solutions were layered, and the organic phase was collected. The aqueous phase was extracted with dichloromethane (100 mL). The product was extracted in 2 M sodium hydroxide (3 x 100 mL). The aqueous phase was acidified with conc. HCl until precipitation was completed. The precipitate was collected by filtration to give a crude product. The crude product was suspended in boiling water (100 mL) and filtered to give 1b (14.9 g, 72%) as a dark red powder. Melting Point: 178 – 179 °C (Lit. [15] 179 °C). 1H NMR (400 mHz, acetone-d6, ppm): δ 2.60 – 2.64 (t, 4 H, 3J = 6.4 Hz), 3.02 – 3.05 (t, 4H, 3J = 6 Hz), 4.62 – 4.63 (t, 4H, 3J = 3.6 Hz), 4.89 – 4.90 (t, 4 H, 3J = 2.4 Hz).
Synthesis of C14H16FeO2 (2a). In a 250 mL flask, zinc (6.0 g, 91.8 mmol), mercuric chloride (0.60 g, 2.21mmol), DI water (20 mL), and concentrated HCl (0.30 mL) were mixed for five minutes by hand, then five minutes with a stir bar. The suspension was allowed to settle, the supernatant was decanted, washed with deionized water, then transferred into a 500 mL flask along with concentrated HCl (8.8 mL), toluene (20 mL), and compound 1a (2.0 g, 7.00 mmol). The reaction was refluxed for 6 h. The reaction was cooled to room temperature and diluted with H2O (20 mL). The product was extracted in ethyl ether (3 × 10 mL), then the organic layer was washed with H2O (2 × 10 mL) and dried with anhydrous magnesium sulfate. The volatiles were removed in vacuo. The crude product was triturated with petroleum ether to give 2a (1.03 g, 54%). Melting Point: 115 – 116 °C (Lit. [14] 115 – 116 °C). 1H NMR (400 MHz, CDCl3, ppm): δ 1.80 – 1.87 (m, 4 H), 2.35 – 2.40 (m, 8 H), 3.98 – 4.09 (m, 8 H).
Synthesis of C18H22FeO4 (2b). In a 250 mL flask, zinc (12.0 g, 184 mmol), mercuric chloride (1.20 g, 4.42mmol), DI water (20 mL), and concentrated HCl (0.60 mL) were mixed for five minutes by hand, then five minutes with a stir bar. The suspension was allowed to settle, the supernatant was decanted, washed with DI water, then transferred into a 500 mL flask along with H2O (8 mL), concentrated HCl (17.6 mL), toluene (20 mL), and compound 1b (4.0 g, 10.4 mmol). The reaction was refluxed for 19 hours. The reaction was cooled to room temperature and diluted with H2O (20 mL). Unreacted zinc was separated by decantation and the residue was washed with diethyl ether (30 mL). The organic layer was collected, and the aqueous phase was extracted with diethyl ether (2 x 50 mL). The combined organic phase was dried with anhydrous magnesium sulfate. Volatiles were removed in vacuo. The final product was purified by trituration with petroleum ether to give 2b (3.36 g, 90%) as a yellow powder. Melting Point: 72 – 73 °C (Lit. [15] 73 °C). 1H NMR (400 mHz, CDCl3, ppm): δ 1.80 – 1.87 (m, 4H), 2.35 – 2.40 (m, 8H), 43.98 – 4.09 (m, 8H).13C{1H} NMR (100 MHz, CDCl3, ppm): δ 25.95, 28.58, 33.64, 67.89, 68.88, 77.32, 88.08, 179.97. IR (ATR, cm–1): 1703 (C=O), 3000 – 3200 (OH).
Synthesis of C14H14FeO (3a). In a 100 mL flask, trifluoroacetic anhydride (0.620 mL) was mixed with dichloromethane (33 mL). A mixture of compound 2a (1.00 g, 3.67 mmol) and dichloromethane (33 mL) was added dropwise to the flask via a dropping funnel and the reaction was allowed to stir for 8 hours at room temperature. A saturated solution of NaHCO3 (10 mL) was added and the mixture was layered in a separatory funnel. The organic layer was collected, dried with anhydrous MgSO4 and volatiles were removed in vacuo to give 3a (0.758 g, 81%). Melting Point: 82 – 83 °C (Lit. [16] 83 – 85 °C) 1H NMR (400 MHz, CDCl3, ppm): δ 2.02 – 2.22 (m, 2H), 2.25 – 2.45 (m, 2H), 2.59 – 2.67 (m, 2H), 4.17 (s, 5H, Cp), 4.45 – 4.47 (m, 2H, Cp), 4.81 – 4.82 (m, 1H, Cp). 13C{ NMR (100 MHz, acetone-d6, ppm): δ 23.4, 23.9, 38.8, 64.6, 70.0, 75.8, 92.5, 202.4. IR (ATR, cm−1): 1664 (C=O). The product was analyzed by single crystal X-ray analysis.
Synthesis of C18H18FeO2 (3b and 3b’). In a 100 mL flask, 8 (0.501 g, 1.40 mmol) was dissolved in dichloromethane (15 mL). Trifluoroacetic anhydride (0.778 mL, 5.59 mmol) in dichloromethane (15 mL) was added dropwise. The reaction was stirred for 20 hours at room temperature. Sodium bicarbonate solution (10%, 100 mL) was added to the mixture and layered with dichloromethane. The organic layer was collected, washed with water (2 × 20 mL), dried with magnesium sulfate, and filtered. The filtrate was evaporated to dryness to give 3b and 3b’ (0.190 g, 42%) as a bright red powder. The two diastereomers were separated by column chromatography in silica using a mixture of ethyl acetate and dichloromethane (2:1 by volume). The racemic mixture (dark red), 3b’ was followed by the meso (dark orange), 3b in the column. 3b (Meso): Melting Pont: 168 -189 °C (Lit. [17] 161 -167 °C. 1H NMR (400 mHz, CDCl3): δ 2.00 -2.11 (m, 4H, CH2), 2.15 – 2.40 (m, 6H, CH2), 2.56 – 2.66 (m 4H CH2), 4.42 – 4.45 (m, 2H, Cp), 4.73 (b, 2H, Cp). 13C{ NMR (100 MHz, CDCl3, ppm): δ 22.92, 23.66, 39.27, 67.38, 72.37, 73.19, 76.58, 93.42, 204.33. IR (ATR, cm−1): 1661 (C=O). The product was characterized by single crystal X-ray analysis. 3b’(Racemic): Melting Point: 156 - 157 °C (Lit. [17] 153 – 161 °C). 1H NMR (400 mHz, CDCl3): δ 2.01 – 2.15 (m, 2 H), 2.21 – 2.47 (m, 6H), 2.61 (dt, 2J = 15.6 Hz, 3J = 4.4 Hz, 4 H), 4.39 (d, 3J = 1.2 Hz, 2H), 4.42 – 4.43 (m, 4 H), 4.65 (t, 3J = 1.2 Hz, 2 H). 13C{ NMR (100 MHz, CDCl3, ppm): δ 23.32, 23.52, 39.20, 68.03, 70.84, 71.85, 77.32, 94.00, 203.40 IR (ATR, cm – 1): 1663 (C=O). The product was analyzed by single crystal X-ray analysis.
Synthesis of C14H16Fe (4a). In a 250 mL flask, zinc (5.0 g, 76.4 mmol), mercuric chloride (0.5 g, 1.84 mmol), and DI water (20 mL) were mixed for 10 min. The suspension was allowed to settle, the supernatant was decanted, and the residue was washed with DI water. To the amalgamated zinc, concentrated HCl (2.0 mL), toluene (20 mL), and compound 3 (200 mg, 0.787 mmol). The reaction was refluxed for 2 h and an additional amount of conc. HCl (2.0 mL) was added and continued reflux for another 3 h. The reaction was cooled to room temperature, the clear solution was decanted, and the unreacted zinc was washed with ethyl acetate (40 mL). The combined organic layer was washed with water (3 x 100 mL), dried with anhydrous MgSO4, and filtered. The volatiles were removed in vacuo to give 4a (166 mg, 87%) as a yellow solid. Melting point: 39 °C (Lit. [18] 39 – 41 °C) 1H NMR (400 MHz, Acetone-d6, ppm): δ 1.53 – 1.64 (m, 2H), 1.85 – 1.92 (m, 2H), 2.17 – 2.26 (m, 2H), 2.60 – 2.67 (m, 2H), 3.85 (t, 3J = 2.4 Hz, 1H, CHCHCH), 3.94 (d, 3J = 2.4 Hz, 2H, CHCHCH), 3.95 (s, 5H, Cp). 13C{1H}, acetone-d6, ppm): δ 23.5, 24.6, 64.8, 65.1, 69.4, 84.9.
Synthesis of C18H22Fe (4b). In a 100 mL flask, a mixture of zinc (5.0 g, 76.47 mmol), mercuric chloride (0.50 g, 1.84 mmol), and DI water (20 mL) was stirred for 10 min. The clear solution was decanted and the residue was washed with DI water (20 mL). To the activated Zn, the diastereomeric mixture of 3b and 3b’ (200 mg, 0.62 mmol), toluene (20 mL), and conc. HCl (2.0 mL) was added. The reaction was refluxed for 3 h and an additional amount of conc. HCl (4.0 mL) was added. The reaction was again refluxed for 2 h. The mixture was cooled to RT and the soluble product was decanted. The residue was washed with ethyl acetate (2 x 10 mL). The extract was washed with water (2 x 80 mL); the organic layer was collected, dried over anhydrous MgSO4, and filtered. Volatiles were removed in vacuo. The crude product was purified by silica gel column chromatography using petroleum ether as eluent to give 4b (107 mg, 59%) as yellow gum. 1H NMR (400 mHz, acetone-d6): δ 1.53 – 1.57 (m, 4H), 1.85 – 1.88 (m, 4H), 2.16 – 2.23 (m, 4H), 2.57 – 2.64 (m, 4H), 3.65 (t, 3J = 2.4 Hz, 2H, CHCHCH), 3.76 (d, 3J = 2.4 Hz, 4H, CHCHCH).13C{ NMR (100 MHz, acetone-d6, ppm): δ 23.6, 23.9, 67.5, 68.3, 84.2.
3. Results and discussion
3.1. Synthesis and Structural Elucidation of Compounds 1a, 1b – 4a, 4b
Synthesis of the target compounds (4a and 4b) was performed by following the reaction sequence as shown in Scheme 1. Our attempts to reproduce the procedures reported in the literature [19,20] resulted in the formation of a mixture of both mono-and bis(3-carboxypropenoyl)ferrocene in a ca. 1:1 ratio. To avoid the tedious separation of these two keto-carboxylic acids, we synthesized (3-carboxypropenoyl)ferrocene, 1a (38%) by slowly adding a suspension of succinic anhydride and aluminum chloride in dichloroethane to the solution of ferrocene in the same solvent. The procedure left some unreacted ferrocene, but the product was purified by solvent extraction using NaOH solution. Synthesis of bis(3-carboxypropenoyl)ferrocene, 1b (72%) was performed by adding ferrocene into the suspension of succinic anhydride and aluminum chloride and stirring the solution at room temperature for 48 hours. After the base extraction, the product was found to contain 1a as an impurity which was removed by washing it with hot water. The Clemmensen reduction of both keto carboxylic acids was performed by following the literature procedures [14,15] with slight modifications to give mono- and bis(3-carboxypropyl)ferrocene 2a (54%) and 2b (90%), respectively.
The ring closing of 2a and 2b was performed in the presence of trifluoroacetic anhydride [17,21] to give homoannularly cyclized products: 1,2(α-ketotetramethylene)ferrocene, 3a (81%) and 1,2,1’,2’-bis(α-ketotetramethylene)ferrocene, 3b (42%) respectively. Although Nesmeyanov[22] and Rinehart [3,17] independently reported the synthesis of 3a and 3b, their full characterization data were not available. The diketone 3b must exist as two geometric isomers (racemic and meso) but the authors were unable to assign the configurations of the diastereomers [17]. Based on 1H NMR analysis of the crude product, we observed the formation of these isomers in ca. 1:1 ratio. Assignment of the stereochemistry of these geometrical isomers (3b and 3b’) was performed by single crystal X-ray analysis (vide infra). The 1H NMR of 3a, 3b and 3b’ exhibited a distinctive ABC pattern of substituted cyclopentadienyl rings. Similarly, the diastereotopic nature of exo- and endo-protons of the methylene groups exhibited a complex coupling pattern giving a set of multiplets.
The keto groups of 3a, 3b, and 3b’ were reduced to methylene under Clemmensen conditions to give 1,2-(tetramethylene)-ferrocene 4a (87%) and 1,2,1’,2’-bis(tetramethylene)-ferrocene 4b (59%), respectively. The loss of carbonyl stretching in IR and the appearance of triplets (1H) and doublets (2H) on the substituted cyclopentadienyl rings in 1H NMR indicated the formation of desired products. Like ketones (3a, 3b, and 3b’), the diastereotopic nature of the exo- and endo-hydrogens of the methylene group gave a complicated coupling pattern. Compound 4b exhibited four sets of CH2 protons and two sets of cyclopentadienyl protons indicating the free rotation of two Cp rings with respect to the Fe(II) center. Moreover, both signals of cyclopentadienyl rings are shifted upfield for both 4a and 4b but the shift is more prominent for 4b (triplet at 3.65 and doublet at 3.75 ppm). Compounds 4a and 4b were previously synthesized by the catalytic hydration of (cyclopentadienyl)(indenyl)iron [18] or bis(indenyl)iron [23]. Synthesis of 4b was also reported from the condensation of spiro[4·4]nona-1,3-diene with iron[24] as well as the treatment of the lithium salt of 4,5,6,7-tetrahydroindene with FeCl2 [25].
3.2. X-ray Crystal Structures
The structures of ferrocene derivatives 3a, 3b, 3b’, and 4a were determined by X-ray crystallographic methods. All crystals were grown by slow evaporation of concentrated ethyl ether solution in air at room temperature. Thermal ellipsoid plots with numbering schemes are shown in Figure 1, Figure 2, Figure 3 and Figure 4. The crystal structure and refinement data for these compounds are given in Table 1. The crystal structure of 3a has been reported by Fleischer, et al. at room temperature [26]. We have redetermined its crystal structure at 90 K with a much higher precision level. The compound 3a exhibits planar chirality since two different substituents are connected to the cyclopentadienyl ring [27]. As the compound crystallizes in centrosymmetric space group (P−1), the two enantiomers Rp/Sp are present in equal numbers within the crystals. The iron atom in compound 3b lies on a crystallographic inversion center. Compound 3b’ crystallizes with Z’ = 2 in which one of the molecules has a disorder of O atom in two positions. The 3b’ crystallizes as a racemic mixture in a centrosymmetric space group P21/n. The two molecules have a roughly perpendicular orientation. Each molecule possesses an approximate crystallographic two-fold rotational axis (Figure 3). Compound 4a crystallizes in the monoclinic space group P21/c with one molecule per asymmetric unit.
In the molecules of 3a, 3b, and 3b’ the ferrocenyl moiety is fused with α-keto tetramethylene rings with one of both cyclopentadienyl rings while in the molecules of 4a, the ferrocenyl moiety is fused with tetramethylene ring. In all molecules, the angle between Fe and two Cp ring centroids is nearly 180° with a maximum deviation of 4.85° from linear geometry in 3b’. The Cp rings in the ferrocene system are almost parallel; as the dihedral angle between the planes of two Cp rings in 3a, 3b’, and 4a are 1.84°, 3.46° (average of two), and 1.92°, respectively. The Cp rings display nearly eclipsed conformation on 3a, 3b’and 4a, as demonstrated by C – Cg1 – Cg2 – C average torsion angles of 9.12°, 0.26°, and 6.12° respectively. The Cp rings in 3b are in perfectly staggered conformation (torsional angle 36.00°) as required by the inversion center located at Fe.
Within the ferrocenyl moieties, the mean Fe – C bond distances for unsubstituted Cp are unremarkable. However, the Fe–C distance for the ipso carbon-bearing keto group next to it is slightly shorter than the rest of the distances. For instance, Fe1 – C1 bond in compound 3a is 2.0397(9) Å while the average distance of Fe1 with C2 to C5 is 2.0505(7) Å. The slippage of the Fe center towards C1 has been observed in other α-keto ferrocenyl derivatives [28,29]. The slipping of Fe1 towards C1 presumably occurs to maximize the interaction of the Fe center with the exocyclic double bond in the resonance of such ketones [30]. The attached carbonyl group lies almost co-planar with the plane of the substituted cyclopentadienyl ring in 3a 3b and 3b’ as given by the torsional angle [C5-C1-C6-O1 = 1.52(11)°] in 3a. The C=O bond length in 3a 1.2265(9) Å, 3b 1.2230(9) Å, and 3b’ 1.225 (17) Å are in normal range of similar α-ferrocenyl ketones.[31,32] In 4a, the average of Fe1-C1 and Fe1-C2 distances is higher by 0.023(3) Å than the rest of Fe-C bonds of the substituted cyclopentadienyl ring. The distance of the Fe center from the centroid of cyclopentadienyl rings ranges from 1.647 Å to 1.660 Å.
The six-membered rings in all structures adopt half-chair conformations. The Cremer & People puckering parameters [33] of the six-membered rings, namely, 3a, 3b, 3b’ and 4a are QT = 0.4340(8) Å, θ = 131.43(11)° and φ = 2.47(14)°; QT =0.4227(8) Å, θ = 129.68(10)° and φ = 358.42(13)°; QT = 0.4356(12) Å, θ = 53.54(14)° and φ = 178.20(18)°; and QT = 0.521(3) Å, θ = 51.7(3)° and φ = 219.0(4)°, respectively. In 3a, atoms C6/C7/C9 lie on the same plane of the substituted cyclopentadienyl ring with a dihedral angle of 1.80°. The C8 projects inward from the main plain of Cp by 0.582 Å. Similar folding of six-membered rings can be seen in 3b and 3b’ as well. The two α-ketotetramethylene groups are oriented with dihedral angles of 180° and 72° in 3b and 3b’, respectively. The six-membered ring of 4a is more twisted than in 3a or 3b. In this molecule, C6 and C9 are almost coplanar with the substituted Cp [deviation: C6 = 0.018 Å, C9 = 0.033 Å]. The C7 is projected down by 0.498 Å and C8 is projected up by 0.305 Å from the Cp plane.
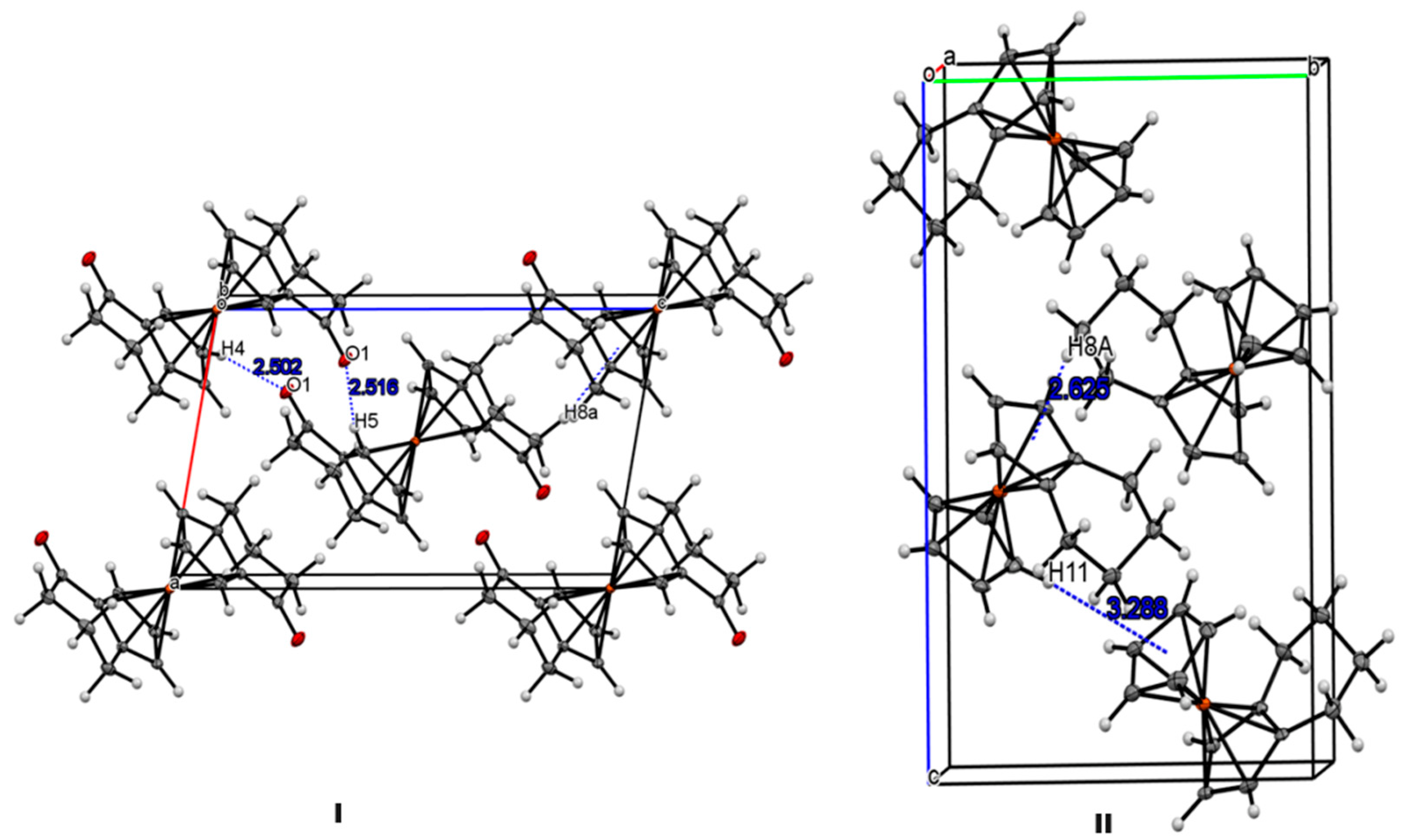
In their crystal structures, compounds 3a, 3b, and 3b’ display similar intermolecular interactions. In these molecules, the most prominent interactions are weak intermolecular C-H···O hydrogen bonds and C-H···π interactions. For example, molecules of 3b exhibit intermolecular C4-H4···O1, C-H5···O1 interactions along the crystallographic b-axis. Similarly, atom H8A is positioned almost perpendicular above the cyclopentadienyl ring centroid of the adjacent molecule (Figure 5 (I). In the crystal structure of 4a, there are C-H···π interactions between methylene hydrogen and the cyclopentadienyl rings (Figure 5 (II)).
Figure 5.
(I) Packing diagram of 3b along the b axis; (II) packing of 4a along a axis (II).
3.3. Electrochemical studies
To investigate the effects of tetramethylene substituents on the oxidation potential of the ferrocene moiety, we measured the half-wave redox potentials (E1/2) of 4a and 4b by cyclic voltammetry using 0.1 M tetrabutylammonium hexafluorophosphate in dichloromethane as a supporting electrolyte at a scan rate of 50 mV s−1 in 10−3 M concentration. All measurements were carried out at room temperature under a dry nitrogen atmosphere by the use of a three electrode system: glassy carbon as the working electrode, Ag/AgCl as the reference electrode, and platinum wire as a counter electrode.
Figure 5.
Cyclic voltammogram of Ferrocene (red), 4a (blue), and 4b (purple).
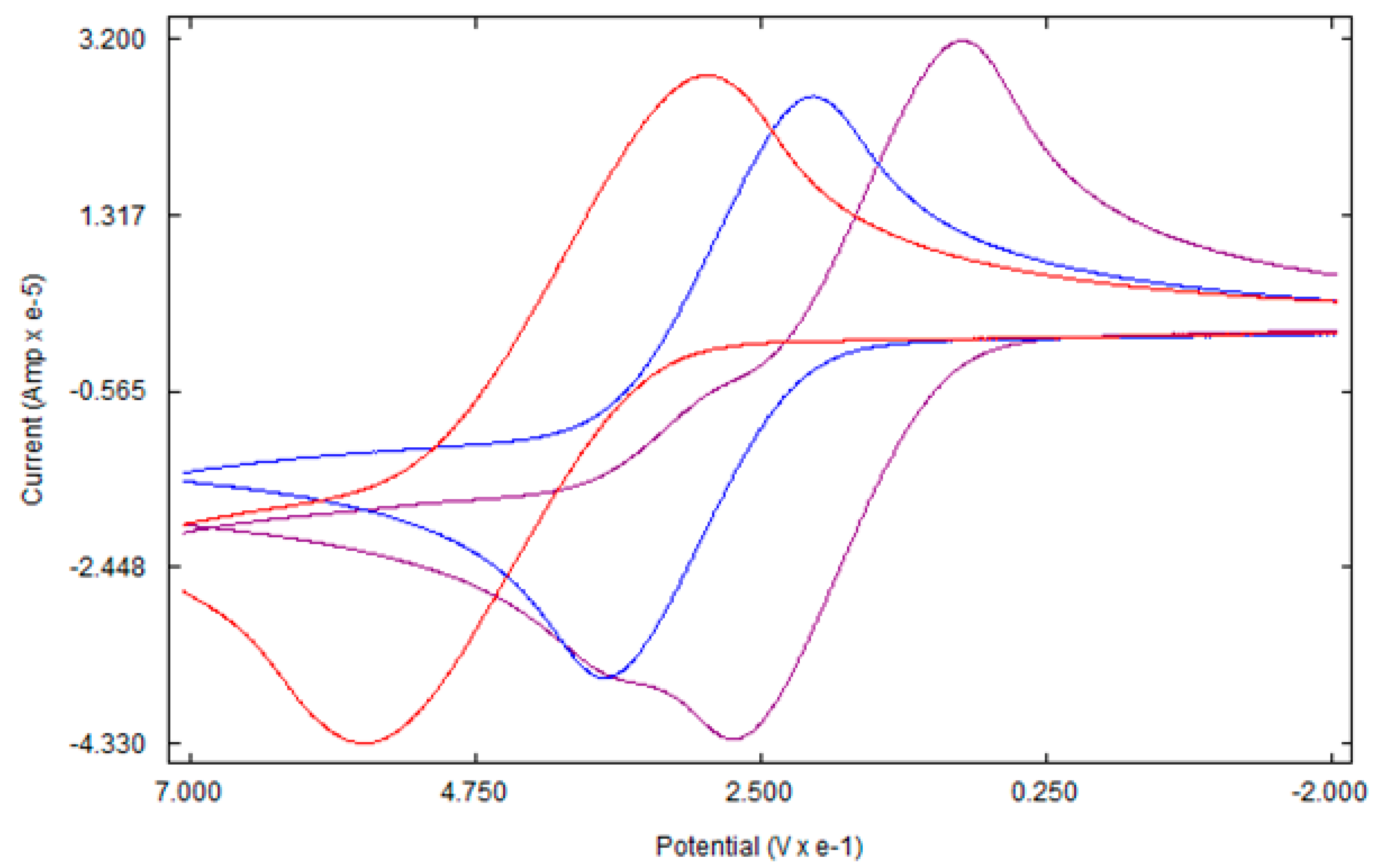
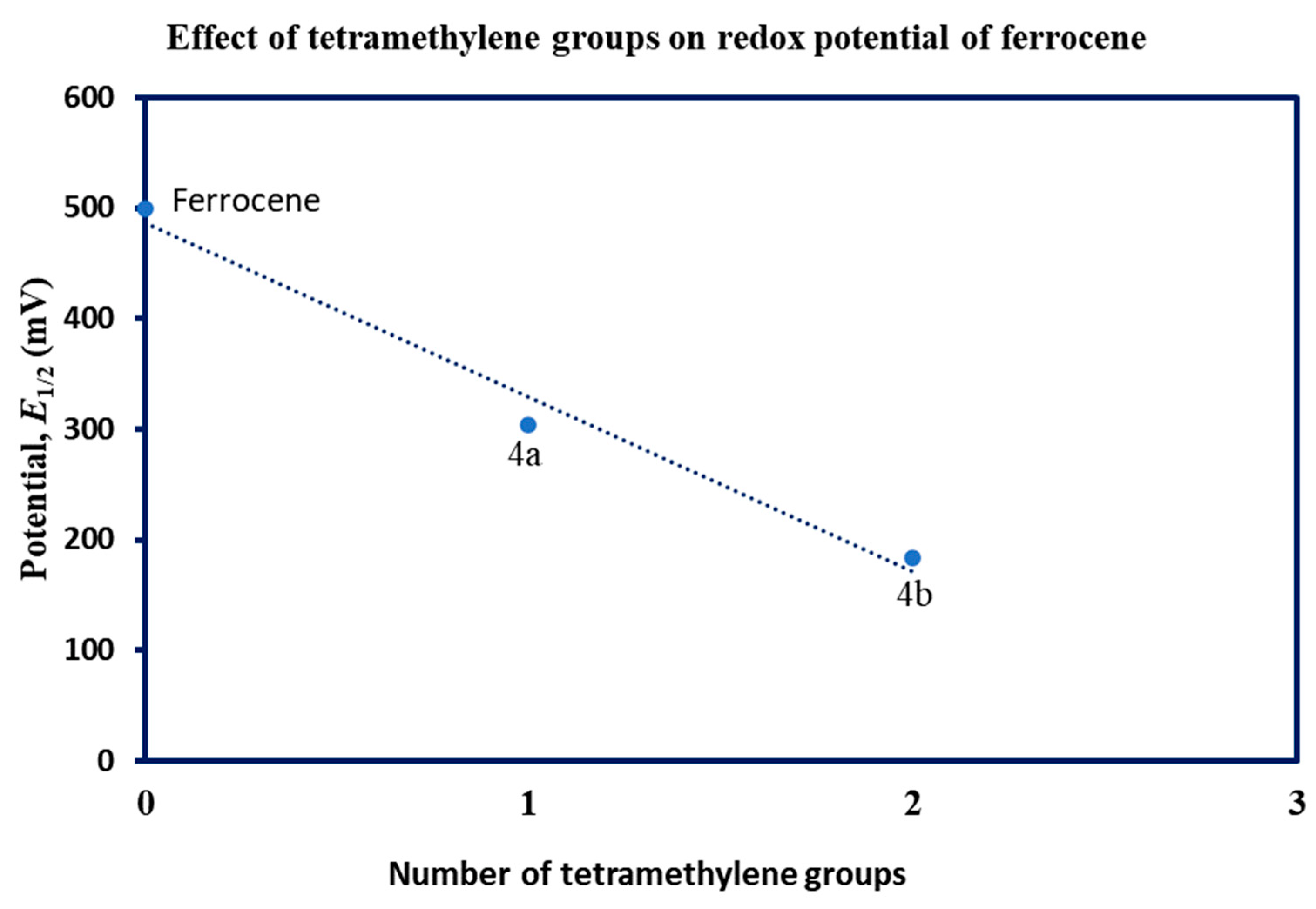
Like ferrocene, the cyclic voltammetry of 4a and 4b shows a single-electron reversible redox process (Figure 5). As expected, the redox potentials of both complexes were lower than that of ferrocene due to the electron-releasing ability of tetramethylene rings. The higher electron density at the iron center due to the tetramethylene ring causes the iron center to lose an electron more easily in comparison to ferrocene [34]. Figure 6 shows a plot of E1/2 vs. the number of tetramethylene groups. Under the experimental conditions, the E1/2 of ferrocene, 4a and 4b are 499 mV, 303 mV, and 183 mV, respectively versus Ag/Ag+. Although there is a sharp decrease in the potential, the relationship between the number of substituents and oxidation potential is not linear. The difference in potential between ferrocene and 4a is 196 mV, while the difference between 4a and 4b is just 120 mV. The number of tetramethylene bridges in 4b is double that of 4a. However, the expected decrease in potential is less than half. A slightly higher potential of 4b than its predicted value might be due to the steric bulk of additional tetramethylene rings around the iron center that render the interaction of the iron atom with the electrode difficult [35].
Figure 6.
Effects of substituents in the redox potential of ferrocene derivatives.
4. Conclusions
We have synthesized and characterized mono- and bis-tetramethyleneferrocenes as viable precursors of π-extended ferrocene derivatives. Four compounds of the reaction sequence have been studied with single-crystal X-ray analysis. The effects of tetramethylene groups on the half-wave oxidation potential have been studied by cyclic voltammetry. Our attempts to dehydrogenate the final products of this reaction sequence with 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) were unsuccessful. Currently, we are working on alternative methods to aromatize the ferrocene-bound aliphatic rings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H NMR of 3a; Figure S2: 13C NMR of 3a; Figure S3: 1H NMR of 3b; Figure S4: 13C NMR of 3b; Figure S5: 1H NMR of 3b’; Figure S6: 13C NMR of 3b’; Figure S7: 1H NMR of 4a; Figure S8: 13C NMR of 4a; Figure S9: 1H NMR of 4b.
Author Contributions
Conceptualization, U. P. synthesis, and spectroscopic characterization, D. D., S. N., G. E. X-ray crystallography, F. F. electrochemistry, M. L., U. P. writing – original draft preparation, U. P. writing – review and editing, U. P. and F. F. visualization, U. P. supervision, U. P. project administration, U. P. funding acquisition, U. P.
Funding
The work was funded by the Louisiana Board of Regents. Contract number: LEOSF (2017-18)-RD-A-28.
Data Availability Statement
Data are contained within the article and Supplementary materials.
Acknowledgments
The authors extend their appreciation to the Department of Chemistry and Physical Sciences, Nicholls State University for providing funds for purchasing chemicals, and the Department of Chemistry, Louisiana State University for providing X-ray crystallography services free of charge.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Astruc, D. Why is ferrocene so exceptional? European Journal of Inorganic Chemistry 2017, 2017, 6–29. [Google Scholar] [CrossRef]
- Kealy, T.J.; Pauson, P.L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039–1040. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Curby, R.J., Jr. Ferrocene bridging and homoannular cyclizations. Journal of the American Chemical Society 1957, 79, 3290–3291. [Google Scholar] [CrossRef]
- Schlögl, K. Stereochemistry of metallocenes. Topics in stereochemistry 1967, 39–91. [Google Scholar] [CrossRef]
- Pokharel, U.R. Organometallic heterocycles and acene-quinone complexes of ruthenium, iron and manganese. PhD dissertation, University of Kentucky, 2012.
- Pokharel, U.R.; Selegue, J.P.; Parkin, S. Ruthenocene 1,2-dicarboxylic acid, carboxylic anhydride, and acid chloride: A facile route to metallocene-fused acenequinones. Organometallics 2011, 30, 3254–3256. [Google Scholar] [CrossRef]
- Batterjee, S.M.; Marzouk, M.I.; Aazab, M.E.; El-Hashash, M.A. The electrochemistry of some ferrocene derivatives: redox potential and substituent effects. Applied Organometallic Chemistry 2003, 17, 291–297. [Google Scholar] [CrossRef]
- Emília, M.; Silva, N.P.R.A.; Pombeiro, A.J.L.; da Silva, J.J.R.F.; Herrmann, R.; Deus, N.; Castilho, T.J.; Silva, M.F.C.G. Redox potential and substituent effects at ferrocene derivatives. Estimates of Hammett σp and Taft polar σ substituent constants. Journal of Organometallic Chemistry 1991, 421, 75–90. [Google Scholar] [CrossRef]
- Hoh, G.L.K.; McEwen, W.E.; Kleinberg, J. Substituent Effects in the Chronopotentiometric Oxidation of Ferrocenes. Journal of the American Chemical Society 1961, 83, 3949–3953. [Google Scholar] [CrossRef]
- Hall, D.W.; Russell, C.D. Substituent effects in the chronopotentiometric oxidation of ferrocene derivatives. Internal solvation of certain substituted ferricenium ions. Journal of the American Chemical Society 1967, 89, 2316–2322. [Google Scholar] [CrossRef]
- N.P.R.A. Silva, M.E.; Pombeiro, A.J.L.; Fraústo da Silva, J.J.R.; Herrmann, R.; Deus, N.; E.Bozak, R. Redox potential and substituent effects in ferrocene derivatives: II. Journal of Organometallic Chemistry 1994, 480, 81-90. [CrossRef]
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallographica Section A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallographica Section C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Patwa, A.N.; Gupta, S.; Gonnade, R.G.; Kumar, V.A.; Bhadbhade, M.M.; Ganesh, K.N. Ferrocene-linked thymine/uracil conjugates: base pairing directed self-assembly and supramolecular packing. Journal of Organic Chemistry 2008, 73, 1508–1515. [Google Scholar] [CrossRef]
- Apreutesei, D.; Lisa, G.; Hurduc, N.; Scutaru, D. Synthesis and un-isotherm kinetic study of some ferrocene acids. Central European Journal of Chemistry 2004, 2, 553–562. [Google Scholar] [CrossRef]
- Huffman, J.; Rabb, D. Notes- The Preparation of 1,2-(α-Ketotetramethylene)ferrocene. The Journal of Organic Chemistry 1961, 26, 3588–3589. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Curby, R.J., Jr.; Gustafson, D.H.; Harrison, K.G.; Bozak, R.E.; Bublitz, D.E. Organic chemistry of ferrocene. V. Cyclization of ω-ferrocenylaliphatic acids. Journal of the American Chemical Society 1962, 84, 3263. [Google Scholar] [CrossRef]
- King, R.B.; Bisnette, M.B. π-Cyclopentadienyl-π-indenyliron. Angewandte Chemie International 1963, 75, 642. [Google Scholar] [CrossRef]
- Bernhard, Y.; Gilbert, J.; Bousquet, T.; Favrelle-Huret, A.; Zinck, P.; Pellegrini, S.; Pelinski, L. One-Pot Synthesis of 2,5-Disubstituted Furans through In Situ Formation of Allenes and Enolization Cascade. European Journal of Organic Chemistry 2019, 2019, 7870–7873. [Google Scholar] [CrossRef]
- Liu, G.; He, H.; Wang, J. Ferrocene redox controlled reversible immobilization of ruthenium carbene in ionic liquid: a versatile catalyst for ring-closing metathesis. Advanced Synthesis & Catalysis 2009, 351, 1610–1620. [Google Scholar] [CrossRef]
- Weißenbacher, M.; Sturm, T.; Kalchhauser, H.; Kratky, C.; Weissensteiner, W. Synthesis and characterization of novel aminophosphine ligands based on ferrocenodecaline backbones. Monatshefte für Chemie 2002, 133, 991–1009. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Vol'kenau, N.A.; Vil'chevskaya, V.D. Intramolecular acylation in the ferrocene series. Cyclization of γ-ferrocenyl substituted acids and oxo acids. Doklady Akademii Nauk SSSR 1958, 118, 512. [Google Scholar]
- Osiecki, J.H.; Hoffman, C.J.; Hollis, D.P. Organometallic compounds. Ruthenium and iron derivatives of indene. Journal of Organometallic Chemistry 1965, 3, 107. [Google Scholar] [CrossRef]
- Hanlan, A.J.L.; Ugolick, R.C.; Fulcher, J.G.; Togashi, S.; Bocarsly, A.B.; Gladysz, J.A. Chemistry via metal atom cocondensation: isomerization and complexation reactions of organocyclopropanes and spirocycles. Inorganic Chemistry 1980, 19, 1543–1551. [Google Scholar] [CrossRef]
- Austin, R.N.; Clark, T.J.; Dickson, T.E.; Killian, C.M.; Nile, T.A.; Schabacker, D.J.; McPhail, A.T. Synthesis and properties of novel substituted 4,5,6,7-tetrahydroindenes and selected metal complexes. Journal of Organometallic Chemistry 1995, 491, 11–18. [Google Scholar] [CrossRef]
- Fleischer, E.B.; Hawkinson, S.W. The structure of [alpha]-keto-1,5-tetramethyleneferrocene. Acta Crystallographica 1967, 22, 376–381. [Google Scholar] [CrossRef]
- Schlögl, K. Stereochemistry of Metallocenes. In Topics in Stereochemistry; 1967; pp. 39-91.
- Paramasivam, S.; Purushothaman, S.; Seshadri, P.R.; Raghunathan, R. (E)-1-Ferrocenyl-3-[2-(2-hydroxyethoxy)phenyl]prop-2-en-1-one. Acta Crystallographica Section E 2013, 69, m144. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, S.; He, Z.; Xu, L.; Huang, S. 2-Amino-4-(4-chlorophenyl)-6-ferrocenylpyridine-3-carbonitrile. Acta Crystallographica Section E 2008, 64, m730. [Google Scholar] [CrossRef]
- Bratych, N.; Hassall, K.; White, J. Redetermination of the structure of diferrocenyl ketone at low temperature. Acta Crystallographica Section E 2003, 59, m33–m35. [Google Scholar] [CrossRef]
- Erben, M.; Ruzicka, A.; Vinklarek, J.; Stava, V.; Handlir, K. 1'-Acetylferrocene-1-carbonitrile. Acta Crystallographica Section E 2007, 63, m2145–m2146. [Google Scholar] [CrossRef]
- Erben, M.; Vinklarek, J.; Ruzicka, A. Acetylferrocene-2-chloro-1-ferrocenylethanone (1/1). Acta Crystallographica Section E 2011, 67, m1447–m1448. [Google Scholar] [CrossRef]
- Cremer, D.t.; Pople, J. General definition of ring puckering coordinates. Journal of the American Chemical Society 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Okuda, J.; Albach, R.W.; Herdtweck, E.; Wagner, F.E. Substituent effects in multiply trimethylsilyl-substituted ferrocenes. Molecular structure of 1,1′,2,2′,4,4′-hexakis(trimethylsilyl)ferrocenium tetrafluoroborate. Polyhedron 1991, 10, 1741–1748. [Google Scholar] [CrossRef]
- Tateaki, O.; Kazuo, O.; Tadashi, F.; Shunsuke, M.; Taeko, I.; Akira, K.; Nobuyuki, T. Electrochemical properties of ferrocenophanes. I. Voltammetric studies on the oxidation of mono-, di-, and tri-bridged ferrocenophanes in acetonitrile. Bulletin of the Chemical Society of Japan 1981, 54, 3723–3726. [Google Scholar] [CrossRef]
Scheme 1.
Scheme 1. Synthesis of homoannular ferrocene derivatives.
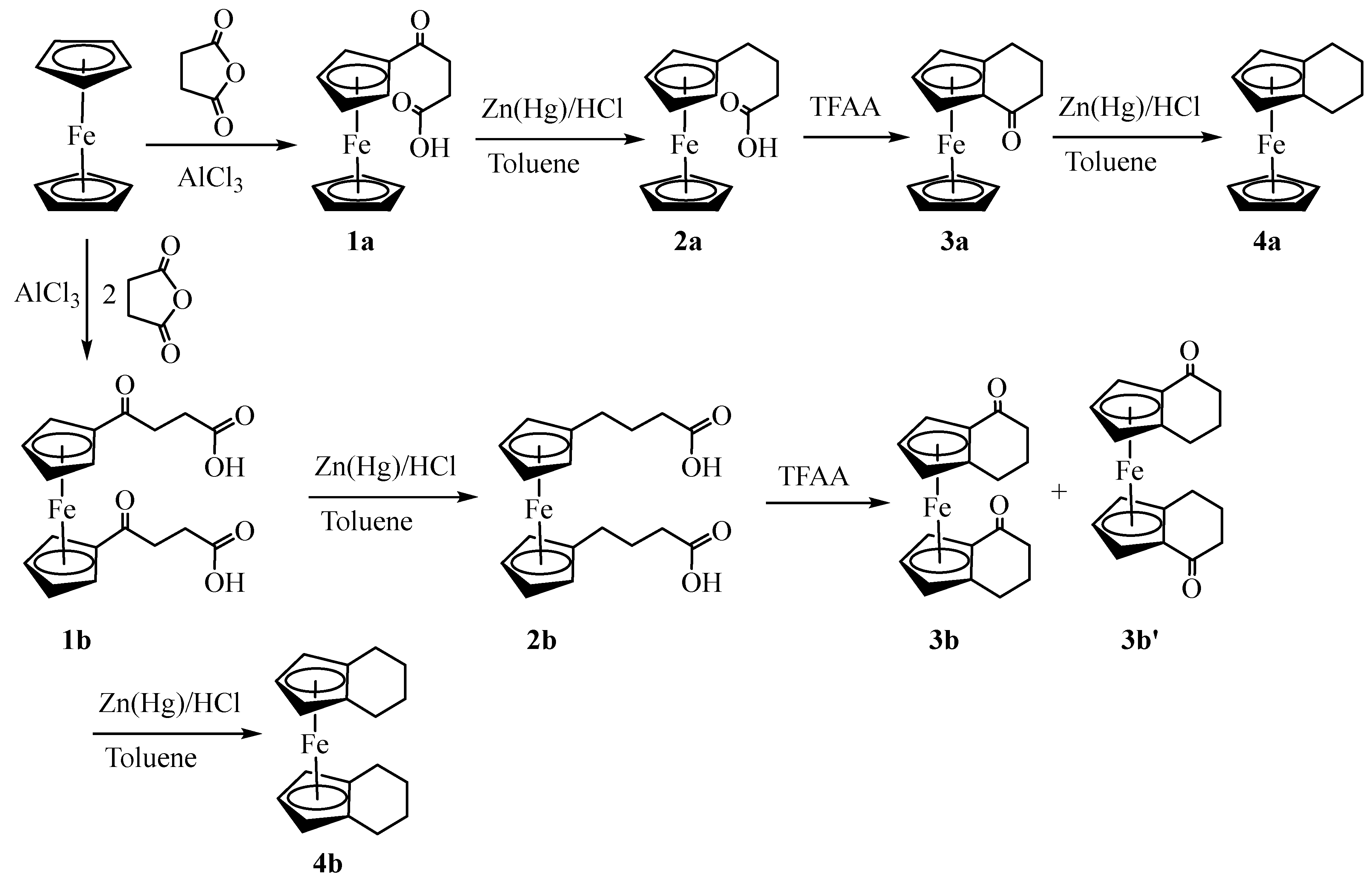
Figure 1.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 3a. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0397(9), Fe1–C2 2.0508(8), Fe1–C3 2.0526(6), Fe1–C4 2.0527(7), Fe1–C5 2.0456(9), Fe1–C10 2.0553(7), Fe1–C11 2.0530(8), Fe1–C12 2.045(1), Fe1–C13 2.0548(9), Fe1–C14 2.0554(6), C1–C6 1.465(1), C6–O1 1.2265(9), Cp (centroid, substituted) – Fe 1.647, Cp (centroid, unsubstituted) 1.655.
Figure 1.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 3a. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0397(9), Fe1–C2 2.0508(8), Fe1–C3 2.0526(6), Fe1–C4 2.0527(7), Fe1–C5 2.0456(9), Fe1–C10 2.0553(7), Fe1–C11 2.0530(8), Fe1–C12 2.045(1), Fe1–C13 2.0548(9), Fe1–C14 2.0554(6), C1–C6 1.465(1), C6–O1 1.2265(9), Cp (centroid, substituted) – Fe 1.647, Cp (centroid, unsubstituted) 1.655.
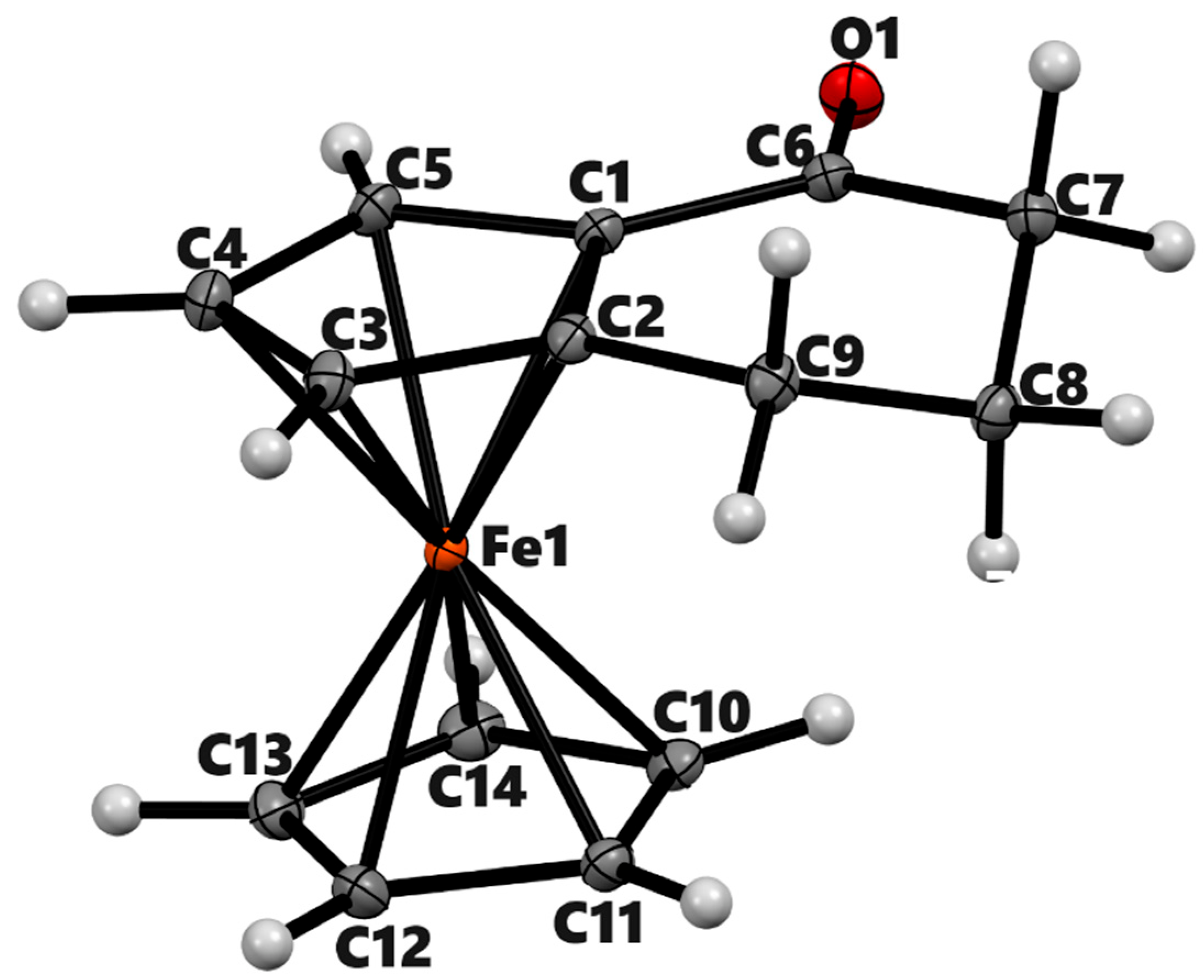
Figure 2.
ORTEP diagram of the solid-state structure showing the atom-numbering scheme of compound 3b (meso). Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0397(8), Fe1–C2 2.0627(7), Fe1–C3 2.0605(8), Fe1–C4 2.0664(8), Fe1–C5 2.0511(7), O1–C9 1.2230(9), Cp (centroid)–Fe 1.660.
Figure 2.
ORTEP diagram of the solid-state structure showing the atom-numbering scheme of compound 3b (meso). Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0397(8), Fe1–C2 2.0627(7), Fe1–C3 2.0605(8), Fe1–C4 2.0664(8), Fe1–C5 2.0511(7), O1–C9 1.2230(9), Cp (centroid)–Fe 1.660.
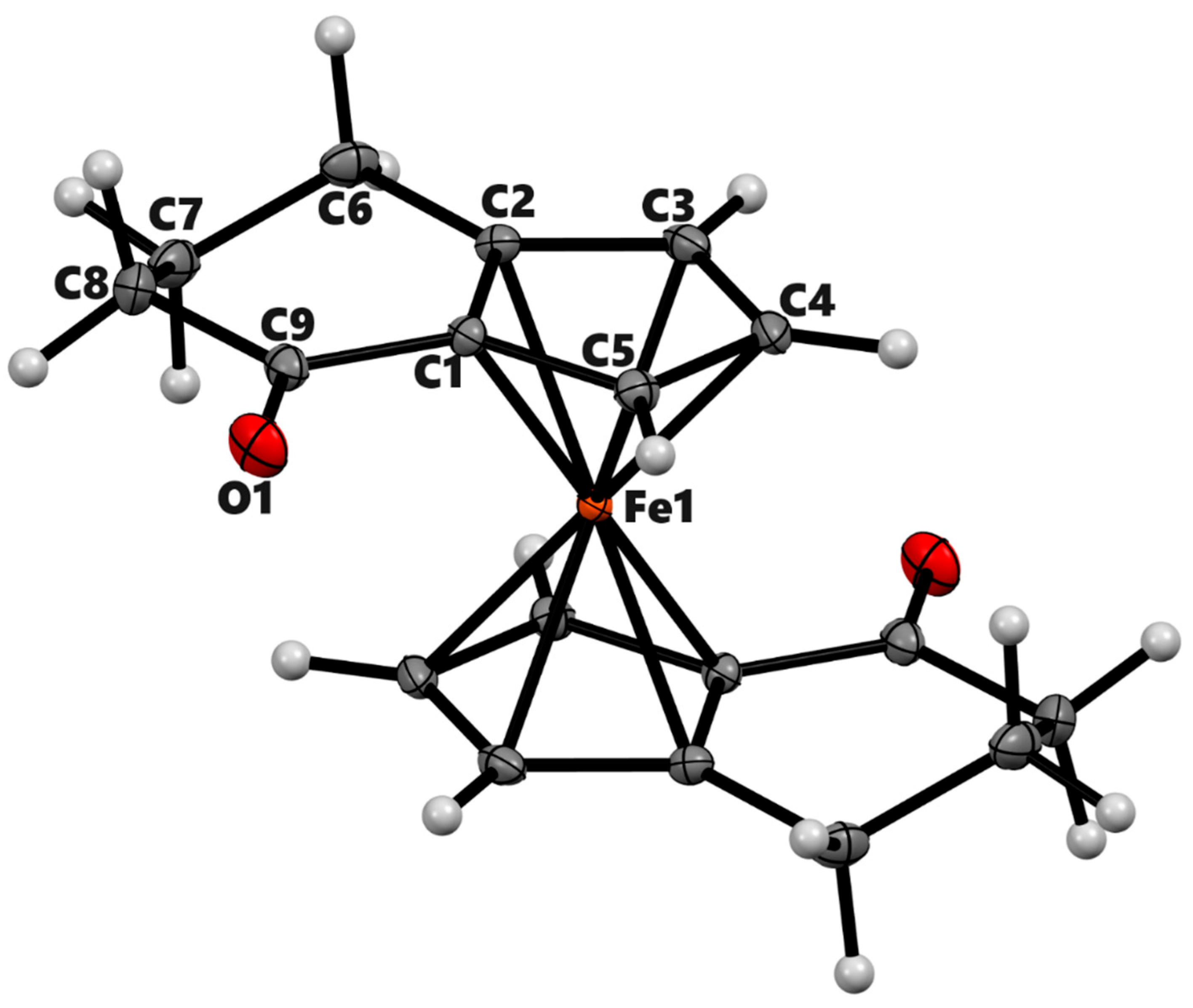
Figure 3.
ORTEP diagram of the solid-state structure showing the atom-numbering scheme of compound 3b’. Thermal ellipsoids are drawn at the 50% probability level. The minor component of disordered O-atom is not shown. Selected bond lengths (Å) for the complexes: Fe1–C1 2.0409(7), Fe1–C2 2.0575(7), Fe1–C3 2.0681(7), Fe1–C4 2.0534(7), Fe1–C5 2.0442(7), Fe1–C10 2.0469(6), Fe1–C11 2.0688(6), Fe1–C12 2.0615(7), Fe1–C13 2.0566(7), Fe1–C4 2.0534(7), Fe1–C4 2.0534(7), O2–C9 1.2212(17), Average Cp(Centroid)–Fe 1.652.
Figure 3.
ORTEP diagram of the solid-state structure showing the atom-numbering scheme of compound 3b’. Thermal ellipsoids are drawn at the 50% probability level. The minor component of disordered O-atom is not shown. Selected bond lengths (Å) for the complexes: Fe1–C1 2.0409(7), Fe1–C2 2.0575(7), Fe1–C3 2.0681(7), Fe1–C4 2.0534(7), Fe1–C5 2.0442(7), Fe1–C10 2.0469(6), Fe1–C11 2.0688(6), Fe1–C12 2.0615(7), Fe1–C13 2.0566(7), Fe1–C4 2.0534(7), Fe1–C4 2.0534(7), O2–C9 1.2212(17), Average Cp(Centroid)–Fe 1.652.
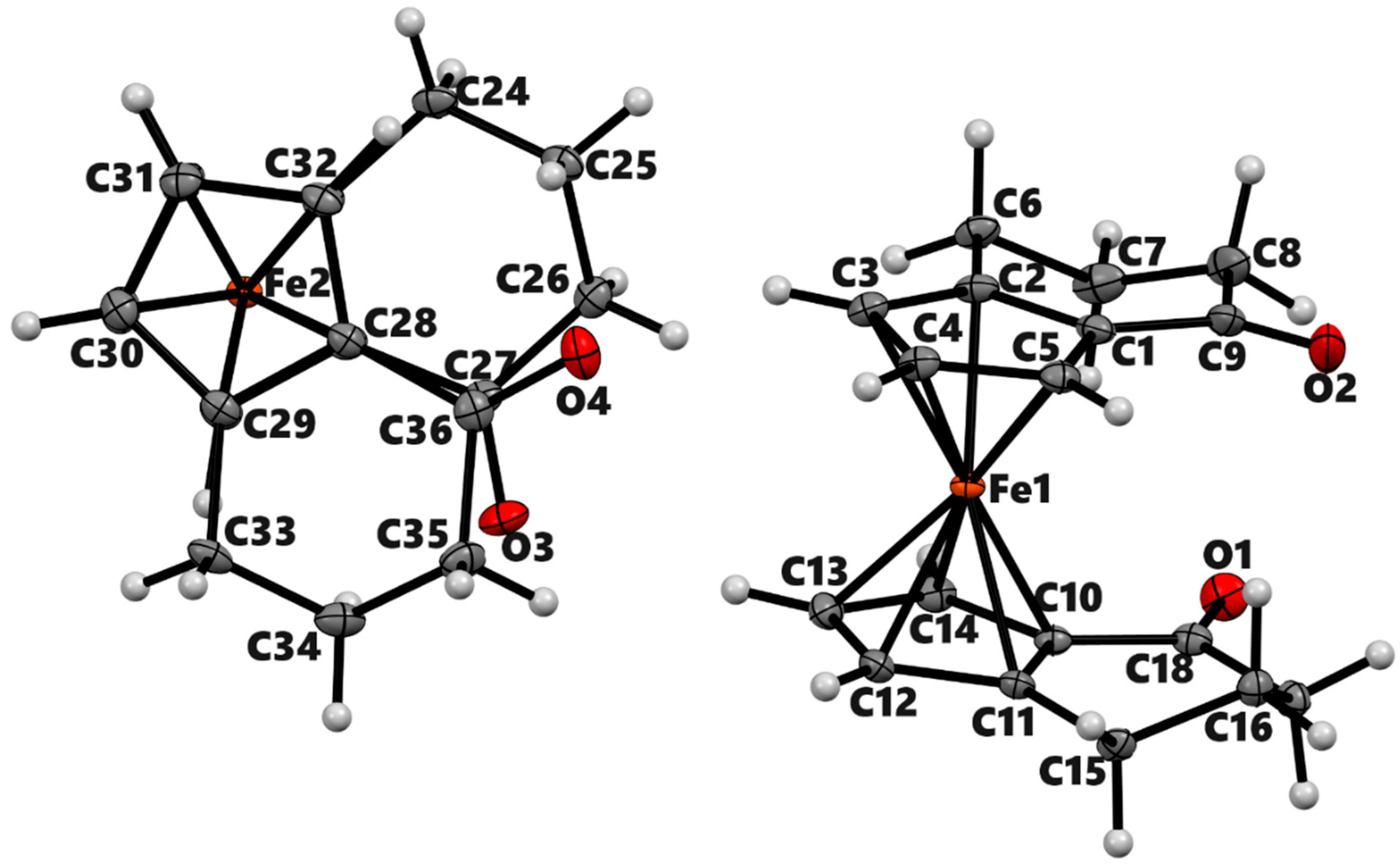
Figure 4.
ORTEP diagram of solid-state structure showing atom-numbering scheme of compound 4a. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complexes: Fe1–C1 2.049(3), Fe1–C2 2.064(3), Fe1–C3 2.040(3), Fe1–C4 2.034(2), Fe1–C5 2.031(3), Fe1–C10 2.043(3), Fe1–C11 2.036(3), Fe1–C12 2.045(3), Fe1–C13 2.051(3), Fe1–C14 2.042(3), Cp (centroid, substituted) – Fe 1.647, Cp (centroid, unsubstituted) 1.648.
Figure 4.
ORTEP diagram of solid-state structure showing atom-numbering scheme of compound 4a. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complexes: Fe1–C1 2.049(3), Fe1–C2 2.064(3), Fe1–C3 2.040(3), Fe1–C4 2.034(2), Fe1–C5 2.031(3), Fe1–C10 2.043(3), Fe1–C11 2.036(3), Fe1–C12 2.045(3), Fe1–C13 2.051(3), Fe1–C14 2.042(3), Cp (centroid, substituted) – Fe 1.647, Cp (centroid, unsubstituted) 1.648.
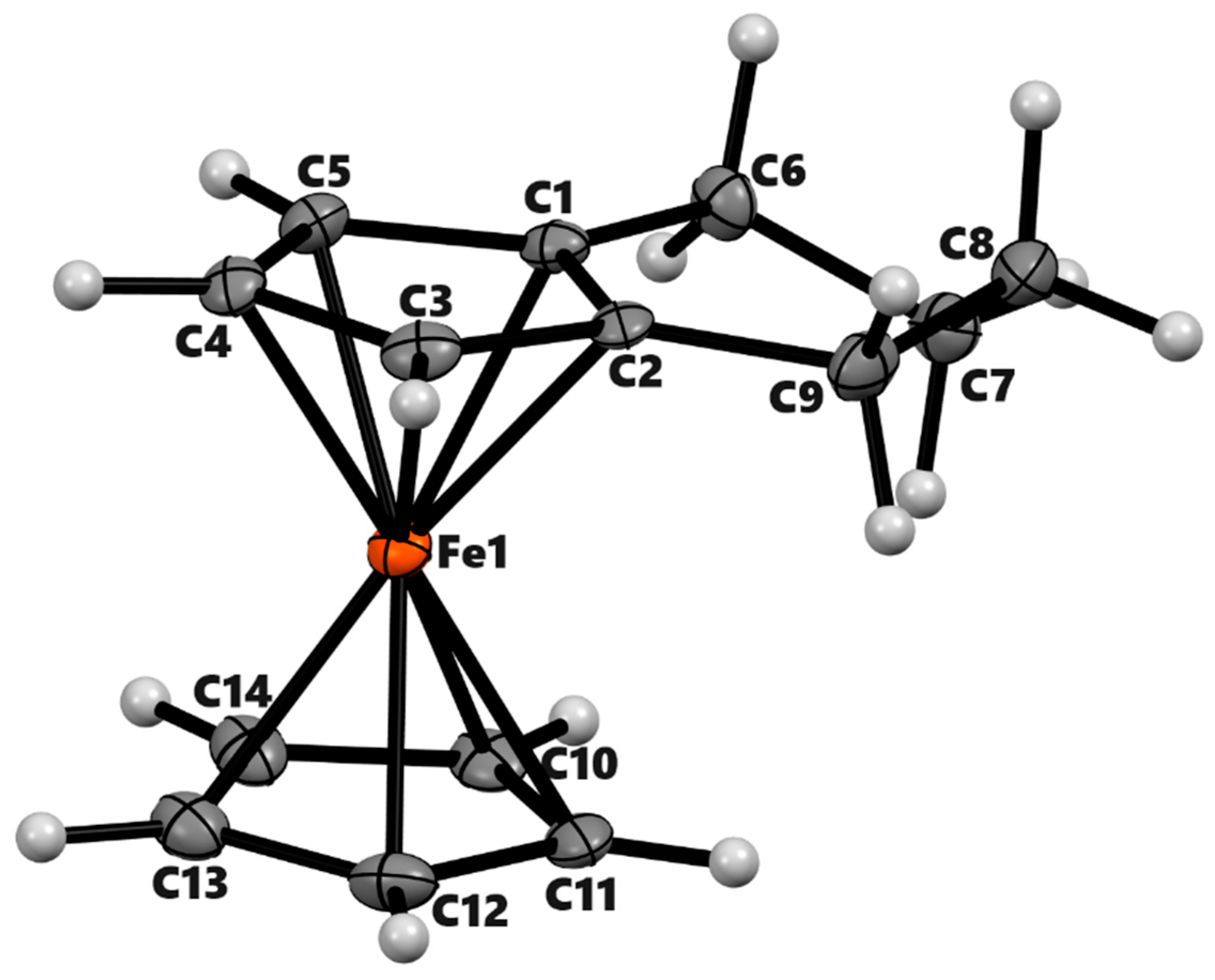

Table 1.
Crystal data and refinement.
| 3a | 3b (meso) | 3b'(racemic) | 4a | ||
| Chemical formula | C14H14FeO | C18H18FeO2 | C18H18FeO2 | C14H16Fe | |
| Mr | 254.1 | 322.17 | 322.17 | 240.12 | |
| Deposition No. | CCDC 2322201 | CCDC 2322202 | CCDC 2322203 | CCDC 2322204 | |
| Crystal system, space group | Triclinic, P-1 | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/c | |
| Temperature (K) | 90 | 90 | 90 | 90 | |
| a, b, c (Å) | 6.5983 (4), 7.7105 (4), 11.8843 (7) | 7.422 (2), 7.551 (2), 12.366 (4) | 13.6414 (6), 14.7516 (6), 13.8763 (6) | 7.661 (3), 9.506 (4), 14.642 (6) | |
| α, β, γ (°) | 108.140 (3), 90.461 (3), 108.897 (3) | 90, 100.397 (14), 90 | 90, 99.432 (2), 90 | 90, 95.574 (6), 90 | |
| V (Å3) | 539.61 (6) | 681.6 (3) | 2754.6 (2) | 1061.4 (7) | |
| Z | 2 | 2 | 8 | 4 | |
| Radiation type | Mo Kα | Mo Kα | Mo Kα | Mo Kα | |
| µ (mm−1) | 1.37 | 1.11 | 1.10 | 1.38 | |
| Crystal size (mm) | 0.16 × 0.15 × 0.06 | 0.36 × 0.20 × 0.11 | 0.44 × 0.39 × 0.36 | 0.15 × 0.11 × 0.06 | |
| Diffractometer | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II DUO | |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan | |
| Tmin, Tmax | 0.863, 0.922 | 0.750, 0.888 | 0.657, 0.749 | 0.753, 0.922 | |
| No. of measured, independent and | 22162, 9304, 7739 | 22653, 5593, 4567 | 74998, 21250, 16339 | 16912, 3242, 1949 | |
| observed [I > 2σ(I)] reflections | |||||
| Rint | 0.025 | 0.026 | 0.028 | 0.139 | |
| (sin θ/λ)max (Å−1) | 1.042 | 1.018 | 0.974 | 0.716 | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.034, 0.081, 1.04 | 0.030, 0.084, 1.07 | 0.033, 0.090, 1.05 | 0.052, 0.100, 1.00 | |
| No. of reflections | 9304 | 5593 | 21250 | 3242 | |
| No. of parameters | 187 | 97 | 400 | 136 | |
| No. of restraints | 0 | 0 | 9 | 0 | |
| H-atom treatment | Only H-atom coordinates refined | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained | |
| Δρmax, Δρmin (e Å−3) | 1.38, −0.75 | 1.05, −0.77 | 1.47, −0.53 | 0.66, −0.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



