Submitted:
11 January 2024
Posted:
12 January 2024
You are already at the latest version
Abstract
Cardiovascular disease (CVD), a group of disorders affecting the heart or blood vessels, are the primary cause of death worldwide, with an immense impact on patient quality of life and disa-bility. According to the World Health Organization, CVD takes an estimated 17.9 million lives each year, where more than four out of five CVD deaths are due to heart attacks and strokes. In the decades to come, increased prevalence of age-related CVD, such as atherosclerosis, coronary artery stenosis, myocardial infarction (MI), valvular heart disease, and heart failure (HF) will contribute to even greater health and economic burden as the global average life expectancy increases and consequently the world’s population continues to age. Considering this, it is important to focus our research efforts on understanding the fundamental mechanisms underlying CVD. In this review, we focus on cellular senescence and mitochondrial dysfunction, which have long been established to contribute to CVD. We also assess the recent advances in targeting mitochondrial dysfunction and senescence with a focus on therapies that influence both and therefore perhaps represent strategies with the most clinical potential, range, and utility.
Keywords:
mitochondrial dysfunction
; senescence
; cardiac
; cardiomyocyte
; cardiac ischemia reperfusion
1. Cellular Senescence
Cellular senescence was originally defined as the irreversible exit from the cell cycle [1]. However, over recent years the definition of senescence has evolved and now includes many other characteristics such, mitochondrial dysfunction, resistance to apoptosis and activation of a hypersecretory phenotype termed the senescence-associated secretory phenotype (SASP) [2]. These characteristics arguably more accurately define senescence given the substantive evidence that non-proliferative, post-mitotic cells, such as cardiomyocytes, can become senescent [3,4,5]. Similarly, while originally described as a consequence of telomere attrition following extensive proliferation [6], it is now accepted that numerous types of stresses which result in DNA damage within the genome or within the telomeres can activate pathways controlling senescence [7]. However, in both proliferative and post-mitotic cell populations, senescence induction is associated with the activation of either or both p21 and p16, cyclin-dependent kinase inhibitors that are components of the tumour suppressor pathways governed by the transcription factor p53 and the retinoblastoma protein (RB), respectively [8]. The traditional view that cellular senescence evolved as a tumour-suppressive mechanism has been recently challenged, as evidence suggests that senescent cells contribute to several important physiological processes throughout life including tissue development, wound healing, and tissue repair [9]. In addition to these beneficial roles, cellular senescence has also been shown to be crucial in tissue pathophysiology, representing a key driver of ageing and age-related diseases [10,11,12]. Therefore, cellular senescence can be viewed as an example of antagonistic pleiotropy, that is, a cellular program which is beneficial in one setting but deleterious in another [2].
Within the cardiovascular system, models of induced and attenuated senescence have implicated senescence in the pathophysiology of myocardial remodelling (age-related, chemotherapy-induced, and post-injury), hypertension, atherosclerosis, and the development of aortic aneurysms [13,14], and have been extensively reviewed [13,14,15,16,17,18].
2. Mitochondrial Abnormalities in Cardiovascular Diseases
Mitochondria are double-membraned organelles with their own circular genome, mitochondrial DNA (mtDNA), which is replicated independently of the host genome [19]. Mitochondria are involved in diverse yet interconnected functions, including the production of adenosyl triphosphate (ATP), and the regulation of nutritional metabolism, calcium homeostasis and programmed cell death [20,21,22]. They are found in the cytoplasm of nearly all eukaryotic cells as highly dynamic networks, undergoing coordinated cycles of biogenesis, fusion, fission, and degradation (mitophagy) to sustain their homeostasis and to adapt energy production based on the cell’s needs [23]. Proper mitochondrial function and dynamics are particularly necessary in tissues and cells with high energy demand such as the heart, and particularly in cardiomyocytes, which continuously require ATP to sustain cardiac activity. In adult cardiomyocytes, mitochondria occupy nearly one-third of the total intracellular volume [24] and provide approximately 95% of the ATP consumed by the heart [25]. It is therefore unsurprising that functional abnormalities of cardiac mitochondria have emerged as a key factor in cardiovascular disease (CVD) leading to decreased ATP production and energy supply, increased reactive oxygen species (ROS) production, cell apoptosis, and mitochondrial dynamic imbalance [26].
2.1. Energy Starvation and Oxidative Stress
Decreased energy supply is considered a leading consequence of mitochondrial dysfunction. The heart’s voracious requirement for energy, in the form of ATP, mainly relies on oxidative phosphorylation (OXPHOS) or β-oxidation of fatty acids and the tricarboxylic acid (TCA) cycle in the mitochondria. During pathological myocardial remodelling, there is a reduction in the levels of carnitine in the heart [27,28], an essential cofactor for mediating the entry of fatty acids into the mitochondria to the site of β-oxidation [29]. Due to this reduction of fatty acids availability within the mitochondria, cardiac metabolism is reprogrammed towards increased reliance on glucose as the energy resource with a significant increase of glycolysis, to maintain ATP production [24]. However, ATP generated from glycolysis contributes less than 5% of the total ATP consumed [28], which is not enough to compensate the reduction of fatty acid oxidation, and therefore, cardiac ATP is progressively depleted. The role of energy deprivation in the induction and pathogenesis of heart failure (HF) is well supported by clinical evidence, in which therapeutic measures to reduce energy consumption have been demonstrated to improve survival while treatment increasing energy demand is detrimental [24]. In the mitochondria, the synthesis of ATP takes place in the electron transport chain (ETC) [30]. Reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2) generated from the Krebs cycle, and from β-oxidation, transfer protons and electrons through the ETC, creating an electrochemical gradient that is then used to activate ATP-synthase and produces ATP. Alterations in mtDNA genes such as NADH-dehydrogenase genes (MT-ND1, MT-ND5 and MT-ND6), cytochrome b (MT-CYB), cytochrome c oxidase I and II (MT-CO1 and MT-CO2) and ATP synthase 6 (MT-ATP6), have been described in dilated cardiomyopathies [31]. Reduced activities of complexes I and IV, as well as of the NADH phosphate (NADPH)-transhydrogenase and the Krebs cycle enzymes have been also observed in patients with HF [26]. Interestingly, some studies suggest that mtDNA mutations induce cardiovascular senescence and CVD, as demonstrated by the observation that Polgm/m mice, which are prone to accumulation of mitochondrial DNA mutations, have increased expression of senescent markers p16ink4a and display early onset cardiomyopathy [32,33]. It remains unclear what mechanism mediates this induction, but an increase in mitochondrial ROS (mtROS) has been proposed as causal.
Mitochondrial ATP production is accompanied by the generation of ROS, a generic term for an array of short-lived and unstable free radicals that contain oxygen with vastly different properties and biological functions that range from signalling (when strictly regulated) to causing cell damage [34]. Physiologically, ROS-mediated signalling pathways are associated with cell survival and proliferation, combatting infectious agents, and have mitogenic effects on cells [35,36]. However, excessive ROS production drives oxidative stress, a deleterious process that potentially causes irreversible damage to various molecules and structures within the cell [37], leading to further mitochondrial dysfunction, oxidative stress, and cell death [21,38]. Increased ROS appears to be capable of inducing senescence through several mechanisms. Telomeres are particularly sensitive to ROS-induced damage, possibly due to their guanine-rich regions, which increase susceptibility to oxidation [39] and increased ROS can accelerate telomere attrition contributing to telomere dysfunction, premature senescence, and accelerated ageing [40]. ROS also generates DNA lesions in the form of single-stranded DNA and/or double-stranded breaks (DSBs) within the genomic or telomeric DNA. Eventually, as a result of telomere shortening or DNA damage, activation of the DNA damage response (DDR) occurs [41]. The DDR is an evolutionarily conserved signal transduction pathway required for genome integrity preservation. It coordinates cellular efforts to repair DNA damage, which if unsuccessful directs cell fate towards apoptosis or senescence thereby impeding the propagation of corrupted genetic information [42,43]. The DDR is characterised by the recruitment and activation of two large protein sensor kinases at the site of the lesion: ataxia telangiectasia and Rad3-related (ATR) when single-stranded DNA is exposed, and ataxia-telangiectasia mutated (ATM) at DSBs. The recruitment of ATR or ATM to the lesion causes the local formation of DNA damage foci containing the phosphorylated form of the histone H2AX (γH2AX) and ultimately induces cell-cycle arrest through the activation of checkpoint proteins, including p53 [41]. Furthermore, once senescent, cells exhibit a decreased mitochondrial membrane potential, increased proton leak and enhanced production of mtROS [44]. As such, the elevated ROS observed in senescent cells may drive mtDNA damage creating a positive feedback loop leading to further increases in ROS and DNA damage highlighting the cyclical interactions between mitochondrial dysfunction, oxidative stress, and senescence, and illustrates how the initiation of any of these processes could lead to a downward spiral in tissue function.
Perhaps unsurprisingly, given the high-volume density of mitochondria required to fulfil the heart’s energy demand, the heart has both high mtROS production and elevated mtROS, which has been shown to contribute to the pathophysiology of a variety of CVDs, including atherosclerosis, cardiac ischemia/reperfusion (IR) injury, HF, cardiac hypertrophy, and degenerative aortic valve disease [21,25,38,45]. ROS has also been shown to be a powerful inducer of senescence in multiple tissues and cell types, including the heart. Monoamine oxidase A (MAO-A) is a protein linked with driving oxidative stress - it is located at the outer mitochondrial membrane, involved in catalysing the oxidative deamination of monoamines, and produces hydrogen peroxide as one of its by-products [46]. Interestingly, cardiomyocyte-specific overexpression of MAO-A results in elevated ROS, increased senescence and mice display a dilated cardiomyopathy and myocardial dysfunction [47]. All of these can be rescued by treatment with antioxidants [4]. Similarly, the accelerated ageing mouse model nfkb1−/− showed increased ROS, telomere dysfunction and cardiomyocyte hypertrophy [48,49]. Similar observations have been reported in more clinically relevant models: aged mice treated with the mitochondrial-targeted peptide SS-31 elamipretide had reduced myocardial ROS and improved cardiac function, which was associated with reduced senescence [50]. Furthermore, aged, senescent cardiomyocytes demonstrated an overall decline in expression of most mitochondrial genes—particularly those genes involved in the ETC, and mitochondrial ultrastructural defects by transmission electron microscopy [50]. Myocardial infarction (MI) results in alterations to mitochondrial dynamics, culminating in increased ROS production and increased oxidative stress - particularly when followed by the clinical gold-standard treatment of reperfusion [51]. Several studies have shown that even in this acute setting of increased oxidative stress, senescence is induced in multiple cell populations, including cardiomyocytes, and that these cells are active participants in post-MI myocardial remodelling since their elimination attenuates, inflammation remodelling and improves functional outcomes [52,53].
Being so closely associated with cardiomyocyte dysfunction, mitochondrial damage is of major interest when exploring the mechanisms underpinning the cardiotoxicity of many otherwise beneficial therapeutics. This is relevant to both preclinical drug development, where cardiac liabilities remain a leading cause of drug attrition [54,55], but also to therapies approved for clinical use today which risk future withdrawal from the market due to cardiac adverse drug reactions (ADRs) [56]. Both traditional and new-generation oncology treatments are plagued by off-target cardiotoxic effects [57]. With cardiovascular disease being a leading noncancer cause of death in an ever-growing population of cancer survivors, understanding the mechanisms behind these cardiotoxicities is increasingly important [58]. As a case in point, anthracycline chemotherapies are notoriously chronically cardiotoxic and have been shown to deleteriously affect mitochondrial function in many ways.
Doxorubicin (DOX), an anthracycline commonly used in the clinic, was historically shown to redox cycle via interactions with mitochondrial complex I, generating excessive ROS as a result [59,60]. Studies have subsequently demonstrated that DOX has a high affinity for cardiolipin, a lipid housed in the inner mitochondrial membrane which is essential for effective energy metabolism and proper mitochondrial function [61,62,63]. Notably, DOX becomes concentrated in the mitochondria of isolated neonatal rat cardiomyocytes, supporting the notion that this organelle is particularly vulnerable to off-target anthracycline toxicity [64]. Zhang and colleagues showed that mitochondrial function and oxidative phosphorylation pathways were disturbed in cardiomyocytes isolated from DOX-dosed mice, and that this was dependent on the topoisomerase IIβ (TopIIβ) enzyme which is thought to be crucial in the cardiotoxicity of this drug [65]. It has since been shown that DOX intercalates into mtDNA, which aids its accumulation in cardiomyocyte mitochondria in the same model [66]. Furthermore, Ichikawa and colleagues showed that DOX causes iron accumulation in cardiomyocyte mitochondria, leading to downstream toxicity. DOX treatment is associated with a depletion and mutation of mtDNA, as identified in hearts of cancer patients [67]. The interplay between DOX-induced mitochondrial damage and cardiomyocyte senescence within this cardiotoxicity is less well-understood, but it has been shown that the two phenomena go hand-in-hand using in vitro and in vivo studies, as evidenced by Mitry et al., amongst others [40,68,69]. As a long-established therapy, the impact of DOX upon cardiomyocyte mitochondria has been well reviewed [70], but newer oncology therapies are far less well-understood. For example, though tyrosine kinase inhibitors (TKIs) provide more targeted anticancer actions, the TKI sorafenib has historically been shown to impair cardiomyocyte mitochondrial function at clinically relevant doses in vitro, and more recent reports highlight that sunitinib may also induce cardiomyocyte mitochondrial damage via ROS accumulation [71,72]. Several other oncology therapies display off-target cardiovascular effects and mitochondrial toxicities [73] and it is clear the changing landscape in cancer survivorship necessitates thorough investigations into the long-term cardiac effects of both established and emerging antineoplastic therapies, and mitochondrial toxicity remains an attractive avenue of research.
2.2. Mitochondria Dynamics Imbalance
In physiological conditions, mitochondria constantly undergo co-ordinated cycles of fusion and fission, also referred to as mitochondrial dynamics [74]. Mitochondrial fusion is characterised by the union of two mitochondria resulting in one elongated mitochondrion, which allows a dynamic repair of reversibly damaged mitochondria. Conversely, mitochondrial fission is characterised by the fragmentation of one irreversibly damaged and potentially harmful mitochondrion into small and spherical mitochondria that can be isolated and removed by mitophagy [75,76]. The coordination of these events is essential for the maintenance of mitochondrial quantity and quality and therefore, the balance between them plays a vital role in the normal function of the cardiovascular system. Indeed, accumulating evidence has confirmed the influence of mitochondrial dynamics on the pathogenesis of CVD [74].
Mitochondrial fusion is first mediated by the transmembrane guanine triphosphatase (GTPase) proteins, mitofusin 1 (MFN1) and MFN2 in the outer mitochondrial membrane, and then by the optic atrophy protein 1 (OPA1) in the inner membrane [77]. Decreased levels of MFN1 and MFN2 have been found in animal models of atherosclerosis [78], and a decreased expression of OPA1 has been observed in post-MI hearts, which correlated with the downregulation of mtDNA and antioxidant genes [79]. Suggesting a causal role of fusion in CVD, ablation of the murine Mfn1 and Mfn2 genes in adult hearts induced mitochondrial fragmentation and dysfunction, and rapidly progressive and lethal dilated cardiomyopathy [80,81]. Different cardiac pathologies have also been associated with the formation of giant mitochondria or megamitochondria [26], which evolve by fusion of the membranes of numerous large individual organelles due to overexpression of protein fusion [82]. The opposing process, mitochondrial fission, is controlled by Mitochondrial fission protein 1 (Fis1) and Dynamin-related protein 1 (Drp1). It has been reported that Drp1 activation during cardiac IR results in left ventricular dysfunction and that Drp1 inhibition reduces cell death, preserves mitochondrial morphology, and inhibits mitochondrial permeability transition pore [83,84]. While the relationship between mitochondrial fusion and myocardial senescence has yet to be investigated, elongated mitochondria have been observed in senescent cells in vitro [85,86] and depletion of Fis1 mRNA levels leads to mitochondrial elongation, induces senescence, and increases ROS production [87].
Fusion and fission events control mitochondria biogenesis [88], a process that increases the number of mitochondria, improves the replication and repair of mtDNA, and induces the synthesis of mitochondrial enzymes and proteins [21]. The co-transcriptional regulator factor peroxisome-proliferator-activated receptor γ co-activator-1α (PGC-1α) induces mitochondrial biogenesis by activating the mitochondrial transcription factor A (TFAM), which drives transcription and replication of mtDNA [89]. Reduced gene expression of PGC-1α has been associated with failing human hearts [90] and there is evidence that sirtuin-1 (SIRT1), a protein involved in metabolic regulation, delays molecular characteristics of myocardial ageing by mediating deacetylation of PGC-1α and activation of mitochondrial biogenesis [91]. The PGC-1α+/-/ApoE-/- mouse model has shown that PGC-1α deficiency promotes vascular senescence, which is associated with increased oxidative stress, mitochondrial abnormalities, and reduced telomerase activity [92]. Mitochondrial biogenesis is also accompanied by variations in mitochondrial morphology [89]. Generally, various aspects of cardiovascular biology, including cardiac development, the response to cardiac IR injury and HF, are related to morphological and structural changes in mitochondria [93]. Dramatic changes in mitochondrial morphology have been also found in senescent cells, where mitochondria exist in a state of hyper-fusion as a response to reduced expression of mediators of the fission process and an overall reduction in the frequency of the fission and fusion events [40].
2.3. Cell Apoptosis and Mitophagy
Mitochondria are pivotal in controlling apoptosis, including the release of caspase activators and participation of B-cell lymphoma-2 (BCL-2) family proteins [94]. Cardiomyocyte apoptosis plays a critical role in the pathogenesis and progression of all types of heart disease, particularly in ischemic heart disease and HF of various aetiologies [95]. For example, cardiac IR injury is related to the apoptotic death of cardiac muscle cells by activating the pro-apoptotic BCL-2 regulators BAX and BAK to change the integrity of the mitochondrial membrane and the cytosolic release of pro-apoptotic factors, which triggers caspase-dependent cell death [96]. In hypertension, the hormone angiotensin II, which plays an important role in volume and blood pressure control, has been linked to cardiomyocyte apoptosis in rats, and treatment with losartan has been associated with a reduction of cardiomyocyte apoptosis in both spontaneous hypertensive rats and hypertensive patients [97]. The subfamily of pro-apoptotic BCL-2 homology (BH) BH3-only proteins, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and its homologue BNIP3-like (BNIP3L or Nix), also induce apoptosis [98] and the forced expression of these genes is sufficient to induce cardiomyopathy in murine models [26,99,100].
To prevent cardiomyocytes containing damaged mitochondria from undergoing apoptosis, mitophagy, a cargo-specific form of autophagy selectively targets the degradation of dysfunctional and damaged, and hence potentially cytotoxic, mitochondria within a cell [21]. There are two described mechanisms for mitophagy: adaptor-mediated and receptor-mediated. The former pathway functions via Phosphatase and Tensin Homolog (PTEN)-induced putative kinase 1 (PINK1) and Parkin-mediated mitophagy [101]. PINK1 is a serine/threonine kinase that continuously monitors mitochondrial health and provides a rapid response when mitochondrial function collapses [102]. PINK1 phosphorylates MFN2, which, in turn, interacts with the E3 ubiquitin ligase Parkin [103]. Parkin conjugates ubiquitin onto key mitochondria-associated proteins, amplifying the signalling cascade involved in the recruitment of autophagosomes to target the damaged mitochondria. The mitochondria-containing autophagosome is trafficked to, and fused with, a lysosome and degraded [104]. In healthy young hearts, there is an underlying level of baseline mitophagy essential for maintaining the cellular homeostasis in an energy-efficient heart, and for responding and adapting to stress [105]. However, decreased mitophagy is associated with CVD, as an accumulation of “old” defective mitochondria may reduce the heart’s potential to adapt to stress. Indeed, multiple animal studies have linked the deletion of mitophagy-related genes at the whole-body level or cardiomyocytes with the spontaneous development of cardiovascular disorders [106]. For example, mice bearing a cardiomyocyte-specific deletion of Mnf2 prematurely succumbed to progressive cardiomyopathy, which could be partially reversed by restoring mitophagy in cardiomyocytes via the expression of the antioxidant enzyme catalase [107]. The whole-body Pink1-/- mice caused left ventricular dysfunction and pathological cardiac hypertrophy by 2 months of age [108]. Mitophagy is also essential for reducing cardiac injury following MI. Under baseline conditions, Parkin-deficient mice hearts shown smaller and disorganised mitochondria as revealed by ultrastructural analysis, but mitochondrial and cardiac function were unaffected [109]. However, after MI, these mice had reduced survival and developed larger infarcts when compared to control mice, which was associated with rapid accumulation of dysfunctional mitochondria in the infarct border zone [110]. In patients with late-stage heart disease, a low number of autophagosomes in cardiomyocytes is associated with a poor prognosis [111]. Damaging events (e.g., acute cardiac IR injury) lead to the reduction of the autophagy flux, and in consequence, damaged dysfunctional mitochondria accumulate in cardiomyocytes, leading to severe oxidative stress and apoptosis [112]. The destabilisation of atherosclerotic plaques has also been associated with deficient mitophagy [113,114]. Furthermore, a reduced expression of autophagic markers p62 and microtubule-associated protein light chain (LC3)-II has been detected within atherosclerotic plaques from human samples and mouse models [115,116,117]. Activation of mitophagy through antioxidant therapeutic strategies has been explored to stabilise atherosclerotic plaques [118].
Interestingly, despite and perhaps because of mitochondrial dysfunction, senescent cells express pro-survival pathways, enhancing survival and increasing resistance to apoptosis. Senescent cells are more resistant to apoptosis in response to stimuli, including serum withdrawal, ultraviolet damage, oxidative stress and treatment with cytotoxic drugs [119]. While there is heterogeneity between cell types and senescence stimuli, enhanced activation of several pathways including BCL-2 family members, p53/p21Cip, ephrins (EFNB1 or 3), the phosphatidylinositol-4,5-bisphosphate 3-kinase delta catalytic subunit (PI3KCD), plasminogen-activated inhibitor-1 and 2 (PAI1 and 2) and hypoxia-inducible factor-1α (HIF1α) can be involved [120,121,122] and are referred to as senescent cell anti-apoptotic pathways (SCAPs). As discussed below, activation of these pathways may contribute to the proinflammatory nature of senescent cells.
3. Mitochondria Dysfunction, Senescence, and Inflammation in CVD
The role of inflammation in promoting CVD is increasingly recognised. Recent discoveries have demonstrated that mitochondria are key elements that stimulate innate immune signalling cascades which triggers inflammation and promotes pathology in an expanding list of diseases, including cardiac pathologies [123]. Many investigations have revealed that, when mitochondrial integrity is compromised, mtROS and mtDNA act as damage-associated molecular patterns (DAMPs), endogenous molecules that are isolated within intracellular compartments and discharged to the extracellular space in response to damage or dying cells [124], promoting pathological inflammatory responses by binding with pattern-recognition receptors (PRRs). For example, a study on mice found that mtDNA released by dying ischemic cells during MI activates the Interferon regulatory factor 3 (IRF3)-dependent innate immune response, which has a harmful effect on ventricular remodelling after MI [125]. The Stimulator of interferon genes (STING)-IRF3 pathway might also facilitate chronic inflammation and dysfunction of endothelial cells via sensing mtDNA [126], which are key events in the development of atherosclerosis and are associated with an elevated risk of many cardiovascular events [127]. Further, circulating mtDNA has been linked with the activation of the immune system via Toll-like receptor 9 (TLR9), which has been associated with elevated arterial pressure and vascular dysfunction in spontaneously hypertensive rats [128], and with exacerbated HF in mice [129]. Accumulating evidence has also shown that mtROS and mtDNA contribute to the molecular inflammation events during pathogenesis of CVD, activating the Nod-like receptor (NLR) family, pyrin domain containing 3 (NLRP3) inflammasome [130,131], although how this unfolds remains unknown. For example, excessive mtROS and dysfunctional mitochondria are considered critical drivers responsible for NLRP3 activation during the progression of atherosclerosis [132,133], and the level of the inflammasome has been found to be highly associated with the severity of disease [134]. Upon activation, NLRP3 inflammasome activates caspase-1, which cleaves and matures the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 [135], which contribute to cardiac fibrosis and HF [136,137]. Elevated IL-1β levels have been also correlated with age-related CVD [131]. Furthermore, suppression of NLRP3 extends lifespan of adult obese mice by reducing liver steatosis and cardiac damage [138]. In turn, PRRs might also modulate mitochondrial dysfunction and apoptosis, protecting against mortality as occurs with the receptor NLR family member X1 (NLRX1) during IR injury [139].
A significant characteristic of senescent cells is the acquisition of a hypersecretory phenotype or SASP, a collection of many biologically active factors, such as inflammatory cytokines, chemokines, matrix remodelling proteases, extracellular vesicles, and growth factors [140]. This heterogeneous group of secreted proteins self-reinforce and spread senescence in an autocrine and paracrine manner, respectively, or affect the local tissue environment of senescent cells and possibly, the entire organism [7]. Although some SASP factors are common to all senescent cells, its composition varies depending on the cell type and the nature of the stimulus [141]. In senescent cardiomyocytes, increased expression of SASP factors such as cellular communication network protein family member 1 (CCN1), interleukins (IL1α, IL1β, and IL6), tumour necrosis factor-alpha (TNFα), monocyte chemoattractant protein-1 (MCP1), endothelin 3 (Edn3), tumour growth factor-beta (TGFβ), and growth and differentiation factor 15 (GDF15) have been clinically linked with age-related myocardial ischemia and infarction [142,143]. mtROS are a component of the SASP [144], and functional mitochondria are critical for SASP production. As would be expected, senescent cells with depleted mitochondria have reduced ROS generation but also lose their proinflammatory phenotype yet remain in cell cycle arrest [69]. SASP production appears to involve mitochondria through several interconnected mechanisms. mtROS can induce the c-Jun N-terminal kinase (JNK) signalling and the release of cytoplasmic chromatin fragments, triggering the innate immunity cytosolic DNA-sensing cyclic GMP-AMP synthase (cGAS)-STING pathway [145]. This in turn activates the nuclear factor-κB (NFκB) signalling, switching on the transcription of proinflammatory genes and the SASP [145]. Recent studies suggest that expression of pro-survival pathways in senescent cells leads to sublethal apoptosis and minority mitochondrial outer membrane permeabilization (miMOMP). This miMOMP allows the release of mtDNA into the cytosol which activates the cGAS–STING pathway, resulting in increased expression of inflammatory mediators and SASP [146]. The sublethal release of cytochrome c and caspase activation, associated with miMOMP may also contribute to further DNA damage, increased genetic instability and perhaps deeper senescence [147].
4. Mitochondrial and Senescent Cells Targeted Therapies for CVD
4.1. Therapeutically Targeting Mitochondrial Dysfunction
In recent years, an increasing number of cardiac mitochondrial targets have shown their cardioprotective effects in experimental and clinical studies. While mitochondria are not only the site of OXPHOS, but their dysfunction is also commonly associated with ATP deficiency and excessive ROS generation. Therefore, restoring the ATP-producing capacity and counteracting the damaging effects of ROS to reduce oxidative stress and chronic inflammation have been suggested as primary therapeutic targets to improve mitochondrial dysfunction [22].
AMP-activated protein kinase (AMPK) is an exclusive kinase of eukaryotes that plays a major role in regulating energy balance by monitoring changes in the level of intracellular ATP and coupling these changes to phosphorylation of downstream substrates, leading to an increase of ATP synthesis and/or a restriction of ATP depletion [148]. Thus, targeting the AMPK pathway has attracted widespread interest [21]. A well-known AMPK agonist is the first-line drug for treating Type 2 diabetes mellitus (T2DM), metformin [149]. Mechanistically, metformin exerts beneficial effects mainly through the inhibition of the respiratory chain at the level of Complex 1, leading to an increased AMP/ATP ratio and activation of the signalling kinase AMPK [150]. This in turn, induces muscles to take up glucose from the blood. Metformin also improves mitochondrial function and quality, as AMPK activation phosphorylates a range of target proteins involved in the regulation of mitochondrial biogenesis (PCG-1α), mitochondrial dynamics (Drp1, MFF), and mitophagy (PINK1-Parkin pathway) [151,152,153,154]. Interestingly, metformin has been described as senomorphic, being able to modulate SASP secretion from senescent cells and improve senescent cell function [155,156]. Metformin is inhibitory to SASP expression due to inhibition of IκB kinase and IKKα/β phosphorylation, thereby preventing the NF-κB nuclear translocation [157]. Furthermore, metformin can attenuate senescence in human diploid fibroblasts and mesenchymal stem cells [158] and in healthy mice, metformin has been observed to extend health span and lifespan [159].
Indicating that metformin may have similar effects clinically, a recent meta-analysis has identified that diabetic patients taking metformin have a significantly increased survival rate and a reduced incidence of age-related diseases, including CVD [160]. Several clinical studies have shown the beneficial effects of metformin in diabetes-related atherosclerosis, IR injury and arrhythmia (as discussed by Bu et al.) [161]. For example, in the Reversing with MetfOrmin Vascular Adverse Lesions (REMOVAL) trial, a double-blind placebo-controlled randomized controlled trial to evaluate the cardiovascular effects of metformin in adults with T1DM, atherosclerosis progression was significantly reduced in metformin-treated patients [162]. In another randomized, placebo-controlled trial involving 390 patients with T2DM treated with insulin, metformin treatment improved endothelial function [163]. In non-diabetes patients, clinical studies are more controversial when evaluating the benefits of metformin in CVD. In a small clinical study consisting of 33 non-diabetic women, metformin reduced myocardial ischemia and improved endothelium-dependent microvascular responses in patients with angina, compared to placebo [164]. However, in a subsequent clinical study consisting of 173 non-diabetic patients with coronary heart disease treated with statins, Preiss et al. [165] found that metformin had no effect on disease progression and little or no effect on several surrogate markers of CVD. Although it remains unclear if the senomorphic activity of metformin contributes to any of these cardioprotective effects, in patients with carotid artery atherosclerosis metformin ameliorates the proinflammatory state, which includes the reduction of serval SASP related proteins including IL-6 and TNF-α [166]. Metformin also influences other hallmarks of biological ageing. For example, nutrient-signalling pathways both through AMPK and SIRT1 activation, as well as downregulating insulin/insulin-like growth factor 1 (IGF-1) signalling and mechanistic target of rapamycin complex 1 (mTORC1). Metformin also attenuates oxidative damage and genome instability by enhancing DNA damage response and repair mechanisms, improving proteostasis by enhancing autophagy and inhibiting protein synthesis, and ameliorating mitochondrial dysfunction via mitochondrial complex I inhibition and PGC-1α upregulation. Furthermore, metformin treatment reduces telomere shortening by activating telomeric repeat-containing RNA [167]. Metformin is now being evaluated for its age-targeting effects in the TAME (Targeting Ageing with Metformin) clinical trial [168].
Because of the role of ROS in CVD, reduction of oxidative stress through the supplementation with antioxidants such as Coenzyme Q10 (CoQ10), vitamin E, vitamin C, and β-carotene, have been clinically studied in humans for the treatment of CVD, including HF, atherosclerosis, and acute MI [169,170,171,172]. However, with the sole exception of CoQ10, no clinically significant benefits were reported. CoQ10 is a lipid-soluble and biologically active quinone whose principal role is to participate in the ETC, where it functions as an electron carrier [173]. In a study including 420 patients with moderate to severe HF, Svend et al. [169] reported that long-term treatment with CoQ10, in addition to standard therapy, was safe, and associated with an improvement in symptoms and a reduction in mortality from cardiovascular events. Zeb et al. [170] reported beneficial effects on inflammatory markers and reduced progression of coronary atherosclerosis in patients treated with a capsule containing aged garlic extract and CoQ10 daily for 1 year. In a meta-analysis of 14 studies with 2149 enrolled subjects Lei et al. [174] found that patients with HF who used coQ10 had lower mortality.
One possible explanation for the ineffectiveness of common antioxidants to show beneficial effects is their inability to enter the mitochondria, the primary source of ROS [175]. There are several approaches to targeting molecules to the mitochondria and one of the most versatile is to develop synthetically modified antioxidants with lipophilic cationic compounds, such as triphenylphosphonium (TPP+) [176]. TPP+ is a membrane-permeant cation that is accumulated within the mitochondria up to several 100-fold because of the negative potential (-140 to –180 mV) generated across the inner mitochondrial membrane by the proton pumping action of the ETC [177]. Mitoquinone (MitoQ) is CoQ10 conjugated to TPP+ and has been shown to display impressive benefits in the treatment of CVD. It has been reported that 100 µM MitoQ in drinking water rescues the cardiac function of pressure-overloaded HF in a mouse model by decreasing hydrogen peroxide formation, improving mitochondrial respiration and mitochondrial permeability transition pore opening [178]. In mice and rat studies, MitoQ protected against IR injury by blocking oxidative damage within the mitochondria [179,180]. Treatment with MitoQ also prevented development of hypertension, improved endothelial function, and limited cardiac hypertrophy in eight-week-old male spontaneously hypertensive rats [181]. Furthermore, MitoQ controls the expression levels of cardiac hypertrophy-associated transcript (Chast) and myosin heavy chain associated transcript (Mhrt), two long non-coding RNAs involved in cardiac remodelling. It also attenuates adverse cardiac remodelling, and prevents HF in mice, by inhibiting the interplay between TGF-β1 and mitochondrial associated redox signalling [182]. In humans, a randomized, placebo-controlled, double-blind, crossover study of 20 healthy adults (60-79 years) with endothelial dysfunction demonstrated that oral supplementation with 20 mg/day of MitoQ was well tolerated and significantly improved endothelial function and reduced arterial stiffness and plasma oxidized low-density lipoprotein (LDL), a marker of oxidative stress, by reduction of mtROS [183]. Currently, in the USA, there are two clinical trials ongoing focused on the effects of MitoQ on cardiac function: the MitoQ Supplementation and Cardiovascular Function in Healthy Men and Women study (NCT03960073), and the Chronic Kidney Disease and Heart Failure With Preserved Ejection Fraction: The Role of Mitochondrial Dysfunction study (NCT03586414). Given that ROS is both an inducer and a consequence of senescence, it is perhaps unsurprising that in a wide range of diseases and models, anti-oxidants (including those that are mitochondrial targeted) are demonstrated to attenuate senescence and SASP [184].
4.2. Senolytics, Senomorphics and Future Approaches
Despite active DNA damage responses, increased mitochondrial dysfunction, increased mitochondrial membrane permeability, and increased ROS production, senescent cells remain resistant to apoptosis. Based on these findings, Zhu and colleagues hypothesized that pharmacologically inhibiting the pro-survival networks could eliminate senescent cells [120]. This gave rise to the advent of compounds collectively termed senolytics, which target various components of anti-apoptotic pathways, including BCL-2 family members, to promote senescent cell apoptosis.
In the context of heart health, studies have largely investigated the senolytic effects of the combination therapy dasatinib and quercetin (D&Q), and navitoclax (ABT-263). Dasatinib is a second-generation tyrosine kinase inhibitor, shown to inhibit ephrins, disrupting the pro-survival network that includes BCL-XL, PI3K, p21Cip, PAI1, and PAI2 [120,185,186]. Quercetin, a natural flavanol, inhibits multiple pro-survival proteins, including PAIs and PI3K, ultimately reducing BCL-W expression [187,188]. Navitoclax, a BH3 mimetic, induces senescent cell apoptosis by inhibiting anti-apoptotic proteins BCL-2, BCL-XL, and BCL-W [189,190].
Use of these senolytics in several animal models of CVD, such as age-related myocardial dysfunction, MI, anthracycline-induced cardiotoxicity and atherosclerosis, have provided proof of principle data that promoting mitochondrial mediated apoptosis in senescent cells reduces inflammation and attenuates disease pathophysiology [4,52,53,120,191,192,193]. On the other hand, as increased apoptosis has been implicated in age-related myocardial dysfunction, and after MI the primary objective of reperfusion therapy is to save as much myocardium as possible, concerns have been raised regarding the long-term outcomes of increased cell death [3]. However, recent studies have demonstrated that senescent cardiomyocytes are indeed detrimental to outcome, and that inhibition/modulation of the senescent phenotype may be beneficial in the senescent cardiomyocyte context: inhibition of p16 in murine cardiomyocytes improved outcome following MI with reperfusion [3].
In future modulation/inhibition of the senescent phenotype may be a more feasible route to successful intervention in this disease context, rather than promotion of senescent cardiomyocyte apoptosis. As such, a senomorphic approach, modulating the senescent phenotype and attenuating the SASP may consequently have more translational potential than senotherapies. Alternatively, specific inhibition of miMOMP-induced inflammation [146], may have therapeutic utility. For instance, inhibition of the mitochondrial membrane BAX and BAK nanopores with small-molecule BAX inhibitor BAI1 was shown to decrease systemic inflammation and improved health span in aged mice [146]. Underpinning all these findings, evaluation of a drug’s senolytic and/or senomorphic capabilities must be approached with rigour and caution, as emphasised in Niedernhofer and Robbins’ 2018 correspondence [194].
5. Conclusions
In the complex realm of cardiovascular diseases, mitochondrial dysfunction, and oxidative stress (in isolation or within the context of senescence induction and senescent cell function) play a key role in pathophysiology (summary Figure 1). A better understanding of these interconnected phenomena will enable the development of novel therapies for CVD. Ongoing research and clinical trials signal a new frontier in cardiovascular medicine, promising innovative treatments that could transform patient outcomes. These developments mark a significant stride towards enhancing both the quality and duration of life for those affected by cardiovascular diseases.
Author Contributions
M.C.-E and G.R conceptualized and supervised the draft, and M.C.-E, L.B, G.R and O.F looked for, read, and made a first selection of the most relevant articles, and M.C.-E, L.B and G.R wrote the draft, and L.B and R.R reviewed and edited the draft. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been funded by the British Heart Foundation (PG/22/10788 and PG/21/10761) and the MRC.
Acknowledgments
Figures made in Biorender.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp Cell Res 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, R.; Dookun, E.; Booth, L.; Folaranm, O.; Tual-Chalot, S.; Gill, J.; Owens, A.; Spyridopoulos, I.; Passos, J.; Richardson, G. Senescent cardiomyocytes contribute to cardiac dysfunction following myocardial infarction. Res Sq 2023. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo j 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres. Trends Biochem Sci 1991, 16, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021, 22, 75–95. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Richardson, G.D.; Passos, J.F. Mechanisms driving the ageing heart. Exp Gerontol 2018, 109, 5–15. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Booth, L.K.; Redgrave, R.E.; Tual-Chalot, S.; Spyridopoulos, I.; Phillips, H.M.; Richardson, G.D. Heart Disease and Ageing: The Roles of Senescence, Mitochondria, and Telomerase in Cardiovascular Disease. Subcell Biochem 2023, 103, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech Ageing Dev 2021, 198, 111540. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Granic, A.; Miwa, S.; Passos, J.F.; Richardson, G.D.; Sayer, A.A. New Horizons in cellular senescence for clinicians. Age Ageing 2023, 52. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.K.; Redgrave, R.E.; Folaranmi, O.; Gill, J.H.; Richardson, G.D. Anthracycline-induced cardiotoxicity and senescence. Front Aging 2022, 3, 1058435. [Google Scholar] [CrossRef] [PubMed]
- Dookun, E.; Passos, J.F.; Arthur, H.M.; Richardson, G.D. Therapeutic Potential of Senolytics in Cardiovascular Disease. Cardiovasc Drugs Ther 2022, 36, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Richardson, G.; Haendeler, J.; Altschmied, J.; Andrés, V.; Spyridopoulos, I. Telomerase as a Therapeutic Target in Cardiovascular Disease. Arterioscler Thromb Vasc Biol 2021, 41, 1047–1061. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach. 2nd edition; Sinauer Associates 2000: 2000.
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front Cell Dev Biol 2020, 8, 624216. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front Cell Dev Biol 2022, 10, 841523. [Google Scholar] [CrossRef]
- Stamerra, C.A.; Di Giosia, P.; Giorgini, P.; Ferri, C.; Sukhorukov, V.N.; Sahebkar, A. Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies. Oxid Med Cell Longev 2022, 2022, 9530007. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- el Alaoui-Talibi, Z.; Landormy, S.; Loireau, A.; Moravec, J. Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am J Physiol 1992, 262, H1068–H1074. [Google Scholar] [CrossRef]
- Allard, M.F.; Schönekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 1994, 267, H742–H750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, Y.Y.; Liu, G.H.; Lu, H.B.; Mao, C.Y. l-Carnitine and heart disease. Life Sci 2018, 194, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol 2015, 13, 89. [Google Scholar] [CrossRef]
- Casademont, J.; Miró, O. Electron transport chain defects in heart failure. Heart Fail Rev 2002, 7, 131–139. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Louboutin, J.P.; Datta, P.; Landel, C.P.; Martinez, D.; Zervos, A.S.; Strayer, D.S.; Fernandes-Alnemri, T.; Alnemri, E.S. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell Death Differ 2013, 20, 259–269. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React Oxyg Species (Apex) 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 2019, 593, 1566–1579. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front Genet 2015, 6, 94. [Google Scholar] [CrossRef]
- Fred, C.L. The DNA damage response - from cell biology to human disease. Journal of Translational Genetics and Genomics 2022, 6, 204–222. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular senescence: all roads lead to mitochondria. Febs j 2023, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.G. Trauma and nostalgia: new aspects on the coping of aging Holocaust survivors. Isr J Psychiatry Relat Sci 1990, 27, 189–198. [Google Scholar]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol 2006, 147 Suppl 1, S287–296. [Google Scholar] [CrossRef]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal 2013, 18, 5–18. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun 2014, 2, 4172. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin improves healthspan but not inflammaging in nfκb1(-/-) mice. Aging Cell 2019, 18, e12882. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife 2020, 9. [Google Scholar] [CrossRef]
- Windecker, S. Chapter 30 - Acute Coronary Syndromes. In Clinical Critical Care Medicine, Albert, R.K., Slutsky, A.S., Ranieri, V.M., Takala, J., Torres, A., Eds.; Mosby: Philadelphia, 2006; pp. 301–318. [Google Scholar]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech Ageing Dev 2022, 208, 111740. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.; Sager, P.T.; Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 2016, 15, 457–471. [Google Scholar] [CrossRef]
- Laverty, H.; Benson, C.; Cartwright, E.; Cross, M.; Garland, C.; Hammond, T.; Holloway, C.; McMahon, N.; Milligan, J.; Park, B.; et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol 2011, 163, 675–693. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of analgesic medications because of adverse drug reactions: a systematic review. Expert Opin Drug Saf 2018, 17, 63–72. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 2015, 12, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.J.; Leger, K.J.; Bhatt, N.S.; Mulrooney, D.A.; Ross, C.J.; Aggarwal, S.; Bansal, N.; Ehrhardt, M.J.; Armenian, S.H.; Scott, J.M.; et al. Paediatric cardio-oncology: epidemiology, screening, prevention, and treatment. Cardiovasc Res 2019, 115, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 1986, 261, 3068–3074. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys Chem 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Dolder, M.; Gerber, I.; Speer, O.; Wallimann, T.; Schlattner, U. Reduced creatine-stimulated respiration in doxorubicin challenged mitochondria: particular sensitivity of the heart. Biochim Biophys Acta 2007, 1767, 1276–1284. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci 2008, 65, 2493–2506. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Furusawa, S.; Tsutsui, Y.; Miyake, R.; Ishimaru, K.; Watanabe, M.; et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci Signal 2022, 15, eabn8017. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Kokkori, A.; Ketelsen, U.P.; Setzer, B.; Walker, U.A. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol 2005, 207, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am J Physiol Cell Physiol 2020, 318, C380–c391. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. Embo j 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci 2008, 106, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krähenbühl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef]
- Rocca, C.; De Francesco, E.M.; Pasqua, T.; Granieri, M.C.; De Bartolo, A.; Gallo Cantafio, M.E.; Muoio, M.G.; Gentile, M.; Neri, A.; Angelone, T.; et al. Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int 2019, 2019, 9757201. [Google Scholar] [CrossRef] [PubMed]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr Opin Cell Biol 2021, 68, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Ren, K.D.; Luan, Y.; Chen, X.; Yang, Y. Mitochondrial Dynamics: Pathogenesis and Therapeutic Targets of Vascular Diseases. Front Cardiovasc Med 2021, 8, 770574. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal 2008, 10, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 2012, 111, 1012–1026. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. Faseb j 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J Biomed Sci 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Honet, J.C.; Wajszczuk, W.J.; Rubenfire, M.; Kantrowitz, A.; Raikes, J.A. Neurological abnormalities in the leg(s) after use of intraaortic balloon pump: report of six cases. Arch Phys Med Rehabil 1975, 56, 346–352. [Google Scholar] [PubMed]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front Endocrinol (Lausanne) 2020, 11, 374. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol 2009, 46, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Wang, J.; Wu, F.; Chen, Y.; Zhang, H.; Guo, Y.; Lin, Y.; Li, L.; Yu, X.; et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging (Albany NY) 2020, 12, 650–671. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; Patrushev, N.; Ma, M.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol 2013, 33, 988–998. [Google Scholar] [CrossRef]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Kanoh, M.; Minatoguchi, S.; Fujiwara, H. Cardiomyocyte apoptosis in the failing heart--a critical review from definition and classification of cell death. Int J Cardiol 2013, 167, 2373–2386. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Fortuño, M.A.; Querejeta, R.; Ravassa, S.; López, B.; López, N.; Díez, J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc Res 2003, 59, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 2010, 3, 374–383. [Google Scholar] [CrossRef]
- Diwan, A.; Wansapura, J.; Syed, F.M.; Matkovich, S.J.; Lorenz, J.N.; Dorn, G.W., 2nd. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 2008, 117, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, G.; Shen, H.M. The Long and the Short of PTEN in the Regulation of Mitophagy. Front Cell Dev Biol 2020, 8, 299. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson Å, B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res 2015, 117, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Song, M.; Chen, Y.; Gong, G.; Murphy, E.; Rabinovitch, P.S.; Dorn, G.W., 2nd. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 2014, 115, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Quinsay, M.N.; Gustafsson, A.B. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol 2013, 6, e24511. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Asai, K.; Sato, S.; Hayashi, M.; Adachi, A.; Sasaki, Y.; Takano, H.; Mizuno, K.; Shimizu, W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 2016, 12, 579–587. [Google Scholar] [CrossRef]
- Campos, J.C.; Bozi, L.H.; Bechara, L.R.; Lima, V.M.; Ferreira, J.C. Mitochondrial Quality Control in Cardiac Diseases. Front Physiol 2016, 7, 479. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ Res 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid Med Cell Longev 2018, 2018, 7687083. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal 2016, 9, ra2. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; López Medina, A.; Freijo Mdel, M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS One 2014, 9, e115176. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid Med Cell Longev 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J Mol Biol 2019, 431, 2629–2643. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Hof, P.R.; Martin, J.L. Adenosine stimulates glycogenolysis in mouse cerebral cortex: a possible coupling mechanism between neuronal activity and energy metabolism. J Neurosci 1986, 6, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P., Jr.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 2015, 107, 119–130. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Yu, Y.; Li, M.; Chen, R. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Transl Med 2020, 10, 91–106. [Google Scholar] [CrossRef]
- Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells 2020, 9. [Google Scholar] [CrossRef]
- Victor, V.M.; Apostolova, N.; Herance, R.; Hernandez-Mijares, A.; Rocha, M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy. Curr Med Chem 2009, 16, 4654–4667. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Heart Vessels 2016, 31, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The Importance of NLRP3 Inflammasome in Heart Failure. J Card Fail 2015, 21, 586–593. [Google Scholar] [CrossRef]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front Physiol 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Cordero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. Geroscience 2020, 42, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med 2017, 214, 2405–2420. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. A common signature of cellular senescence; does it exist? Ageing Res Rev 2021, 71, 101458. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front Endocrinol (Lausanne) 2020, 11, 280. [Google Scholar] [CrossRef]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev 2018, 170, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Passos, J.F. Targeting the SASP to combat ageing: Mitochondria as possible intracellular allies? Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr Opin Cell Biol 2017, 45, 31–37. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Núñez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 2015, 27, 978–988. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ Res 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.E.; Kragh-Hansen, U.; Sheikh, M.I. Transport of leucine, isoleucine and valine by luminal membrane vesicles from rabbit proximal tubule. J Physiol 1990, 422, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat Commun 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res Rev 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Peng, M.; Tang, X.; Xu, X.; Wu, Y.; Chen, A.F.; Yang, X. Protective effects of metformin in various cardiovascular diseases: Clinical evidence and AMPK-dependent mechanisms. J Cell Mol Med 2022, 26, 4886–4903. [Google Scholar] [CrossRef]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol 2017, 5, 597–609. [Google Scholar] [CrossRef]
- De Jager, J.; Kooy, A.; Lehert, P.; Bets, D.; Wulffelé, M.G.; Teerlink, T.; Scheffer, P.G.; Schalkwijk, C.G.; Donker, A.J.; Stehouwer, C.D. Effects of short-term treatment with metformin on markers of endothelial function and inflammatory activity in type 2 diabetes mellitus: a randomized, placebo-controlled trial. J Intern Med 2005, 257, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Ferrell, W.; Greer, I.A.; Petrie, J.R.; Cobbe, S.M.; Sattar, N. Effects of metformin on microvascular function and exercise tolerance in women with angina and normal coronary arteries: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol 2006, 48, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Preiss, D.; Lloyd, S.M.; Ford, I.; McMurray, J.J.; Holman, R.R.; Welsh, P.; Fisher, M.; Packard, C.J.; Sattar, N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol 2014, 2, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Deng, Y.Y.; Yang, L.; Zhao, S.; Liu, J.; Zhao, Z.; Wang, L.; Maharjan, P.; Gao, S.; Tian, Y.; et al. Metformin ameliorates the proinflammatory state in patients with carotid artery atherosclerosis through sirtuin 1 induction. Transl Res 2015, 166, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. Febs j 2023, 290, 1362–1383. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail 2014, 2, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Zeb, I.; Ahmadi, N.; Nasir, K.; Kadakia, J.; Larijani, V.N.; Flores, F.; Li, D.; Budoff, M.J. Aged garlic extract and coenzyme Q10 have favorable effect on inflammatory markers and coronary atherosclerosis progression: A randomized clinical trial. J Cardiovasc Dis Res 2012, 3, 185–190. [Google Scholar] [CrossRef] [PubMed]
- MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [CrossRef]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. Jama 2008, 300, 2123–2133. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; de la Cruz-Ares, S.; Torres-Peña, J.D.; Alcalá-Diaz, J.F.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q(10) and Cardiovascular Diseases. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Liu, Y. Efficacy of coenzyme Q10 in patients with cardiac failure: a meta-analysis of clinical trials. BMC Cardiovasc Disord 2017, 17, 196. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol 2013, 1, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A Novel Triphenylphosphonium Carrier to Target Mitochondria without Uncoupling Oxidative Phosphorylation. J Med Chem 2021, 64, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb j 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.M.; Bradley, J.A.; Pettigrew, G.J.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J Heart Lung Transplant 2015, 34, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef]
- Goh, K.Y.; He, L.; Song, J.; Jinno, M.; Rogers, A.J.; Sethu, P.; Halade, G.V.; Rajasekaran, N.S.; Liu, X.; Prabhu, S.D.; et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol 2019, 21, 101100. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Varesi, A.; Chirumbolo, S.; Campagnoli, L.I.M.; Pierella, E.; Piccini, G.B.; Carrara, A.; Ricevuti, G.; Scassellati, C.; Bonvicini, C.; Pascale, A. The Role of Antioxidants in the Interplay between Oxidative Stress and Senescence. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Breccia, M.; Alimena, G. Activity and safety of dasatinib as second-line treatment or in newly diagnosed chronic phase chronic myeloid leukemia patients. BioDrugs 2011, 25, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin Cancer Res 2012, 18, 3580–3591. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anticancer Agents Med Chem 2013, 13, 1025–1031. [Google Scholar] [CrossRef]
- Olave, N.C.; Grenett, M.H.; Cadeiras, M.; Grenett, H.E.; Higgins, P.J. Upstream stimulatory factor-2 mediates quercetin-induced suppression of PAI-1 gene expression in human endothelial cells. J Cell Biochem 2010, 111, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Lérida-Viso, A.; Estepa-Fernández, A.; Morellá-Aucejo, Á.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepúlveda, P.; García-Fernández, A.; Orzáez, M.; et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol Res 2022, 183, 106356. [Google Scholar] [CrossRef]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc Res 2022, 118, 1713–1727. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat Rev Drug Discov 2018, 17, 377. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Common characteristics that define mitotic and post-mitotic senescent cells.
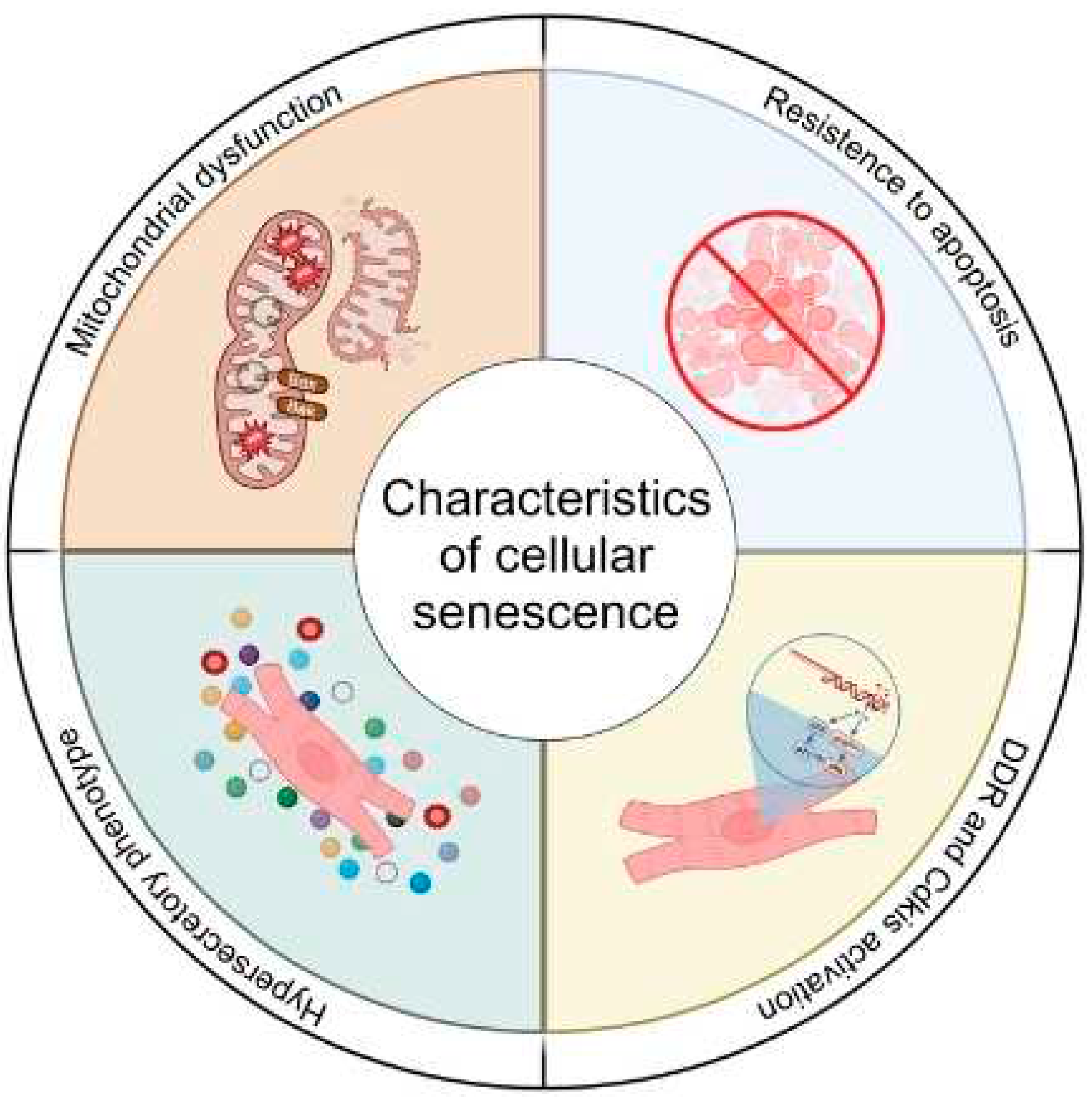
Figure 2.
Mitochondrial dysfunction contributes to cardiovascular disease through apoptosis and senescence. Mitochondrial dysfunction and increased ROS production promotes DNA damage, both chromosomal and mitochondrial. DNA damage leads to p53 activation and a cell fate decision between apoptosis and senescence. In apoptotic cells p53 induces mitochondrial outer membrane permeabilization via formation of the apoptotic pore which allows cytochrome c release, activation of the caspase cascade and cell death. While not yet completely understood, but perhaps as a result as less severe stress, DNA damage and p53 can lead to expression of the p21 (a negative regulator of apoptosis and the cell cycle), activation of the p16 pathway, or activation of both p21 and p16 pathways resulting in cellular senescence. Upregulation of pro-survival pathways in senescent cells suppresses apoptotic pore formation leading to miMOMP, sublethal apoptosis and the release of mtDNA into the cytoplasm. mtDNA fragments are sensed by the cGAS-STING pathway, upregulating expression of inflammatory mediators. Sublethal activation of the caspase cascade may also promote additional DNA damage. A combination of apoptosis and senescence will drive pathological myocardial remodelling.
Figure 2.
Mitochondrial dysfunction contributes to cardiovascular disease through apoptosis and senescence. Mitochondrial dysfunction and increased ROS production promotes DNA damage, both chromosomal and mitochondrial. DNA damage leads to p53 activation and a cell fate decision between apoptosis and senescence. In apoptotic cells p53 induces mitochondrial outer membrane permeabilization via formation of the apoptotic pore which allows cytochrome c release, activation of the caspase cascade and cell death. While not yet completely understood, but perhaps as a result as less severe stress, DNA damage and p53 can lead to expression of the p21 (a negative regulator of apoptosis and the cell cycle), activation of the p16 pathway, or activation of both p21 and p16 pathways resulting in cellular senescence. Upregulation of pro-survival pathways in senescent cells suppresses apoptotic pore formation leading to miMOMP, sublethal apoptosis and the release of mtDNA into the cytoplasm. mtDNA fragments are sensed by the cGAS-STING pathway, upregulating expression of inflammatory mediators. Sublethal activation of the caspase cascade may also promote additional DNA damage. A combination of apoptosis and senescence will drive pathological myocardial remodelling.
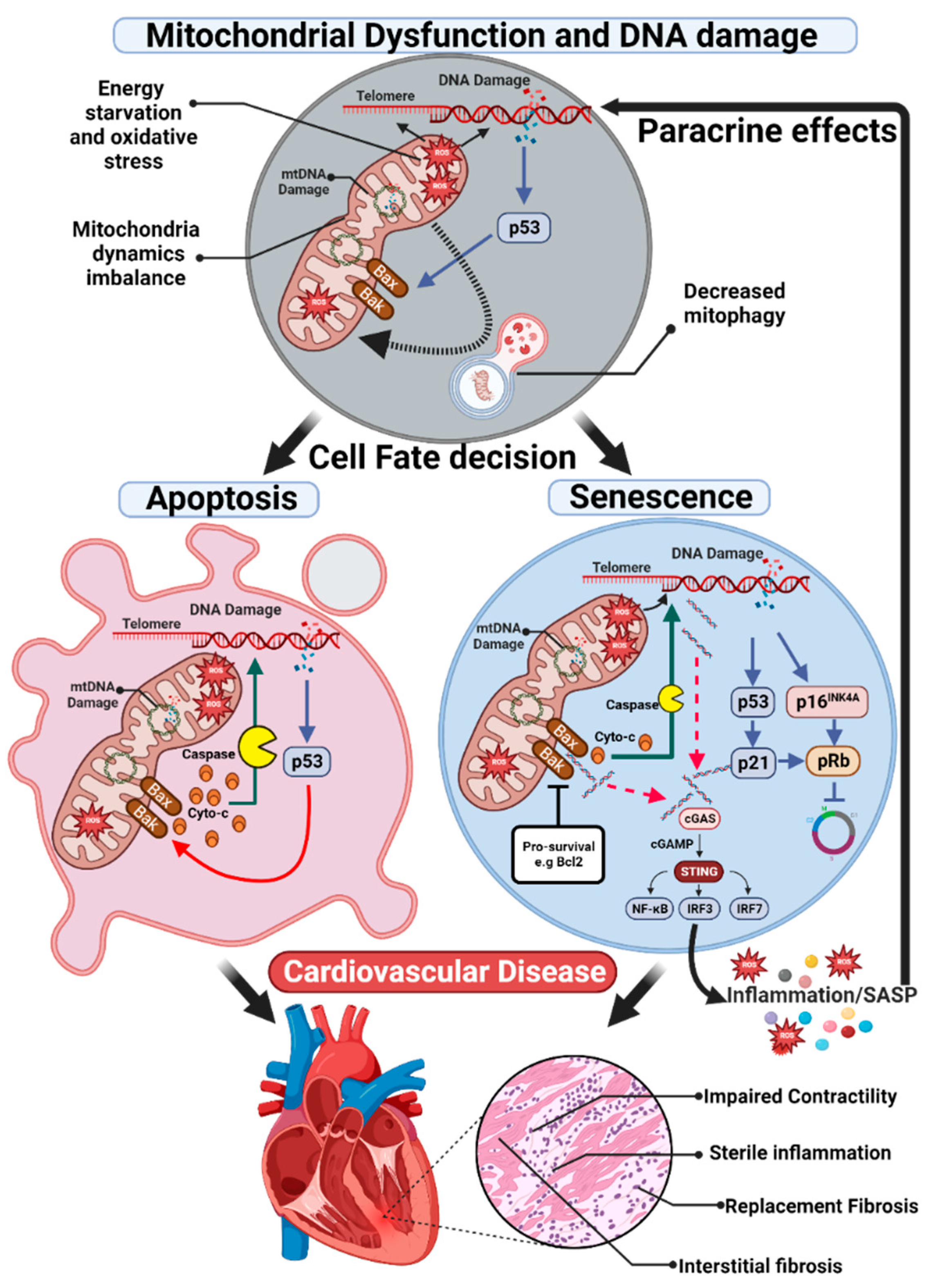
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



