
Submitted:
11 January 2024
Posted:
12 January 2024
You are already at the latest version
Abstract
There is remarkable morphologic and genetic heterogeneity in acute myeloid leukemia (AML). In a small percentage of cases of AML, increased eosinophils and/or basophils are present in the bone marrow and sometimes in the peripheral blood. This is often a puzzling diagnostic situation but also an important finding that requires special investigation. Unique chromosomal rearrangements have been correlated with an increased number of eosinophils and basophils in AML. Identification of the underlying genetic lesion that promotes eosinophilia and basophilia can dramatically change both the prognosis and the treatment of the patient. Thus clinicians must be vigilant in searching for the cause of eosinophilia and/or basophilia in patients with AML, since the different causes may lead to different treatments and survival outcomes. In this article we examine the significance of increased eosinophils and/or basophils in the context of AML, provide guidance that simplifies the differential diagnosis, and prognostic and therapeutic information about specific subtypes of AML associated with eosinophilia and/or basophilia. Evidence supporting personalized (molecularly targeted) therapy for these patients is also presented.
Keywords:
acute myeloid leukemia
; AML
; eosinophil
; eosinophilia
; basophil
; basophilia
; morphology
; CBF
; PDGFRA
; PDGFRB
1. Introduction
There is remarkable morphologic and genetic heterogeneity in acute myeloid leukemia (AML). Specific chromosomal abnormalities correlate with particular subtypes of AML that have characteristic morphologic and clinical features. Conversely, certain morphologic findings may serve as a basis for the identification of specific genetic subtypes. The presence of eosinophilia and/or basophilia in the context of a myeloid cancer such as AML usually indicates a clonal origin. These eosinophils and basophils are usually part of the neoplastic process, and may be morphologically abnormal with abnormal granulation, abnormal cytochemical reactions or nuclear hyposegmentation. Eosinophilia and/or basophilia are usually found in the bone-marrow aspirates of patients with AML and less frequently in the peripheral blood smears.
Although eosinophilia and/or basophilia may occur as a nonspecific finding in AML, this finding may also indicate specific subtypes of immediate therapeutic and prognostic relevance. Therefore, the presence of increased eosinophils and basophils may be an important diagnostic lead in a patient with AML. The aim of this article is to review the significance of increased eosinophils and basophils in the context of AML, putting emphasis on the appropriate diagnostic evaluation, prognostic significance, and treatment.
2. Physiologic Features of Eosinophils and Basophils
2.1. Physiologic Features of Eosinophils
Eosinophils are produced in the bone marrow from granulocyte–macrophage colony-forming units (CFU-GM). The latter cells differentiate first into hybrid precursors with properties of basophils and eosinophils (CFU-Bas/Eo) and then into a separate eosinophil lineage. Three cytokines — interleukin-3, interleukin-5, and granulocyte–macrophage colony-stimulating factor (GM-CSF) — are particularly important in the development of eosinophil granulocytes [1, 2]. These cytokines are encoded by closely linked genes on chromosome 5q31 and bind to receptors that have a common beta chain (common beta [CD131] subunit) and different alpha chains. The regulation of genes that drive eosinophil differentiation is controlled by a network of transcription factors including C/EBPα, C/EBPε, GATA-1, GATA-2, FOG-1, and PU.1 [2, 3]. After maturing in the bone marrow, eosinophils migrate from the circulation into tissues [4, 5]. Eosinophils, unlike neutrophils, can survive in tissues for extended periods (perhaps weeks), depending on the cytokines in the microenvironment.
The granules of eosinophils contain a crystalloid core composed of major basic protein (MBP) and a matrix composed of eosinophil cationic protein (ECP), eosinophil peroxidase (EPX), and eosinophil-derived neurotoxin (EDN). In addition, eosinophils produce hydrolytic enzymes such as phospholipase A2 (lysophospholipase), cytokines (some of which are stored in the granules), and large amounts of lipid mediators (leukotriene C4, which is metabolized to leukotriene D4 and E4) that are generated after cellular activation. These three lipid mediators are the slow-reacting substances of anaphylaxis that increase vascular permeability, mucus secretion and are potent stimulators of smooth-muscle contraction [6, 7].
Eosinophils normally account for 1-5% of peripheral-blood leukocytes, and the upper limit of the normal range is 0.5×109/L. A marked accumulation of eosinophils (>1.5 × 109/L) is referred to as hypereosinophilia. The normal range for eosinophils and their precursors in the bone marrow is 1-5%. Therefore, the cut-off value for increased bone-marrow eosinophilic granulocytes and their precursors is 6%. A definition of severe marrow eosinophilia has been proposed that requires ≥20% of marrow cells to be eosinophils, with or without peripheral-blood eosinophilia [8, 9]. There as many causes of reactive (non-clonal) eosinophilia [10].
2.1.1. Charcot-Leyden Crystals
Charcot-Leyden crystals occur in association with marked eosinophilia. Charcot-Leyden crystals were first reported in 1853 by Jean-Martin Charcot who found tiny crystals in the cardiac blood and spleen of a patient who died from leukemia. In 1872, Ernst Viktor von Leyden also described colorless crystals in the sputum of patients with asthma. Following the discovery of Charcot-Leyden crystals many conflicting reports appeared as to the chemical nature and significance of these crystals. At that time, it was thought that lysolecithin acylhydrolase, which has lysophospholipase activity, was the constituent protein of Charcot-Leyden crystals. Subsequent investigation revealed galectin-10, a lysophospholipase binding protein, as the constituent protein of Charcot-Leyden crystals. Figure 1 illustrates Charcot-Leyden crystals in association with AML.
2.2. Physiologic Features of Basophils
Basophils normally account for ≤2% of peripheral-blood leukocytes, and the upper limit of the normal range is 0.1×109/L. The normal range for eosinophils and their precursors in the bone marrow is 1-5%. Therefore, the cut-off value for increased bone-marrow eosinophilic granulocytes and their precursors is 6%. Hyperbasophilia occurs when the peripheral-blood basophil count is >1.0×109/L. There are many causes of reactive (non-clonal) basophilia [11]. Persistent basophilia (defined as lasting >8 months) is always suggestive of a neoplastic hematologic disorder. The normal range for basophils and their precursors in the bone marrow is 0-2%.
CD123, commonly expressed in basophils, accounts for the α-subunit of the interleukin-3 receptor (IL-3Rα). Interleukin-3 is the main cytokine for basophilic differentiation and maturation. Other important regulators are GM-CSF, TGF-β, interleukin-5 and thymic stromal lymphopoietin (TSLP) [11]. STAT5 is the effector of interleukin-3 signaling and induces GATA-2 dependent transcriptional activation [12]. Other important transcription factors for basophil development include GATA-1 [13], IRF8 [14], and RUNX1 [15]. Basophils have a short life span (<3 days).
Basophils, like mast cells, carry the high affinity IgE receptor (FcεRI). Basophils, unlike mast cells, do not contain tryptase. Histamine is the most important constituent of basophilic granules [11]. Their metachromatic granules are also rich in proteoglycans such as chondroitins and heparin and a variety of enzymes. They are also able to produce a variety of growth factors such as VEGF and HGF and inflammatory cytokines, namely interleukin-4 and inteleukin-13 [16].
2.2.1. Metachromatic Granules
The presence of granules taking a metachromatic stain means that they are stained pink or red using blue dyes (tolouidine blue or methylene blue). This is due to the large amount of proteoglycans in basophil and mast-cell granules. Toluidine blue stain, which is necessary to reveal the metachromatic character of the granules, stains both the basophilic and mastocytic granules and, therefore, it cannot differentiate between the two [17]. The periodic-acid Schiff stain may aid in the differential diagnosis: basophils typically show a speckled pattern, whereas mast cells exhibit negativity or a very weak reaction [18]. The normal counterpart of the metachromatic blast is thought to be the common progenitor of both the basophilic and mastocytic cell differentiation.
3. AML with Increased Eosinophils (Table 1)
3.1. AML with inv(16) or t(16;16)
Of the chromosomal abnormalities associated with AML with increased marrow eosinophils (i.e. >5% marrow eosinophils), the best example is the inv(16)(p13.1q22) or t(16;16)(p13.1;q22), resulting in the fusion of CBFB at 16q22 to MYH11 at 16p13.1 (CBFB‒MYH11). MYH11 encodes for smooth muscle myosin heavy chain and CBFB codes for the beta subunit of core-binding factor (CBF), a heterodimeric transcription factor that controls the transcription of genes involved in hematopoiesis. The CBFB subunit normally heterodimerizes with RUNX1 (CBFA2), the gene product of RUNX1 and increases its DNA-binding affinity. CBFB‒MYH11 mutant protein does not bind RUNX1 leading to RUNX1 sequestration on cytoskeletal structures in the cytoplasm and rendering it inactive [19, 20]. The exact mechanism by which these alterations cause eosinophilia remains unclear [21, 22].

Table 1.
Subtypes of AML with increased eosinophils.
| AML subtype | Epidemiology | Clinical features | Diagnostic methods | Treatment | Prognosis |
|---|---|---|---|---|---|
| AML with inv(16)(p13.1q22)/ t(16;16)(p13.1;q22); CBFB‒MYH11 | 5-8% of AML cases; more common in young patients | Myelomonocytic (M4) differentiation; abnormal eosinophil morphology (basophilic granules, hypolobated); peripheral eosinophilia uncommon |
RT-PCR Karyotype FISH |
Standard intensive regimen plus gemtuzumab ozogamicin | Favorable |
| AML with t(8;21)(q22;q22); RUNX1‒RUNX1T1 | 1-5% of AML cases; more common in young patients |
Myeloblastic differentiation (M2 or M1); normal eosinophil morphology; excess basophils; peripheral eosinophilia uncommon | RT-PCR Karyotype FISH | Standard intensive regimen plus gemtuzumab ozogamicin | Favorable |
| AML with t(9;12)(q34;p13); ETV6‒ABL1 | Rare; most common in young men | Typically accompanied by peripheral-blood eosinophilia; abnormal eosinophil morphology (coarse eosinophilic and/or basophilic granules) | FISH RT-PCR Karyotype |
Standard chemotherapy plus second generation TKI | Poor prognosis; aggressive disease |
| AML with FIP1L1‒PDGFRA | Most common in young men | Typically accompanied by peripheral-blood eosinophilia; dysplastic eosinophils; frequent mast cells and reticulin fibrosis | FISH RT-PCR |
Standard chemotherapy plus imatinib | High rate of complete remission; increased risk of relapse |
| AML with PDGFRB (5q31-q33) rearrangement | Most common in men | Typically accompanied by peripheral-blood eosinophilia (but less prominent than FIP1L1‒PDGFRA); dysplastic eosinophils | FISH Karyotype | Standard chemotherapy plus imatinib | High rate of complete remission; increased risk of relapse |
| AML with FGFR1 (8p11) rearrangement | Wide range of ages (3-84 years) | Frequent hepatosplenomegaly | FISH Karyotype | TKI, midostaurin, pemigatinib | Poor prognosis |
Abbreviations: AML, acute myeloid leukemia; FISH, fluorescent in situ hybridization; RT-PCR, reverse-transcriptase–polymerase-chain-reaction; TKI, tyrosine kinase inhibitor.
3.1.1. Morphology
AML with inv(16) or t(16;16) morphologically corresponds to acute myelomonocytic leukemia with eosinophilia (M4Eo); rarely, cells with these features can be identified as acute myeloblastic leukemia with maturation (M2). The bone marrow usually shows an increased number of eosinophils at all stages of maturation; they usually amount to ≥5% of non-erythroid cells (Figure 2).
Most importantly, these eosinophils are abnormal, and some have, in addition to the characteristic specific eosinophilic granules, large basophilic (immature) granules and may have a single unsegmented nucleus. The basophilic granules are larger and more irregular, as well as more numerous, than those occasionally seen in immature eosinophils. The anomalous immature granules are mostly evident at the late promyelocyte and myelocyte stages; however, they tend to persist in some band and segmented forms. Unlike normal eosinophils, these cells stain with chloroacetate esterase and periodic-acid Schiff. The peripheral blood is not different from that in other cases of acute myelomonocytic leukemia; eosinophils are not increased in peripheral blood. AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22) is associated with a high rate of complete remission and favorable overall survival rate when treated with intensive induction and consolidation therapy.
3.1.2. Confirmation of Diagnosis
By conventional cytogenetic analysis, inv(16)(p13.1 q22) is a subtle rearrangement that may be overlooked when metaphase preparations are suboptimal. In addition, occasionally, cytological features of AML with abnormal eosinophils may be present without karyotypic evidence of a chromosome 16 abnormality, but with CBFB‒MYH11 nevertheless demonstrated by molecular studies such as reverse-transcriptase–polymerase-chain-reaction (RT-PCR) or fluorescent in situ hybridization (FISH), as shown in Figure 3. Therefore, RT-PCR and/or FISH for CBFB‒MYH11 should be requested in AML cases (typically M4) with proliferation of abnormal eosinophils.
3.2. AML with t(8;21)
AML with t(8;21)(q22;q22) translocation may also present with marrow eosinophilia. The t(8;21)(q22;q22.1) involves RUNX1 (AML1) which encodes the alpha subunit of CBF, and RUNX1T1 (ETO); RUNX1‒RUNX1T1 (AML1‒ETO). The CBF transcription factor is essential for hematopoiesis; transformation by RUNX1‒RUNX1T1 results from transcriptional repression of normal RUNX1 target genes via aberrant recruitment of nuclear transcriptional co-repressor complexes.
3.2.1. Morphology
Most cases of t(8;21) are classified as M2, rarely as M1 or M4. Eosinophils and eosinophil precursors are frequently increased but they lack the cytological or cytochemical abnormalities seen in AML with inv(16) or t(16;16). Thus, cases of M2 with t(8;21) and marrow eosinophilia lack the abnormal granules seen in M4 with eosinophilia (Figure 4).
An excess of basophils is commonly present in AML with t(8;21). Like AML with inv(16) and t(16;16), eosinophils are not increased in the peripheral blood. AML with t(8;21) is associated with a high complete remission rate and favorable overall survival when treated with intensive induction and consolidation therapy.
3.2.2. Confirmation of Diagnosis
Usually, t(8;21)(q22;q22) is readily detected by means of conventional cytogenetic analysis. RT-PCR and FISH may allow earlier detection of RUNX1‒RUNX1T1 in cases of AML with an eosinophilic component without abnormal granules.
3.3. AML with ETV6‒ABL1 (ΤΕL‒ABL1)
The t(9;12)(q34;p13) involves the ETV6 (TEL) gene at 12p13, a transcription factor frequently rearranged in myeloid and lymphoid leukemias, and ABL1 at 9q34. The ETV6‒ABL1 fusion gene leads to the activation ABL1 kinase. ETV6‒ABL1 is a notable rearrangement involved in AML with eosinophilia [23-25]. ETV6‒ABL1 AML is more common in men than women and occurs predominantly between the ages of 20 and 50 years. It generally has an adverse prognosis with poor response to traditional AML therapy or imatinib [26, 27]. Recent reports suggest higher response rates and improved survival outcomes survival with second generation tyrosine-kinase-inhibitor therapy such as dasatinib and nilotinib followed by allogeneic hematopoietic-cell transplantation [27, 28].
3.3.1. Morphology
The ETV6‒ABL1 fusion gene seems to have BCR‒ABL1 like activity and, therefore, patients usually present with a picture that resembles atypical chronic myeloid leukemia (aCML) with marked peripheral-blood eosinophilia. However, this translocation has also been reported in AML (usually M1 or M2) with peripheral eosinophilia [24, 29]. The presence of peripheral eosinophilia and abnormal bone-marrow eosinophils with coarse eosinophilic and/or basophilic granules is the morphologic hallmark of this type of AML.
3.3.2. Confirmation of Diagnosis
The t(9;12)(q34;p13) translocation is very difficult to be identified by conventional cytogenetics since it might result from cryptic translocation or complex chromosomal translocations involving more than two chromosomes at more than two breakpoints and usually requires FISH with a combination of ETV6 and ABL1 probes. However, there are cases in which FISH is not diagnostic (ETV6‒ABL1 occult fusion) and require targeted RT-PCR or next-generation sequencing techniques (e.g. RNA sequencing [RNAseq] or whole-genome sequencing [WGS] for detection of cryptic rearrangements) to be identified [24]. Other techniques such as spectral karyotyping (SKY) have also been used to detect abnormalities involving ETV6 otherwise missed by conventional karyotyping [30, 31].
3.4. AML with PDGFRA, PDGFRΒ, and FGFR1 Rearrangements
These translocations create fusion tyrosine kinases with ligand-independent tyrosine-kinase activity leading to uncontrolled cell proliferation, and stimulation of downstream signaling pathways, including those involving phosphatidylinositol 3-kinase and mitogen-activated protein kinases that promote proliferation and survival. The clinical and hematological features are influenced by the partner gene involved. These disorders typically present as chronic myeloproliferative neoplasms with high-grade peripheral eosinophilia, but may also present as de novo AML.
3.4.1. AML with PDGFRA Rearrangements
PDGFRA related disorders usually present as hypereosinophilic syndrome (HES) of chronic eosinophilic leukemia (CEL). Rarely, they may present as AML with peripheral-blood eosinophilia. The most common abnormality is a fusion of the Fip1-like 1 (FIP1L1) gene to the platelet-derived growth factor receptor α (PDGFRA) gene generated by an interstitial deletion on chromosome 4q12 between the two genes known as CHIC2 locus or LNX locus, with resultant juxtaposition of FIP1L1 and PDGFRA [32]. FIP1L1‒PDGFRA encodes a constitutively activated tyrosine kinase that transforms hematopoietic cells. Because the deletion is small (~800 kb) it is not visible by conventional karyotype and should be sought after by FISH for CHIC2 or LNX deletion as shown in Figure 5, or RT-PCR for identification of FIP1L1‒PDGFRA transcripts. Most patients with FIP1L1‒PDGFRA are men (male:female ratio, 9:1) [9]. PDGFRA translocations may rarely occur with other partners such as BCR and ETV6 [33].
Peripheral-blood morphology is useful in FIP1L1‒PDGFRA myeloid neoplasms because it may show many abnormal eosinophils [10], including:
- eosinophils with a trilobed nucleus or hypersegmented eosinophils;
- unilobed eosinophils (nuclear hyposegmentation);
- eosinophils with reduced or sparse granulation (abnormal granulation);
- eosinophils with smaller than normal granules (microgranulation);
- eosinophils with many cytoplasmic vacuoles due to degranulation (Figure 6).
The FIP1L1‒PDGFRA fusion gene leads to eosinophil proliferation mediated by phosphatidylinositol 3-kinase (PI3K), ERK1/2 and STAT5 signaling pathways [34].
3.4.2. AML with PDGFRB Rearrangements
Chromosomal translocations involving PDGFRB on chromosome 5q33 are much more heterogeneous than PDFGRA. Over 30 partner genes for PDGFRB have been identified, of which ETV6 is the commonest producing the ETV6‒PDGFRB from the t(5;12)(q31-33;p13) translocation [33, 35]. The ETV6‒PDGFRB fusion gene leads to proliferation mediated by STAT5, NF-κB and ERK1/2 signaling [36]. Moreover, a number of genes that are important in the proliferation and differentiation of eosinophils are found in the same 5q31-q35 chromosomal region, including interleukin-3, interleukin-4, interleukin-5, interleukin-13 and granulocyte–macrophage colony-stimulating factor. The morphologic features of PDGFRB related disease are variable but are often those of chronic myelomonocytic leukemia, aCML, or chronic myeloproliferative disease with prominent peripheral-blood eosinophilia. Blastic transformation is usually myeloid, and several de novo cases of AML with ETV6‒PDGFRB and eosinophilia have also been reported. Similar to FIP1L1‒PDGFRA, eosinophils are often dysmorphic in ETV6‒PDGFRB. The most common findings are marked hypogranulation (agranular cytoplasm ≥25%), marked cytoplasmic vacuolization (vacuoles ≥25% of cytoplasm), and unilobed nuclei [37]. PDGFRB rearrangements are usually easy to detect with cytogenetic analysis; however, some cases require FISH for identification (Figure 7) [38].
3.4.3. AML with FGFR1 Rearrangements
Fibroblast growth factor receptor 1 (FGFR1) rearrangements involving the 8p11 locus were first described in 1992 [39, 40]. The most common translocation is t(8;13)(p11;q12) resulting in ZNF198‒FGFR1 [41]. Many other partners have been identified, also resulting in constitutive activation of FGFR1 tyrosine kinase [42 -44]. These patients present a wide variety of myeloid and lymphoid neoplasms with peripheral-blood eosinophilia (so-called 8p11 myeloid/lymphoid neoplasms) [45].
Recognizing AML with eosinophilia due to PDGFRA and PDGFRΒ rearrangement is important, because the aberrant tyrosine kinase activity can make the disease responsive to tyrosine kinase inhibitors such as imatinib. Therefore, we suggest that molecular genetic analysis (FISH and RT-PCR) for PDGFRA and PDGFRΒ should be carried out in cases of AML with peripheral eosinophilia.
Imatinib can be used effectively to treat patients carrying PDGFRA and PDGFRB translocations. The dosage ranges from 100 mg (for PDGFRA translocations) to 400 mg (for PDGFRB translocations) per day. The study by Metzgeroth and colleagues [46] reported encouraging results with imatinib in 17 patients with AML carrying these translocations: complete remission was achieved in all patients and median overall survival was 88% at 65 months. Complete molecular remissions were achieved after a median of 5 months of imatinib treatment (range 3-32). Other studies, however, have shown worse outcomes in AML with PDGFRA and PDGFRB translocations treated with imatinib, particularly in patients with additional cytogenetic (e.g. complex karyotype) or molecular abnormalities, who had a median overall survival of 12.5 months (range, 2-20) [32, 47-49]. Treatment-emergent imatinib resistance was mediated by mutations in the PDGFRα or PDGFRβ kinase domain. Treatment of AML with PDGFRA and PDGFRB rearrangement should involve a combination of imatinib plus chemotherapy, with consideration of allogeneic stem-cell transplantation. In patients with high-grade peripheral eosinophilia, glucocorticoids should be used for 7-10 days in order to avoid rapid eosinophil degranulation and associated risk of inflammatory cardiac injury after initiation of imatinib, especially in those with a history of cardiovascular disease and/or elevated cardiac troponin levels [33].
The treatment of AML with FGFR1 rearrangement is far from standardized owing to the poor results obtained with present-day therapy. First and second generation tyrosine kinase inhibitors have been used with suboptimal results [50]. Recently, pemigatinib, an FGFR2 inhibitor used in patients with FGFR2-rearranged cholangiocarcinoma, received FDA approval for relapsed/refractory myeloid and/or lymphoid neoplasms with FGFR1 rearrangements. Allogeneic hematopoietic-cell transplantation is indicated for these patients.
3.5. Rare Translocations Involved in AML with Eosinophilia
Several other gene rearrangements involving JAK2 (located on chromosome 9p24) and FLT3 (located on chromosome 13q12) with may rarely be manifested as AML with clonal eosinophilia [51]. These JAK2 translocations (e.g. PCM1‒JAK2, ETV6‒JAK2, and BCR‒JAK2) and FLT3 translocations (e.g. ETV6‒FLT3, BCR‒FLT3, and FLT3‒TRIP11) are less common than PDGFRB translocations, generally manifest as chronic myeloproliferative disease (e.g. CEL, aCML or CMML with eosinophilia), acute lymphoblastic leukemia, and very rarely as AML with eosinophilia. These translocations may be identified by conventional cytogenetic analysis but cryptic rearrangements require FISH or sequencing assays. The presence of +9/+9p chromosomal abnormalities in the context of AML with eosinophilia may be a clue for cryptic JAK2 structural rearrangements. Cases of AML with JAK2 rearrangements e.g. PCM1‒JAK2 resulting from t(8;9)(p22;p24.1) may respond to JAK inhibitors such as ruxolitinib. There have even been reports of treatment-free remission with ruxolitinib [52]. Cases with FLT3 rearrangement may respond to FLT3 inhibitors. Allogeneic hematopoietic-cell transplantation seems to be the best choice for these patients.
3.6. The Authors’ Recommendation for a Practical Approach to AML with Increased Eosinophils
It is not uncommon to find increased marrow eosinophils in patients with AML in the absence of peripheral-blood eosinophilia, particularly in patients with the myelomonocytic subtype. In many cases this is a non-specific finding occurring in association with non-specific cytogenetic lesions such as 7q deletion or normal-karyotype, but this finding may also indicate recurrent translocations with immediate therapeutic and prognostic relevance such as CBFB‒MYH11 or RUNX1‒RUNX1T1. Of note, the likelihood of AML with CBFB‒MYH11 is increased in the presence of abnormal eosinophils containing both eosinophilic and basophilic staining granules. Also, myeloid blast phase of CML or BCR‒ABL1 positive AML with an eosinophilic component should always be borne in mind since about 5% of CML cases are diagnosed in blast phase without a recognized chronic phase. Obviously, the identification of BCR‒ABL1 has important therapeutic implications. Therefore, we recommend RT-PCR (or FISH) testing for CBFB‒MYH11, RUNX1‒RUNX1T1, and BCR‒ABL1 in all cases of AML with increased eosinophils.
If these translocations are negative or there is peripheral eosinophilia or a history of leukocytosis with eosinophilia, we recommend FISH for ETV6‒ABL1, FIP1L1‒PDGFRA, PDGFRB, or FGFR1 rearrangements. If FISH testing is negative but clinical suspicion is high, we recommend RT-PCR for FIP1L1‒PDGFRA and ETV6‒ABL1. In cases in which none of the tests described above is positive, additional testing with JAK2 FISH, FLT3 FISH, and RNAseq for cryptic translocations (if available) may be considered.
4. AML with Increased Basophils
In assessing leukemias with a prominent basophilic component, it is important to distinguish whether the basophils constitute a mature or immature population. The best example of the latter is acute basophilic leukemia, a separate subtype of AML in which the primary differentiation is to basophils. AML with t(6;9) is associated with basophilia in 42-62% of the cases. Core-binding factor AML can also produce basophilia. Moreover, many myeloid cancers can transform to AML with hyperbasophilia including CML, aCML, Ph-negative myeloproliferative neoplasms, and myelodysplastic syndromes.
4.1. Differential Diagnosis of Leukemias with Basophilic Granules (Table 2)
As mentioned, the presence of blast cells with dark basophilic granules should raise suspicion for acute basophilic leukemia. This condition should always be distinguished from the basophilic myeloid blast crisis of CML (“secondary acute basophilic leukemia”) as well as from AML with DEK‒NUP214 and AML with RUNX1‒RUNX1T1. It should also be differentiated from acute mast-cell leukemia. Acute promyelocytic leukemia (APL) should always be borne in mind when one sees a case of AML with blasts containing dark granules.
4.1.1. Basophilic Blast Phase of CML
In most cases of blast-phase CML, the blast lineage is myeloblastic, but may also be monocytic, megakaryocytic, erythroid, eosinophilic, or basophilic (or any combination thereof). Rarely, patients may present in blast phase with a morphologic picture identical to de novo acute basophilic leukemia. Thus, RT-PCR for BCR‒ABL1 should always be performed in any case of AML with a prominent basophilic component.
Basophils of blast phase of CML may display dysplastic features such as reduced granulation [53] and tryptase production [54]. The source of the basophilic blast phase of CML may be the CFU-Bas/Eo hybrid progenitor. IKZF1 mutations that produce either loss of IKAROS or dominant negative isoforms, have been described in CML lymphoblastic crisis [55, 56]. Disruption of IKAROS activity in primitive CML cells mimics myeloid disease progression with enhanced STAT5 activation and shifted granulopoiesis to the basophilic lineage [57].
4.1.2. AML with t(6;9)(p23;q34.1); DEK‒NUP214
AML with t(6;9)(p23;q34.1) shows primarily myeloblastic or myelomonocytic differentiation (M2 or M4). It is accompanied by marrow basophilia consisting of mature, dysplastic basophils in 42-62% of the cases, and it is often associated with prominent multilineage dysplasia [58]. From a flow-cytometry point of view, the blasts have a non-specific myeloid immuno-phenotype (MPO+, CD34+, CD13+, CD33+, CD117+, CD38+, CD123+, and HLA-DR+), whereas basophils can be seen as separate clusters of cells positive for CD123, CD33, and CD38 but negative for HLA-DR. TdT expression is common in AML with t(6;9) (50%) [59]. Notably, the presence of FLT3-ITD mutations is very common in AML with t(6;9)(p23;q34.1), occurring in approximately 75% of patients [60]. The t(6;9)(p23;q34.1) occurs as a sole karyotypic abnormality in most cases but, rarely, it occurs in association with a complex karyotype. AML with t(6;9)(p23;q34.1) has a poor prognosis and allogeneic stem cell transplantation is indicated in first complete remission. Given the high frequency of FLT3-ITD in AML with t(6;9), patients may benefit from FLT3 inhibitors.
4.1.3. AML with t(8;21)(q22;q22.1); RUNX1‒RUNX1T1
Core-binding factor AML can produce marrow basophilia: AML with inv(16) or t(16;16) is characterized by the presence of the abnormal eosinophils with mixed eosinophilic and basophilic granules but AML with t(8;21) often has a true basophilic component [58]. In fact, AML with t(8;21) can mimic or masquerade as acute basophilic leukemia [61]. Therefore, RT-PCR (or FISH) testing for RUNX1‒RUNX1T1 should be performed in all cases of AML with prominent basophils.
4.1.4. Acute Mast-Cell Leukemia
Acute mast-cell leukemia is the leukemic variant of systemic mastocytosis in which peripheral-blood smears contain ≥10% and bone-marrow aspirates ≥20% mast cells. A diagnosis of aleukemic mast-cell leukemia is made if the percentage of mast cells in the peripheral-blood smear is <10% (Figure 8) [58]. Unlike in indolent systemic mastocytosis, the mast cells in mast-cell leukemia are often round rather than spindle-shaped. Some of these mast cells may exhibit bilobed nuclei (“promastocytes”). Rare cases in which the mast cells are mature-appearing and the clinical course less aggressive, constitute chronic mast-cell leukemia.
Strong expression of CD117 and positive staining for tryptase are of great diagnostic value in the diagnosis of acute mast-cell leukemia. Acute mast-cell leukemia may also express myeloid antigens such as CD13 and CD33 and sometimes CD203c, CD30 and CD38 [17]. Typically, in systemic mastocytosis, the neoplastic mast cells show dual expression of CD2 and CD25. In mast-cell leukemia, however, there is often loss of one or both antigens: loss of CD25 occurs in 25%, loss of CD2 in 42%, and 30% of patients are negative for both CD2 and CD25 [62]. Absence of C-KIT D816V is more common in CD2 and/or CD25 negative cases [62]. Bone marrow biopsy shows a diffuse, dense infiltration with atypical mast cells that are tryptase-positive. The majority of patients with mast-cell leukemia have no skin lesions. Acute mast-cell leukemia is an aggressive disease with poor prognosis (median overall survival is ≤12 months). Features helpful in distinguishing patients with acute basophilic leukemia from those with acute mast-cell leukemia are given in Table 3.
4.1.5. Basophilic Variant of APL; PML‒RARA
The basophilic subtype of APL (M3b) was first described in 1982 as a “hyperbasophilic microgranular variant” of APL [63]. It is characterized by neoplastic promyelocytes with unusual nuclear lobulation and deeply basophilic cytoplasm containing few or dark granules. The cytoplasm is scanty and may have cytoplasmic projections or blebs. Like the microgranular subtype of APL, CD2 is positive in the subtype with basophilic cells. In addition, APL with basophil differentiation with blasts containing large basophilic granules has been described [18, 64]. Aberrations of the 12p13 locus―in addition to the t(15;17)(q22;q11-12)―have been described in M3b [18, 64]. Relapse of APL after all-trans retinoic acid (ATRA) therapy with M3b morphology may occur. APL with eosinophilic differentiation has also been described. Recognition of the less common, atypical forms of APL with basophilic blasts is important because of the unique response of this disease to retinoic acid and arsenic trioxide.
4.1.6. Acute Basophilic Leukemia
Although acute basophilic leukemia has long been recognized [65], it was not until the 2008 edition of WHO classification of myeloid neoplasms that it became a distinct clinicopathologic entity classified within the category of AML, not otherwise specified. It is a very rare disease, accounting for <1% of AML cases [58].
The characteristic morphologic feature of acute basophilic leukemia is the presence of blasts carrying coarse basophilic granules (see Figure 9 for an example).
The most striking feature is the presence of blast cells with coarse basophilic granules, raising suspicion for ABL. ABL can be identified by expression of either CD123 or CD203c by cells that do not express CD117. The absence of myeloperoxidase (MPO) rules out the possibility of acute promyelocytic leukemia (M3b). In this case, positivity for CD11b and CD123 and the absence of CD117, strongly suggested a diagnosis of ABL. PML-RARA, BCR-ABL1, RUNX1-RUNX1T1, CBFβ-MYH11, and C-KIT D816V were negative. NPM1 and FLT3-ITD, FLT3-TKD mutations were also negative. A peripheral-blood sample for cytogenetic analysis showed no abnormalities (46, XY). The patient’s symptoms can be explained on the basis of hyperhistaminemia. Accordingly, the histamine levels were found to be 710 pg/ml (normal, 0-90 pg/ml). The patient was treated with high doses of two H1 inhibitors, an H2 inhibitor, esomeprazole, and methylprednisone, before initiation of induction chemotherapy.
According to WHO classification [51], there are three diagnostic requirements for acute basophilic leukemia: (1) blasts and mature/immature basophils with metachromatic granules (as shown by tolouidine blue stain); (2) blast cytochemistry negative for myeloperoxidase (MPO), Sudan Black B, and non-specific esterase (ANAE); and (3) absence of strong CD117 expression. Valent and co-workers have proposed simpler diagnostic criteria for acute basophilic leukemia, including the presence of myeloid blasts and metachromatic blasts ≥20% and basophils ≥40% of total nucleated bone-marrow or peripheral-blood cells.
Leukemic cells express antigens of myeloid differentiation such as CD13 and CD33 but are MPO negative [66, 67]. CD34 and HLA-DR show variable expression [68]. Antigens of basophilic differentiation include CD123, CD203c, CD9, IgE receptor (FceRI), and CD11b [69]. Aberrant expression of CD4, TdT, and CD22 has also been reported in acute basophilic leukemia [58, 70]. CD25 is usually negative or weakly expressed [66].
On morphological grounds it is not possible to make a differential diagnosis of acute basophilic leukemia from acute mast-cell leukemia, since blast cells with metachromatic granules are present in both disorders. Specific esterase (chloroacetate esterase [ChlorE]) staining is helpful in distinguishing basophilic from mastocytic granules. It reacts with mastocytic granules but does not react with basophilic granules [71]. Electron microscopy, if available, is also helpful in distinguishing the lineage of the metachromatic blasts: the presence of Θ granules, i.e. electron dense particles that carry a single fold of their membrane (“theta” character), is typical of basophilic differentiation. In contrast, mast cells carry four different types of granules (crystal-rich, with multiple membrane folds, solid-dense granules, and non-dense granules) without theta character [67]. Expression of CD123 and/or CD11b indicates basophil differentiation, whereas tryptase and/or CD117 indicate mast-cell origin. Markers of systemic mastocytosis i.e. CD2 and CD25 are less helpful [62].
Owing to its rarity, little is known about cytogenetic lesions in acute basophilic leukemia. One abnormality occurring in male infants with acute basophilic leukemia is t(X;6)(p11;q23) [72]. This translocation leads to MYB‒GATA1 fusion which disrupts the translational regulation by GATA1. Since male patients have only one copy of GATA1 on their X chromosome, GATA1 dependent cellular differentiation is completely abrogated. On the other hand, the chimeric protein shows great intracellular stability and retains MYB function. MYB‒GATA1 mutant protein is a positive transcriptional regulator of both interleukin 1 receptor-like 1 (IL1RL1) and NTRK1, which promote basophilic differentiation. Other chromosomal alterations that have been reported include t(16;21)(p11;q22), which creates the FUS‒ERG fusion gene and chromosomal aberrations of the 12p13 locus involving ETV6 e.g. 12p13 deletion [73, 74]. Chromosome 17 abnormalities as well as mutations in TP53, TET2 and NPM1 have also been reported [75].
Like other AML subtypes, patients with acute basophilic leukemia present with features related to bone-marrow failure and may or may not have circulating blasts. Skin involvement, hepatosplenomegaly, lytic lesions, and symptoms related to hyperhistaminemia may be present. Histamine, an autocrine and paracrine vasoactive hormone, is found in abundance in the metachromatic granules of basophils. Thus, erythematous cutaneous reactions are frequently seen in acute basophilic leukemia. Moreover, histamine promotes hydrochloric acid production in the stomach, causing peptic ulcers and gastritis. Lytic and osteoporotic lesions are also common (histamine affects osteoblasts, inducing RANKL expression which directly activates osteoclasts [76]). Other symptoms suggestive of hyperhistaminemia include diarrhea, malabsorption, abdominal pain, marked bronchospasm, nausea, and migraine. Induction chemotherapy can worsen or elicit such symptoms as a consequence of the chemotherapy-induced cell lysis and release of large amounts of histamine in the circulation. Serious complications of hyperhistaminemia during induction chemotherapy include anaphylactic shock, status asthmaticus, pulmonary edema, capillary leak syndrome, arrhythmias, heart failure, gastrointestinal bleeding, hepatic and coagulation abnormalities [77-80]. Whereas the anti-leukemic regimens do not differ from standard AML, it is crucial that patients with acute basophilic leukemia be given H1 and H2 inhibitors, proton-pump inhibitors (PPIs), and corticosteroids to abrogate or treat the effects or hyperhistaminemia. The cases observed have generally been associated with a poor prognosis due to both the occurrence of allergic reactions during induction chemotherapy and disease refractoriness.
5. Conclusions
In a small percentage of cases of AML, increased eosinophils and/or basophils are present in the cytologic material, usually in the bone marrow and sometimes in the peripheral blood. This is often a puzzling diagnostic situation that requires special investigation with RT-PCR and FISH for specific genetic abnormalities of clinical relevance.
Author Contributions
All authors have contributed to the writing of the manuscript. All authors have read and agreed to the final version of the manuscript.
Funding
There was no external funding for this research from any funding agency in the public, commercial or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Metcalf D. Hematopoietic regulators: redundancy or subtlety? Blood. 1993, 82, 3515-3523.
- Mack E.A.; Pear W.S. Transcription factor and cytokine regulation of eosinophil lineage commitment. Curr Opin Hematol. 2020, 27, 27-33. [CrossRef]
- Tenen D.G. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003, 3, 89-101. [CrossRef]
- Kato M.; Kephart G.M.; Talley N.J.; Wagner J.M.; Sarr M.G.; Bonno M.; McGovern T.W.; Gleich G.J. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec. 1998, 252, 418-425. [CrossRef]
- Hoffman R.; Benz E.; Silberstein L.; Heslop H.; Weitz J.; Anastasi J.; Salama M. Hematology: Basic Principles and Practice. Elsevier: Philadelphia, United States, 2018; pp. 330-331. [CrossRef]
- Hoffbrand V.; Higgs R.; Keeling D.; Mehta A. Postgraduate haematology. John Wiley & Sons: Chichester, England, 2016; pp. 260-262.
- Acharya K.R.; Ackerman S.J. Eosinophil granule proteins: form and function. J Biol Chem. 2014, 289, 17406-17415. [CrossRef]
- Valent P.; Klion A.D.; Horny HP.; Roufosse F.; Gotlib J.; Weller P.F.; Hellmann A.; Metzgeroth G.; Leiferman K.M.; Arock M.; Butterfield J.H.; Sperr W.R.; Sotlar K.; Vandenberghe P.; Haferlach T.; Simon H.U.; Reiter A.; Gleich G.J. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012, 130, 607-612. [CrossRef]
- Shomali W.; Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022, 97, 129-148. [CrossRef]
- Liapis K. Approach to eosinophilia. Haema. 2019, 10, 118‒128.
- Valent P.; Sotlar K.; Blatt K.; Hartmann K.; Reiter A.; Sadovnik I.; Sperr W.R.; Bettelheim P.; Akin C.; Bauer K.; George T.I.; Hadzijusufovic E.; Wolf D.; Gotlib J.; Mahon F.X.; Metcalfe D.D.; Horny HP.; Arock M. Proposed diagnostic criteria and classification of basophilic leukemias and related disorders. Leukemia. 2017, 31, 788-797. [CrossRef]
- Sasaki H.; Kurotaki D.; Tamura T. Regulation of basophil and mast cell development by transcription factors. Allergol Int. 2016, 65, 127-134. [CrossRef]
- Nei Y.; Obata-Ninomiya K.; Tsutsui H.; Ishiwata K.; Miyasaka M.; Matsumoto K.; Nakae S.; Kanuka H.; Inase N.; Karasuyama H. GATA-1 regulates the generation and function of basophils. Proc Natl Acad Sci U S A. 2013, 110, 18620-18625. [CrossRef]
- Sasaki H.; Kurotaki D.; Osato N.; Sato H.; Sasaki I.; Koizumi S.; Wang H.; Kaneda C.; Nishiyama A.; Kaisho T.; Aburatani H.; Morse H.C. 3rd.; Ozato K.; Tamura T. Transcription factor IRF8 plays a critical role in the development of murine basophils and mast cells. Blood. 2015, 125, 358-69. [CrossRef]
- Mukai K.; BenBarak M.J.; Tachibana M.; Nishida K.; Karasuyama H.; Taniuchi I.; Galli SJ. Critical role of P1-Runx1 in mouse basophil development. Blood. 2012, 120, 76-85. [CrossRef]
- Chirumbolo S. State-of-the-art review about basophil research in immunology and allergy: is the time right to treat these cells with the respect they deserve? Blood Transfus. 2012, 10, 148-164. [CrossRef]
- Lichtman M.A.; Segel G.B. Uncommon phenotypes of acute myelogenous leukemia: basophilic, mast cell, eosinophilic, and myeloid dendritic cell subtypes: a review. Blood Cells Mol Dis. 2005, 35, 370-383. [CrossRef]
- Tallman M.S.; Hakimian D.; Snower D.; Rubin C.M.; Reisel H.; Variakojis D. Basophilic differentiation in acute promyelocytic leukemia. Leukemia. 1993, 7, 521-526.
- Adya N.; Stacy T.; Speck N.A.; Liu P.P. The leukemic protein core binding factor beta (CBFbeta)-smooth-muscle myosin heavy chain sequesters CBFalpha2 into cytoskeletal filaments and aggregates. Mol Cell Biol. 1998, 18, 7432-7443. [CrossRef]
- Kanno Y.; Kanno T.; Sakakura C.; Bae S.C.; Ito Y. Cytoplasmic sequestration of the polyomavirus enhancer binding protein 2 (PEBP2)/core binding factor alpha (CBFalpha) subunit by the leukemia-related PEBP2/CBFbeta-SMMHC fusion protein inhibits PEBP2/CBF-mediated transactivation. Mol Cell Biol. 1998, 18, 4252-4261. [CrossRef]
- Haferlach T.; Winkemann M.; Löffler H.; Schoch R.; Gassmann W.; Fonatsch C.; Schoch C.; Poetsch M.; Weber-Matthiesen K.; Schlegelberger B. The abnormal eosinophils are part of the leukemic cell population in acute myelomonocytic leukemia with abnormal eosinophils (AML M4Eo) and carry the pericentric inversion 16: a combination of May-Grünwald-Giemsa staining and fluorescence in situ hybridization. Blood. 1996, 87, 2459-2463. [CrossRef]
- Xiao W.; Yabe M.; Offin M.; Khattar P.; Baik J.; Daley R.J.; Pappacena J.J.; Roshal M.; Zhang Y.; Tallman M.S.; Cai SF. Evolution of a chemosensitive core-binding factor AML into an aggressive leukemia with eosinophilic differentiation. Blood Adv. 2018, 2, 1517-1521. [CrossRef]
- La Starza R.; Trubia M.; Testoni N.; Ottaviani E.; Belloni E.; Crescenzi B.; Martelli M.; Flandrin G.; Pelicci P.G.; Mecucci C. Clonal eosinophils are a morphologic hallmark of ETV6/ABL1 positive acute myeloid leukemia. Haematologica. 2002, 87, 789-794.
- Zaliova M.; Moorman A.V.; Cazzaniga G.; Stanulla M.; Harvey R.C.; Roberts K.G.; Heatley S.L.; Loh M.L.; Konopleva M.; Chen I.M.; Zimmermannova O, Schwab C.; Smith O.; Mozziconacci M.J.; Chabannon C.; Kim M.; Frederik Falkenburg J.H.; Norton A.; Marshall K.; Haas O.A.; Starkova J.;Stuchly J.; Hunger SP.; White D.; Mullighan C.G.; Willman CL.; Stary J.; Trka J.; Zuna J. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica. 2016, 101, 1082-1093. [CrossRef]
- Tirado C.A.; Siangchin K.; Shabsovich D.S.; Sharifian M.; Schiller G. A novel three-way rearrangement involving ETV6 (12p13) and ABL1 (9q34) with an unknown partner on 3p25 resulting in a possible ETV6-ABL1 fusion in a patient with acute myeloid leukemia: a case report and a review of the literature. Biomark Res. 2016, 4, 16. [CrossRef]
- Park J.; Kim M.; Lim J.; Kim Y.; Han K.; Kim J.S.; Lee S.; Kim H.J.; Min W.S. Variant of ETV6/ABL1 gene is associated with leukemia phenotype. Acta Haematol. 2013, 129, 78-82. [CrossRef]
- Yao J.; Xu L.; Aypar U.; Meyerson H.J.; Londono D.; Gao Q.; Baik J.; Dietz J.; Benayed R.; Sigler A.; Yabe M.; Dogan A.; Arcila M.E.; Roshal M.; Zhang Y.; Mauro M.J.; Xiao W. Myeloid/lymphoid neoplasms with eosinophilia/ basophilia and ETV6-ABL1 fusion: cell-of-origin and response to tyrosine kinase inhibition. Haematologica. 2021, 106, 614-618. [CrossRef]
- Schwaab J.; Naumann N.; Luebke J.; Jawhar M.; Somervaille TCP.; Williams M.S.; Frewin R.; Jost P.J.; Lichtenegger F.S.; La Rosée P.; Storch N.; Haferlach T.; Horny HP.; Fabarius A.; Haferlach C.; Burchert A.; Hofmann W.K.; Cross NCP.; Hochhaus A.; Reiter A.; Metzgeroth G. Response to tyrosine kinase inhibitors in myeloid neoplasms associated with PCM1-JAK2, BCR-JAK2 and ETV6-ABL1 fusion genes. Am J Hematol. 2020, 95, 824-833. [CrossRef]
- Golub T.R.; Goga A.; Barker G.F.; Afar D.E.; McLaughlin J.; Bohlander S.K.; Rowley J.D.; Witte O.N.; Gilliland D.G. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol. 1996, 16, 4107-4116. [CrossRef]
- Odero M.D.; Carlson K.; Calasanz M.J.; Lahortiga I.; Chinwalla V.; Rowley J.D. Identification of new translocations involving ETV6 in hematologic malignancies by fluorescence in situ hybridization and spectral karyotyping. Genes Chromosomes Cancer. 2001, 31, 134-142. [CrossRef]
- Odero M.D.; Carlson K.M.; Calasanz M.J.; Rowley J.D. Further characterization of complex chromosomal rearrangements in myeloid malignancies: spectral karyotyping adds precision in defining abnormalities associated with poor prognosis. Leukemia. 2001, 15, 1133-1136. [CrossRef]
- Cools J.; DeAngelo D.J.; Gotlib J.; Stover E.H.; Legare R.D.; Cortes J.; Kutok J.; Clark J.; Galinsky I.; Griffin J.D.; Cross N.C.; Tefferi A.; Malone J.; Alam R.; Schrier S.L.; Schmid J.; Rose M.; Vandenberghe P.; Verhoef G.; Boogaerts M.; Wlodarska I.; Kantarjian H.; Marynen P.; Coutre S.E.; Stone R.; Gilliland D.G. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003, 348, 1201-1214. [CrossRef]
- Gotlib J.; Cools J. Five years since the discovery of FIP1L1-PDGFRA: what we have learned about the fusion and other molecularly defined eosinophilias. Leukemia. 2008, 22, 1999-2010. [CrossRef]
- Buitenhuis M.; Verhagen L.P.; Cools J.; Coffer P.J. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer Res. 2007, 67, 3759-3766. [CrossRef]
- Maccaferri M.; Pierini V.; Di Giacomo D.; Zucchini P.; Forghieri F.; Bonacorsi G.; Paolini A.; Quadrelli C.; Giacobbi F.; Fontana F.; Cappelli G.; Potenza L.; Marasca R.; Luppi M.; Mecucci C. The importance of cytogenetic and molecular analyses in eosinophilia-associated myeloproliferative neoplasms: an unusual case with normal karyotype and TNIP1- PDGFRB rearrangement and overview of PDGFRB partner genes. Leuk Lymphoma. 2017, 58, 489-493. [CrossRef]
- Montano-Almendras C.P.; Essaghir A.; Schoemans H.; Varis I.; Noël L.A.; Velghe A.I.; Latinne D.; Knoops L.; Demoulin JB. ETV6-PDGFRB and FIP1L1-PDGFRA stimulate human hematopoietic progenitor cell proliferation and differentiation into eosinophils: the role of nuclear factor-κB. Haematologica. 2012, 97, 1064-1072. [CrossRef]
- Goasguen J.E.; Bennett J.M.; Bain B.J.; Brunning R.; Zini G.; Vallespi M.T.; Tomonaga M.; Locher C. International Working Group on Morphology of MDS. The role of eosinophil morphology in distinguishing between reactive eosinophilia and eosinophilia as a feature of a myeloid neoplasm. Br J Haematol. 2020, 191, 497-504. [CrossRef]
- Savage N.; George T.I.; Gotlib J. Myeloid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, and FGFR1: a review. Int J Lab Hematol. 2013, 35, 491-500. [CrossRef]
- Abruzzo L.V.; Jaffe E.S.; Cotelingam J.D.; Whang-Peng J.; Del Duca V. Jr.; Medeiros L.J. T-cell lymphoblastic lymphoma with eosinophilia associated with subsequent myeloid malignancy. Am J Surg Pathol. 1992, 16, 236-245. [CrossRef]
- Bain J.; Horny HP.; Arber A.; Tefferi A.; Hasserjian P. Myeloid/Lymphoid Neoplasms with Eosinophilia and Rearrangement of PDGFRA, PDGFRB or FGFR1, or with PCM1-JAK2. In book WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Rev. 4th ed.; Swerdlow S.; Campo E.; Harris NL.; Jaffe E.; Pileri S.; Stein H.; Thiele J, Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, pp 72-79.
- Jackson C.C.; Medeiros L.J.; Miranda R.N. 8p11 myeloproliferative syndrome: a review. Hum Pathol. 2010, 41, 461-476. [CrossRef]
- Baer C.; Muehlbacher V.; Kern W.; Haferlach C.; Haferlach T. Molecular genetic characterization of myeloid/lymphoid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. Haematologica. 2018, 103, 348-350. [CrossRef]
- Lee H.; Kim M.; Lim J.; Kim Y.; Han K.; Cho B.S.; Kim H.J. Acute myeloid leukemia associated with FGFR1 abnormalities. Int J Hematol. 2013, 97, 808-812. [CrossRef]
- McKeague S.J.; O'Rourke K.; Fanning S.; Joy C.; Throp D.; Adams R.; Harvey Y.; Keng T.B. Acute leukemia with cytogenetically cryptic FGFR1 rearrangement and lineage switch during therapy: A case report and literature review. Am J Clin Pathol. 2023, 19, 135. [CrossRef]
- Liapis K.; Kousiafes D.; Papanikolaou A.; Pagratis P.; Kokkini G. The 8p11 myeloid and lymphoid neoplasm. Eur J Haematol. 2011, 87, 471-472. [CrossRef]
- Metzgeroth G.; Walz C.; Score J.; Siebert R.; Schnittger S.; Haferlach C.; Popp H.; Haferlach T.; Erben P.; Mix J.; Müller M.C.; Beneke H.; Müller L.; Del Valle F.; Aulitzky W.E.; Wittkowsky G.; Schmitz N.; Schulte C.; Müller-Hermelink K.; Hodges E.; Whittaker S.J.; Diecker F.; Döhner H.; Schuld P.; Hehlmann R.; Hochhaus A.; Cross NC.; Reiter A. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophilia-associated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia. 2007, 21, 1183-1188. [CrossRef]
- Sorour Y.; Dalley C.D.; Snowden J.A.; Cross N.C.; Reilly J.T. Acute myeloid leukaemia with associated eosinophilia: justification for FIP1L1-PDGFRA screening in cases lacking the CBFB-MYH11 fusion gene. Br J Haematol. 2009, 146, 225-227. [CrossRef]
- Lierman E.; Michaux L.; Beullens E.; Pierre P.; Marynen P.; Cools J.; Vandenberghe P. FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009, 23, 845-851. [CrossRef]
- Jawhar M.; Naumann N.; Schwaab J.; Baurmann H.; Casper J.; Dang T.A.; Dietze L.; Döhner K.; Hänel A.; Lathan B.; Link H.; Lotfi S.; Maywald O.; Mielke S.; Müller L.; Platzbecker U.; Prümmer O.; Thomssen H.; Töpelt K.; Panse J.; Vieler T.; Hofmann WK.; Haferlach T.; Haferlach C.; Fabarius A.; Hochhaus A.; Cross NCP.; Reiter A.; Metzgeroth G. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann Hematol. 2017, 96, 1463-1470. [CrossRef]
- Arber D.A.; Orazi A.; Hasserjian R.P.; Borowitz M.J.; Calvo K.R.; Kvasnicka H.M.; Wang S.A.; Bagg A.; Barbui T.; Branford S.; Bueso-Ramos C.E.; Cortes J.E.; Dal Cin P.; DiNardo C.D.; Dombret H.; Duncavage E.J.; Ebert B.L.; Estey E.H.; Facchetti F.; Foucar K.; Gangat N.; Gianelli U.; Godley L.A.; Gökbuget N.; Gotlib J.; Hellström-Lindberg E.; Hobbs G.S.; Hoffman R.; Jabbour E.J.; Kiladjian J.J.; Larson R.A.; Le Beau M.M.; Loh M.L.; Löwenberg B.; Macintyre E.; Malcovati L.; Mullighan C.G.; Niemeyer C.; Odenike O.M.; Ogawa S.; Orfao A.; Papaemmanuil E.; Passamonti F.; Porkka K.; Pui C.H.; Radich J.P.; Reiter A.; Rozman M.; Rudelius M.; Savona M.R.; Schiffer C.A.; Schmitt-Graeff A.; Shimamura A.; Sierra J.; Stock W.A.; Stone R.M.; Tallman M.S.; Thiele J.; Tien H.F.; Tzankov A.; Vannucchi A.M.; Vyas P.; Wei A.H.; Weinberg O.K.; Wierzbowska A.; Cazzola M.; Döhner H.; Tefferi A. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022, 140, 1200-1228. [CrossRef]
- Khoury J.D.; Solary E.; Abla O.; Akkari Y.; Alaggio R.; Apperley J.F.; Bejar R.; Berti E.; Busque L.; Chan JKC.; Chen W.; Chen X.; Chng W.J.; Choi J.K.; Colmenero I.; Coupland S.E.; Cross NCP.; De Jong D.; Elghetany M.T.; Takahashi E.; Emile J.F.; Ferry J.; Fogelstrand L.; Fontenay M.; Germing U.; Gujral S.; Haferlach T.; Harrison C.; Hodge JC.; Hu S.; Jansen J.H.; Kanagal-Shamanna R.; Kantarjian H.M.; Kratz C.P.; Li X.Q.; Lim M.S.; Loeb K.; Loghavi S.; Marcogliese A.; Meshinchi S.; Michaels P.; Naresh K.N.; Natkunam Y.; Nejati R.; Ott G.; Padron E.; Patel K.P.; Patkar N.; Picarsic J.; Platzbecker U.; Roberts I.; Schuh A.; Sewell W.; Siebert R.; Tembhare P.; Tyner J.; Verstovsek S.; Wang W.; Wood B.; Xiao W.; Yeung C.; Hochhaus A. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022, 36, 1703-1719. [CrossRef]
- Metzgeroth G.; Schwaab J.; Naumann N.; Jawhar M.; Haferlach T.; Fabarius A.; Hochhaus A.; Hofmann W.K.; Cross NCP.; Reiter A. Treatment-free remission in FIP1L1-PDGFRA-positive myeloid/lymphoid neoplasms with eosinophilia after imatinib discontinuation. Blood Adv. 2020, 4, 440-443. [CrossRef]
- Valent P.; Horny HP.; Arock M. The underestimated role of basophils in Ph+ chronic myeloid leukaemia. Eur J Clin Invest. 2018, 48, e13000. [CrossRef]
- Samorapoompichit P.; Kiener H.P.; Schernthaner G.H.; Jordan J.H.; Agis H.; Wimazal F.; Baghestanian M.; Rezaie-Majd A.; Sperr W.R.; Lechner K.; Valent P. Detection of tryptase in cytoplasmic granules of basophils in patients with chronic myeloid leukemia and other myeloid neoplasms. Blood. 2001, 98, 2580-2583. [CrossRef]
- Nakayama H.; Ishimaru F.; Avitahl N.; Sezaki N.; Fujii N.; Nakase K.; Ninomiya Y.; Harashima A.; Minowada J.; Tsuchiyama J.; Imajoh K.; Tsubota T.; Fukuda S.; Sezaki T.; Kojima K.; Hara M.; Takimoto H.; Yorimitsu S.; Takahashi I.; Miyata A.; Taniguchi S.; Tokunaga Y.; Gondo H.; Niho Y.; Harada M.; et al. Decreases in Ikaros activity correlate with blast crisis in patients with chronic myelogenous leukemia. Cancer Res. 1999, 59, 3931-3934.
- Mullighan C.G.; Miller C.B.; Radtke I.; Phillips L.A.; Dalton J.; Ma J.; White D.; Hughes T.P.; Le Beau M.M.; Pui C.H.; Relling M.V.; Shurtleff S.A.; Downing J.R. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008, 453, 110-114. [CrossRef]
- Beer PA.; Knapp D.J.; Miller P.H.; Kannan N.; Sloma I.; Heel K.; Babovic S.; Bulaeva E.; Rabu G.; Terry J.; Druker B.J.; Loriaux M.M.; Loeb K.R.; Radich J.P.; Erber W.N.; Eaves C.J. Disruption of IKAROS activity in primitive chronic-phase CML cells mimics myeloid disease progression. Blood. 2015, 125, 504-515. [CrossRef]
- Arber A.; Brunning D.; Orazi A.; Porwit A.; Peterson C.; Thiele J.; Le Beau M.; Hasserjian P. Acute myeloid leukaemia, NOS. In book WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Rev. 4th ed.; Swerdlow S.; Campo E.; Harris NL.; Jaffe E.; Pileri S.; Stein H.; Thiele J, Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, pp 164-165.
- Drexler H.G .; Sperling C .; Ludwig W.D. Terminal deoxynucleotidyl transferase (TdT) expression in acute myeloid leukemia. Leukemia. 1993, 7, 1142-1150.
- Thiede C.; Steudel C.; Mohr B.; Schaich M.; Schäkel U.; Platzbecker U.; Wermke M.; Bornhäuser M.; Ritter M.; Neubauer A.; Ehninger G.; Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002, 99, 4326-4335. [CrossRef]
- Seth T.; Vora A.; Bhutani M.; Ganessan K.; Jain P.; Kochupillai V. Acute basophilic leukemia with t(8;21). Leuk Lymphoma. 2004, 45, 605-608. [CrossRef]
- Georgin-Lavialle S.; Lhermitte L.; Dubreuil P.; Chandesris M.O.; Hermine O.; Damaj G. Mast cell leukemia. Blood. 2013, 121, 1285-1295. [CrossRef]
- McKenna R.W.; Parkin J.; Bloomfield C.D.; Sundberg R.D.; Brunning R.D. Acute promyelocytic leukaemia: a study of 39 cases with identification of a hyperbasophilic microgranular variant. Br J Haematol. 1982, 50, 201-214. [CrossRef]
- Ghimire A.; Liesveld J.;Wallace D.; Zhao J.; Burack R.; Bennett J. Case of acute promyelocytic leukemia with basophilic differentiation and an ETV6 mutation. J. Hematop. 2021, 14, 333–336.
- Joachim, G. Uber Mastzellenleukamien. Dtsch. Arch. Kiln. Med. 1906, 87, 437.
- Horny HP.; Sotlar K.; Reiter A.; Valent P. Myelomastocytic leukemia: histopathological features, diagnostic criteria and differential diagnosis. Expert Rev Hematol. 2014, 7, 431-437. [CrossRef]
- Duchayne E.; Demur C.; Rubie H.; Robert A.; Dastugue N. Diagnosis of acute basophilic leukemia. Leuk Lymphoma. 1999, 32, 269-278. [CrossRef]
- Staal-Viliare A.; Latger-Cannard V.; Didion J.; Grégoire M.J.; Lecompte T.; Jonveaux P.; Rio Y. CD203c /CD117-, an useful phenotype profile for acute basophilic leukaemia diagnosis in cases of undifferentiated blasts. Leuk Lymphoma. 2007, 48, 439-441. [CrossRef]
- Giagounidis A.A.; Hildebrandt B.; Heinsch M.; Germing U.; Aivado M.; Aul C. Acute basophilic leukemia. Eur J Haematol. 2001, 67, 72-76. [CrossRef]
- Scolyer R.A.; Brun M.; D'Rozario J.; Webb M. Acute basophilic leukemia presenting with abnormal liver function tests and the absence of blast cells in the peripheral blood. Pathology. 2000, 32, 52-55. [CrossRef]
- Liso V.; Troccoli G.; Specchia G. Mast cell leukemia and acute basophilic leukemia. Cytochemical studies. Bibl Haematol. 1978, 45, 142-146. [CrossRef]
- Quelen C.; Lippert E.; Struski S.; Demur C.; Soler G.; Prade N.; Delabesse E.; Broccardo C.; Dastugue N.; Mahon FX.; Brousset P. Identification of a transforming MYB-GATA1 fusion gene in acute basophilic leukemia: a new entity in male infants. Blood. 2011, 117, 5719-5722. [CrossRef]
- Toda Y.; Nagai Y.; Shimomura D.; Kishimori C.; Tsuda K.; Fukutsuka K.; Hayashida M.; Ohno H. Acute basophilic leukemia associated with the t(16;21)(p11;q22)/FUS-ERG fusion gene. Clin Case Rep. 2017, 5, 1938-1944. [CrossRef]
- Kritharis A.; Brody J.; Koduru P.; Teichberg S.; Allen SL. Acute basophilic leukemia associated with loss of gene ETV6 and protean complications. J Clin Oncol. 2011, 29, 623-626. [CrossRef]
- Shimizu T.; Kondo T.; Nannya Y.; Watanabe M.; Kitawaki T.; Shindo T.; Hishizawa M.; Yamashita K.; Ogawa S.; Takaori-Kondo A. Next-generation sequencing in two cases of de novo acute basophilic leukaemia. J Cell Mol Med. 2021, 25, 7095-7099. [CrossRef]
- Greene L.W.; Asadipooya K.; Corradi P.F.; Akin C. Endocrine manifestations of systemic mastocytosis in bone. Rev Endocr Metab Disord. 2016, 17, 419-431. [CrossRef]
- Shah I.; Lewkow L.M.; Koppitch F. Acute basophilic leukemia. Am J Med. 1984, 76, 1097-1099. [CrossRef]
- Bernini J.C.; Timmons C.F.; Sandler E.S. Acute basophilic leukemia in a child. Anaphylactoid reaction and coagulopathy secondary to vincristine-mediated degranulation. Cancer. 1995, 75, 110-114.
- Anderson W.; Helman C.A.; Hirschowitz B.I. Basophilic leukemia and the hypersecretion of gastric acid and pepsin. Gastroenterology. 1988, 95, 195-198. [CrossRef]
- Luo X.H.; Zhu Y.; Tang X.Q. Acute basophilic leukemia presenting with maculopapular rashes and a gastric ulcer: A case report. Oncol Lett. 2014, 8, 2513-2516. [CrossRef]
Figure 1.
Charcot-Leyden crystals in association with AML. These bizarre bipyramidal crystal structures were noted in the bone marrow aspirate smears of a 75-year-old woman with pancytopenia.
Figure 1.
Charcot-Leyden crystals in association with AML. These bizarre bipyramidal crystal structures were noted in the bone marrow aspirate smears of a 75-year-old woman with pancytopenia.
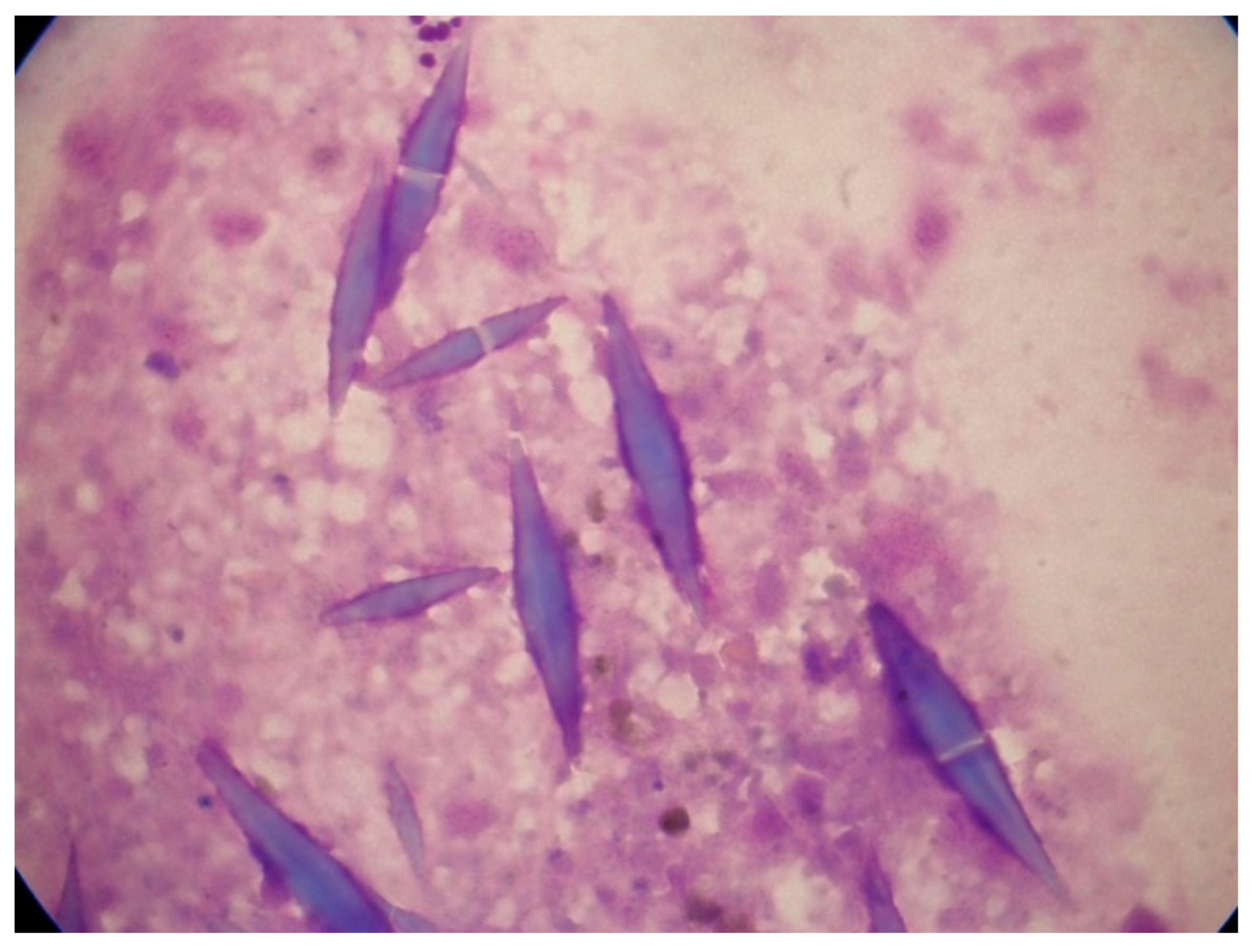
Figure 2.
Abnormal eosinophils in the bone-marrow aspirates of a patient with AML with inv(16)(p13.1q22). These micrographs showing abnormal eosinophils containing both eosinophilic and basophilic staining granules are representative of myelomonocytic leukemia with eosinophilia (May-Grünwald-Giemsa, ×1000).
Figure 2.
Abnormal eosinophils in the bone-marrow aspirates of a patient with AML with inv(16)(p13.1q22). These micrographs showing abnormal eosinophils containing both eosinophilic and basophilic staining granules are representative of myelomonocytic leukemia with eosinophilia (May-Grünwald-Giemsa, ×1000).
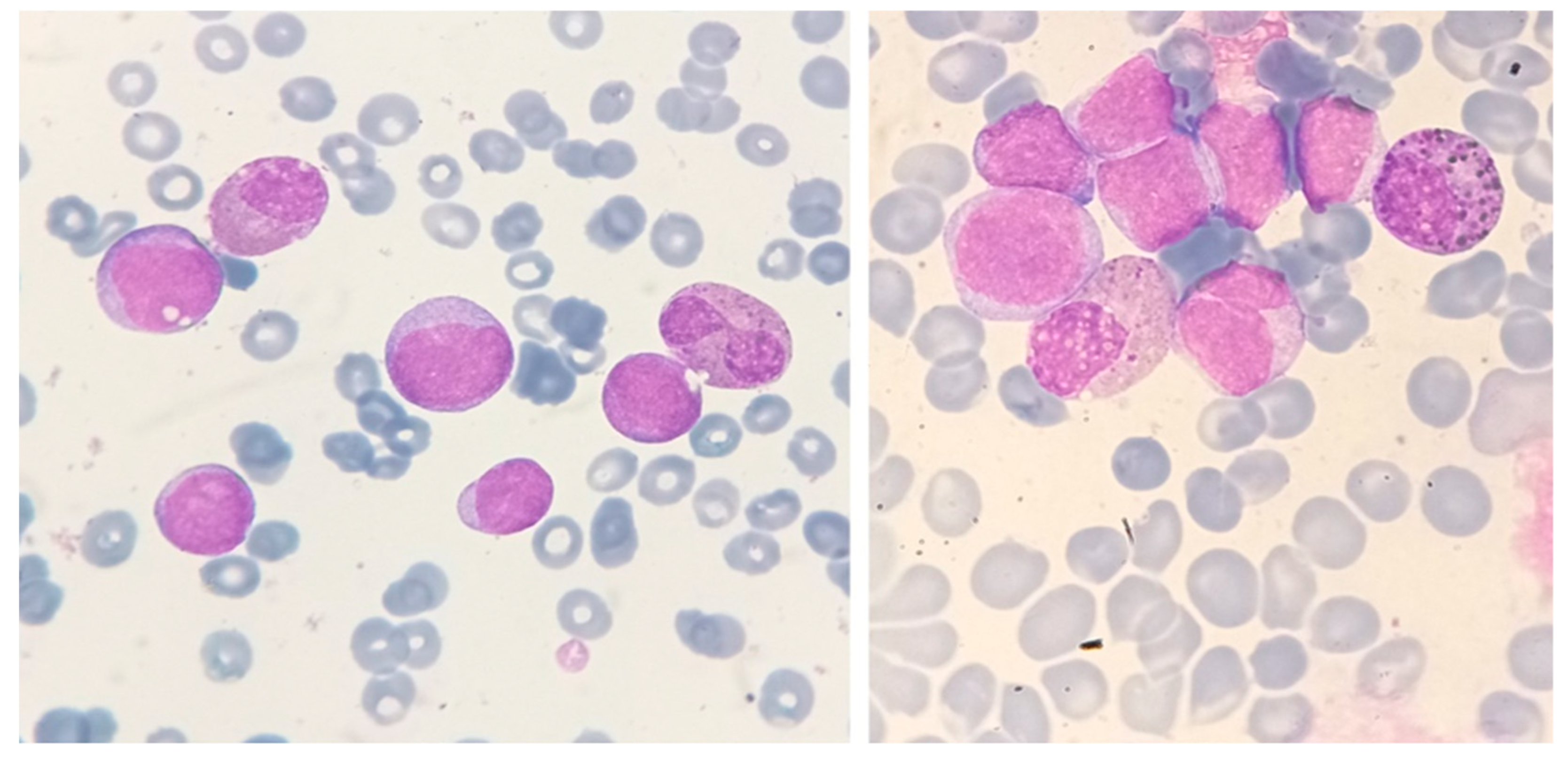
Figure 3.
A fluorescence in situ hybridization (FISH) study with specific probes mapping on both sides of the chromosome 16p breakpoint region, showing a nucleus with inv(16)(p13.1 q22). The presence of CBFB rearrangement is indicated by separate red and green signals.
Figure 3.
A fluorescence in situ hybridization (FISH) study with specific probes mapping on both sides of the chromosome 16p breakpoint region, showing a nucleus with inv(16)(p13.1 q22). The presence of CBFB rearrangement is indicated by separate red and green signals.
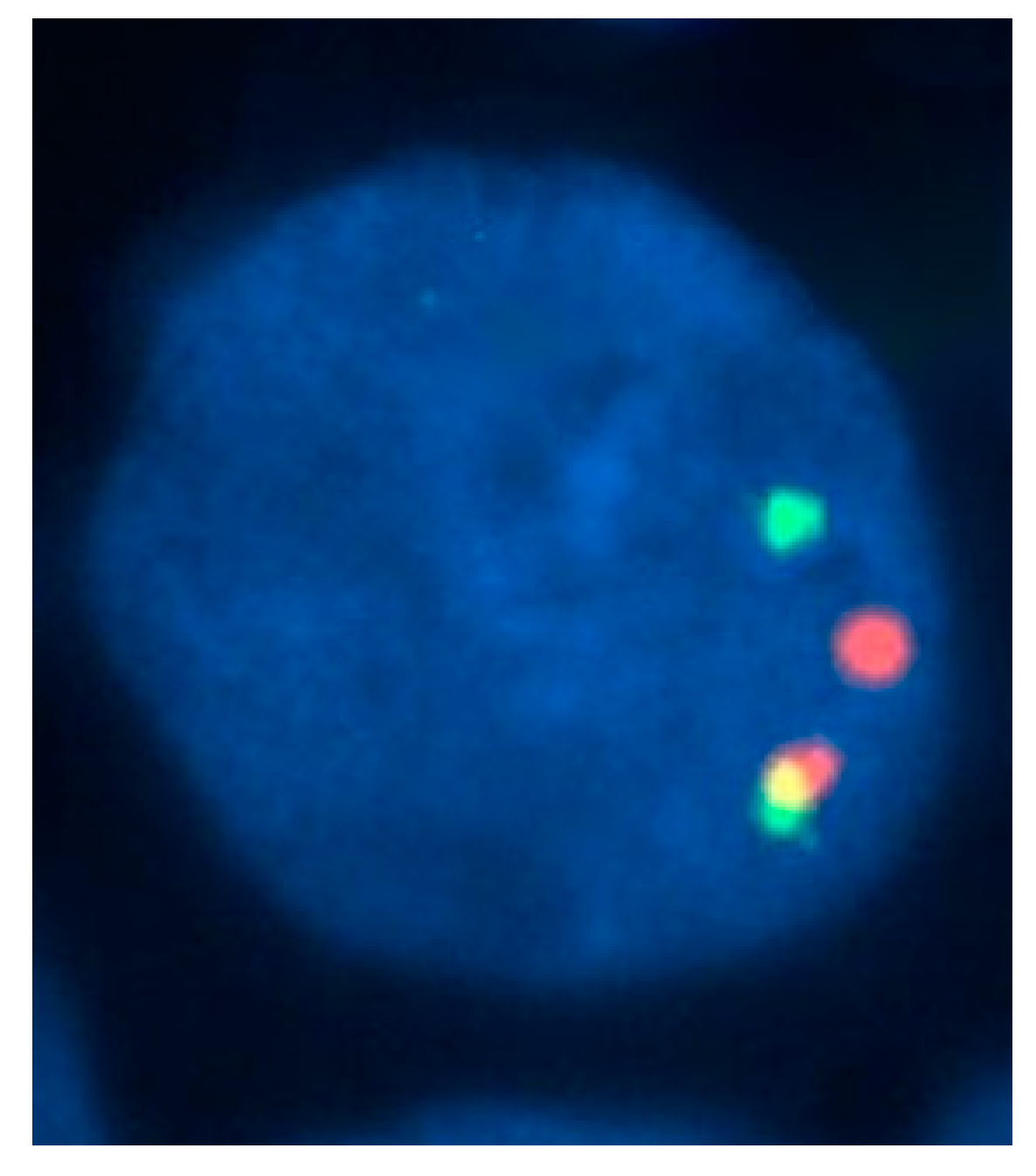
Figure 4.
Bone marrow aspirate smear of a 54-year-old man with AML with t(8;21)(q22;q22) translocation and eosinophilia. The white-cell count was 2.0 × 109/L, without increased eosinophils in the peripheral blood. In the bone marrow, however, eosinophils and eosinophil precursors constituted 22% of cells, without abnormal basophilic granules (May-Grünwald-Giemsa, ×1000).
Figure 4.
Bone marrow aspirate smear of a 54-year-old man with AML with t(8;21)(q22;q22) translocation and eosinophilia. The white-cell count was 2.0 × 109/L, without increased eosinophils in the peripheral blood. In the bone marrow, however, eosinophils and eosinophil precursors constituted 22% of cells, without abnormal basophilic granules (May-Grünwald-Giemsa, ×1000).
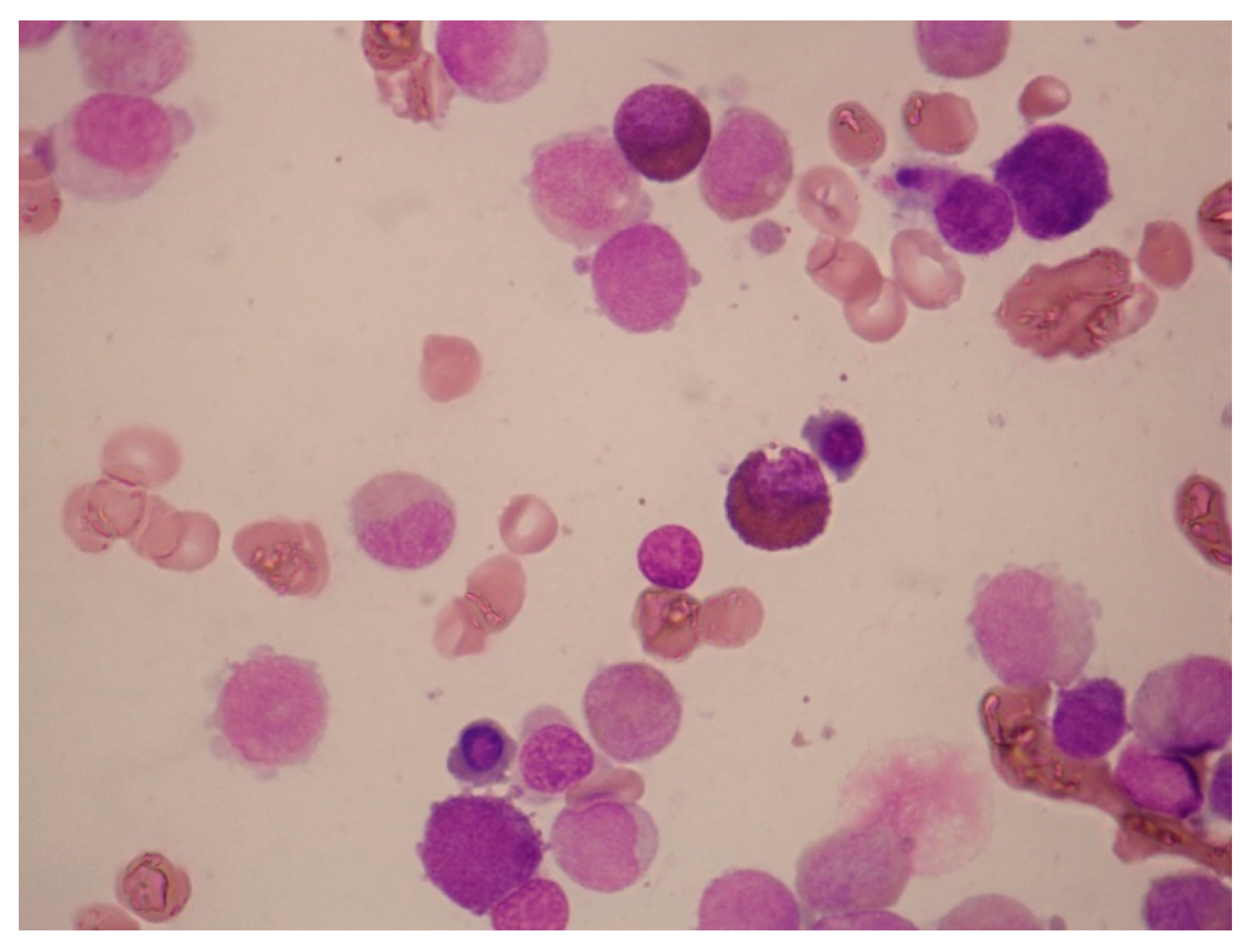
Figure 5.
Detection of FIP1L1-PDGFRA in a case of AML with eosinophilia, using a three-color probe strategy. On interphase fluorescence in-situ hybridization performed with the use of probes to LNX (in red), FIP1L1 (in green), and PDGFRA (in aqua), the nucleus shows one chromosome with all three signals intact and another chromosome with intact FIP1L1 and PDGFRA signals but without the LNX signal (i.e. loss of red signal). This finding indicates a deletion of the region between FIP1L1 and PDGFRA on chromosome 4q12 consistent with the FIP1L1-PDGFRA fusion.
Figure 5.
Detection of FIP1L1-PDGFRA in a case of AML with eosinophilia, using a three-color probe strategy. On interphase fluorescence in-situ hybridization performed with the use of probes to LNX (in red), FIP1L1 (in green), and PDGFRA (in aqua), the nucleus shows one chromosome with all three signals intact and another chromosome with intact FIP1L1 and PDGFRA signals but without the LNX signal (i.e. loss of red signal). This finding indicates a deletion of the region between FIP1L1 and PDGFRA on chromosome 4q12 consistent with the FIP1L1-PDGFRA fusion.
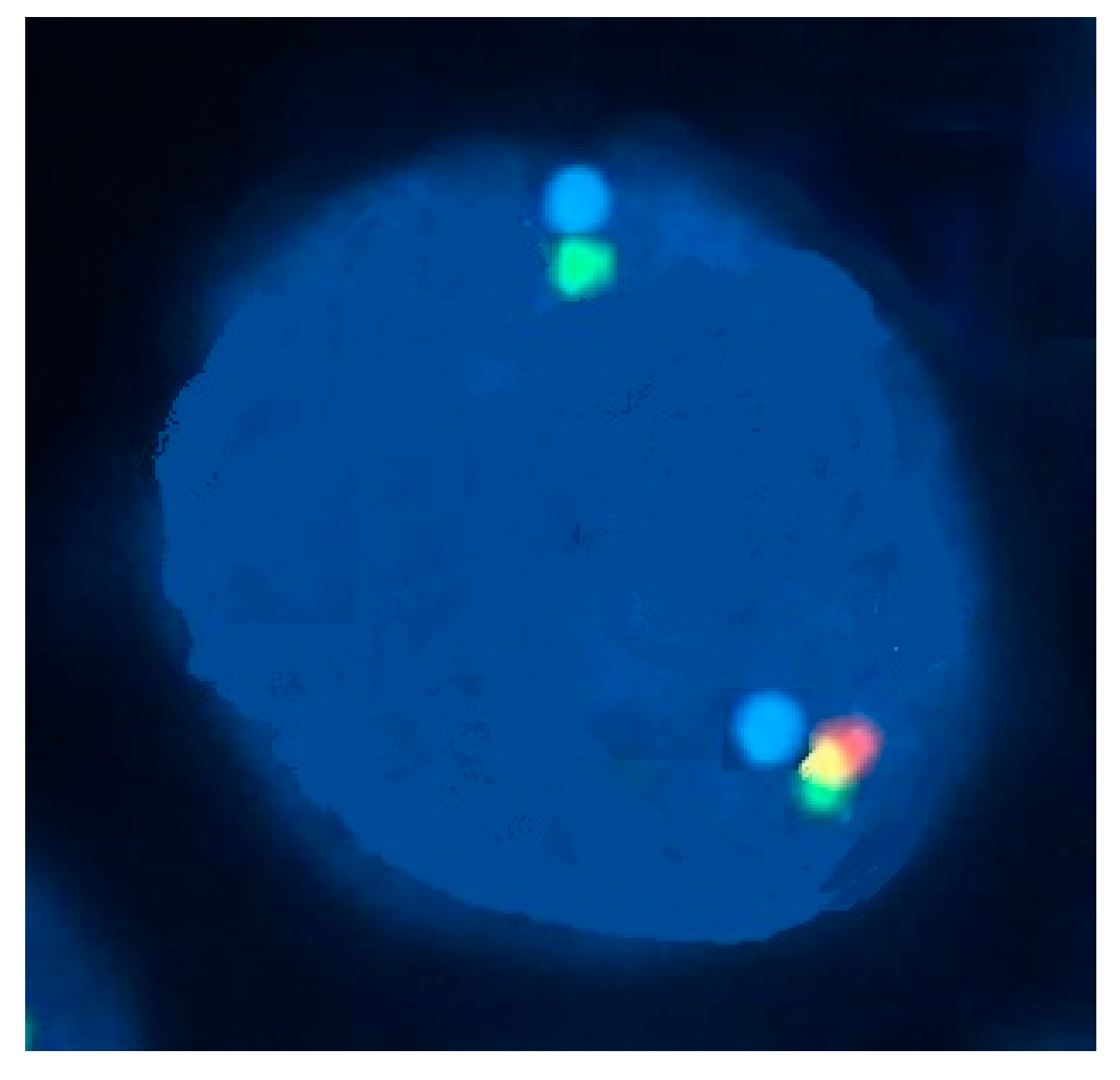
Figure 6.
Abnormal eosinophil morphology associated with FIP1L1‒PDGFRA rearrangement. This peripheral-blood smear shows eosinophils with trilobed nuclei or hypersegmented eosinophils as well as eosinophils with many cytoplasmic vacuoles due to degranulation (May-Grünwald-Giemsa stain, ×1000).
Figure 6.
Abnormal eosinophil morphology associated with FIP1L1‒PDGFRA rearrangement. This peripheral-blood smear shows eosinophils with trilobed nuclei or hypersegmented eosinophils as well as eosinophils with many cytoplasmic vacuoles due to degranulation (May-Grünwald-Giemsa stain, ×1000).
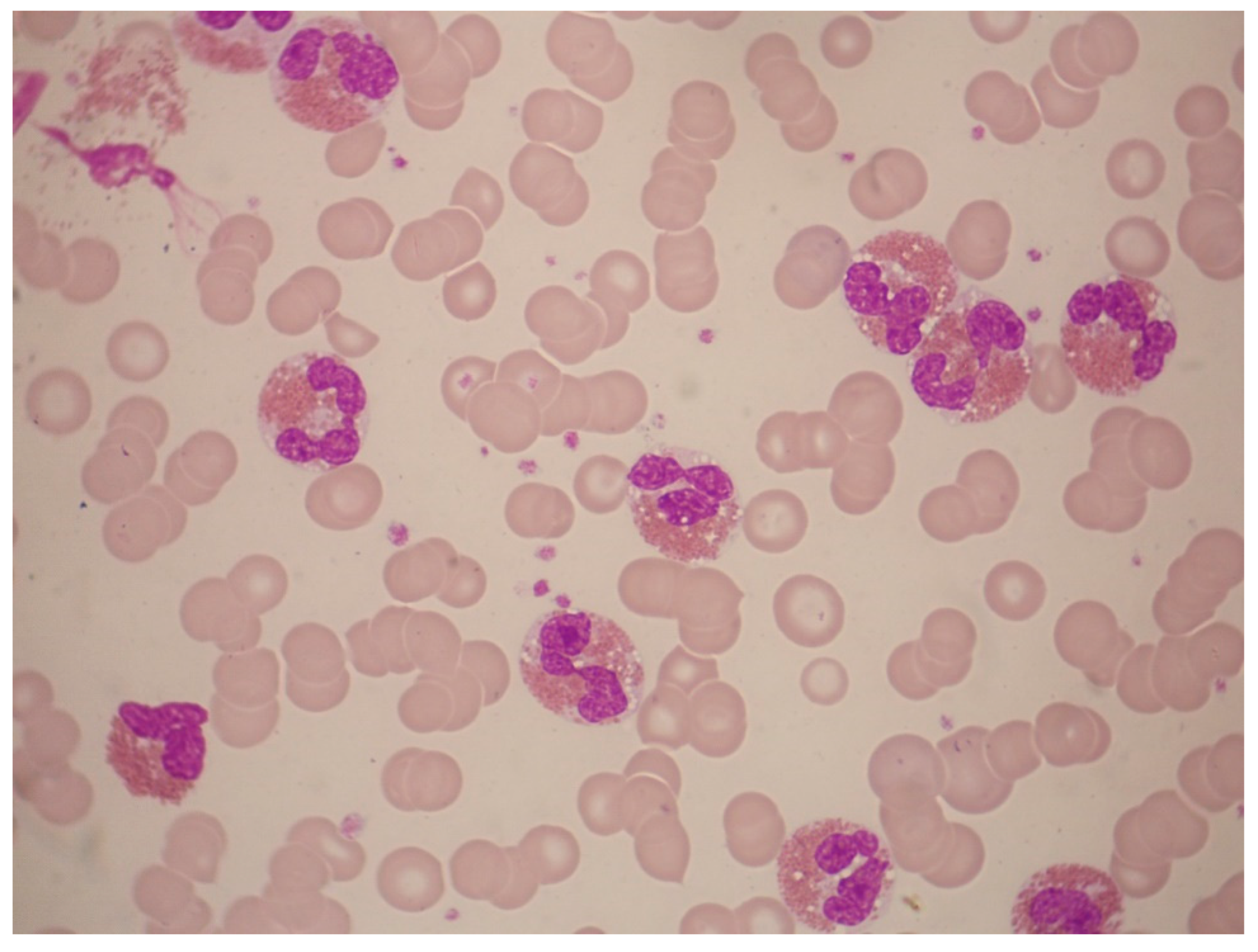
Figure 7.
A 70-year-old man with monocytosis and eosinophilia due to PDGFRB rearrangement. Two-color fluorescence in situ hybridization for the PDGFRB rearrangement with two flanking cosmid probes for chromosome 5 showed that more than 80% of cells had one fused signal and separate red and green signals, indicating disruption of the PDGFRB gene.
Figure 7.
A 70-year-old man with monocytosis and eosinophilia due to PDGFRB rearrangement. Two-color fluorescence in situ hybridization for the PDGFRB rearrangement with two flanking cosmid probes for chromosome 5 showed that more than 80% of cells had one fused signal and separate red and green signals, indicating disruption of the PDGFRB gene.
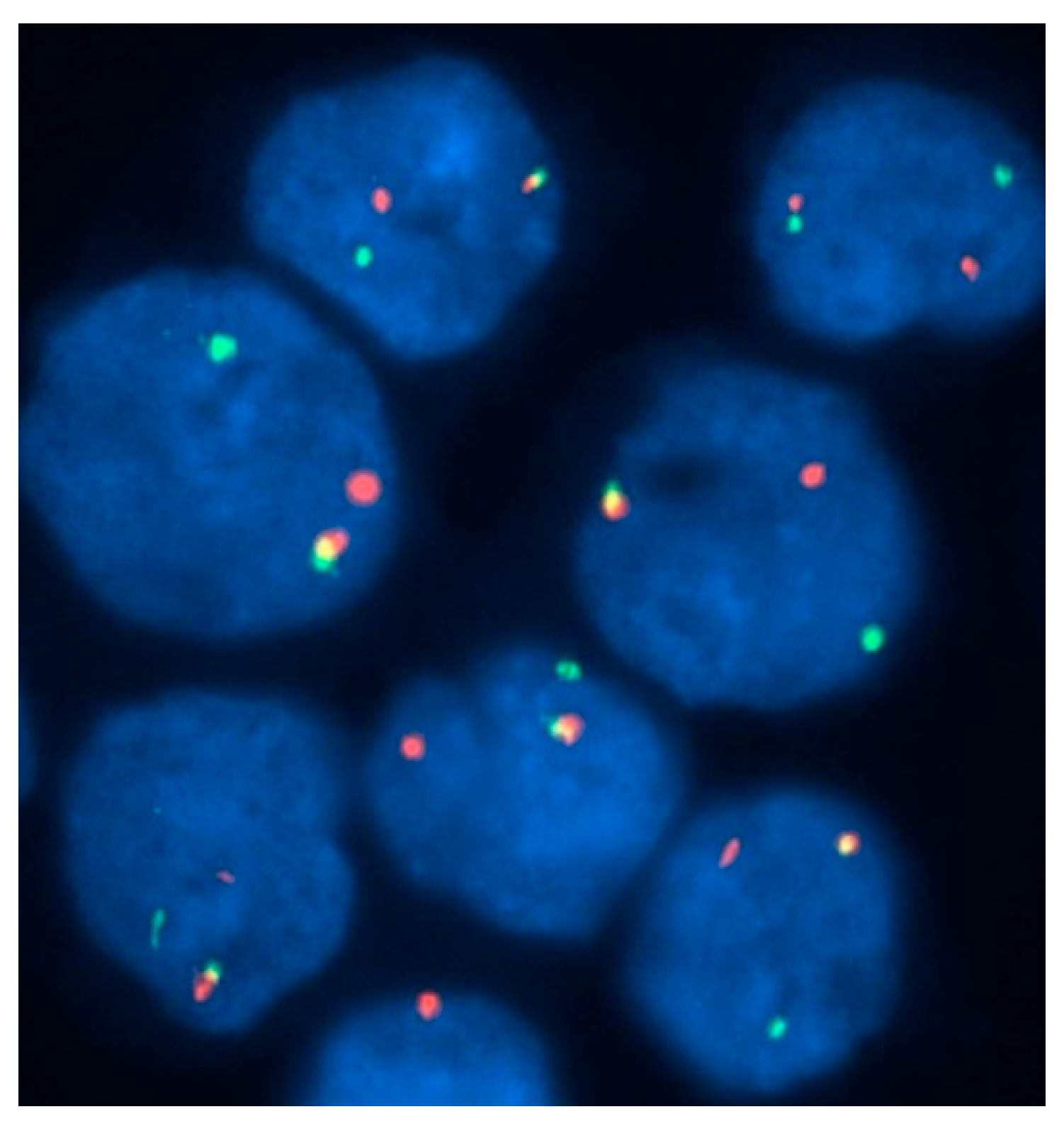
Figure 8.
Acute mast-cell leukemia. Bone marrow aspirate smears of a 79-year-old man with aleukemic acute mast-cell leukemia who presented with pancytopenia and abnormal liver function tests. The photomicrographs show round neoplastic cells with dark cytoplasmic granules. The cells were CD13+, HLA-DR+, CD33+, CD203c+, CD38+, CD2-, CD9-, CD123-, CD11b-, FcεRI+, CD117+, and CD25+. PCR was positive for C-KIT D816V mutation.
Figure 8.
Acute mast-cell leukemia. Bone marrow aspirate smears of a 79-year-old man with aleukemic acute mast-cell leukemia who presented with pancytopenia and abnormal liver function tests. The photomicrographs show round neoplastic cells with dark cytoplasmic granules. The cells were CD13+, HLA-DR+, CD33+, CD203c+, CD38+, CD2-, CD9-, CD123-, CD11b-, FcεRI+, CD117+, and CD25+. PCR was positive for C-KIT D816V mutation.
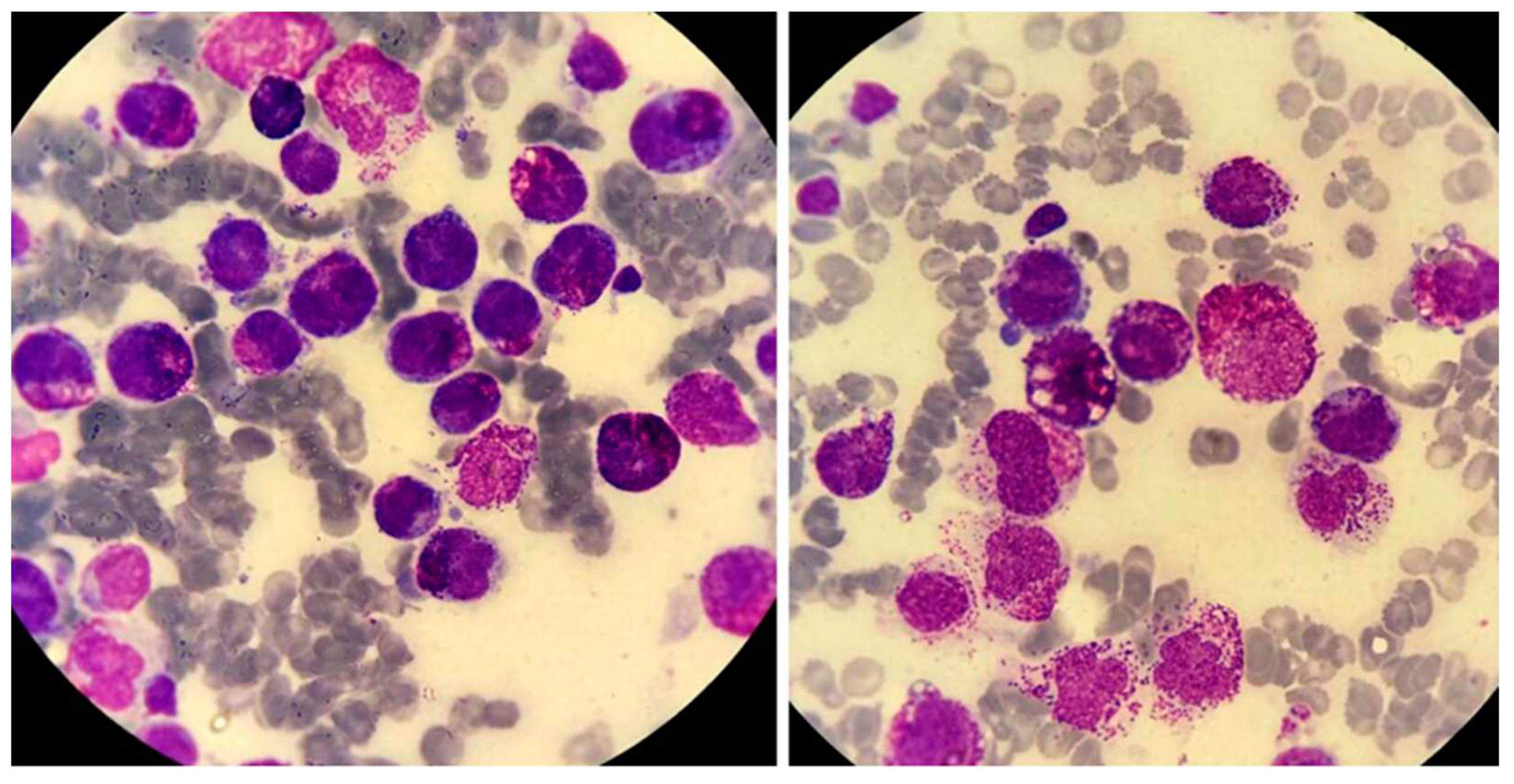
Figure 9.
Illustrative case of acute basophilic leukemia (ABL). A 67-year-old man presented for evaluation of general weakness and lower back pain. Over the previous 8 months he had had postprandial epigastric pain. He had undergone esophagogastroduodenoscopy, which showed the presence of three simultaneous gastric ulcers. Biopsies were negative for Helicobacter pylori infection. His past medical history was otherwise unremarkable. He smoked 20 cigarettes daily, drank alcohol rarely, and took no medications. On examination, there was mild hepatomegaly (2 cm below right coastal margin), small-volume peripheral lymphadenopathy (diameter, ≤2 cm) and no splenomegaly. Multiple skin lesions were noted on the trunk and extremities, measuring a few centimeters. They were reddish, reddish brown or purple, plaque-like and produced significant itching and discomfort. A complete blood count revealed anemia (hemoglobin, 9.5 g/dL; ΜCV 80 fL), white-cell count 5.1×109/L and platelet count 45×109/L. May-Grünwald-Giemsa staining of a peripheral-blood smear revealed 41% neutrophils, 36% lymphocytes, 1% monocytes, 8% nucleated red blood cells, and 22% “atypical” cells with basophilic granules, easily identifiable under oil immersion (×1000) (Figure). Coagulation studies and hepatic biochemistry were normal, but there was renal dysfunction (urea 65 mg/dL, creatinine 2.2 mg/dL) and hypocalcemia (6.2 mg/dL). Lactate dehydrogenase level was 1,376 U/L (<246 U/L). Flow cytometry showed that the immature cell population in the CD45weak/SSClow gate was CD34+, MPO-, CD11b+, CD13+, CD33+, CD9+, CD123+, CD203c+, HLA-DR-, CD2-, CD3-, CD5-, CD16-, CD10-, CD22weak, CD25weak, CD15-, TdT-, and CD117-. An attempted aspiration of the bone marrow yielded a "dry tap”.
Figure 9.
Illustrative case of acute basophilic leukemia (ABL). A 67-year-old man presented for evaluation of general weakness and lower back pain. Over the previous 8 months he had had postprandial epigastric pain. He had undergone esophagogastroduodenoscopy, which showed the presence of three simultaneous gastric ulcers. Biopsies were negative for Helicobacter pylori infection. His past medical history was otherwise unremarkable. He smoked 20 cigarettes daily, drank alcohol rarely, and took no medications. On examination, there was mild hepatomegaly (2 cm below right coastal margin), small-volume peripheral lymphadenopathy (diameter, ≤2 cm) and no splenomegaly. Multiple skin lesions were noted on the trunk and extremities, measuring a few centimeters. They were reddish, reddish brown or purple, plaque-like and produced significant itching and discomfort. A complete blood count revealed anemia (hemoglobin, 9.5 g/dL; ΜCV 80 fL), white-cell count 5.1×109/L and platelet count 45×109/L. May-Grünwald-Giemsa staining of a peripheral-blood smear revealed 41% neutrophils, 36% lymphocytes, 1% monocytes, 8% nucleated red blood cells, and 22% “atypical” cells with basophilic granules, easily identifiable under oil immersion (×1000) (Figure). Coagulation studies and hepatic biochemistry were normal, but there was renal dysfunction (urea 65 mg/dL, creatinine 2.2 mg/dL) and hypocalcemia (6.2 mg/dL). Lactate dehydrogenase level was 1,376 U/L (<246 U/L). Flow cytometry showed that the immature cell population in the CD45weak/SSClow gate was CD34+, MPO-, CD11b+, CD13+, CD33+, CD9+, CD123+, CD203c+, HLA-DR-, CD2-, CD3-, CD5-, CD16-, CD10-, CD22weak, CD25weak, CD15-, TdT-, and CD117-. An attempted aspiration of the bone marrow yielded a "dry tap”.
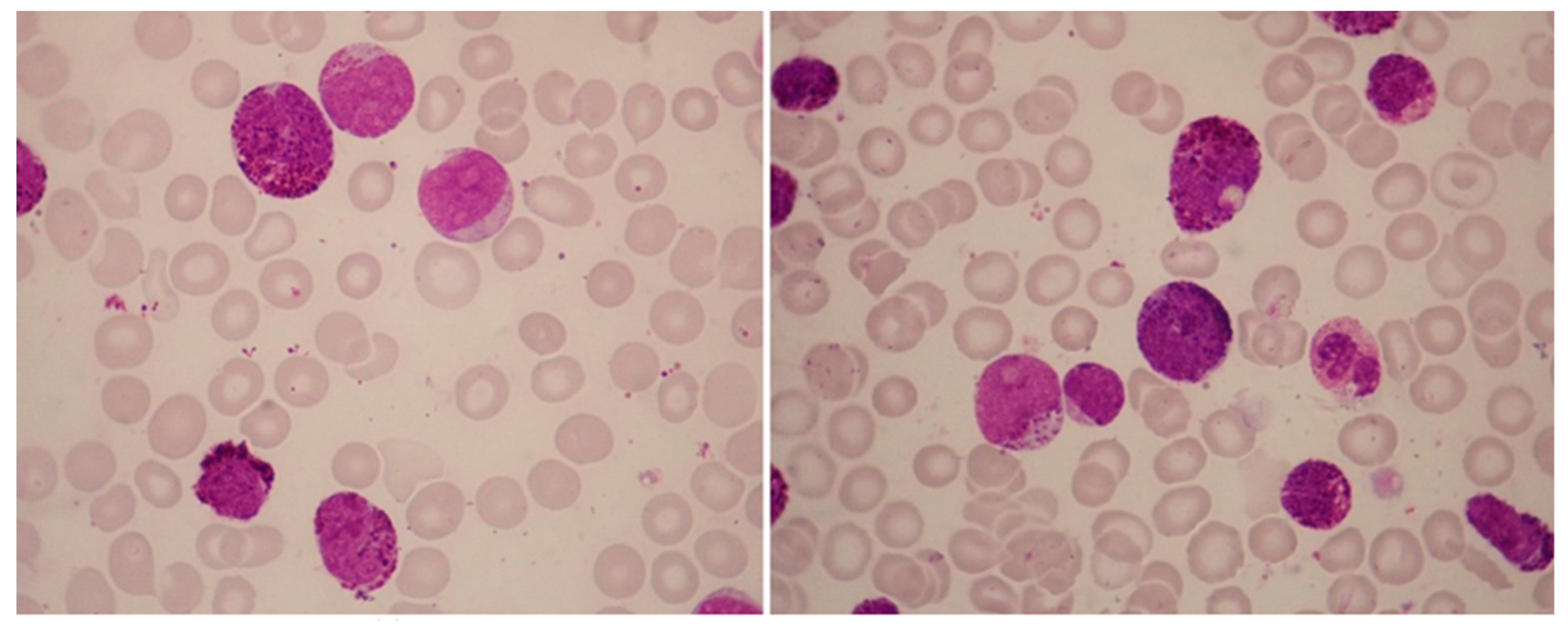
Table 2.
Differential diagnosis of leukemias with prominent basophilic granules.
|
Abbreviations: AML, acute myeloid leukemia; CML, chronic myeloid leukemia; APL, acute promyelocytic leukemia; MPN, myeloproliferative neoplasm.
Table 3.
Acute basophilic leukemia, as compared with acute mast-cell leukemia.
| Acute basophilic leukemia | Acute mast-cell leukemia | |
|---|---|---|
| Clinical features | ||
| Hyperhistaminemia Skin involvement Hepatosplenomegaly Lymphadenopathy |
Common Common Common Uncommon |
Common Uncommon Common Common |
| Special stains | ||
| Tolouidine Blue | Positive | Positive |
| Periodic-acid Schiff (P.A.S) | Positive | Negative or weak |
| Chloracetate esterase (ChorE) | Negative | Positive |
| Tryptase | Negative or weak | Positive |
| Myeloperoxidase (MPO) | Negative | Negative |
| Immunophenotypic studies | ||
| CD34 | Negative or weakly positive | Negative or weakly positive |
| CD45weak/CD13/CD33 expression | Positive | Positive |
| CD14/CD15/CD64 expression | Negative | Negative |
| CD11b | Positive | Negative |
| CD17 | Positive | Negative |
| CD123 | Positive | Negative |
| CD203c | Positive | Negative or weakly positive |
| CD2 | Negative | Positive or negative |
| CD25 | Positive | Positive or negative |
| CD117 | Negative | Strongly positive |
| Cytogenetic and molecular studies | ||
| t(X;6)(p11;q23) | C-KIT mutations (e.g. C-KIT D816V) | |
| t(16;21)(p11;q22) | ||
| del(12p) | TET2 mutations | |
| TP53, TET2 and NPM1 mutations | SRSF2, ASXL1, RUNX1 (“S/A/R”) mutations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



