
Submitted:
15 January 2024
Posted:
15 January 2024
You are already at the latest version
Abstract
Redox balance is increasingly identified as a major player in cellular signaling. A fundamentally simple reaction of oxidation and reduction of cysteine residues in cellular proteins is the central concept in this complex mode of protein function regulation. Oxidation of key cysteine residues occurs at the physiological levels of reactive oxygen species (ROS) but are reduced by a supply of thiol antioxidant molecules including glutathione, glutaredoxin and thioredoxin. While these molecules show complex compensatory roles in experimental conditions, transgenic animal models provide a comprehensive picture to pinpoint the role of each antioxidant. In this review we have specifically focused on the available literature on Thioredoxin-1 system transgenic models that includes Thioredoxin and Thioredoxin reductase proteins. As identification of Thioredoxin protein targets is technically challenging the true contribution of this system in maintaining cellular balance remains unidentified, including the role of this system in the brain.
Keywords:
Redox balance
; thiol antioxidants
; transgenic models
; brain
; heart
; glutaredoxin
; thioredoxin reductase
1. Introduction
The importance of cellular redox status in cell physiology is increasingly identified. Reactive oxygen species (ROS), such as superoxide anion and H2O2, are critical secondary messengers in cells during health and disease. Their messenger role is achieved by the oxidation of key cysteine residues in different signaling proteins, a highly efficient way to regulate protein activity. These oxidized residues must be quickly reduced to keep the cell in an optimal state. The reducing equivalents are provided by the reduced nicotinamide adenine dinucleotide phosphate (NADPH). These are transferred to Glutathione (GSH)/Glutaredoxin and Thioredoxin (Trx) systems through GSH-Reductase (GR) and Trx-Reductase (TrxR). Despite seemingly overlapping functions of GSH and Trx systems, their mode of action is quite unique. Reduction of oxidized substrates by GSH requires the formation of a glutathione adduct. These adducts are stable and can be detected using several mass spectrophotometry methods, hence the importance of GSH in neurobiology is well documented. Trx reduces its substrate through the formation of a transient mixed disulfide intermediate compound, followed by a fast thiol–disulfide exchange reaction with oxidized cysteines in the target protein. This results in oxidation of Trx and reduction of the substrate. Since there are no stable Trx-adducts in this reaction, the identification of Trx targets is a very challenging task, therefore the extent of Trx contribution to cell signaling systems remains vastly unknown. Early attempts in creating whole-body knockout (constitutive) of Trx system resulted in the death of embryos before gastrulation [1,2], therefore several cell-specific and organ-specific knockout mouse models have been generated to better understand this protein. These models generally have a short life but have provided a glimpse of Trx1's contribution to cell biology. Here, we aim to review the available information on these Trx1 system knockout models.
1.1. The Trx family of proteins
Thioredoxins are small antioxidant proteins with a conserved active site (Cys32-Pro-Gly-Cys35) in all organisms and work as general oxidoreductases. These proteins react with a wide range of proteins and control their stability and function. The name “thioredoxin” originated from the molecular mechanism that underlies their redox function. Two readily accessible Cysteine residues at Trx active site (Cys32-Gly-Pro-Cys35) are used as electron sources for its redox activity (Figure 1). Trx1 has a low affinity for H2O2, however, it is an important oxidoreductase protein and can directly reduce the oxidized proteins through a kiss-and-run reaction. During this reaction, Trx first donates an electron from its Cys32 to the oxidized substrate, forming a transient disulfide bond with the target. The second electron at Cys35 is then donated to the oxidized substrate, fully reducing it [3]. This results in oxidation of Trx by forming an intracellular disulfide bond between Cys35 and Cys32. The oxidized Trx must be reduced by TrxR using two electrons from NADPH, before resuming the redox regulation activity. The reduced Trx can also act as an electron donor for Peroxiredoxins (Prx). This family of Trx-dependent peroxidases is responsible for peroxide scavenging [4] and will be discussed briefly below. Trx can be inactivated by binding to its natural endogenous inhibitor, Thioredoxin Inhibiting protein (TXNIP) also known as Vitamin D3 Upregulated protein (VDUP). TXNIP binding to Trx results in exacerbating oxidative stress [5] as shown in Type-1 Diabetes, resulting in β-cell death [6].
There are several other proteins in eukaryotes that are evolutionally related to Trx and share structural similarities with Trx. These are mostly members of a group of proteins known as Protein Disulfide Isomerase that are involved in proper protein folding in the Endoplasmic reticulum. These proteins have been reviewed recently [7,8] and will not be discussed here.
1.2. Isoforms of mammalian Trx
To date, three Trx isoforms have been identified in mammals: Trx1, Trx2, and Trx3 (Table 1). While Trx1 and Trx2 are encoded by separate genes, Txn1 and Txn2, while Trx3 results from agenetic duplication of the Txn1 gene [9]. Despite their relatively similar structures with a conserved disulfide active site sequence (Trp-Cys-Gly-Pro-Cys) [10], their distinct subcellular and tissue-specific localizations determine their specific functions. Thioredoxin isoforms can also be categorized into two subgroups: a) those ubiquitously expressed in all cell types (Trx1 and Trx2) and b) testes-specific (Trx3) [11]. Trx3 is predominantly localized in the Golgi apparatus of mammalian spermatids and spermatocytes [9]. Interestingly, expression of Trx3 is notably elevated in defective spermatozoa making this isoform a promising di-agnostic marker for detection of aberrant spermatogenesis and infertility in males [9].
- Cysteines located within the active center of Thioredoxin-1 (Trx-1), Thioredoxin-2 (Trx-2), and Thioredoxin-3 (Trx-3) are highlighted with yellow, Cysteines outside of the active center are highlighted with Cyan.
Trx1 and Trx2 are the most researched isoforms of this family. We will first briefly discuss Trx2 and then will focus majorly on Trx1.
Trx2 is predominantly localized in mitochondria [12]. Mitochondria, as the primary producers of reactive oxygen species (ROS) in mammalian cells, heavily rely on redox control through mitochondrial antioxidant systems. Trx2's general function in mitochondria involves maintaining membrane potential by reducing the protein disulfides, which impacts mitochondrial function. Downregulation of Trx2 leads to decreased ATP/ADP ratio, reduced oxygen consumption, elevated lactate production, and activation of caspase 3 and caspase 7 [13]. Trx2 also plays a role in Tumor Necrosis Alpha/Apoptosis Signal-Regulating Kinase 1 (TNF-α/ASK-1)-stimulated release of Cytochrome C during apoptosis [14]. Thus, Trx2 binds to Cys-30 in the N-terminal domain of ASK1 and prevents its activation. To illustrate, Zhang et al. demonstrated that overexpression of Trx2 in human umbilical vein endothelial cells prevents ASK1-induced apoptosis without significant effect on ASK1-mediated c-Jun N-terminal kinase activation [15]. Other works confirm that Trx2 depletion in vitro has a pro-apoptotic effect [14,16]. Trx2 expression is higher in tissues with heavy metabolic rate, e.g., stomach, testis, ovary, liver, brain, heart, and adrenal gland [16]. Furthermore, Trx2 plays an important role in embryogenesis, as constitutional Trx2 knockout in mice is embryonically lethal [17]. In this work, mutant animals displayed defects in neural tube closure and dramatic activation of apoptosis.
Trx1 is found mainly in the cytoplasm of all cells and tissues, but it also translocates to the nucleus in response to oxidative or nitrosative stress [18] or it can be secreted to the extracellular space [19]. Both Trx1 and its truncated form (Trx-80) are secreted by immune cells (e.g., monocytes, lymphocytes, and neutrophils), which plays a role in cell-to-cell communication and facilitation of chemotaxis [20]. This mechanism is especially important in infection, inflammation, and neoplastic changes in the hematopoietic system [21]. These features make the Trx1 protein a unique member cellular redox system.
Trx1 was first isolated from E. coli in 1964 [22] and was sequenced by the late Dr. Arne Holmgren [23]. The contributions of Dr. Holmgren have had a major part in shaping the existing knowledge of the Trx field. Prior to its discovery, Trx1 had been identified under various names as a hydrogen donor facilitating the reduction of methionine sulfoxide and sulfate by NADPH in yeast [11,20]. Trx1 system is conserved across all botanic and animal species [22] and extensive structural data exist with examples of crystal structure for yeasts (Saccharomyces cerevisiae, Schizosaccharomyces pombe) plants (Arabidopsis thaliana), bacteria (E. coli, M. tuberculosis), protozoa (T. vaginalis, P. falciparum, Dictyostelium discoideum), nematodes (Caenorhabditis elegans), and mammals (Rattus norvegicus, Mus musculus, Bos taurus, Equus caballus, Ovis aries, Sus scrofa, Homo sapiens) [20,24]. The active site of the Trx1 molecule (Trp-Cys-Gly-Pro-Cys) [11] is conserved across all the species described to date, indicating its essential role [22].
Interestingly, mammalian Trx1 has a slightly different structure compared to other species [11]. In addition to Cys32 and Cys35 located in its active site, it contains three other Cysteine residues (Cys62, Cys69, and Cys73) [11]. To date, there is no clear opinion regarding the function of these additional cysteines, however, it has been shown that oxidation of cysteines localized outside of the active center leads to loss of Trx1 enzymatic activity (Packer 1995). While both Cys62 and Cys69 undergo only S- nitrosylation [25,26,27], Cys73 is a multimodification site that can undergo dimerization [26], nitrosylation [27,29], gluthathionylation [31] or 4-hydroxy-2-nonenal modification [30]. In comparison, Trx2 has only one Cys residue outside its active site, therefore it is less susceptible to stress induced posttranslational modifications [32].
Trx1 is a structurally and functionally unique protein with multiple roles in cellular homeostasis. While its disulfide reductase activity is well known, Trx1 can also modify its substrates by transnitrosylation or denitrosylation of specific proteins, therefore it is involved in numerous vital cellular processes, such as cell proliferation, differentiation, migration, apoptosis, autophagy, inflammation, and metabolism [33]. Trx1 mediated reactions are fast and the Trx- target intermediate complexes are not easily detectable; hence this protein plays the role of an invisible conductor for the protein orchestra. The roles of Trx1 in vital cellular processes are demonstrated in Table 2.
Reduction of oxidized Trx1 by TrxR1 is essential for ensuring Trx1 availability and because TrxR1 is the only reducing enzyme for Trx1, it is generally expected that mutations affecting TrxR1 activity will have a similar outcome as the Trx1 null mutation [62]. In contrast, Du et al. reported that in some cellular models, cell viability is not affected after pharmacological inhibition of TrxR1, as glutathione and glutaredoxin systems were able to functionally replace TrxR1 reducing action of Trx1 in HeLa cells [63]. However, our group has reported that inhibition of TrxR1 [64] or Trx1 [41] significantly affected cell survival in neuroblastoma cells. This may reflect the difference between the cellular models used in these studies.
In this review, we aim to summarize the molecular, morphological, and functional characteristics of animal models with abolished Trx1/TrxR1 system in various organs. By comparing the described effects of Trx1/TrxR1 loss in organ- and cell-type-specific knockouts, we aimed to dissect the common functions of Trx1/TrxR1 system from those restricted to particular organ or cell type. The present review provides a valuable resource for those researchers who investigate tissue-specific roles of the Trx1 system or aim to generate specific Trx1/TrxR1 knockout strains.
2. Review of murine Trx1 system knockout models
2.1. Constitutive Trx1 and TrxR1 knockout
Global inborn Trx1 deletion is reported to be embryonically lethal [1]. While the heterozygote mice appear to be normal and fertile, embryos with complete Trx1 knockoutshow rapid growth retardation at the stage of a blastocyst (approximately 3.5 days of gestation). Trx1- null embryos had significant deficits in trophoblast formation. Ex-vivo experiments revealed defective hatching from zona pellucida which was due to significant loss of proliferative capacity. The authors linked these findings to the role of Trx1 in DNA synthesis previously shown in E. coli [1]. These findings emphasized the indispensable biological role of Trx1 and evoked interest in the investigation of underlying pathological mechanisms associated with Trx1 deficiency in mammals. The importance of Trx system in embryogenesis was further confirmed by Jakupoglu et al., who used the Cre-LocP re-combination technique [65] for excision of exon 15 in TrxR1 [66]. Similar to Trx1 general knockouts, these animals exhibited early embryonic lethality. However, the mean survival of these embryos was longer than in Trx1 knockouts with average resorption of knockout embryos from 9.5 to 10.5 gestational days [66]. TrxR1 deficient embryos displayed growth retardation with a significantly shortened anterior-posterior axis, failure of neural tube closure, and other signs of impaired somatogenesis. Interestingly, despite dramatic dysmorphogenesis in most vital organs, heart development showed no significant deviations from controls. Histological examination revealed no apparent signs of apoptosis activation in TrxR1- deficient embryos, while cell proliferation was strikingly reduced in all cell types except cardiomyocytes [66].
Bondareva et al. further elaborated on the characterization of TrxR1-null phenotype [2]. This group generated TrxR1-deficient mice by excising exons 1 and 2 of TrxR1 gene. Consequently, the mutant protein lacked both N-terminal active site cysteines (Cys59 and Cys64) and was functionally null. Unlike the TrxR1-null embryos described by Jakupoglu [66], these embryos typically stopped development earlier (at 8.5 embryonic day) failing to gastrulate. Pathohistological assessment revealed that embryonic development was interrupted pre-streak stage and there was no formation of mesoderm and ectoderm layers. The pattern of embryonic tissues was either arrested or incorrect. Authors associate reduction in embryo size with disruptions in differentiation rather than proliferation as mutant embryos were capable of generating several thousands of cells with intact intracellular coefficient (an indicator of cell size). Of note, several markers of early differentiation were found in TrxR1 knockout embryos (e.g., Cripto, Fgf8, Snail1, Mash2, and Pl1) suggesting some extent of differentiation in these embryos [2].
2.2. General inducible Trx1 knockout and dominant negative Trx1
To avoid embryonic lethality in Trx1 deficient animals. Jabbar et al. were the first to employ a Tamoxifen-inducible model of general Trx1 knockout [67]. Upon induction of Trx1 depletion, these animals experienced significant deterioration in their health condition and could survive only up to 30 days post-injection with a mean survival of 15 days. The group reported robust changes in the spleen, with approximately 30% decrease in wet mass and a 50% decrease in splenocyte count compared to the control. Flow cytometry revealed a significantly increased number of apoptotic spleen cells. Histopathologic assessment revealed no signs of inflammation, necrosis, cell degeneration or embolism in other organs (e.g., brain, heart, lung, liver, adrenal glands, or kidney). However, some changes in the morphology of the gastrointestinal tract (disorganized intestinal villi and inflammatory cell infiltration) were observed [67].
Using the dominant negative form of Trx1 is an alternative approach to disrupt the activity of the wild-type Trx1. This was achieved using site-directed mutagenesis in human Trx1 and the active Cys 32 and 35 residues were mutated to Serine, leading to loss of Trx1 activity. The transgene vector was injected into the pronuclei of fertilized eggs. These animals expressed high levels of non-functional human Trx1 and low levels of wild-type mouse Trx1 [68]. Although it is not specifically stated in this article, the animals were seemingly able to survive under normal conditions. This study mainly focused on the protective role of Trx1 in hyperoxic lung injury lungs. When exposed to hyperoxia, 100% of mice died within 72 hours of recovery period. Histopathologic assessments revealed increased alveolar damage with focal hemorrhages in alveoli and hyaline membrane deposits. On a cellular level, a decrease in ATP production and non-mitochondrial respiration was observed in both normoxic and hyperoxic conditions. Despite an increase in the expression of cell cycle regulator and DNA damage sensor p53 in non-functional Trx1 animals in both normoxic and hyperoxic conditions, apoptosis was not found to be the main mechanism of cell death in this animal after exposure to toxic concentrations of oxygen. Moreover, neither of the pro-apoptotic markers (cleaved caspase 3, cleaved PARP, Bax, BCL2, JNK, pASK, etc.) analyzed by Das showed a significant increase compared to control. Consequently, the author suggested necrosis to be the underlying mechanism of severe and rapid lung damage after hyperoxic injury. The involvement of Trx1 in this study was confirmed by the generation of Trx1 overexpressing mice. These mice were able to better handle the effect of hyperoxic injury [68].
2.3. Organ-specific Trx1/TrxR1 knockouts
2.3.1. Heart-specific depletion of Trx1 activity
The role of Trx1 in cardiac muscle has been previously shown in several in vitro studies and reviewed here [69]. A model of cardiac-specific Trx1functional depletion was first generated by Yamamoto et al. [70]. A redox inactive isoform of human-Trx1 (hTrx1) was expressed in cardiomyocytes using α-myosin heavy chain-Cre mice strain. In this model, disulfide oxidoreductase activity of endogenous Trx1 is suppressed by mutant hTrx1. When kept in basal conditions, these mice developed a concentric form of cardiac hypertrophy with a significantly enlarged septum and left ventricle wall as assessed by histology and echocardiography. Of note, these morphological abnormalities did not reflect in systolic or diastolic dysfunction. The authors link observed changes in myocardium with severe oxidative distress caused by loss of Trx1 function. Thus, cardiomyocytes in these animals displayed signs of oxidative stress-induced DNA damage, evidenced by 8-OHdG staining, and enhanced lipid peroxidation that was shown by 4HAE staining. There were no signs of altered cell proliferation in these mice suggesting the sole contribution of cardiomyocyte hypertrophy to enlargement of the cardiac muscle. Mechanistically, loss of Trx1 function resulted in activation of the ERK pathway, a well-known player in concentric heart hypertrophy and protection against pro-apoptotic stimuli [71].
One of the latest advances in dissecting the role of Trx1 in the metabolism and function of the heart was the generation of Trx1 heart-specific knockout model by Oka et al. This group was the first to report heart-specific loss of function model [43]. Cardiomyocyte-specific Trx1 deletion was achieved by breeding Trx1fl/fl animal with Myh6-Cre mouse. These animals had dramatically shortened lifespans (median survival 25.5 days) due to significant functional deficits in heart function. Thus, cardiac dilatation hypertrophy with decreased left ventricle ejection fraction in these mice resulted in chronic heart failure. At the cellular level, Trx1-deficient cardiomyocytes showed a two-fold increase in size compared to controls with a large proportion of apoptotic cells as demonstrated by the TUNEL assay. Furthermore, a nearly twofold activation of caspase 3 levels confirmed the domination of pro-apoptotic response in Trx1-null heart muscle cells. In addition to involvement of Trx1 in regulation of cell proliferation that was observed in other studies, Trx1 protective function has also been linked to regulation of autophagy. This highly regulated recycling program is responsible for removing damaged proteins and organelles. Accordingly, interruption of autophagy in Trx1 heart-specific knockout was shown by oxidation of mTOR, the master regulator of autophagy in the cell. Activation of autophagy is assessed by increased levels of p62 and LC3II/LC3I ratio. Authors showed that lack of Trx1 resulted in oxidation of mTOR at Cys 1483, however, mTOR inactivation did not result in changes in p62 or LC I/II levels. Surprisingly, mTOR inhibition in this model contributed to the dysregulation of metabolic genes and mitochondria dysfunction, which are critical for the health of highly specialized cell types with enhanced metabolic turnover such as cardiomyocytes [72]. Similar to other cardiac-specific gene knockouts that impair mitochondria health, disruption of mitochondria metabolism is viewed as the dominant cause of cardiomyopathy and heart failure in Trx1 deficient mice [43].
2.3.2. Liver-specific Trx1 and TrxR1 knockouts
Metabolism of hepatocytes is associated with excessive production of ROS; therefore, these cells heavily rely on cellular antioxidants [73]. Interestingly liver-specific downregulation of Trx1 and TrxR1 is not lethal [62]. Despite some lethality of 15%, the adult mice were fertile in both sexes. The adult mice have larger livers due to hepatocyte proliferation. The hepatocytes maintained their redox balance through de novo GSH synthesis using methionine, however as expected the hepatocytes were not able to reduce the oxidized GSH (GSSG). Trx1 was shown to be used as an electron source in the reduction of cysteine and GSH synthesis. The group then attempted a triple liver-specific knockout mice model of null Trx1/TrxR1/GR [62]. Trx1 depletion in hepatocytes did not cause overall liver pathology and it did not decrease the lifespan of these animals. Despite the absence of obvious morphological changes, ultrastructural analysis of Trx1-null hepatocytes showed a significant decrease in mitochondria cross-sectional area compared to controls with mitochondrial networks composed of thinner branches. However, this did not result in any dysfunctions of mitochondrial metabolism. Another finding was the increased size of nuclei in Trx1 deficient hepatocytes. However, due to a slight but not significant increase in hepatocyte volume, the nucleus/cytoplasm ratio was not altered [62]. Examination of serum from wildtypes with mice with TrxR1/GR null livers showed evidence of chronic liver damage but overall hepatic functions were unaffected.
Another study on TrxR1 deficiency in hepatocytes used Cre-LoxP recombinase under the control of Albumin promoter [74]. For this purpose, they bred TrxR1fl/fl and Alb- Cre animals. The offspring was fully viable with a mean survivance of more than 1 year. Despite anticipated disruption in the redox system, this group did not detect any evidence of oxidative distress in TrxR1-deficient livers. Transcriptome assessment showed evidence of increased oxidative stress in this model as 21 out of 56 upregulated genes contained antioxidant response elements (AREs) and belonged to the Nrf2 pathway. The proposed activation of the Nrf2 pathway was supported by an increased Nrf2 signal in nuclei of TrxR1-depleted hepatocytes indicating increased cytoplasmic to nuclear translocation. Canonically, Nrf2 activation is triggered by oxidative stress. The possibility of Nrf2 pathway upregulation in the absence of oxidative damage and the role of the Trx1 system in it remains unexplored. Overall, normal lifespan, morphology and absence of oxidative stress markers indicate the presence of robust compensatory mechanisms in TrxR1- depleted hepatocytes [74].
This group further continued dissecting the role of TrxR1 in liver metabolism. Thus, using the previously described model, they investigated the impact of TrxR1 deficiency on the replicative potential of hepatocytes during development and regeneration [75]. In this work, they demonstrated no significant difference in liver growth rate and number of proliferating hepatocytes (as evidenced by PCNA and phosphohistone H3 staining, and BrdU in-corporation). These findings led authors to the conclusion that the Trx-R1 is not indispensable for DNA replication and normal proliferative growth in the liver [75].
Later publication from this group describes the outcomes of liver-specific TrxR1 ablation for hepatic lipogenesis, glycogen synthesis and detoxification [76]. They showed that TrxR1-deficient livers exhibit repression of lipogenic genes reflecting in glycogen accumulation (specifically in periportal hepatocytes). Additionally, as a mechanism compensatory to TrxR1 depletion, mutant hepatocytes upregulated the machinery for glutathione biosynthesis, glutathionylation and glucuronidation. These modifications resulted in the preconditioning of mutant hepatocytes to even more robust elimination of xenobiotics compared to wild-type littermates. For instance, TrxR1-deficient liver cells were refractory to classic acetaminophen-induced pathology (APAP-pathology). In this experiment, wild types showed signs of classic APAP-induced pathology, from centrilobular vacuolization at 2 hours after APAP challenge to profound sinusoidal congestion at 8 hours upon exposure. Excitingly, TrxR1-null livers displayed minimal centrilobular necrosis with no sinusoidal congestion. Overall, in these animals, the APAP challenge provoked only slight and transient changes in liver morphology with reversion to normal morphology as early as 8-12 hours after the challenge [76].
A recent publication from this group elucidates the role of TrxR1 during acute cholestatic liver injury [77]. In this study, they subjected mice with liver-specific depletion of TrxR1 to bile duct ligation surgery. Unlike wild-type, TrxR1 livers did not display histological signs of necrosis or enhanced fibrogenesis. TrxR1-depleted hepatocytes also had significantly lower expression of NLRP3 inflammasome complex along with less prominent expression of proinflammatory cytokines. These findings highlight the role of TrxR1 in the regulation of pro-inflammatory responses in acute cholestasis [77].
2.3.3. Pancreatic β-cell-specific TrxR1 knockout
Oxidative distress is a mediator of pancreatic β-cell damage [78]. ROS overproduction has been documented in pancreatic tissue derived from diabetic patients and animal models [79,80,81,82,83,84]. Additionally, antioxidant proteins are expressed in lower concentrations compared to other tissue types, e.g., liver or kidney [85,86]. In vitro reports suggest that inhibition of Trx1 system sensitizes β-cells to oxidative damage and activates apoptosis [87]. Thus, it has been shown that TrxR1 Is essential for the β-cell defence against continuous exposure to hydrogen peroxide and peroxynitrite [88].
Importantly, there was no evidence of any secondary mechanism to protect β-cells from such oxidative damage. Stancill et al. questioned whether these findings could be replicated in in vitro systems [89]. In their study, Insulin-Cre (Ins-Cre) mice were crossed with TrxR1fl/fl to generate β-a cell-specific TrxR1 knockout. Functionally, there was no difference in glucose tolerance after a 4-hour fasting period or in plasma insulin levels 15 min after glucose challenge in mutants of both sexes compared to controls. Despite this fact, male knockouts demonstrated slightly but significantly increased blood glucose after 4 hours of fasting, whereas in females’ glucose concentration was similar to controls. Ex-vivo assessment of pancreatic islands derived from the knockouts showed a significant reduction of glucose-stimulated and membrane depolarization-stimulated insulin secretion, highlighting the importance of TrxR1 in β-cell function. Intriguingly, the reduction of β-cell function in this model was not due to apoptosis but rather because of disturbances in the maturation of TrxR1-deficient β-cells. Identification of factors involved in β-cell maturation involved in the regulation of glucose sensing (e.g., Mafa, Pdx1, Ucn3, Slc2a2, and Gpd2) may explain the observed deficits [89].
2.3.4. T-Cell-specific TrxR1 knockout
Previous studies have demonstrated the role of Txnip/Trx1 system in the regulation of glucose metabolism [90] Txnip-deficient mice display hyperglycemia, hyperinsulinemia, and liver steatosis [90]. Txnip is reported to be significantly reduced in activated T-cells, while Trx1 and TrxR1 are upregulated. This mechanism is crucial for DNA synthesis during metabolic reprogramming of T-cells [91]. Additionally, activated T-cells require high amounts of glucose and amino acids and therefore upregulate glycolysis and glutaminolysis [91,92]. To elucidate the role of Trx1/TrxR1 system in T-cell mediated immunity, Muri et al. developed T-cell specific TrxR1 knockout [93]. They crossed TrxR1fl/fl with a CD4-Cre animal, which drives Cre-recombinase expression to CD4 and CD8 double-positive thymocytes. Consistent with the role of Trx1/TrxR1 system in providing reducing equivalents for ribonucleotide reductase [94], TrxR1-depleted thymocytes had improper purine and pyrimidine synthesis. Muri et al. also reported an accumulation of metabolites in glutamate and aspartate metabolic pathways that the authors identified as another sign of impaired nucleotide biosynthesis. Morphologically, TrxR1-deficient thymuses were significantly reduced in size. This was typically associated with a dramatic decrease in both the number and size of thymocytes. Further investigation failed to detect any signs of apoptosis activation. In contrast, the decreased expression of CD4 and CD8 T-cell population was aligned with a reduced count of EDU+ thymocytes and a simultaneous increase in cell cycle inhibitor Cdkn1a. These findings showed a decrease in CD4 and CD8 cell pools due to proliferation deficits rather than because of cell death [93].
2.3.5. Brain-specific Trx1 null-mutation
Brain tissue is highly sensitive to redox balance and therefore is more vulnerable to oxidative stress compared to liver or kidney [95]. Due to higher levels of oxygen consumption, increased content of lipids with unsaturated fatty acids, elevated iron content, and lower activities of redox scavengers (e.g., superoxide dismutase, catalase, and glutathione peroxidase), brain metabolism is associated with increased ROS production [96]. Oxidative distress has been linked to several neurodegenerative disorders such as amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and Huntington’s disease [95,97,98]. The role of Trx1 in neuronal homeostasis is extensively described in in vitro models [41,99]. However, to the best of our knowledge, no neuron/glial-specific Trx1 knockdown animal model has been reported. A recent report has linked Trx1 deficiency in the brain to epilepsy [100,101].
This study focused on a rare rat strain with frequent running seizures generated by N-ethyl-N- nitrosourea (ENU) mutation [102]. To identify the potential mutation, the animals with the desired phenotype were first backcrossed for more than 10 generations to achieve a higher frequency of mutation in offspring. Using genetic locus mapping methods, they identified a mutation in chromosome 5, which was then sequenced resulting in the identification of a single heterozygous missense mutation (Phenylalanine replaced by Leucine) in the exon region c.T160C of Trx1 gene. Intriguingly, this mutation was remote from the active site that was predicted to impair Trx1 functions beyond dithiol/disulfide exchange. These rats presented with the highest frequency of seizures at 5 weeks of age. The histopathological assessment showed vacuolar degeneration in the midbrain (inferior colliculus and thalamus). Interestingly, no changes were observed in the hippocampus, the brain region responsible for the onset of epileptic activity in classic temporal lobe epilepsy [103]. Degeneration in the midbrain was also associated with decreased neuronal and oligodendrocyte count, myelin loss, astro-gliosis, and increased expression of 8-OHd6, the marker of ROS-mediated DNA damage. Trx1 depletion also had an impact on energy metabolism in the thalamus as shown by absolute quantitative values of NAD+, NADP+, UPD-glucose, etc. Despite the performed genetic mapping, the authors could not completely exclude other mutations in the gene resulting in such phenotype. Therefore, they used the CRISPR- Cas approach to reproduce the previously identified mutations in wild-type rats. The behavioral and histopathologic features in this knock-in model were similar to those found in previously studied mutants. Successful phenotype reproduction confirmed the major contribution of c.T160C mutation in the exon of the Trx1 gene to this pathology. Interestingly, the severity of behavioral abnormalities was observed to decrease after the peak at 5 weeks with a complete absence of seizure activity by week 9. Similarly, histological assessment showed no signs of vacuolar degeneration which was evident at 5 weeks. The nature of this spontaneous recovery remains to be investigated [100,101].
2.3.6. Brain- and neuron-specific knockouts of TrxR1
The depletion of the Trx1/TrxR1 system has been linked to several neurodegenerative diseases, e.g., Alzheimer’s disease and Parkinson’s disease [98,104]. Defects in neural tube closure in embryos with constitutive TrxR1 suggest the involvement of this antioxidant protein in the proper development of the neural system [66]. Soerensen et al. were the first to investigate the role of TrxR1 in mice brains using a gene knockout approach [105]. This group used TrxR1tm1Marc to generate two TrxR1 knockout strains: 1) by crossing with Nestin-Cre mouse which drove Cre-recombinase to precursor cells of both neuronal and glial lineages; 2) by crossing with Tα1-Cre mouse which drove Cre-recombinase specifically to neurons [105]. Interestingly, these two mutant strains displayed drastically different phenotypes. Mice with TrxR1 depletion in neuronal and glial lineages had significant growth retardation reaching only 50% of the body mass compared to controls by 4 weeks. Additionally, they displayed notable impairments in coordinated movement (e.g., ataxic gate, balance problems, and tremor) suggesting primary involvement of the cerebellum. Histologically, these mice displayed striking cerebellar hypoplasia with reduced proliferation in the external granular layer. Additionally, a strong reduction of fissure formation and laminar organization of the cerebellar cortex was present in this mutant strain. On the cellular level, Soerensen et al. observed disruption of the Bergmann glia network along with ectopic localization and reduced arborization of Purkinje cells. According to the authors, reduction in size of the cerebellum and decreased cell count were not associated with apoptosis, but rather with reduced proliferation of granule cell precursors in the external granule layer [105].
In contrast, the mice with neuron-specific ablation of TrxR1 did not present any significant phenotype. There were no significant behavioural or histological abnormalities [105]. Taken together, these findings may suggest that TrxR1 expression has more impact on neural precursors and glial cells rather than on mature neurons.
3. Concluding remarks
The generation of animal strains lacking Trx1 or TrxR1 activity has proven invaluable for exploring the roles of this redox system in maintaining cellular homeostasis. Despite a limited number of characterized animal models, current findings underscore the indispensable role of the Trx1 system in the brain and heart, but liver or pancreas-specific deletion of these genes seems to be normal, and the mice had an overall healthy life span. This perhaps reflects the differential dependency of various cell types to different antioxidants. For example, a higher dependency of rat brain mitochondria on Trx system for per-oxide scavenging has been shown by the addition of specific inhibitors Trx and GSH [106]. These observations suggest that the Trx1/TrxR1 system's specific functions in different organs likely depend on organ-specific roles. While it exhibits a general cytoprotective effect across various cell types, it also influences cell proliferation and differentiation in mitotic cells. In post-mitotic cells, the Trx1/TrxR1 system predominantly has an antiapoptotic effect. However, in cell types with high metabolic activity, such as hepatocytes or cardiomyocytes, it plays a crucial role in regulating mitochondrial metabolism. It is also plausible that different stress conditions will require differential involvement of cellular antioxidants.
Although theoretically, the effects of Trx1 and TrxR1 knockouts are expected to be identical, experimental models of Trx1 deficiency tend to have more pronounced effects compared to TrxR1 deficiency. Studies with hepatocyte Trx1 vs. TrxR1 knockout models showed that Trx1 plays other roles than simply being a substrate of TrxR1. In fact, Trx1 has been shown to regulate the activity of many other proteins in both the cytoplasm and the nucleus. It is therefore expected that functional deletion of Trx1 will have more devastating effects than TrxR1 depletion. While there is substantial literature on Trx system in the heart, we have just started to understand the impact of Trx system on the brain. Novel Trx1 deletion models are currently available that will highlight the multifaceted functions of Trx1 that extend beyond its antioxidant capacity. The summarized characterization of the discussed Trx1 and TrxR1 knockout mice strains is provided in Table S1.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Summary of murine Trx1 system knockout models.
Author Contributions
TS: conceptualization, writing—original draft preparation and editing; EE: writing— original draft, review and editing, supervision. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
n/a.
Data Availability Statement
n/a.
Acknowledgments
n/a.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Matsui M, Oshima M, Oshima H, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178(1):179-185. [CrossRef]
- Bondareva AA, Capecchi MR, Iverson SV, et al. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43(6):911-923. [CrossRef]
- Holmgren, A. Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure. 1995;3(3):239-243. [CrossRef]
- Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci. 2015;40(8):435-445. [CrossRef]
- Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. Published 2014 Jan 9. [CrossRef]
- Borowiec AM, Właszczuk A, Olakowska E, Lewin-Kowalik J. TXNIP inhibition in the treatment of diabetes. Verapamil as a novel therapeutic modality in diabetic patients. Med Pharm Rep. 2022;95(3):243-250. [CrossRef]
- Medinas DB, Rozas P, Hetz C. Critical roles of protein disulfide isomerases in balancing proteostasis in the nervous system. J Biol Chem. 2022;298(7):102087. [CrossRef]
- Wang L, Wang CC. Oxidative protein folding fidelity and redoxtasis in the endoplasmic reticulum. Trends Biochem Sci. 2023;48(1):40-52. [CrossRef]
- Jiménez A, Zu W, Rawe VY, et al. Spermatocyte/spermatid-specific thioredoxin-3, a novel Golgi apparatus-associated thioredoxin, is a specific marker of aberrant spermatogenesis. J Biol Chem. 2004;279(33):34971-34982. [CrossRef]
- Ouyang Y, Peng Y, Li J, Holmgren A, Lu J. Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems. Metallomics. 2018;10(2):218-228. [CrossRef]
- Hasan AA, Kalinina E, Tatarskiy V, Shtil A. The Thioredoxin System of Mammalian Cells and Its Modulators. Biomedicines. 2022;10(7):1757. Published 2022 Jul 21. [CrossRef]
- Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem. 1997;272(5):2936-2941. [CrossRef]
- Lowes DA, Galley HF. Mitochondrial protection by the thioredoxin-2 and glutathione systems in an in vitro endothelial model of sepsis. Biochem J. 2011;436(1):123-132. [CrossRef]
- Tanaka T, Hosoi F, Yamaguchi-Iwai Y, et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002;21(7):1695-1703. [CrossRef]
- Zhang R, Al-Lamki R, Bai L, et al. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res. 2004;94(11):1483-1491. [CrossRef]
- Damdimopoulos AE, Miranda-Vizuete A, Pelto-Huikko M, Gustafsson JA, Spyrou G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J Biol Chem. 2002;277(36):33249-33257. [CrossRef]
- Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23(3):916-922. [CrossRef]
- Monteiro HP, Ogata FT, Stern A. Thioredoxin promotes survival signaling events under nitrosative/oxidative stress associated with cancer development. Biomed J. 2017;40(4):189-199. [CrossRef]
- Mougiakakos D, Johansson CC, Jitschin R, Böttcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. 2011;117(3):857-861. [CrossRef]
- Bertini R, Howard OM, Dong HF, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189(11):1783-1789. [CrossRef]
- Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267(34):24161-24164.
- Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102-6109. [CrossRef]
- Holmgren, A. Thioredoxin. 6. The amino acid sequence of the protein from escherichia coli B. Eur J Biochem. 1968;6(4):475-484. [CrossRef]
- Andoh T, Chock PB, Chiueh CC. The roles of thioredoxin in protection against oxidative stress-induced apoptosis in SH-SY5Y cells. J Biol Chem. 2002;277(12):9655-9660. [CrossRef]
- Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4(10):743-749. [CrossRef]
- Tao L, Gao E, Bryan NS, et al. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: role of S-nitrosation [corrected] [published correction appears in Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13694]. Proc Natl Acad Sci U S A. 2004;101(31):11471-11476. [CrossRef]
- Weichsel A, Brailey JL, Montfort WR. Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Biochemistry. 2007;46(5):1219-1227. [CrossRef]
- Wang Y, Liu T, Wu C, Li H. A strategy for direct identification of protein S-nitrosylation sites by quadrupole time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2008;19(9):1353-1360. [CrossRef]
- Wu C, Liu T, Chen W, et al. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics. 2010;9(10):2262-2275. [CrossRef]
- Casagrande S, Bonetto V, Fratelli M, et al. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A. 2002;99(15):9745-9749. [CrossRef]
- Go YM, Halvey PJ, Hansen JM, Reed M, Pohl J, Jones DP. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am J Pathol. 2007;171(5):1670-1681. [CrossRef]
- Kim MK, Zhao L, Jeong S, et al. Structural and Biochemical Characterization of Thioredoxin-2 from Deinococcus radiodurans. Antioxidants (Basel). 2021;10(11):1843. Published 2021 Nov 20. [CrossRef]
- Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2013;18(10):1165-1207. [CrossRef]
- Ueno M, Masutani H, Arai RJ, et al. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem. 1999;274(50):35809-35815. [CrossRef]
- Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90(12):1259-1266. [CrossRef]
- Hwang J, Suh HW, Jeon YH, et al. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat Commun. 2014;5:2958. [CrossRef]
- Akterin S, Cowburn RF, Miranda-Vizuete A, et al. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 2006;13(9):1454-1465. [CrossRef]
- Nakamura, H. Extracellular functions of thioredoxin. Novartis Found Symp. 2008;291:184-224. [CrossRef]
- Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596-2606. [CrossRef]
- Chae JS, Gil Hwang S, Lim DS, Choi EJ. Thioredoxin-1 functions as a molecular switch regulating the oxidative stress-induced activation of MST1. Free Radic Biol Med. 2012;53(12):2335-2343. [CrossRef]
- Islam MI, Nagakannan P, Ogungbola O, Djordjevic J, Albensi BC, Eftekharpour E. Thioredoxin system as a gatekeeper in caspase-6 activation and nuclear lamina integrity: Implications for Alzheimer's disease. Free Radic Biol Med. 2019;134:567-580. [CrossRef]
- Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A. 2007;104(28):11609-11614. [CrossRef]
- Oka SI, Chin A, Park JY, et al. Thioredoxin-1 maintains mitochondrial function via mechanistic target of rapamycin signalling in the heart. Cardiovasc Res. 2020;116(10):1742-1755. [CrossRef]
- Nagarajan N, Oka SI, Nah J, et al. Thioredoxin 1 promotes autophagy through transnitrosylation of Atg7 during myocardial ischemia. J Clin Invest. 2023;133(3):e162326. Published 2023 Feb 1. [CrossRef]
- Pérez-Pérez ME, Zaffagnini M, Marchand CH, Crespo JL, Lemaire SD. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy. 2014;10(11):1953-1964. [CrossRef]
- Hu J, Liu J, Chen S, et al. Thioredoxin-1 regulates the autophagy induced by oxidative stress through LC3-II in human lens epithelial cells. Clin Exp Pharmacol Physiol. 2023;50(6):476-485. [CrossRef]
- Wang X, Ling S, Zhao D, et al. Redox regulation of actin by thioredoxin-1 is mediated by the interaction of the proteins via cysteine 62. Antioxid Redox Signal. 2010;13(5):565-573. [CrossRef]
- Landino LM, Iwig JS, Kennett KL, Moynihan KL. Repair of peroxynitrite damage to tubulin by the thioredoxin reductase system. Free Radic Biol Med. 2004;36(4):497-506. [CrossRef]
- Morinaka A, Yamada M, Itofusa R, et al. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci Signal. 2011;4(170):ra26. Published 2011 Apr 26. [CrossRef]
- Bai J, Nakamura H, Kwon YW, et al. Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J Neurosci. 2003;23(2):503-509. [CrossRef]
- Sartelet H, Rougemont AL, Fabre M, et al. Activation of the phosphatidylinositol 3'-kinase/AKT pathway in neuroblastoma and its regulation by thioredoxin 1. Hum Pathol. 2011;42(11):1727-1739. [CrossRef]
- Chen B, Guan D, Cui ZJ, Wang X, Shen X. Thioredoxin 1 downregulates MCP-1 secretion and expression in human endothelial cells by suppressing nuclear translocation of activator protein 1 and redox factor-1. Am J Physiol Cell Physiol. 2010;298(5):C1170-C1179. [CrossRef]
- Schenk H, Klein M, Erdbrügger W, Dröge W, Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci U S A. 1994;91(5):1672-1676. [CrossRef]
- Naranjo-Suarez S, Carlson BA, Tobe R, et al. Regulation of HIF-1α activity by overexpression of thioredoxin is independent of thioredoxin reductase status. Mol Cells. 2013;36(2):151-157. [CrossRef]
- Guo Y, Einhorn L, Kelley M, et al. Redox regulation of the embryonic stem cell transcription factor oct-4 by thioredoxin. Stem Cells. 2004;22(3):259-264. [CrossRef]
- Ago T, Liu T, Zhai P, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133(6):978-993. [CrossRef]
- Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82(1):308-317. [CrossRef]
- Makino Y, Yoshikawa N, Okamoto K, et al. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J Biol Chem. 1999;274(5):3182-3188. [CrossRef]
- Rao AK, Ziegler YS, McLeod IX, Yates JR, Nardulli AM. Thioredoxin and thioredoxin reductase influence estrogen receptor alpha-mediated gene expression in human breast cancer cells. J Mol Endocrinol. 2009;43(6):251-261. [CrossRef]
- King BC, Nowakowska J, Karsten CM, Köhl J, Renström E, Blom AM. Truncated and full-length thioredoxin-1 have opposing activating and inhibitory properties for human complement with relevance to endothelial surfaces. J Immunol. 2012;188(8):4103-4112. [CrossRef]
- Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31(11):1287-1312. [CrossRef]
- Prigge JR, Coppo L, Martin SS, et al. Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase. Cell Rep. 2017;19(13):2771-2781. [CrossRef]
- Du Y, Zhang H, Lu J, Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem. 2012;287(45):38210-38219. [CrossRef]
- Nagakannan P, Iqbal MA, Yeung A, et al. Perturbation of redox balance after thioredoxin reductase deficiency interrupts autophagy-lysosomal degradation pathway and enhances cell death in nutritionally stressed SH-SY5Y cells. Free Radic Biol Med. 2016;101:53-70. [CrossRef]
- Shcholok T, Eftekharpour E. Cre-recombinase systems for induction of neuron-specific knockout models: a guide for biomedical researchers. Neural Regen Res. 2023;18(2):273-279. [CrossRef]
- Jakupoglu C, Przemeck GK, Schneider M, et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25(5):1980-1988. [CrossRef]
- Jabbar S, Mathews P, Wang X, et al. Thioredoxin-1 regulates self-renewal and differentiation of murine hematopoietic stem cells through p53 tumor suppressor. Exp Hematol Oncol. 2022;11(1):83. Published 2022 Oct 31. [CrossRef]
- Das, KC. Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;308(5):L429-L442. [CrossRef]
- Nagarajan N, Oka S, Sadoshima J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic Biol Med. 2017;109:125-131. [CrossRef]
- Yamamoto M, Yang G, Hong C, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112(9):1395-1406. [CrossRef]
- Mutlak M, Kehat I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front Pharmacol. 2015;6:149. Published 2015 Jul 24. [CrossRef]
- Chandra M, Escalante-Alcalde D, Bhuiyan MS, et al. Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol. 2018;14:261-271. [CrossRef]
- Mello T, Zanieri F, Ceni E, Galli A. Oxidative Stress in the Healthy and Wounded Hepatocyte: A Cellular Organelles Perspective. Oxid Med Cell Longev. 2016;2016:8327410. [CrossRef]
- Suvorova ES, Lucas O, Weisend CM, et al. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4(7):e6158. Published 2009 Jul 7. [CrossRef]
- Rollins MF, van der Heide DM, Weisend CM, et al. Hepatocytes lacking thioredoxin reductase 1 have normal replicative potential during development and regeneration. J Cell Sci. 2010;123(Pt 14):2402-2412. [CrossRef]
- Iverson SV, Eriksson S, Xu J, et al. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic Biol Med. 2013;63:369-380. [CrossRef]
- Shearn CT, Anderson AL, Miller CG, et al. Thioredoxin reductase 1 regulates hepatic inflammation and macrophage activation during acute cholestatic liver injury. Hepatol Commun. 2023;7(1):e0020. Published 2023 Jan 10. [CrossRef]
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic β-cells. Biochim Biophys Acta Gen Subj. 2017;1861(8):1929-1942. [CrossRef]
- Ghiselli A, Laurenti O, De Mattia G, Maiani G, Ferro-Luzzi A. Salicylate hydroxylation as an early marker of in vivo oxidative stress in diabetic patients. Free Radic Biol Med. 1992;13(6):621-626. [CrossRef]
- Gopaul NK, Anggård EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS Lett. 1995;368(2):225-229. [CrossRef]
- Nourooz-Zadeh J, Tajaddini-Sarmadi J, McCarthy S, Betteridge DJ, Wolff SP. Elevated levels of authentic plasma hydroperoxides in NIDDM. Diabetes. 1995;44(9):1054-1058. [CrossRef]
- Ihara Y, Toyokuni S, Uchida K, et al. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48(4):927-932. [CrossRef]
- Shin CS, Moon BS, Park KS, et al. Serum 8-hydroxy-guanine levels are increased in diabetic patients. Diabetes Care. 2001;24(4):733-737. [CrossRef]
- Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45(1):85-96. [CrossRef]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20(3):463-466. [CrossRef]
- Lenzen, S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36(Pt 3):343-347. [CrossRef]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146(5):2397-2405. [CrossRef]
- Stancill JS, Broniowska KA, Oleson BJ, Naatz A, Corbett JA. Pancreatic β-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J Biol Chem. 2019;294(13):4843-4853. [CrossRef]
- Stancill JS, Hansen PA, Mathison AJ, Schmidt EE, Corbett JA. Deletion of Thioredoxin Reductase Disrupts Redox Homeostasis and Impairs β-Cell Function. Function (Oxf). 2022;3(4):zqac034. Published 2022 Jul 4. [CrossRef]
- Oka S, Yoshihara E, Bizen-Abe A, et al. Thioredoxin binding protein-2/thioredoxin-interacting protein is a critical regulator of insulin secretion and peroxisome proliferator-activated receptor function. Endocrinology. 2009;150(3):1225-1234. [CrossRef]
- Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345-1360. [CrossRef]
- Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871-882. [CrossRef]
- Muri J, Heer S, Matsushita M, et al. The thioredoxin-1 system is essential for fueling DNA synthesis during T-cell metabolic reprogramming and proliferation. Nat Commun. 2018;9(1):1851. Published 2018 May 10. [CrossRef]
- Holmgren A, Sengupta R. The use of thiols by ribonucleotide reductase. Free Radic Biol Med. 2010;49(11):1617-1628. [CrossRef]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65-74. [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62(6):649-671. [CrossRef]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787-795. [CrossRef]
- Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019;218:165-184. [CrossRef]
- Islam MI, Nagakannan P, Shcholok T, et al. Regulatory role of cathepsin L in induction of nuclear laminopathy in Alzheimer's disease. Aging Cell. 2022;21(1):e13531. [CrossRef]
- Ohmori I, Ouchida M, Imai H, Ishida S, Toyokuni S, Mashimo T. Thioredoxin deficiency increases oxidative stress and causes bilateral symmetrical degeneration in rat midbrain. Neurobiol Dis. 2022;175:105921. [CrossRef]
- Ohmori I, Ouchida M, Shinohara M, Kobayashi K, Ishida S, Mashimo T. Novel animal model of combined generalized and focal epilepsy. Epilepsia. 2022;63(7):e80-e85. [CrossRef]
- Mashimo T, Yanagihara K, Tokuda S, et al. An ENU-induced mutant archive for gene targeting in rats. Nat Genet. 2008;40(5):514-515. [CrossRef]
- Chatzikonstantinou, A. Epilepsy and the hippocampus. Front Neurol Neurosci. 2014;34:121-142. [CrossRef]
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103(2):373-383. [CrossRef]
- Soerensen J, Jakupoglu C, Beck H, et al. The role of thioredoxin reductases in brain development. PLoS One. 2008;3(3):e1813. Published 2008 Mar 19. [CrossRef]
- Drechsel DA, Patel M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J Biol Chem. 2010;285(36):27850-27858. [CrossRef]
Figure 1.
Thioredoxin system. Thioredoxins (Trx) donate Hydrogen to resolve disulfide bonds (S-S) in protein Cysteines (Cys). Oxidized Trx is reduced by Thioredoxin reductases (TrxR) with the help of Reduced Nicotinamide Adenine Dinucleotide Phosphate (NADPH). Thioredoxin-1 (Trx-1) is typically localized in cytoplasm but can also undergo nuclear translocation, membrane binding or secretion. Thioredoxin-2 (Trx-2) and Thioredoxin reductase-2 (TrxR2) are localized in mitochondria.
Figure 1.
Thioredoxin system. Thioredoxins (Trx) donate Hydrogen to resolve disulfide bonds (S-S) in protein Cysteines (Cys). Oxidized Trx is reduced by Thioredoxin reductases (TrxR) with the help of Reduced Nicotinamide Adenine Dinucleotide Phosphate (NADPH). Thioredoxin-1 (Trx-1) is typically localized in cytoplasm but can also undergo nuclear translocation, membrane binding or secretion. Thioredoxin-2 (Trx-2) and Thioredoxin reductase-2 (TrxR2) are localized in mitochondria.
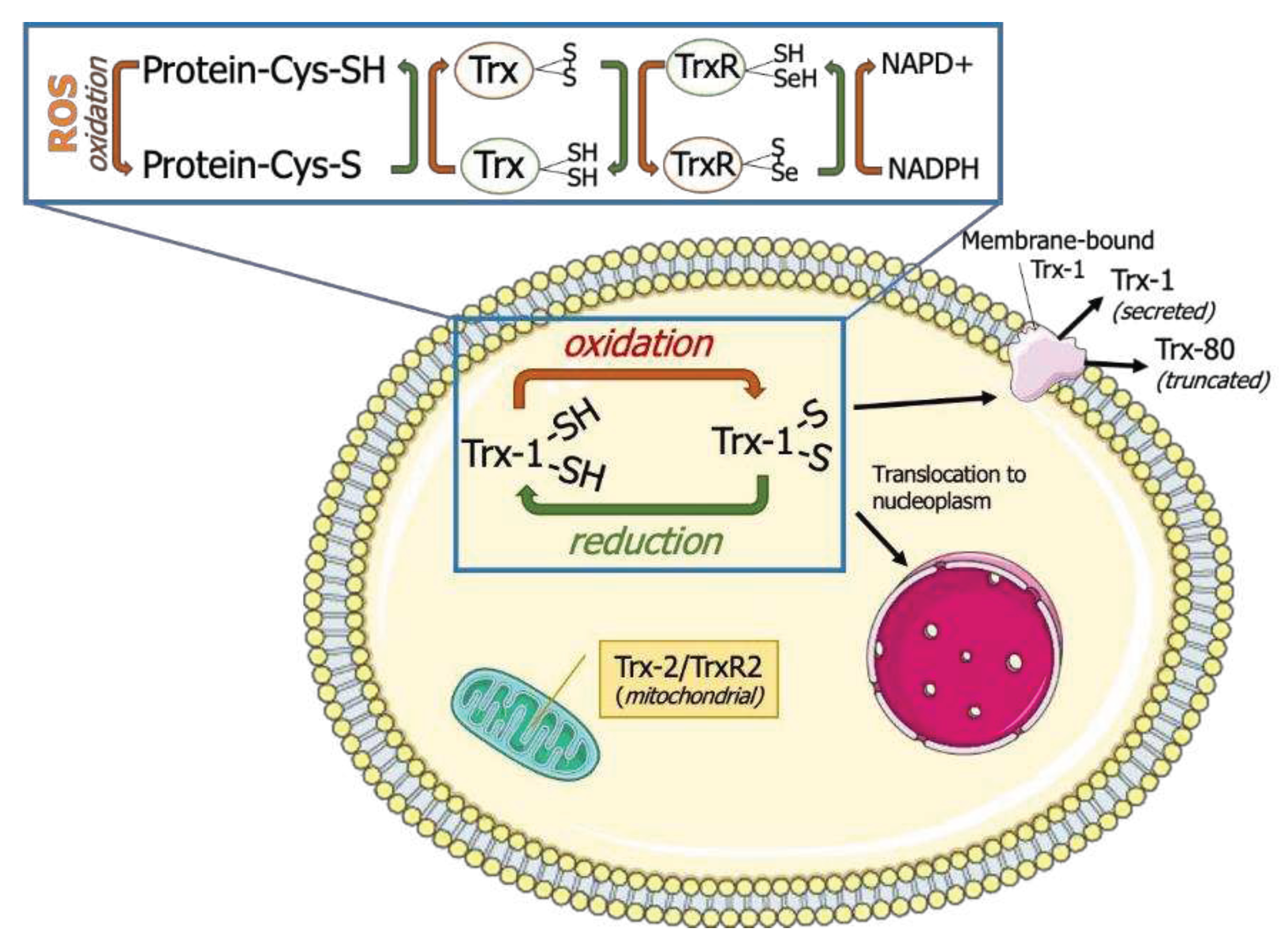

Table 1.
Characterization of Trx isoforms.
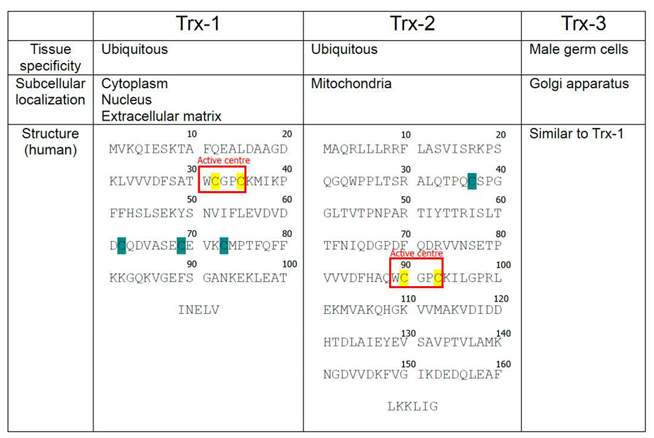 |
Table 2.
The roles of Trx1 in cellular processes in vitro.
| Subcellular localization | Cell Function | Target | Cell type | Reference |
|---|---|---|---|---|
| Cytoplasm | Antioxidative defence | Oxidized proteins |
All | Bertini et al. [20], Ueno et al. [34], Liu et al. [35], Hwang et al. [36], Akterin et al. [37], Nakamura et al. [38], Tao et al. [25], Arner et al. [22] |
| Apoptosis | ASK-1 | Mv1Lu (Mink Lung Epithelial Cells), L929 (mouse fibroblast cell line) and 293 (mouse fibroblast cell line) | Saitoh et al. [39] | |
| MST-1 | MEF (murine embryonic fibroblasts) | Chae et al. [40] | ||
| Casp-6 | SH-SY5Y (human neuroblastoma cells) | Islam et al. [41] | ||
| Casp-3 | Jurkat (immortalized human T-lymphocytes) | Mitchell et al. [42] | ||
| Autophagy | mTOR | Rat cardiomyocytes | Oka et al. [43] | |
| ATG7 | Rat cardiomyocytes | Nagarajan et al. [44] | ||
| ATG4 | Yeast Saccharomyces cerevisiae | Perez et al. [45] | ||
| LC III-B | HLE-B3 (human lens epithelial cells) | Hu et al. [46] | ||
| Cytoskeleton organization | Actin | SH-SY5Y (human neuroblastoma cells) | Wang et al. [47] | |
| Tubulin | Purified porcine tubulin | Landino et al. [48] | ||
| CRMP-2 | Embryonic DRG neurons from Sprague-Dawley rats | Morinaka et al. [49] | ||
| NGF | PC-12 (adrenal phaeochromocytoma cells) | Bai et al. [50] | ||
| Signalling transduction | AKT and PTEN | Neuroblastic neoplasms, and neuroblastoma cell lines | Sartelet et al. [51] | |
| Inflammation | Monocyte chemoattractant protein-1 (MCP-1) | Trx-1 Overexpressing/ knockdown EA.hy 926 and bovine aortic endothelial cells | Chen et al. [52] | |
| Immunomodulatory | Regulatory T-cells | Tregs (regulatory T-lymphocytes) | Mougiakakos et al. [19] | |
| Nucleus | Gene regulation | NFk-B, AP-1, and Ref-1 | L929 (mouse fibroblast cell line), HeLa (cervical cancer cells) COS-7 (African green monkey kidney fibroblast-like cells) | Chen et al., [52] Schenk et al. [53] |
| HIF-1a | HeLa (cervical cancer cells) | Naranjo-Suarez et al. [54] | ||
| Oct-4 | Embryonic stem cells | Guo et al. [55] | ||
| HDAC4 | Cardiac myocytes | Ago et al. [56] | ||
| DNA-binding | NRF-2 | HeLa (cervical cancer cells) | Hansen et al. [57] | |
| Transportation to nucleus | Glucocorticoid receptor | COS7 and CV-1 (African green monkey kidney fibroblast-like cells), HeLa (cervical cancer cells) | Makino et al. [58] | |
| Estrogen receptor | MCF-7 (breast cancer cells) | Rao et al. [59] | ||
| Cell membrane | Immunomodulatory | Complement deposition | HUVEC (human umbilical vein endothelial cells) | King et al. [60] |
| Extracellular matrix | Chemokine-like | Chemoattraction | human monocytes, PMNs, leucocytes | Bertini et al. [20] Nordberg et al. [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



