
Submitted:
15 January 2024
Posted:
16 January 2024
You are already at the latest version
Abstract
Lymphedema refers to a group of disorders characterized by the accumulation of protein-rich fluid in the interstitial tissues, resulting from an imbalance between lymph production and it’s drainage via lymphatic vessels. More than 10 types of isolated primary LD is described, and among them CELSR1-associated lymphedema are one of the less studied. A family with two sibs, a 17-year-old boy and an 8-year-old girl, both diagnosed with primary LD, presented to the clinic. Both patients were observed with primary symptoms of LD at 2-3 months of age, presented by the swelling of right foot. Lymphatic pathologies were not diagnosed in parents or close relatives. The rare c.7664A>C variant, resulting in the H2555P missense mutation in the intracellular loop of CELSR1 transmembrane domain, was discovered in a patients. The clinical signs, MRL scanning data, in silico predictors and the absence of known pathogenic mutations or rare variants of unknown significance in other genes, associated with the development of primary lymphedema, allow us to assume that c.7664A>C variant of CELSR1 may be responsible for pathogenesis of the disease in the presented family case.
Keywords:
lymphedema
; CELSR1
; family case
1. Introduction
Primary lymphedema (PLD), including isolated, syndromic, and systemic forms, occurs at a frequency of approximately 1-1.15 per 100,000 individuals under the age of 20 [1,2]. PLD arises from congenital anomalies and is associated with dysplasia, hypoplasia, or hyperplasia of components of the lymphatic system, caused by genetic mutations. In 1998, the first association between mutations in the FLT4 (VEGFR3) gene and primary congenital lymphedema was demonstrated, followed by the discovery that lymphedema-distichiasis can be caused by FOXC2 mutations in 2000 [3].
Mutations in seven genes, FLT4, GJC2, FOXC2, SOX18, GATA2, CCBE1, and PTPN14, were shown to be responsible for the development of 36% of inherited and 8% of sporadic lymphedema cases[4]. Most of them are associated with the VEGFR3 signaling pathway, the key player of embryonic lymphangiogenesis. There are increasing evidence of an association between pathways involved in vascular morphology and the development of PLD. The examples include the adhesion receptor CELSR1 and lymphatic malformation 9 (OMIM: 619319), atypical cadherin FAT4 and Hennekam lymphangiectasia-lymphedema syndrome 2 (OMIM: 616006), connexin GJC2 and lymphatic malformation 3 (OMIM: 613480), and the mechanosensitive ion channel component PIEZO1 and lymphatic malformation 6 (OMIM: 616843) [5].
2. Materials and Methods
Patient’s recruitment
Patients were treated at the Lymphology Center of the Kazan Federal University in 2021-2023. Study was approved by the local ethical committee (2020-12-28, #27) of the Kazan Federal University. Written informed consent was obtained for all patients.
MRI
Magnetic resonance lymphography (MRL) was performed using a 3.0 T MR Siemens Magnetom Verio instrument. 3D-TSE heavily T2-weighted magnetic resonance imaging was performed, data were analyzed by Syngo 3D for MIP transformation (Siemens Medical Solutions, USA).
Exome sequencing and analysis
DNA was isolated from blood samples using the QIAAmp DNA mini kit (Qiagen, USA) according to the manufacturer's instructions. DNA libraries were prepared using Agilent SureSelectXT Human All Exon v8 (Agilent Technologies, USA) and sequenced on the NextSeq 500 platform (Illumina Inc., USA). Raw reads were mapped to the human reference genome hg38 using BWA v. 0.7.17 [6]. The improvement of reads alignment and whole exome variant calling were performed using the Genome Analysis Tool Kit v.4 [7] according to the GATK Best Practices recommendations [8]. For single nucleotide variant (SNV) calling, the Haplotype Caller module was used. High-quality SNVs were filtered using vcf-annotate [9] with the following parameters: MinDP=20, Qual=10, and MinMQ=10, and annotated using wANNOVAR [10]. Synonymous variants and variants with MAF ≥1% according to the GnomAD (v. 3.1.2, total) [11] and RUSeq [12] databases were excluded from variant analysis. PolyPhen2, SIFT, MutationTaster, and CADD algorithms were used to predict the contribution of the detected variants to the functional activity of CELSR1. Clinical significance was assessed according to the ACMG criteria [13].
Sanger sequencing
Direct sequencing of exon 25 of CELSR1 were performed using ABI 3730 DNA Analyzer (Thermo Fisher Scientific, USA). Primers for PCR and sequencing were designed with PrimerQuest Tool (Integrated DNA Technologies, USA) and synthesized by Genera, LLC (Russia). Primer’s sequences were the follows (5’→3’):
Forward: GGGACTGGGTTTGTGTTCAT
Reverse: GGTCTGTTGGAAATCTGTGGT
CELSR1 target region was amplified by Q5 DNA polymerase (NEB, USA) according to the manufacturer’s guide with Ta=65°C.
3. Results
3.1. Family case
Children were born in a family of non-consanguineous parents; lymphatic pathologies were not diagnosed in parents or closer relatives in 3 generations. Mother was diagnosed with chronic pyelonephritis, toxoplasmosis, herpes simplex virus, and cytomegalovirus infection. Patient’s aunt (mother’s sister) has frequent recurrent erysipelas without visual signs of foot edema.
Male patient was delivered by natural vaginal childbirth as the first child of a first gravida with birth weight of 3454 g and Apgar score 8/9. Intrauterine left-sided hydronephrosis was diagnosed using routine antenatal ultrasound examination. Surgical treatment of vesicoureteral reflux was performed after birth, and secondary shrinkage of the left kidney and chronic pyelonephritis developed. At two months increased volume of the right foot was observed, at three months lymphostasis was confirmed by medical examination. Complex Physical Decongestive Therapy (CPDT) was carried out irregularly from the age of 11; positive result was achieved after 4 courses of therapy at 13 years, whereas intradermal thrombosis of the right foot was diagnosed at 15. At the age 17 patient had II-III stage lymphedema of right lower limb, including foot and lower third of the leg, with recurrent erysipelas, lymphocysts, papillomas. After CPDT symptoms decreased significantly (Figure 1 A-C).
His younger sister was delivered by natural vaginal childbirth as the second child of a second gravida with birth weight of 3994 g and Apgar score 8/9. Mixed CNS lesion, congenital adrenal hyperplasia and virilization was diagnosed after birth, whether her karyotype test showed no deviations. Prenatal lesions associated with lymphedema or other disorders were not discovered, edema of right lower limb was detected at 2-3 months, left lower limb edema was developed at 10 months (Figure 1D). Patient underwent treatment with CPFT 1-2 times a year with positive results from 2 years, and at the age 8 she had II stage lymphedema of both lower limbs, including foot and leg, without skin lesions.
3.2. Family case
Both patients were examined by MRL without contrast agent. No pathogenic changes were revealed in the thoracic duct, whereas hypoplastic and aplastic lymphatic components in lower extremities were discovered (Figure 2).
In boy on the right thigh in the upper third of the lower extremities, the femoral lymphatic vessel was not visualized, proximally and distally on the thigh vessels were not changed, deep lymphatic vessels of the muscles in the right leg were 2 times smaller than in the left. The medial and lateral subcutaneous lymphatic collectors of the right lower limb were not visualized, and subcutaneous lymphatic vessels were represented as diffuse small vessels.
In girl hypoplastic deep lymphatic vessels of the lower legs and thighs were visualized. The medial and lateral subcutaneous lymphatic collectors of both lower extremities were aplastic and not visualized. The subcutaneous lymphatic vessels were represented as diffuse small vessels.
3.3. Exome analysis
Exome variants were sorted by the frequency according to GnomAD 3.1 database and those with frequency ≤ 1% were selected for further analysis. 467 common variants in 360 genes were identified, and among them only CELSR1 was shown to be associated with PLD. No pathogenic or even rare variants with uncertain/unknown significance were found in the 87 other genes associated with isolated or syndromic PLD, including FLT4, GJC2, FOXC2, SOX18, GATA2, CCBE1, and PTPN14, which have been linked with the most cases of the disease [4].
Genomic c.7664A>C variant was identified as a joint variant for two siblings. Variant was additionally analyzed by Sanger sequencing in children and their parents. Heterozygous change was confirmed in both children and their mother, whereas father was unaffected (Figure 3).
In silico predictors SIFT, Plyphen2 and Mutation tester, as well as the evolutionary conservation assessment instruments alone, were used to estimate the possible influence of c.7664A>C variant on the protein function, and all of them predicted this variant to be deleterious (Table 1).
4. Discussion
CELSR, planar cell polarity organizer, forms trans-dimers and is involved in the organization of polarized structures between neighboring cells [14,15] and have a coordinating effect in the propagation of a signal from cell to cell [16,17]. Animal models revealed, that these process are necessary for the proper formation of the neural tube and its derivatives[18,19], stereocilia and neurons in the inner ear[20,21], ciliated cells of the fallopian tube[22] and other tubular organs, hair[23], and endothelial valve cells[24]. Nevertheless, the functioning of CELSR family proteins at the molecular level remains largely unclear. Like most adhesion GPCRs, they are orphan receptors with an unknown activation mechanism. Even the assumption of a connection between CELSR and heterotrimeric G-protein is based only on the presence of a 7-TM domain characteristic of GPCRs, and the site that interacts with any G-protein has not yet been identified. Experimental data indicate that adhesion G protein–coupled receptors (ADGRs) can activate signaling events unrelated to G-proteins but based on homophilic trans-interactions necessary for planar cell polarity[15].
Disruptions in CELSR1 in humans are associated with the development of lymphatic malformation 9 (LM9, OMIM: 619319). The expression of Celsr1 in mouse vessels begins during the formation of valves at embryonic day 16 (E16-E16.5) [24], which is approximately equivalent to 23 weeks of gestation in humans [25]. During this process, endothelial cells elongate, undergo reorientation, and coordinated migration into the lumen of the vessel, where they initiate valve leaflet formation. Knockout of Celsr1 in mice leads to disruption of intercellular contacts, endothelial cells are unable to rearrange and adopt a perpendicular orientation for valve formation, leading to aplasia and impeding the ability of the lymphatic system to collect and drain fluid from tissues, ultimately resulting in lymphedema[26]. However, Celsr1 is necessary for development but not for maintaining the structure of lymphatic vessels - knockouts after valve formation do not have a significant impact on vessel structure[24].
CELSR proteins are non-classical cadherins that do not interact with catenins, unlike classical representatives. The structure of CELSRs includes a large ectodomain, with 8 or 9 N-terminal cadherin repeats, up to 8 epidermal growth factor-like domains, two laminin G-type repeats, an EGF-like laminin, one hormone receptor motif (HRM), and a GPCR Autoproteolysis Induction (GAIN) domain. The membrane part consists of a 7-transmembrane domain (7-TM), and the intracellular cytoplasmic tail has 300-600 amino acid residues [15]. The GPS region in the GAIN domain of most ADGRs is responsible for autoproteolysis in the extracellular domain, which occurs in the ER during receptor biosynthesis. Mature ADGRs are usually cleaved and exist as a non-covalently bound complex of N- and C-terminal fragments, but some of them, including CELSR1, do not cleave due to the absence of a consensus catalytic sequence (Leu↓Thr/Ser/Cys) in GPS [15]. Cadherin-like, EGF-like, and laminin-like repeats confer adhesive properties and facilitate intercellular communication, while GAIN and laminin domains can bind unidentified ligands.
Transmembrane domain of CELSR1 is relatively small (~8% of all amino acids), and contains few known mutations, especially those associated with diseases. We discovered rare variant, c.7664A>C, resulted in the substitution of histidine to proline at position 2555 (H2555P) of the intracellular loop in the CELSR1 transmembrane domain in a brother and sister with lower extremity lymphedema caused by aplasia and hypoplasia of lymphatic structures. The oedema in both children appeared in the first year of life, and LD was not diagnosed in of other family members. Despite the low/unknown frequency of the detected polymorphism in the population, which is one of the evidences of pathogenicity [13], identified variant cannot be confidently classified as pathogenic or neutral. The possible pathogenicity of H2555P, in addition to its frequency of occurrence, is evidenced by its presence in two close relatives with primary lymphedema and by the results of in silico analysis predicting a damaging/negative effect of the mutation on protein function. However, no missense mutation in the transmembrane domain of CELSR1 has been previously classified as pathogenic in LM, and no functional analysis of such alterations has been performed. The pathogenic/likely pathogenic findings closest to p.H2555 include p.A2510P/V and p.D2601N, revealed in the patients with congenital heart defects and primary angle closure glaucoma [27,28,29].
To date, less than 10 experimental studies have been published describing the genetic features and clinical-phenotypic characteristics of CELSR1-associated lymphedema (Table 2). It has been shown that the disease belongs to forms with early manifestation (first decade of life), characterized by incomplete penetrance and variable expression, predominantly affecting females.
Both our patients demonstrate hypoplasia and aplasia of lymphatic vessels in lower extremities. Such a structure of the lymphatic system, onset of lymphedema at birth or in infancy without syndromic features and systemic involvement is associated with the number of conditions, including Lymphatic malformation 1 (Milroy’s lymphedema, OMIM 153100, 5q35.3, FLT4), Lymphatic malformation 2 (OMIM 611944, 6q16.2-q22.1), Lymphatic malformation 3 (OMIM 615907, 1q42.13, GJC2), Lymphatic malformation 4 (OMIM 615907, 4q34.3, VEGFC), Lymphatic malformation 5 (OMIM 153200, not mapped), Lymphatic malformation 9 (OMIM 619319, 22q13.31 CELSR1), Lymphatic malformation 10 (OMIM 619369, 8p23.1, ANGPT2).
Aplasia or hypoplasia of initial lymphatic vessels is well-known clinical feature of Milroy’s lymphedema, as well as blood vessel abnormalities, including prominent leg veins and saphenous venous reflux [35,36,37]. In our study, both siblings have been presented by aplasia of lymphatic vessels according to MRL data (Figure 2). Additionally, male patient had prominent vein on left leg, however nail abnormalities or venous insufficiency were not identified in any of the children. Clinical features of another PLD types are much less studied, so it’s not possible to make a more accurate comparison in phenotypes. However, we did not find any pathogenic changes in coding sequences of genes, associated with primary lymphedema, except of CELSR1, in our patients.
5. Conclusions
Advancements in genetic testing technologies have revolutionized the diagnosis of changes in various genes associated with lymphedema development. The identification of molecular genetic features of primary lymphedema subtypes has enabled healthcare professionals to provide patients with information about their disease and how to manage potential complications effectively[3,38,39]. At the same time, the use of next-generation sequencing technologies in biomedical and clinical research has led to the discovery of numerous variants of uncertain significance, making the interpretation of results challenging and reducing the number of successful genetic diagnoses. In the case of CELSR1, analyzing the functional and clinical significance of genetic variations is complicated by the fact that the mechanisms of activation and regulation of the receptor remain poorly understood, and the frequency of detectable mutations is very low. Several pathogenic and likely pathogenic variants of CELSR1 have been previously associated with primary lymphedema progression. In our study, for the first time, a substitution in the 7-TM domain of the CELSR1 was revealed in the familiar case of primary lymphedema. To date variant cannot be confidently classified as pathogenic and requires further investigations but might be considered during molecular genetic examination of DNA samples of patients with primary lymphedema.
Author Contributions
Conceptualization, R.M., Yu.F. and A.R.; methodology, Yu.F. and R.M..; validation, A.R. and R.M.; formal analysis, A.I., E.B., M. Zh.; investigation, Yu.F., A.F., A.I..; resources, A.R.; writing—original draft preparation, Yu.F., A.I.; writing—review and editing, R.M., M.Zh. and A.R; visualization, A.F.; supervision, R.M. and A.R.; funding acquisition, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
The work was carried out at the expense of a subsidy allocated to the Kazan Federal University to fulfill the state task in the field of scientific activity (project № FZSM-2023-0011).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the local ethical committee of the Kazan Federal University (2020-12-28, #27).”
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Acknowledgments
This work was done within the framework of the Strategic Academic Leadership Program (PRIORITY-2030) of Kazan Federal University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rockson, S. G., and K. K. Rivera. Estimating the Population Burden of Lymphedema. Ann N Y Acad Sci 1131 (2008): 147-54. [CrossRef]
- Greene, Arin K. Epidemiology and Morbidity of Lymphedema. In Lymphedema: Presentation, Diagnosis, and Treatment, edited by Arin K. Greene, Sumner A. Slavin and Håkan Brorson, 33-44. Cham: Springer International Publishing, 2015. [CrossRef]
- Brouillard, P., L. Boon, and M. Vikkula. Genetics of Lymphatic Anomalies. J Clin Invest 124, no. 3 (2014): 898-904. [CrossRef]
- Mendola, A., M. J. Schlögel, A. Ghalamkarpour, A. Irrthum, H. L. Nguyen, E. Fastré, A. Bygum, C. van der Vleuten, C. Fagerberg, E. Baselga, I. Quere, J. B. Mulliken, L. M. Boon, P. Brouillard, and M. Vikkula. Mutations in the Vegfr3 Signaling Pathway Explain 36% of Familial Lymphedema. Mol Syndromol 4, no. 6 (2013): 257-66. [CrossRef]
- Bonetti, G., S. Paolacci, M. Samaja, P. E. Maltese, S. Michelini, S. Michelini, S. Michelini, M. Ricci, M. Cestari, A. Dautaj, M. C. Medori, and M. Bertelli. Low Efficacy of Genetic Tests for the Diagnosis of Primary Lymphedema Prompts Novel Insights into the Underlying Molecular Pathways. Int J Mol Sci 23, no. 13 (2022). [CrossRef]
- Li, H., and R. Durbin. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26, no. 5 (2010): 589-95. [CrossRef]
- McKenna, A., M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, A. Kernytsky, K. Garimella, D. Altshuler, S. Gabriel, M. Daly, and M. A. DePristo. The Genome Analysis Toolkit: A Mapreduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res 20, no. 9 (2010): 1297-303. [CrossRef]
- Van der Auwera, G. A., M. O. Carneiro, C. Hartl, R. Poplin, G. Del Angel, A. Levy-Moonshine, T. Jordan, K. Shakir, D. Roazen, J. Thibault, E. Banks, K. V. Garimella, D. Altshuler, S. Gabriel, and M. A. DePristo. From Fastq Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinformatics 43, no. 1110 (2013): 11 10 1-11 10 33. [CrossRef]
- Danecek, P., A. Auton, G. Abecasis, C. A. Albers, E. Banks, M. A. DePristo, R. E. Handsaker, G. Lunter, G. T. Marth, S. T. Sherry, G. McVean, R. Durbin, and Group Genomes Project Analysis. The Variant Call Format and Vcftools. Bioinformatics 27, no. 15 (2011): 2156-8. [CrossRef]
- Wang, K., M. Li, and H. Hakonarson. Annovar: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res 38, no. 16 (2010): e164. [CrossRef]
- Karczewski, K. J., L. C. Francioli, G. Tiao, B. B. Cummings, J. Alfoldi, Q. Wang, R. L. Collins, K. M. Laricchia, A. Ganna, D. P. Birnbaum, L. D. Gauthier, H. Brand, M. Solomonson, N. A. Watts, D. Rhodes, M. Singer-Berk, E. M. England, E. G. Seaby, J. A. Kosmicki, R. K. Walters, K. Tashman, Y. Farjoun, E. Banks, T. Poterba, A. Wang, C. Seed, N. Whiffin, J. X. Chong, K. E. Samocha, E. Pierce-Hoffman, Z. Zappala, A. H. O'Donnell-Luria, E. V. Minikel, B. Weisburd, M. Lek, J. S. Ware, C. Vittal, I. M. Armean, L. Bergelson, K. Cibulskis, K. M. Connolly, M. Covarrubias, S. Donnelly, S. Ferriera, S. Gabriel, J. Gentry, N. Gupta, T. Jeandet, D. Kaplan, C. Llanwarne, R. Munshi, S. Novod, N. Petrillo, D. Roazen, V. Ruano-Rubio, A. Saltzman, M. Schleicher, J. Soto, K. Tibbetts, C. Tolonen, G. Wade, M. E. Talkowski, Consortium Genome Aggregation Database, B. M. Neale, M. J. Daly, and D. G. MacArthur. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 581, no. 7809 (2020): 434-43. [CrossRef]
- Barbitoff, Yury A., Darya N. Khmelkova, Ekaterina A. Pomerantseva, Aleksandr V. Slepchenkov, Nikita A. Zubashenko, Irina V. Mironova, Vladimir S. Kaimonov, Dmitrii E. Polev, Victoria V. Tsay, Andrey S. Glotov, Mikhail V. Aseev, Sergey G. Scherbak, Oleg S. Glotov, Arthur A. Isaev, and Alexander V. Predeus. Expanding the Russian Allele Frequency Reference Via Cross-Laboratory Data Integration: Insights from 7,452 Exome Samples. medRxiv (2022): 2021.11.02.21265801. [CrossRef]
- Richards, S., N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding, H. L. Rehm, and Acmg Laboratory Quality Assurance Committee. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, no. 5 (2015): 405-24. [CrossRef]
- Simons, M., and M. Mlodzik. Planar Cell Polarity Signaling: From Fly Development to Human Disease. Annu Rev Genet 42 (2008): 517-40. [CrossRef]
- Hamann, J., G. Aust, D. Arac, F. B. Engel, C. Formstone, R. Fredriksson, R. A. Hall, B. L. Harty, C. Kirchhoff, B. Knapp, A. Krishnan, I. Liebscher, H. H. Lin, D. C. Martinelli, K. R. Monk, M. C. Peeters, X. Piao, S. Promel, T. Schoneberg, T. W. Schwartz, K. Singer, M. Stacey, Y. A. Ushkaryov, M. Vallon, U. Wolfrum, M. W. Wright, L. Xu, T. Langenhan, and H. B. Schioth. International Union of Basic and Clinical Pharmacology. Xciv. Adhesion G Protein-Coupled Receptors. Pharmacol Rev 67, no. 2 (2015): 338-67. [CrossRef]
- Formstone, C. J. 7tm-Cadherins: Developmental Roles and Future Challenges. Adv Exp Med Biol 706 (2010): 14-36. [CrossRef]
- Chen, W. S., D. Antic, M. Matis, C. Y. Logan, M. Povelones, G. A. Anderson, R. Nusse, and J. D. Axelrod. Asymmetric Homotypic Interactions of the Atypical Cadherin Flamingo Mediate Intercellular Polarity Signaling. Cell 133, no. 6 (2008): 1093-105. [CrossRef]
- Curtin, J. A., E. Quint, V. Tsipouri, R. M. Arkell, B. Cattanach, A. J. Copp, D. J. Henderson, N. Spurr, P. Stanier, E. M. Fisher, P. M. Nolan, K. P. Steel, S. D. Brown, I. C. Gray, and J. N. Murdoch. Mutation of Celsr1 Disrupts Planar Polarity of Inner Ear Hair Cells and Causes Severe Neural Tube Defects in the Mouse. Curr Biol 13, no. 13 (2003): 1129-33. [CrossRef]
- Formstone, C. J., C. Moxon, J. Murdoch, P. Little, and I. Mason. Basal Enrichment within Neuroepithelia Suggests Novel Function(S) for Celsr1 Protein. Mol Cell Neurosci 44, no. 3 (2010): 210-22. [CrossRef]
- Ghimire, S. R., E. M. Ratzan, and M. R. Deans. A Non-Autonomous Function of the Core Pcp Protein Vangl2 Directs Peripheral Axon Turning in the Developing Cochlea. Development 145, no. 12 (2018). [CrossRef]
- Simon, F., F. Tissir, V. Michel, G. Lahlou, M. Deans, and M. Beraneck. Implication of Vestibular Hair Cell Loss of Planar Polarity for the Canal and Otolith-Dependent Vestibulo-Ocular Reflexes in Celsr1(-/-) Mice. Front Neurosci 15 (2021): 750596. [CrossRef]
- Usami, F. M., M. Arata, D. Shi, S. Oka, Y. Higuchi, F. Tissir, M. Takeichi, and T. Fujimori. Intercellular and Intracellular Cilia Orientation Is Coordinated by Celsr1 and Camsap3 in Oviduct Multi-Ciliated Cells. J Cell Sci 134, no. 4 (2021). [CrossRef]
- Devenport, D., and E. Fuchs. Planar Polarization in Embryonic Epidermis Orchestrates Global Asymmetric Morphogenesis of Hair Follicles. Nat Cell Biol 10, no. 11 (2008): 1257-68. [CrossRef]
- Tatin, F., A. Taddei, A. Weston, E. Fuchs, D. Devenport, F. Tissir, and T. Makinen. Planar Cell Polarity Protein Celsr1 Regulates Endothelial Adherens Junctions and Directed Cell Rearrangements During Valve Morphogenesis. Dev Cell 26, no. 1 (2013): 31-44. [CrossRef]
- Krishnan, A., R. Samtani, P. Dhanantwari, E. Lee, S. Yamada, K. Shiota, M. T. Donofrio, L. Leatherbury, and C. W. Lo. A Detailed Comparison of Mouse and Human Cardiac Development. Pediatr Res 76, no. 6 (2014): 500-7. [CrossRef]
- Alitalo, K. The Lymphatic Vasculature in Disease. Nat Med 17, no. 11 (2011): 1371-80. [CrossRef]
- Qiao, X., Y. Liu, P. Li, Z. Chen, H. Li, X. Yang, R. H. Finnell, Z. Yang, T. Zhang, B. Qiao, Y. Zheng, and H. Wang. Genetic Analysis of Rare Coding Mutations of Celsr1-3 in Congenital Heart and Neural Tube Defects in Chinese People. Clin Sci (Lond) 130, no. 24 (2016): 2329-40. [CrossRef]
- Li, R., F. Fu, Q. Yu, D. Wang, X. Jing, Y. Zhang, F. Li, F. Li, J. Han, M. Pan, L. Zhen, D. Li, and C. Liao. Prenatal Exome Sequencing in Fetuses with Congenital Heart Defects. Clin Genet 98, no. 3 (2020): 215-30. [CrossRef]
- Narta, K., M. R. Teltumbade, M. Vishal, S. Sadaf, M. Faruq, H. Jama, N. Waseem, A. Rao, A. Sen, K. Ray, and A. Mukhopadhyay. Whole Exome Sequencing Reveals Novel Candidate Genes in Familial Forms of Glaucomatous Neurodegeneration. Genes (Basel) 14, no. 2 (2023). [CrossRef]
- Gonzalez-Garay, M. L., M. B. Aldrich, J. C. Rasmussen, R. Guilliod, P. E. Lapinski, P. D. King, and E. M. Sevick-Muraca. A Novel Mutation in Celsr1 Is Associated with Hereditary Lymphedema. Vasc Cell 8 (2016): 1. [CrossRef]
- Erickson, R. P., L. W. Lai, D. J. Mustacich, M. J. Bernas, P. H. Kuo, and M. H. Witte. Sex-Limited Penetrance of Lymphedema to Females with Celsr1 Haploinsufficiency: A Second Family. Clin Genet 96, no. 5 (2019): 478-82. [CrossRef]
- Maltese, P. E., S. Michelini, M. Ricci, S. Maitz, A. Fiorentino, R. Serrani, A. Lazzerotti, A. Bruson, S. Paolacci, S. Benedetti, and M. Bertelli. Increasing Evidence of Hereditary Lymphedema Caused by Celsr1 Loss-of-Function Variants. Am J Med Genet A 179, no. 9 (2019): 1718-24. [CrossRef]
- Seo, S. H., S. Lee, J. K. Park, E. J. Yang, B. Kim, J. S. Lee, M. J. Kim, S. S. Park, M. W. Seong, S. Y. Nam, C. Y. Heo, and Y. Myung. Clinical Staging and Genetic Profiling of Korean Patients with Primary Lymphedema Using Targeted Gene Sequencing. Sci Rep 12, no. 1 (2022): 13591. [CrossRef]
- Sudduth, Christopher L., Patrick J. Smits, Yu Sheng Cheng, Klaus Schmitz-Abe, Pankaj Agrawal, and Arin K. Greene. Primary Upper Extremity Lymphedema Caused by a Celsr1 Variant. Journal of Vascular Anomalies 3, no. 2 (2022): e041. [CrossRef]
- Brice, G, A H Child, A Evans, R Bell, S Mansour, K Burnand, M Sarfarazi, S Jeffery, and P Mortimer. Milroy Disease and the <Em>Vegfr-3</Em> Mutation Phenotype. Journal of Medical Genetics 42, no. 2 (2005): 98-102. [CrossRef]
- Connell, F. C., P. Ostergaard, C. Carver, G. Brice, N. Williams, S. Mansour, P. S. Mortimer, S. Jeffery, and Consortium Lymphoedema. Analysis of the Coding Regions of Vegfr3 and Vegfc in Milroy Disease and Other Primary Lymphoedemas. Hum Genet 124, no. 6 (2009): 625-31. [CrossRef]
- Mellor, R. H., C. E. Hubert, A. W. Stanton, N. Tate, V. Akhras, A. Smith, K. G. Burnand, S. Jeffery, T. Makinen, J. R. Levick, and P. S. Mortimer. Lymphatic Dysfunction, Not Aplasia, Underlies Milroy Disease. Microcirculation 17, no. 4 (2010): 281-96. [CrossRef]
- Klinner, J., M. Kruger, T. Brunet, C. Makowski, K. M. Riedhammer, A. Mollweide, M. Wagner, and J. Hoefele. Congenital Lymphedema as a Rare and First Symptom of Tuberous Sclerosis Complex. Gene 753 (2020): 144815. [CrossRef]
- Gordon, K., P. S. Mortimer, M. van Zanten, S. Jeffery, P. Ostergaard, and S. Mansour. The St George's Classification Algorithm of Primary Lymphatic Anomalies. Lymphat Res Biol 19, no. 1 (2021): 25-30. [CrossRef]
Figure 1.
Photographs of foot of male patients (A-B), male patient after thrombosis (C) and female patient (D).
Figure 1.
Photographs of foot of male patients (A-B), male patient after thrombosis (C) and female patient (D).
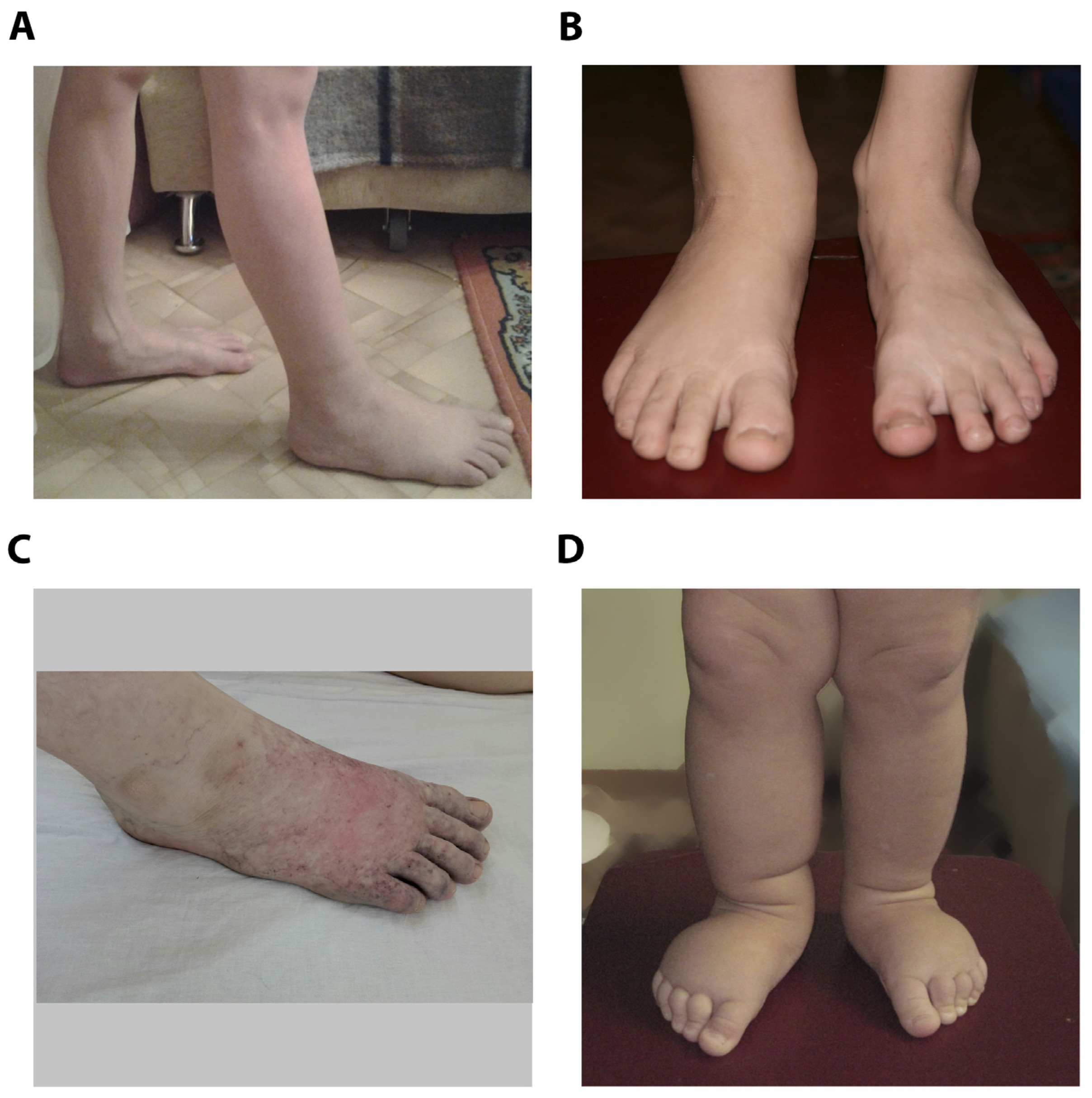
Figure 2.
MRL images of male (A) and female (B) patients indicating reduction of lymphatic vasculature (white arrows), subcutaneous thickness (yellow arrows) and edema (blue arrows).
Figure 2.
MRL images of male (A) and female (B) patients indicating reduction of lymphatic vasculature (white arrows), subcutaneous thickness (yellow arrows) and edema (blue arrows).
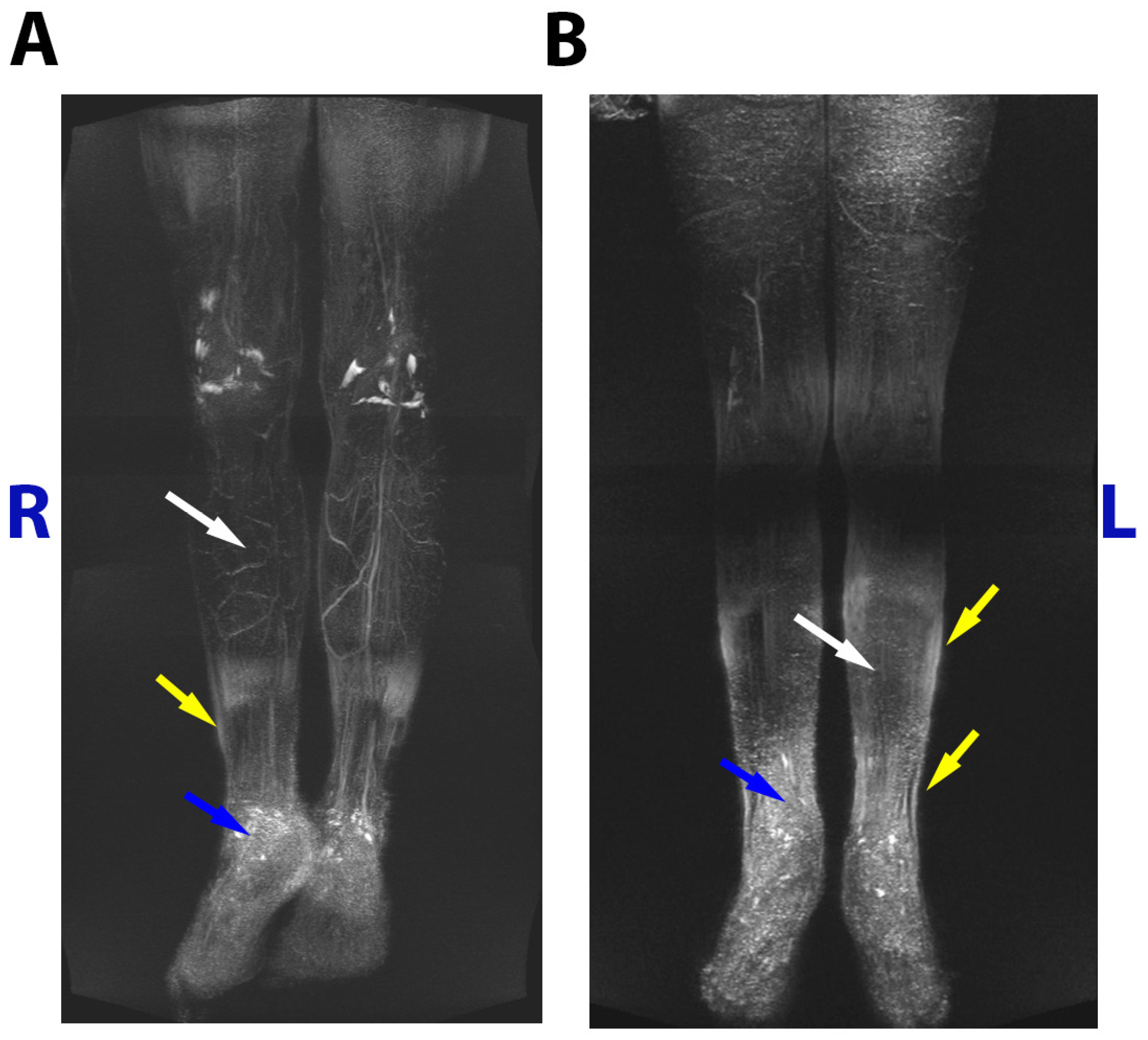
Figure 3.
The family tree showing the c.7664A>C carriers without swellings (gray), LD affected (black), and unaffected members with wild type CELSR1 (white).
Figure 3.
The family tree showing the c.7664A>C carriers without swellings (gray), LD affected (black), and unaffected members with wild type CELSR1 (white).
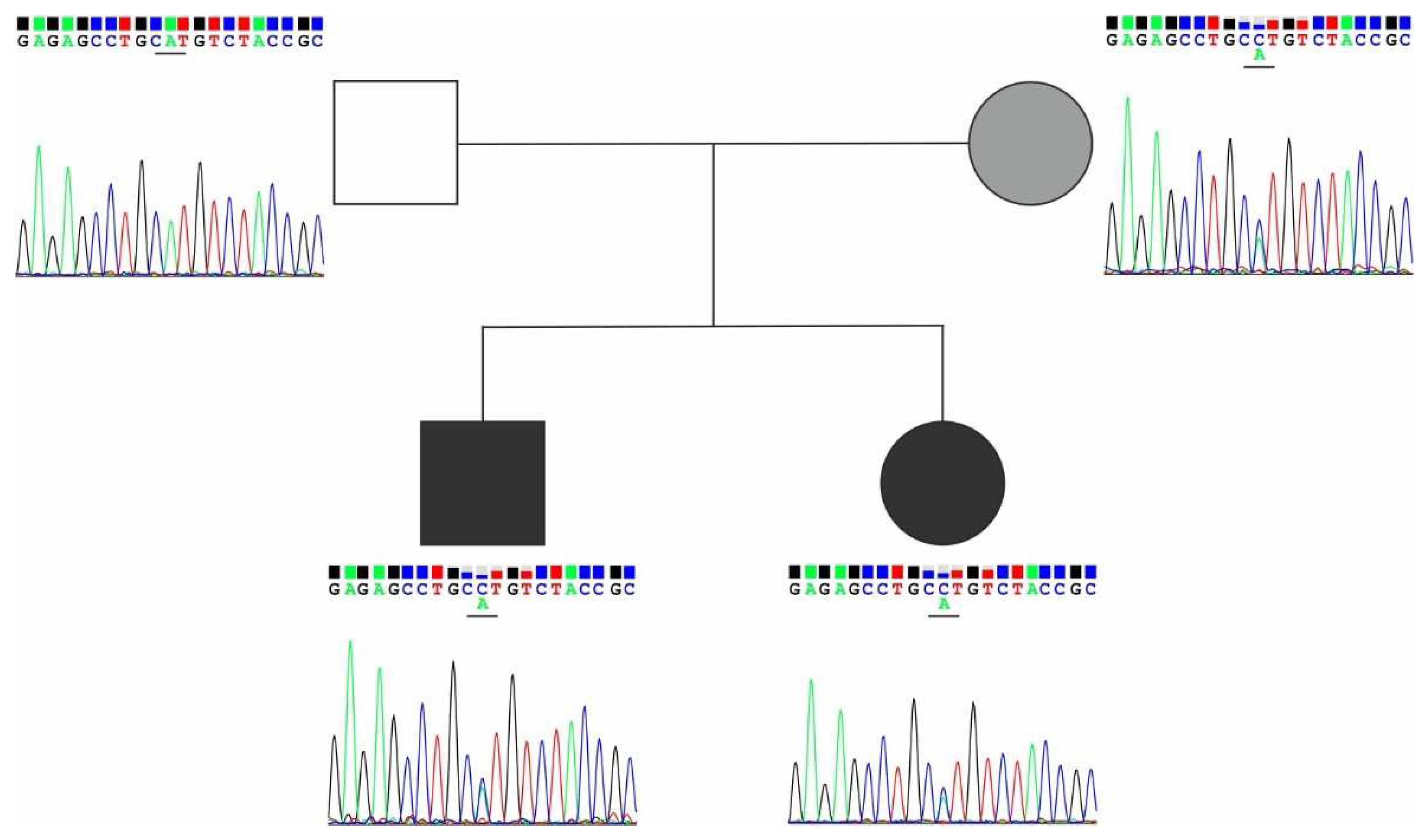

Table 1.
Characterization of the c.7664A>C (p.H2555P) variant by in silico prediction tools.
| Variant | Title 2 |
|---|---|
| Exon | 25 |
| Protein domain | Transmembrane |
| SIFT prediction | Deleterious (D) |
| Polyphen2 prediction | Probably damaging (D) |
| Mutation taster prediction | Disease causing (D) |
| 100 vertebrates conservation by PhastCons | 1.0 |
Table 2.
Experimental studies describing the genetic features and clinical-phenotypic characteristics of CELSR1-associated lymphedema.
Table 2.
Experimental studies describing the genetic features and clinical-phenotypic characteristics of CELSR1-associated lymphedema.
| DNA change | Protein change | Main features | Reference |
|---|---|---|---|
| c.5871G>A | p.Trp1957* | Inactivating variant of CELSR1 was identified in 5 family members with confirmed lower limb lymphedema and was absent in 6 healthy individuals. Proband had leg swelling appeared at ten years, a tortuous structure of vessels, extensive retrograde flow of lymph, and a unique "sheet flow" in both legs. |
[30] |
| c.5121dupC | p.Ile1708fs*44 | Inactivating mutation of CELSR1 in two patients with lymphatic disorders and their asymptomatic father. Lymphoscintigraphy of the lower limbs showed lymphatic abnormalities correlated with the disease burden. Sanger sequencing revealed carriers of this variant to be 5 women and 4 men, but clinical manifestations were only found in women. |
[31] |
| c.5526+2T>A | NA | 5 individuals with inactivating CELSR1 variants were identified in 95 probands with primary lymphedema. Four women with inactivating mutations had disease onset in childhood and adolescence, characterized by unilateral or bilateral lower limb edema. A man with the c.5702-1G>C mutation developed the disease at the age of 77, his mother and daughter had lymphedema. In probands’ families lymphedema was present in 5 out of 6 women with mutations (83%) and in 1 out of 4 men (25%). |
[32] |
| c.6739+1G>A | NA | ||
| c.868G>T | p.Glu290* | ||
| c.2042del | p.Asn681Metfs*16 | ||
| c.5702-1G>C | NA | ||
| c.8446C>T | p.Gln2816* | Among 60 patients with primary lymphedema, CELSR1 was found to be the most frequently mutated gene in 27 patients; c.8446C>T (p.Q2816X) was detected in a mother and daughter with lymphedema in the upper and lower limbs and classified as “likely pathogenic”; c.8871_8872del (p.C2957X) was found in the daughter, and may account for a more severe course of the disease |
[33] |
| c.8871_8872del | p.Cys2957* | ||
| c.4326_4332del | p.Thr1443Glyfs*14 | Non-syndromic lymphedema in a young male: right arm oedema appeared at 9 months and right leg oedema developed at 19 years. Patient and his mother with lower limb lymphedema, had a deletion in the CELSR1 gene resulting in a reading frame shift and premature stop codon formation. | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



