
Submitted:
02 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
In recent decades, considerable attention has been focused on the design and development of surfaces with defined or tunable properties for a wide range of applications and fields. To this end, self-assembled monolayers (SAMs) of organic compounds offer a unique and straightforward route of modifying and engineering the surface properties of any substrate. Thus, alkane-based self-assembled monolayers constitute one of the most extensively studied organic thin-film nanomaterials, which have found wide application in antifouling surfaces, control of wettability or cell adhesion, sensors, optical devices, corrosion protection, organic electronics, among many other applications. Some of which have led technological transfer to industry. Nevertheless, recently, aromatic-based SAMs have gained in importance as functional components, particularly in molecular electronics, bioelectronics, sensors, etc., due to their intrinsic electrical conductivity and optical properties, opening up new perspectives in these fields. However, some key issues affecting device performances still need to be lifted off to ensure their full use and access to novel functionalities such as memories, sensors, or active layers in optoelectronic devices. In this context, we will present herein recent advances of p-conjugated systems-based self-assembled monolayers (e.g., push-pull chromophores) as active layer and their applications.
Keywords:
self-assembled monolayers
; push-pull chromophores
; self-assembly
; active layers
; optoelectronics
1. Introduction
Self-assembly is a ubiquitous phenomenon in nature, in which building units composed of atoms, (bio)molecules, polymers, colloids or particles are capable of self-organizing into ordered and/or functional patterns or superstructures [1,2]. Self-assembling proceeds randomly or directionally and is governed by local interactions (repulsive or attractive forces) between the monomer units themselves, with or without external direction [3]. Nanoscale self-assembling occurs at interfacial or solution and constitutes the easiest bottom-up approaches [4].
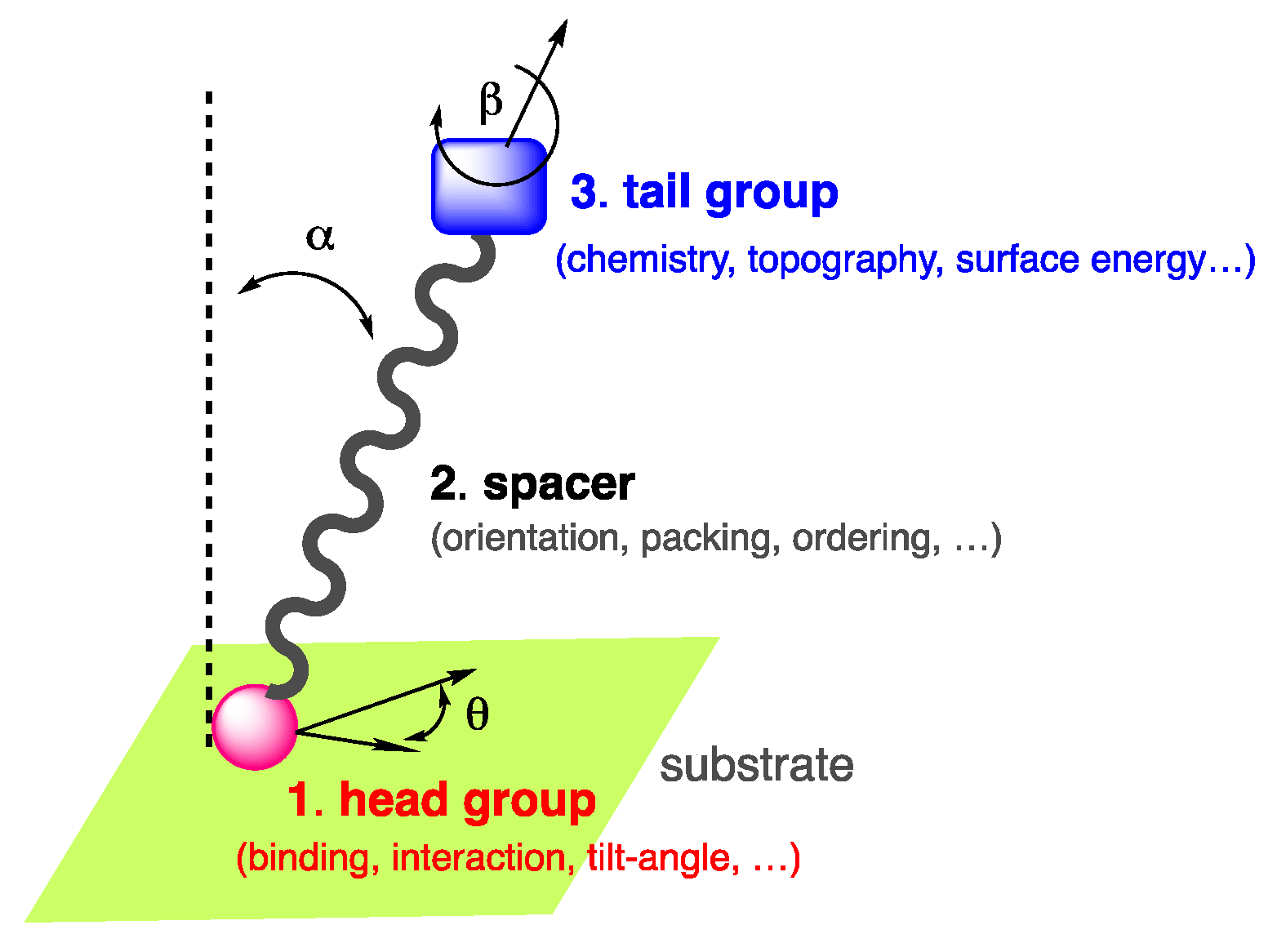
Among these self-assemblies, self-assembled monolayers, classically referred to as SAMs [5,6], constitute an interesting approach for surface functionalization, fine-tune the properties of a surface of interest and are suitable for industry. The concept of SAMs was first introduced at the liquid-gas interfaces [7,8] prior to be reported in the late 40’s by Bigelow et al. [9,10] then successively by Sagiv et al. [11] and Nuzzo et al. [12] on substrates and suggested later for applications by Whitesides et al. [13]. Due to the wide range of applications of SAMs, the pioneers of the field were awarded in 2022 by the Kavli prize in nanoscience [14]. SAMs are long-range ordered two dimensional single-molecular layers of well-oriented chemisorbed or physisorbed organic compounds, which assemble spontaneously on various surfaces, in the gas phase or in solution. The compounds used for the fabrication of SAMs are generally amphiphilic molecules composed of three parts [15]: (i) the anchoring or head group which interacts and binds to the surface, (ii) a spacer typically a molecular backbone made of aliphatic chain or aromatic oligomer imparting the molecular packing and order, (iii) the end or tail group which is responsible of the surface energy, chemistry, and topography of the outer interface [16,17] (Figure 1).
Figure 1.
Amphiphilic self-assembling molecule showing the end group, spacer, and tail group in interaction with a substrate.
Figure 1.
Amphiphilic self-assembling molecule showing the end group, spacer, and tail group in interaction with a substrate.
Based on the molecular deposition techniques used, uniform homomolecular or heteromolecular SAMs can be obtained at the liquid/liquid, liquid/solid, air/liquid or air/solid interfaces with the existence of two monolayer types namely the Gibbs and Langmuir monolayers [18,19]. However, the development and performance of such SAMs can be hampered by some hurdles. Indeed, the quality and properties of the SAMs depend on several parameters among them the thickness of the monolayer, the molecular orientation and ordering, the uniformity and coverage, the chemical composition, odd-even effects of the linker [20], electrostatic effects [21] the thermal and chemical stabilities [22]. In addition, several factors affect the SAMs formation [23] i.e., temperature [24,25], immersion time [26], solvent [27,28], concentration, humidity and O2 contains [29,30] and need to be mastered and controlled to attain the desired properties.
SAMs based on alkanethiols [14,31], alkanesilanes [32], alkanecarboxylates [33], alkanephosphonates [34] etc. on metallic or metal oxides [35] substrates have been extensively studied, both experimentally and theoretically along with the development of novel efficient headgroups such as N-heterocyclic carbenes (NHCs) [36,37] or multidentate adsorbates [38]. Indeed, SAMs are a versatile and inexpensive surface coatings that have been used either in a static or dynamic way (by using various stimulus) for a variety of applications including micro- and nanofabrication [39,40], sensors [41], batteries [42], biological [43], energy [44], electronics [45] among many other applications.
Recently an interest to SAMs made from π-conjugated systems have gained a wide attention due to their intrinsic optical and electrical properties, which can be used in molecular electronic devices, molecular (bio)sensors, bioelectronics, photovoltaics and so on [46]. For instance, in molecular electronics, these SAMs are commonly used to fine tune the work functions of metallic or inorganic electrodes to minimizing the energy barriers for holes, electrons injection or extraction from an active organic layer [47]. Such highly ordered π-conjugated chromophores are also often encountered in nature and act either as photon absorber, electron donor and acceptors (chlorophyll, pheophytins, quinines etc.), or as eye photoreceptors in human retina [48] resulting in efficient photoinduced charge separation and electron transfer. Thereby, self-assembling of π-conjugated chromophores constitutes a key-point in organic nanodevices to improve their properties and operation [49]. Elaboration of SAMs made from organic π-conjugated molecules will follow the same recipes as described for alkane-based self-assembled monolayers considering additional parameters that could influence the SAMs like the dipole effect [50,51], the alkyl spacer influence between the aromatic backbone and surface [52], the number of aromatic rings, and so on.
Π-conjugated systems are versatile organic materials for optoelectronics whose optical and electrical properties are strongly impacted when self-assembled as thin films [53] and need to be well controlled and oriented to optimize the device performances in which they are implemented [54,55]. Among them, push-pull chromophores (referred classically as donor-π−bridge-acceptor or D-π-A) are a class of peculiar importance [56] with various shapes linear, branched, twisted, planar, nonplanar etc. [57,58], exhibiting linear and/or nonlinear optical properties, electrical properties that are useful for instance in the design, conception, and manufacture of electronic devices. However, in most cases these push-pull chromophores are embedded and used in their final form in polymeric or composite matrixes (as doped or grafted, poled or not) [59] and rarely as SAMs. Thus, we will report herein some recent results and applications dealing with push-pull chromophores-based SAMs and their applications.
2. SAMs from push-pull chromophores for dye-sensitized solar cell applications
Push-pull chromophores have been extensively studied during last years with the aim to incorporate them within the conception of organic solar cells [60], mainly as donor materials. Among various solar cells technology, their use in dye-sensitized solar cells (DSSC) revealed interesting since such donor–π–acceptor (D–π–A) sensitizers exhibit strong molar absorption making it possible to use thin oxide films compatible with the improvement of the open circuit voltage [61,62,63,64,65]. Operating principle of DSSC incorporating push-pull assemblies at inorganic oxide surface is depicted in Figure 2 extracted from the recent article of D’Annibale et al. [66].
For DSSC good operation, it is mandatory that dyes should be grafted onto the inorganic mesoporous semiconductor oxide film in a controlled organized stacks enabling to determine energy alignments and high charge-transfer kinetics to improve light harvesting [67]. Such grafting as SAMs can be carried out using push-pull with adequate anchoring group [68] that could be profitably optimized [69]. As for incorporating such assemblies within DSSC one can cite for example the work of Gholamrezaie et al. [70,71] using π-conjugated quinquethiophene derivatives chromophores. Therefore, it is necessary to develop energy-level engineering of chromophores on the metal oxide surface, which can be achieved by the right design of push-pull chromophore through the nature and length of the π -bridge groups, together with using various acceptor and donor groups with different electron affinities. Among the studies on various acceptors one can cite the work of Keremane and co-workers [72] (Figure 3), Paul and Sarkar on PCM-based acceptor [73], and Mustafa et al. [74] dealing on a theoretical study of the influence of the acceptor nature.
Donor nature has also been extensively studied for instance with auxiliary methoxy as a donor to improve metal-free organic dyes performance for DSSC [75]. Some works have been devoted to the effect of both donor and π-spacer [76,77,78], or solely on the latter for instance with unconventional helical push-pull enabling internal charge transfer leading to good injection into the conduction band of TiO2 [79], or π-bridge extension to decrease the gap and widen the light absorption range [80]. Such spectral absorption broadening has also been obtained by the co-adsorption of two push-pull dyes [81]. Moreover, donor-π-bridge-acceptor (D-π-A) structure represents a convenient configuration for high charge separation rate on the organic sensitizer. In D-π-A molecule, intramolecular charge transfer (ICT) occurs efficiently between the donor and the acceptor parts, and the intramolecular electronic relaxation has been shown to play a role in the injection process [82]. Improvement of the intramolecular charge transfer rate is indeed a key issue to improve DSSC efficiency [83,84]. To fulfill requirement of a broader spectral response, it is also possible to add an internal electron-withdrawing unit within some new donor-acceptor-π-acceptor (D-A-π-A) dyes by further grafting benzothiadiazole, benzotriazole, diketopyrrolopyrrole, or quinoxaline to the usual D-π-A structure. For instance, Demirak et al. [85] worked on a novel unsymmetrical push-pull sensitizers based on triarylamine-substituted quinoxaline push-pull dyes with the aim to improve the performance of DSSC. Furthermore, such a strategy can also be completed by proper choice of side chains. Indeed, unsymmetrical push-pull porphyrazine have also been reported by Fernandez-Ariza et al. [86]. With the aim to improve absorption features of porphyrins and phthalocyanines, the authors proposed a molecule with a phthalocyanine core incorporating a specific design of the peripheral substituents (Figure 4), thus enabling increased absorption in the red, improved solubility and the possibility to tune the electronic parameters. With such a seminal molecule they obtained a PCE of 3.42% and opened the way to the improvement of panchromatic light harvesting by tuning properly the peripheral functions.
Particularly, in a more recent work they studied the effect on tuning of the electron-donating unit of push-pull porphyrazines [87] (Figure 5) showing its effect on both adsorption and electron injection processes thus highlighting the design of porphyrazine is a delicate process with strong effect on the electronic properties of the organic dyes and therefore on the DSSC functioning.
Playing on the structure of the push-pull has been studied on other several chromophores such as distyryl boron dipyrromethenes as near-infrared sensitizers [88] exhibiting a wide absorption range from UV-visible to near infrared. Interestingly the authors also demonstrated that the size of the TiO2 nanoparticles is also a parameter to tune according to the molecular size of the dye to improve the DSSC performance. As seen with porphyrazine example, macrocycle-based push-pull chromophores such as phthalocyanines [89,90,91] (Figure 6) and porphyrins [92,93,94] are good candidates for being used as organic dyes within DSSC and have been extensively studied during the last decades.
Concerning porphyrins, push-pull type porphyrin-based dyes have shown the best results [95,96,97,98,99,100,101,102] and notably with such compounds Grätzel et al. managed to obtain an efficiency of about 13% [103,104]. Panagiotakis et al. [105] have reported increased efficiency with PCE ranging from 5 to 7.6% using carefully designed zinc porphyrin push-pull derivatives grafted via cyanoacrylic acid on TiO2. Particularly they showed using a π-conjugated spacer between the chromophore and the anchoring group enhanced electron transfer and hindered undesirable aggregation on TiO2 surface. Cheema et al. [106] have obtained near-infrared absorption by conjugating the porphyrin to indolizine as planar strong electron donor thus inducing π−π interactions such as head-to-tail dye aggregation (Figure 7).
As for phthalocyanines, they exhibit very good photo- and electrochemical stability and high light harvesting capability in the red/NIR spectral regions [89]. Interestingly, the optoelectronic properties of phthalocyanines can be tuned through the proper choice of the organic substituents around the core, since it has a direct impact on the HOMO-LUMO energy levels as well as on the electron density, but also by the metalation nature of the core to reach long-living excited states. As it is the case for porphyrin-based DSSC, it is necessary to avoid phthalocyanine aggregation at the oxide surface. Because phthalocyanines easily exhibit aggregation at titania surface, Milan et al. studied unsymmetrically substituted push-pull Zn phthalocyanines on original SnO2-based DSSC [107].
3. SAMs of push-pull chromophores to improve perovskite solar cell performances
Beside DSSC which represents the main solar cell type incorporating push-pull self-assembly, recent works have also been reported with beneficial use of push-pull chromophores within perovskite solar cells. Indeed, in addition to energy band alignment and improvement of electron transfer at the interface such solar cells undergo a drastic lack of stability and push-pull chromophores have been shown to be able to address all these critical issues. Liu and co-workers have designed an acceptor-donor-acceptor chromophore as an interfacial organic layer whose push-pull effect promotes the charge transfer between organic and inorganic layers in 2D perovskite solar cells by lowering the bandgap of the organic spacer of the perovskite [108]. The push-pull is made of dithienyl diketopyrrolopyrrole (DPP-2T) with two ammonium cations attached onto both sides of the DPP unit, thus allowing hydrogen bonds with inorganic [PbI6]4- sheet within the perovskite cell. Incorporating such DPP-2T push-pull enables to improve the current (Figure 8) and the performance of the cell with a PCE as high as 18.6%.
Still within the use of push-pull as interfacial layer, the push-pull nature has also been exploited with layer deposition on top of the metal oxide to improve electron injection from the photoactive absorber to the metal oxide resulting in the enhancement of the device photocurrent. For example, Gkini and coworkers [109] have successfully used bodypi-porphyrin dyad with this aim, but with a spin-coated layer. Regarding the key issue of passivation and stability improvement of perovskite cells, some studies have also shown the successful use of push-pull SAM. For instance, bi-phenyl-based SAMs have been successfully used recently as an interfacial layer between ZnO electron transport layer and CH3NH3PbI3 hybrid perovskite to improve their stability [110] as shown in Figure 9.
Alagumali and co-workers [111] highlighted the role of push-pull in passivation of the defects within perovskite active materials. Indeed, they used push−pull D−π−A organic small molecule to passivate the undercoordinated Pb2+ defects, and to both align the bands and increase hydrophobicity which results in the improvement of the solar cell stability by hindering moisture effect (Figure 10).
Still with push-pull compounds attached to the perovskite material, Liu et al. [112] addressed the passivation issue. With this aim, they used 3D polydendate complexing agents to achieve defect passivation and crystal growth modulation. The 3D complexing agents are phytic acid (PA) and phytate dipotassium (PAD), and the core of PA material is a six-membered carbon ring surrounded by six phosphate groups which have been shown to perform 3D structural stability for PA materials. The six branches of the PA material undergo multiple chemical complexation which results in 3D skeleton templates enabling passivate defects and regulate perovskite crystallization. Another example of passivation is presented in the article by Zhang et al. [113] where they used polyaromatic molecules based on naphthalene-1,8-dicarboximide (NMI) (Figure 11) and perylene-3,4-dicarboximide (PMI) with different molecular dipoles.
It is shown that such push-pull chromophores provide passivation of defects and, notably, NMI enables energy band alignment. Particularly, NMI passivation leads to the reduction of grain boundaries (Figure 12) and the defect density about three times which allows to reduce non radiative recombination rate and to increase the carrier lifetimes, thus resulting in an increase of nearly 24% of the perovskite solar cell efficiency (PCE).
Moreover, NMI-modified perovskite cells exhibit noticeably higher stability upon exposure to N2 as well as oxygen and humidity (Figure 13).
4. SAMs of push-pull chromophores as dielectric materials.
Self-assembled monolayer of push-pull is widely used in the design of self-assembled multilayer nanodielectric (SAND). The major applications of these SAND are in capacitors and in gate insulator of OFET. In the past several years, Fachetti’s group [114] was an early leader in investigation into special type of self-assembled nanodielectrics (SAND) grown by depositing an alternating σ (Alk) and π (STB) molecular layers and having an octochlorotrisiloxane derived capping layer to stabilize/planarize the assembly and to regenerate a reactive hydroxyl surface for subsequent monolayer deposition. (Figure 14).
From capacitance measurements at 102 Hz, they found that these depend on the constituent molecules. The higher value was obtained for Type II (710 nF.cm−2), for Type I and III it’s 400 and 390 nF.cm−2 respectively. These results show the importance of the highly polarizable dipolar layer in improving dielectric constant k, and hence increasing capacitance which gives SANDs excellent insulating properties. These structures were integrated as gate dielectrics in both p-type and n-type OFET’s [115]. The carrier mobilities are comparable to OFETs obtained with SiO2 dielectrics but at lower operating voltage allowing to reduce the power consumption of device.
Several groups have shown that the turn on characteristics of Organic Thin Film Transistor (OTFT) are related to the dipole moment of the molecule used in the SAM. The permanent dipole feature of push pull chromophore well oriented into the SAM on the surface generates the formation of an electrostatic potential which modulates the densities of carrier charge in the semiconductor channel. [116,117,118] . In 2012, Salinas et al described and correlated the dipole moment of SAM molecules with the threshold voltage of OTFT [119]. They investigated a set of functionalized n-alkane phosphonic acid molecules with various dipole moments that was deposited on a thin aluminum oxide (Al2O3) layer to form a hybrid gate dielectric. They show the Vth is shifted from negative to positive values with increasing dipole moment of the SAM molecule. Although he has demonstrated that the polarization of SAM can play an important role on the charge injection into the channel and can therefore impact Vth, other parameters like charge trapping or impurities can also affect it. An optimum interface between dielectrics and semiconductor is fundamental for efficient device function.
A major part of SAND studied in the literature was composed of an alternating monolayer of polarized molecules such as phosphonic acid derivatives of stilbazolium salts and high-k dielectric metal oxide (ZnO2; HfOx), deposited on metal or semiconductor substrate. These organic-inorganic assemblies provide high gate capacitances, lower gate leakage currents than inorganic film, limited trapped charges, chemical and thermal stability.
For SAND fabrication, the step requiring annealing and such thermal annealing limits SAND compatibility with many plastic or biocompatible substrates and restricts applications such as in biointegrated electronics. This is associated with the metal oxide layer densification. In this contribution, the growth, nanostructural, and dielectric properties as well as implementation in TFTs of zirconium oxide-based SANDs self-assembled using UV radiation processing to make ZrOx thin-film was done by Huang et al [120]. The very high UV photon decomposes the metal oxide precursor and significantly densifies the film (vide infra) [121].
The assembly of the PAE layers between the two ZrOx is known to enhance the stack orientational stability and durability (Figure 15) [115]. To further enhance the insulating properties, repetition of PAE-ZrOx bilayer (self-assembling of PAE in methanol and UV irradiation of ZrOx) deposition n times achieves the fabrication of (Zr-SAND)n superlattices, where n = 1 to 4 (Figure 15d).
An important question in this fabrication process is whether the stilbazolium unit of the PAE layer is affected by exposure to the strong UV irradiation. The data from optical absorption evolution suggest that the overlying ZrOx layer stabilizes the PEA layer against UV damage, likely because PAE is not only sandwiched between oxide layers but also chemically locked on them by the chemistry of the chromophore head and tail. Furthermore, in this way the PAE layer is also essentially encapsulated and protected from ambient air. The XPS spectra argues that oxide precursor decomposition by UV irradiation is more effective than thermal annealing. The data indicate that upon UV exposure, the ZrOx films become thinner and plausibly denser.
The new UV irradiated Zr-SANDs (UVD-ZrSANDs) were imaged by AFM to quantify surface characteristics. All AFM images are essentially featureless, and such exceptionally smooth surfaces are ideal for the fabrication of back-gated transistor devices where even moderate interface roughness can detrimentally affect TFT properties, such as carrier mobility, and illustrate the precise level of control afforded by the present processing methodology. MIS sandwich structures containing (UVD-ZrSAND)n layers were fabricated. The results indicate that combining UV irradiated ZrOx and PAE at room temperature is an efficient route to replace the conventional thermal processing method, thereby realizing high-performance SAND dielectric layers at room temperature.
The molecular dipolar orientation affects thin-film transistor (TFT) threshold and turn-on voltages for devices based on either p-channel pentacene or n-channel copper perfluorophthalocyanine.
Inverted SAND made from inverted PAE units, namely IPAE (Figure 16) [122] and affects the principal OTFT parameters relevant to circuit design and fabrication. Note that, although the π-conjugated azastilbazolium cores of PAE and IPAE are identical, there are minor differences in the structures such as larger distance between the phenylphosphonic acid portion and the core (1 atom in PAE and 2 atoms in IPAE) and, more evident, two hydroxylethyl fragments in the latter versus one in the former structure. However, the hydroxyethyl group is not the anchoring point of the chromophore to the surface but is simply used to achieve good chemical adhesion to the overlying ZrOx layer. More importantly, it does not drive the self-assembly process as judged from the kinetics of PAE/IPAE absorption, which are governed by the phosphonic acid fragment and are identical for the two systems.
Even if the main structure of the push-pull chromophore is the pyridinium (+) and aromatic amine (-), the two different anchors have different impacts on the electronic density along the molecular backbone. The phosphonic acid is electron-withdrawing and aliphatic alcohol is electron-donating. Thus, the push-pull characteristic is more significant with the case if the phosphoric acid is connected with pyridinium, and aliphatic alcohol is connected with aromatic amine (the case of PAE). And for IPAE, the hypsochromic shift is due to diminished intrinsic electric dipole strength inside the molecule. Specifically, Zr-SAND shifts the threshold and turn-on voltages to more positive values, whereas IZr-SAND shifts them in the opposite direction. Capping these SANDs with −SiMe3 groups enhances the effect, affording a 1.3 V difference in turn-on voltage for IZr-SAND vs. Zr-SAND-gated organic TFTs. Such tunability should facilitate the engineering of more complex circuits. This type of junction metal/SAM/dielectric can also be used to modulate the interface thermal conductance.
Lu et al. [123] demonstrated that, by using PAE or IPAE chromophores and mixtures of the two, as organic linkers between Au and SiO2 thin films, the interface thermal conductance of the molecular junction can be tuned based on the relative density of the PAE and IPAE chromophores. The PAE chromophore has two CH2CH2OH terminal groups compared to one for the IPAE, and these terminal groups control the weak hydrogen bonding between the organic molecule and the Au film. The transition from PAE to IPAE SAMs leads to a 20% decrease in the cross-plane thermal conductance of the junction. Furthermore, the thermal conductance of the mixed PAE−IPAE SAMs (50%:50% molar ratio) is close to a linear combination of the PAE and IPAE SAMs, suggesting that the chromophores act as independent channels for heat conduction.
In the SAND-n samples, on the other hand, we predict that the conductance of the PAE−ZrO2 contact is enhanced compared to the conductance of the Au−PAE−SiO2 contacts, possibly due to a stronger chemical affinity between the phosphonic acid headgroup and ZrO2 compared to SiO2, in addition to the stronger adhesion between the hydroxylate tail group with ZrO2 versus Au. Although the cross-plane thermal conductance of the SAND-n decreases monotonically with an increasing number of PAE−ZrO2 layers, the cross plane thermal conductivity increases with n. Heat buildup at the organic/inorganic interfaces in the SAND-n resulting from the low thermal conductivity of the ZrO2 layers and the interface thermal resistance at the PAE−ZrO2 interface can lead to increased temperatures in these films beyond their suitable operational limits, leading to thermoelastic strain and reconfiguration of the PAE molecules.
A new hafnium oxide-organic self-assembled nanodielectric (Hf-SAND) material consisting of regular, alternating π-electron layers of 4-[[4-[bis(2-hydroxyethyl)amino]phenyl]diazenyl]-1-[4-(diethoxyphosphoryl) benzyl]pyridinium bromide) (PAE) and HfO2 nanolayers is reported [124]. The goal of this research is to develop enhanced performance hybrid superlattice dielectrics using alternative oxides as the SAND oxide component. The motivation for extending to hafnia is based on reports indicating differential affinity of phosphonic acids for various oxides versus ZrO2, along with HfO2 thermodynamic and surface chemical differences that may beneficially affect the dielectric properties at low process temperatures [125].
Figure 17 shows the fabrication scheme for the new Hf-SAND-n films. Multilayer variants can be prepared by repeating the indicated self-assembly steps in an iterative fashion, where the n index indicates the number of π-electron/HfOx bilayers grown on top of the initial HfOx priming layer. The resulting HfOx/π-electron bilayer is then “capped” with a second layer of HfOx by spin-coating and baking. This process regenerates the metal oxide surface for additional layers (if desired) of phosphonic acid PAE SAM to initiate the next nanodielectric repeat unit.
Hf-SAND-n variants ranging from one to four bilayers (Hf-SAND-1∼4) were imaged by AFM to quantify the surface roughness, conformality, and contiguity. These films exhibiting RMS roughness values ranking from 1.3 Å for a single layer to 1.7 Å for the four-layer variant, highlighting a negligible additional roughness compared to the native Si oxide surface [126]. This modest increase in roughness is consistent with the deposition of additional dielectric layers. The exceptionally smooth surfaces are ideal for the fabrication of back-gated transistor devices where even moderate interface roughness can detrimentally affects TFT properties [127]. To analyse the elemental and chemical composition of the completed Hf-SAND multilayers, XPS was employed. It is concluded that the O 1s signal observed after the present 150 °C processing is qualitatively like metal oxide spectra of samples processed at much higher temperatures (300 °C) [128].
In all electronic circuits, it is critical to limit TFT gate dielectric leakage currents for efficient switching and to minimize power consumption during device operation. In the case of the present capacitor structures, Hf-SAND-n dielectric layers’ leakage is several orders of magnitude lower than that of native SiO2 capacitors (1 A/cm2 at ±2 MV/cm) and is comparable to previous reports utilizing either solution phase self-assembly or vacuum deposition dielectric growth techniques, such as ALD [129], which typically afford optimized leakage current densities of ∼10−8 A/cm2 or less. The thicker, multilayer Hf-SAND variants exhibit significantly lower electric field normalized leakages which should allow larger voltage biasing windows, useful for several transistor applications. A single layer of Hf-SAND can achieve a capacitance density greater than 1 μF/cm2 (1.1 μF/cm2 measured) versus 0.75 μF/cm2 for a single layer of Zr-SAND, and 0.71 μF/cm2 for the silane-based SAND type II structure reported in the literature [114]. This represents a capacitance enhancement of nearly 50% over current-generation SAND materials and enables microFarad capacitance densities for the first time from a solid-state SAM-metal oxide hybrid dielectrics which is processable at low temperatures in ambient. In the literature, the one used ZrP as the solid support for the PAE SAM showed 700 nF/cm2 for 1p-SAND and 520 nF/cm2 for 2p-SAND in the accumulation regime (0 to +1.0 V). Figure 18 graphically illustrates Hf-SAND dielectric trends in terms of capacitance density versus the bilayer n number, inverse capacitance versus n, EOT, and overall dielectric permittivity (k) of Hf-SAND versus thickness. Also note that as n increases, the overall dielectric constant (keff) also increases. This implies that the PAE layer, which is the majority component in subsequent layers, has a larger k than that of the inorganic oxide. This supports the conclusion of Yoon et al [114]. that the stilbazolium group has very high permittivity, generally greater than what can normally be achieved with low temperature sol−gel oxides.
Recently, this type of SAND has been deposited on IGZO semiconductor as the underlying channel layer. This device exhibits impressive electron mobilities (µsat= 19.4 cm2 V-1 s-1) and low threshold voltage (Vth= 0.83 V) compared to the similar device without push-pull layer combined with Hf oxide. [130]
5. Various electrical and photonic properties generated by push-pull chromophores assemblies
Ordered assembly of push-pull chromophores at surface is mandatory to settle the expected right functionalization property. Obviously, the nature of the linker between the chromophore and the surface plays a key-role on the organization. Hupfer and coworkers [131] have studied the role of arylic versus alkylic linker on the supramolecular structure and the optoelectronic properties of tripodal push-pull thiazoles. Although its more insulating property, the alkyl linker has been found to give higher conductivity to the assembly presumably because it promotes more degrees of freedom enabling supramolecular rearrangement upon electrical measurement. There are very few studies on push-pull electrical properties within single push-pull molecule thick junction [132]. Some works dealing with single push-pull junction are noticeable, such as the use of mechanically break junction to study the electrical conductance modification activated by an external electric field, such as resonance features with oligo(phenyleneethynylene) wires with donor–acceptor substitution on the central ring [133], or electrical bistability of FeII-bis-terpyridine push-pull complexes activated by an external electric field which triggers a spin-cross-over transition [134] (Figure 19).
In the literature, self-assembled push-pull chromophores is mainly used to form supramolecular ordered layers. For instance, Li et al. [135] have processed layer-by-layer stacking of self-assembled push-pull derivatives to form 2D organic crystals, by layering amphiphilic-like stacking with alternating attractive layers (AL) and repulsive layers (RL) to build supramolecular “push–pull” assemblies within a liquid surface-assisted solution self-assembly strategy (Figure 20). A monolayer (~1.5 nm thick) is made of two outside RLs and a sandwiched AL with such high packing density that, interestingly, the stacks exhibit outstanding photoelectric integrated property, with high mobility, high crystalline state, and superior deep-blue laser characteristics.
Organization-dependent photoelectric properties have also been studied on intermolecular charge transfer push-pull derivatives, such as dithiole-nitrofluorene strong push-pull dyads [136]. On such dyad assembly controlled by symmetrical or asymmetrical dipole-dipole interaction, the authors have reported aggregation induced emission, and particularly red-emitting behavior of the well-formed hierarchical micro- and nanostructures which could find interest in OLEDs. Aggregation induced emission enhancement and strong intermolecular charge transfer have also been used within a solution-processed non-doped orange-red emitting multifunctional organic fluorophore made of two terminal attachments of a push–pull moiety separated by a biphenyl free rotor and its copolymers (Figure 21) [137]. Still controlling the interaction, Kim and co-workers have developed 2D single-crystal down-to two monolayers of (2E,2′E)-3,3′-(2,5-difluoro-1,4-phenylene) bis (2-(5-(4-(trifluoromethyl)phenyl) thiophen-2-yl)acrylonitrile) which exhibit field-effect electron mobility and photoresponsivity of 3.6 × 103 A W−1 under green light-emitting diode irradiation [138]. Such results constitute the first example of green-sensitive 2D organic phototransistors. All these studies point out the role of the organization within self-assembled push-pull multilayers that could be profitably further developed within a self-assembled monolayer strategy.
6. Conclusions
In this paper we have reviewed the beneficial use of organized layers of push-pull chromophores to generate photoelectrical and electrical properties, mainly focusing on self-assembled monolayer strategy to arrange push-pull assemblies. Regarding electrical properties, self-assembled monolayers have mostly been devoted to high k dielectric thin layers for possible application towards nanoelectronic devices such as nanotransistors. Push-pull assemblies have also been extensively studied for improving DSSC and perovskite solar cell operation. Within DSSC push-pull assemblies could behave as efficient sensitizers, whereas for perovskite solar cells they have been shown to be able to address the critical issues of energy band alignment, improvement of electron transfer at the interface, and increase of stability as a passivating interfacial layer. At last, some other photo-electrical properties such as high mobility, high crystalline state, and superior deep-blue laser characteristics, bistability, aggregation induced emission, particularly red-emitting behavior finding interest in OLEDs or green-sensitive 2D organic phototransistors, have also been shown to arise from ordered assemblies of push-pull chromophores. In all these studies, beside the intrinsic push-pull properties, their organization appeared to play a key-point to generate the desired properties. We believe that further developing self-assembled monolayer strategy in all the above-mentioned fields should even improve the beneficial effects of push-pull assemblies.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors read and gave valuable suggestions on the manuscript. J. W. review, writing, V. G. review, writing, L. P. conceptualization, review, writing, J. M. R. conceptualization, review, writing, editing.
Funding
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the ministère de l’Enseignement Supérieur et de la Recherche (MESR). J. W. acknowledges the MESR for his PhD fellowship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kelsall, R.; Hamley, I.W.; Geoghegan, M. Nanoscale science and technology, Wiley, 2005.
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science, 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Pelesko, J.A. Self-assembly: the science of things that put themselves together, Chapman & Hall/CRC, London, 2007.
- Chai, Z.; Childress, A.; Busnaina, A.A. Directed assembly of nanomaterials for making nanoscale devices and structures: mechanism and applications. ACS Nano, 2022, 16, 17641–17686. [Google Scholar] [CrossRef] [PubMed]
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev., 1996, 96, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-M. Supramolecular chemistry. Science 1993, 260, 1762–1763. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, K.B. Films built by depositing successive monomolecular layers on a solid surface. J. Am. Chem. Soc., 1935, 57, 1007–1022. [Google Scholar] [CrossRef]
- . Blodgett, K.B.; Langmuir, I. Built-up films of barium stearate and their optical properties. Phys. Rev., 1937, 51, 0964–0982. [Google Scholar] [CrossRef]
- Bigelow, W.C.; Pickett, D.L.; Zisman, W.A. Oleophobic monolayers: films adsorbed from solution in non-polar liquids. J. Colloid Sci., 1946, 1, 513–538. [Google Scholar] [CrossRef]
- Bigelow, W.C.; Glass, E.; Zisman, W.A. Oleophobic monolayers: temperature effects and energy of adsorption. J. Colloid Sci., 1947, 2, 563–591. [Google Scholar] [CrossRef]
- Sagiv, J. Organized monolayers by adsorption. Formation and structure of oleophobic mixed monolayers on solid surfaces. J. Am. Chem. Soc., 1980, 102, 92–98. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Allara, D.L. Adsorption of bifunctional organic disulfides on gold surfaces, J. Am. Chem. Soc., 1983, 105, 4481–4483. [Google Scholar] [CrossRef]
- Xia, Y.; Whitesides, G.M. Soft litography. Annu. Rev. Mater. Sci., 1998, 28, 153–184. [Google Scholar] [CrossRef]
- Savig, J.; Nuzzo, R.G.; Allara, D.L.; Whitesides, G.M. Self-assembled monolayers (SAMs) on solid substrates: molecular coating to control surface properties, Kavli prize in Nanoscience, 2022, https://www.kavliprize.org/prizes/nanoscience/2022.
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev., 2005, 105, 1103–1170. [Google Scholar] [CrossRef]
- Bain, C.D.; Whitesides, G.M. Formation of monolayers by the coadsorption of thiols on gold: variation in the length of the alkyl chain. J. Am. Chem. Soc., 1989, 111, 7164–7175. [Google Scholar] [CrossRef]
- Claridge, S.A.; Liao, W.-S.; Thomas, J.C.; Zhao, Y.; Cao, H.H.; Cheunkar, S.; Serino, A.C.; Andrews, A.M.; Weiss, P.S. From the bottom up: dimensional control and characterization in molecular monolayers. Chem. Soc. Rev. 2013, 42, 2725–2745. [Google Scholar] [CrossRef]
- Ariga, K. Organized Organic Ultrathin Films: Fundamentals and Applications, John Wiley & Sons, New Jersy, Ariga, Kat., 2012.
- Rogalska, E.; Bilewicz, R.; Brigaud, T.; El Moujahid, C.; Foulard, G.; Portella, C.; Stébé, M.J. Formation and properties of Langmuir and Gibbs monolayers: a comparative study using hydrogenated and partially fluorinated amphiphilic derivatives of mannitol, Chem. Phys. Lipids, 2000, 105, 71–91. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Oyola-Reynoso, S.; Thuo, M. The Porter-Whitesides discrepancy: revisiting odd-even effects in wetting properties of n-alkanethiolate SAMs. Coatings, 2015, 5, 1034–1055. [Google Scholar] [CrossRef]
- Liu, Y.; Katzbach, S.; Asyuda, A.; Das, S.; Terfort, A.; Zharnikov, M. Effect of substitution on the charge transport properties of oligophenylenethiolate self-assembled monolayers. Phys. Chem. Chem. Phys., 2022, 24, 27693–27704. [Google Scholar] [CrossRef]
- Aswal, D.K.; Lenfant, S.; Guerin, D.; Yakhmi, J.V.; Vuillaume, D. Self-assembled monolayers on silicon for molecular electronics, Anal. Chim. Acta, 2006, 568, 84–108. [Google Scholar] [CrossRef]
- Cegiełka, D.M.; Kozieł, K.; Zharnikov, M.; Cyganik, P. Molecular engineering of technologically relevant surfaces- carboxylic acids on naturally oxidized aluminium, Appl. Surf. Sci., 2023, 636, 157798. [Google Scholar] [CrossRef]
- Mamun, A.H.A.; Hahn, J.R. Effect of immersion temperature on self-assembled monolayers of octanethiol on Au(111). Surf. Sci., 2012, 606, 664–669. [Google Scholar] [CrossRef]
- Azzam, W. Effect of immersion time temperature on structure of the self-assembled monolayer of 1,2-diphenyldiselenide on gold surface, Mater. Chem. Phys., 2016, 180, 432–439. [Google Scholar]
- Lee, N.-S.; Kang, H.; Seong, S.; Noh, J. Effect of immersion time on the structure of octanethiol self-assembled monolayers on Au(111) at elevated solution temperature. Bull. Korean Chem. Soc., 2019, 40, 1152–1153. [Google Scholar] [CrossRef]
- Chen, X.; Luais, E.; Darwish, N.; Ciampi, S.; Thordarson, P.; Gooding, J.J. Studies on the effect of solvents on self-assembled monolayers formed from organophosphonic acids on indium tin oxide. Langmuir 2012, 28, 9487–9495. [Google Scholar] [CrossRef]
- Chen, G.-S.; Chang, W.-H.; Chang, C.-C.; Chien, Y.-H.; Fang, J.-S.; Cheng, Y.-L. Structural models and barrier properties of amine-terminated trialkoxysilane monolayers incubated in nonpolar vs. polar protic solvents. Mater. Chem. Phys., 2021, 274, 125113. [Google Scholar] [CrossRef]
- Bunker, B.C.; Carpick, R.W.; Assink, R.A.; Thomas, M.L.; Hankins, M.G.; Voigt, J.A.; Sipola, D.; de Boer, M.P.; Gulley, G.L. The impact of solution agglomeration on the deposition of self-assembled monolayers. Langmuir, 2000, 16, 7742–7751. [Google Scholar] [CrossRef]
- Rozlosnik, N.; Gerstenberg, M.; Larsen, N.B. Effect of solvents and concentration on the formation of a self-assembled monolayer of octodecylsiloxane on silicon(001). Langmuir 2003, 19, 1182–1188. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Corthey, G.; Pensa, E.; Cortés, E.; Fonticelli, M.H.; Ibañez, F.; Benitez, G.E.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiolates on metals: a review article on sulfur-metal chemistry and surface structures. RSC Adv., 2014, 4, 27730–27754. [Google Scholar] [CrossRef]
- Wang, L.; Schubert, U.S.; Hoeppener, S. Surface chemical reactions on self-assembled silane-based monolayers. Chem. Soc. Rev., 2021, 50, 6507–6540. [Google Scholar] [CrossRef]
- Jadhav, S.A. Self-assembled monolayers (SAMs) of carboxylic acids: an overview. Cent. Eur. J. Chem., 2011, 9, 369–378. [Google Scholar] [CrossRef]
- Bauer, T.; Schmaltz, T.; Lenz, T.; Halik, M.; Meyer, B.; Clark, T. Phosphonate- and carboxylate-based self-assembled monolayers for organic devices: a theoretical study of surface binding on aluminium oxide with experimental support. ACS Appl. Mater. Interfaces, 2013, 5, 6073–6080. [Google Scholar] [CrossRef] [PubMed]
- Pujari, S.P.; Scheres, L.; Marcelis, A.T.M.; Zuilhof, H. Covalent surface modification of oxide surfaces. Angew. Chem. Int. Ed, 2014, 53, 2–36. [Google Scholar] [CrossRef]
- Engel, S.; Fritz, E.-C.; Ravoo, B.J. New trends in the functionalization of metallic gold: from organosulfur ligands to N-heterocyclic carbenes. Chem. Soc. Rev., 2017, 46, 2057–2075. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Thimes, R.L.; Camden, J.P.; Jenkins, D.M. Fundamentals and applications of N-heterocyclic carbene functionalized gold surfaces and nanoparticles. Chem. Commun., 2022, 58, 13188–13197. [Google Scholar] [CrossRef] [PubMed]
- Chinwangso, P.; Jamison, A.C.; Lee, T.R. Multidente adsorbates for self-assembled monolayer films. Acc. Chem. Res., 2011, 44, 511–519. [Google Scholar] [CrossRef]
- Wilbur, J.L.; Whitesides, G.M. Self-assembly and self-assembled monolayers in micro- and nanofabrication, in Nanotechnology, edited by Timp, G., New York: Springer-Verlag, 1999, 331-369.
- Lee, S.H.; Rho, W.-Y.; Park, S.J.; Kim, J.; Kwon, O.S.; Jun, B.-H. Multifunctional self-assembled monolayers via microcontact printing and degas-driven flow guided patterning. Sci. Rep., 2018, 8, 16763. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Bennett, R.K.A.; Xu, Y.; Rather, A.M.; Li, S.; Cheung, T.C.; Bhanji, A.; Kreder, M.J.; Daniel, D.; Adera, S.; Aizenberg, J.; Wang, X. Wettability-based ultrasensitive detection of amphiphiles through directed concentration at disordered regions in self-assembled monolayers. Proc. Natl. Acad. Sci. U.S.A, 2022, 119, e2211042119. [Google Scholar] [CrossRef]
- Yi, R.; Mao, Y.; Shen, Y.; Chen, L. Self-assembled monolayers for batteries. J. Am. Chem. Soc., 2021, 143, 12897–12912. [Google Scholar] [CrossRef]
- Carrara, S.; Rouvier, F.; Auditto, S.; Brunel, F.; Jeanneau, C.; Camplo, M.; Sergent, M.; About, I.; Bolla, J.-M.; Raimundo, J.-M. Nanoarchitectonics of electrically activable phosphonium self-assembled monolayers to efficiently kill and tackle bacterial infections on demand. Int. J. Mol. Sci., 2022, 23, 2183. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Roldán-Carmona, C.; Sohail, M.; Nazeeruddin, M.K. Applications of self-assembled monolayers for perovskite solar cells interface engineering to address efficiency and stability. Adv. Energy Mater., 2020, 10, 2002989. [Google Scholar] [CrossRef]
- He, J.; Tao, L.; Xian, W.; Arbaugh, T.; Li, Y. Molecular self-assembled monolayers anomalously enhance thermal conductance across polymer-semiconductor interfaces. Nanoscale, 2022, 14, 17681–17693. [Google Scholar] [CrossRef]
- Casalini, S.; Bortolotti, C.A.; Leonardi, F.; Biscarini, F. Self-assembled monolayers in organic electronics, Chem. Soc. Rev., 2017, 46, 40–71. [Google Scholar] [CrossRef] [PubMed]
- .Singh, M.; Kaur, N.; Comini, E. Role of self-assembled monolayers in electronic devices. J. Mater. Chem. C, 2020, 8, 3938–3955. [Google Scholar] [CrossRef]
- Hussey, K.A.; Hadyniak, S.E.; Johnston Jr, R.J. Patterning and development of photoreceptors in the human retina. Front. Cell Dev. Biol., 2022, 10, 878350. [Google Scholar] [CrossRef]
- Borchert, J.W.; Peng, B.Y.; Letzkus, F.; Burghartz, J.N.; Chan, P.K.L.; Zojer, K.; Ludwigs, S.; Klauk, H. Small contact resistance and high-frequency operation of flexible low-voltage inverted coplanar organic transistors. Nat. Commun., 2019, 10, 1119. [Google Scholar] [CrossRef]
- Sauter, E.; Terfort, A.; Zharnikov, M. Pronounced solvent effect on the composition of binary self-assembled monolayers with embedded dipole moments. J. Phys. Chem. C, 2020, 124, 28596–28604. [Google Scholar] [CrossRef]
- Abu-Husein, T.; Schuster, S.; Egger, D.A.; Kind, M.; Santowski, T.; Wiesner, A.; Chiechi, R.; Zojer, E.; Terfort, A.; Zharnikov, M. The effect of embedded dipoles in aromatic self-assembled monolayers. Adv. Func. Mater., 2015, 25, 3943–3957. [Google Scholar] [CrossRef]
- Azzam, W.; Subaihi, A. Influence of an alkyl spacer on the formation and structure of 4-fluorobenthiol and 4-fluorobenzenemethanethiol self-assembled monolayers on Au (111). Surf. Interfaces, 2020, 20, 100544. [Google Scholar] [CrossRef]
- Khasbaatar, A.; Xu, Z.; Lee, J.-H.; Campillo-Alvarado, G.; Hwang, C.; Onusaitis, B.N.; Diao, Y. From solution to thin film: molecular assembly of p-conjugated systems ans impact on (opto-electronic properties. Chem. Rev., 2023, 123, 8395–8487. [Google Scholar] [CrossRef]
- Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Progress in the enhancement of electro-optic coefficients and orientation stability for organic second-order nonlinear optical materials. Dyes Pigm., 2020, 181, 108509. [Google Scholar] [CrossRef]
- Gadenne, V.; Grenier, B.; Praveen, C.; Marsal, P.; Valmalette, J.C.; Patrone, L.; Raimundo, J.-M. Combined SERS/DFT studies of push-pull chromophores self-assembled monolayers: insights of their surface orientation. Phys. Chem. Chem. Phys., 2019, 21, 25865–25871. [Google Scholar] [CrossRef]
- Burěs, F. Fundamental aspects of property tuning in push-pull molecules. RSC Adv., 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Klikar, M.; Solanke, P.; Tydlitát; Burěs, F. Alphabet-inspired design of (hetero)aromatic push-pull chromophores. Chem. Rec., 2016, 16, 1886–1905. [Google Scholar] [CrossRef]
- Gámez-Valenzuela, S.; Neusser, D.; Benitez-Martin, C.; Najera, F.; Guadix, J.A.; Moreno-Yruela, C.; Villacampa, B.; Ponce Ortiz, R.; Ludwigs, S.; Andreu, R.; Ruiz Delgado, M.C. V-shaped pyranylidene/triphenylamine-based chromophores with enhanced photophysical, electrochemical and nonlinear optical properties. Mater. Adv., 2021, 2, 4255–4263. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Islamova, L.N.; Sharipova, S.M.; Fazleeva, G.M.; Gaysin, A.I.; Shmelev, A.G.; Simanchuk, A.E.; Turgeneva, S.A.; Sharipova, A.V.; Mukhtarov, A.S.; Vakhonina, T.A.; Forminykh, O.D.; Mikerin, S.L.; Balakina, M.Y. Quadratic nonlinear optical response of composite polymer materials based on push-pull quinoxaline chromophores with various groups in the aniline donor moiety. New J. Chem., 2023, 47, 2296–2306. [Google Scholar] [CrossRef]
- Malytskyi, V.; Simon, J.J.; Patrone, L.; Raimundo, J.-M. Thiophene-based push–pull chromophores for small molecule organic solar cells (SMOSCs). RSC Adv., 2015, 5, 354–397. [Google Scholar] [CrossRef]
- Raymakers, J.; Krysova, H.; Artemenko, A.; Cermak, J.; Nicley, S.S.; Verstappen, P.; Gielen, S.; Kromka, A.; Haenen, K.; Kavan, L.; Maes, W.; Rezek, B. Functionalization of boron-doped diamond with a push–pull chromophore via Sonogashira and CuAAC chemistry. RSC Adv., 2018, 8, 33276–33290. [Google Scholar] [CrossRef] [PubMed]
- Unny, D.; Kandregula, G.R.; Sivanadanam, J.; Ramanujam, K. Molecular engineering of pyrene carbazole dyes with a single bond and double bond as the mode of linkage. New J. Chem., 2020, 44, 16511–16525. [Google Scholar] [CrossRef]
- Lindh, L.; Gordivska, O.; Persson, S.; Michaels, H.; Fan, H.; Chábera, P.; Rosemann, N.W.; Kumar Gupta, A.; Benesperi, I.; Uhlig, J.; Prakash, O.; Sheibani, E.; Kjaer, K.S.; Boschloo, G.; Yartsev, A.; Freitag, M.; Lomoth, R.; Persson, P.; Wärnmark, K. Dye-sensitized solar cells based on Fe N-heterocyclic carbene photosensitizers with improved rod-like push-pull functionality. Chem. Sci., 2021, 12, 16035–16053. [Google Scholar] [CrossRef]
- James, R.D.; Algahtani, L.S.; Mallows, J.; Flint, H.V.; Waddell, P.G.; Woodford, O.J.; Gibson, E.A. Pentafluorosulfanyl-functionalised BODIPY push–pull dyes for p-type dye-sensitized solar cells. Sustain. Energy Fuels, 2023, 7, 1494–1501. [Google Scholar] [CrossRef]
- Fauvel, S.; Riquelme, A.J.; Castán, J.-M. A.; Mwalukuku, W.M.; Kervella, Y.; Challuri, V.K.; Sauvage, F.; Narbey, S.; Maldivi, P.; Demadrille, R. Push-pull photochromic dyes for semi-transparent solar cells with light-adjustable optical properties and high color-rendering index. Chem. Sci., 2023, 14, 8497–8506. [Google Scholar] [CrossRef]
- D'Annibale, V.; Chen, C.G.; Bonomo, M.; Dini, D.; D'Abramo, M. P1 push-pull dye as a case study in QM/MM theoretical characterization for dye-sensitized solar cell organic chromophores. ChemistrySelect, 2023, 8, e20220490. [Google Scholar] [CrossRef]
- Ashwell, G.J.; Tyrell, W.D.; Whittam, A.J. Molecular rectification: seld-assembled monoloayers in which donor-(p-bridge)-acceptor moities are centrally located and symmetrically coupled to both gold electrodes. J. Am. Chem. Soc., 2004, 126, 7102–7110. [Google Scholar] [CrossRef] [PubMed]
- . Malytskyi, V.; Gadenne, V.; Ksari, Y.; Patrone, L.; Raimundo, J.-M. Synthesis and characterization of thiophene-based push-pull chromophores for tuning the electrical and optical properties of surfaces with controlled SAM formation. Tetrahedron 2017, 73, 5738–5744. [Google Scholar] [CrossRef]
- Abdellaha, I.M.; El-Shafei, A. Synthesis and characterization of novel tetra anchoring A2-D-D-D-A2 architecture sensitizers for efficient dye-sensitized solar cells. Sol. Energy, 2020, 198, 25–35. [Google Scholar] [CrossRef]
- Gholamrezaie, F.; Kirkus, M.; Mathijissen, S.G.J.; de Leeuw, D.M.; Meskers, S.C.J. Photophysics of self-assembled monolayers of a π-conjugated quinquethiophene derivative. J. Phys. Chem. A, 2012, 116, 7645–7650. [Google Scholar] [CrossRef]
- Vijayaraghavan, R.K.; Gholamrezaie, F.; Meskers, S.C.J. Photovoltaic effect in self-assembled molecular monolayers on gold: influence of orbital energy level alignment on short-circuits current generation. J. Phys. Chem. C, 2013, 117, 16820–16829. [Google Scholar] [CrossRef]
- Keremane, K.S.; Planchat, A.; Pellegrin, Y.; Jacquemin, D.; Odobel, F.; Adhikari, A.V. Push-pull phenoxazine-based sensitizers for p-type DSSCs: effect of acceptor units on photovoltaic performance. Chem. Sus. Chem.; 2022, 15, e202200520. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Sarkar, U. Designing of PC31BM-based acceptors for dye-sensitized solar cell. J. Phys. Org. Chem. 2022, e4419. [Google Scholar] [CrossRef]
- Mustafa, F.M.; Latif, M.K.A.; Khalek, A.A.A. Theoretical investigation on the influence of electron-accepting substitutions in modulating the optoelectronic properties of dye-sensitized solar cells. Mol. Phys. 2023, e2245060. [Google Scholar] [CrossRef]
- Avhad, K.; Jadhav, M.; Patil, D.; Chowdhury, T.H.; Bedja, I.; Sekar, N. Rhodanine-3-acetic acid containing D-π-A push-pull chromophores: Effect of methoxy group on the performance of dye-sensitized solar cells. Org. Electron., 2019, 65, 386–393. [Google Scholar] [CrossRef]
- Belen Marco, A.; Martinez de Baroja, N.; Andres-Castan, J.M.; Franco, S.; Andreu, R.; Villacampa, B.; Orduna, J.; Garin, J. Pyranylidene/thienothiophene-based organic sensitizers for dye-sensitized solar cells. Dyes Pigm., 2019, 161, 205–213. [Google Scholar] [CrossRef]
- Gümüşgöz Çelik, G.; Tunç, G.; Lafzi, F.; Saracoglu, N.; Seçkin Arslan, B.; Nebioğlu, M.; Şişman, I.; Gül Gürek, A. Influence of spacer and donor groups as tetraphenylethylene or triphenylamine in asymmetric zinc phthalocyanine dyes for dye-sensitized solar cells. J. Photochem. Photobiol. A, 2023, 444, 114962. [Google Scholar] [CrossRef]
- Trabelsi, S.; Kouki, N.; Seydou, M.; Maurel, F.; Tangour, B. Intramolecular path determination of active electrons on push-pull oligocarbazole dyes-sensitized solar cells. ChemistryOpen, 2019, 8, 580–588. [Google Scholar] [CrossRef]
- Dovaa, D.; Cauteruccioa, S.; Manfredib, N.; Pragerc, S.; Dreuwc, A.; Arnaboldia, S.; Mussinia, P.R.; Licandroa, E.; Abbotto, A. An unconventional helical push-pull system for solar cells. Dyes Pigm., 2019, 161, 382–388. [Google Scholar] [CrossRef]
- Lee, M.W.; Kim, J.Y.; Lee, H.G.; Gil Cha, H.; Lee, D.H.; Jae Ko, M. Effects of structure and electronic properties of D-π-A organic dyes on photovoltaic performance of dye-sensitized solar cells. J. Energy Chem., 2021, 54, 208–216. [Google Scholar] [CrossRef]
- An, J.; Tian, Z.; Zhang, L.; Yang, X.; Cai, B.; Yu, Z.; Zhang, L.; Hagfeldt, A.; Sun, L. Supramolecular Co-adsorption on TiO2 to enhance the efficiency of dye-sensitized solar cells. J. Mater. Chem. A, 2021, 9, 13697–13703. [Google Scholar] [CrossRef]
- Maffeis, V.; Dogan, H.; Cassette, E.; Jousselme, B.; Gustavsson, T. Role of electronic relaxation in the injection process of organic push–pull dyes in complete dye-sensitized solar cells. J. Phys. Chem. Lett., 2019, 10, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, T.W.; Kim, S.; Oh, I.; Wonneberger, H.; Yoon, K.; Kwak, M.; Kim, J.; Kim, J.; Li, C.; Müllen, K.; Ihee, H. Molecular-level understanding of excited states of N-annulated rylene dye for dye-sensitized solar sells. J. Phys. Chem. C, 2020, 124, 22993–23003. [Google Scholar] [CrossRef]
- Prakash, K.; Alsaleh, A.Z.; Neeraj; Rathi, P.; Sharma, A.; Sankar, M.; D’Souza, F. Synthesis, spectral, electrochemical and photovoltaic studies of A3B porphyrinic dyes having peripheral donors. Chem. Phys. Chem., 2019, 20, 2627–2634. [Google Scholar] [CrossRef]
- Demirak, K.; Can, M.; Özsoy, C.; Yiğit, M.Z.; Gültekin, B. Synthesis and photovoltaic characterization of triarylamine-substituted quinoxaline push-pull push-pull dyes to improve the performance of dye-sensitized solar cells. Turk. J. Chem., 2017, 41, 309–322. [Google Scholar] [CrossRef]
- Fernandez-Ariza, J.; Urbani, M.; Graetzel, M.; Rodriguez-Morgade, M.S.; Nazeeruddin, M.K.; Torres, T. An unsymmetrical, push–pull porphyrazine for dye-sensitized solar cells. ChemPhotoChem., 2017, 1, 164–166. [Google Scholar] [CrossRef]
- Medina, D.-P.; Fernández-Ariza, J.; Urbani, M.; Sauvage, F.; Torres, T.; Rodríguez-Morgade, M.S. Tuning the acceptor unit of push–pull porphyrazines for dye-sensitized solar cells. Molecules, 2021, 26, 2129. [Google Scholar] [CrossRef]
- Shi, W.J.; Kinoshita, T.; Ng, D.K.P. Push–pull distyryl boron dipyrromethenes as near-infrared sensitizers for dye-sensitized solar cells. Asian J. Org. Chem., 2017, 6, 1476–1485. [Google Scholar] [CrossRef]
- Urbani, M.; Ragoussi, M.E.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines for dye-sensitized solar cells. Coord. Chem. Rev., 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Ghazal, B.; Azizi, K.; Ewies, E.F.; Youssef, A.S.A.; Mwalukuku, V.M.; Demadrille, R.; Torres, T.; Makhseed, S. Push–pull zinc phthalocyanine bearing hexa-tertiary substituted carbazolyl donor groups for dye-sensitized solar cells. Molecules, 2020, 25, 1692. [Google Scholar] [CrossRef]
- Chindeka, F.; Mashazi, P.; Britton, J.; Oluwole, D.O.; Mapukata, S.; Nyokong, T. Fabrication of dye-sensitized solar cells based on push-pull asymmetrical substituted zinc and copper phthalocyanines and reduced graphene oxide nanosheets. J. Photochem. Photobiol. A, 2020, 399, 112612. [Google Scholar] [CrossRef]
- Urbani, M.; Gratzel, M.; Nazeeruddin, M.K.; Torres, T. Meso-substituted porphyrins for dye-sensitized solar cells. Chem. Rev., 2014, 114, 12330–12396. [Google Scholar] [CrossRef]
- Birel, O.; Nadeem, S.; Duman, H. Porphyrin-based dye-sensitized solar cells (DSSCs): a review. J. Fluoresc., 2017, 27, 1075–1085. [Google Scholar] [CrossRef]
- Lu, J.; Liu, S.; Wang, M. Push-pull zinc porphyrins as light-harvesters for efficient dye-sensitized solar cells. Front. Chem. 2018, 6, 541. [Google Scholar] [CrossRef]
- Higashino, T.; Imahori, H. Porphyrins as excellent dyes for dye-sensitized solar cells: recent developments and insights. Dalton Trans., 2015, 44, 448–463. [Google Scholar] [CrossRef]
- Song, H.; Li, X.; Ågren, H.; Xie, Y. Branched and linear alkoxy chains-wrapped push-pull porphyrins for developing efficient dye-sensitized solar cells. Dyes Pigm., 2017, 137, 421–429. [Google Scholar] [CrossRef]
- Higashino, T.; Kawamoto, K.; Sugiura, K.; Fujimori, Y.; Tsuji, Y.; Kurotobi, K.; Ito, S.; Imahori, H. Effects of bulky substituents of push–pull porphyrins on photovoltaic properties of dye-sensitized solar cells. ACS Appl. Mater. Interfaces, 2016, 8, 15379–15390. [Google Scholar] [CrossRef]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Gratzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)–based redox electrolyte exceed 12 percent efficiency. Science, 2011, 334, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Iiyama, H.; Nishimura, I.; Imahori, H. Exploration on the combination of push-pull porphyrin dyes and copper(I/II) redox shuttles toward high-performance dye-sensitized solar cells. Chem. Lett., 2020, 49, 936–939. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, J.C.; Li, R.; Ci, C.G. Different metal upper porphyrin based self-assembly sensitizers for application in efficient dye-sensitized solar cells. Polyhedron, 2022, 211, 115573. [Google Scholar] [CrossRef]
- Higashino, T.; Sugiura, K.; Namikawa, K.; Imahori, H. Energy level tuning of push-pull porphyrin sensitizer by trifluoromethyl group for dye-sensitized solar cells. J. Porphyr. Phthalocyanines 2023, 27, 145–156. [Google Scholar] [CrossRef]
- Hu, Y.; Webre, W.A.; Thomas, M.B.; Moss, A.; Hancock, S.N.; Schaffner, J.; D'Souza, F.; Wang, H. β-Functionalized push–pull opp-dibenzoporphyrins as sensitizers for dye-sensitized solar cells: the role of the phenylethynyl bridge. J. Mater. Chem. A, 2019, 7, 10712–10722. [Google Scholar] [CrossRef]
- . Mattew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Gratzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem., 2014, 6, 242–247. [Google Scholar] [CrossRef]
- Yella, A.; Mai, C.-L.; Zakeeruddin, S.M.; Chang, S.-N.; Hsieh, C.-H.; Yeh, C.-Y.; Gratzel, M. Molecular engineering of push–pull porphyrin dyes for highly efficient dye-sensitized solar cells: the role of benzene spacers. Angew. Chem. Int. Ed,. 2014, 53, 2973–2977. [Google Scholar] [CrossRef]
- Panagiotakis, S.; Giannoudis, E.; Charisiadis, A.; Paravatou, R.; Lazaridi, M.-E.; Kandyli, M.; Ladomenou, K.; Angaridis, P.A.; Bertrand, H.; Sharma, G.D.; Coutsolelos, A.G. Increased efficiency of dye-sensitized solar cells by incorporation of a π-spacer in donor–acceptor zinc porphyrins bearing cyanoacrylic acid as an anchoring group. Eur. J. Inorg. Chem. 2018, 20-21, 2369–2379. [Google Scholar] [CrossRef]
- Cheema, H.; Baumann, A.; Loya, E.K.; Brogdon, P.; McNamara, L.E.; Carpenter, C.A.; Hammer, N.I.; Mathew, S.; Risko, C.; Delcamp, J.H. Near-infrared-absorbing indolizine-porphyrin push–pull dye for dye-sensitized solar cells. ACS Appl. Mater. Interfaces, 2019, 11, 16474–16489. [Google Scholar] [CrossRef] [PubMed]
- Milan, R.; Selopal, G.S.; Cavazzini, M.; Orlandi, S.; Boaretto, R.; Caramori, S.; Concina, I.; Pozzi, G. Zinc phthalocyanines as light harvesters for SnO2-based solar cells: a case study. Sci. Rep. 2020, 10, 1176. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, Z.; Sun, J.; Shang, M.; Zheng, K.; Yang, W.; Ge, Z. Donor-acceptor-donor type organic spacer for regulating the quantum wells of Dion-Jacobson 2D perovskites. Nano Energy, 2022, 93, 106800. [Google Scholar] [CrossRef]
- Gkini, K.; Verykios, A.; Balis, N.; Kaltzoglou, A.; Papadakis, M.; Adamis, K.S.; Armadorou, K.-K.; Soultati, A.; Drivas, C.; Gardelis, S.; Petsalakis, I.D.; Palilis, L.C.; Fakharuddin, A.; Haider, M.I.; Bao, X.; Kennou, S.; Argitis, P.; Schmidt-Mende, L.; Coutsolelos, A.G.; Falaras, P.; Vasilopoulou, M. Enhanced Organic and Perovskite Solar Cell Performance through Modification of the Electron-Selective Contact with a Bodipy−Porphyrin Dyad, ACS Appl. Mater. Interfaces, 2020, 12, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Kouki, H.; Pitié, S.; Torkhani, A.; Mamèche, F.; Decorse, P.; Seydou, M.; Kouki, F.; Lang, P. Tailor-Made Amino-Based Self-Assembled Monolayers Grafted on Electron Transport ZnO Layers: Perovskite Solar Cell Performance and Modified Interface Relationship, ACS Appl. Energy Mater., 2022, 5, 1635–1645. [Google Scholar] [CrossRef]
- Alagumalai, A.; Rajendran, M.V.; Ganesan, S.; Menon, V.S.; Raman, R.K.; Chelli, S.M.; Vijayasayee, S.M.; Gurusamy Thangavelu, S.A.; Krishnamoorthy, A. Interface Modification with Holistically Designed Push−Pull D−π−A Organic Small Molecule Facilitates Band Alignment Engineering, Efficient Defect Passivation, and Enhanced Hydrophobicity in Mixed Cation Planar Perovskite Solar Cells. ACS Appl. Energy Mater., 2022, 5, 6783–6796. [Google Scholar] [CrossRef]
- Liu, C.; Su, H.; Pu, Y.; Guo, M.; Zhai, P.; Liu, L.; Fu, H. 3D Polydentate Complexing Agents for Passivating Defects and Modulating Crystallinity for High-Performance Perovskite Solar Cells. Adv. Funct. Mater., 2023, 33, 2212577. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, Y.; Li, Z.; Qiao, L.; Xiong, Q.; Deng, L.; Zhang, Z.; Long, R.; Zhou, Q.; Du, Y.; Lan, Z.; Zhao, Y.; Li, C.; Müllen, K.; Gao, P. Marked Passivation Effect of Naphthalene-1,8-Dicarboximides in High-Performance Perovskite Solar Cells. Adv. Mater., 2021, 33, 2008405. [Google Scholar] [CrossRef]
- Yoon, M.-H.; Facchetti, A.; Marks, T.J. σ-π Molecular Dielectric Multilayers for Low-Voltage Organic Thin-Film Transistors. Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 4678–4682. [Google Scholar] [CrossRef]
- DiBenedetto, S.A.; Frattarelli, D.; Ratner, M.A.; Facchetti, A.; Marks, T.J. Vapor Phase Self-Assembly of Molecular Gate Dielectrics for Thin Film Transistors. J. Am. Chem. Soc., 2008, 130, 7528–7529. [Google Scholar] [CrossRef]
- Li, L.; Lu, N.; Liu, M. Effect of dipole layer on the density-of-states and charge transport in organic thin film transistors. Appl. Phys. Lett., 2013, 103, 253303. [Google Scholar] [CrossRef]
- Ashkenasy, G.; Cahen, D.; Cohen, R.; Shanzer, A.; Vilan, A. Molecular engineering of semiconductor surfaces and devices. Acc Chem Res., 2002, 35, 121–128. [Google Scholar] [CrossRef]
- Cahen, D.; Naaman, R.; Vager, Z. The Cooperative Molecular Field Effect. Adv. Funct. Mater., 2005, 15, 1571–1578. [Google Scholar] [CrossRef]
- .Salinas, M.; Jäger, C.M.; Amin, A.Y.; Dral, P.O.; Meyer-Friedrichsen, T.; Hirsch, A.; Clark, T.; Halik, M. The Relationship between Threshold Voltage and Dipolar Character of Self-Assembled Monolayers in Organic Thin-Film Transistors. J. Am. Chem. Soc., 2012, 134, 12648–12652. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yu, X.; Zeng, L.; Wang, B.; Takai, A.; Di Carlo, G.; Bedzyk, M.J.; Marks, T.J.; Facchetti, A. Ultraviolet Light-Densified Oxide-Organic Self-Assembled Dielectrics: Processing Thin-Film Transistors at Room Temperature. ACS Appl. Mater. Interfaces, 2021, 13, 3445–3453. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Heo, J.-S.; Kim, T.-H.; Park, S.; Yoon, M.-H.; Kim, J.; Oh, M.S.; Yi, G.-R.; Noh, Y.-Y.; Park, S.K. Flexible Metal-Oxide Devices Made by Room-Temperature Photochemical Activation of Sol–Gel Films. Nature, 2012, 489, 128–132. [Google Scholar] [CrossRef]
- Wang, B.; Di Carlo, G.; Turrisi, R.; Zeng, L.; Stallings, K.; Huang, W.; Bedzyk, M.J.; Beverina, L.; Marks, T.J.; Facchetti, A. The Dipole Moment Inversion Effects in Self-Assembled Nanodielectrics for Organic Transistors. Chem. Mater., 2017, 29, 9974–9980. [Google Scholar] [CrossRef]
- Lu, B.; Wang, B.; Chen, Y.; Facchetti, A.; Marks, T.J.; Balogun, O. Cross-Plane Thermal Conductance of Phosphonate-Based Self-Assembled Monolayers and Self-Assembled Nanodielectrics. ACS Appl. Mater. Interfaces, 2020, 12, 34901–34909. [Google Scholar] [CrossRef] [PubMed]
- Everaerts, K.; Emery, J.D.; Jariwala, D.; Karmel, H.J.; Sangwan, V.K.; Prabhumirashi, P.L.; Geier, M.L.; McMorrow, J.J.; Bedzyk, M.J.; Facchetti, A.; Hersam, M.C.; Marks, T.J. Ambient-Processable High Capacitance Hafnia-Organic Self-Assembled Nanodielectrics. J. Am. Chem. Soc., 2013, 135, 8926–8939. [Google Scholar] [CrossRef] [PubMed]
- Veres, J.; Ogier, S.; Lloyd, G.; de Leeuw, D. Gate Insulators in Organic Field-Effect Transistors. Chem. Mater., 2004, 16, 4543–4555. [Google Scholar] [CrossRef]
- Ting, G.G.; Acton, O.; Ma, H.; Ka, J.W.; Jen, A.K.-Y. Study on the Formation of Self-Assembled Monolayers on Sol−Gel Processed Hafnium Oxide as Dielectric Layers. Langmuir, 2009, 25, 2140–2147. [Google Scholar] [CrossRef]
- Jung, Y.; Kline, R.J.; Fischer, D.A.; Lin, E.K.; Heeney, M.; McCulloch, I.; DeLongchamp, D.M. The Effect of Interfacial Roughness on the Thin Film Morphology and Charge Transport of High-Performance Polythiophenes. Adv. Funct. Mater., 2008, 18, 742–750. [Google Scholar] [CrossRef]
- Kim, M.-G.; Kim, H.S.; Ha, Y.-G.; He, J.; Kanatzidis, M.G.; Facchetti, A.; Marks, T.J. High-Performance Solution-Processed Amorphous Zinc−Indium−Tin Oxide Thin-Film Transistors. J. Am. Chem. Soc., 2010, 132, 10352–10364. [Google Scholar] [CrossRef]
- Alaboson, J.M.P.; Wang, Q.H.; Emery, J.D.; Lipson, A.L.; Bedzyk, M.J.; Elam, J.W.; Pellin, M.J.; Hersam, M.C. Seeding Atomic Layer Deposition of High- k Dielectrics on Epitaxial Graphene with Organic Self-Assembled Monolayers. ACS Nano, 2011, 5, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Stallings, K.; Smith, J.; Chen, Y.; Zeng, L.; Wang, B.; Di Carlo, G.; Bedzyk, M.J.; Facchetti, A.; Marks, T.J. Self-Assembled Nanodielectrics for Solution-Processed Top-Gate Amorphous IGZO Thin-Film Transistors. ACS App. Mater. Interfaces, 2021, 13, 15399–15408. [Google Scholar] [CrossRef] [PubMed]
- Hupfer, M.L.; Kaufmann, M.; Roussille, L.; Preiß, J.; Weiß, D.; Hinrichs, K.; Deckert, V.; Dietzek, B.; Beckert, R.; Presselt, M. Arylic ve rsus Alkylic-Hydrophobic Linkers Determine the Supramolecular Structure and Optoelectronic Properties of Tripodal Amphiphilic Push−Pull Thiazoles. Langmuir, 2019, 35, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Malytskyi, V.; Simon, J.-J.; Patrone, L.; Raimundo, J.-M. Synthesis, self-assembly and characterization of a novel push–pull thiophene-based chromophore on a gold surface. RSC Adv., 2015, 5, 26308. [Google Scholar] [CrossRef]
- Lissau, H.; Frisenda, R.; Olsen, S.T.; Jevric, M.; Parker, C.R.; Kadziola, A.; Hansen, T.; van der Zant, H.S.J.; Nielsen, M.B.; Mikkelsen, K.V. Tracking molecular resonance forms of donor–acceptor push–pull molecules by single-molecule conductance experiments. Nature Comm. 2015, 6, 10233. [Google Scholar] [CrossRef] [PubMed]
- Brandl, T.; El Abbassi, M.; Stefani, D.; Frisenda, R.; Harzmann, G.D.; van der Zant, H.S.J.; Mayor, M. Enhanced Separation Concept (ESC): Removing the Functional Subunit from the Electrode by Molecular Design. Eur. J. Org. Chem., 2019, 5334–5343. [Google Scholar] [CrossRef]
- Li, Y.-X.; Dong, X.-M.; Yu, M.-N.; Liu, W.; Nie, Y.-J.; Zhang, J.; Xie, L.-H.; Xu, C.-X.; Liu, J.-Q.; Huang, W. A Bio-Inspired Molecular Design Strategy toward 2D Organic Semiconductor Crystals with Superior Integrated Optoelectronic Properties. Small, 2021, 17, 2102060. [Google Scholar] [CrossRef]
- Xu, J.; Wen, L.; Zhou, W.; Lv, J.; Guo, Y.; Zhu, M.; Liu, H.; Li, Y.; Jiang, L. Asymmetric and Symmetric Dipole-Dipole Interactions Drive Distinct Aggregation and Emission Behavior of Intramolecular Charge-Transfer Molecules. J. Phys. Chem. C, 2009, 113, 5924–5932. [Google Scholar] [CrossRef]
- Ravindran, E.; Somanathan, N. Efficient white-light emission from a single polymer system with ‘‘spring-like’’ self-assemblies induced emission enhancement and intramolecular charge transfer characteristics. J. Mater. Chem. C, 2017, 5, 4763. [Google Scholar] [CrossRef]
- Kim, J.H.; Oh, S.; Park, S.K.; Park, S.Y. Green-Sensitive Phototransistor Based on Solution-Processed 2D n-Type Organic Single Crystal. Adv. Electron. Mater., 2019, 5, 1900478. [Google Scholar] [CrossRef]
Figure 2.
a) Structure of p-DSSCs dye (green: electron donor; violet: π-bridge; red: electron acceptor), b) electron pathway in an illuminated p-DSSCs. From reference [66].
Figure 2.
a) Structure of p-DSSCs dye (green: electron donor; violet: π-bridge; red: electron acceptor), b) electron pathway in an illuminated p-DSSCs. From reference [66].
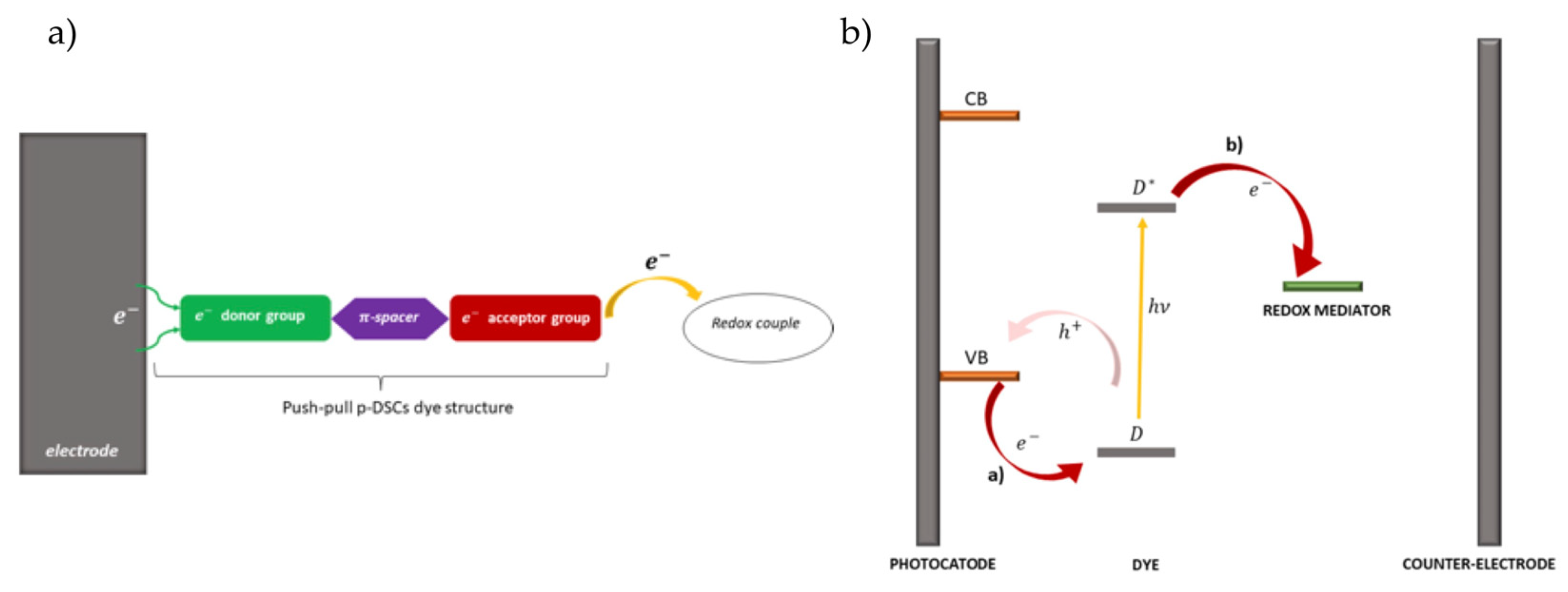
Figure 3.
a) Molecular design of D-A new dyes PO1-6 whose studied structures with carboxylic acid anchoring group and different acceptors are presented in b). The benchmark reference dye P1 is shown in c). From reference [72].
Figure 3.
a) Molecular design of D-A new dyes PO1-6 whose studied structures with carboxylic acid anchoring group and different acceptors are presented in b). The benchmark reference dye P1 is shown in c). From reference [72].
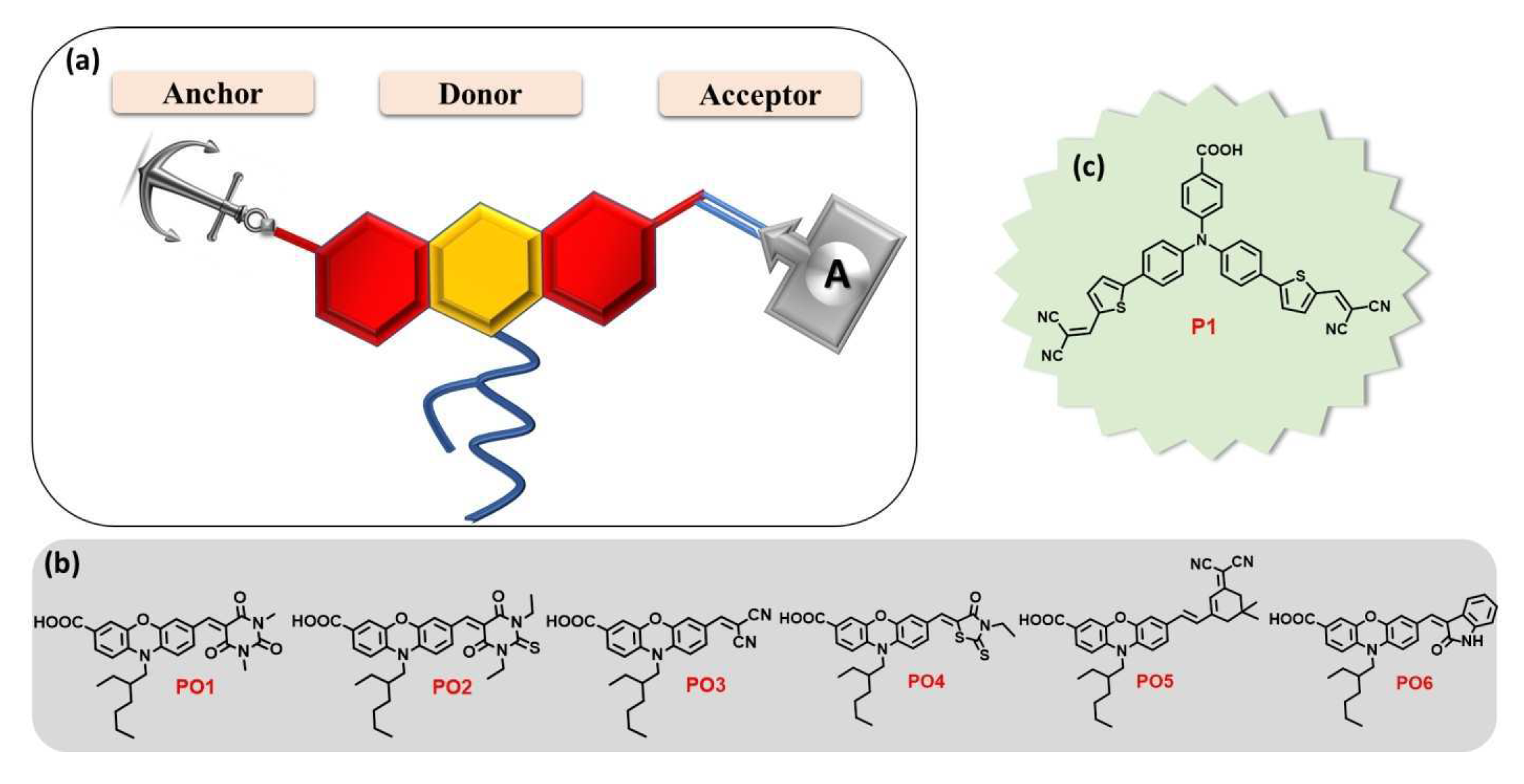
Figure 4.
Chemical structures of Pc TT1 and Pz TT112 porphyrazine-based sensitizers and IPCE measured with TT112, from Reference [86].
Figure 4.
Chemical structures of Pc TT1 and Pz TT112 porphyrazine-based sensitizers and IPCE measured with TT112, from Reference [86].
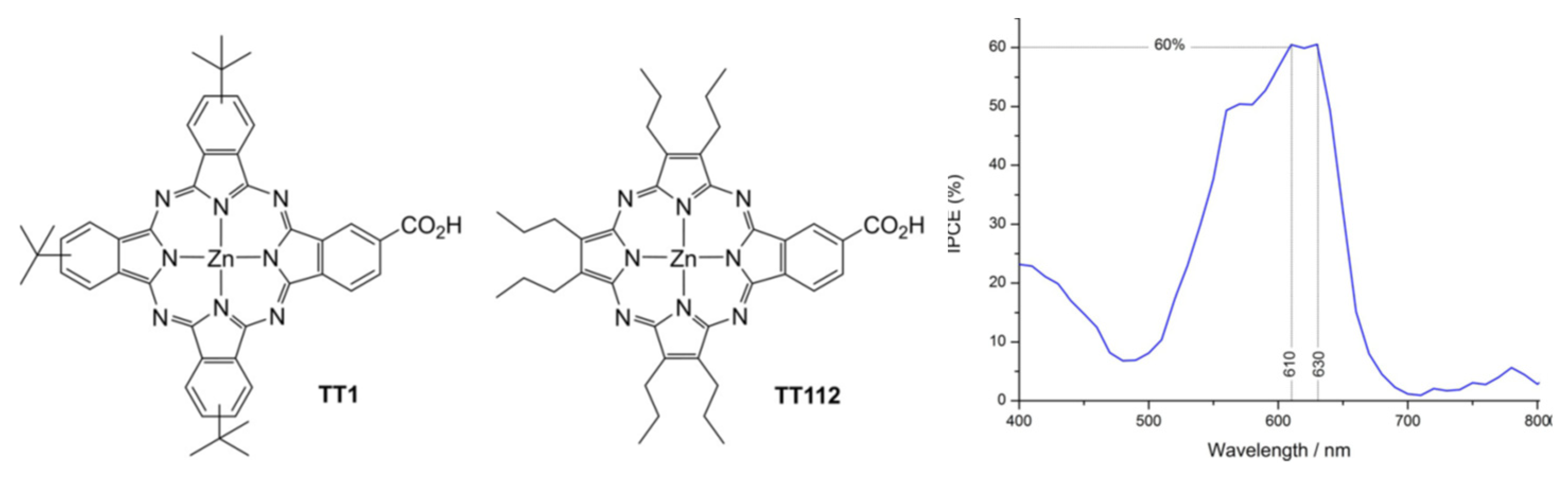
Figure 5.
Chemical structures of A3B panchromatic push-pull porphyrazines studied in reference [87] still incorporating the electron-withdrawing B moiety of T112 presented in Figure 4, but with subunits made of pyrrole rings substituted with different donor groups.
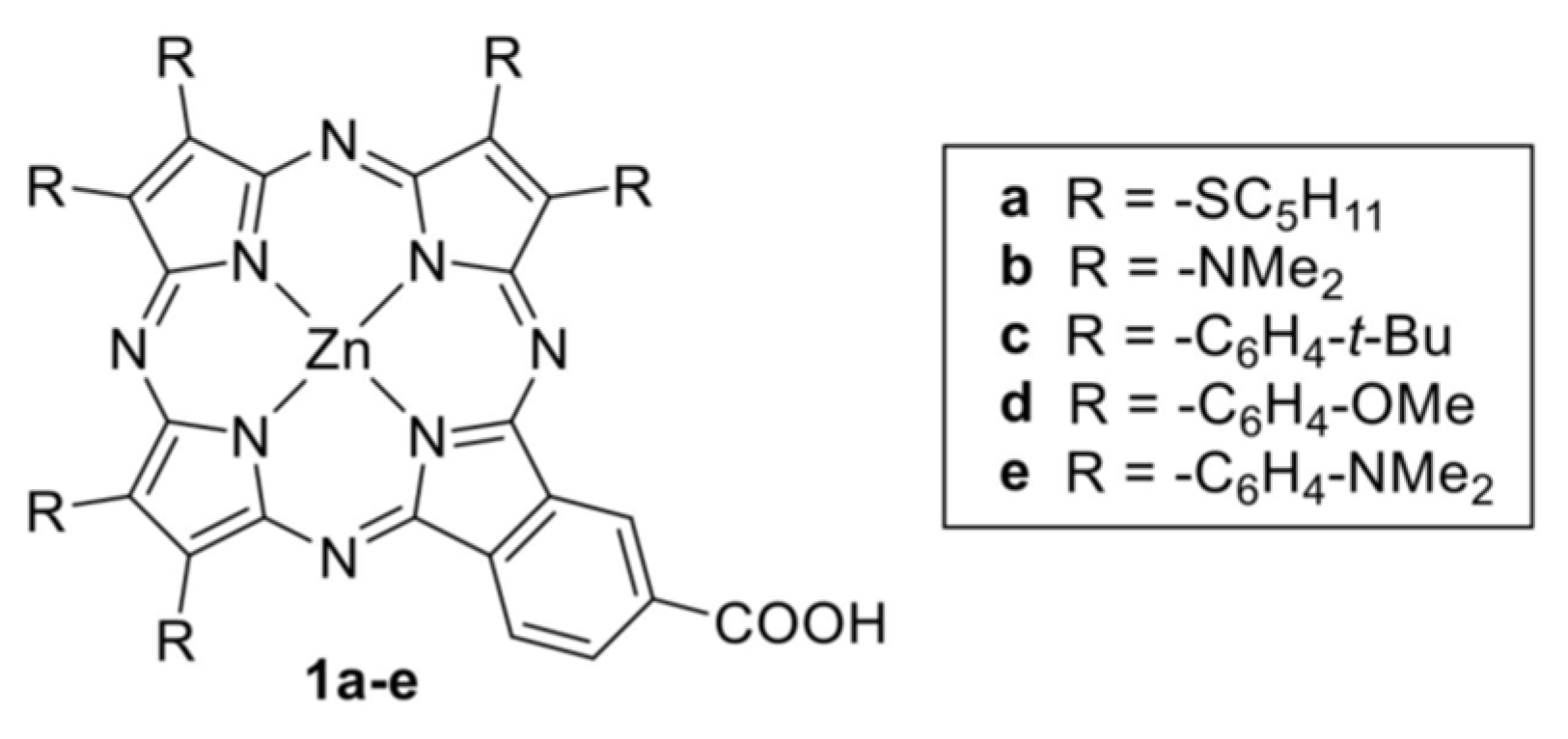
Figure 6.
Asymmetric CuPc synthesized and scheme of phthalocyanine (ZnPc) based DSCC. From reference [91].
Figure 6.
Asymmetric CuPc synthesized and scheme of phthalocyanine (ZnPc) based DSCC. From reference [91].

Figure 7.
Indolizine-Porphyrin Push−Pull Dye proposed in reference [106] and evidence of its absorption in the near-infrared.
Figure 7.
Indolizine-Porphyrin Push−Pull Dye proposed in reference [106] and evidence of its absorption in the near-infrared.
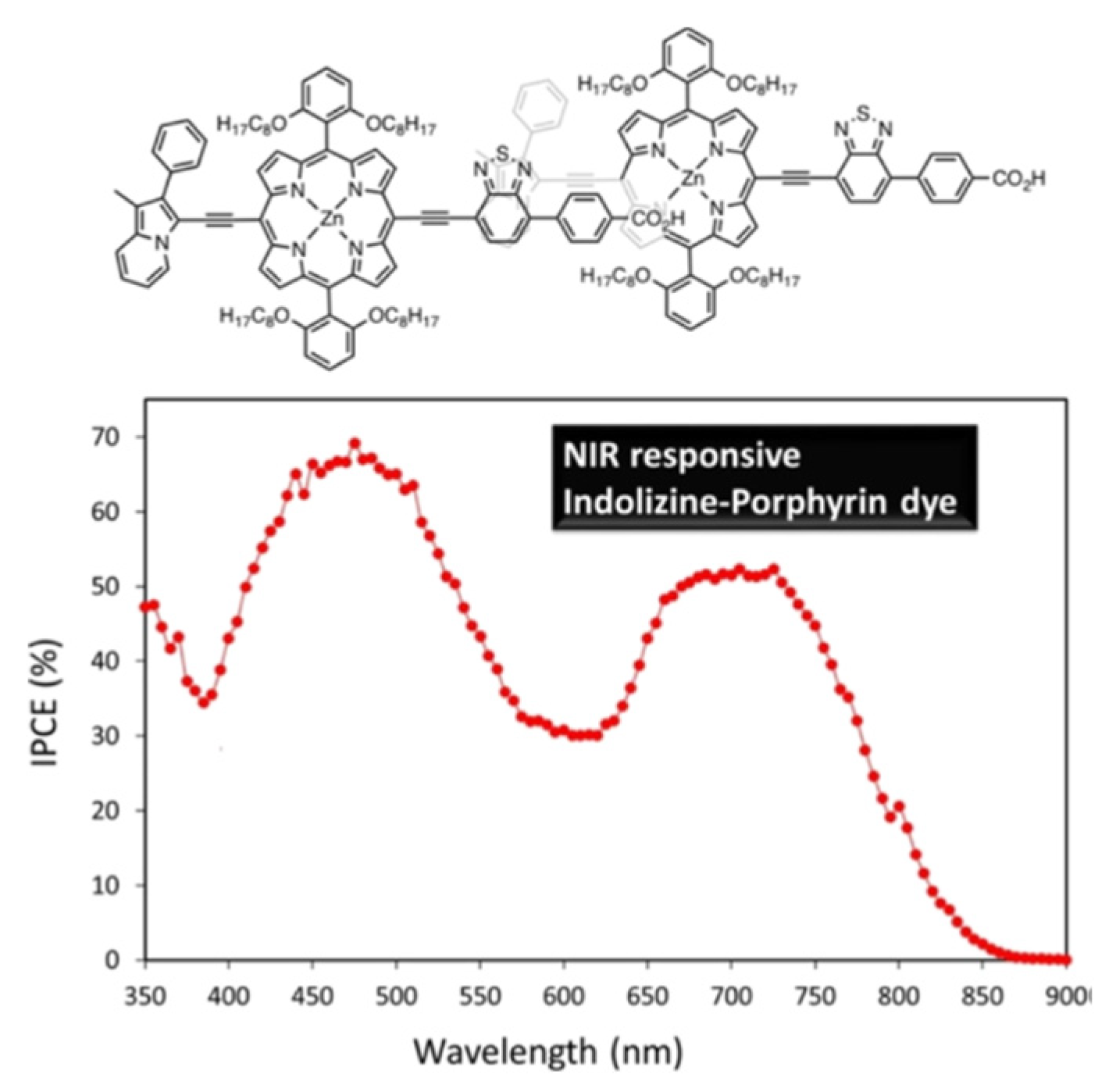
Figure 8.
Hole and electron current measured on perovskite solar cell showing higher values for the cell incorporating DPP-2T push-pull. From reference [108].
Figure 8.
Hole and electron current measured on perovskite solar cell showing higher values for the cell incorporating DPP-2T push-pull. From reference [108].
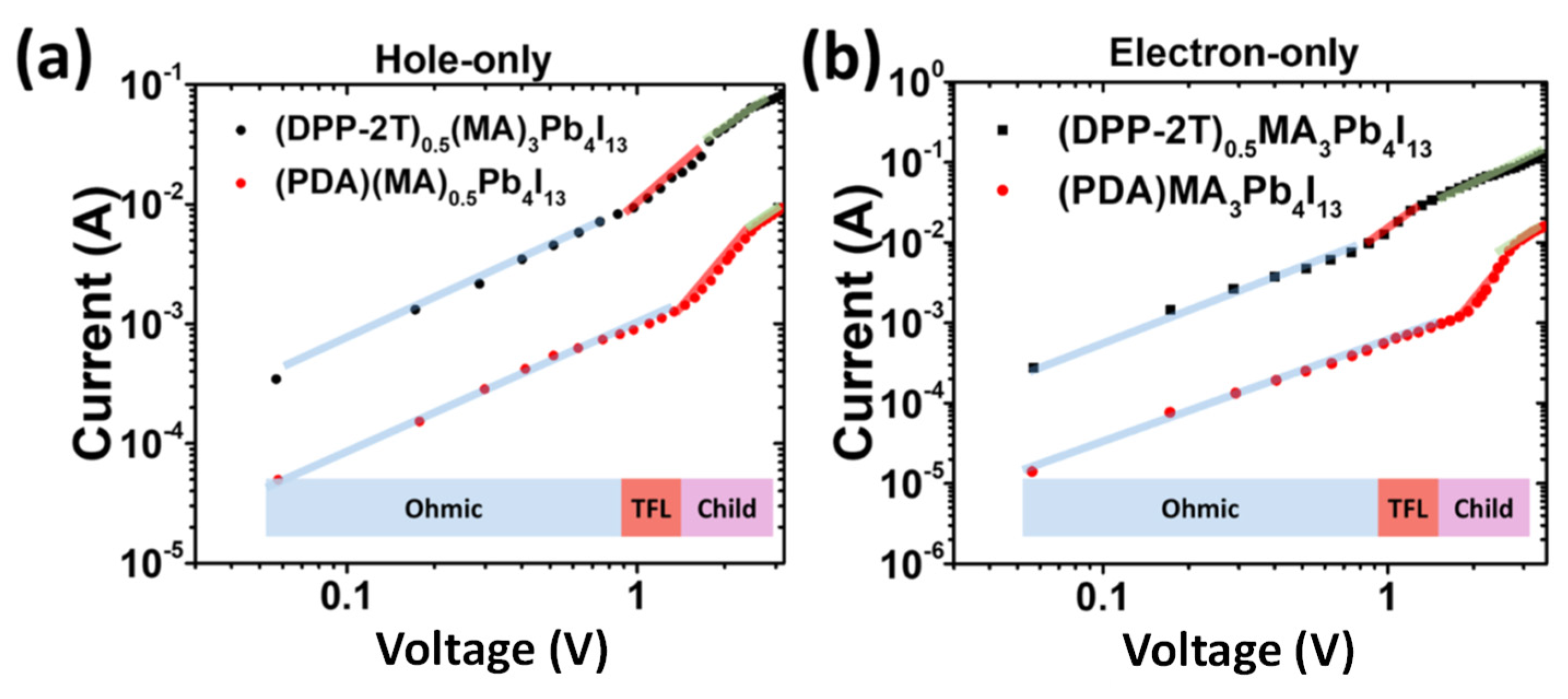
Figure 9.
Interfacial SAM between ZnO electron transport layer and perovskite (left) and their effect on the solar cell stability (right). From reference [110].
Figure 9.
Interfacial SAM between ZnO electron transport layer and perovskite (left) and their effect on the solar cell stability (right). From reference [110].
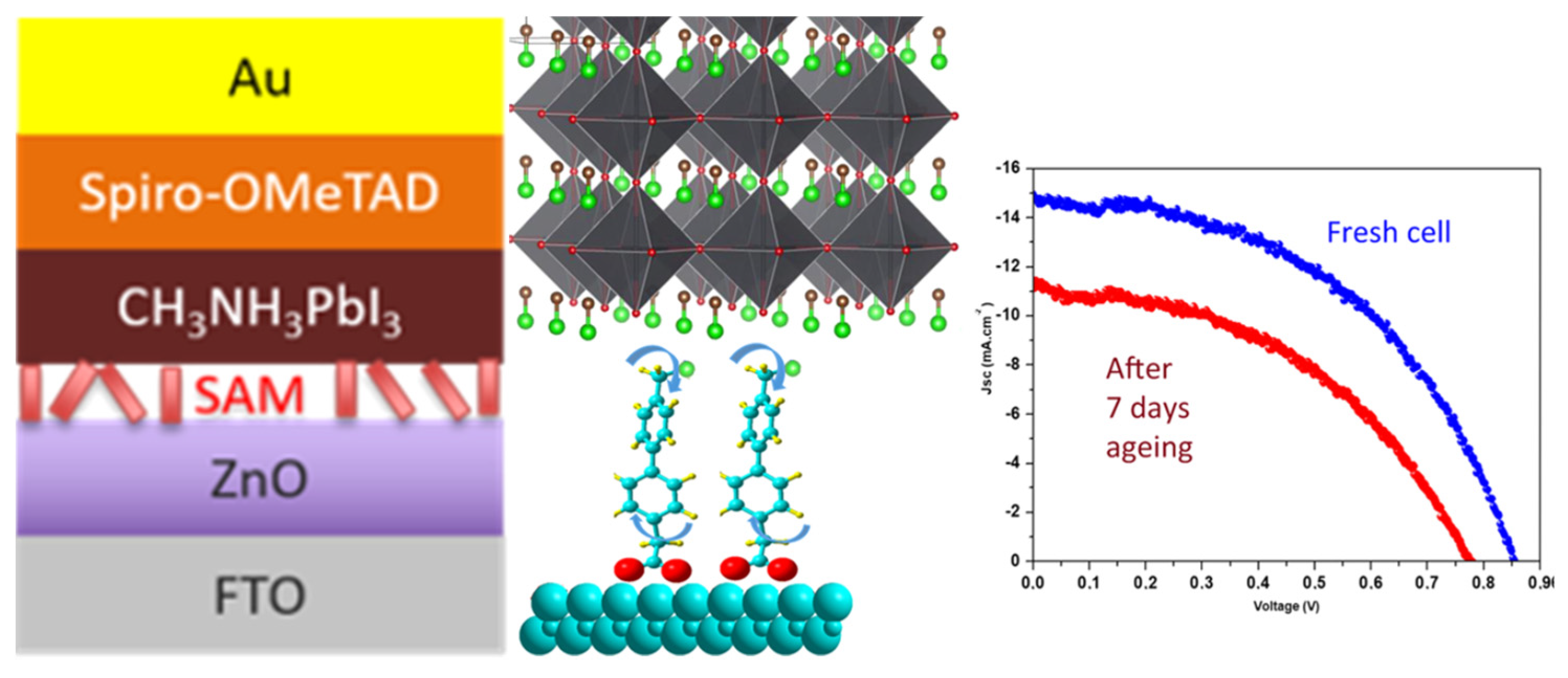
Figure 10.
Scheme of the push-pull incorporation within perovskite materials, induced band alignment, and evidence of the increase of open circuit voltage and hydrophobicity. From reference [111].
Figure 10.
Scheme of the push-pull incorporation within perovskite materials, induced band alignment, and evidence of the increase of open circuit voltage and hydrophobicity. From reference [111].
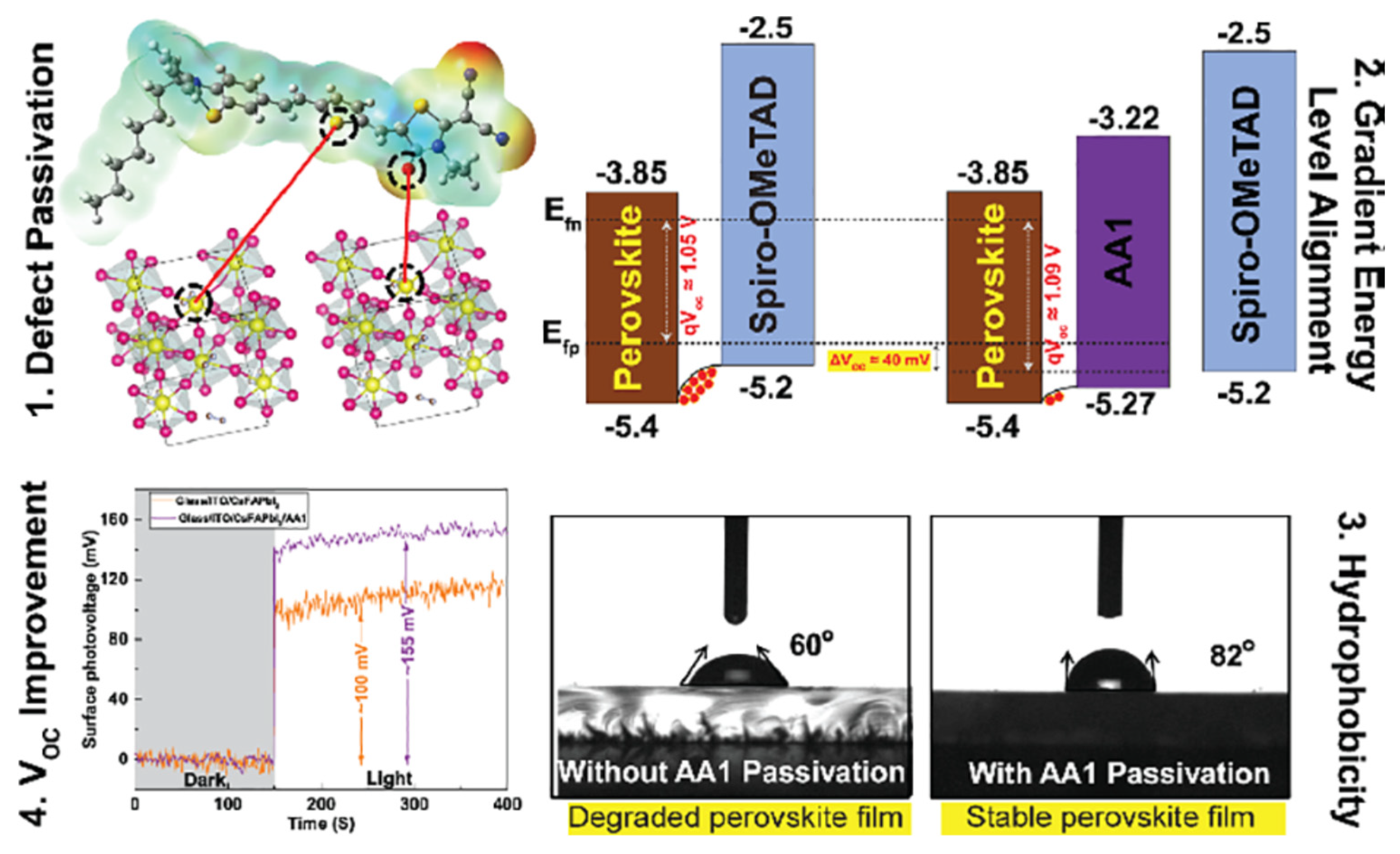
Figure 11.
Scheme of the different chemical passivation functions of 4OH-NMI with corresponding defects in perovskite. From reference [113].
Figure 11.
Scheme of the different chemical passivation functions of 4OH-NMI with corresponding defects in perovskite. From reference [113].
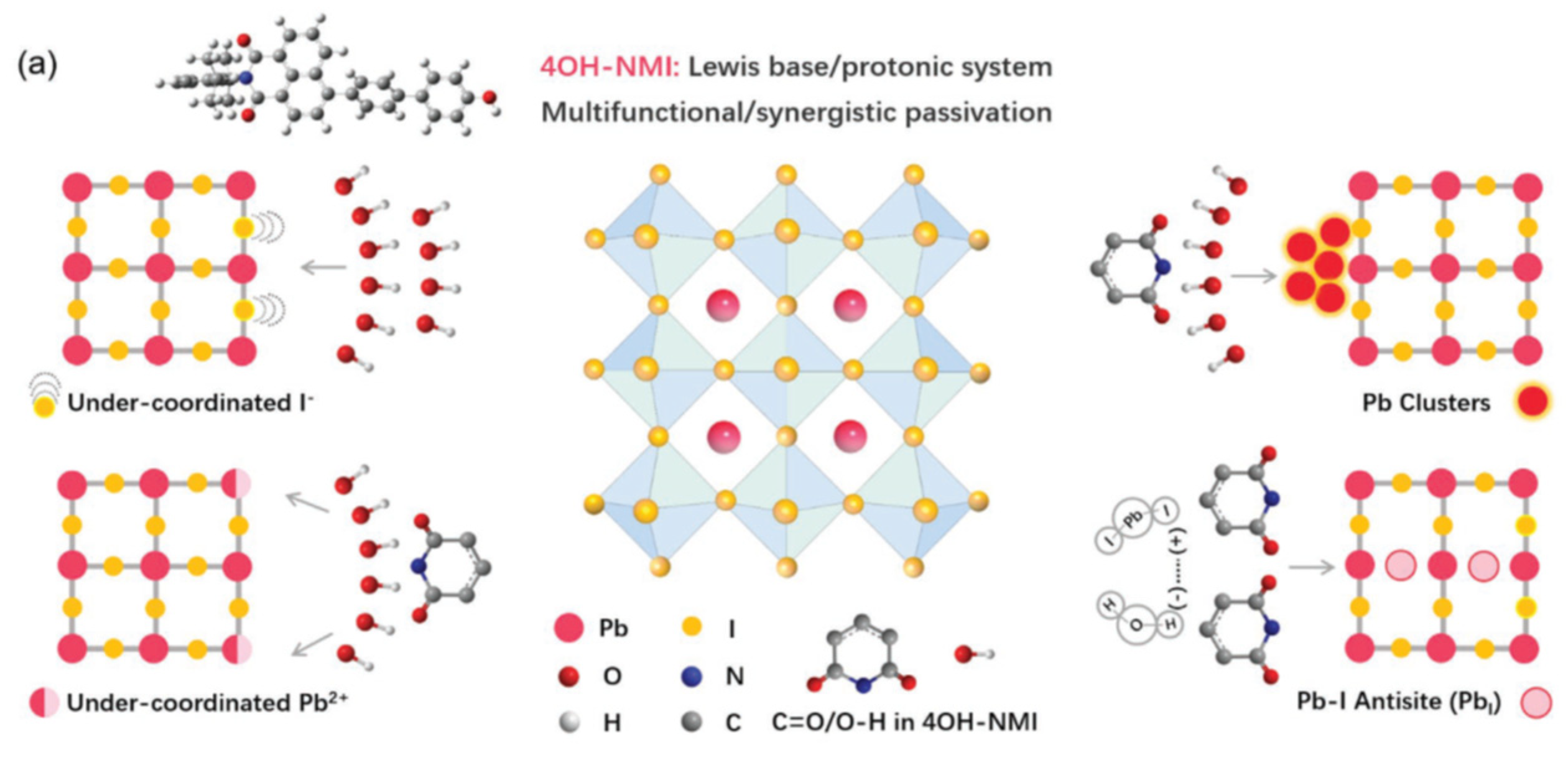
Figure 12.
Thermal admittance spectroscopy images showing the higher grain boundaries reduction for NMI-modified than for PMI modidified perovskite solar cell in comparison to the non-modified cell (control image). From reference [113].
Figure 12.
Thermal admittance spectroscopy images showing the higher grain boundaries reduction for NMI-modified than for PMI modidified perovskite solar cell in comparison to the non-modified cell (control image). From reference [113].

Figure 13.
Stability test of PMI/NMI/non-modified perovskite solar cell upon exposure to N2 atmosphere, and oxygen and 20-30% of relative humidity. From reference [113].
Figure 13.
Stability test of PMI/NMI/non-modified perovskite solar cell upon exposure to N2 atmosphere, and oxygen and 20-30% of relative humidity. From reference [113].
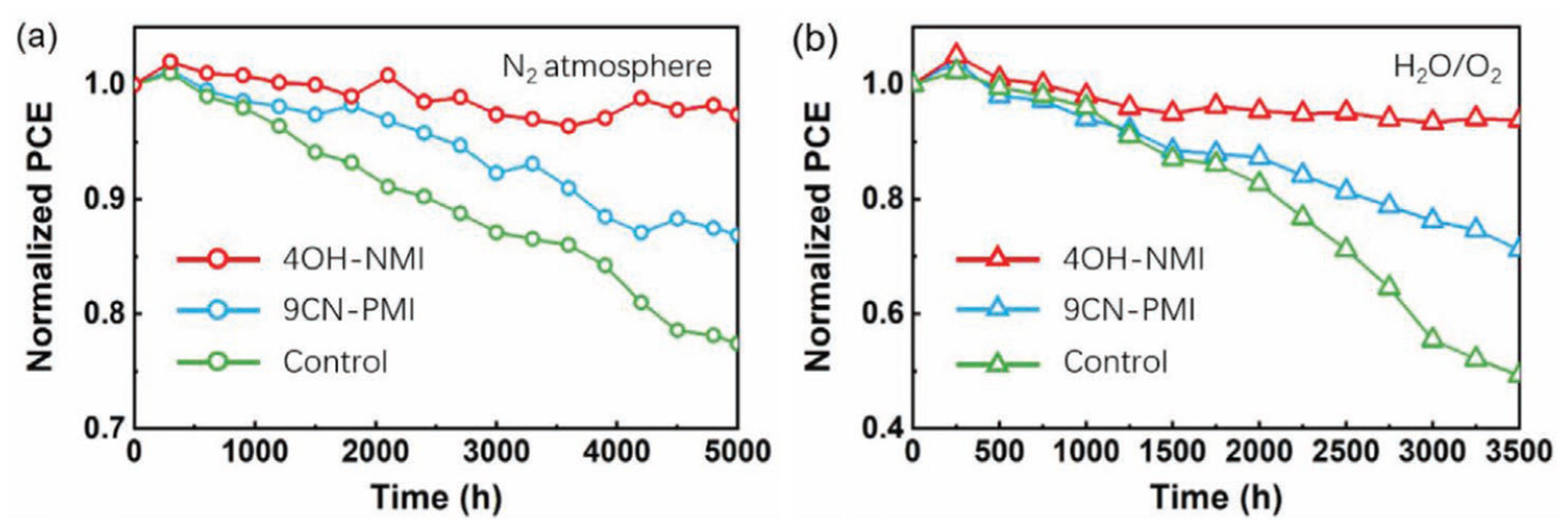
Figure 14.
Schematic depiction of SAND studied by Fachetti’s group. From reference [114].
Figure 14.
Schematic depiction of SAND studied by Fachetti’s group. From reference [114].
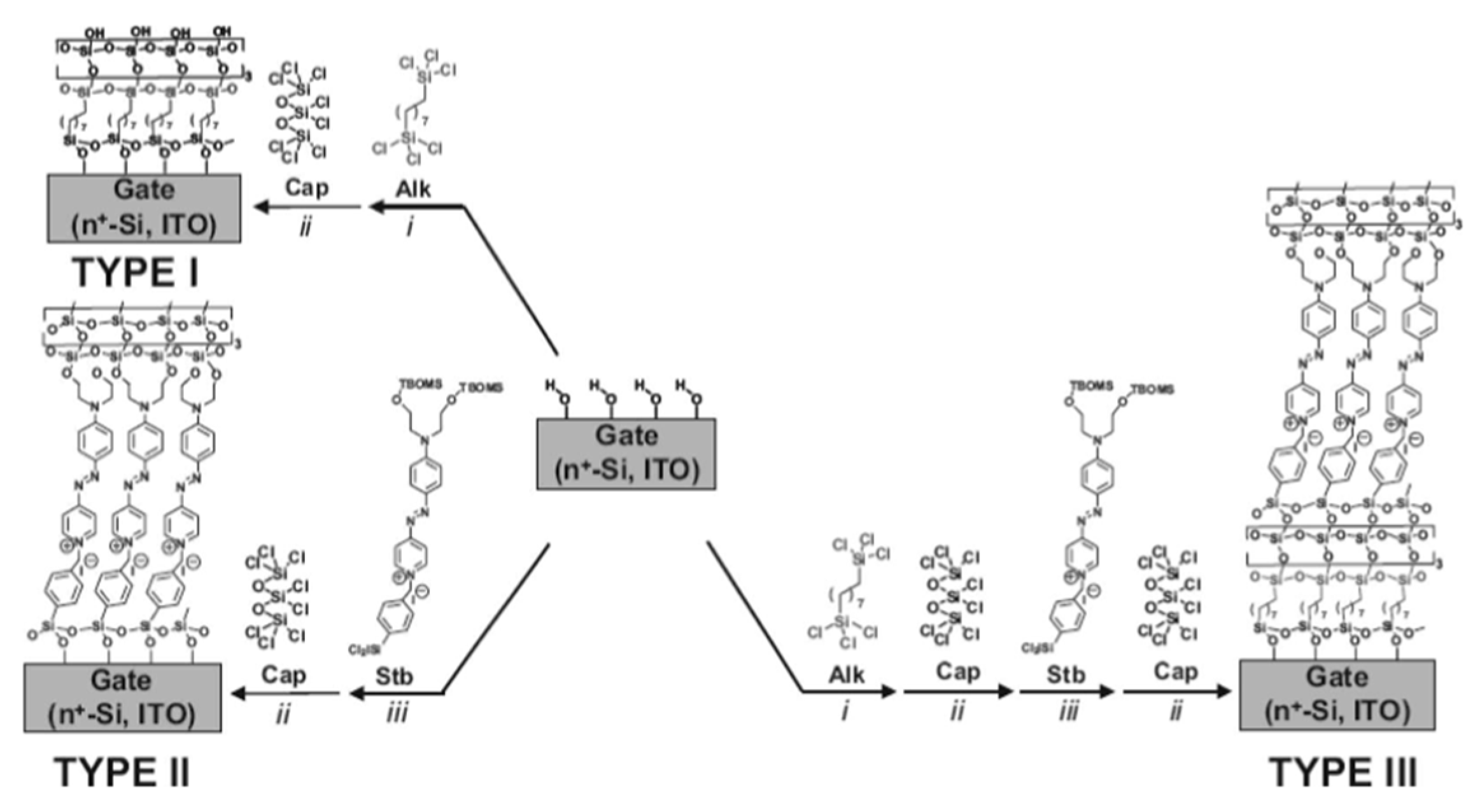
Figure 15.
(a) Schematic representation of ZrOx film fabrication by UV irradiation. (d) Fabrication procedure for UV-densified zirconia self-assembled nanodielectric (UVD-Zr-SAND)n multilayers. (e) Molecular structure of the PEI organic component of SAND. (f) Absorbance spectra of a primer-ZrOx-PAE film without the ZrOx capping layer before and after irradiation.
Figure 15.
(a) Schematic representation of ZrOx film fabrication by UV irradiation. (d) Fabrication procedure for UV-densified zirconia self-assembled nanodielectric (UVD-Zr-SAND)n multilayers. (e) Molecular structure of the PEI organic component of SAND. (f) Absorbance spectra of a primer-ZrOx-PAE film without the ZrOx capping layer before and after irradiation.
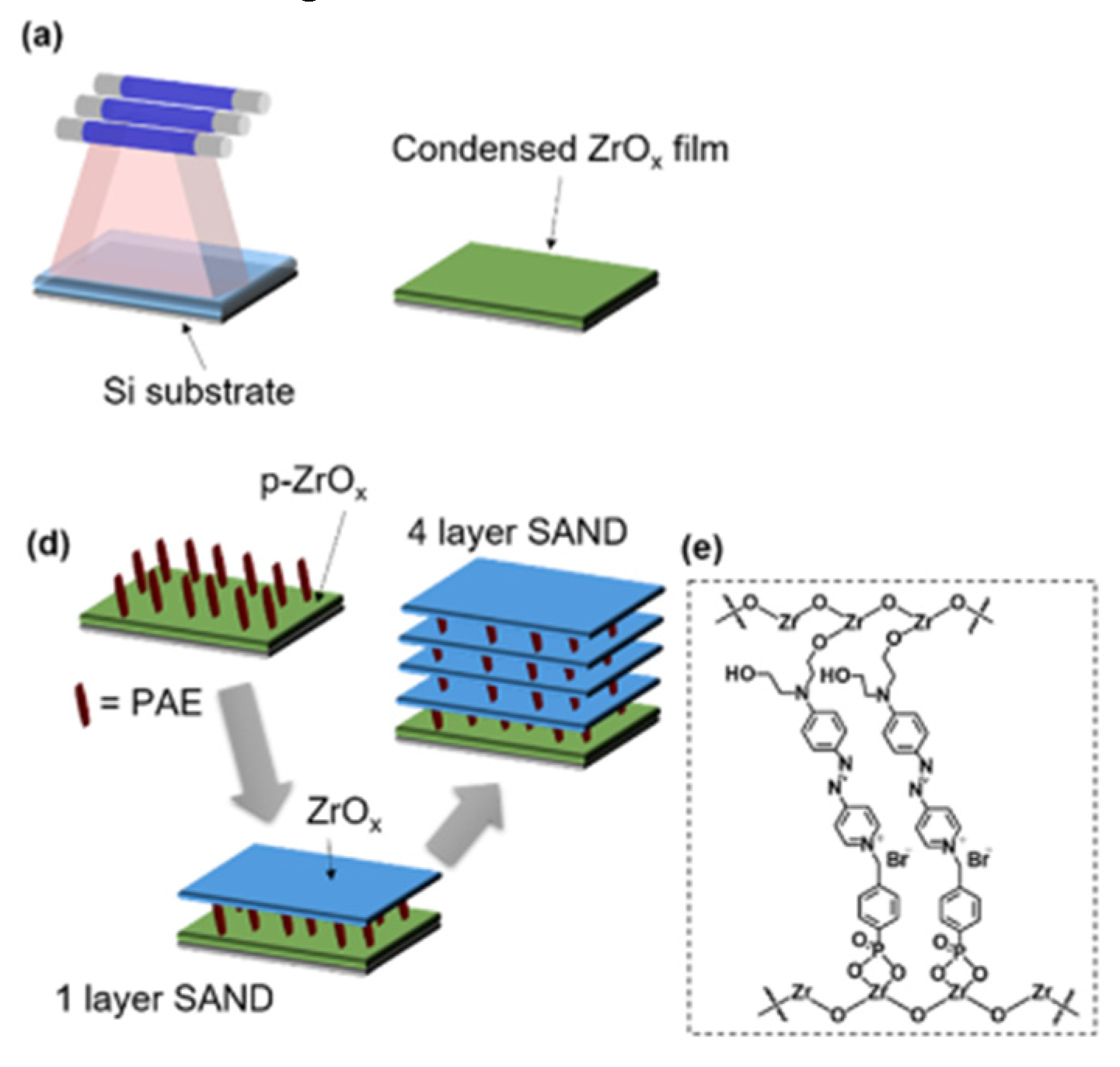
Figure 16.
Dielectric stacks comprising four-chromophore/ZrOx layers on top of the ZrOx (p-ZrOx) primer film. (a) Conventional Zr-SAND with a phosphonate π-electron (PAE) unit. (b) Inverted IZr-SAND with an inverted PAE (IPAE) π-unit.
Figure 16.
Dielectric stacks comprising four-chromophore/ZrOx layers on top of the ZrOx (p-ZrOx) primer film. (a) Conventional Zr-SAND with a phosphonate π-electron (PAE) unit. (b) Inverted IZr-SAND with an inverted PAE (IPAE) π-unit.
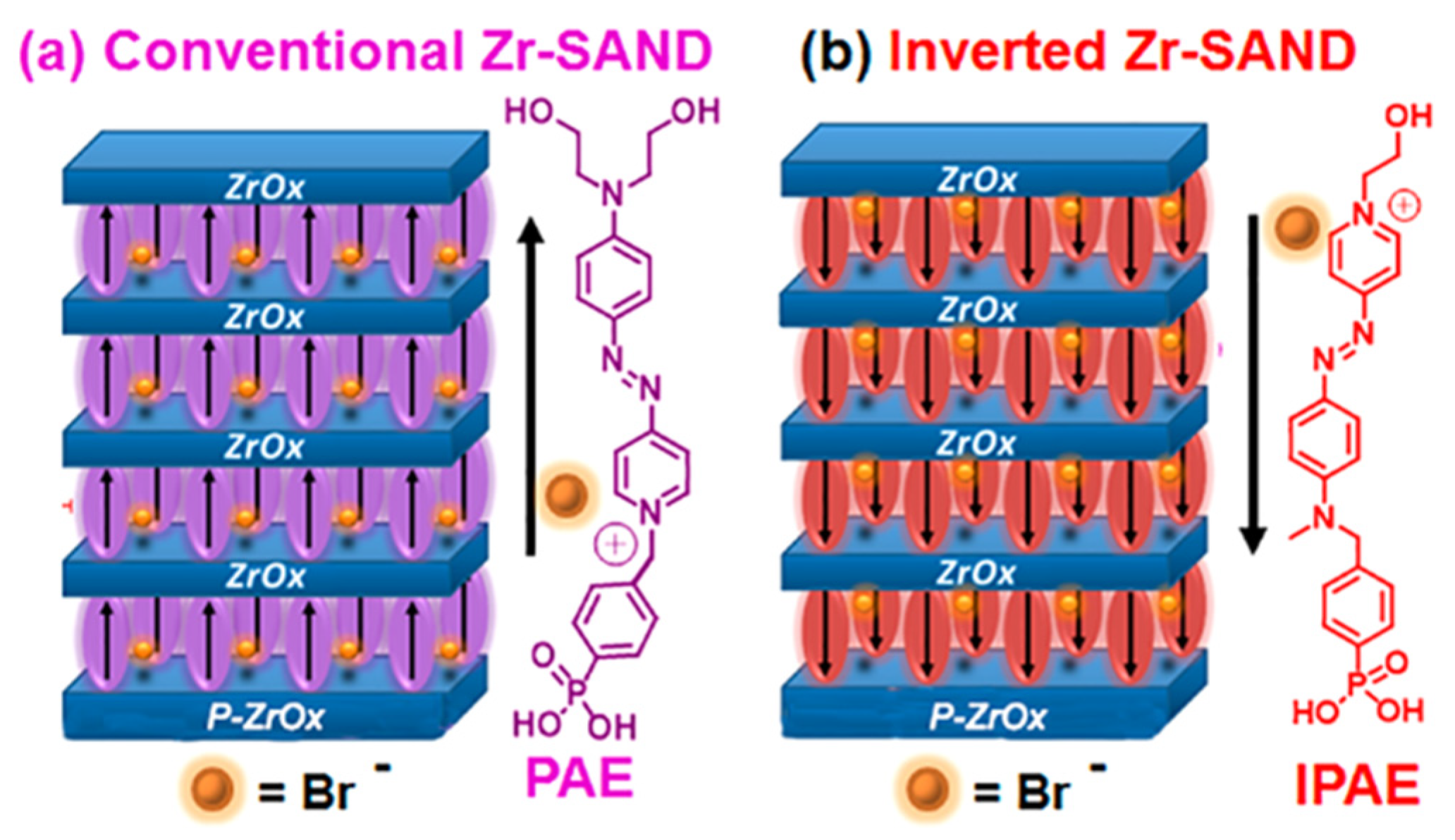
Figure 17.
(a) Solution-based Hf-SAND self-assembly procedure employed in this study. (b) Schematic of the various Hf-SAND multilayers produced including the corresponding X-ray reflectivity derived thicknesses (Hf-SAND-1, -4) and estimated thicknesses (HfSAND-2, -3).
Figure 17.
(a) Solution-based Hf-SAND self-assembly procedure employed in this study. (b) Schematic of the various Hf-SAND multilayers produced including the corresponding X-ray reflectivity derived thicknesses (Hf-SAND-1, -4) and estimated thicknesses (HfSAND-2, -3).
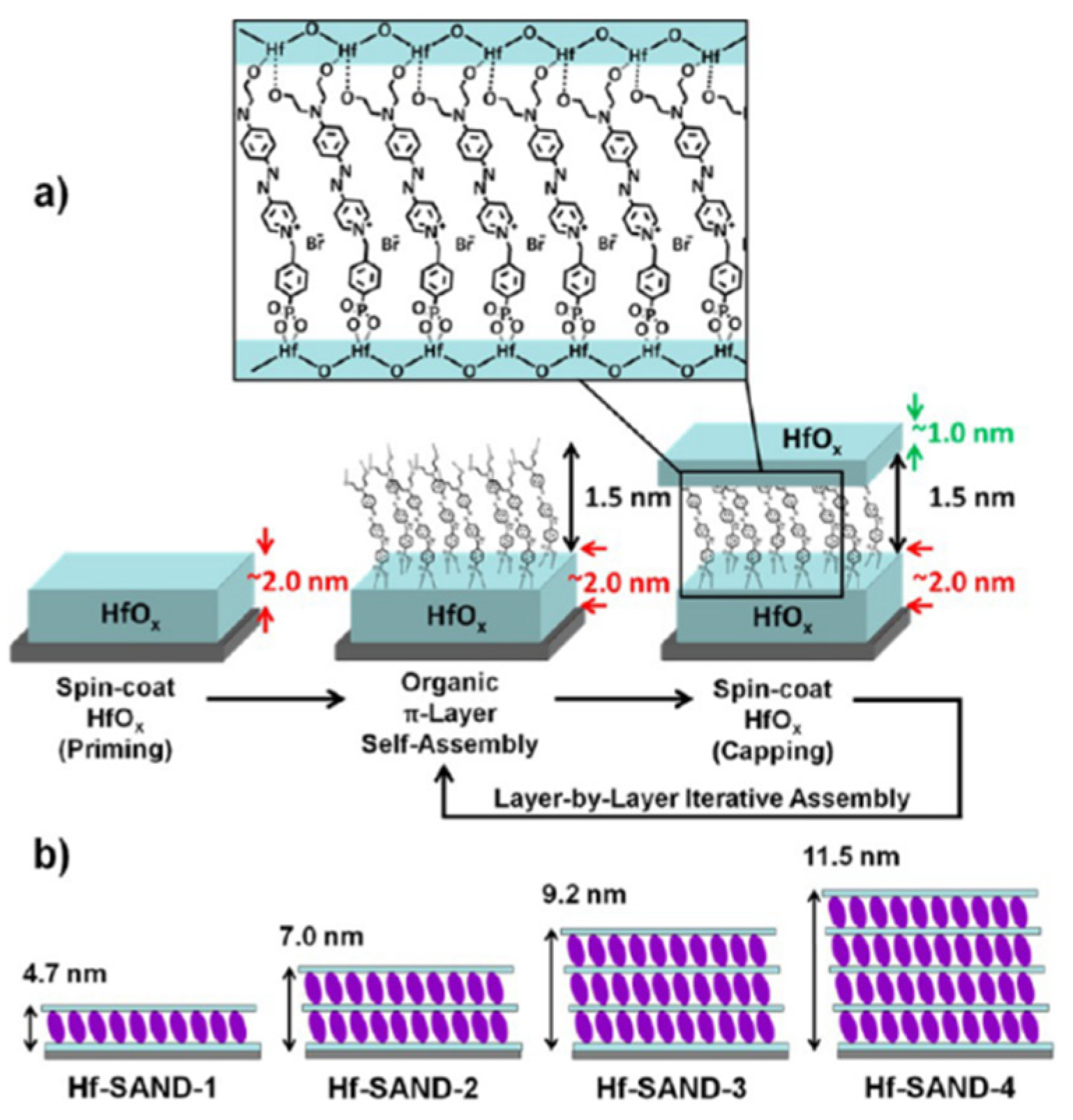
Figure 18.
Plots with best fits of Hf-SAND-n MIS properties. (a) Assuming 1/x decay dependence of capacitance density versus Hf SAND layer number n. (b) Inverse capacitance versus layer number linear relationship. (c) Increasing effective dielectric constant keff versus layer number n. (d) EOT versus layer number n.
Figure 18.
Plots with best fits of Hf-SAND-n MIS properties. (a) Assuming 1/x decay dependence of capacitance density versus Hf SAND layer number n. (b) Inverse capacitance versus layer number linear relationship. (c) Increasing effective dielectric constant keff versus layer number n. (d) EOT versus layer number n.

Figure 19.
(a) FeII-bis-terpyridine complexes 1–3 as E-field sensitive spin cross-over switching junctions. (b) Scheme of the E-field triggered switching using the distortion of the FeII-pyridine ligand sphere upon applying of a large enough voltage. From Ref [134].
Figure 19.
(a) FeII-bis-terpyridine complexes 1–3 as E-field sensitive spin cross-over switching junctions. (b) Scheme of the E-field triggered switching using the distortion of the FeII-pyridine ligand sphere upon applying of a large enough voltage. From Ref [134].
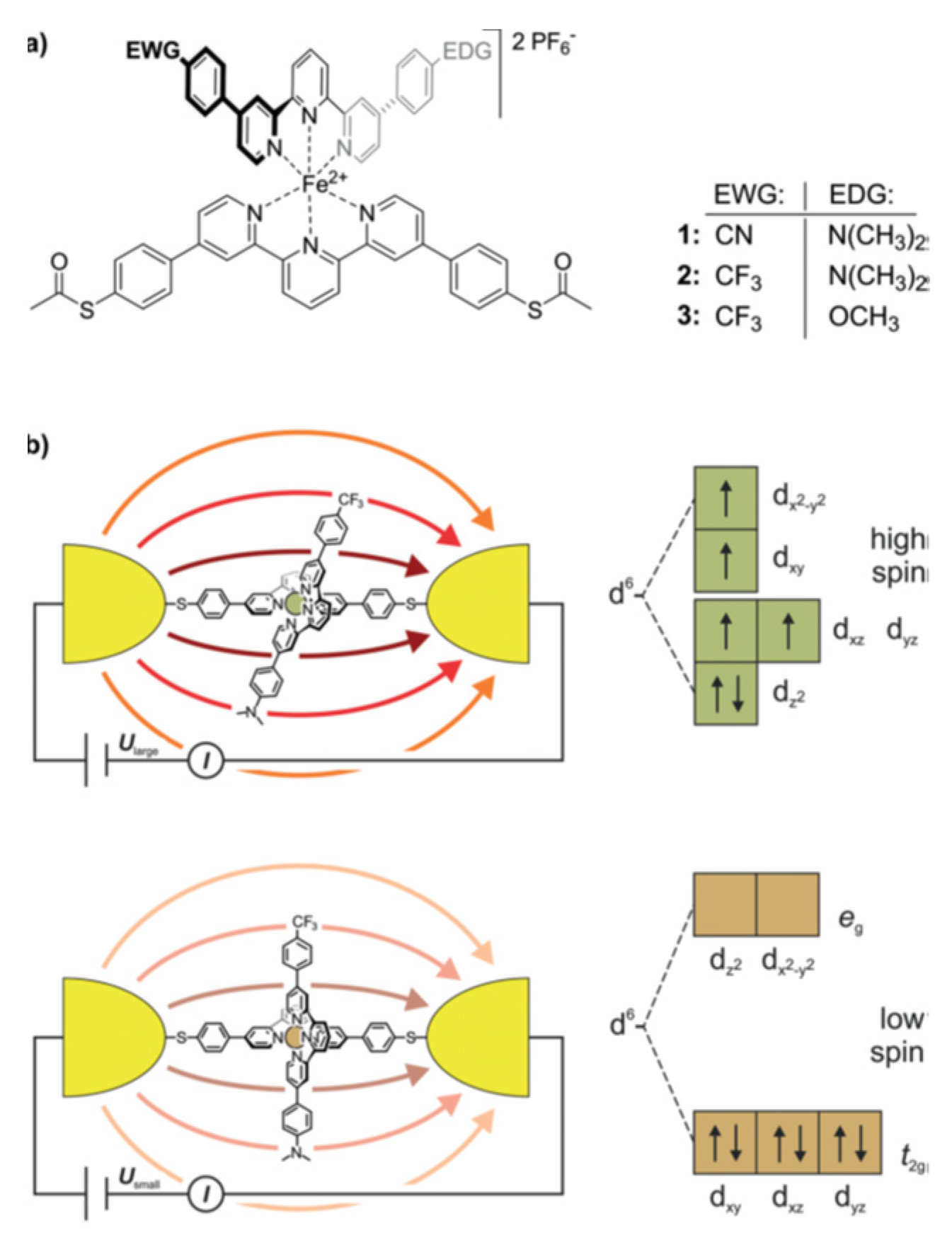
Figure 20.
Schematic representation of the 2D amphiphile-like packing layer similar to 2D lipid bilayer in nature (left), and the AL/RL alternate stacks developed (right). From Ref [135].
Figure 20.
Schematic representation of the 2D amphiphile-like packing layer similar to 2D lipid bilayer in nature (left), and the AL/RL alternate stacks developed (right). From Ref [135].
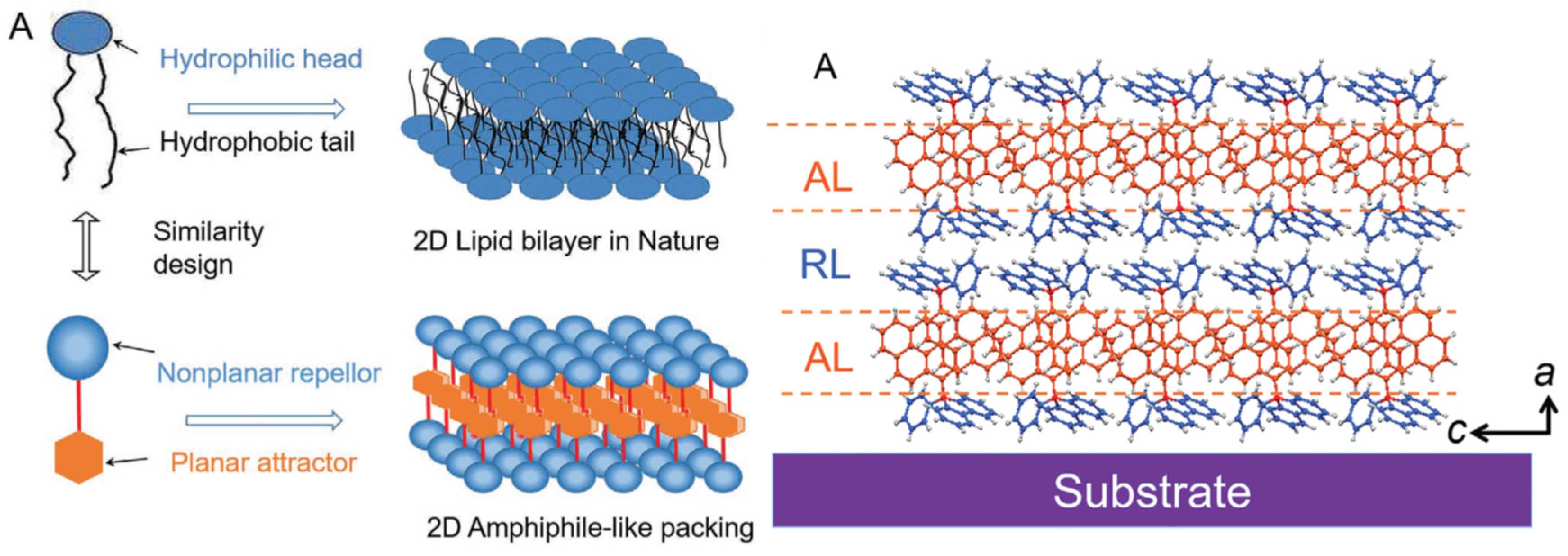
Figure 21.
Optimized structure of BPPTA & various molecular compositions of BPPTA–fluorene copolymers (top) ; Absorption spectra in (a) solution, (b) thin film of the copolymer and BPPTA under irradiation at ~360 nm; PL spectra in (c) solution, (d) thin film of BPPTA and the copolymers. From Ref [137].
Figure 21.
Optimized structure of BPPTA & various molecular compositions of BPPTA–fluorene copolymers (top) ; Absorption spectra in (a) solution, (b) thin film of the copolymer and BPPTA under irradiation at ~360 nm; PL spectra in (c) solution, (d) thin film of BPPTA and the copolymers. From Ref [137].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



