
Submitted:
17 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Pancreatic Ductal Adenocarcinoma (PDAC) is the most frequent infiltrating type of pancreatic cancer. The poor prognosis is due to the absence of specific biomarkers, aggressiveness and treatment resistance. PDAC is a deadly malignancy bearing distinct genetic alterations, the most common being those that cause cancer-causing versions of the KRAS gene. Cannabigerol (CBG) is a non-psychomimetic cannabinoid with anti-inflammatory property. Regarding CBG anticancer ef-fect, up to now there are only few evidence in human cancers. We investigated CBG effects on PDAC cell lines, PANC-1 and MIAPaCa-2. It was investigated CBG activity on cell viability, cell death and its effect of EGFR-RAS-associated signalling. Moreover, the potential synergistic effect of CBG in combination with gemcitabine (GEM) and paclitaxel (PTX) was investigated. MTT was applied to investigate CBG effect on PDAC cell lines viability. Annexin-V and Acridine Orange staining followed by cytofluorimetric analysis and Western blot were used to evaluate CBG effect on cell death. Modulation of EGFR-RAS-associated pathways was determined by Western Blot analysis and Milliplex multiplex assay. Moreover, by MTT data and Synergy finder software analysis the effect of the combination of CBG and chemotherapeutic drugs, was determined.
Keywords:
cannabigerol
; pancreatic cancer
; autophagy
; chemo-resistance
; RAS pathways
1. Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is an infiltrating neoplasm derived from the pancreatic ductal tree, and its 5-years overall survival rate increased from 3% to 9% in the last 50 years [1]. The worst prognosis is related to the absence of specific symptoms, diagnosis biomarkers, high invasiveness, and treatment resistance [2]. The PI3K/AKT/mTOR and RAF/MEK/ERK pathways, frequently activated in PDAC, are involved in mediating autophagy, apoptosis and chemoresistance, and they are known to drive PDAC development and tumor maintenance [3]. While pharmacological approaches to block RAS signalling were clinically unsuccessful, as demonstrated by no clinical benefit as monotherapy in trials [4], efforts are focusing on downstream proteins, such as RAF, MEK and Akt [5]. Different PI3K/Akt/mTOR inhibitors are in clinical evaluation, but their antitumoral effects may be limited by the over-activation of the RAS/RAF/MEK signalling [6]. Novel strategy to combine inhibitors of these pathways could be more effective, and some preclinical studies have evidenced improved efficacy in various tumour types [7]. Regarding PDAC, this adjuvant therapy was demonstrated to improve overall survival but in a limited number of patients [8], thus new pharmacological and combination therapies are needed. Phytocannabinoids as Tetrahydrocannabinol (THC) and Cannabidiol (CBD), compounds derived from Cannabis sativa L., as well as medical cannabis, were evidenced to exert an antitumoral activity when in combination therapies in glioblastoma, as reported in one clinical trial and in one case report [9,10]. Moreover, many in vitro and in vivo preclinical studies evidenced as phytocannabinoids show antitumoral effects in different human cancers, and synergistic effects with approved chemotherapeutic drugs, as recently reviewed [11].
The non-psychomimetic phytocannabinoid, cannabigerol (CBG) was evaluated for its potential anticancer effect in human cancer cell lines, including melanoma and glioblastoma [12,13]. Although the pathways activated by CBG were not yet clarified, recently CBG was demonstrated to bind the kinase active site of EGFR and inhibit its intracellular kinase activity, inducing apoptosis in EGFR-overexpressing A549 and A431 cell lines [14]. This is particularly important because EGFR protein family is involved in initialization and progression of PDAC and EGFR inhibitors are used in clinical trials as anticancer therapy [15].
In this study, CBG anticancer effect was investigated in two human PDAC cell lines, PANC-1 and MIAPaCa-2, in order to analyse its interactions with the EGFR-RAS related pathways involved in sustaining cell viability and chemoresistance.
2. Results
2.1. CBG inhibits cell growth of PDAC cell lines
PANC-1 and MIAPaCa-2 PDAC cell lines, and immortalized NCM460D and NHFA-12 cell lines were treated with different doses of CBG (up to 31.648 μg/mL) as single administration and cell viability was determined by MTT assay at 72 h post-treatment. Results showed that CBG reduces cell viability in both cell lines with an IC50 of 15.64 μg/mL for PANC-1 and 13.77 μg/mL for MIAPaCa-2 (Figure 1), while lower cytotoxicity was observed in normal human NCM460D and NHFA12 cell lines (Supplementary Figure 1). For the next experiments, we selected two doses of CBG (11.08 and 12.66 µg/mL), as lower cytotoxic doses for studying molecular and biological mechanisms in PDAC cell lines and with no cytotoxic effects on normal cells. CBG was always used as single administration.
2.2. CBG induces autophagy by inhibition of EGFR and Akt/mTOR pathway in PDAC cell lines
EGFR and Akt/mTOR protein levels were evaluated by Western blot in PDAC cell lines at 48 h post-treatment with CBG 11.08 and 12.66 µg/mL. Immunoblots evidenced a decrease of EGFR expression in PANC-1 with CBG highest dose, while for MIAPaCa-2 the reduction was significant with both doses (Figure 2). Then, total Akt and its phosphorylation form were investigated. Data showed for PANC-1 cells a slight modulation of total Akt protein and a reduction of phospho-Akt levels after the administration of CBG. In MIAPaCa-2 cells, the highest dose of CBG induced a marked reduction of total Akt and also a significant decrease of its phosphorylation. Similar results were obtained for mTOR protein expression, that was reduced especially after the treatment with the highest dose of CBG (Figure 2).
Since CBG reduced the Akt/mTOR signalling pathway, also autophagy induction was evaluated. We observed the conversion of microtubule-associated protein 1A/1B-light chain 3 (LC3-I) to LC3-II form at 48 h post-treatment with CBG (11.08 and 12.66 µg/mL). CBG induced in both cell lines a significant increase of LC3-II lipidated form suggesting the activation of the early steps of autophagy; in particular MIAPaCa-2 cells showed the highest levels of LC3-II at the highest dose of CBG (Figure 3A).
To confirm the induction of autophagy, acridine orange dye (AO) analysis, which accumulates in acidic spaces and emits bright red fluorescence related to the degree of the acidity and the volume of acidic vesicular organelles (AVOs), was applied. AVOs are indicative of autophagy, therefore we evaluated AVOs under confocal fluorescence microscope. As shown (Figure 3B), an increase in the level of red fluorescent signals reflecting an AVO formation was observed in CBG-treated cell lines, 48 h post-treatment.
2.3. CBG reduces RAS downstream pathway in PDAC cell lines
To further investigate the CBG-induced effect in inhibiting EGFR pathway, the expression of total RAS (Pan-RAS) was assessed by Western blot analysis. As evidenced in Figure 4A, Pan-RAS levels were statistically reduced at 48 hr post-CBG treatment. Due to the relevance of the RAS/RAF/MEK pathway in PDAC, we measured the signalling proteins downstream of RAS in the lysate of PDAC cells treated with CBG at the most effective dose of 12.66 mg/ml. By MILLIPLEX® RAS-RAF Oncoprotein Magnetic Bead Panel 6-plex, we evaluated changes in phosphorylated BRAF (Ser446, pBRAF), CRAF (Ser338, pCRAF), and MEK1 (Ser217/Ser221, pMEK1), as well as the relative total protein levels (Figure 4B). The results confirmed that CBG treatment reduced total Pan-RAS levels in both cell lines. It also promotes the downregulation of BRAF, CRAF and MEK1 phosphorylation in PANC-1, and of BRAF in the MIAPaCa-2 cell line. Summarizing, these data further confirm that CBG was able to interfere with the EGFR-RAS pathways by reducing also downstream effectors often aberrantly activated in PDAC.
2.4. CBG induces apoptosis in PDAC cell lines
To further investigate whether reduction of cell viability and activation of autophagy was associated to cell death, PANC-1 and MIAPaCa-2 cells were treated with CBG 11.08 and 12.66 µg/mL and Annexin V-FITC staining analysis were performed 48 h post-treatment. Results showed that CBG increases Annexin V+ cells in a dose-dependent mode (Figure 5A), suggesting the induction of apoptotic cell death. The activation of ProCaspase-3 (ProCasp-3) and the presence of Caspase-3 (Casp-3) cleaved form was investigated by Western blot analysis. Blots showed an increase in Casp-3/ProCasp-3 ratio after 48 h post-treatment with CBG 12.66 µg/mL, in both cell lines (Figure 5B).
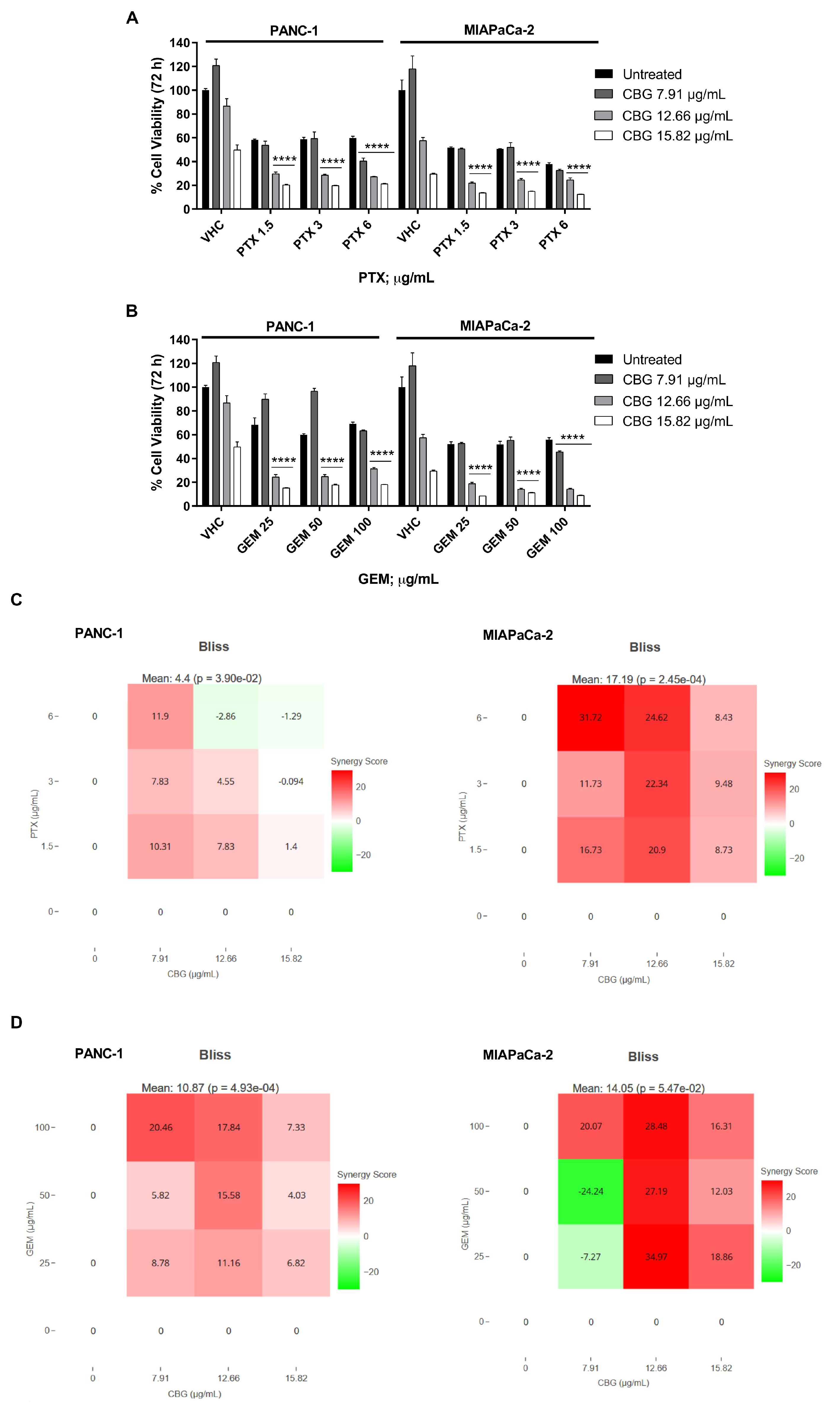
2.5. The combination of CBG with PTX or GEM induces higher cytotoxicity compared to administration of single drugs in PDAC cell lines
To support the potential use of CBG as integrative therapy in PDAC, we evaluated the combination effects of CBG with PTX and GEM, by MTT assay. Both cell lines were treated with CBG 7.91, 12.66 and 15.82 µg/mL in combination with three cytotoxic doses of PTX (1.5, 3 and 6 µg/mL) or GEM (25, 50 and 100 µg/mL) and cell viability was evaluated by MTT assay at 72 h post-treatment. Results showed that the combinations with the two higher doses of CBG resulted in greater cytotoxicity when compared to PTX or GEM alone, in both cell lines (Figure 6 A, B). Then drugs interaction was evaluated with SynergyFinder, using the Bliss model. We obtained a Bliss synergy score of 4.40 for PTX and CBG, and 10.87 for GEM and CBG, on PANC-1 cell line, indicating an additive and synergistic effect, respectively. In MIAPaCa-2 cell line, the Bliss synergy score was 17.19 for PTX and CBG and 14.05 for GEM and CBG combination, suggesting a synergistic effect (Figure 6 C, D).
3. Discussion
PDAC poor prognosis is mainly due to an aggressive behaviour associated with chemoresistance. The search of integrative therapies that can ameliorate chemotherapy side effects or improve their effect are constantly under study [18]. Cannabis sativa L. contains more than 100 phytocannabinoids and, for some of them, several biological properties are well known. Beside a direct anticancer effect, mainly demonstrated for CBD, in in vitro and in vivo experiments, phytocannabinoids were suggested to ameliorate numerous important side effects induced by chemotherapeutics [19]. In this study, CBG showed a dose-dependent cytotoxicity in PDAC cells, as also evidenced in other human preclinical cancer models as in glioblastoma multiforme (GBM) [13]. Further, THC, CBD and synthetic cannabinoids reduced PANC-1 and MIAPaCa-2 cell growth and viability, as reported in several studies and, in line with our evidence, MIAPaCa-2 was more sensitive than PANC-1 to treatments with phytocannabinoids [20,21]. In several pathologies, including cancer such as in glioma cells, cannabinoids have been demonstrated to activate autophagy and apoptotic cell death, through the interaction between apoptosis and autophagy signalling mechanisms [16,22]. Several pathways are mediated through the multiprotein complex involved in EGF/EGFR, including RAS and mTOR. These pathways suppress autophagy and promote proliferation and resistance to chemotherapy [23]. The simultaneous inhibition of EGFR and RAS/mTOR was demonstrated to provide a synergistic antitumor effect in various human cancers, indeed the PI3K/AKT/mTOR axis, a frequently dysregulated pathway in PDAC, is responsible for the control of cell proliferation and resistance [24], and these pathways can be inhibited by cannabinoids [16,25]. The present data evidenced as CBG induced autophagy by reducing the Akt-mTOR pathways with consequent LC-3 conversion and autophagic vesicle formation, as previously observed with others phytocannabinoids. For example, THC inhibits AKT/mTOR [22], reducing the proliferation of glioma cells. A similar finding was observed in carcinoma cells, where THC inhibits AKT/mTORC1 through ER stress-dependent activation of AMPK [26]. In our results, CBG reduced mTOR protein expression and, in line with this, some studies demonstrated that CBD inhibits mTOR signalling pathway in breast cancer and in human glioma [25,27]. Autophagy mechanism can be mediated by EGFR and Akt/mTOR signalling axis. We investigated the modulation of pro-autophagic markers and results showed that CBG increased LC3-II expression and in particular, for the more sensitive MIAPaCa-2, the increase has been very noticeable. Autophagy has a double function: to induce cancer resistance to chemotherapy and protect cancer cells from death, or to be correlated with cancer cell death. Herein, we evidenced that CBG induced autophagy and also apoptosis in both cell lines. Cannabinoids are also involved in reducing cancer cell growth, by the EGFR-RAS-RAF-MAPK pathway. In pancreatic cancer KRAS mutated upregulates EGFR endogenous expression and hyperactivation request in acinar to ductal metaplasia [28]. Herein, we showed that CBG suppresses EGFR expression in PANC-1 and MIAPaCa-2. Up to now, there are no data about the ability of CBG to reduce the EGFR expression; however, some studies demonstrated that CBD and THC can reduce EGFR expression in A549, H460 and H1792 cells and suppress EGF/EGFR signalling pathways in breast cancer [29]. Moreover, our data evidenced as CBG was able to reduce downstream RAS signalling, suggesting a specific role in decreasing the RAS oncogenic pathways in PDAC. In our studies, Annexin V positive cells and Caspase-3 cleavage confirmed CBG induction of apoptosis. In line with our results, CBG induced Caspase-3/-7 dependent apoptosis in glioblastoma and Caco-2 cells [13,14]. Moreover, in PANC-1 and MIAPaCa-2 cells also CBD induced apoptosis and Caspase-3 activation [20]. Lastly, many studies demonstrated that cannabinoids could increase the chemotherapeutic drugs efficacy, reducing tumour growth and overcoming drugs resistance [11]. Herein, the combination of CBG with GEM or PTX, increased the cytotoxicity compared to the administration of the drug alone, also showing a synergistic effect for some combinations. In GBM cells, CBD and CBG plus temozolomide did not show additive effect, but in cholangiocarcinoma cells, CBG synergized with GEM and cisplatin [13,30]. Moreover, in PDAC, CBD showed the ability to increase GEM and PTX efficacy in in vitro tests and KPC mice treated with CBD and GEM showed a survival three times longer than mice treated with GEM [20,21]. Overall, our data evidenced the ability of CBG to induce autophagy, reduce EGFR/AKT/RAS pathways, promote apoptotic cell death, and increase the sensitivity of PDAC cell lines to chemotherapeutic drugs.
4. Materials and Methods
4.1. Cell Lines
PANC-1 and MIAPaCa-2, human pancreatic ductal adenocarcinoma cell lines purchased from Sigma Aldrich (Milan, Italy), were cultured in DMEM high glucose medium (EuroClone, Milan, Italy) supplemented with 10% of fetal bovine serum (FBS), 2 mM L-glutamine, 100 IU/mL penicillin, 100 mg streptomycin and 1 mM sodium pyruvate. Epithelial cell line derived from the human normal colon (NCM460D), and human normal fibroblast (NHFA12) were used as normal cell for cytotoxicity assay. NCM460D was cultured in RPMI1640 supplemented with 10% of fetal bovine serum (FBS), 2 mM L-glutamine, 100 IU/mL penicillin, 100 mg streptomycin. NHFA12 was cultured as for PANC-1 cell line. Cell lines were maintained at 37 °C with 5% CO2 and 95% of humidity.
4.2. Reagents
Pure CBG (≥98% purity) was purchased (Cayman Chemical, cat. No. 36975). CBG was solubilised in ethanol 70% (Et-OH) at 50 mM (15.8 mg/mL) concentration. Paclitaxel (PTX, 6 mg/mL; cat. No. 33069-62-4) and Gemcitabine (GEM; 50 mg/mL; cat. No. 122111-03-9) supplied by (Sigma Aldrich) were dissolved in water. All the compounds were aliquoted, stored at -20°C and each aliquot was used one time.
4.3. MTT Assay
3 x 104 cells/ml were seeded in 96-wells plate. The following day, compounds or vehicles were added, and six replicates were used for each treatment. At 72 h post-treatment, cell viability was analysed as previously reported [16]. The absorbance of the sample was measured at 570 nm using an ELISA reader microliter plate (BioTek Instruments, Winooski,VT).
4.4. Annexin V Staining
Annexin V-FITC staining and cytofluorimetric analysis was applied for detection of apoptotic cells on PDAC cell lines. 3x104 cells/ml were treated with different doses of CBG for 48 h. After treatment, cells were stained with Annexin V-FITC (5 ml; Adipogen, No. AG-40B-0005F) and the percentage of positive cells analysed by FACScan flow cytometer using the CellQuest software.
4.5. Western Blot Analysis
PDAC cell lines were treated with CBG and harvested at 48 h post-treatments. Lysates were obtained with lysis buffer (TRIS 1M pH 7.4, NaCl 1M, EGTA 10 mM, NaF 100 mM, Deoxycholate 2%, EDTA 100 mM, TritonX-100 10%, Glycerol, SDS 10%, Na2P2O7 1M, Na3VO4 100 mM, PMSF 100 mM, Cocktail of enzyme inhibitors and H2O), separated on a SDS polyacrylamide gel, and Western blot were performed as previously reported (Marinelli et al., 2020). The following antibodies were used: mouse anti-mTOR (1:1000, GeneTex), mouse anti-EGFR (1:5000, GeneTex), rabbit anti-pAkt (1:1000, Cell Signaling), rabbit anti-Akt (1:1000, Cell Signaling), rabbit anti-LC3 (1:1000, Cell Signaling), rabbit anti-Caspase-3 (1:1000, Cell Signaling), mouse pan-RAS (1:500, Santa Cruz Biotechnology), mouse β-actin (1:500, Santa Cruz Biotechnology) and mouse anti-glyceraldehydes-3-phosphate dehydrogenase (GAPDH, 1:1000, Santa Cruz Biotechnology). Abs were incubated overnight or 1 h according to manufacturer’s protocol and then incubated with their respective HRP-cojugated anti-rabbit or anti-mouse (1:2000, Cell Signaling, Danvers, MA, USA) Abs for 1 h. Peroxidase activity was visualized with the LiteAblot®PLUS or TURBO (EuroClone, Milan, Italy) kit and densitometric analysis was carried out by a Chemidoc using the Quantity One software version 4.6 (Bio-Rad, Milan, Italy).
4.6. Acridine Orange Staining
To detect acidic vesicular organelles, the vital staining of cells with acridine orange (AO, Sigma-Aldrich) was performed. 3x104 cells/mL were seeded in 12-well plates and treated with CBG at 11.08 and 12.66 µg/mL for 48 h. Then, cells were stained with 1 μg/mL AO, washed in PBS, immobilized on slides using the cytospin centrifuge and analysed with C2 Plus confocal laser scanning microscope (Nikon Instruments, Firenze, Italy). Optimized emission detection bandwidth was configured by Zeiss Zen control software. Images were processed using NIS Element Imaging Software (Nikon Instruments, Firenze, Italy). Cytoplasm and nuclei of AO-stained cells fluoresced bright green, whereas the acidic autophagic vacuoles fluoresced bright red.
4.7. Milliplex multiplex assay
The levels of Total RAS, pBRAF, pCRAF, pMEK1 in treated cells were measured using RAS-RAF Oncoprotein Panel 6-Plex Magnetic Bead Kit 96-well Plate (EMD Millipore Corporation, Billerica, MA, USA) following the manufacturer's protocol. Data were analysed using Luminex® MAGPIX® instrument with xPONENT® software (Luminex Corporation, Austin, TX, USA).
4.8. Statistical Analysis
The data presented represent the mean with standard deviation (SD) of at least 3 independent experiments. The statistical significance was determined by Two Way-Anova followed by Dunnett's or Sidak’s multicomparison test, using GraphPad software.
4.9. Drug interaction
Drugs interaction was evaluated with SynergyFinder, using the Bliss model [17]. Bliss independence reference model was used for multiplicative effect of single drugs as if they acted independently. Bliss synergy score larger than 10 is considered synergistic, from -10 to 10 is considered additive and less than -10 antagonistic.
5. Conclusions
In conclusion, our results showed that CBG, a non-psychomimetic cannabinoid from Cannabis Sativa L., can induce an anticancer effect on two human PDAC cell lines, supporting the ability of cannabinoids in interfering with several pro-tumoral pathways. Further study in pre-clinical in vivo models will be performed to better understand CBG effects on PDAC progression.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: CBG effect on cell viability in normal human NCM460D and NHFA12 cell lines.
Author Contributions
Conceptualization, M.N.; Methodology, M.B.M.; Software, L.Z., C.A.; Validation, M.B.M., O.M., M.L. and M.N.; Formal Analysis, M.B.M. and M.N.; Investigation, L.Z., C.A., O.M. and M.G.; Resources, M.N.; Data Curation, L.Z., C.A. and M.G.; Writing – Original Draft Preparation, L.Z. and C.A.; Writing – Review & Editing, O.M., M.L., C.Am., G.S. and M.N.; Visualization, L.Z, C.A. and M.B.M.; Supervision, M.B.M.; Project Administration, M.N.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
This study doesn’t involve human samples.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We are very grateful to the School of Pharmacy of the University of Camerino, for the contribution for the purchase of the Milliplex instrument.
Conflicts of Interest
“The authors declare no conflicts of interest.
References
- Siegel, R. L.; Miller, K. D.; Jemal, A. Cancer statistics. CA Cancer J Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. IJMS. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Collins, M. A.; Bednar, F.; Zhang, Y.; Brisset, J. C.; Galbán, S.; Galbán, C. J.; Rakshit, S.; Flannagan, K. S.; Adsay, N. V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D. R.; La Stella, P. J.; Ciuleanu, T. E.; Pover, G.; Tebbutt, N. C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012, 30, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Cox, A. D.; Fesik, S. W.; Kimmelman, A. C.; Luo, J.; Der, C. J. Drugging the undruggable RAS: Mission possible? Nature reviews. Drug discovery. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Soares, H. P.; Ming, M.; Mellon, M.; Young, S. H.; Han, L.; Sinnet-Smith, J.; Rozengurt, E. Dual PI3K/mTOR Inhibitors Induce Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic Cancer Cells through Suppression of mTORC2. Mol Cancer Ther. 2015, 14, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, E.; Koivunen, J. P. MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol. 2015, 7, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A. A.; Philip, P. A. Adjuvant treatment of surgically resectable pancreatic ductal adenocarcinoma. Clin Adv Hematol Oncol. 2019, 17, 54–63. [Google Scholar]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S. C. GWCA1208 study group. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br J Cancer. 2021, 124, 1379–1387. [Google Scholar] [CrossRef]
- Antonini, M.; Aguzzi, C.; Fanelli, A.; Frassineti, A.; Zeppa, L.; Morelli, M.; Pastore, G.; Nabissi, M.; Luongo, M. The Effects of a Combination of Medical Cannabis, Melatonin, and Oxygen–Ozone Therapy on Glioblastoma Multiforme: A Case Report. Reports. 2023, 6, 22. [Google Scholar] [CrossRef]
- Yan, C.; Li, Y.; Liu, H.; Chen, D.; Wu, J. Antitumor mechanism of cannabidiol hidden behind cancer hallmarks. Biochim Biophys Acta Rev Cancer. 2023, 1878, 188905. [Google Scholar] [CrossRef] [PubMed]
- Łuczaj, W.; Dobrzyńska, I.; Skrzydlewska, E. Differences in the phospholipid profile of melanocytes and melanoma cells irradiated with UVA and treated with cannabigerol and cannabidiol. Sci Rep. 2023, 13, 16121. [Google Scholar] [CrossRef]
- Lah, T. T.; Novak, M.; Pena Almidon, M. A.; Marinelli, O.; Žvar Baškovič, B.; Majc, B.; Mlinar, M.; Bošnjak, R.; Breznik, B.; Zomer, R.; Nabissi, M. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells. 2021, 10, 340. [Google Scholar] [CrossRef]
- Lamtha, T.; Tabtimmai, L.; Songtawee, N.; Tansakul, N.; Choowongkomon, K. Structural analysis of cannabinoids against EGFR-TK leads a novel target against EGFR-driven cell lines. Curr Res Pharmacol Drug Discov. 2022, 3, 100132. [Google Scholar] [CrossRef]
- Kaushik, G.; Seshacharyulu, P.; Rauth, S.; Nallasamy, P.; Rachagani, S.; Nimmakayala, R. K.; Vengoji, R.; Mallya, K.; Chirravuri-Venkata, R.; Singh, A. B.; et al. Selective inhibition of stemness through EGFR/FOXA2/SOX9 axis reduces pancreatic cancer metastasis. Oncogene. 2021, 40, 848–862. [Google Scholar] [CrossRef]
- Marinelli, O.; Morelli, M. B.; Annibali, D.; Aguzzi, C.; Zeppa, L.; Tuyaerts, S.; Amantini, C.; Amant, F.; Ferretti, B.; Maggi, F.; et al. The Effects of Cannabidiol and Prognostic Role of TRPV2 in Human Endometrial Cancer. IJMS. 2020, 21, 5409. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A. K.; Aittokallio, T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020, 48(W1), W488–W493. [Google Scholar] [CrossRef]
- Frenkel, M.; David, A.; Sapire, K.; Hausner, D. Complementary and Integrative Medicine in Pancreatic Cancer. Curr Oncol Rep. 2023, 25, 231–242. [Google Scholar] [CrossRef]
- Afrin, F.; Chi, M.; Eamens, A. L.; Duchatel, R. J.; Douglas, A. M.; Schneider, J.; Gedye, C.; Woldu, A. S.; Dun, M. D. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers. 2020, 12, 1033. [Google Scholar] [CrossRef]
- Luongo, M.; Marinelli, O.; Zeppa, L.; Aguzzi, C.; Morelli, M. B.; Amantini, C.; Frassineti, A.; di Costanzo, M.; Fanelli, A.; Santoni, G.; Nabissi, M. Cannabidiol and Oxygen-Ozone Combination Induce Cytotoxicity in Human Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers. 2020, 12, 2774. [Google Scholar] [CrossRef] [PubMed]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C. E.; Arifin, S. A.; Fyffe, C. A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene. 2018, 37, 6368–6382. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I. J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, P. EGFR-mediated autophagy in tumourigenesis and therapeutic resistance. Cancer letters. 2020, 469, 207–216. [Google Scholar] [CrossRef]
- Stanciu, S.; Ionita-Radu, F.; Stefani, C.; Miricescu, D.; Stanescu-Spinu, I. I.; Greabu, M.; Ripszky Totan, A.; Jinga, M. Targeting PI3K/AKT/mTOR Signaling Pathway in Pancreatic Cancer: From Molecular to Clinical Aspects. IJMS. 2022, 23, 10132. [Google Scholar] [CrossRef]
- Huang, T.; Xu, T.; Wang, Y.; Zhou, Y.; Yu, D.; Wang, Z.; He, L.; Chen, Z.; Zhang, Y.; Davidson, D.; et al. Cannabidiol inhibits human glioma by induction of lethal mitophagy through activating TRPV4. Autophagy. 2021, 17, 3592–3606. [Google Scholar] [CrossRef]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzmán, M.; Velasco, G.; Díaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P. M.; Groopman, J. E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Blasco, M. T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C. G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer cell. 2019, 35, 573–587.e6. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E. 3rd; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol Oncol. 2015, 9, 906–19. [Google Scholar] [CrossRef]
- Viereckl, M. J.; Krutsinger, K.; Apawu, A.; Gu, J.; Cardona, B.; Barratt, D.; Han, Y. Cannabidiol and Cannabigerol Inhibit Cholangiocarcinoma Growth In Vitro via Divergent Cell Death Pathways. Biomolecules. 2022, 12, 854. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
CBG reduces cell viability in PDAC cell lines. Cell viability was determined by MTT assay. PANC-1 and MIAPaCa-2 cells were treated with vehicle (Vhc; Et-OH 70%) or different concentrations of CBG and cell viability was evaluated at 72 h post-treatment. Data shown are the mean ± SD of three separate experiments. *p<0.05, ****p<0.0001 vs Vhc.
Figure 1.
CBG reduces cell viability in PDAC cell lines. Cell viability was determined by MTT assay. PANC-1 and MIAPaCa-2 cells were treated with vehicle (Vhc; Et-OH 70%) or different concentrations of CBG and cell viability was evaluated at 72 h post-treatment. Data shown are the mean ± SD of three separate experiments. *p<0.05, ****p<0.0001 vs Vhc.
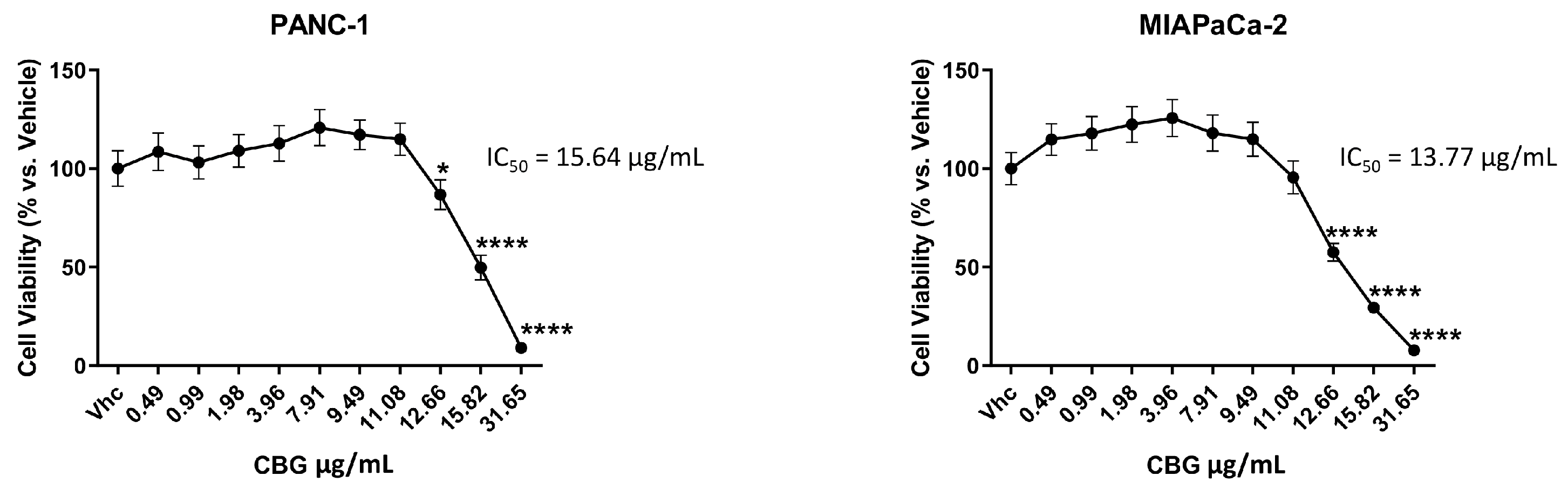
Figure 2.
CBG treatment reduces EGFR and Akt/mTOR protein expression in PDAC cell lines. PDAC cell lines were treated with CBG and the expression levels of EGFR, Akt, pAkt and mTOR was evaluated by Western blot, at 48 h post-treatment. EGFR, Akt, and mTOR densitometric values were normalized to β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used as loading control; pAkt densitometric values were normalized to Akt. Images are representative of one of three separate experiments. Data are expressed as mean ± SD of three separate experiments. *p<0.05, **p<0.01, ****p<0.0001 treated cells vs VHC.
Figure 2.
CBG treatment reduces EGFR and Akt/mTOR protein expression in PDAC cell lines. PDAC cell lines were treated with CBG and the expression levels of EGFR, Akt, pAkt and mTOR was evaluated by Western blot, at 48 h post-treatment. EGFR, Akt, and mTOR densitometric values were normalized to β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used as loading control; pAkt densitometric values were normalized to Akt. Images are representative of one of three separate experiments. Data are expressed as mean ± SD of three separate experiments. *p<0.05, **p<0.01, ****p<0.0001 treated cells vs VHC.
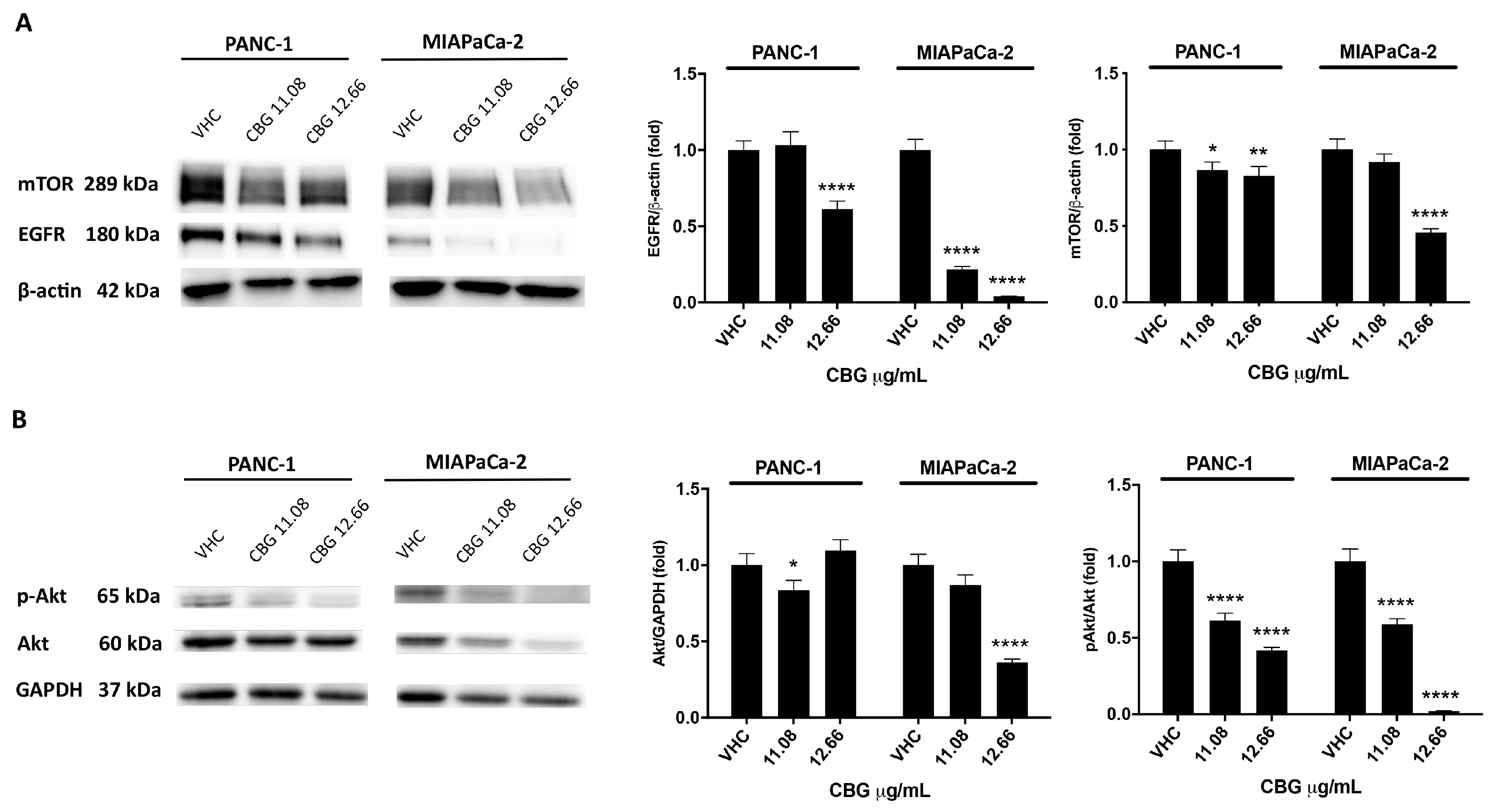
Figure 3.
CBG stimulates autophagy in PDAC cell lines. PDAC cell lines were treated with CBG for 48 h. A) The expression of LC3-I and LC3-II was assessed by Western blot. Densitometric values of LC3-II were normalized to GAPDH used as loading control. Densitometric values of LC3-II were also normalized to LC3-I. Images are representative of one of three separate experiments and data are expressed as the mean ± SD of three separate experiments. **p<0.01, ****p<0.0001 CBG- vs VHC-treated cells. B) Fluorescent microscope was used to visualize AVOs (red fluorescence) as well as the cytoplasm and nucleus (green fluorescence) after the vital staining of the cells with AO. Representative images of PANC-1 and MIAPaCa-2 cells stained with AO. Rapamycin was used as positive control of autophagic induction.
Figure 3.
CBG stimulates autophagy in PDAC cell lines. PDAC cell lines were treated with CBG for 48 h. A) The expression of LC3-I and LC3-II was assessed by Western blot. Densitometric values of LC3-II were normalized to GAPDH used as loading control. Densitometric values of LC3-II were also normalized to LC3-I. Images are representative of one of three separate experiments and data are expressed as the mean ± SD of three separate experiments. **p<0.01, ****p<0.0001 CBG- vs VHC-treated cells. B) Fluorescent microscope was used to visualize AVOs (red fluorescence) as well as the cytoplasm and nucleus (green fluorescence) after the vital staining of the cells with AO. Representative images of PANC-1 and MIAPaCa-2 cells stained with AO. Rapamycin was used as positive control of autophagic induction.
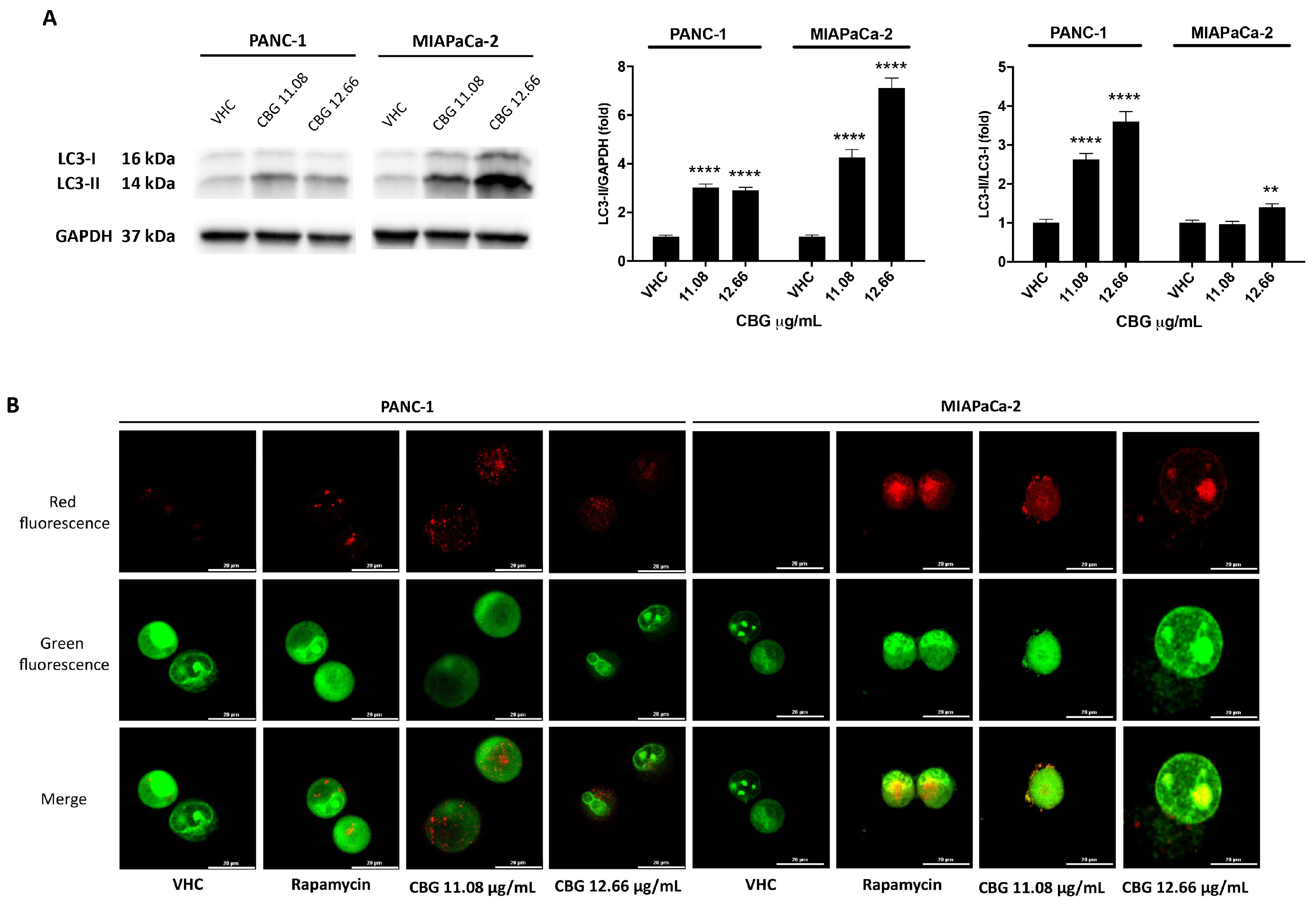
Figure 4.
CBG reduces the total Pan RAS expression and the phosphorylated forms of BRAF, CRAF and MEK1 in PDAC cell lines. A) PDAC cell lines were treated with CBG and the expression of pan RAS was determined with Western blot at 48 h post-treatment. The densitometric values were normalized to GAPDH used as loading control. Images are representative of one of three separate experiments and data are expressed as the mean ± SD of three separate experiments. **p<0.01, ***p<0.001, ****p<0.0001 treated cells vs VHC. B) PDAC cell lines were treated with CBG 12.66 µg/mL for 48 h and the expression of RAS, pBRAF, pCRAF, pMEK1 was evaluated by Milliplex multiplex assay and the Median Fluorescence Intensity (MFI) was measured with the Luminex ® system. Data shown are expressed as the mean ± SD of three separate experiments. **p <0.01, ***p<0.001, ****p<0.0001 treated cells vs VHC.
Figure 4.
CBG reduces the total Pan RAS expression and the phosphorylated forms of BRAF, CRAF and MEK1 in PDAC cell lines. A) PDAC cell lines were treated with CBG and the expression of pan RAS was determined with Western blot at 48 h post-treatment. The densitometric values were normalized to GAPDH used as loading control. Images are representative of one of three separate experiments and data are expressed as the mean ± SD of three separate experiments. **p<0.01, ***p<0.001, ****p<0.0001 treated cells vs VHC. B) PDAC cell lines were treated with CBG 12.66 µg/mL for 48 h and the expression of RAS, pBRAF, pCRAF, pMEK1 was evaluated by Milliplex multiplex assay and the Median Fluorescence Intensity (MFI) was measured with the Luminex ® system. Data shown are expressed as the mean ± SD of three separate experiments. **p <0.01, ***p<0.001, ****p<0.0001 treated cells vs VHC.
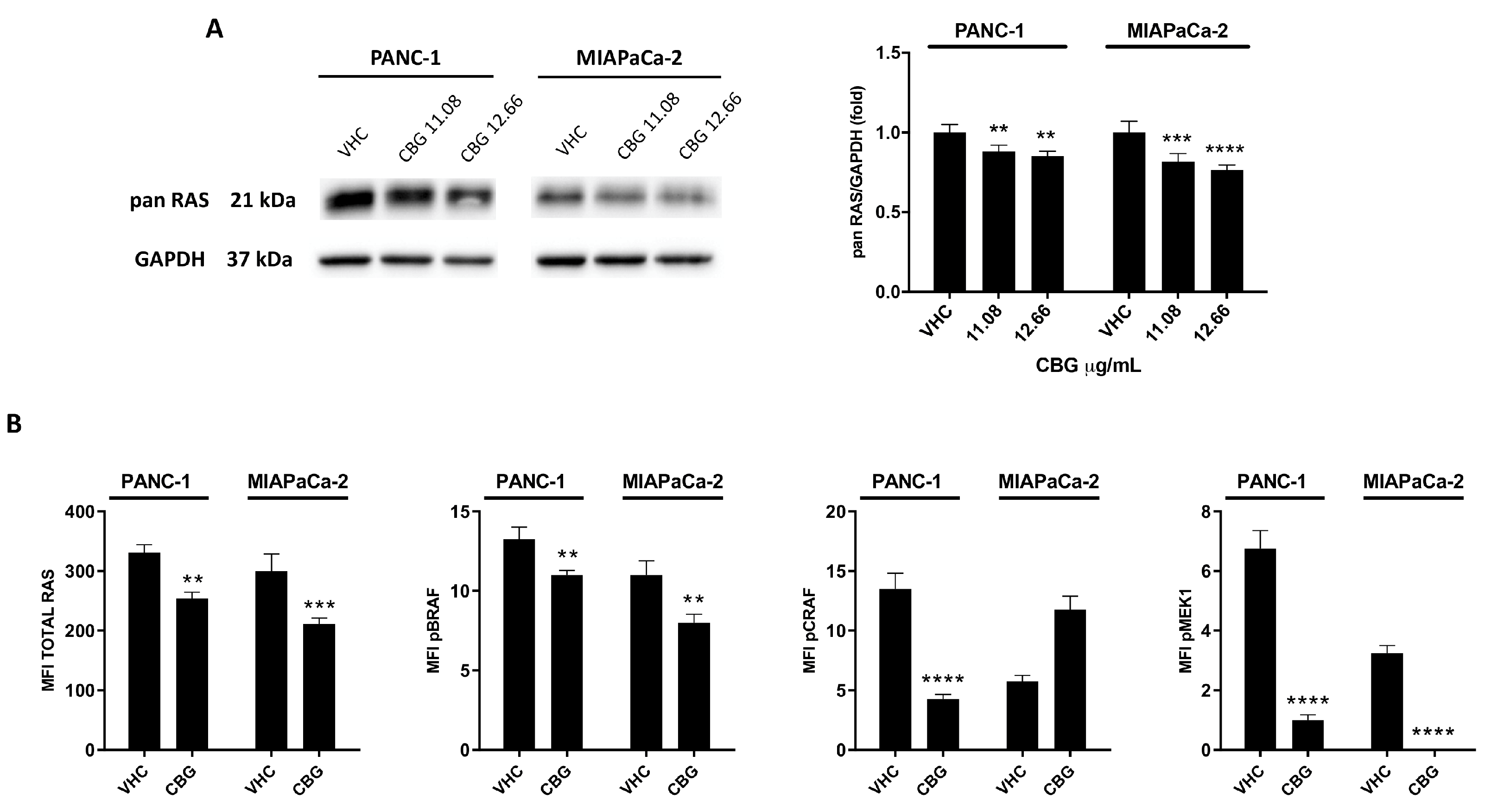
Figure 5.
CBG induced apoptotic cell death in PDAC cell lines. PDAC cell lines were treated with CBG for 48 h. A) Flow cytometric analysis was performed after Annexin V staining. Images are representative of one of three separate experiments and histograms are the mean ± SD of three separate experiments. The percentage refers to Annexin V+ cells of CBG- respect to VHC-treated cells. B) The expression of Caspase-3 was determined by Western blot. Caspase-3 densitometric values were normalized to proCaspase-3. Image is representative of one of three separate experiments and bars represent the mean ± SD of three separate experiments. ****p<0.0001 treated cells vs VHC.
Figure 5.
CBG induced apoptotic cell death in PDAC cell lines. PDAC cell lines were treated with CBG for 48 h. A) Flow cytometric analysis was performed after Annexin V staining. Images are representative of one of three separate experiments and histograms are the mean ± SD of three separate experiments. The percentage refers to Annexin V+ cells of CBG- respect to VHC-treated cells. B) The expression of Caspase-3 was determined by Western blot. Caspase-3 densitometric values were normalized to proCaspase-3. Image is representative of one of three separate experiments and bars represent the mean ± SD of three separate experiments. ****p<0.0001 treated cells vs VHC.
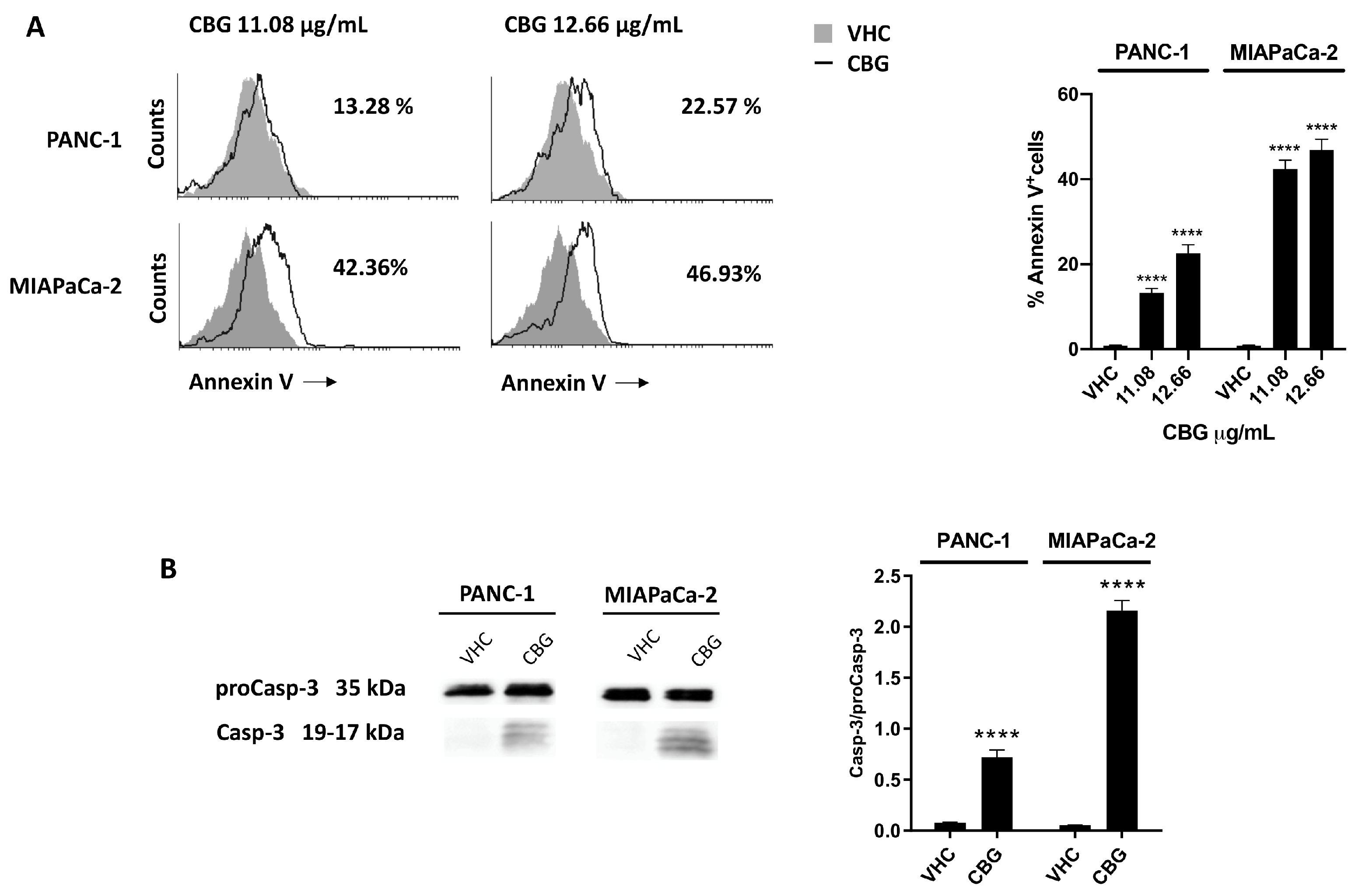
Figure 6.
CBG increased the cytotoxic effect of chemotherapeutic drugs in PDAC cell lines. A, B) Cell viability was determined in PDAC cell lines by MTT assay. Cells were treated for 72 h with CBG alone or in combination with different doses of GEM and PTX. Data shown are expressed as mean ± SD of three separate experiments. ****p < 0.0001 CBG combined to chemotherapeutic drug vs chemotherapeutic drug alone. C, D) Drug interaction of CBG with PTX and GEM in PDAC cell lines. Effect of single and combined treatments with CBG (7.91, 12.66, 15.82 µg/mL), PTX (1.5, 3, 6 µg/mL), GEM (25, 50, 100 µg/mL) on cell viability of PDAC cell lines. Drugs interaction was evaluated with SynergyFinder software, using the Bliss model. Bliss synergy score larger than 10 is considered synergistic, from -10 to 10 is considered additive and less than -10 antagonistic.
Figure 6.
CBG increased the cytotoxic effect of chemotherapeutic drugs in PDAC cell lines. A, B) Cell viability was determined in PDAC cell lines by MTT assay. Cells were treated for 72 h with CBG alone or in combination with different doses of GEM and PTX. Data shown are expressed as mean ± SD of three separate experiments. ****p < 0.0001 CBG combined to chemotherapeutic drug vs chemotherapeutic drug alone. C, D) Drug interaction of CBG with PTX and GEM in PDAC cell lines. Effect of single and combined treatments with CBG (7.91, 12.66, 15.82 µg/mL), PTX (1.5, 3, 6 µg/mL), GEM (25, 50, 100 µg/mL) on cell viability of PDAC cell lines. Drugs interaction was evaluated with SynergyFinder software, using the Bliss model. Bliss synergy score larger than 10 is considered synergistic, from -10 to 10 is considered additive and less than -10 antagonistic.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



