
Submitted:
17 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Tumor-associated macrophages (TAMs) are the major component of the tumor microenvironment (TME), where they sustain tumour progression and anti-tumor immunity. Due to their plasticity, macrophages can exhibit anti- or pro-tumor functions through the expression of different gene sets leading to distinct macrophages phenotypes: M1-like or pro-inflammatory and M2-like or anti-inflammatory. NF-κB transcription factors are central regulators of TAMs in cancers, where they often drive macrophage polarization towards M2-like phenotype. Therefore, the NF-κB pathway is an attractive therapeutic target for cancer immunotherapy in a wide range of human cancers. Hence, targeting NF-κB pathways in the myeloid compartment is a potential clinically strategy to overcome microenvironment-induced immunosuppression and increase anti-tumor immunity. In this review, we discuss the role of NF-κB as key driver of macrophage functions in tumours as well as the principal strategies to overcome tumour immunosuppression by targeting NF-κB pathway.
Keywords:
NF-κB
; Tumor-associated macrophages (TAMs)
; Tumor microenvironment (TME)
1. Introduction
Macrophages represent one of the major lines of host defense in the innate immune system, able to kill pathogens and induce inflammatory [1]. They control tissue repair and homeostasis via the extracellular matrix remodeling and scavenging of cellular debris and apoptotic cells [2]. Macrophages are highly versatile and can exert several different functions by changing their transcriptional profile based on the anatomic location and physiologic or pathophysiologic [1,3].
In the tumor microenvironment (TME), the complex interactions between tumour cells, immune cells, endothelial cells, fibroblasts and extracellular matrix shape tumour clinical behavior through the release and uptake of a number of angiogenic, mitogenic, immunosuppressive or pro-migratory factors that may either inhibit or stimulate tumour progression [4]. Chemoattractants produced by malignant cells and the stromal tumour compartment, such as C-C Motif Chemokine Ligand 2 (CCL2), vascular endothelial growth factor (VEGF), CXCL12 (SDF1) and Colony-stimulating factor-1 (CSF-1), recruit monocytes from the bloodstream that migrate into tumour site [5,6]. Different studies showed that the tissue-resident macrophages (MTR) (e.g., microglia, Kupffer cells, Alveolar macrophages) are responsible for regulating tissues homeostasis and inflammation, and during tumorigenesis they developed progressively into pro-tumoral phenotype within the TME in presence of various molecules such as interleukin-10 (IL-10) [7,8,9]. Macrophages recruited in tumour site, called tumor-associated macrophages (TAMs), are the major population of leukocytes in the TME, and display a high phenotype plasticity [10]. TAMs acquire a distinct phenotype and activation status and can exert anti- or pro-tumor activities through the expression of different functional programs [11].
There is a common agreement that macrophages can predominantly polarize into two different subtypes, the so-called M1-like and M2-like macrophages according to different stimuli. The M1 macrophages are “classically” activated by microbial products or soluble cytokines (e.g., lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and interferon-gamma (IFN-γ) produced by activated CD4+ T helper (Th) 1 cells, CD8+ T cytotoxic cells, and Natural Killer (NK) cells, and show specific surface markers such as toll-like receptor(TLR)-2 (TLR-2), TLR-4, CD80, CD86, inducible nitric oxide synthase (iNOS), and major histocompatibility complex-II (MHC-II) [12]. M1 macrophages secrete nitric oxide (NO), reactive oxygen species (ROS) and various cytokines and chemokines (e.g., TNF-α, interleukin(IL)-1α (IL-1α), IL-1β, IL-6, IL-12, C-X-C motif chemokine ligand(CXCL) 9 (CXCL9), and CXCL10) which trigger the activity of NK and cytotoxic T cells [13]. In addition, these secreted factors activate unpolarized macrophages promoting the M1 state, in a positive feedback [14].
The M2 macrophages are “alternatively” activated by anti-inflammatory molecules such as glucocorticoid hormones and Th2 cytokines (interleukin (IL)-4 (IL–4), IL-10, and IL-13), as well as apoptotic cells and immune complexes [15]. As for M1, specific surface markers such as CD206, CD163, CD209, mannitol receptor, Ym1/2 and FIZZ1 characterize M2 macrophages. They also express anti-inflammatory cytokines such as IL-10, transforming growth factor-beta (TGF-β), and chemokines (e.g., Chemokine (C-C motif) Ligand(CCL) 1 (CCL1), CCL17, CCL22, CCL24) that contribute to dampen the inflammatory response and maintain macrophages into M2 phenotype by acting [16,17].
Given the high plasticity of macrophages, M2 phenotype has been subdivided into M2a, M2b, M2c and M2d subtypes, that differ both for stimuli and exerted functions. M2a, activated by IL-4 and IL-13, express higher levels of IL-10, TGF-β, CCL17 and CCL22 and promote cell growth, tissue repair and endocytic activity. Immune complexes, TLR ligands and Interleukin-1 receptor (IL-1R) agonists activate M2b macrophages that regulate the intensity of inflammatory response and immune reaction via releasing TNF-α, IL-1β, IL-6 and IL-10. M2c macrophages, induced by IL-10, suppress immune responses and are responsible for tissue remodeling. Finally, M2d macrophages release IL-10 and VEGF and thus promote tumour progression and angiogenesis [10,17,18,19,20].
Recently the transcriptomic and proteomic analysis of TAMs has identified different macrophage subpopulations in the TME, that goes beyond the simple dichotomy “M1-M2” system, highlighting the presence of a more complex population of macrophages, highly plastic and heterogeneous [13,14]. Hence, several evidence indicated that microRNAs (miRNAs) [21], non-coding RNAs [22], extracellular vescicles (EVs) [23,24] and epigenetic modification [25] contribute to shape TAM phenotype in the TME, suggesting that the M1/M2 model has numerous limitations [13]. Although the current classification of macrophages is challenging as several heterogeneous subsets have been identified within the TME, here we use the common M1 and M2 classification to describe the role of NF-κB in TAMs polarization.
TAMs respond to the local signals provided by TME depending on tumour type and stage [26,27,28]. Although in the TME M2 macrophages are the most abundant population, M1 macrophages can be present in the TME during pathological conditions [29,30].
The balance between M1/M2 phenotypes and the switch between these two extreme borders of macrophage polarization is finely regulated by an intricate network of receptors and signaling pathways (e.g., JAK/STATs, MAPK, PI3K/AKT, NOTCH and NF-κB [31,32].
Among these crucial signaling, NF-κB is a central regulator of macrophage function in cancers, tipping the balance between the immunosuppressive, pro-tumoral activity and the pro-inflammatory, protective functions of TAMs [15]. Given the central role during tumorigenesis, TAMs represent a potential target for cancer treatment. NF-κB family of transcription factors plays a critical role in most physiological and pathological processes such as cell proliferation, survival, apoptosis, [33] inflammation [34], immune response [35] tumour progression, invasion, metastasis and angiogenesis [36]. NF-κB is also responsible for the activation and differentiation of innate immune cells and T cells [37] and the regulation of macrophage gene expression pattern [15].
The NF-κB family is composed of five structurally related DNA-binding subunits, consisting of the homo- and heterodimers of p50, p52, c-Rel, RelA (p65), RelB and c-Rel, which mediate transcription of target genes by binding specific DNA elements, called κB enhancers [38]. In resting cells, the NF-κB complexes are retained into the cytoplasm, bound to the inhibitory proteins of IκB family [39]. A wide range of stimuli, including microbial and viral infections products, stress, pro-inflammatory cytokines, and antigen receptors can trigger NF-κB activation [40] leading to the phosphorylation of IκBs by the IκB kinase (IKK) complex, which in turn, triggers the polyubiquitination and proteolysis of the IκB inhibitors, leading to the translocation of the NF-κB complexes in the nucleus, where they drive the transcription of several target genes [41]. Based on stimuli, NF-κB activation involves different signaling pathways including the canonical, the non-canonical and the atypical pathways [42].
The canonical pathway is activated by microbial products, IL-1β, damage-associated molecular patterns (DAMPs), pattern-recognition receptors (PRRs), T-cell receptor (TCR) and B-cell receptor (BCR) [43,44] and leads to the phosphorylation of the IκB-family members (IκBα, IκBβ and IκBɛ) by the IκB kinase (IKK), complex, which is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also named NF-κB essential modulator, or NEMO). The IκB kinase IKKβ phosphorylates the IκB inhibitor molecules via an IKKγ/NEMO mechanism leading to their proteasomal degradation, via ubiquitin-proteasome system. The active NF-κB dimers, predominantly the heterodimer p50/p65, translocate to the nucleus and induce the expression of several target genes [38,45].
The non-canonical pathway responds to different stimuli, such as LTβR, BAFFR, CD40 (belong to a TNFR superfamily members) and RANK, and actives a different response signaling. The non-canonical pathway relies on NF-κB-inducing kinase (NIK) for the phosphorylation of IKKα and the subsequent processing of the NF-κB2 precursor protein (p100) that lead to the release of mature NF-κB2/p52-RelB heterodimers. The NF-κB2/p52-RelB complex translocates to the nucleus and regulates the transcription of non-canonical NF-κB target genes [46].
The atypical NF-κB activation pathway is triggered by factors involved in aging process, such as endoplasmic stress response, oxidative stress, mitochondrial dysfunction, and DNA damage. In the atypical pathway, NF-κB induces the transcription of pro-survival genes and activates genes responsible of ROS scavenging and inhibition of calcium release from the ER, to protect organelles from stress [47].
In this review, we will discuss the role of NF-κB in TAMs and how targeting NF-κB pathway could be a promising strategy to overcome tumour immunosuppression.
2. The NF-κB Pathway in TAMs
NF-κB plays a key role in TAM polarization during tumorigenesis (Figure 1) [15,36]. Accordingly, NF-κB, in response to activating stimuli such as TLR ligands, IL-1β, TNF-α, can directly regulate the transition of macrophages toward M1 phenotype, usually exerting a tumour suppressor function, while, in different contexts, NF-κB activation can induce the transcription of many genes responsible for M2 polarization, thus promoting tumour growth [14,48,49,50].
Weigert and collaborators demonstrated that sphingosine-1-phosphate (S1P) produced by apoptotic tumour cells suppresses TNF-a production and increases interleukin-8 (IL-8) and IL-10 levels thus promoting macrophage polarization toward an alternative activated (M2) phenotype in vitro. They showed that in response to LPS, S1P and apoptotic cancer cells inhibit the activation of NF-κB in macrophages. Accordingly, the reduced levels of S1P after genetic inhibition of Sphingosine kinase 2 (Sphk2), restored M1 macrophages in vitro [51,52,53]. Recently, Shan and colleagues demonstrated that mechanical stretch (MS) (e.g., Flexcell Tension system) promotes M1 macrophage phenotype in a NF-κB-dependent manner thus increasing tumouricidal effects in vitro and reducing tumour growth in vivo. The authors demonstrated that macrophages stretched with Flexcell Tension system increase the levels of M1-related genes such as iNOS, TNF-α, IL-1β, IL-6, as the well as the release of M1 cytokines. The MS induces the up-regulation of Focal Adhesion Kinase (FAK) that, in turn, activates NF-κB signaling, thus promoting the transcription of genes responsible of the M1 phenotype activity. Accordingly, NF-κB inhibition reduces the expression of M1 target genes in vitro [54]. In vivo study demonstrated that intratumoral injection of macrophages treated with MS enhances M1 macrophage polarization within the TME and increases apoptosis of cancer cells thus reducing melanoma growth [55].
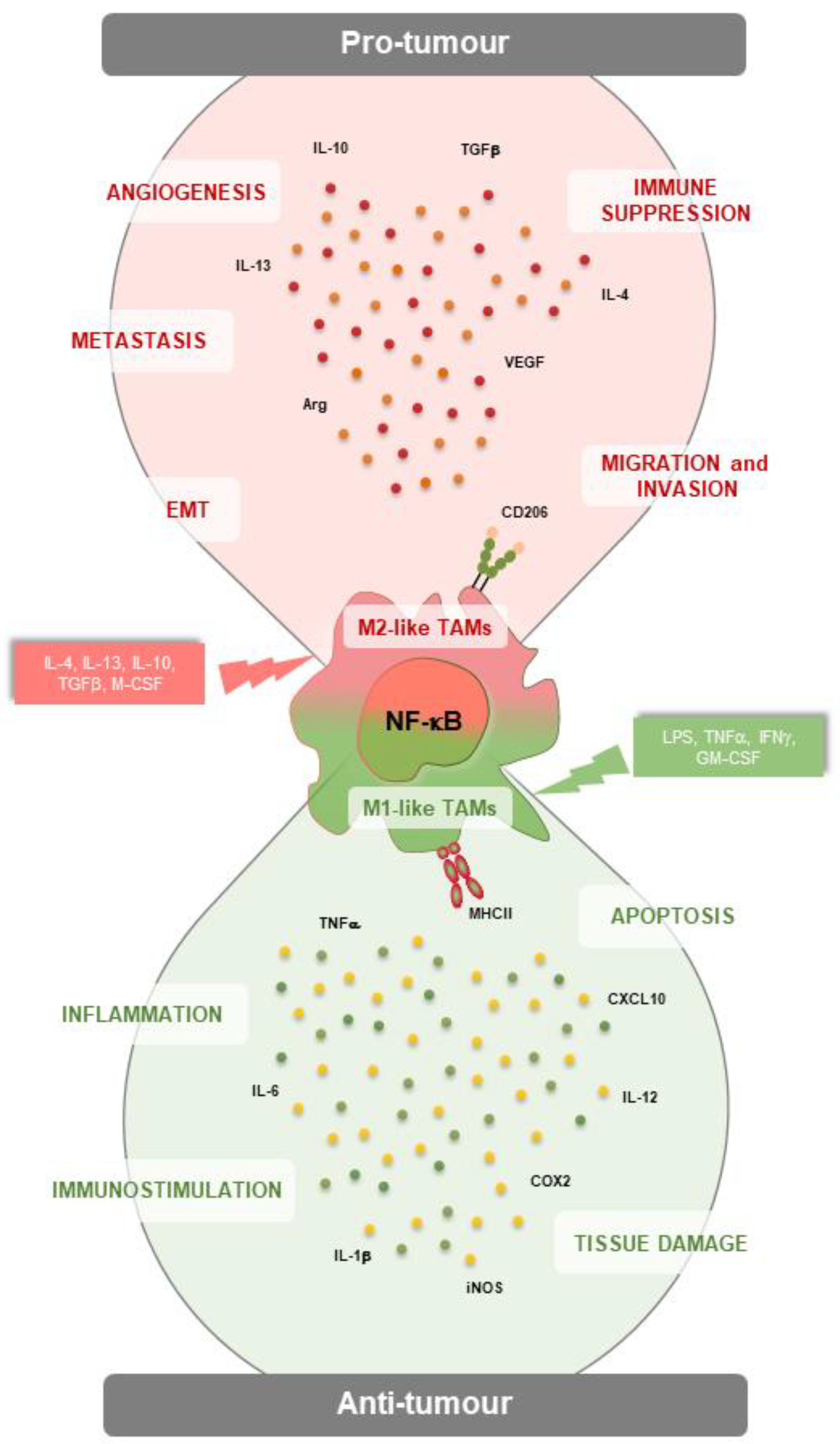
Figure 1.
NF-κB signalling in M1- and M2-like tumor-associated macrophages (TAMs) in the tumor microenvironment (TME). NF-κB activation can polarize myeloid cells towards M1-like macrophages, which counteract tumorigenesis by promoting inflammation, immunostimulation, tissue damage and apoptosis of cancer cells by secreting several molecules such as TNF-a, IL-12, iNOS, COX2 and IL-6. By contrast, NF-κB activation can shift macrophages towards M2-like anti-inflammatory TAMs, which promote tumour growth, angiogenesis, metastasis, EMT, as well as the establishment of an immunosuppressive TME.
Figure 1.
NF-κB signalling in M1- and M2-like tumor-associated macrophages (TAMs) in the tumor microenvironment (TME). NF-κB activation can polarize myeloid cells towards M1-like macrophages, which counteract tumorigenesis by promoting inflammation, immunostimulation, tissue damage and apoptosis of cancer cells by secreting several molecules such as TNF-a, IL-12, iNOS, COX2 and IL-6. By contrast, NF-κB activation can shift macrophages towards M2-like anti-inflammatory TAMs, which promote tumour growth, angiogenesis, metastasis, EMT, as well as the establishment of an immunosuppressive TME.
In according with the role of MS to promote M1 polarization, Gao and collaborators demonstrated that tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) plays an important role in re-educating macrophages towards an antitumour phenotype by inducing the activation of NF-κB as well as the expression of pro-inflammatory cytokines such as IL-1b, IL-6 and TNF-a that in turn promote a cytotoxic effects in the tumour cells [56]. The authors showed that TRAIL also enhanced the expression of miR-146a via NF-κB and its overexpression blocked the production of pro-inflammatory cytokines suggesting that miR146a negatively controls the immunosuppressive phenotype. The modulation of this immune response regulated by the TRAIL/NF-κB/miR-146a axis identified TRAIL as a potential target to re-educate macrophages in tumour tissues.
Studies conducted by Lee and collaborators pointed out how NF-κB activation plays an important role in controlling the communication between tumour cells and TAMs through TLR4, reporting how NF-κB activation on one hand sustains cancer cell proliferation and invasion, and on the other hand induces TAMs to release inflammatory cytokines and angiogenic factors in the TME, that in turn, support tumour proliferation, thus creating a vicious circle. They demonstrated that TLR4 signaling is the mediator of the NF-κB activation in TAMs. Consistently, TLR4 deficient TAMs showed a decreased NF-κB activity, a reduced production of inflammatory and angiogenic factors, thus limiting tumour growth in vivo. Furthermore, TLR4 KO TAMs were not able to induce the activation of NF-κB in tumour cells. In contrast, macrophages TLR4 wild-type adoptive transferred in TLR4-deficient mice bearing tumour, showed a significantly higher NF-κB activity, enhanced release of inflammatory factors such as TNF-a and VEGF, thus prompting an increased NF-κB activity in tumour cells and tumour growth in vivo [57]. Therefore, targeting TLR4 in TAMs could be an attractive therapeutic strategy to counteract tumour growth in cancer patients.
It is recognized that NF-κB can exert both anti-tumor and pro-tumor functions within the TME (Figure 1). In fact, the NF-κB activation can either promote the polarization of macrophages toward a pro-inflammatory anti-tumor phenotype, or, at the same time, sustain the immunosuppressive activity of other cells such as Treg, macrophages and dendritic cells, leading to tumour growth [58]. Yang and collaborators demonstrated that M-CSF stimulation increased the expression of c-Jun, a member of the AP-1 family, which in turn induces the macrophages polarization toward the M2 phenotype. Additionally, they showed that NF-κB synergize with c-Jun to promote macrophages transformation from M1 to M2. Immunoprecipitation experiments confirmed the interaction between c-Jun and p50 after M-CSF stimulation, interaction that weakened in absence of Macrophage Colony-Stimulating Factor (M-CSF) or after treatment with p50 inhibitor, andrographolide [59].
Although the activation of NF-κB is important for inducing the M2 phenotype, isolated TAMs from several well-established tumours, have reduced NF-κB [15,60,61]. Saccani and collaborators demonstrated that high expression of the p50 NF-κB inhibitory homodimer inhibits M1 activation of TAMs and fosters tumour progression. Accordingly, TAMs isolated from mice knockout for p50 showed normal M1 activation with secretion of inflammatory cytokines and reduced tumour growth [62].
Kühnemuth and Michl demonstrated that the Cut-like homeobox 1 (CUX1), a homeodomain transcription factor expressed in different tumour types, act as an antagonist of NF-κB signaling in TAMs. CUX1, that is the transcriptional target of the immunosuppressive cytokine TGFβ, exerts its action by displacing RelA from the promoters of several genes (e.g., CXCL10, CCL5) related with the M1-phenotype and by mediating the de-acetylation of RelA through the recruitment of Histone Deacetylase 1 (HDAC1) to the promoters of NF-κB target genes. Furthermore, CUX1 inhibits the secretion of pro-inflammatory factors and supports tumorigenesis [63]. The inactivation of NF-κB by CUX1 inhibits the transactivation of inflammatory cytokines regulated by this transcription factor in established tumours [63]. Another mechanism that inhibits TAM antitumour activities is the degradation of NF-κB via selective autophagy. In vitro studies demonstrated that TLR2 signaling induces the accumulation of ubiquitinated NF-κB p65, that in turn forms aggresome-like structures (ALS) in the cytoplasm of M2 but not in M1 polarized macrophages. These structures are then recognized by the ubiquitin-binding proteins p62/SQSTM1 (sequestosome 1) and degraded via lysosomes. In addition, the authors showed that autophagy-dependent NF-κB p65 degradation is supported by sustained ERK1/2 phosphorylation that is triggered by TLR signaling [64].
NF-κB is also involved in the sophisticated mechanisms that regulate TAMs’ action in the processes of cancer cells invasion and metastasis [65]. A characteristic feature of the TME is the crosstalk between pericytes (PCs), cancer-associated fibroblasts (CAFs) and TAMs, that together coordinate the molecular mechanisms responsible of metastasis [65]. TAMs play important roles in promoting cancer cell dissemination [66]. Accordingly, interleukin-33 (IL-33) produced by CAFs drives the release of Th2-associated cytokines that polarize macrophages toward M2 phenotype. IL-33-stimulated TAMs show an increase of NF-κB-mediated Matrix metalloproteinase-9 (MMP9) expression, that in turn degrades the extracellular matrix protein laminin and allow the extravasation and dissemination of tumour cells, suggesting that the IL-33-NF-κB-MMP9-laminin axis moderates the CAF-TAM crosstalk to foster cancer metastasis [65].
It is well known that TME is characterized by low levels of oxygen (hypoxia) which promotes tumour progression and resistance to therapy. In addition, hypoxia enhances macrophage recruitment, thus conferring aggressiveness [67,68]. In this scenario, tumour cells and macrophages activate pro-angiogenic programs mediated by NF-κB-regulated Hypoxia Inducible Factor 1 (HIF-1) that promote tumour cells adaptation and proliferation as well as TAM recruitment and oncogenic activities [69,70].
Studies showed that TAMs together with other immune cells (e.g., myeloid-derived suppressor cells (MDSCs), T-regulatory cells (Treg)) infiltrate the hypoxic regions within the tumour and inhibit their anti-tumor function [68,71]. A study conducted by Delprat and colleagues indicated that cycling hypoxia (cyH), also called intermittent hypoxia, promotes the M1-like phenotype of macrophages via activation of JNK/p65 signalling pathway [72]. In this study the authors demonstrated that cyH promotes and amplifies a pro-inflammatory phenotype in non-activated (M0) and M1 macrophages by increasing the expression of M1 markers such as TNFa, IL-8, CXCL10, Macrophage inflammatory protein 2 (MIP-2). This pro-inflammatory phenotype in human M0 and M1 macrophages was due to an increase activation of c-jun/NF-κB signalling. Accordingly, p65 and JNK ablation inhibit the pro-inflammatory phenotype induced by cyH, suggesting that c-jun-p65 axis regulates the cyH-mediated M1 macrophages [72].
TAMs support tumour resistance by regulating drug metabolism and/or secreting cytokines such as IL-6 in several cancer types. Additionally, M2-TAMs promote angiogenesis and tumour relapse [73]. A recent work showed that NF-κB is involved in the development of chemo and radiotherapy resistance, as well as in tumour response to therapy. In particular, several chemotherapeutic agents, such as taxol, cyclophosphamide and cisplatin induce the up-regulation of proinflammatory cytokine such as TNF-a, IL-12, INOS, cyclooxygenase-2 (COX2), and the downregulation of anti-inflammatory factors like IL-10 and TGFb via NF-κB activation. In addition, cisplatin and carboplatin treatment enhance the activation of the NF-κB pathway through the chemotherapy-induced DNA damage response (DDR), thus sustaining the polarization of monocytes toward M2-like macrophages in the TME [74]. Although radiotherapy affects TAM recruitment and phenotype in cancer, its role in reprogramming TAMs towards an anti-tumor phenotype remains unclear [75]. It is known that NF-κB plays an opposite role in TAM response during radiotherapy depending on the dose irradiation. Indeed, low radiation doses polarize macrophages towards a pro-tumoral phenotype by reducing the expression of IL-1b through the increase of the nuclear translocation of p50-p50 homodimer and inhibition of p65 translocation [76]. On the contrary, moderate doses of radiations reprogram macrophages into M1 phenotype by inducing higher p65-p50 transcriptional activity, which in turn result in increased TNF-α, IL-6 and IL-8 production [77]. The immunosuppressive phenotype is maintained at high irradiation doses, where sustained activation of p50 keeps TAMs in a M2 polarization state [78].
3. NF-κB signalling in TAMs: the lesson from different human cancer types
3.1. Hepatocellular Carcinoma
According to the Global cancer statistic 2020, hepatocellular carcinoma (HCC) is the third leading cause of cancer death and the sixth most common cancer worldwide [79]. Many patients cannot benefit from the most common effective treatments, such as surgical resection, local tumour ablation and liver transplant due to tumour size or the invasion and spreading of cancerous cells in other tissues and sites [80]. TME plays an important role in HCC progression and TAMs can mediate the acquisition and maintenance of tumour cells stemness conferring chemo and radiotherapy tolerance and resistance [81]. It is known that high density of TAMs in HCC has been associated to unfavorable prognosis [82]. This relationship is due to the mutual influence that tumour cells and TAMs exert on each other. In fact, the release of several factors such as M-CSF, CCL2, VEGF, and TGF-b and the expression of surface markers (e.g., glypican-3) on cancer cells are responsible of TAM recruitment and polarization. On the other hand, recruited and activated TAMs in HCC release many cytokines, chemokines and growth factors that are responsible for tumour cell proliferation, angiogenesis, extravasation, invasion and metastasis, as well as the suppression of anti-tumor immune response [69]. The NF-κB pathway plays a fundamental role in the establishment of HCC. It has been demonstrated that it exerts opposite functions based on the stage and development of the disease as well as the cell-type where NF-κB is activated. NF-κB activation showed a tumor suppressor function in cancerous epithelial and parenchymal cells, while explained a tumour promoting function if activated in Kupffer cells, the resident macrophages of the liver. In this scenario, the inflammatory cytokines released by activated Kupffer cells provide a pro-survival and proliferative stimulus to malignant hepatocytes mediated by Myeloid-Differentiation-Factor-88 (MyD-88)/NF-κB pathway, thus sustaining tumorigenesis. The inhibition of NF-κB signalling in macrophages but not in hepatocytes reduces the production of pro-inflammatory cytokines and tumour growth [83,84,85,86].
A comparative analysis of RNA sequencing and whole-genome expression profiling of TAMs and M0 macrophages in HCC identified the upregulation in TAMs of the S100 calcium-binding protein A9 (S100A9) gene, which has a potential impact on HCC prognosis. In fact, the expression of S100A9 correlated with poor clinical outcome in patients with HCC [87]. It has been found that the S100A9 stimulates the NF-κB-dependent secretion of CCL2, the major chemoattractant for TAMs, thus promoting the recruitment, infiltration, and activation of TAMs. Furthermore, S100A9 secreted by TAMs induces a strong NF-κB activation and an increased expression of stemness-associated genes in HCC cells in a dose-dependent manner resulting in an enhanced sphere formation ability and self-renewal in vitro. The knockdown of advanced glycosylation end product-specific receptor (AGER), the receptor of S100A9, eradicates the NF-κB-mediated pro-tumorigenic effects of S100A9, suggesting that S100A9 controls the crosstalk between tumour cells and TAMs and could be a potential target for treating HCC patients [87,88].
The oxidored nitrodomain containing protein1 (NOR1) is overexpressed in human HCC tissues and correlates with advance clinical stage and poor prognosis [89]. In vivo study demonstrated that in diethylnitrosamine (DEN)-induced HCC, oxidored-nitro domain containing protein 1 (NOR1) is overexpressed in F4/80 positive macrophages and decreases M1-like markers (e.g., iNOS) and increases M2-like markers, such as arginase-1 (Arg1) and IL-10, thus promoting M2 polarization. Consistently, loss of NOR1 ablates NF-κB p65 expression, impairs the production of inflammatory cytokines such as IL-6 and TNF-a in mice and reduces DEN-induced HCC, suggesting that NOR1/NF-κB plays a role in TAMs polarization and HCC development [89].
Another protein involved in promoting tumorigenesis and M2-like polarization is Growth Arrest and DNA Damage beta (GADD45β), a NF-κB-regulated anti-apoptotic protein, able to suppress the JNK-mediated pro-apototic signalling by targeting MKK7 [90,91]. Recently GADD45β has been identified as an essential modulator of TAMs reprogramming and of the CD8+ T-cell trafficking in HCC tumours [92]. The authors demonstrated that Gadd45b −/− mice displayed a reduced number of HCCs compared to WT mice. Indeed, Gadd45b −/− HCC tumours, but not Gadd45b +/+ tumours, showed an enhanced F4/80+ and IBA1+ TAMs and T-cells infiltration as well as a higher number of tertiary lymphoid structures (TLSs), which are correlated to a favorable clinical outcome in most human cancers. Strikingly, M1-like macrophages was observed in Gadd45b-deficient HCC tumours, characterized by high expression of inflammatory markers, such as iNOS, COX-2 and MHC-II than Gadd45b+/+ HCCs, suggesting that Gadd45b loss increases the M1-like polarization state via upregulation of p38 signalling. These findings show that Gadd45b supports anti-inflammatory TAMs activation and blocks TLS formation and lymphocyte infiltration within tumours thus sustaining tumour growth [92]. Although in other tumours, the authors also demonstrated that macrophage specific Gadd45b loss blocks oncogenesis, indicating that Gadd45b governs the immunosuppressive activity of the TME across multiple cancer types [92]. Therefore, targeting Gadd45b, in combination with conventional immunotherapies, could be a potential new therapeutic approach to counterstain Gadd45b-mediated tumorigenesis in human cancers.
Receptor-interacting protein 140 (RIP140) is a protein widely expressed in macrophages, responsible for the regulation of TAMs energy metabolism and inflammatory response that has been associated with NF-κB pathway [93]. In HCC, NF-κB/IL-6 axis induces the alternative polarization of TAMs. In contrast, the overexpression of RIP140 in TAMs can reduce the expression of M2-like polarized markers (Arg-1, Peroxisome proliferator-activated receptors (PPAR), CD206) and enhance the expression of TNF-α, a M1-like marker. In vivo studies demonstrated that the inhibition of M2 polarization reduces tumour burden and invasiveness (Proliferating Cell Nuclear Antigen (PCNA) and VEGF ratio) and increases HCC cells apoptosis. In addition, they observed a decrease of p-p65, p-c-Jun and Tumour necrosis factor receptor-associated factor 3 (TRAF3) expression levels, as well as of IL-6. These findings suggest that the overexpression of RIP140 inhibits NF-κB activation and influences TAMs polarization in HCC [93].
Contrastingly, Sharen et al. demonstrated that the infiltration of M1-TAM in HCCs induces a reduction of the efficacy of postoperative transcatheter arterial chemoembolization (TACE) in patients with liver cancer. In particular, the infiltration of M1 TAMs promotes the iper-activation of NF-κB pathway, the increase of anti-apoptotic activity, the upregulation of the expression of cyclin-dependent kinase (CDK)1, CDK2 and cyclin D1, and the reduction of p21 expression in HCC cells, sustaining tumour proliferation. The pro-tumoral effect of M1-like TAMs was reverted by the administration of the p65 inhibitor, JSH-23, underlying the controversial role of NF-κB in M1 TAMs in HCC progression [94].
The use of immunotherapy for HCC treatment is still far of achieving its intended effect in the clinical setting, even if increasing number of patients have greatly benefited from this therapeutic approach [95,96,97,98,99,100].
The inhibitors of the programmed cell death protein-1 (PD-1)/programmed death-ligand 1 (PD-L1) pathway are one of the most used immune checkpoint inhibitors able to re-modulate T-cells and TAMs immune response [101]. Recently, Xu and collaborators demonstrated that the combination therapy with Listeria monocytogenes-based tumour vaccine, Lmdd-MPFG, and anti-PD-1 reduced HCC tumour volume and increased survival rates, by inducing CD8 T cells activation and IFN-γ signals. No effects are shown following monotherapy with anti-PD-1. In addition, they observed a shift of TAMs from M2 to M1 phenotype, an increased autophagy, driven by enhanced activation of the NF-κB pathway. This evidence highlighted that the combination therapy synergizes to affect TAM polarization and TME composition in a NF-κB dependent manner [102].
Liver is the preferential metastatic site to several tumours, such as colorectal cancer (CRC), lung cancer, melanoma, and gastric carcinoma. TAMs promote tumour progression by increasing tumour metastasis and stabilizing a pre-metastatic microenvironment [103,104]. Studies conducted by Li and collaborators demonstrated that N-myc downstream-regulated gene 2 (NDRG2), a protein that binds PTEN via protein phosphatase 2A (PP2A) recruitment, controls TME remodeling and TAMs polarization during metastatic processes through activation of the NF-κB pathway. Interestingly, in a liver metastasis model, they found that Ndrg2−/− macrophages have a tumor-suppressor phenotype compared to WT macrophages, and the M1–like polarization is driven by an enhanced activation of the NF-κB pathway. Accordingly, the inhibition of IκBα phosphorylation abolishes the tumor-suppressor function of Ndrg2−/− macrophages, thus inhibiting cancer liver metastasis [105].
3.2. Breast cancer
The role of M2 TAMs in promoting proliferation, invasion, migration and angiogenesis in human breast cancer seems to be related to the NF-κB-mediated expression and secretion of galectin-3, a member of the β-galactoside binding proteins. Galectin-3 results particularly expressed in hypoxic tumour regions, where it promotes the migration and invasiveness of breast cancer cells and enhances angiogenesis and vascular mimicry. The expression of galectin-3 in TAMs is dramatically reduced in presence of the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), both in normoxia and hypoxia conditions. In contrast, the presence of ROS induces the nuclear accumulation of NF-κB and the consequent upregulation of galectin-3 [106]
The chemokine profiling of murine and human breast cancer models indicated that CXCL1 is one of the most abundant chemokines secreted by TAMs. The levels of CXCL1 result significantly higher in metastatic lung cancer specimens compared to primary breast cancer tumour, suggesting the active role of CXCL1 in the promotion of breast cancer cell migration and invasiveness. Indeed, the presence of CXCL1 significantly increased breast cancer 4T1 cell migration and invasiveness ability, matrix metalloproteinase-2 (MMP-2) and MMP-9 secretion, as well as the expression of EMT-related proteins in vitro. TAMs-secreted CXCL1 mediates the EMT in breast cancer cells and induces the NF-κB-mediated expression of several metastatic genes, such as SRY-Box Transcription Factor 4 (SOX4). ChIP assay showed that NF-κB actively binds the predicted binding region on the SOX4 promoter after CXCL1 treatment. Accordingly, the inhibition of NF-κB blocks CXCL1-induced SOX4 overexpression, increases the E-cadherin and reduces both vimentin and β-catenin expression, underlying the importance of CXCL1/SOX4/NF-κB axis in sustaining breast cancer [107].
Annexin 1 (ANXA1), is a protein member of the Annexin family with multifunctional roles in cancer development and progression. ANXA1 is highly expressed in metastatic and triple negative (estrogen, progesterone and HER2 receptor) breast cancer (TNBC). Studies conducted on ANXA1+/+ and ANXA1−/− mice indicate that ANXA1 is necessary for a macrophage phenotypic switch from M1 to M2. Interestingly, ANXA1 directly induces ERK and NF-κB activation via the formyl peptide receptors 2 (FPR2), leading to a macrophage polarization and tumour cell proliferation [108].
The physical structure of the tumoural extracellular matrix (tECM) in breast cancer has an important role in the protection of the tumour niche and in the penetrance of the immune system. TAMs redefine the composition of the tECM by enhancing the deposition of the heparan sulfate proteoglycan 2 (HSPG2), known as perlecan, that confers mechanical rigidity to the ECM and generates a favorable gradient for cancer cells development. NF-κB p65/p52 complex is responsible of the transcriptional regulation of HSPG2 in M2 TAMs and is associated with the altered expression of perlecan. Accordingly, the treatment with QNZ, the NF-κB inhibitor, on TAMs significantly downregulates the expression of HSPG2, indicating that NF-κB pathway is involved in the remodeling of the tECM and in the tumour escaping from the immune system [109].
The acquisition of properties associated with mammary stem cells (MaSCs) and cancer stem cells (CSCs) is one of the key passages that enables breast cancer cells to perform the epithelial mesenchymal transition (EMT) and develop malignancy, invasiveness and therapy resistance [110,111,112]. Interestingly, human monocytes or TAMs fail to promote tumour-initiation or progression in breast cancer cells expressing an NF-κB super-repressor, indicating that NF-κB activation and cytokines production have a critical role in the CSCs and in the tumourigenic effects of the TAMs [112].
Human mammary epithelial cells responsible of EMT were characterized as CD44+ and CD24− [113]. Interestingly, mammary epithelial cells cocultured with M2 TAMs exhibit increased percentage of CD44+/CD24− and elevated levels of HIF-1α, α-catenin, NF-κB and twist family bHLH transcription factor 1 (Twist 1). In fact, the silencing of NF-κB in the M2-like MDA-MB-231 activated macrophages decrease the migration and invasion abilities of breast cancer cells. Mechanistically, NF-κB directly downregulates the anti-metastatic miR-488 and enhances special AT-rich sequence-binding protein-1 (SATB1) and Twist1 expression, the major initiators of the EMT, suggesting that the NF-κB/miR-488 axis is responsible for cancer cells migration and metastasis in presence of M2-TAMs [114].
It is known that the intercellular communication between TAMs, TME and several cancer cells, including breast cancer cells, is also mediated by EVs [115]. A recent study uncovered that TAM-derived EVs enriched with miR-660 inhibit Kelch-like Protein 21 (KLHL21) in breast cancer cells leading to the activation of NF-κB p65 signaling pathway. In turn, the activation of NF-κB controls the cancer cells invasiveness, migration, and metastasis by modulating the expression of E-cadherin, N-cadherin, MMP-9, MMP-3 and β-actin [116].
3.3. Colon cancer
Several evidence showed that TAMs sustain the initiation and progression of CRC by regulating several processes including tumour proliferation, metabolism, immunosuppression, angiogenesis and metastasis [117]. In CRC intratumoral macrophages exert tumor-promoting functions while TAMs found in the tumour invasive front correlate with better prognosis [115,116]. However, mixed macrophage populations are observed in human colorectal cancer tumours [118].
Studies in colitis-associated cancer (CAC) and in genetically driven ApcMin mouse models showed that NF-κB pathway regulates the shift from M1 to M2 macrophages and the transition from colitis to colon cancer. While the accumulation of p50 induces the activation of the M2-like transcriptional program, p50 deficiency reduces inflammation and number and size of neoplastic lesions in chemically induced CAC. Similar results were found in genetically driven CRC, where tumour incidence and growth are reduced in ApcMinp50–/– mice compared to ApcMin mice. Accordingly, tumour inhibition was observed in immunogenic MC38 transplantable CRC model in p50–/– mice compared to WT mice, as well as an increased expression of M1 genes and cytokines. Moreover, p50 ablation reduces TAMs accumulation indicating the fundamental role of NF-κB in modulating macrophages infiltration [119]. Consistently, the inhibition of NF-κB (p50) in M2-TAMs by siRNAs induces M2 macrophages to switch towards M1-like phenotype, thus promoting the production of pro-inflammatory and tumoricidal factors, as well as growth factors and NO. These findings suggest that NF-κB plays a central role in the assessment and maintenance of TAMs phenotype in colon cancer [120].
Prolyl hydroxylases domain proteins (PHDs) are dioxygenases activated in response to oxygen availability. The overexpression of PHD2 isoform is associated with the suppression of colon cancer growth and invasiveness [121]. Interestingly, the reduced p65 phosphorylation and the downregulation of NF-κB-related downstream genes involved in cell cycle, EMT, and inflammation (e.g., CyclinD1, E-cadherin, and TNF-α) as well as in the recruitment of M2-like TAMs within the TME, has been found in colon cancer cells expressing PHD2 and in PHD2-overexpressing colon cancer xenografts, suggesting that NF-κB mediates the anti-inflammatory and anti-cancer effects of PHD2 in colon cancers [122].
Recent studies demonstrated that prolonged and enhanced NF-κB signalling activation sustains TAMs F4/80-positive recruitment and angiogenesis via P2X purinoceptor 7 (P2X7R) in CRC cells [123]. It is known that NF-κB sustains cancer cells survival and progression by regulating metabolic switch and adaptation to low nutrient conditions [33,124]. In vivo studies demonstrated that ketogenic diet (KD) induces a strong p65 inhibition in both TAMs and CRC cells, that in turn promotes TAMs M1 polarization and reduces tumour growth and metastasis, suggesting that NF-κB plays a role in the modulation of tumour metabolic adaptation and inflammatory response [125]. It has been demonstrated that metabolism is also associated to the TAMs ability to migrate with cancer cells to metastatic sites [126,127].
In CRC, macrophages have distinct metabolic profile based on their migratory abilities. In fact, migration-active macrophages express lower abhydrolase domain containing 5 (ABHD5), a catalysator of triglyceride hydrolysis, compared to the nonmigratory TAMs. Interestingly, ABHD5 deficiency and consequently triglyceride accumulation, stimulate NLRP3 inflammasome activation, in a ROS-dependent manner, and induce IL1b secretion that in turn stimulates p65 signalling. Conversely, gene microarray showed that ABHD5 suppresses NF-κB-dependent MMPs expression and activity, fundamental for cancer cells migration. In addition, the authors demonstrated that ABHD5-mediated knockdown (ABHD5-KD) macrophages have an enrichment of JAK–STAT and TLR pathway, that stimulates the NF-κB and JNK signalling compared to controls. In conclusion, TAMs expressing ABHD5 exert an inhibitory effect on colon cancer cell migration, while ABHD5low macrophages promote NF-κB-dependent colon cancer cells invasiveness [128].
3.4. Glioblastoma
It is known that up to 30% of the total Glioblastoma (GBM) composition is represented by peripheral macrophages and microglia [129,130]. In a GBM syngeneic immune competent mouse model, it has been shown that the LysM-Cre-mediated conditional deletion of p65 in myeloid lineage induces the TAMs polarization from the M2 to M1 phenotype. Indeed, p65 deficiency enhances CD8+T cells proliferation and increases levels of IFN-γ, TNF-α and IL1-β, which lead to a decreased GBM tumour growth. In contrast, where p65 KO and control donor mouse bone marrow were transplanted in immune-deficient mice, no reduction in tumour growth was observed in p65 KO chimera mice compared to control mice. These finding suggest that the anti-tumor immunity in GBM is inextricably linked to T cells activity and its negatively regulated by the NF-κB pathway [131]. Interestingly, in a GL261-implanted mouse model the treatment with curcumin (CC) induced an overall TAM repolarization from the M2 to the M1 phenotype via STAT1 and NF-κB signalling activation. Indeed, the CC treatment suppresses p50 homodimers in the microglia and promotes the activation of the p50/p65 NF-κB pathway, which leads to the M1 tumoricidal phenotype activation. Moreover, the concerted activation of STAT1 and NF-κB pathways induces apoptosis of GBM cancer cells and increases iNOS and Iba1+ expression in the microglia, indicating the presence of activated M1 macrophages which eliminate the remaining GBM cells and apoptotic debris, and a tumour remission in 50–60% of treated mice [132].
GBM growth is significantly accelerated by IL-1β, a cytokine highly expressed in TAMs and neutrophils localized in the perinecrotic tumour areas. IL-1β expression in TAMs is synergistically activated by CXCL8 and IL-6, which in turn activate NF-κB and STAT3 pathways. TAMs-derived IL-1β stimulates GBM tumour cells and induces the expression of IL-1β itself and CCL2, which actively recruits TAMs in the tumour site. Furthermore, damage-associated molecular patterns (DAMPs) released from necrotic cells stimulate IL-1β expression in TAMs with an autocrine/paracrine feedback loop that accelerates TAMs migration and tumour growth [133]. Consistently, higher IL-1β expression is associated with a worse clinical course in GBM [33,124] suggesting that NF-κB /IL-8 β axis in TAMs has a key role in GBM growth and progression.
3.5. Gynecologic cancer
As for HCC, NF-κB/GADD45b axis govern the immunosuppressive activity of the TME and the M2 macrophage polarization in ovarian cancer (OC) [92]. Therefore, the Gadd45b ablation in macrophages re-establishes proinflammatory TAM activation and CD8+ T-cell infiltration into the tumour, thus inhibiting ovarian adenocarcinoma growth [92].
The crosstalk between endothelial ovarian cancer cells (EOC) and TAMs promotes the progression of OC [134], and exosomes have been identified as principal mediators of this interplay [126]. In fact, exosomes isolated from TAMs in the ascites of EOC patients and carrying high levels of miR-146b-5p are then internalized in the cytoplasm of EOCs as endosome-like vesicles. Once internalized in EOCs, miR-146b-5p directly targets and inhibits the TRAF6/NF-κB/MMP2 axis, suppressing endothelial cell migration. In contrast, exosomes derived from EOCs, that transport 2 lncRNAs, restore the endothelial cell migration by activating NF-κB pathway in the recipient cells [135].
Mitochondrial transcription factor B2 (TFB2M), responsible for the mitochondrial DNA (mtDNA) transcription and compacting, is expressed in ovarian cancer patients, and is associated with poor prognosis, immunosuppressive TME and macrophages polarization. In fact, IHC analysis on primary OC specimens indicates a striking correlation between TFB2M expression and M2 TAM infiltration. Interestingly, the overexpression of TFB2M increases cytosolic mtDNA and IL-6 expression in OC cells, via TLR9/NF-κB signalling pathway activation, contributing to M2 macrophage polarization and infiltration in the TME [127]. Accordingly, the inhibition of cytosolic mtDNA, TLR9 or NF-κB pathway blocks the TFB2M-induced IL-6 expression and abrogates the M2 macrophage polarization and the immunosuppressive TME in OC.
In other types of gynecologic cancers, TLR9 pathway activates lymphocyte T cell shift towards Th1 profile and reduces the number of MDSCs, TAMs and Tregs in the TME, displaying anti-tumor properties [126]. However, TLR9 pathway stimulated by radiotherapy, cell death and CpG release, can also induce tumour progression, angiogenesis and invasiveness in cervical cancers through the activation of the NF-κB and STAT3 transcription factors [136].
As extensively reported, hypoxia plays a fundamental role in cancer progression, and hypoxic regions are often infiltrated by immunosuppressive cells, such as TAMs, MDSCs and Tregs [137,138] and are strongly associated with poor patient outcome [139]. It has been demonstrated that hypoxia induces the binding of HIF-1α to the proximal promoter of zinc finger E-box binding homeobox 1 (ZEB1), which is highly expressed in cervical cancer hypoxic cells islets and is associated with a stronger pro-tumor phenotype. ZEB1-driven cancer cells produce CCL8, attract and promote macrophages activity through NF-κB activation. Interestingly, TCGA data showed that both ZEB1 and CCL8 overexpression correlate with poor prognosis in human cervical cancer. These findings suggest that the NF-κB pathway is responsible for hypoxia-induced ZEB1-mediated TAM infiltration, and targeting ZEB1-CCL8-NF-κB axis could be a promising strategy for the cervical cancer treatment [140].
3.6. Other solid tumours
Other solid tumours in which TAMs have a crucial role in mediating tumour progression and immune escape are neuroblastoma (NB), bladder cancer, renal carcinoma, gastric cancer (GC), basal cell carcinoma (BCC), oral squamous cell carcinoma (OSCC), prostate cancer (PCa), nasopharyngeal carcinoma (NPC), multiple myeloma (MM), sarcoma, pancreatic cancer (PDA) and lung cancer [141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156]. In addition, it is known that in these tumours TAMs correlates with worse prognosis and drug resistance [157].
NB is the most frequently pediatric cancer with a heterogeneous clinical outcomes based on tumour stage [158]. Recently, it has been demonstrated that higher levels of lipid metabolism-related genes are associated with unfavorable histology and advanced stage of NB compared to the early stage. Fatty acid binding protein 4 (FABP4), a lipid chaperone that facilitate lipid distribution and response in cells, is highly expressed in NB macrophages compared to other cell types of the TME, leading to tumour progression [130]. In NB orthotopic and metastatic mouse models with FABP4 KO macrophages the tumour growth is repressed. Accordingly, the authors observed an increased tumour growth when FABP4 is overexpressed in macrophages. Furthermore, FABP4 inhibits NF-κB activity and IL-1α secretion in TAMs and downregulates ATP production via the ubiquitination of ATPB, maintaining TAMs in an anti-inflammatory state and sustaining the pro-tumorigenic effects on NB cells [130].
Bladder cancer is the fourth most common cancer in men and the overall survival is still low [142]. In vitro studies with HTB-1 and T24 bladder cancer cell lines cocultured with M2-like TAMs, demonstrated that the coculture upregulate the expression of EMT-related genes, such as VEGF, Twist, Vimentin and NF-κB in bladder cancer cells, leading to increased migratory and tumour sphere generation abilities. Interestingly, the treatment with BAY11-7082, a potent NF-κB signalling inhibitor, reduces the expression of the CD206, a M2-macrophage marker, and increases the expression of TNF-α, a M1 marker. In addition, the treatment reverts the EMT-induced phenotype of bladder cancer cells and reduces the expression of CD133+, a marker produced by M2-like TAMs. The authors also showed that BAY11-7082 induces the expression of miR-30a both in bladder cancer cell and in TAMs, reprogramming M2-like macrophages towards M1 phenotype. These findings underly the role of NF-κB in the regulation of TME and cancer cells metastatic potential in bladder cancer [131].
In human renal cell carcinoma (RCC), the most common type of kidney cancer in humans [159], blood derived monocytes display a distinct transcriptional profile characterized by the upregulation of pro-inflammatory cytokines and chemokines, pro-tumor genes, (e.g., COX2, IL-8, VEGFA, MMP19, MMP10, CXCR4, and HIF1A) and polarization-related genes [160]. Interestingly, RCC-conditioned monocytes display enhanced IκBα phosphorylation and p65 NF-κB nuclear translocation, via IL-1/IL-1R/MyD88 signalling activation. In RCC mouse models, the blockade of IL-1/IL-1R pathway with recombinant IL-1RA or Il1r1 knockdown, inhibits tumour growth and TAMs pro-tumor phenotype. Meta-analysis of tumour gene-expression data from RCC patients highlights that the higher expression of IL-1B correlates with pro-tumor genes induces by TAMs and advanced tumour stages, indicating that IL-1/IL-1R signalling is crucial in shaping the TME and the RCC tumour development through MyD88-dependent NF-κB pathway activation [161].
GC is the fifth cause of death in the worldwide [151]. Immunostaining of primary GC tissue samples suggest that TAMs are preferentially localized in the invasive tumour front and correlates with invasion, lymph node metastasis, TNM stage, and poor prognosis [162]. In vitro co-culture of GC cells and macrophages indicate that TAMs actively produce angiogenic and lymphangiogenic growth factors, VEGF and VEGF-C, through the activation of NF-κB signalling pathway. Interestingly, NF-κB inhibition in TAMs decreases the expression of VEGF and VEGF-C in both macrophages and cancer cells, but the molecular mechanism is still unclear. However, this study indicates that NF-κB has a role in the interactions between tumour cells and TAMs in GC [162].
BCC is the most common skin cancer with a complicated pathogenesis [153]. In aggressive (micronodular) BCC, increased tumour invasion and angiogenesis correlates with the number of TAMs [163]. Using an in vitro noncontact co-culture system, Tjiu and colleagues demonstrated that both M2-polarized THP-1 and monocyte-derived M2 macrophages promotes extracellular matrix degradation, invasiveness and angiogenesis of BCC cells by inducing COX2-dependent MMP-9, VEGF-A and basic fibroblast growth factor (bFGF) expression, via the p38 MAPK/NF-κB cascade [163].
Another skin cancer with a considerable tumour mutational burden is melanoma. Melanoma is the most fatal skin cancer and TAMs play an important role in regulating tumour development [154]. In situ immunofluorescence analysis of human primary melanoma samples indicate that there is a strong correlation between the abundance of TAMs expressing CCL20/TNF/VEGF-A and worse prognosis. The study illustrates that this pro-tumoral M2 phenotype of TAMs are driven by a consistent p53 and NF-κB activation [164].
TAMs promote tumour progression also in OSCC, a common head and neck tumour with a low overall survival [155]. It is known that TAMs’ phenotype is related to Axl signalling, an oncogenic pathway activated by autocrine and paracrine release of Growth arrest – specific 6 (Gas6), an antiapoptotic and proliferative protein [165,166]. In vitro studies showed that OSCC cells expressing Axl polarize THP-1 towards M2 phenotype and induce the secretion of pro-tumorigenic factors like MMP2, MMP9 and VEGF, by activating PI3/Akt/NF-κB pathway. Accordingly, the inhibition of Axl, PI3/Akt and NF-κB on OSCC cells with specific single inhibitors, reduces M2 polarization [167]. Furthermore, co-culture of OSCC cells with THP-1 cells enhances the expression of Axl and its ligand Gas6 and NF-κB transcription activity in cancer cells that, in turn, acquire tumour invasion/migration abilities and express EMT related genes [168]. These findings suggest the presence of a bidirectional interaction between TAMs and OSCC that promotes malignancy and tumour growth via Gas6/Axl/ NF-κB signalling [167,168].
The expression of Nephroblastoma Overexpressed (NOV)/ Cellular Communication Network Factor 3 (CCN3) has been associated with M2 macrophage infiltration in PCa. PCa is the second most common tumour in men. NF-κB plays an important role during prostate cancer progression and its overexpression is correlated with worse prognosis [156,169]. CCN3 secreted by PCa cells recruits macrophages and educate TAMs toward M2 phenotype, while PCa cells pre-treated with CCN3-neutralizing antibody attenuate CCN3-induced macrophage migration and polarization. Interestingly, in RAW264.7 cells, CCN3 induces the phosphorylation of Focal adhesion kinase (FAK), Akt and NF-κB, in a time-dependent manner, and enhances p65 binding to the NF-κB element on the VEGF promoter, leading to increased angiogenesis. Correspondingly, pre-treatment with FAK, Akt and NF-κB inhibitors or transfection with dominant-negative (DN) mutant of FAK, Akt, IKKα, or IKKβ, abolishes the VEGF expression. Consistently, knockdown of CCN3 in PCa cells inhibits RAW264.7-promoted angiogenesis and tumour growth in mouse models, suggesting that CCN3 secreted by PCa cells regulates TAMs infiltration and function by modulating the FAK/Akt/NF-κB signalling pathway in macrophages [170].
MM cells can modify the bone marrow (BM) niche and induce M2 phenotype polarization by releasing several soluble factors [171]. A recent study showed that circulating miR-16 in MM patients is associated with a better survival, and MM patients with chromosome 13 deletion (Del13) express lower levels of miR-16 compared to non-Del13 patients. Interestingly, primary basal state spleen MΦ (M0-MΦ) isolated from miR-16 KO mouse showed a more pronounced M2 phenotype compared to WT mice. The treatment of MΦ isolated from MM patients and mice, MΦ-like malignant cell line and human stromal cell line with ds-miR-16 significantly downmodulates the expression of both IKKα and IKKβ. Consistently, miR-16 directly binds IKKβ 3′UTR and subsequently affects protein translation. Indeed, miR-16 has a significant additive effect in inhibiting NF-κB activity in MM cells when combined with bortezomib or BAY11-7082 and sensitizes MM-PCs and resident MΦ of the BM-ME to bortezomib treatment. These findings indicate that miR-16 mediated-NF-κB pathway activation has critical role in the selection and polarization of specific monocytes clones during MM progression [172].
The development and progression of Nasopharyngeal carcinoma (NPC) is associated with Epstein-Barr virus (EBV) infection and sustained inflammation, due to the inhibition of Forkhead box P1 (FOXP1) tumour-suppressive activity driven by one of the 44 EBV miRNAs, EBV-miR-BART11. In fact, EBV-miR-BART11 has been found significantly higher in NPC biopsies compared to non-tumor nasopharyngeal epithelial tissues. Inhibition of FOXP1 induces enhanced NPC cells proliferation and the expression of inflammatory factors (e.g., IL-1β, IL-6, and IL-8) leading to a sustained local inflammation. Moreover, macrophages transfected with EBV-miR-BART11 are hyperresponsive to LPS and promote epithelial cell proliferation. Indeed, FOXP1 overexpression decreases NF-κB p65 protein expression, while EBV-miR-BART11 overexpression or FOXP1 knockdown increases NF-κB p65 expression, indicating the role of NF-κB in the crosstalk between NPC cell and TAMs in response to inflammation and tumour development [173].
In Ewing sarcoma (ES), the regulation of macrophages infiltration is driven by let-7a expression. The let-7a has a tumor suppressive activity and inhibits TAMs recruitment on tumour site. The overexpression of let-7a represses STAT3 signalling pathway, which is constitutively activated in ES cells. Accordingly, STAT3 upregulation suppresses let-7a activity by inducing NF-κB pathway, that inhibits the expression of mature let-7a through lin-28B, leading to enhanced TAMs infiltration and activation [174].
It has been demonstrated that acidic polysaccharide IAPS-2 exhibits an anti-tumor effect on sarcoma by re-educating TAMs towards an anti-tumor phenotype. The IAPS-2 treatment induces NF-κB and STAT1 pathways and inhibits STAT3 signalling, that in turn modulates TAM gene expression and cytokine profile. In S180 tumour-bearing mice, IAPS-2 promotes the secretion of several M1 markers and reduces the concentration of MMP-9 and VEGF in the tumour, restoring the immunosurveillance with an anti-angiogenetic effect [175].
In pancreatic (PDA) cancer, the transcription factor Cut Like Homeobox 1 (CUX1) is an important modulator of TAM phenotype plasticity and functions and it is highly expressed not only in PDA tumour cells but also in PDA TAMs. It is recognized that CUX1 displaces NF-κB p65 binding at the promoter level of CXCL10 and, recruits HDAC1 that decreases the acetylation and transcriptional activity of NF-κB, leading to a reduced expression of M1 phenotype-related cytokines. As a result, CUX1 reduces the recruitment and the activation of M1 TAMs, promoting tumour progression and angiogenesis in PDA. These finding suggest that CUX1 regulates TAMs phenotype by counteracting NF-κB activity [176]. It is established that ApoE mediates cholesterol metabolism, is highly expressed by TAMs and CAFs both in mouse and human PDA and correlates with patient survival. Recently, Kemp and colleagues showed that apolipoprotein E (ApoE) sustains immunosuppressive TME in PDA via NF-κB-mediated CXCL1/5. Specifically, ApoE expressed in tumor macrophages binds LDLR and induces CXCL1/5 expression through NF-κB signalling [177]. Accumulating evidence showed that lung cancer displays alveolar macrophages (AM), the tissue-resident macrophages, which exhibit different functions within the TME. As for other cancers, TAMs sustain proliferation, immunosuppression and invasion in lung cancer via secretion of several cytokines, chemokines and growth factors [148]. Analysis of Non-Small Cell Lung Cancer (NSCLC) tumours from 60 patients indicate that plasminogen activator inhibitor-1 (PAI-1) expression correlates with TGF-β expression and percentage of TAMs. In vitro study demonstrated that PAI-1 on cancer cells binds TLR4 and promotes TGF-β secretion in macrophages by increasing IL-6 production via NF-κB activation. At the same time, TGF-β promotes PAI-1 expression in NSCLC cells in a R1/SMAD3-dependent manner. However, while high concentration of TGF-β can inhibit NF-κB activation, optimal concentration of TGF-β activates NF-κB pathway and IL-6-induced TGF-β production. These findings indicate the presence of an auto-regulatory or auto-inhibitory loop regulated by NF-κB activation responsible for the immunosuppressive TME in NSCLC [178].
4. Targeting NF-κB Pathway in TAMs
It is recognized that TAMs are involved in orchestrating the immune response, tumour growth and progression as well as metastasis and TME remodeling. In particular, the polarization of TAMs to the M1 phenotype exerts important tumor suppressing effects on the TME, while the M2-like macrophages display pro-tumoral and angiogenetic activity [11]. NF-κB plays a key role in the regulation of TAMs as it swings the scale between tumour suppression and tumour progression [15]. Thus, there is a rationale to target NF-κB pathway to counteract TMAs activity within the TME. In this contest, the principal strategies explored are a) re-education of TAMs towards the antitumoral (M1) phenotype; b) TAMs depletion; c) termination/blocking of macrophage recruitment and accumulation into the TME (Table 1). Although all these strategies showed efficacy in early-stage diseases, recent evidence suggests that the TAM reprogramming approach could be the safer and more effective.
4.1. Re-educating TAMs via NF-κB inhibition
A promising and large investigated approach engages toll like receptors (TLRs) to induce an immunostimulatory response and reshape TAMs into M1 phenotype. Several studies using bioactive compounds such as chrysin thiazole derivative (ChR-TD), Anemoside A3 (A3), β-D-(1→6) glucan (AAMP-A70), Aqueous Extract of Cimicifuga dahurica (CRAE) as well as antineoplastic agent (e.g., paclitaxel) demonstrated that these drugs activate TLR2 or TLR4 that in turn directly trigger the NF-κB pathway leading to TAMs reprogramming toward M1-like antitumour phenotype. In vitro studies demonstrated that these compounds induce a significantly increase of TNF-α, IL-12, IL-6 and IL-1β mRNA expression and downregulate M2-like genes like IL-10, Ym-1, CD206 and Arg-1 in several models of cancer (e.g., breast cancer, colon cancer, MM) [179,180,181,182,200]. Accordingly, Homogeneous Polyporus Polysaccharide (HPP) induces M1 polarization via TLR2/NF-κB/NLRP3 signalling activation and reduces the progression of bladder cancer [183].
The PI3K/NF-κB axis is another molecular target investigated for its capability to promotes M1 polarization. He and colleagues demonstrated that the inhibition of PI3Kγ with the natural compound Baicalein potentiates the M1 macrophage polarization and inhibits tumour growth via NF-κB/TNF-α inflammatory signalling in both breast cancer and melanoma mouse [54].
Hoover and collaborators showed that increased IKK2 activity, an enzyme involved in the NF-κB signalling pathway, in macrophages contributes to M1 TAMs polarization and abrogates ovarian cancer growth in vivo [184].
It has been shown that the use of BAY11-7082, a selected inhibitor of the NF-κB pathway, inhibits anti-inflammatory macrophage phenotype and reduces the invasiveness of bladder cancer cells by increasing the expression of miR30a [185]. Additionally, targeting IncRNA DCST1-AS1/NF-κB axis in oral epithelial squamous cell carcinoma (OSCC) blocks the M2 polarization and tumour progression [186].
Recent and innovative approaches exploited nanoparticle technology to restrain TAM activity. An interesting attempt have been made with Glycocalyx-mimicking nanoparticles (GNPs) both in vitro and in vivo. These nanoparticles are specifically internalized by TAMs and can neutralize and reprogram M2 TAMs through the reciprocal STAT6 suppression and NF-κB phosphorylation. In addition, the authors demonstrated that GNPs improve the therapeutic efficacy of anti-PDL1 and reduce Lewis lung carcinoma (LLC) growth [187]. Congruently, mannose modified lipid nanoparticles (M-IMD-LNP), containing the NF-κB inhibitor IMD-0354, as well as hyaluronic acid (HA) nanoparticles, loaded with the micro-RNA miR-125, showed that M2 TAMs are able to repolarize toward M1 phenotype, in an NF-κB dependent manner, both in melanoma cells and in an in vivo model of non-small cell lung cancer (NSCLC) [188,189]. Furthermore, Li and collaborators showed that a modified porous hollow iron oxide nanoparticles (PHNPs) loaded with a P13Kγ small molecule inhibitor (3-methyladenine, 3-MA), (PHNPs@DPA-S-S BSA-MA@3-MA), selectively inhibit the PI3Kγ/Akt signalling in TAMs by inducing a prolonged activation of NF-κB, through the reduction of the P13Kγ protein in macrophages and cancer cells, that in turn promote the switch of TAMs toward pro-inflammatory M1 phenotype and the activation of immune response leading to reduced tumour growth and immunosuppressive TME [190].
The use of cell membrane-coated nanocarrier system such as PLGA-ION-R837@M, is another strategy to repolarize macrophages toward M1 and mitigate immunosuppressive TME by activating interferon regulatory factor 5 (IRF5) and NF-κB pathway using the synergy of Fe3O4 NPs and R837, a TLR7 antagonist [191]. As for PLGA-ION-R837@M, Gd@C82 nanoparticles modified with b-alanines (GF-Ala) showed an anticancer effect in vivo by activating the anti-tumor immune response and relieving the immunosuppressive TME via NF-κB/IRF5 activation [192].
In addition, the reprogramming of M2 macrophage in M1 via NF-κB pathway activation and the consequently tumour inhibition, was also observed in melanoma-bearing mice using copper sulfide nanoparticles (CuS-NP) [193].
Recently, Zhao and collaborators demonstrated that Cetuximab exerts its antitumoural effect in colon cancer attenuating the M2 TAMs pro-tumorigenic activity and triggering the repolarization of TAMs from the M2 to the M1 phenotype. Cetuximab inhibits EGFR signalling and suppresses IL-6 expression in M2 TAMs by directly inhibiting NF-κB and STAT3 pathways. Cetuximab-induced TAM repolarization strongly affects the immunosuppressive TME by reducing tumour burden and inflammation in both xenograft and azoxymethane/dextran sodium sulfate (AOM/DSS)-induced mouse colon cancer model [194]. Another drug able to re-programming TAMs toward a M1 phenotype is cabazitaxel. RNA profiling of bone marrow–derived macrophage (BMDM) treated with cabazitaxel displays a significant TLR/NF-κB signalling activation and consequently the upregulation of NF-κB-mediated cytokines and chemokines expression compared to control. Indeed, rather than exerting a direct cytotoxic effect on breast cancer cells, cabazitaxel appears to induce an NF-κB-mediated macrophage’s polarization toward a M1 state stimulating the Programmed Cell Removal (PrCR) macrophage activity [195].
According to the TAM re-polarization strategies, the treatment with proton irradiation has shown to be effective in the reprogramming of TAMs via modulation of NF-κB signalling. Proton therapy applied on THP1-derived M0, M1 and M2 phenotype macrophages promotes an enhanced nuclear translocation of NF-κB p65 in all three groups after 2h of irradiation. Both gene expression and cytokines analysis showed that moderate doses of proton irradiation promote the reprogramming of M0 and M2 macrophages towards an M1 phenotype. However, the combined treatment of irradiation with the NF-κB inhibitor Bay 11-7082 (IKK inhibitor) produces a clear induction of the macrophages towards the M2 phenotype. In particular, the M0 macrophages shifted to the M2 phenotype rather than M1 polarization, while a reinforcement of the M2 phenotype was observed in M2 macrophages [77].
4.2. Re-educating TAMs via NF-κB inhibition
Recently, it has been demonstrated that CCL2, a pro-inflammatory and NF-κB-mediated chemokine, promotes TAM infiltration. In addition, NF-κB/CCL2 signalling pathways plays a key role in tumour progression, invasion and metastasis in a wide range of human cancers [196]. Therapeutic inhibition of CCL2/CCR2 axis through NF-κB ablation, abrogates inflammatory monocyte recruitment and TAM infiltration in different mice models of several cancer types such as primary and metastatic breast cancer, HCC and lung cancer [197,201]. In this contest, the natural compound total glucosides of paeony (TGP) downregulates the expression of genes involved in the formation and function of TAMs via NF-κB/CCL2 ablation as well as the secretion of CCL2 in primary and metastatic breast cancer. Furthermore, it decreases tumour growth and mitigates the immunosuppressive TME by decreasing CD45+CD11b+F4/80+ TAMs population and promoting CD4+ and CD8+ T cell infiltration in vivo [197].
The knocking down of IκBα protein obtained through the transfection of the IκBα si-RNA encapsulated into mannosylated siRNA-delivering NPs, promotes the recruitment of T-cells within the TME and inhibits macrophage infiltration both in ovarian and in breast cancer [198,202,203].
All trans-retinoic acid (ATRA) is a clinically available molecule able to immune-modulate myeloid cells. In a study on prostate cancer, it was shown that the treatment of primary macrophages with medium conditioned with PC3 prostate cancer cells induces a clear activation of the NF-κB and ERK pathways, and M2 macrophages polarization. Exposure to ATRA suppresses the production of the M2-like released factors (e.g., IL-10, IL-1b, indoleamine-pyrrole 2,3-dioxygenase (IDO) and VEGF) and downregulates MHC class I and II and Fas ligand (FasL) molecules expression, through a specific inhibition of NF-κB p50 without effects on ERK phosphorylation [199].
5. Conclusions
TAMs are the principal component of the TME, and infiltrating macrophages are associated with worse clinical outcome and drug resistance. Although numerous progresses have been made for cancer treatment, targeting macrophages represent a new approach in cancer immunotherapy to improve tumor immune microenvironment. NF-κB is a master regulator of innate immune responses and play a key role in TAM reprogramming within the TME. However, while a great bulk of evidence has been collected, there are still controversies about the functions of NF-κB in myeloid cells within the TME. Hence, NF-κB activation in TAMs has been associated to the acquisition of both immunosuppressive and immunopermissive TAM phenotype, with opposite effects in terms of tumour progression. Therefore, further studies are needed to better understand in which tumoral contexts the inhibition of NF-κB signalling to reprogram TAMs could be of benefit.
Author Contributions
Writing—original draft preparation, J.C.; writing—review and editing, D.V. (Daniela Verzella), D.C., F.Z. and G.F.; figure and table preparation, D.V. (Davide Vecchiotti) and P.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Cancer Research UK, grant number A15115; NIHR Imperial Biomedical Research Centre (BRC), grant number P81135; Medical Research Council (MRC), grant number MR/V027581/1; Imperial College London Joint Translational Fund, Medical Research Council (MRC) & Yuhan Corporation, Imperial Confidence in Concept (ICiC) 2021, grant number RSRO_PA2579, RSRO_P87961 to G.F.; Progetto di Ateneo per la Ricerca di Base 2022, University of L’Aquila, grant number 07_Progetto_Ricerca_Ateneo_Verzella to D.V. (Daniela Verzella); Intramural DISCAB GRANT 2023, Department of Biotechnological and Applied Clinical Sciences, University of L'Aquila , grant number 07_DG_2023_22 to D.V. (Daniela Verzella).
Acknowledgments
D.V. (Daniela Verzella) has been supported by National Operational Programme (PON)-Attraction and International Mobility (AIM) Research and Innovation 2014-2020, Project AIM1855453; D.V. (Davide Vecchiotti) has been supported by National Operational Programme (PON)-Attraction and International Mobility (AIM) Research and Innovation 2014-2020, Project AIM1887574.
Conflicts of Interest
The authors declare no competing financial interests.
References
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19. [Google Scholar] [CrossRef]
- Verschoor, C.P.; Puchta, A.; Bowdish, D.M.E. The Macrophage. Methods Mol. Biol. 2012, 844, 139–156. [Google Scholar] [CrossRef]
- Taylor, P.R.; Gordon, S. Monocyte Heterogeneity and Innate Immunity. Immunity 2003, 19, 2–4. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or Foes - Bipolar Effects of the Tumour Stroma in Cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumour Invasion by Tumour-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers (Basel). 2017, 9. [Google Scholar] [CrossRef]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and Pro-Inflammatory Macrophages in the Colon Represent Alternative Context-Dependent Fates of the Same Ly6Chi Monocyte Precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary Alveolar Macrophages Contribute to the Premetastatic Niche by Suppressing Antitumour T Cell Responses in the Lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of Macrophage Function in Tumours: The Multifaceted Role of NF-ΚB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Biswas, S.K.; Lewis, C.E. NF-ΚB as a Central Regulator of Macrophage Function in Tumours. J. Leukoc. Biol. 2010, 88, 877–884. [Google Scholar] [CrossRef]
- Mulder, R.; Banete, A.; Basta, S. Spleen-Derived Macrophages Are Readily Polarized into Classically Activated (M1) or Alternatively Activated (M2) States. Immunobiology 2014, 219, 737–745. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b Macrophage Polarization and Its Roles in Diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying Functional MicroRNAs in Macrophages with Polarized Phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.Y.; Glass, C.K. Non-Coding RNAs as Regulators of Gene Expression and Epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Q.; Wang, X.; Yao, X.; Zhang, B.; Wu, J.; Sun, C. Exosomal NcRNAs Facilitate Interactive “dialogue” between Tumour Cells and Tumour-Associated Macrophages. Cancer Lett. 2023, 552, 215975. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Rubenich, D.S.; Zaręba, Ł.; Siewiera, J.; Pieper, J.; Braganhol, E.; Reichert, T.E.; Szczepański, M.J. Potential Roles of Tumour Cell- and Stroma Cell-Derived Small Extracellular Vesicles in Promoting a Pro-Angiogenic Tumour Microenvironment. Cancers (Basel). 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Bowdridge, S.; Gause, W.C. Regulation of Alternative Macrophage Activation by Chromatin Remodeling. Nat. Immunol. 2010, 11, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A Cytokine-Mediated Link between Innate Immunity, Inflammation, and Cancer. J. Clin. Invest. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the Chemokine Network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 Promotes Tumour Incidence and Growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J. Neuroinflammation 2013, 10, 35. [Google Scholar] [CrossRef]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.F.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumour-Associated Macrophages in the Cutaneous SCC Microenvironment Are Heterogeneously Activated. J. Invest. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major Pathways Involved in Macrophage Polarization in Cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yang, L.; Cai, J.; Li, H.; Xing, Z.; Hou, Y. Phosphoinositide 3-Kinase/Akt and Its Related Signaling Pathways in the Regulation of Tumour-Associated Macrophages Polarization. Mol. Cell. Biochem. 2022, 477, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-ΚB: Blending Metabolism, Immunity, and Inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-KappaB: A Key Role in Inflammatory Diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Henkel, T. Function and Activation of NF-Kappa B in the Immune System. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-ΚB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-ΚB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IκB Kinase Complex in NF-ΚB Regulation and Beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-KappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-ΚB Subunits in the Control of Transcription. Cells 2016, 5. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S. NF- κ B Signaling in in Fl Ammation. 2017. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-ΚB, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.-C. NF-ΚB in Inflammation and Renal Diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good Cop, Bad Cop: The Different Faces of NF-KappaB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A.; Mayo, K.L. Atypical Pathways of NF-KappaB Activation and Aging. Exp. Gerontol. 2009, 44, 250–255. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. How Toll-like Receptors Signal: What We Know and What We Don’t Know. Curr. Opin. Immunol. 2006, 18, 3–9. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptor Downstream Signaling. Arthritis Res. Ther. 2005, 7, 12–19. [Google Scholar] [CrossRef]
- Ozato, K.; Tsujimura, H.; Tamura, T. Toll-like Receptor Signaling and Regulation of Cytokine Gene Expression in the Immune System. Biotechniques 2002, Suppl, 66–68, 70, 72 passim. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumour Cell Apoptosis Polarizes Macrophages Role of Sphingosine-1-Phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP Represses the Generation of Alternatively Activated Macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A Distinct and Unique Transcriptional Program Expressed by Tumour-Associated Macrophages (Defective NF-KappaB and Enhanced IRF-3/STAT1 Activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, S.; Liu, S.; Li, Z.; Liu, X.; Wu, J. Baicalein Potentiated M1 Macrophage Polarization in Cancer Through Targeting PI3Kγ/ NF-ΚB Signaling. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Fang, B.; Zhang, Y.; Wang, C.; Zhou, J.; Niu, C.; Gao, Y.; Zhao, D.; He, J.; Wang, J.; et al. Mechanical Stretch Promotes Tumouricidal M1 Polarization via the FAK/NF-ΚB Signaling Pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13254–13266. [Google Scholar] [CrossRef]
- Gao, J.; Wang, D.; Liu, D.; Liu, M.; Ge, Y.; Jiang, M.; Liu, Y.; Zheng, D. Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand Induces the Expression of Proinflammatory Cytokines in Macrophages and Re-Educates Tumour-Associated Macrophages to an Antitumour Phenotype. Mol. Biol. Cell 2015, 26, 3178–3189. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Wu, C.-L.; Shiau, A.-L. Toll-like Receptor 4 Signaling Promotes Tumour Growth. J. Immunother. 2010, 33, 73–82. [Google Scholar] [CrossRef]
- He, R.; He, Y.; Du, R.; Liu, C.; Chen, Z.; Zeng, A.; Song, L. Revisiting of TAMs in Tumour Immune Microenvironment: Insight from NF-ΚB Signaling Pathway. Biomed. Pharmacother. 2023, 165. [Google Scholar] [CrossRef]
- Yang, Y.; Qin, J.; Lan, L.; Li, N.; Wang, C.; He, P.; Liu, F.; Ni, H.; Wang, Y. M-CSF Cooperating with NFκB Induces Macrophage Transformation from M1 to M2 by Upregulating c-Jun. Cancer Biol. Ther. 2014, 15, 99–107. [Google Scholar] [CrossRef]
- Mancino, A.; Lawrence, T. Nuclear Factor-KappaB and Tumour-Associated Macrophages. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2010, 16, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Greten, F.R. IKK- and NF-ΚB-Mediated Functions in Carcinogenesis. Curr. Top. Microbiol. Immunol. 2011, 349, 159–169. [Google Scholar] [CrossRef]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 Nuclear Factor-ΚB Overexpression in Tumour-Associated Macrophages Inhibits M1 Inflammatory Responses and Antitumour Resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Kühnemuth, B.; Michl, P. The Role of CUX1 in Antagonizing NF-ΚB Signaling in TAMs. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef]
- Chang, C.P.; Su, Y.C.; Hu, C.W.; Lei, H.Y. TLR2-Dependent Selective Autophagy Regulates NF-ΚB Lysosomal Degradation in Hepatoma-Derived M2 Macrophage Differentiation. Cell Death Differ. 2013, 20, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular Mechanisms of IL-33-Mediated Stromal Interactions in Cancer Metastasis. JCI insight 2018, 3. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Span, P.N.; Bussink, J. Biology of Hypoxia. Semin. Nucl. Med. 2015, 45, 101–109. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.-F. Hypoxia and the Tumour Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumour-Associated Macrophages. Biomed Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-KappaB Links Innate Immunity to the Hypoxic Response through Transcriptional Regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A Key Player in Antitumour Immune Response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–79. [Google Scholar] [CrossRef]
- Delprat, V.; Tellier, C.; Demazy, C.; Raes, M.; Feron, O.; Michiels, C. Cycling Hypoxia Promotes a Pro-Inflammatory Phenotype in Macrophages via JNK/P65 Signaling Pathway. Sci. Rep. 2020, 10, 882. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, G.F.; Kuzmina, D.O.; Nuzhina, J.; Shtil, A.A.; Dukhinova, M.S. How Macrophages Become Transcriptionally Dysregulated: A Hidden Impact of Antitumour Therapy. Int. J. Mol. Sci. 2021, 22, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumour Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Beach, C.; MacLean, D.; Majorova, D.; Arnold, J.N.; Olcina, M.M. The Effects of Radiation Therapy on the Macrophage Response in Cancer. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Lödermann, B.; Wunderlich, R.; Frey, S.; Schorn, C.; Stangl, S.; Rödel, F.; Keilholz, L.; Fietkau, R.; Gaipl, U.S.; Frey, B. Low Dose Ionising Radiation Leads to a NF-ΚB Dependent Decreased Secretion of Active IL-1β by Activated Macrophages with a Discontinuous Dose-Dependency. Int. J. Radiat. Biol. 2012, 88, 727–734. [Google Scholar] [CrossRef]
- Genard, G.; Wera, A.C.; Huart, C.; Le Calve, B.; Penninckx, S.; Fattaccioli, A.; Tabarrant, T.; Demazy, C.; Ninane, N.; Heuskin, A.C.; et al. Proton Irradiation Orchestrates Macrophage Reprogramming through NFκB Signaling. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Crittenden, M.R.; Cottam, B.; Savage, T.; Nguyen, C.; Newell, P.; Gough, M.J. Expression of NF-ΚB P50 in Tumour Stroma Limits the Control of Tumours by Radiation Therapy. PLoS One 2012, 7, e39295. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumour-Associated Macrophages Promote Cancer Stem Cell-like Properties via Transforming Growth Factor-Beta1-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of Venous Metastases, Recurrence, and Prognosis in Hepatocellular Carcinoma Based on a Unique Immune Response Signature of the Liver Microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKbeta Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation That Promotes Chemical Hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-KappaB Functions as a Tumour Promoter in Inflammation-Associated Cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in Liver Parenchymal Cells Causes Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.-Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1 Alpha Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Zhu, W.W.; Yu, G.Y.; Wang, X.; Gao, C.; Zhou, X.; Lin, Z.F.; Shao, W.Q.; Wang, S.H.; Lu, M.; et al. S100 Calcium-Binding Protein A9 from Tumour-Associated Macrophage Enhances Cancer Stem Cell-like Properties of Hepatocellular Carcinoma. Int. J. Cancer 2021, 148, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, L.; Spadaro, F.; Purificato, C.; Cecchetti, S.; Podo, F.; Belardelli, F.; Gessani, S.; Ramoni, C. Phosphatidylcholine-Specific Phospholipase C Activation Is Required for CCR5-Dependent, NF-KB-Driven CCL2 Secretion Elicited in Response to HIV-1 Gp120 in Human Primary Macrophages. Blood 2008, 111, 3355–3363. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, P.; Wang, W.; Yi, M.; Chen, P.; Cai, J.; Li, J.; Peng, Q.; Ban, Y.; Zhou, Y.; et al. Abberent Expression of NOR1 Protein in Tumour Associated Macrophages Contributes to the Development of DEN-Induced Hepatocellular Carcinoma. J. Cell. Physiol. 2018, 233, 5002–5013. [Google Scholar] [CrossRef]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 Beta Mediates the NF-Kappa B Suppression of JNK Signalling by Targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of Gadd45beta by NF-KappaB Downregulates pro-Apoptotic JNK Signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef]
- Verzella, D.; Bennett, J.; Fischietti, M.; Thotakura, A.K.; Recordati, C.; Pasqualini, F.; Capece, D.; Vecchiotti, D.; D’Andrea, D.; Di Francesco, B.; et al. GADD45β Loss Ablates Innate Immunosuppression in Cancer. Cancer Res. 2018, 78, 1275–1292. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.C.; Yi, Z.J.; Zhou, Y.; Li, P.Z.; Liu, Z.J.; Duan, S.G.; Gong, J.P. Overexpression of RIP 140 Suppresses the Malignant Potential of Hepatocellular Carcinoma by Inhibiting NF-KB-Mediated Alternative Polarization of Macrophages. Oncol. Rep. 2017, 37, 2971–2979. [Google Scholar] [CrossRef]
- Sharen, G.; Cheng, H.; Hu, X.; Miao, J.; Zhao, D. M1-like Tumour-Associated Macrophages Enhance Proliferation and Anti-Apoptotic Ability of Liver Cancer Cells via Activating the NF-ΚB Signaling Pathway. Mol. Med. Rep. 2022, 26. [Google Scholar] [CrossRef]
- Wu, S.-P.; Liao, R.-Q.; Tu, H.-Y.; Wang, W.-J.; Dong, Z.-Y.; Huang, S.-M.; Guo, W.-B.; Gou, L.-Y.; Sun, H.-W.; Zhang, Q.; et al. Stromal PD-L1-Positive Regulatory T Cells and PD-1-Positive CD8-Positive T Cells Define the Response of Different Subsets of Non-Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 521–532. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumour Responses with Lambrolizumab (Anti-PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Clark, J.I.; Quinn, D.I. Immune Checkpoint Inhibitors in Advanced Renal Cell Carcinoma: Experience to Date and Future Directions. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Martini, D.J.; Lalani, A.-K.A.; Bossé, D.; Steinharter, J.A.; Harshman, L.C.; Hodi, F.S.; Ott, P.A.; Choueiri, T.K. Response to Single Agent PD-1 Inhibitor after Progression on Previous PD-1/PD-L1 Inhibitors: A Case Series. J. Immunother. cancer 2017, 5, 66. [Google Scholar] [CrossRef]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-Based Hepatocellular Carcinoma Vaccine Facilitates Anti-PD-1 Therapy by Regulating Macrophage Polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef]
- Van den Eynden, G.G.; Majeed, A.W.; Illemann, M.; Vermeulen, P.B.; Bird, N.C.; Høyer-Hansen, G.; Eefsen, R.L.; Reynolds, A.R.; Brodt, P. The Multifaceted Role of the Microenvironment in Liver Metastasis: Biology and Clinical Implications. Cancer Res. 2013, 73, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 Alters BMP Signaling to Promote Colorectal Cancer Cell Metastasis via Activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208. [Google Scholar] [CrossRef]
- Li, M.; Lai, X.; Zhao, Y.; Zhang, Y.; Li, M.; Li, D.; Kong, J.; Zhang, Y.; Jing, P.; Li, H.; et al. Loss of NDRG2 in Liver Microenvironment Inhibits Cancer Liver Metastasis by Regulating Tumour Associate Macrophages Polarization Article /13/1 /13/21 /13/31 /13/95 /14/5 /14/19 /38/77 /38/109 /64/60 /82/80. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.S.; Yu, L.G.; Zhang, X.K.; Zhao, L.; Gong, F.L.; Yang, X.X.; Guo, X.L. Galectin-3 Expression and Secretion by Tumour-Associated Macrophages in Hypoxia Promotes Breast Cancer Progression. Biochem. Pharmacol. 2020, 178. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 Derived from Tumour-Associated Macrophages Promotes Breast Cancer Metastasis via Activating NF-ΚB/SOX4 Signaling. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 Enhances Breast Cancer Growth and Migration by Promoting Alternative Macrophage Polarization in the Tumour Microenvironment. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- De Paolis, V.; Maiullari, F.; Chirivì, M.; Milan, M.; Cordiglieri, C.; Pagano, F.; La Manna, A.R.; De Falco, E.; Bearzi, C.; Rizzi, R.; et al. Unusual Association of NF-ΚB Components in Tumour-Associated Macrophages (TAMs) Promotes HSPG2-Mediated Immune-Escaping Mechanism in Breast Cancer. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS One 2008, 3, e2888. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A Breast Cancer Stem Cell Niche Supported by Juxtacrine Signalling from Monocytes and Macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and Mesenchymal Subpopulations within Normal Basal Breast Cell Lines Exhibit Distinct Stem Cell/Progenitor Properties. Stem Cells 2012, 30, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Di Vito Nolfi, M.; Vecchiotti, D.; Flati, I.; Verzella, D.; Di Padova, M.; Alesse, E.; Capece, D.; Zazzeroni, F. EV-Mediated Chemoresistance in the Tumour Microenvironment: Is NF-ΚB a Player? Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Li, C.; Li, R.; Hu, X.; Zhou, G.; Jiang, G. Tumour-Promoting Mechanisms of Macrophage-Derived Extracellular Vesicles-Enclosed MicroRNA-660 in Breast Cancer Progression. Breast Cancer Res. Treat. 2022, 192, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, T.; Zhang, J. Tumour-Associated Macrophages (Tams) in Colorectal Cancer (Crc): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High Macrophage Infiltration along the Tumour Front Correlates with Improved Survival in Colon Cancer. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-C.; Chen, J.-S.; Lee, C.-H.; Chang, J.-J.; Shieh, Y.-S. Intratumoural Macrophage Counts Correlate with Tumour Progression in Colorectal Cancer. J. Surg. Oncol. 2010, 102, 242–248. [Google Scholar] [CrossRef]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.-A.; Palmqvist, R. The Distribution of Macrophages with a M1 or M2 Phenotype in Relation to Prognosis and the Molecular Characteristics of Colorectal Cancer. PLoS One 2012, 7, e47045. [Google Scholar] [CrossRef]
- Porta, C.; Ippolito, A.; Consonni, F.M.; Carraro, L.; Celesti, G.; Correale, C.; Grizzi, F.; Pasqualini, F.; Tartari, S.; Rinaldi, M.; et al. Protumour Steering of Cancer Inflammation by P50 Nf-Kb Enhances Colorectal Cancer Progression. Cancer Immunol. Res. 2018, 6, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Rawluszko, A.A.; Bujnicka, K.E.; Horbacka, K.; Krokowicz, P.; Jagodziński, P.P. Expression and DNA Methylation Levels of Prolyl Hydroxylases PHD1, PHD2, PHD3 and Asparaginyl Hydroxylase FIH in Colorectal Cancer. BMC Cancer 2013, 13, 526. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Niu, Z.; Wang, X.; Li, Z.; Liu, Y.; Luo, F.; Yan, X. PHD2 Exerts Anti-Cancer and Anti-Inflammatory Effects in Colon Cancer Xenografts Mice via Attenuating NF-ΚB Activity. Life Sci. 2020, 242. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; D’Andrea, D.; Begalli, F.; Goracci, L.; Tornatore, L.; Alexander, J.L.; Di Veroli, A.; Leow, S.-C.; Vaiyapuri, T.S.; Ellis, J.K.; et al. Enhanced Triacylglycerol Catabolism by Carboxylesterase 1 Promotes Aggressive Colorectal Carcinoma. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Ji, X.; Zhang, L.; Chen, J.; Li, C.; Shi, R.; Xiang, W.; Kang, X.; Zhang, D.; Yang, F.; et al. Macrophage ABHD5 Suppresses NFκB-Dependent Matrix Metalloproteinase Expression and Cancer Metastasis. Cancer Res. 2019, 79, 5513–5526. [Google Scholar] [CrossRef] [PubMed]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg Metabolism in Tumour-Conditioned Macrophages Promotes Metastasis in Human Pancreatic Ductal Adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/MTOR Inhibitors: Rationale and Importance to Inhibiting These Pathways in Human Health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in Experimental and Human Brain Tumours. Part 2: Studies of the Macrophage Content of Human Brain Tumours. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Chen, G.G.; Chu, Y.S.; Chak, E.C.W.; Leung, B.C.S.; Poon, W.S. Induction of Apoptosis in Glioma Cells by Molecules Released from Activated Macrophages. J. Neurooncol. 2002, 57, 179–186. [Google Scholar] [CrossRef]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFκB Signaling in Myeloid Cells Is Required for the Glioblastoma Growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef]
- Liang, F.; Liang, J.; Wang, W.-Q.; Sun, J.-P.; Udho, E.; Zhang, Z.-Y. PRL3 Promotes Cell Invasion and Proliferation by Down-Regulation of Csk Leading to Src Activation. J. Biol. Chem. 2007, 282, 5413–5419. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, L.; Lai, W.; Zeng, Y.; Xu, H.; Lan, Q.; Su, P.; Chu, Z. Interaction with Tumour-associated Macrophages Promotes PRL-3-induced Invasion of Colorectal Cancer Cells via MAPK Pathway-induced EMT and NF-κB Signaling-induced Angiogenesis. Oncol. Rep. 2019, 41, 2790–2802. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Q.; Chen, J.; Zhang, N.; Liu, C.; Wang, T.; Yang, F.; Siebert, H.C.; Zheng, X. Ketogenic Diet Elicits Antitumour Properties through Inducing Oxidative Stress, Inhibiting MMP-9 Expression, and Rebalancing M1/ M2 Tumour-Associated Macrophage Phenotype in a Mouse Model of Colon Cancer. J. Agric. Food Chem. 2020, 68, 11182–11196. [Google Scholar] [CrossRef]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumour Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Fehri, E.; Ennaifer, E.; Bel Haj Rhouma, R.; Guizani-Tabbane, L.; Guizani, I.; Boubaker, S. The Role of Toll-like Receptor 9 in Gynecologic Cancer. Curr. Res. Transl. Med. 2016, 64, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Regulation of Immunity and Inflammation by Hypoxia in Immunological Niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-Dependent Regulation of Inflammatory Pathways in Immune Cells. J. Clin. Invest. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular Profiling Reveals a Tumour-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Deng, Y.R.; Wang, Z.C.; Wei, W.F.; Zhou, C.F.; Zhang, Y.M.; Yan, R.M.; Liang, L.J.; Zhong, M.; Liang, L.; et al. Correction: Hypoxia-Induced ZEB1 Promotes Cervical Cancer Progression via CCL8-Dependent Tumour-Associated Macrophage Recruitment (Cell Death & Disease, (2019), 10, 7, (508), 10.1038/S41419-019-1748-1). Cell Death Dis. 2022, 13. [Google Scholar] [CrossRef]
- Vitale, C.; Bottino, C.; Castriconi, R. Monocyte and Macrophage in Neuroblastoma: Blocking Their Pro-Tumoral Functions and Strengthening Their Crosstalk with Natural Killer Cells. Cells 2023, 12. [Google Scholar] [CrossRef]
- Leblond, M.M.; Zdimerova, H.; Desponds, E.; Verdeil, G. Tumour-Associated Macrophages in Bladder Cancer: Biological Role, Impact on Therapeutic Response and Perspectives for Immunotherapy. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef]
- Sun, J.; Park, C.; Guenthner, N.; Gurley, S.; Zhang, L.; Lubben, B.; Adebayo, O.; Bash, H.; Chen, Y.; Maksimos, M.; et al. Tumour-Associated Macrophages in Multiple Myeloma: Advances in Biology and Therapy. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef]
- Chen, Y.L. Prognostic Significance of Tumour-Associated Macrophages in Patients with Nasopharyngeal Carcinoma: A Meta-Analysis. Med. (United States) 2020, 99, E21999. [Google Scholar] [CrossRef]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumour-Associated Macrophages in Sarcomas. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumour-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing Pancreatic Cancer: Combat with a Double Edged Sword. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- XU, F.; WEI, Y.; TANG, Z.; LIU, B.; DONG, J. Tumour-Associated Macrophages in Lung Cancer: Friend or Foe? Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, J.; Chen, S.; Ma, X.; Ying, Y.; Li, J.; Wang, W.; Wang, X.; Xie, L. Prognostic Value of Tumour-Associated Macrophages in Clear Cell Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 657318. [Google Scholar] [CrossRef]
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumour-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Gambardella, V.; Castillo, J.; Tarazona, N.; Gimeno-Valiente, F.; Martínez-Ciarpaglini, C.; Cabeza-Segura, M.; Roselló, S.; Roda, D.; Huerta, M.; Cervantes, A.; et al. The Role of Tumour-Associated Macrophages in Gastric Cancer Development and Their Potential as a Therapeutic Target. Cancer Treat. Rev. 2020, 86. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumour-Associated Macrophages: Therapeutic Targets for Skin Cancer. Front. Oncol. 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.; Stafford, H.; Buell, J.; Ramesh, U.; Amit, M.; Nagarajan, P.; Migden, M.; Yaniv, D. Review of the Tumour Microenvironment in Basal and Squamous Cell Carcinoma. Cancers (Basel). 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Fang, T.; Wei, S.; Chai, S.; Yang, H.; Tao, M.; Cao, Y. Macrophages in Melanoma: A Double-edged Sword and Targeted Therapy Strategies (Review). Exp. Ther. Med. 2022, 24, 640. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Song, X.; Fan, S.; Deng, R. The Role of Tumour-Associated Macrophages in Oral Squamous Cell Carcinoma. Front. Physiol. 2022, 13, 959747. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumour-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Choi, S.H. Tumour-Associated Macrophages in Cancer: Recent Advancements in Cancer Nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41. [Google Scholar] [CrossRef]
- Qiu, B.; Matthay, K.K. Advancing Therapy for Neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef]
- Garcia, J.A.; Cowey, C.L.; Godley, P.A. Renal Cell Carcinoma. Curr. Opin. Oncol. 2009, 21, 266–271. [Google Scholar] [CrossRef]
- Mukherjee, S.; Baidoo, J.; Fried, A.; Atwi, D.; Dolai, S.; Boockvar, J.; Symons, M.; Ruggieri, R.; Raja, K.; Banerjee, P. Curcumin Changes the Polarity of Tumour-Associated Microglia and Eliminates Glioblastoma. Int. J. cancer 2016, 139, 2838–2849. [Google Scholar] [CrossRef]
- Kai, K.; Komohara, Y.; Esumi, S.; Fujiwara, Y.; Yamamoto, T.; Uekawa, K.; Ohta, K.; Takezaki, T.; Kuroda, J.; Shinojima, N.; et al. Macrophage/Microglia-Derived IL-1β Induces Glioblastoma Growth via the STAT3/NF-ΚB Pathway. Hum. Cell 2022, 35, 226–237. [Google Scholar] [CrossRef]
- Wu, H.; Xu, J.B.; He, Y.L.; Peng, J.J.; Zhang, X.H.; Chen, C.Q.; Li, W.; Cai, S.R. Tumour-Associated Macrophages Promote Angiogenesis and Lymphangiogenesis of Gastric Cancer. J. Surg. Oncol. 2012, 106, 462–468. [Google Scholar] [CrossRef]
- Tjiu, J.W.; Chen, J.S.; Shun, C.T.; Lin, S.J.; Liao, Y.H.; Chu, C.Y.; Tsai, T.F.; Chiu, H.C.; Dai, Y.S.; Inoue, H.; et al. Tumour-Associated Macrophage-Induced Invasion and Angiogenesis of Human Basal Cell Carcinoma Cells by Cyclooxygenase-2 Induction. J. Invest. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef]
- Gutiérrez-Seijo, A.; García-Martínez, E.; Barrio-Alonso, C.; Pareja-Malagón, M.; Acosta-Ocampo, A.; Fernández-Santos, M.E.; Puig-Kröger, A.; Parra-Blanco, V.; Mercader, E.; Márquez-Rodas, I.; et al. Ccl20/Tnf/Vegfa Cytokine Secretory Phenotype of Tumour-Associated Macrophages Is a Negative Prognostic Factor in Cutaneous Melanoma. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef]
- Paccez, J.D.; Vogelsang, M.; Parker, M.I.; Zerbini, L.F. The Receptor Tyrosine Kinase Axl in Cancer: Biological Functions and Therapeutic Implications. Int. J. cancer 2014, 134, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, V.A. Axl-Dependent Signalling: A Clinical Update. Clin. Sci. (Lond). 2012, 122, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of Tumour-Associated Macrophages and Gas6/Axl Signaling in Oral Squamous Cell Carcinoma. Oral Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Liu, S.Y.; Chou, K.C.; Yeh, C.T.; Shiah, S.G.; Huang, R.Y.; Cheng, J.C.; Yen, C.Y.; Shieh, Y.S. Tumour-Associated Macrophages Promote Oral Cancer Progression through Activation of the Axl Signaling Pathway. Ann. Surg. Oncol. 2014, 21, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Vecchiotti, D.; Verzella, D.; Di Vito Nolfi, M.; D’andrea, D.; Flati, I.; Di Francesco, B.; Cornice, J.; Alesse, E.; Capece, D.; Zazzeroni, F. Elevated NF-ΚB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Cheng, H.-C.; Wang, J.; Wang, S.-W.; Tai, H.-C.; Lin, C.-W.; Tang, C.-H. Prostate Cancer-Derived CCN3 Induces M2 Macrophage Infiltration and Contributes to Angiogenesis in Prostate Cancer Microenvironment; 2014; Vol. 5. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple Myeloma Cells Recruit Tumour-Supportive Macrophages through the CXCR4/CXCL12 Axis and Promote Their Polarization toward the M2 Phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef] [PubMed]
- Khalife, J.; Ghose, J.; Martella, M.; Viola, D.; Rocci, A.; Troadec, E.; Terrazas, C.; Satoskar, A.R.; Gunes, E.G.; Dona, A.; et al. MiR-16 Regulates Crosstalk in NF-ΚB Tolerogenic Inflammatory Signaling between Myeloma Cells and Bone Marrow Macrophages. JCI insight 2019, 4. [Google Scholar] [CrossRef]
- Song, Y.; Li, X.; Zeng, Z.; Li, Q.; Gong, Z.; Liao, Q.; Li, X.; Chen, P.; Xiang, B.; Zhang, W.; et al. Epstein-Barr Virus Encoded MiR-BART11 Promotes Inflammation-Induced Carcinogenesis by Targeting FOXP1; 2016; Vol. 7. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An Epigenetic Switch Involving NF-KappaB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Li, Q.; Hao, Z.; Hong, Y.; He, W.; Zhao, W. Reprogramming Tumour Associated Macrophage Phenotype by a Polysaccharide from Ilex Asprella for Sarcoma Immunotherapy. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Kühnemuth, B.; Mühlberg, L.; Schipper, M.; Griesmann, H.; Neesse, A.; Milosevic, N.; Wissniowski, T.; Buchholz, M.; Gress, T.M.; Michl, P. CUX1 Modulates Polarization of Tumour-Associated Macrophages by Antagonizing NF-ΚB Signaling. Oncogene 2015, 34, 177–187. [Google Scholar] [CrossRef]
- Kemp, S.B.; Carpenter, E.S.; Steele, N.G.; Donahue, K.L.; Nwosu, Z.C.; Pacheco, A.; Velez-Delgado, A.; Menjivar, R.E.; Lima, F.; The, S.; et al. Apolipoprotein E Promotes Immune Suppression in Pancreatic Cancer through NF-ΚB-Mediated Production of CXCL1. Cancer Res. 2021, 81, 4305–4318. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Shen, H.; Zhu, L.; Zhao, F.; Shu, Y. Plasminogen Activator Inhibitor 1 Promotes Immunosuppression in Human Non-Small Cell Lung Cancers by Enhancing TGF-Β1 Expression in Macrophage. Cell. Physiol. Biochem. 2018, 44, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yu, W.; Cao, L.; Meng, F.; Cong, M. A Novel Chrysin Thiazole Derivative Polarizes Macrophages to an M1 Phenotype via Targeting TLR4. Int. Immunopharmacol. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Fan, Z.; Liu, P.; Chen, L.; Guan, Z.; Liu, Y.; Luo, Y. Anemoside A3 Activates TLR4-Dependent M1-Phenotype Macrophage Polarization to Represses Breast Tumour Growth and Angiogenesis. Toxicol. Appl. Pharmacol. 2021, 432. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Sun, L.; Shen, D.; Ren, A.; Ma, F.; Tai, G.; Fan, L.; Zhou, Y. Beta-1,6 Glucan Converts Tumour-Associated Macrophages into an M1-like Phenotype. Carbohydr. Polym. 2020, 247. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Han, X.; Sha, X.; Tian, F.; Huang, H.; Jiang, P.; Huang, G.; Ma, B.; Zhang, H.; Zhu, Y.; et al. Aqueous Extract of Cimicifuga Dahurica Reprogramming Macrophage Polarization by Activating TLR4-NF-ΚB Signaling Pathway. J. Inflamm. Res. 2022, 15, 1027–1046. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, D.; Zhang, S.; Chen, H.; Zhao, J.; Li, X.; Zeng, X. Homogeneous Polyporus Polysaccharide Inhibit Bladder Cancer by Resetting Tumour-Associated Macrophages Toward M1 Through NF-ΚB/NLRP3 Signaling. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Hoover, A.A.; Hufnagel, D.H.; Harris, W.; Bullock, K.; Glass, E.B.; Liu, E.; Barham, W.; Crispens, M.A.; Khabele, D.; Giorgio, T.D.; et al. Increased Canonical NF-KappaB Signaling Specifically in Macrophages Is Sufficient to Limit Tumour Progression in Syngeneic Murine Models of Ovarian Cancer. BMC Cancer 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Mao, Z.; Sun, J. NF-ΚB Inhibitor, BAY11-7082, Suppresses M2 Tumour-Associated Macrophage Induced EMT Potential via MiR-30a/NF-ΚB/Snail Signaling in Bladder Cancer Cells. Gene 2019, 710, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Liu, S.; Luo, H.; Wu, S.; Wei, H.; Tang, Z.; Li, X.; Zou, C. LncRNA DCST1-AS1 Facilitates Oral Squamous Cell Carcinoma by Promoting M2 Macrophage Polarization through Activating NF- κ B Signaling. J. Immunol. Res. 2021, 2021. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumour-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, S.; Jiang, D.; Gao, T.; Fang, Y.; Fu, S.; Guan, L.; Zhang, Z.; Mu, W.; Chu, Q.; et al. Manipulation of TAMs Functions to Facilitate the Immune Therapy Effects of Immune Checkpoint Antibodies. J. Control. Release 2021, 336, 621–634. [Google Scholar] [CrossRef]
- Parayath, N.N.; Parikh, A.; Amiji, M.M. Repolarization of Tumour-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018, 18, 3571–3579. [Google Scholar] [CrossRef]
- Li, K.; Lu, L.; Xue, C.; Liu, J.; He, Y.; Zhou, J.; Xia, Z.; Dai, L.; Luo, Z.; Mao, Y.; et al. Polarization of Tumour-Associated Macrophage Phenotype: Via Porous Hollow Iron Nanoparticles for Tumour Immunotherapy in Vivo. Nanoscale 2020, 12, 130–144. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Guo, X.; Zhao, J.; Zhou, S. A Biomimetic Polymer Magnetic Nanocarrier Polarizing Tumour-Associated Macrophages for Potentiating Immunotherapy. Small 2020, 16. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Zhen, M.; Zhen, M.; Wang, H.; Wang, H.; Sun, Z.; Sun, Z.; Jia, W.; Jia, W.; et al. Functional Gadofullerene Nanoparticles Trigger Robust Cancer Immunotherapy Based on Rebuilding an Immunosuppressive Tumour Microenvironment. Nano Lett. 2020, 20, 4487–4496. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, B.; Zhang, S.; Liao, X.; Tong, Q.; Wei, G.; Yu, S.; Chen, G.; Wu, A.; Gao, S.; et al. Copper Sulfide Nanoparticle-Redirected Macrophages for Adoptive Transfer Therapy of Melanoma. Adv. Funct. Mater. 2021, 31, 1–12. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Huo, M.; Wang, Y.; Li, Y.; Xu, N.; Zhu, H. Cetuximab Enhances the Anti-Tumor Function of Macrophages in an IL-6 Dependent Manner. Life Sci. 2021, 267. [Google Scholar] [CrossRef]
- Cao, X.; Li, B.; Chen, J.; Dang, J.; Chen, S.; Gunes, E.G.; Xu, B.; Tian, L.; Muend, S.; Raoof, M.; et al. Effect of Cabazitaxel on Macrophages Improves CD47-Targeted Immunotherapy for Triple-Negative Breast Cancer. J. Immunother. cancer 2021, 9. [Google Scholar] [CrossRef]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumour Cells and Host Cells in Tumour Microenvironment. Front. Oncol. 2021, 11, 722916. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Guo, Y.; Mao, W.; Wang, J.; Jin, L.; Liu, X.; Shou, Q.; Fu, H. Total Glucosides of Paeony Inhibit Breast Cancer Growth by Inhibiting TAMs Infiltration through NF-ΚB/CCL2 Signaling. Phytomedicine 2022, 104. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.A.; Barham, W.; Sharman, K.; Tikhomirov, O.; Giorgio, T.D.; Yull, F.E. Manipulating the NF-ΚB Pathway in Macrophages Using Mannosylated, SiRNA-Delivering Nanoparticles Can Induce Immunostimulatory and Tumour Cytotoxic Functions. Int. J. Nanomedicine 2016, 11, 2163–2177. [Google Scholar] [CrossRef]
- Tsagozis, P.; Augsten, M.; Pisa, P. All Trans-Retinoic Acid Abrogates the pro-Tumorigenic Phenotype of Prostate Cancer Tumour-Associated Macrophages. Int. Immunopharmacol. 2014, 23, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces Tumour Growth by Reprogramming Tumour-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of Tumour-Infiltrating Macrophages via CCL2/CCR2 Signalling as a Therapeutic Strategy against Hepatocellular Carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Glass, E.B.; Hoover, A.A.; Bullock, K.K.; Madden, M.Z.; Reinfeld, B.I.; Harris, W.; Parker, D.; Hufnagel, D.H.; Crispens, M.A.; Khabele, D.; et al. Stimulating TAM-Mediated Anti-Tumor Immunity with Mannose-Decorated Nanoparticles in Ovarian Cancer. BMC Cancer 2022, 22. [Google Scholar] [CrossRef]
- Liu, M.; Sakamaki, T.; Casimiro, M.C.; Willmarth, N.E.; Quong, A.A.; Ju, X.; Ojeifo, J.; Jiao, X.; Yeow, W.-S.; Katiyar, S.; et al. The Canonical NF-KappaB Pathway Governs Mammary in Transgenic Mice and Tumour Stem Cell Expansion. Cancer Res. 2010, 70, 10464–10473. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Molecules/drugs targeting NF-κB pathways to reshape tumor microenvironment via termination of macrophage recruitment, TAMs depletion, and TAMs repolarization.
Table 1.
Molecules/drugs targeting NF-κB pathways to reshape tumor microenvironment via termination of macrophage recruitment, TAMs depletion, and TAMs repolarization.
| TAM REPROGRAMMING | |||
| Molecules/drug | Target | Cancers | ref |
| Chrysin thiazole derivative (ChR-TD) |
TLR4/NF-κB | Breast cancer cell line (4T1) | [179] |
| Anemoside A3 (A3) | TLR4/NF-κB | Breast cancer | [180] |
| β-D-(1→6) glucan (AAMP-A70) |
TLR2/Akt/NF-κB | Colon cancer cells | [181] |
| Aqueous Extract of Cimicifuga dahurica (CRAE) | TLR4/MyD88/TAK1/NF-κB | MM | [182] |
| Homogeneous Polyporus Polysaccharide (HPP) | TLR2/NF-κB/NLRP3 | Bladder cancer | [183] |
| Baicalein | PI3K/NF-κB | Breast cancer, melanoma | [54] |
| IKK2 | NF-κB | OC | [184] |
| BAY11-7082 | NF-κB/miR30a | Bladder cancer cells | [185] |
| IncRNA DCST1-AS1 | NF-κB | OSCC | [186] |
| Glycocalyx-mimicking nanoparticles (GNPs) | STAT6 and NF-κB | LLC | [187] |
| Mannose modified lipid nanoparticles (M-IMD-LNP) with IMD-0354 |
NF-κB | Melanoma cells (B16) | [188] |
| Hyaluronic acid (HA) nanoparticles, loaded with micro-RNA miR-125 |
NF-κB | NSCLC | [189] |
| Porous hollow iron oxide nanoparticles (PHNPs) loaded with 3-methyladenine (3-MA) | PI3Kγ/Akt/NF-κB |
Breast cancer cell line (MDA-MB-231) | [190] |
| PLGA-ION-R837@M | TLR7/IRF5/NF-κB | Breast cancer cell line (4T1) | [191] |
| Gd@C82 nanoparticles modified with b-alanines (GF-Ala) | NF-κB/IRF5 | Breast cancer cell line (4T1) | [192] |
| Copper sulfide nanoparticles (CuS-NP) | NF-κB | Melanoma | [193] |
| Cetuximab | NF-κB and STAT3 | CRC | [194] |
| Cabazitaxel | TLR/NF-κB | Breast cancer cells | [195] |
| Proton irradiation | NF-κB | THP1 cells | [77] |
| TAM DEPLETION AND TERMINATION OF RECRUITMENT | |||
| CCR2 antagonist or knocking out of host CCR2 | CCL2/CCR2/NF-κB | HCC | [196] |
| Total glucosides of paeony (TGP) | NF-κB/CCL2 | Breast cancer | [197] |
| IκBα si-RNA encapsulated into mannosylated siRNA-delivering NPs | NF-κB | OC, breast cancer |
[198] |
| Trans-retinoic acid (ATRA) | NF-κB | PCa |
[199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



