
Submitted:
18 January 2024
Posted:
19 January 2024
You are already at the latest version
Abstract
Understanding the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is critical for advancing treatment options. This review explores the novel hypothesis that herpesviruses' infection of endothelial cells (ECs) may underlie ME/CFS symptomatology. We review evidence linking herpesviruses to persistent EC infection and the implications for endothelial dysfunction, encompassing blood flow regulation, coagulation, and cognitive impairment – symptoms consistent with ME/CFS and Long COVID. The paper provides a synthesis of current research on herpesvirus latency and reactivation, detailing the impact on ECs and subsequent systemic complications, including latent modulation and long-term maladaptation. We suggest that the chronicity of ME/CFS symptoms and the multisystemic nature of the disease may be partly attributable to herpesvirus-induced endothelial maladaptation. Our conclusions underscore the necessity for further investigation into the prevalence and load of herpesvirus infection within ECs of ME/CFS patients. This review offers a conceptual advance by proposing an endothelial infection model as a systemic mechanism contributing to ME/CFS, steering future research towards potentially unexplored avenues in understanding and treating this complex syndrome.
Keywords:
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
; Endothelial cells
; Herpesvirus
1. Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a chronic condition characterized by unresolved fatigue, cognitive dysfunction, malaise, orthostatic issues, and post-exertional symptom exacerbation (PESE), among other symptoms [1]. The etiological cause is officially unknown, but viral infection is believed to be a precipitating factor and pathology is very much associated with viral activity [2,3,4,5]..
Herpesviruses are the most implicated in ME/CFS research [2,4,5,6,7,8,9,10], and as such the relationship between herpesviruses and ME/CFS has been reviewed extensively [2,4,10,11,12]. Herpesviruses have the ability to infect a number of different cell types within the body, but exhibit preference for a particular population. For instance, the primary target cells of EBV are B-cells [13], whereas human cytomegalovirus (HCMV) attacks non-lymphoid cells, of which endothelial cells are a favoured cell-type for infection [14,15]. That is not to say, however, that EBV, for example, is unable to infect cell types other than B-cells and cause significant pathological consequences.
Endothelial dysfunction is another prominent characteristic of ME/CFS pathology, and has been repeatedly demonstrated in older and more recent studies [16,17,18,19,20,21,22,23,24]. Blood flow, especially cerebral blood flow is perturbed and reduced in ME/CFS patients (van Campen et al., 2020b, van Campen et al., 2020a, van Campen et al., 2021b, Campen et al., 2022, Staud et al., 2018, Biswal et al., 2011, Boissoneault et al., 2019), as well as the perfusion of various brain regions (COSTA et al., 1995, Yoshiuchi et al., 2006, Shungu et al., 2012, Li et al., 2021). ME/CFS is not typically viewed as a vascular disease, but the aforementioned findings as well as evidence of endothelial and vascular dysfunction in Long COVID (Ambrosino et al., 2021, Haffke et al., 2022, Stahl et al., 2020, Turner et al., 2023), a disease that shares many symptoms with ME/CFS [25,26,27,28], begs the question as to whether or not endothelial and vascular pathology are important factors for ME/CFS pathology and symptom manifestation.
We have recently demonstrated hematological pathology in ME/CFS platelet-poor plasma (PPP) samples, specifically pertaining to platelet and clotting processes [29]. We found significant levels of amyloid fibrin(ogen)/microclots – the clotting material that is implicated in Long COVID [30,31,32] – in ME/CFS PPP samples. These microclots are therapeutically targeted with considerable success in Long COVID patients [33]. Other research groups have also demonstrated platelet abnormalities in ME/CFS cohorts [34,35,36], as well as abnormalities in clot formation and kinetics [37]. It is well-known that the integrity of endothelial cells and normal signalling thereof is paramount for the regulation of coagulation [38,39], and this leads to the recognition that endothelial dysfunction might be, at least in part, responsible for the abnormalities in coagulation and platelet function observed in ME/CFS patients.
The ideas of impaired circulatory function, reduced tissue oxygen supply, and unmet metabolic demands revolving around endothelial dysfunction and its inability to correctly regulate vascular tone have been discussed in the context of ME/CFS pathology and symptom manifestation before, and have even been tied to symptoms including fatigue and cognitive dysfunction (Wirth and Scheibenbogen, 2020, Wirth et al., 2021, Wirth and Scheibenbogen, 2021)[40].
Circling back to viruses, very few or no studies have focussed on herpesvirus infection of the endothelium in ME/CFS and the consequences that this might have for pathology and symptom manifestation. It is acknowledged that herpesviruses can induce pathology independent of the endothelium, phenomena which certainly have relevance to ME/CFS and other diseases. However, here we aim to focus on herpesviruses and the endothelium or associated tissue. There are a number of different herpesviruses, but the focus here will be on the ones previously implicated in ME/CFS, such as HHV-4 (EBV), HHV-5 (HCMV), and HHV-6 [2,3,4,5,41,42]. For an overview of the idea presented in this paper, as well as a mind map for clarification, see Figure 1.
2. Infection of Endothelial Cells By Herpesviruses, Latent Modulation, Systemic Complications, and the Potential for Long-Term Maladaptation
With relevance to the present idea, endothelial cells (ECs) are known to be able to succumb to infection by EBV [43,44,45,46,47,48,49,50], HCMV [14,15,51,52,53,54,55,56], and HHV-6 [57,58,59,60,61,62]. Other herpesviruses also infect ECs [63,64]. Since herpesviruses are intimately associated with ME/CFS, this already provides reason to hypothesize that the endothelium in ME/CFS patients is, to some extent, infected by herpesviruses.
Next, it is important to state that herpesvirus latency, specifically, can occur in ECs. Herpesviruses establish lifelong infection/latency by either integrating their genome into the host genome or, more commonly, having the genetic material exist in the nucleus as an episome [65]. Both HHV-6 [59,60,61,66,67], and HCMV latently infects ECs (and smooth muscle cells) [14,56,68,69], which are the HCMV’s preferred cell type for infection. There are indications that EBV establishes latency in ECs [48,49] and certainly evidence that EBV infects ECs (as will be discussed in the following sections); but the exact mechanisms around EBV latency in this cell type seems to be unestablished.
Active viral infection has been shown in ME/CFS [3,41,70,71], but has not been specifically studied in an endothelial context. Active infection might not necessarily coincide with all symptoms as active infection is not necessarily a continuous process; hence active infection might fall short in providing a unitary explanation for daily symptoms. While active infection will certainly involve the endothelium, it may be true that latency is sufficient enough to induce endothelial dysfunction that brings about ME/CFS symptoms – that is, if herpesvirus latency occurs within ECs (it is possible that latency in non-ECs can indirectly cause endothelial dysfunction too, see later).
As with the development of bacterial dormancy [72,73], herpesvirus latency is not a passive process [65,74,75], especially from the perspective of the host cell. For example, HHV-8 latency causes dysfunction of the endothelium, including increased permeability, disruption of endothelial cell junctions, activation of NF-κB, angiogenic disturbances, and increased protein phosphorylation [76,77,78,79]. There are viral proteins and nucleic material that regulate latency and reactivation, modulate host cell functions and proliferation, and ensure viral subversion of the immune system [65,80]. The activity of herpesvirus latency and all the specific molecular processes that occur within the host nucleus (as well as those that occur in the cytosol and extracellularly) might bring about specific defects at the endothelial layer. The immune and neurological systems are certainly involved, but so might be the endothelium.
ME/CFS is a chronic condition, and the chronicity of symptoms bears an explanation. Herpesvirus infection of ECs might be able to explain the persisting nature of ME/CFS symptoms. Firstly, endothelium has a low turnover, with the entire population being replaced every 6 years in adults [81]. Other studies estimate anywhere from months to decades [82], but note that tissue-specific ECs vary in rates of turnover. Regardless, the idea is that the endothelium is a normally long-lasting tissue, and viral-induced dysfunction, especially caused by persisting viruses that can establish latency, may continue for the rest of an EC’s life. Relevantly, EBV, HCMV, and HHV-8 inhibit apoptosis in ECs [83,84,85] and can increase the persistence of dysfunctional ECs. HHV-6 also inhibits apoptosis of host cells [86].
This ability of herpesviruses to establish latency in (endothelial) host cells, along with the long life span of ECs, means that herpesvirus-infected endothelium and the resulting complications may persist for months to years. This is a timeline which is in accord with the chronicity of ME/CFS symptoms. Hence, it is plausible that ME/CFS ECs undergo chronic maladaptation as a result of (latent) herpesvirus infection, which might be important for the maintenance of pathology and symptom manifestation. Furthermore, this may be an ongoing phenomenon in other diseases too.
A particular significance of this reasoning is its focus on the cell type that we are postulating to be involved and infected. ECs are the interface between blood and tissue. They enable gas exchange, nourishment, and waste clearance, regulate inflammatory processes, secrete bioactive amines that contribute to hematological and vascular homeostasis, and perform many other essential physiological functions. Any dysfunction in these cells and their functional processes are likely to contribute to or induce pathology. Furthermore, the vasculature extends into every organ system, and hence such endothelial dysfunction might account for the multi-organ and systemic nature of ME/CFS pathology.
3. Latent Infection by Herpesviruses is Sufficient to Bring About Cellular Dysfunction, and Might Hold Relevance to Endothelial Dysfunction and Symptom Manifestation in ME/CFS
Herpesviruses are persistent viruses that affect host cells for a lifetime, supporting the notion that latency might be able to cause chronic pathologies like ME/CFS, in susceptible individuals. It is emphasized that persistent, latent infection is not a passive process and in fact exerts pathological effects on the host [74,75,87,88]. Hence, herpesvirus reactivation and active infection may not be a necessity for the manifestation of ME/CFS symptoms, although it is expected to exacerbate any issues. Here, we aim to show that latent proteins from herpesviruses are sufficient to induce cellular dysfunction, and hint at the idea that they and latency-related processes contribute to the endothelial and vascular dysfunction – as well as other pathophysiological characteristics – observed in ME/CFS.
Herpesviruses use a number of proteins and microRNAs to drive latency, evade immune surveillance, regulate host processes, and coordinate the transition to active infection [65,89]. Studies of HHV-8 and ECs have shown significant dysfunction as a result of infection, where multiple cell signalling pathways are disturbed by viral activity [90]. Most notably, Liu et al (2010) demonstrated that latent proteins cause the activation of a Notch ligands and receptors, and induce the expression of endothelial precursor markers prompting a state of differentiation. It has also been shown that HHV-8 latency causes increased permeability, disruption of endothelial junctions, activation of NF-κB, and angiogenic disturbances (Guilluy et al., 2011, DiMaio et al., 2011, Grossmann et al., 2006)[76,77,79]. These studies, although conducted on HHV-8, provide mechanistic evidence for the present idea of herpesvirus-induced endothelial dysfunction, and are represented in Figure 2.
EBV is a heavily studied herpesvirus, most notably due to its ability to transform lymphocytes and epithelial cells into malignant phenotypes [91,92], and for its role in causing infectious mononucleosis. In fact, it is well known that EBV latent genes and their products specifically interrupt cell cycles and promote oncogenesis [89], and both latent and lytic gene products illicit notable immune responses [93]. Epstein-Barr Nuclear Antigens (EBNAs) are a group of proteins encoded by EBV which are essential for viral genome replication and transcription, the establishment and regulation of latency (even though some function for lytic processes), and immune evasion [94,95,96]. Other latency-associated molecules encoded by EBV include latent membrane proteins (LMPs) and EBV-encoded small RNAs (EBERs) [97,98].
EBNA-1 significantly increases ROS production in the host cells that they infect, and, via this mechanism, contributes to DNA damage and the inhibition of the repair thereof [99,100,101]. It inhibits apoptosis and enhances cell survival, contributing to its recognition as an oncoprotein [102]. Expression of this latent protein in ECs is associated with higher IL-6 production [48], and hints at the potential of an EBV-infected endothelium to adopt a proinflammatory phenotype with a range of downstream consequences, including immune activation, increased clotting propensity, and vascular dysregulation. Immune responses against EBNA-1 also lead to the production of autoantibodies [103,104], which might have relevance to autoimmunity in ME/CFS [105,106,107]. EBNA-1 is also associated with an upregulation of IL-8, hypoxia-inducible factor-1 alpha, and vascular endothelial growth factor (VEGF) [108], of which the latter two promote angiogenesis and influence vascular integrity and dynamics.
EBV’s LMP-1, via NF-κB, increases the expression of cyclooxygenase (COX) 2, prostaglandin E2, and VEGF [109]. In fact, LMP-1 is capable of activating several forms of NF-κB involving a number of different signalling mechanisms [110], and also activates JAK3, p38, mitogen-activated protein kinases, and several STAT-related proteins [111,112]. Importantly, in ECs LMP-1 leads to NF-κB activation and increased expression of IL-1β, IL-6, IL-8, monocyte chemotactic protein-1, RANTES, ICAM-1, VCAM-1, and E-selectin; and the inhibition of caspase-3 and hence the reduction of apoptotic tendencies [113]. This study emphasizes the potential cellular dysregulation that occurs in ECs as a result of herpesvirus latency, as well as localized and potentially systemic physiological disturbances, including the activation and binding of immune cells and platelets to the endothelium.
Endocan is upregulated by LMP-1, again via NF-κB, and levels of endocan and LMP-1 are positively correlated in patient tissue samples [114]. Endocan can promote endothelial dysfunction and cardiovascular disease by increasing inflammation, oxidative stress, and the expression of adhesion molecules [115]. LMP-1 increases the sumoylation of proteins related to cellular migration and transcriptional activity [116] and also increases glycolytic processes and interferes with host metabolism [117,118,119]. LMP-1 also modulates host epigenetic processes [120,121,122] and hinders DNA repair mechanisms [123].
Host protein synthesis is inhibited and autophagy and the regulation thereof is interrupted by LMP-1 [124], and this latency protein also activates the unfolded protein response [125]. Lastly, LMP-1 also interferes with mitochondrial regulation and cell metabolism by altering the phosphorylation of the mitochondrial dynamin-related protein 1 [126]. LMP-2A exerts potent anti-apoptotic effects and aids in immune evasion by reducing the reactivity of CD8+ T cells to cells infected by EBV [127,128]. RNAs from EBV, EBERs, are also associated with cellular dysfunction and proinflammatory processes [129], and represent other mechanisms by which EBV latency can bring about cellular dysfunction. Figure 3 represents the mechanisms discussed by which EBV latency proteins EBNA-1 and LMP-1 can induce cellular dysfunction in endothelial cells.
UL138 is a latency-associated protein produced by HCMV [130,131] and is expressed in infected ECs [132]. UL138 upregulates host EGFR/PI3K/AKT signalling to maintain latency and in turn increases its own expression via EGR-1 [132], and interferes with the function and expression of transport proteins, such as the multidrug resistance-associated protein-1 [133]. It increases levels of TNF-α bound to cell membranes and leads to tumour necrosis factor receptor 1 hyperresponsiveness [134], which prompts immune activation and proinflammatory processes.
US28 from HCMV, while not considered a latency-associated protein, is expressed during both latent and lytic infection and is a potential target for therapy in vascular disease as it increases inflammation and modulates the activity and migration of smooth muscle cells [135,136]. US28 interferes with a variety of cell signalling pathways in ECs including STAT, phospholipase C, GPCR, and NO pathways, and can increase cellular NO expression [137,138]. It binds chemokines including monocyte chemotactic protein 1 and RANTES [139]. US28 increases leukocyte adhesion to the endothelium and aids viral dissemination [140]. Furthermore, antibodies directed against US28 bind to ECs and induce endothelial damage and apoptosis [141]. ECs exposed to these antibodies increase expression and release of HSP60 which can go on to stimulate TLR4 and lead to proinflammatory sequelae in circulation and in the vasculature [142]. It must be noted that US28 itself, not the antibodies directed against it, acts in an anti-apoptotic manner [143]. Importantly, HMCV-infected ECs express US28 on their cell membranes [143].
In ECs, U94, a latency-associated protein from HHV-6 [144], reduces cell migration and angiogenic potential (by desensitizing the response to VEGF), and thus leads to prolonged wound healing [66,145]. U94 increases the expression of human leukocyte antigen G, which is believed to underlie its effects on angiogenesis [58]. This latent protein from HHV-6 also shows anticancer potential as it inhibits DNA repair genes and aspects of the cell cycle, and leads to apoptosis via the intrinsic pathway [146]. While U94 shows potential in cancer therapy, its effects on DNA repair and cell function might not be so favourable in non-malignant, healthy cells.
Ultimately, latency-related molecules and processes are sufficient to induce (endothelial) cellular dysfunction. Latency of herpesviruses, especially when carried out in a particular cell type (such as ECs), might have consequences for systemic physiology. The extent to which this is an ongoing process in ME/CFS warrants further investigation.
4. Evidence for Herpesvirus-Induced Endothelial Dysfunction
We next present the links between herpesvirus infection and endothelial pathology, specifically (Table 1). This will focus on direct and indirect mechanisms, but will not be exclusive to latency-related molecules and processes.
5. Herpesvirus-Induced Endothelial Dysfunction and Its Relevance To ME/CFS
We have discussed the potential of herpesviruses to infect and establish latency in ECs, how their latent proteins and processes are sufficient to induce (endothelial) cell dysfunction, and some of the evidence of herpesvirus-induced endothelial dysfunction. Now, we want to touch on some of the pathophysiological characteristics of ME/CFS and how they might relate to the present discussion thus far.
6. Endothelial Cells, Smooth Muscle Cells, Substance Exchange, Vascular Dysregulation, and Perfusion: What Role Might Herpesviruses Have to Play in the Dysregulation of Blood Flow Observed in ME/CFS?
One of the most important findings in ME/CFS research is that of reduced cerebral blood flow in patients, even in those without tachycardia and hypotension [202,203,204,205]. The orthostatic symptoms associated with ME/CFS are not due to deconditioning [206,207], suggesting an underlying defect in blood flow regulation, perhaps related to autonomic dysfunction [208]. Viral infection of vascular cells, such as ECs and smooth muscle cells, and neurons might contribute to these blood flow and perfusion abnormalities of ME/CFS.
As we have seen, herpesviruses can significantly affect ECs and result in structural and functional changes, which have consequences for the physiological roles of ECs. Endothelial dysfunction is associated with and contributes to impaired tissue perfusion [209,210,211,212,213], and there is even evidence demonstrating an association between herpesviruses and impaired perfusion of tissues [50,174,214].
EBV and HHV-6 are associated with reduced cerebral blood flow and perfusion of particular regions [214,215,216]. Furthermore, Farina et al (2021) demonstrated that skin perfusion is significantly reduced in patients with higher EBV loads in the blood compared to patients with low or undetectable viral loads (hence, EBV load is inversely associated with blood perfusion; refer to the adopted Figure 4). Bentz et al (2006) also demonstrated that HCMV disrupted junction proteins of ECs and increased the permeability of an in vitro endothelial layer [174].
HHV-6 encephalopathy is associated with reductions in cerebral blood flow and perfusion of the frontal lobe [215], as well as perturbations in coronary microcirculation [181]. Similarly, EBV encephalitis is also associated with reduced cerebral blood flow [214]. Caruso et al (2002) showed that large vessel ECs, specifically aortic ECs are more susceptible to infection by HHV-6 than are ECs of the microvasculature [57], and might have relevance to reduced cerebral blood flow in ME/CFS. Latent HCMV infection has been documented in smooth muscle cells from patients suffering from atherosclerosis [217] and abdominal aortic aneurysm with resulting functional changes including enhanced cell proliferation [218]. Patients with a history of HCMV infection display signs of endothelial dysfunction and blood flow dysregulation [219].
Nitric oxide signalling, which is essential for blood flow regulation, is hindered by HCMV, as it upregulates the P38-MAPK pathway which in turn inhibits endothelial nitric oxide synthase (eNOS) [220]. Also necessary for vascular control, specifically vasoconstriction, are endothelins, of which HCMV decreases endothelin-1 in ECs and smooth muscle cells [221], but increases the receptor for endothelin-1 in ECs [222]. HCMV modulates smooth muscle cell migration, proliferation, and expression of platelet derived growth factor receptor beta [223]. Apoptosis of smooth muscles cells are inhibited via a p53-related mechanism by immediate-early proteins produced by HCMV and is proposed to contribute to the accumulation of cells in atherogenic lesions [224]. Furthermore, smooth muscle cells grown from such lesions express HCMV immediate-early proteins [225]. Consequently, HCMV features can occupy a central role in aetiology of dysregulated blood flow and pressure due to its infection of and effect on ECs and vascular smooth muscle cells – namely, its ability to interfere with NO and endothelin signalling, induce proinflammatory sequelae, and activate the renin-angiotensin system [220,221,226].
In a histopathological examination of penile tissue from two males with and two males without a history of COVID-19 infection, viral RNA was detected and virus particles were found in the proximity of ECs in the COVID-positive patients [227]. Furthermore, eNOS expression was also decreased in the COVID-positive samples. The researchers inferred that systemic (and of course localized) COVID-19-induced endothelial dysfunction can result in erectile dysfunction. If this is the case, then it emphasizes the extent to which virus-infected/affected ECs can impair tissue perfusion.
Viruses (and bacterial LPS) can damage the glycocalyx of the endothelial layer [228,229,230,231,232]. A damaged glycocalyx impairs perfusion and also increases risk of mortality in hospitalised patients [213,233,234,235,236,237]. As mentioned earlier, these herpesviruses can also damage endothelial cellular junctions and hence endothelial barriers [153]. HHV-6 can cause fibrosis in ECs [238], and may have implications for perfusion and the ability to exchange substances across vessel walls. These are possible mechanisms by which herpesviruses might bring about impairments in vessel regulation, blood flow, and perfusion.
Hence, infection of ECs and smooth muscle cells by herpesviruses might play a contributory role to the blood flow deficits and vascular dysregulation observed in ME/CFS [202,203,204,205,239]. Investigation of vascular tissue from patients can further inform our understanding of vascular dysfunction and impaired tissue perfusion in ME/CFS. Furthermore, herpesviruses might also contribute to this issue by infecting neurons and causing dysregulation of autonomic control [107,208,240].
7. Herpesviruses, Endothelial Cells, Platelets & Coagulation
Related to the platelet-related abnormalities of a procoagulant phenotype found in ME/CFS patients [29,34,35,36,37], there have been cases where EBV infection caused/was associated with severe cardiac and vascular issues, including myocarditis, vasculitis, disseminated intravascular coagulation, venous thromboembolism, thrombotic thrombocytopenic purpura, deep vein thrombosis, and stroke [241,242,243,244,245,246,247,248,249], emphasizing EBV’s role in hematological and vascular pathology. EBV infection of ECs causes a significant increase in von-Willebrand factor (VWF), VEGF, and platelet endothelial cell adhesion molecule-1 (PECAM) levels [152], which contribute to procoagulant processes. ECs participate in coagulation processes and the regulation thereof, and hence minor cellular disturbances such as an increase in endothelial vWF expression caused by EBV [152] will have significant effects on clotting processes.
Thrombotic processes and events are associated with HCMV infection [175,250,251,252,253,254,255,256,257,258,259,260,261]. It has been proposed that HCMV brings about thrombotic complications due to its modulation of the endothelium and dysfunction thereof [54,262]. HCMV can greatly influence the phenotype of ECs and bring about procoagulant effects [263]. ECs infected with HCMV increase surface expression of adhesion molecules, with some related to coagulation [170]. In a patient suffering from thrombosis without predisposing risk factors, HCMV infection was present and ECs from the patient were found to be abnormal and containing viral inclusion bodies [175]. These endothelial viral inclusion bodies contain virions and HCMV-specific proteins, along with the lysosomal marker LAMP-1 [159]. They are believed to aid in viral maturation in ECs, and lead to the release of virus particle/protein-containing exosomes in fibroblasts. Another such case with simultaneous HCMV infection, along with the presence of antiphospholipid syndrome, was treated successfully with anticoagulants [255]. Antiphospholipid syndrome may result due to antigen similarities between viral and human phospholipids [256,264]. Acute CMV infection has also been documented alongside venous and pulmonary embolisms [259,265], and thromboembolic processes in patients with AIDS [266].
HMCV directly binds to platelets via toll-like receptor 2 (TLR2) and induces platelet activation, leukocyte adhesion and recruitment, VEGF expression, and proinflammatory sequelae [267]. HMCV can induce platelet adhesion and aggregation within infected ECs too [54]. A mechanism by which HMCV-infected ECs induce platelet activation and aggregation occurs involves vWF, as HCMV-infected cells increase intracellular and extracellular expression of this molecule [54]. Platelet aggregation was inhibited when vWF or the platelet receptor for vWF was blocked, but not when the GPIIIb/IIa receptor (which binds fibrinogen) was blocked. Blocking of ICAM-1 also decreased the extent of platelet aggregation, highlighting its role in thrombogenic processes. Rahbar et al (2005) also noticed an increase in P-selectin on non-infected cells treated with the extracellular fluid of HCMV-infected ECs, which is in accord with our findings of increased P-selectin expression on platelets in an ME/CFS cohort [29]. Treatment with ganciclovir inhibited this procoagulant effect of HCMV-infected ECs [54]. Ultimately, Rahbar et al (2005) concluded: ‘Hence, HCMV may promote thrombosis and disseminated intravascular coagulation as a result of endothelial injury—or, as we have found, possibly by increasing the surface expression of vWF, perhaps by causing the translocation of already formed vWF present in Weibel-Palade bodies to the cell surface’. This is in accord with the current hypothesis of virus-induced endothelial maladaptation, and also provides reason for the coagulation and platelet abnormalities observed in ME/CFS.
The HCMV membrane contains phospholipids that enable the assembly of prothrombinase [268], a procoagulant protein that leads to thrombin generation. HCMV infection is also associated with a decrease in protein S and subsequent thrombosis [257]. We have recently discovered reduced protein S levels in an ME/CFS cohort via DIA LC-MS/MS techniques. Whether this finding is related to HCMV infection in the ME/CFS population is undetermined, but seems plausible as ECs – which succumb to HCMV infection – are responsible for regulating protein S expression.
HHV-6 infection is associated with prothrombotic states [70,180]. Specifically, it is associated with thrombotic microangiopathy – whether this is related to fibrinaloid microclots which are found in both Long COVID and ME/CFS cohorts requires further investigation [29,31].
Hence, there is reason to suspect that the clotting and platelet abnormalities noted in ME/CFS arise from the consequences of herpesvirus-infected ECs, as well as other procoagulant effects of herpesviruses independent of endothelial cells. ECs are extremely important for the homeostasis of the coagulation system. Virus-induced endothelial maladaptation may underlie findings of abnormal coagulation, platelet hyperactivity and platelet aggregation in ME/CFS cohorts [29,34,35].
8. Herpesviruses and Neurological Issues in ME/CFS: Implications at the Cerebro-Endothelium?
Endothelial dysfunction has been associated with cognitive dysfunction in vascular dementia [269] coronary artery disease [270], obesity [271], postoperative cognitive dysfunction [272], type II diabetes [273], sleep apnoea [274], and the elderly [275]. Endothelial markers, such as endothelial lipase, positively correlates with cognitive impairment [276]. A systematic review inferred an ‘intrinsic’ relationship between endothelial dysfunction and vascular cognitive impairments [277]. These studies, along with those demonstrating endothelial dysfunction in study cohorts, suggest that endothelial dysfunction might have a significant role to play in the neurological issues suffered by ME/CFS patients.
As we’ve discussed in this paper, many of the herpesviruses are capable of infecting brain microvascular ECs, which act as viral reservoirs from which CNS infection can ensue. Furthermore, a compromised BBB is an expected consequence. Importantly, EBV and HHV-6 have been detected in CNS tissue from deceased ME/CFS patients [5] – cerebrovascular ECs might be the reservoir/latent site for these viruses in patients. EBV infection is associated with cognitive impairments [278,279], and EBV proteins, including EBV dUTPase, are posited to contribute neuroinflammation and subsequent neurological issues in ME/CFS [9]. HMCV is associated with impairments in cognition, with possible mechanisms involving the virus’ immediate-early 2 protein [280,281]. HHV-6 is also associated with cognitive impairments [282,283,284,285]. HHV-6 antigens have been found within ECs from the frontal lobe of a fatal case of herpesvirus infection [191], and in a mice study, HHV-6 infection of the CNS persisted and induced proinflammatory cytokine production via TLR-9 [199].
Infection of brain ECs and other vascular cells (as well as neurons and glial cells) by herpesviruses and subsequent vasculitis in brain blood vessels might lead to inflammation, CNS infection, oxidative and NS damage, and symptom expression including cognitive dysfunction and even fatigue and PESE. Neurological pathology might also ensue as a result of autoimmune processes in the central nervous system, whereby molecular mimicry involving EBV and other herpesviruses antigens result in autoantibodies directed against brain antigens, for example, glial cell adhesion molecule [104]. This may have relevance to ME/CFS, and tie together working hypotheses on endothelial dysfunction and neurological symptoms in ME/CFS [286] with herpesvirus activity.
9. Ways Forward
It is of interest to determine whether ECs from patients suffering from ME/CFS (and controls) are infected with herpesviruses. This would be the first step in confirming this idea as presented in this paper. ECs, and brain microvascular ECs specifically, can be extracted and isolated via a number of techniques [287,288,289,290]. Other vascular tissue cells, such as smooth muscle cells, should also be isolated and tested for herpesvirus infection. Whilst there have been studies that have demonstrated reduced permeability of endothelial linings as a result of herpesvirus infection, and studies associating herpesvirus infection with perfusion deficits, further experiments focussing on these phenomena and the molecular processes involved, from a post-viral perspective, are necessary.
10. Conclusion
We have presented the idea that herpesvirus infection of ECs might be an important, overlooked phenomenon that can, in part, account for the pathophysiology and symptoms of ME/CFS and, potentially, Long COVID. This idea is somewhat novel, but is not unprecedented [71,79,90,162,262]. Endothelial dysfunction and herpesvirus activity are two characteristics of ME/CFS pathology that have yet to be officially linked. Perhaps latent infection of ECs alone (that is, without active infection) is sufficient to bring about pathology and subsequent symptoms, and may provide a explanation for daily symptoms, the chronicity of symptoms, and the multi-organ, systemic nature of ME/CFS.
Importantly, the load of EC infection, i.e., how widespread in the systemic vasculature herpesvirus infection is, and perhaps the tissue- and organ-specific site of infection are likely vital factors that determine the manifestation of symptoms as a result of this hypothesized pathophysiological process. It is acknowledged that herpesviruses infect many cell types, so the infection of ECs by herpesviruses is likely not responsible for all ME/CFS pathology and symptoms. For example, infection of immune cells and manipulation of immune processes by herpesviruses contribute to ME/CFS pathology, and likely account for much of the immune disturbances seen in this population; similarly, infection of cells of the nervous system might account neurological deficits, and even vascular dysregulation. However, we want to bring attention to the possibility of endothelial infection, as this is somewhat of an understudied topic, especially in the context of ME/CFS.
Further studies are required to determine the extent of herpesvirus infection of the endothelium in ME/CFS patients, and also need to take into account the possibility of tissue- or organ-specific sites of infection. This is a difficult phenomenon to prove, especially when considering the ability of herpesviruses to cause pathology in certain individuals and in certain physiological states, and hence requires diligent and elaborative study and experimentation. It is possible that this idea of herpesvirus latency-induced endothelial maladaptation might turn out to be irrelevant, but herpesvirus-induced endothelial dysfunction, with and without direct infection of ECs, will still be relevant for ME/CFS pathology and symptom manifestation. Hence, a more refined focus on herpesviruses and endothelial function and health in ME/CFS (and Long COVID) is warranted.
Author Contributions
Conceptualization, J. M. Nunes; software, Biorender.; validation and editing, E. Pretorius, D. B. Kell.; investigation, J. M. Nunes.; writing—original draft preparation, J. M. Nunes.; writing—review and editing, X.X.; visualization, J. M. Nunes. All authors have read and agreed to the published version of the manuscript.
Funding
E.P.: Funding was provided by NRF of South Africa (grant number 142142) and SA MRC (self-initiated research (SIR) grant), and Balvi Foundation (grant 31). D.B.K.: Funding was provided by Balvi Foundation (grant 18) and the Novo Nordisk Foundation for funding (grant NNF10CC1016517). The content and findings reported and illustrated are the sole deduction, view and responsibility of the researchers and do not reflect the official position and sentiments of the funders.
Conflicts of Interest
The authors have none to disclose.
References
- Lim, E.-J. and C.-G. Son, Review of case definitions for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Journal of Translational Medicine, 2020. 18(1): p. 289. [CrossRef]
- Rasa, S. , et al., Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med, 2018. 16(1): p. 268. [CrossRef]
- Rasa-Dzelzkaleja, S. , et al., The persistent viral infections in the development and severity of myalgic encephalomyelitis/chronic fatigue syndrome. Journal of Translational Medicine, 2023. 21(1): p. 33. [CrossRef]
- Ariza, M.E. , Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules, 2021. 11(2): p. 185.
- Kasimir, F. , et al., Tissue specific signature of HHV-6 infection in ME/CFS. Frontiers in Molecular Biosciences, 2022. 9. [CrossRef]
- Shikova, E. , et al., Cytomegalovirus, Epstein-Barr virus, and human herpesvirus-6 infections in patients with myalgic еncephalomyelitis/chronic fatigue syndrome. Journal of Medical Virology, 2020. 92(12): p. 3682-3688. [CrossRef]
- Cox, B.S. , et al., EBV/HHV-6A dUTPases contribute to myalgic encephalomyelitis/chronic fatigue syndrome pathophysiology by enhancing TFH cell differentiation and extrafollicular activities. JCI Insight, 2022. 7(11). [CrossRef]
- Ruiz-Pablos, M. , et al., Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. Front Immunol, 2021. 12: p. 656797. [CrossRef]
- Williams Ph, D.M. , et al., Epstein-Barr Virus dUTPase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin Ther, 2019. 41(5): p. 848-863.
- Mozhgani, S.-H. , et al., Human Herpesvirus 6 Infection and Risk of Chronic Fatigue Syndrome: A Systematic Review and Meta-Analysis. Intervirology, 2021. 65(1): p. 49-57. [CrossRef]
- Navaneetharaja, N. , et al., A Role for the Intestinal Microbiota and Virome in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)? Journal of Clinical Medicine, 2016. 5(6): p. 55.
- Eriksen, W. , ME/CFS, case definition, and serological response to Epstein-Barr virus. A systematic literature review. Fatigue: Biomedicine, Health & Behavior, 2018. 6(4): p. 220-234. [CrossRef]
- Hatton, O.L. , et al., The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol Res, 2014. 58(2-3): p. 268-76.
- Sinzger, C. , et al., Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. Journal of General Virology, 1995. 76(4): p. 741-750. [CrossRef]
- Adler, B. and C. Sinzger, Endothelial cells in human cytomegalovirus infection: One host cell out of many or a crucial target for virus spread? Thromb Haemost, 2009. 102(12): p. 1057-1063.
- Scherbakov, N. , et al., Peripheral endothelial dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. ESC Heart Failure, 2020. 7(3): p. 1064-1071. [CrossRef]
- Sørland, K. , et al., Reduced Endothelial Function in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome–Results From Open-Label Cyclophosphamide Intervention Study. Frontiers in Medicine, 2021. 8. [CrossRef]
- Sandvik, M.K. , et al., Endothelial dysfunction in ME/CFS patients. PLOS ONE, 2023. 18(2): p. e0280942. [CrossRef]
- Haffke, M. , et al., Endothelial dysfunction and altered endothelial biomarkers in patients with post-COVID-19 syndrome and chronic fatigue syndrome (ME/CFS). Journal of Translational Medicine, 2022. 20(1): p. 138. [CrossRef]
- Blauensteiner, J. , et al., Altered endothelial dysfunction-related miRs in plasma from ME/CFS patients. Sci Rep, 2021. 11(1): p. 10604.
- Bertinat, R. , et al., Decreased NO production in endothelial cells exposed to plasma from ME/CFS patients. Vascular Pharmacology, 2022. 143: p. 106953. [CrossRef]
- Newton, D.J. , et al., Large and small artery endothelial dysfunction in chronic fatigue syndrome. International Journal of Cardiology, 2012. 154(3): p. 335-336. [CrossRef]
- Flaskamp, L. , et al., Serum of Post-COVID-19 Syndrome Patients with or without ME/CFS Differentially Affects Endothelial Cell Function In Vitro. Cells, 2022. 11(15): p. 2376. [CrossRef]
- Cambras, T. , et al., Skin Temperature Circadian Rhythms and Dysautonomia in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Role of Endothelin-1 in the Vascular Tone Dysregulation. International Journal of Molecular Sciences, 2023. 24(5): p. 4835. [CrossRef]
- Kedor, C. , et al., Chronic COVID-19 Syndrome and Chronic Fatigue Syndrome (ME/CFS) following the first pandemic wave in Germany – a first analysis of a prospective observational study. medRxiv, 2021: p. 2021.02.06.21249256.
- Poenaru, S. , et al., COVID-19 and post-infectious myalgic encephalomyelitis/chronic fatigue syndrome: a narrative review. Ther Adv Infect Dis, 2021. 8: p. 20499361211009385. [CrossRef]
- Wong, T.L. and D.J. Weitzer, Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina, 2021. 57(5): p. 418.
- Tate, W. , et al., Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Frontiers in Neurology, 2022. 13. [CrossRef]
- Nunes, J.M. , et al., The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals, 2022. 15(8): p. 931. [CrossRef]
- Turner, S. , et al., Long COVID: pathophysiological factors and abnormalities of coagulation. Trends in Endocrinology & Metabolism, 2023. 34(6): p. 321-344. [CrossRef]
- Pretorius, E. , et al., Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol, 2021. 20(1): p. 172. [CrossRef]
- Kell, D.B. and E. Pretorius, The potential role of ischaemia–reperfusion injury in chronic, relapsing diseases such as rheumatoid arthritis, Long COVID, and ME/CFS: evidence, mechanisms, and therapeutic implications. Biochemical Journal, 2022. 479(16): p. 1653-1708.
- Laubshder, G. , et al., Treatment of Long COVID symptoms with triple anticoagulant therapy. 2023.
- Bonilla, H. , et al., Comparative Analysis of Extracellular Vesicles in Patients with Severe and Mild Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Frontiers in Immunology, 2022. 13. [CrossRef]
- Jahanbani, F. , et al., Phenotypic characteristics of peripheral immune cells of Myalgic encephalomyelitis/chronic fatigue syndrome via transmission electron microscopy: A pilot study. PLOS ONE, 2022. 17(8): p. e0272703. [CrossRef]
- Ahmed, F. , et al., Single-cell transcriptomics of the immune system in ME/CFS at baseline and following symptom provocation. bioRxiv, 2022: p. 2022.10.13.512091. [CrossRef]
- Berg, D. , et al., Chronic fatigue syndrome and/or fibromyalgia as a variation of antiphospholipid antibody syndrome: an explanatory model and approach to laboratory diagnosis. Blood Coagul Fibrinolysis, 1999. 10(7): p. 435-8.
- van Hinsbergh, V.W.M. , Endothelium—role in regulation of coagulation and inflammation. Seminars in Immunopathology, 2012. 34(1): p. 93-106.
- Neubauer, K. and B. Zieger, Endothelial cells and coagulation. Cell and Tissue Research, 2022. 387(3): p. 391-398. [CrossRef]
- Sfera, A. , et al., Endothelial Senescence and Chronic Fatigue Syndrome, a COVID-19 Based Hypothesis. Frontiers in Cellular Neuroscience, 2021. 15. [CrossRef]
- Chapenko, S. , et al., Association of Active Human Herpesvirus-6, -7 and Parvovirus B19 Infection with Clinical Outcomes in Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Advances in Virology, 2012. 2012: p. 205085. [CrossRef]
- Blomberg, J. , et al., Antibodies to Human Herpesviruses in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Frontiers in Immunology, 2019. 10. [CrossRef]
- Ban, S. , et al., Systemic granulomatous arteritis associated with Epstein-Barr virus infection. Virchows Arch, 1999. 434(3): p. 249-54. [CrossRef]
- Guarner, J. and E.R. Unger, Association of Epstein-Barr virus in epithelioid angiomatosis of AIDS patients. Am J Surg Pathol, 1990. 14(10): p. 956-60. [CrossRef]
- Farina, A. , et al., Epstein–Barr Virus Infection Induces Aberrant TLR Activation Pathway and Fibroblast–Myofibroblast Conversion in Scleroderma. Journal of Investigative Dermatology, 2014. 134(4): p. 954-964. [CrossRef]
- Kanai, K. , et al., Leukocytoclastic-vasculitic neuropathy associated with chronic Epstein–Barr virus infection. Muscle & Nerve, 2003. 27(1): p. 113-116. [CrossRef]
- Truszewska, A. , et al., EBV load is associated with cfDNA fragmentation and renal damage in SLE patients. Lupus, 2021. 30(8): p. 1214-1225. [CrossRef]
- Jones, K. , et al., Infection of human endothelial cells with Epstein-Barr virus. Journal of Experimental Medicine, 1995. 182(5): p. 1213-1221. [CrossRef]
- Casiraghi, C., K. Dorovini-Zis, and M.S. Horwitz, Epstein-Barr virus infection of human brain microvessel endothelial cells: A novel role in multiple sclerosis. Journal of Neuroimmunology, 2011. 230(1): p. 173-177. [CrossRef]
- Farina, A. , et al., Innate Immune Modulation Induced by EBV Lytic Infection Promotes Endothelial Cell Inflammation and Vascular Injury in Scleroderma. Frontiers in Immunology, 2021. 12. [CrossRef]
- Yamamoto, S. and Y. Sakai, Acute gastritis caused by concurrent infection with Epstein–Barr virus and cytomegalovirus in an immunocompetent adult. Clinical Journal of Gastroenterology, 2019. 12(3): p. 274-278. [CrossRef]
- Grefte, A. , et al., Circulating Cytomegalovirus (CMV)-Infected Endothelial Cells in Patients with an Active CMV Infection. The Journal of Infectious Diseases, 1993. 167(2): p. 270-277. [CrossRef]
- Percivalle, E. , et al., Circulating endothelial giant cells permissive for human cytomegalovirus (HCMV) are detected in disseminated HCMV infections with organ involvement. J Clin Invest, 1993. 92(2): p. 663-70. [CrossRef]
- Rahbar, A. and C. Söderberg-Nauclér, Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol, 2005. 79(4): p. 2211-20. [CrossRef]
- Ho, D.D. , et al., Replication of Human Cytomegalovirus in Endothelial Cells. The Journal of Infectious Diseases, 1984. 150(6): p. 956-957. [CrossRef]
- Smiley, M.L., E. -C. Mar, and E.-S. Huang, Cytomegalovirus infection and viral-induced transformation of human endothelial cells. Journal of Medical Virology, 1988. 25(2): p. 213-226. [CrossRef]
- Caruso, A. , et al., HHV-6 infects human aortic and heart microvascular endothelial cells, increasing their ability to secrete proinflammatory chemokines. Journal of Medical Virology, 2002. 67(4): p. 528-533.
- Rizzo, R. , et al., Human Herpesvirus 6A and 6B inhibit in vitro angiogenesis by induction of Human Leukocyte Antigen G. Sci Rep, 2018. 8(1): p. 17683. [CrossRef]
- Caruso, A. , et al., Human herpesvirus-6 modulates RANTES production in primary human endothelial cell cultures. J Med Virol, 2003. 70(3): p. 451-8. [CrossRef]
- Rotola, A. , et al., Human herpesvirus 6 infects and replicates in aortic endothelium. J Clin Microbiol, 2000. 38(8): p. 3135-6. [CrossRef]
- Wu, C.A. and J.D. Shanley, Chronic infection of human umbilical vein endothelial cells by human herpesvirus-6. Journal of General Virology, 1998. 79(5): p. 1247-1256. [CrossRef]
- Reddy, S. , et al., Human herpesvirus 6-induced inflammatory cardiomyopathy in immunocompetent children. Ann Pediatr Cardiol, 2017. 10(3): p. 259-268. [CrossRef]
- Visser, M.R. , et al., Enhanced thrombin generation and platelet binding on herpes simplex virus-infected endothelium. Proceedings of the National Academy of Sciences, 1988. 85(21): p. 8227-8230. [CrossRef]
- Fonsato, V. , et al., PAX2 expression by HHV-8–infected endothelial cells induced a proangiogenic and proinvasive phenotype. Blood, 2008. 111(5): p. 2806-2815. [CrossRef]
- Cohen, J.I. , Herpesvirus latency. J Clin Invest, 2020. 130(7): p. 3361-3369.
- Caruso, A. , et al., U94 of human herpesvirus 6 inhibits in vitro angiogenesis and lymphangiogenesis. Proc Natl Acad Sci U S A, 2009. 106(48): p. 20446-51. [CrossRef]
- Shioda, S. , et al., The human vascular endothelial cell line HUV-EC-C harbors the integrated HHV-6B genome which remains stable in long term culture. Cytotechnology, 2018. 70(1): p. 141-152. [CrossRef]
- Jarvis, M.A. and J.A. Nelson, Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Current Opinion in Microbiology, 2002. 5(4): p. 403-407. [CrossRef]
- Jarvis, M.A. and J.A. Nelson, Human Cytomegalovirus Tropism for Endothelial Cells: Not All Endothelial Cells Are Created Equal. Journal of Virology, 2007. 81(5): p. 2095-2101. [CrossRef]
- Brewer, J.H. and D. Berg, Hypercoaguable State Associated with Active Human Herpesvirus-6 (HHV-6) Viremia in Patients with Chronic Fatigue Syndrome. Journal of Chronic Fatigue Syndrome, 2001. 8(3-4): p. 111-116. [CrossRef]
- Liu, Z. , et al., Increased circulating fibronectin, depletion of natural IgM and heightened EBV, HSV-1 reactivation in ME/CFS and long COVID. medRxiv, 2023: p. 2023.06.23.23291827.
- Kaprelyants, A.S., J. C. Gottschal, and D.B. Kell, Dormancy in non-sporulating bacteria. FEMS Microbiol Rev, 1993. 10(3-4): p. 271-85.
- Kell, D.B. , et al., Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Van Leeuwenhoek, 1998. 73(2): p. 169-87. [CrossRef]
- Nikolich-Zugich, J. , et al., Known unknowns: how might the persistent herpesvirome shape immunity and aging? Curr Opin Immunol, 2017. 48: p. 23-30.
- Simanek, A.M. , et al., Seropositivity to cytomegalovirus, inflammation, all-cause and cardiovascular disease-related mortality in the United States. PLoS One, 2011. 6(2): p. e16103. [CrossRef]
- Guilluy, C. , et al., Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood, 2011. 118(19): p. 5344-5354. [CrossRef]
- DiMaio, T.A., K. D. Gutierrez, and M. Lagunoff, Latent KSHV Infection of Endothelial Cells Induces Integrin Beta3 to Activate Angiogenic Phenotypes. PLOS Pathogens, 2011. 7(12): p. e1002424. [CrossRef]
- Grossmann, C. , et al., Activation of NF-kappaB by the latent vFLIP gene of Kaposi's sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol, 2006. 80(14): p. 7179-85.
- Qian, L.-W. , et al., Kaposi's Sarcoma-Associated Herpesvirus Disrupts Adherens Junctions and Increases Endothelial Permeability by Inducing Degradation of VE-Cadherin. Journal of Virology, 2008. 82(23): p. 11902-11912.
- Griffin, B.D., M. C. Verweij, and E.J.H.J. Wiertz, Herpesviruses and immunity: The art of evasion. Veterinary Microbiology, 2010. 143(1): p. 89-100. [CrossRef]
- Bergmann, O. , et al., Dynamics of Cell Generation and Turnover in the Human Heart. Cell, 2015. 161(7): p. 1566-1575. [CrossRef]
- Hobson, B. and J. Denekamp, Endothelial proliferation in tumours and normal tissues: continuous labelling studies. Br J Cancer, 1984. 49(4): p. 405-13. [CrossRef]
- Wang, L. and B. Damania, Kaposi's Sarcoma–Associated Herpesvirus Confers a Survival Advantage to Endothelial Cells. Cancer Research, 2008. 68(12): p. 4640-4648.
- Xiong, A. , et al., Epstein-Barr virus latent membrane protein 1 activates nuclear factor-kappa B in human endothelial cells and inhibits apoptosis. Transplantation, 2004. 78(1): p. 41-9.
- Kovacs, A. , et al., Cytoplasmic sequestration of p53 in cytomegalovirus-infected human endothelial cells. Am J Pathol, 1996. 149(5): p. 1531-9.
- Takemoto, M. , et al., Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis. Journal of General Virology, 2004. 85(4): p. 869-879. [CrossRef]
- Pawelec, G. , et al., Immunosenescence and Cytomegalovirus: where do we stand after a decade? Immun Ageing, 2010. 7: p. 13.
- Hogestyn, J.M., D. J. Mock, and M. Mayer-Proschel, Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen Res, 2018. 13(2): p. 211-221. [CrossRef]
- Yin, H. , et al., Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Medical Microbiology and Immunology, 2019. 208(5): p. 573-583. [CrossRef]
- Liu, R. , et al., KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood, 2010. 115(4): p. 887-895. [CrossRef]
- Elgui de Oliveira, D., B. G. Müller-Coan, and J.S. Pagano, Viral Carcinogenesis Beyond Malignant Transformation: EBV in the Progression of Human Cancers. Trends in Microbiology, 2016. 24(8): p. 649-664. [CrossRef]
- Kanda, T., M. Yajima, and K. Ikuta, Epstein-Barr virus strain variation and cancer. Cancer Science, 2019. 110(4): p. 1132-1139. [CrossRef]
- Niller, H.H. , et al., Epigenetic dysregulation of epstein-barr virus latency and development of autoimmune disease. Adv Exp Med Biol, 2011. 711: p. 82-102.
- Lee, M.-A., E. Diamond Margaret, and L. Yates John, Genetic Evidence that EBNA-1 Is Needed for Efficient, Stable Latent Infection by Epstein-Barr Virus. Journal of Virology, 1999. 73(4): p. 2974-2982.
- Leight, E.R. and B. Sugden, EBNA-1: a protein pivotal to latent infection by Epstein–Barr virus. Reviews in Medical Virology, 2000. 10(2): p. 83-100.
- Szymula, A. , et al., Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naïve B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog, 2018. 14(2): p. e1006890.
- Young, L.S. and P.G. Murray, Epstein–Barr virus and oncogenesis: from latent genes to tumours. Oncogene, 2003. 22(33): p. 5108-5121. [CrossRef]
- Kang, M.-S. and E. Kieff, Epstein–Barr virus latent genes. Experimental & Molecular Medicine, 2015. 47(1): p. e131-e131.
- Gruhne, B. , et al., The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A, 2009. 106(7): p. 2313-8. [CrossRef]
- Kamranvar, S.A. and M.G. Masucci, The Epstein–Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia, 2011. 25(6): p. 1017-1025. [CrossRef]
- Gruhne, B., R. Sompallae, and M.G. Masucci, Three Epstein–Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene, 2009. 28(45): p. 3997-4008.
- Frappier, L. , Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses, 2012. 4(9): p. 1537-1547. [CrossRef]
- Poole, B.D. , et al., Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity, 2006. 39(1): p. 63-70. [CrossRef]
- Lanz, T.V. , et al., Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature, 2022. 603(7900): p. 321-327. [CrossRef]
- De Bellis, A. , et al., Hypothalamic-Pituitary Autoimmunity and Related Impairment of Hormone Secretions in Chronic Fatigue Syndrome. The Journal of Clinical Endocrinology & Metabolism, 2021. 106(12): p. e5147-e5155.
- Sotzny, F. , et al., Myalgic Encephalomyelitis/Chronic Fatigue Syndrome – Evidence for an autoimmune disease. Autoimmunity Reviews, 2018. 17(6): p. 601-609. [CrossRef]
- Wirth, K. and C. Scheibenbogen, A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ß2-adrenergic receptors. Autoimmunity Reviews, 2020. 19(6): p. 102527. [CrossRef]
- O'Neil, J.D. , et al., Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J Gen Virol, 2008. 89(Pt 11): p. 2833-2842. [CrossRef]
- Murono, S. , et al., Induction of cyclooxygenase-2 by Epstein–Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proceedings of the National Academy of Sciences, 2001. 98(12): p. 6905-6910. [CrossRef]
- Kung, C.-P. and N. Raab-Traub, Epstein-Barr Virus Latent Membrane Protein 1 Induces Expression of the Epidermal Growth Factor Receptor through Effects on Bcl-3 and STAT3. Journal of Virology, 2008. 82(11): p. 5486-5493. [CrossRef]
- Gires, O. , et al., Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. Embo j, 1999. 18(11): p. 3064-73. [CrossRef]
- Eliopoulos, A.G. and L.S. Young, LMP1 structure and signal transduction. Seminars in Cancer Biology, 2001. 11(6): p. 435-444. [CrossRef]
- Xiong, A. , et al., Epstein-Barr Virus Latent Membrane Protein 1 Activates Nuclear Factor-κB in Human Endothelial Cells and Inhibits Apoptosis. Transplantation, 2004. 78(1).
- Yu, P.H. , et al., Upregulation of endocan by Epstein-Barr virus latent membrane protein 1 and its clinical significance in nasopharyngeal carcinoma. PLoS One, 2013. 8(12): p. e82254. [CrossRef]
- Chen, J. , et al., Endocan: A Key Player of Cardiovascular Disease. Front Cardiovasc Med, 2021. 8: p. 798699. [CrossRef]
- Bentz, G.L., C. B. Whitehurst, and J.S. Pagano, Epstein-Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J Virol, 2011. 85(19): p. 10144-53. [CrossRef]
- Xiao, L. , et al., Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene, 2014. 33(37): p. 4568-78. [CrossRef]
- Lo, A.K. , et al., Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J Pathol, 2015. 237(2): p. 238-48. [CrossRef]
- Jiang, Y. , et al., Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene, 2015. 34(50): p. 6079-91. [CrossRef]
- Hino, R. , et al., Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res, 2009. 69(7): p. 2766-74.
- Martin, K.A., L. N. Lupey, and I. Tempera, Epstein-Barr Virus Oncoprotein LMP1 Mediates Epigenetic Changes in Host Gene Expression through PARP1. J Virol, 2016. 90(19): p. 8520-30. [CrossRef]
- Chen, S. , et al., LMP1 mediates tumorigenesis through persistent epigenetic modifications and PGC1β upregulation. Oncol Rep, 2023. 49(3): p. 53. [CrossRef]
- Liu, M.T. , et al., Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene, 2004. 23(14): p. 2531-9. [CrossRef]
- Lee, D.Y. and B. Sugden, The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene, 2008. 27(20): p. 2833-42. [CrossRef]
- Lee, D.Y. and B. Sugden, The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood, 2008. 111(4): p. 2280-9. [CrossRef]
- Xie, L. , et al., Drp1-dependent remodeling of mitochondrial morphology triggered by EBV-LMP1 increases cisplatin resistance. Signal Transduction and Targeted Therapy, 2020. 5(1): p. 56. [CrossRef]
- Mancao, C. and W. Hammerschmidt, Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood, 2007. 110(10): p. 3715-3721. [CrossRef]
- Rancan, C. , et al., Latent Membrane Protein LMP2A Impairs Recognition of EBV-Infected Cells by CD8+ T Cells. PLOS Pathogens, 2015. 11(6): p. e1004906. [CrossRef]
- Iwakiri, D. , Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers, 2014. 6(3): p. 1615-1630. [CrossRef]
- Goodrum, F. , et al., Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood, 2007. 110(3): p. 937-45. [CrossRef]
- Albright, E.R., C. K. Mickelson, and R.F. Kalejta, Human Cytomegalovirus UL138 Protein Inhibits the STING Pathway and Reduces Interferon Beta mRNA Accumulation during Lytic and Latent Infections. mBio, 2021. 12(6): p. e0226721. [CrossRef]
- Mlera, L. , et al., The Role of the Human Cytomegalovirus UL133-UL138 Gene Locus in Latency and Reactivation. Viruses, 2020. 12(7). [CrossRef]
- Weekes, M.P. , et al., Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science, 2013. 340(6129): p. 199-202. [CrossRef]
- Le, V.T., M. Trilling, and H. Hengel, The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb'-encoded modulation of TNF-α signaling. J Virol, 2011. 85(24): p. 13260-70. [CrossRef]
- Streblow, D.N., S. L. Orloff, and J.A. Nelson, The HCMV chemokine receptor US28 is a potential target in vascular disease. Curr Drug Targets Infect Disord, 2001. 1(2): p. 151-8. [CrossRef]
- Streblow, D.N. , et al., The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell, 1999. 99(5): p. 511-20. [CrossRef]
- Zhu, D. , et al., Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat Microbiol, 2018. 3(4): p. 503-513. [CrossRef]
- Miller, W.E. , et al., US28 Is a Potent Activator of Phospholipase C during HCMV Infection of Clinically Relevant Target Cells. PLOS ONE, 2012. 7(11): p. e50524. [CrossRef]
- Dolcino, M. , et al., Infections and autoimmunity: role of human cytomegalovirus in autoimmune endothelial cell damage. Lupus, 2015. 24(4-5): p. 419-32. [CrossRef]
- Wu, S.-e. and W.E. Miller, The HCMV US28 vGPCR induces potent Gαq/PLC-β signaling in monocytes leading to increased adhesion to endothelial cells. Virology, 2016. 497: p. 233-243. [CrossRef]
- Lunardi, C. , et al., Induction of endothelial cell damage by hCMV molecular mimicry. Trends in Immunology, 2005. 26(1): p. 19-24. [CrossRef]
- Lunardi, C. , et al., Endothelial Cells' Activation and Apoptosis Induced by a Subset of Antibodies against Human Cytomegalovirus: Relevance to the Pathogenesis of Atherosclerosis. PLOS ONE, 2007. 2(5): p. e473. [CrossRef]
- Billstrom Schroeder, M., R. Christensen, and G.S. Worthen, Human cytomegalovirus protects endothelial cells from apoptosis induced by growth factor withdrawal. Journal of Clinical Virology, 2002. 25: p. 149-157. [CrossRef]
- Rotola, A. , et al., U94 of human herpesvirus 6 is expressed in latently infected peripheral blood mononuclear cells and blocks viral gene expression in transformed lymphocytes in culture. Proc Natl Acad Sci U S A, 1998. 95(23): p. 13911-6. [CrossRef]
- Campbell, A. , et al., Expression of the Human Herpesvirus 6A Latency-Associated Transcript U94A Disrupts Human Oligodendrocyte Progenitor Migration. Sci Rep, 2017. 7(1): p. 3978. [CrossRef]
- Caccuri, F. , et al., Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94. Cancers (Basel), 2019. 11(7). [CrossRef]
- Indari, O. , et al., Early biomolecular changes in brain microvascular endothelial cells under Epstein–Barr virus influence: a Raman microspectroscopic investigation. Integrative Biology, 2022. 14(4): p. 89-97. [CrossRef]
- Wekerle, H. , Epstein-Barr virus sparks brain autoimmunity in multiple sclerosis. Nature, 2022. 603(7900): p. 230-232. [CrossRef]
- Kanno, H. , et al., Adhesion of Epstein–Barr virus-positive natural killer cell lines to cultured endothelial cells stimulated with inflammatory cytokines. Clinical and Experimental Immunology, 2008. 151(3): p. 519-527. [CrossRef]
- Waldman, W.J. , et al., Epstein-Barr virus-encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: Evidence for depression-induced atherosclerotic risk. Brain, Behavior, and Immunity, 2008. 22(2): p. 215-223. [CrossRef]
- Barrett, M.M. , et al., Lymphocytic Arteritis in Epstein–Barr Virus Vulvar Ulceration (Lipschütz Disease): A Report of 7 Cases. The American Journal of Dermatopathology, 2015. 37(9).
- Indari, O., R. Chandramohanadas, and H.C. Jha, Epstein–Barr virus infection modulates blood–brain barrier cells and its co-infection with Plasmodium falciparum induces RBC adhesion. Pathogens and Disease, 2020. 79(1). [CrossRef]
- Li, D.-K. , et al., Exosomal HMGA2 protein from EBV-positive NPC cells destroys vascular endothelial barriers and induces endothelial-to-mesenchymal transition to promote metastasis. Cancer Gene Therapy, 2022. 29(10): p. 1439-1451. [CrossRef]
- Granato, M. , et al., Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol, 2014. 88(21): p. 12715-26. [CrossRef]
- Baglio, S.R. , et al., Sensing of latent EBV infection through exosomal transfer of 5'pppRNA. Proc Natl Acad Sci U S A, 2016. 113(5): p. E587-96. [CrossRef]
- McNamara, R.P. , et al., Extracellular vesicles from Kaposi Sarcoma-associated herpesvirus lymphoma induce long-term endothelial cell reprogramming. PLOS Pathogens, 2019. 15(2): p. e1007536. [CrossRef]
- Gerna, G., A. Kabanova, and D. Lilleri, Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines, 2019. 7(3): p. 70. [CrossRef]
- Vanarsdall, A.L. , et al., CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio, 2018. 9(3). [CrossRef]
- Bughio, F. , et al., Human Cytomegalovirus UL135 and UL136 Genes Are Required for Postentry Tropism in Endothelial Cells. J Virol, 2015. 89(13): p. 6536-50. [CrossRef]
- Sinzger, C. , et al., Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J Gen Virol, 1999. 80 ( Pt 11): p. 2867-2877. [CrossRef]
- Du, Y., G. Zhang, and Z. Liu, Human cytomegalovirus infection and coronary heart disease: a systematic review. Virology Journal, 2018. 15(1): p. 31. [CrossRef]
- Shen, Y.H. , et al., Human cytomegalovirus causes endothelial injury through the ataxia telangiectasia mutant and p53 DNA damage signaling pathways. Circ Res, 2004. 94(10): p. 1310-7. [CrossRef]
- Hosogai, M. , et al., Analysis of human cytomegalovirus replication in primary cultured human corneal endothelial cells. British Journal of Ophthalmology, 2015. 99(11): p. 1583-1590. [CrossRef]
- Shen, K. , et al., Human cytomegalovirus-encoded miR-UL112 contributes to HCMV-mediated vascular diseases by inducing vascular endothelial cell dysfunction. Virus Genes, 2018. 54(2): p. 172-181.
- Heybar, H. , et al., Cytomegalovirus infection and atherosclerosis in candidate of coronary artery bypass graft. Jundishapur J Microbiol, 2015. 8(3): p. e15476. [CrossRef]
- Melnick, J.L. , et al., Cytomegalovirus DNA in arterial walls of patients with atherosclerosis. J Med Virol, 1994. 42(2): p. 170-4. [CrossRef]
- Froberg, M.K. , et al., Cytomegalovirus infection accelerates inflammation in vascular tissue overexpressing monocyte chemoattractant protein-1. Circ Res, 2001. 89(12): p. 1224-30. [CrossRef]
- The, T.H. , et al., Cellular and humoral parameters for vascular damage in blood during cytomegalovirus infections. Transplant Proc, 2001. 33(1-2): p. 1813. [CrossRef]
- Hsich, E. , et al., Cytomegalovirus infection increases development of atherosclerosis in Apolipoprotein-E knockout mice. Atherosclerosis, 2001. 156(1): p. 23-8. [CrossRef]
- Sedmak, D.D. , et al., Divergent patterns of ELAM-1, ICAM-1, and VCAM-1 expression on cytomegalovirus-infected endothelial cells. Transplantation, 1994. 58(12): p. 1379-85.
- SPAN, A.H.M., C. P.A.V. BOVEN, and C.A. BRUGGEMAN, The effect of cytomegalovirus infection on the adherence of polymorphonuclear leucocytes to endothelial cells. European Journal of Clinical Investigation, 1989. 19(6): p. 542-548. [CrossRef]
- Bruns, T. , et al., CMV infection of human sinusoidal endothelium regulates hepatic T cell recruitment and activation. Journal of Hepatology, 2015. 63(1): p. 38-49. [CrossRef]
- Zhao, J. , et al., Human cytomegalovirus infection-induced autophagy was associated with the biological behavioral changes of human umbilical vein endothelial cell (HUVEC). Biomedicine & Pharmacotherapy, 2018. 102: p. 938-946. [CrossRef]
- Bentz, G.L. , et al., Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol, 2006. 80(23): p. 11539-55.
- Ofotokun, I. , et al., Acute Cytomegalovirus Infection Complicated by Vascular Thrombosis: A Case Report. Clinical Infectious Diseases, 2001. 32(6): p. 983-986. [CrossRef]
- Dumortier, J. , et al., Human cytomegalovirus secretome contains factors that induce angiogenesis and wound healing. J Virol, 2008. 82(13): p. 6524-35. [CrossRef]
- Lee, G.C. , et al., Mitochondria-targeted apoptosis in human cytomegalovirus-infected cells. J Microbiol Biotechnol, 2013. 23(11): p. 1627-35. [CrossRef]
- Nakamura, H. , et al., Human cytomegalovirus induces apoptosis in neural stem/progenitor cells derived from induced pluripotent stem cells by generating mitochondrial dysfunction and endoplasmic reticulum stress. Herpesviridae, 2013. 4(1): p. 2. [CrossRef]
- Takatsuka, H. , et al., Endothelial damage caused by cytomegalovirus and human herpesvirus-6. Bone Marrow Transplantation, 2003. 31(6): p. 475-479. [CrossRef]
- Matsuda, Y. , et al., Thrombotic microangiopathy associated with reactivation of human herpesvirus-6 following high-dose chemotherapy with autologous bone marrow transplantation in young children. Bone Marrow Transplantation, 1999. 24(8): p. 919-923. [CrossRef]
- Vallbracht, K.B. , et al., Differential Aspects of Endothelial Function of the Coronary Microcirculation Considering Myocardial Virus Persistence, Endothelial Activation, and Myocardial Leukocyte Infiltrates. Circulation, 2005. 111(14): p. 1784-1791. [CrossRef]
- Maximilian Buja, L. , HHV-6 in Cardiovascular Pathology, in Perspectives in Medical Virology, G. Krueger and D. Ablashi, Editors. 2006, Elsevier. p. 233-241.
- Kühl, U. , et al., Chromosomally integrated human herpesvirus 6 in heart failure: prevalence and treatment. European Journal of Heart Failure, 2015. 17(1): p. 9-19. [CrossRef]
- Comar, M. , et al., Human herpes virus 6 in archival cardiac tissues from children with idiopathic dilated cardiomyopathy or congenital heart disease. Journal of Clinical Pathology, 2009. 62(1): p. 80-83. [CrossRef]
- Wada, A. , et al., Brainstem infarction associated with HHV-6 infection in an infant. Brain and Development, 2018. 40(3): p. 242-246. [CrossRef]
- Brennan, Y. , et al., A fatal case of acute HHV-6 myocarditis following allogeneic haemopoietic stem cell transplantation. Journal of Clinical Virology, 2015. 72: p. 82-84. [CrossRef]
- Leveque, N. , et al., A fatal case of Human Herpesvirus 6 chronic myocarditis in an immunocompetent adult. Journal of Clinical Virology, 2011. 52(2): p. 142-145. [CrossRef]
- Escher, F. , et al., CARDIAC INVOLVEMENT OF HUMAN HERPESVIRUS 6 IN PATIENTS WITH INFLAMMATORY CARDIOMYOPATHY. Journal of the American College of Cardiology, 2015. 65(10, Supplement): p. A945. [CrossRef]
- Mahrholdt, H. , et al., Presentation, Patterns of Myocardial Damage, and Clinical Course of Viral Myocarditis. Circulation, 2006. 114(15): p. 1581-1590.
- Miyahara, H. , et al., Unique cell tropism of HHV-6B in an infantile autopsy case of primary HHV-6B encephalitis. Neuropathology, 2018. 38(4): p. 400-406. [CrossRef]
- Ueda, T. , et al., Distribution of human herpesvirus 6 and varicella-zoster virus in organs of a fatal case with exanthem subitum and varicella. Acta Paediatr Jpn, 1996. 38(6): p. 590-5. [CrossRef]
- Harberts, E. , et al., Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A, 2011. 108(33): p. 13734-9. [CrossRef]
- Yoshikawa, T. and Y. Asano, Central nervous system complications in human herpesvirus-6 infection. Brain Dev, 2000. 22(5): p. 307-14. [CrossRef]
- Eliassen, E., C. C. Hemond, and J.D. Santoro, HHV-6-Associated Neurological Disease in Children: Epidemiologic, Clinical, Diagnostic, and Treatment Considerations. Pediatric Neurology, 2020. 105: p. 10-20. [CrossRef]
- Bartolini, L. , et al., Infection with HHV-6 and its role in epilepsy. Epilepsy Research, 2019. 153: p. 34-39. [CrossRef]
- Santpere, G. , et al., The Presence of Human Herpesvirus 6 in the Brain in Health and Disease. Biomolecules, 2020. 10(11). [CrossRef]
- Romeo, M.A. , et al., HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis, 2020. 1866(3): p. 165647. [CrossRef]
- Romeo, M.A. , et al., Viral Infection and Autophagy Dysregulation: The Case of HHV-6, EBV and KSHV. Cells, 2020. 9(12): p. 2624. [CrossRef]
- Reynaud, J.M. , et al., Human herpesvirus 6A infection in CD46 transgenic mice: viral persistence in the brain and increased production of proinflammatory chemokines via Toll-like receptor 9. J Virol, 2014. 88(10): p. 5421-36. [CrossRef]
- Flamand, L. , et al., Human herpesvirus 6 induces interleukin-1 beta and tumor necrosis factor alpha, but not interleukin-6, in peripheral blood mononuclear cell cultures. J Virol, 1991. 65(9): p. 5105-10. [CrossRef]
- Flamand, L. and J. Menezes, Cyclic AMP-responsive element-dependent activation of Epstein-Barr virus zebra promoter by human herpesvirus 6. J Virol, 1996. 70(3): p. 1784-91. [CrossRef]
- van Campen, C.M.C. , et al., Cerebral blood flow is reduced in ME/CFS during head-up tilt testing even in the absence of hypotension or tachycardia: A quantitative, controlled study using Doppler echography. Clinical Neurophysiology Practice, 2020. 5: p. 50-58. [CrossRef]
- van Campen, C.M.C., P. C. Rowe, and F.C. Visser, Cerebral blood flow remains reduced after tilt testing in myalgic encephalomyelitis/chronic fatigue syndrome patients. Clinical Neurophysiology Practice, 2021. 6: p. 245-255. [CrossRef]
- Campen, C.M.C.v., P. C. Rowe, and F.C. Visser, Orthostatic Symptoms and Reductions in Cerebral Blood Flow in Long-Haul COVID-19 Patients: Similarities with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Medicina, 2022. 58(1): p. 28. [CrossRef]
- Staud, R. , et al., Task related cerebral blood flow changes of patients with chronic fatigue syndrome: an arterial spin labeling study. Fatigue: Biomedicine, Health & Behavior, 2018. 6(2): p. 63-79. [CrossRef]
- van Campen, C., P. C. Rowe, and F.C. Visser, Deconditioning does not explain orthostatic intolerance in ME/CFS (myalgic encephalomyelitis/chronic fatigue syndrome). J Transl Med, 2021. 19(1): p. 193. [CrossRef]
- van Campen, C.M.C. and F.C. Visser, Comparison of the Degree of Deconditioning in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Patients with and without Orthostatic Intolerance. Medical Research Archives, 2022. 10(6).
- Nelson, M.J. , et al., Evidence of altered cardiac autonomic regulation in myalgic encephalomyelitis/chronic fatigue syndrome: A systematic review and meta-analysis. Medicine (Baltimore), 2019. 98(43): p. e17600.
- Vallet, B. , Endothelial cell dysfunction and abnormal tissue perfusion. Crit Care Med, 2002. 30(5 Suppl): p. S229-34.
- Levy, B.I. , et al., Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation, 2008. 118(9): p. 968-76.
- Bauersachs, J. and J.D. Widder, Endothelial dysfunction in heart failure. Pharmacol Rep, 2008. 60(1): p. 119-26.
- Hasdai, D. , et al., Coronary Endothelial Dysfunction in Humans Is Associated With Myocardial Perfusion Defects. Circulation, 1997. 96(10): p. 3390-3395. [CrossRef]
- Machin, D.R. , et al., Advanced age results in a diminished endothelial glycocalyx. American Journal of Physiology-Heart and Circulatory Physiology, 2018. 315(3): p. H531-H539.
- Ozbek, O. , et al., Epstein-Barr virus encephalitis: findings of MRI, MRS, diffusion and perfusion. Turk J Pediatr, 2011. 53(6): p. 680-3.
- Yoshinari, S. , et al., Human Herpesvirus 6 Encephalopathy Predominantly Affecting the Frontal Lobes. Pediatric Neurology, 2007. 36(1): p. 13-16. [CrossRef]
- Kuo, Y.-T. , et al., Cerebral perfusion in children with Alice in Wonderland syndrome. Pediatric Neurology, 1998. 19(2): p. 105-108. [CrossRef]
- Hendrix, M.G. , et al., High prevalence of latently present cytomegalovirus in arterial walls of patients suffering from grade III atherosclerosis. Am J Pathol, 1990. 136(1): p. 23-8.
- Gredmark-Russ, S. , et al., Active cytomegalovirus infection in aortic smooth muscle cells from patients with abdominal aortic aneurysm. Journal of Molecular Medicine, 2009. 87(4): p. 347-356. [CrossRef]
- Petrakopoulou, P. , et al., Cytomegalovirus Infection in Heart Transplant Recipients Is Associated With Impaired Endothelial Function. Circulation, 2004. 110(11_suppl_1): p. II-207-II-212. [CrossRef]
- Zhang, M. , et al., Human cytomegalovirus infection is a novel etiology for essential hypertension. Medical Hypotheses, 2011. 76(5): p. 682-684. [CrossRef]
- Yaiw, K.-C. , et al., Human Cytomegalovirus Reduces Endothelin-1 Expression in Both Endothelial and Vascular Smooth Muscle Cells. Microorganisms, 2021. 9(6): p. 1137. [CrossRef]
- Yaiw, K.-C. , et al., Human Cytomegalovirus Up-Regulates Endothelin Receptor Type B: Implication for Vasculopathies? Open Forum Infectious Diseases, 2015. 2(4).
- Zhou, Y.F. , et al., The immediate early gene products of human cytomegalovirus increase vascular smooth muscle cell migration, proliferation, and expression of PDGF beta-receptor. Biochem Biophys Res Commun, 1999. 256(3): p. 608-13. [CrossRef]
- Tanaka, K. , et al., Effects of human cytomegalovirus immediate-early proteins on p53-mediated apoptosis in coronary artery smooth muscle cells. Circulation, 1999. 99(13): p. 1656-9. [CrossRef]
- Speir, E. , et al., Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science, 1994. 265(5170): p. 391-4.
- Cheng, J. , et al., Cytomegalovirus infection causes an increase of arterial blood pressure. PLoS Pathog, 2009. 5(5): p. e1000427. [CrossRef]
- Kresch, E. , et al., COVID-19 Endothelial Dysfunction Can Cause Erectile Dysfunction: Histopathological, Immunohistochemical, and Ultrastructural Study of the Human Penis. World J Mens Health, 2021. 39(3): p. 466-469. [CrossRef]
- Taghavi, S. , et al., Glycocalyx degradation and the endotheliopathy of viral infection. PLOS ONE, 2022. 17(10): p. e0276232. [CrossRef]
- Wadowski, P.P. , et al., Glycocalyx as Possible Limiting Factor in COVID-19. Frontiers in Immunology, 2021. 12. [CrossRef]
- Stahl, K. , et al., Injury to the Endothelial Glycocalyx in Critically Ill Patients with COVID-19. Am J Respir Crit Care Med, 2020. 202(8): p. 1178-1181. [CrossRef]
- Terasawa, M. , et al., Anti-Inflammatory Activity of Orally Administered Monostroma nitidum Rhamnan Sulfate against Lipopolysaccharide-Induced Damage to Mouse Organs and Vascular Endothelium. Marine Drugs, 2022. 20(2): p. 121. [CrossRef]
- De Pasquale, V. , et al., Heparan Sulfate Proteoglycans in Viral Infection and Treatment: A Special Focus on SARS-CoV-2. International Journal of Molecular Sciences, 2021. 22(12): p. 6574.
- Lee, D.H. , et al., Deeper Penetration of Erythrocytes into the Endothelial Glycocalyx Is Associated with Impaired Microvascular Perfusion. PLOS ONE, 2014. 9(5): p. e96477. [CrossRef]
- Beurskens, D.M. , et al., Decreased endothelial glycocalyx thickness is an early predictor of mortality in sepsis. Anaesth Intensive Care, 2020. 48(3): p. 221-228. [CrossRef]
- Cabrales, P. , et al., Microvascular and capillary perfusion following glycocalyx degradation. Journal of Applied Physiology, 2007. 102(6): p. 2251-2259. [CrossRef]
- Chappell, D., M. Westphal, and M. Jacob, The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Current Opinion in Anesthesiology, 2009. 22(2). [CrossRef]
- Ikonomidis, I. , et al., Endothelial glycocalyx and microvascular perfusion are associated with carotid intima-media thickness and impaired myocardial deformation in psoriatic disease. Journal of Human Hypertension, 2022. 36(12): p. 1113-1120. [CrossRef]
- Caselli, E. , et al., HHV-6A Infection and Systemic Sclerosis: Clues of a Possible Association. Microorganisms, 2020. 8(1): p. 39. [CrossRef]
- van Campen, C.M.C., P. C. Rowe, and F.C. Visser, Cerebral Blood Flow Is Reduced in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients During Mild Orthostatic Stress Testing: An Exploratory Study at 20 Degrees of Head-Up Tilt Testing. Healthcare, 2020. 8(2): p. 169. [CrossRef]
- Wirth, K.J. and C. Scheibenbogen, Pathophysiology of skeletal muscle disturbances in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Journal of Translational Medicine, 2021. 19(1): p. 162. [CrossRef]
- Chimenti, C. , et al., Infarct-like myocarditis with coronary vasculitis and aneurysm formation caused by Epstein–Barr virus infection. ESC Heart Failure, 2020. 7(3): p. 938-941. [CrossRef]
- Jamal, O. , et al., Fatal Systemic Vasculitis Associated with Chronic Active Epstein-Barr Virus Infection. Mo Med, 2021. 118(3): p. 226-232.
- Gatto, A. , et al., Pediatric cerebral stroke induced by Epstein-Barr virus infection: Role of Interelukin overexpression. Acta Biomed, 2021. 92(S1): p. e2021135. [CrossRef]
- Bader, M.S. , et al., Disseminated Intravascular Coagulation and Venous Thromboembolism Due to Acute Epstein-Barr Virus Infection. Infectious Diseases in Clinical Practice, 2011. 19(4). [CrossRef]
- O'Connor, D.S. , et al., Disseminated Intravascular Coagulation Complicating Epstein-Barr Virus Infection in a Cardiac Transplant Recipient: A Case Report. Transplantation Proceedings, 2010. 42(5): p. 1973-1975. [CrossRef]
- Plummer, M.P. , et al., Probable Catastrophic Antiphospholipid Syndrome with Intracerebral Hemorrhage Secondary to Epstein–Barr Viral Infection. Neurocritical Care, 2018. 28(1): p. 127-132. [CrossRef]
- Oka, S. and M. Nohgawa, EB virus reactivation triggers thrombotic thrombocytopenic purpura in a healthy adult. Leukemia Research Reports, 2017. 8: p. 1-3. [CrossRef]
- Mashav, N. , et al., Epstein-Barr virus-associated venous thromboembolism: A case report and review of the literature. Thrombosis Research, 2008. 122(4): p. 570-571. [CrossRef]
- Hal, S.V., S. Senanayake, and R. Hardiman, Splenic infarction due to transient antiphospholipid antibodies induced by acute Epstein-Barr virus infection. Journal of Clinical Virology, 2005. 32(3): p. 245-247. [CrossRef]
- Neumann, F.J. , et al., Previous cytomegalovirus infection and risk of coronary thrombotic events after stent placement. Circulation, 2000. 101(1): p. 11-3. [CrossRef]
- Inacio, C. , et al., Case report: cytomegalovirus infection as a cause of acute portal vein thrombosis. J Gastroenterol Hepatol, 1997. 12(4): p. 287-8. [CrossRef]
- Uthman, I., Z. Tabbarah, and A.E. Gharavi, Hughes syndrome associated with cytomegalovirus infection. Lupus, 1999. 8(9): p. 775-7. [CrossRef]
- Ailani, R.K. , et al., Extensive mesenteric inflammatory veno-occlusive disease of unknown etiology after primary cytomegalovirus infection: first case. Am J Gastroenterol, 1997. 92(7): p. 1216-8.
- Lanari, M. , et al., Neonatal aortic arch thrombosis as a result of congenital cytomegalovirus infection. Pediatrics, 2001. 108(6): p. E114. [CrossRef]
- Labarca, J.A. , et al., Antiphospholipid syndrome associated with cytomegalovirus infection: case report and review. Clin Infect Dis, 1997. 24(2): p. 197-200. [CrossRef]
- Labarca, J.A. , et al., Thrombosis, Vasculitis, and Cytomegalovirus Infection. Clinical Infectious Diseases, 2002. 34(12): p. 1658-1659.
- Arav-Boger, R., S. Reif, and Y. Bujanover, Portal vein thrombosis caused by protein C and protein S deficiency associated with cytomegalovirus infection. J Pediatr, 1995. 126(4): p. 586-8. [CrossRef]
- Madalosso, C. , et al., Cytomegalovirus and its association with hepatic artery thrombosis after liver transplantation. Transplantation, 1998. 66(3): p. 294-7. [CrossRef]
- Abgueguen, P. , et al., Vascular Thrombosis and Acute Cytomegalovirus Infection in Immunocompetent Patients: Report of 2 Cases and Literature Review. Clinical Infectious Diseases, 2003. 36(11): p. e134-e139. [CrossRef]
- Popović, M. , et al., Human cytomegalovirus infection and atherothrombosis. Journal of Thrombosis and Thrombolysis, 2012. 33(2): p. 160-172. [CrossRef]
- Delbos, V. , et al., Acute cytomegalovirus infection and venous thrombosis: role of antiphospholipid antibodies. J Infect, 2007. 54(1): p. e47-50. [CrossRef]
- Vercellotti, G.M. , Effects of viral activation of the vessel wall on inflammation and thrombosis. Blood Coagul Fibrinolysis, 1998. 9 Suppl 2: p. S3-6.
- van Geelen, A.G. , et al., Membrane related effects in endothelial cells induced by human cytomegalovirus. Arch Virol, 1995. 140(9): p. 1601-12. [CrossRef]
- Gharavi, E.E. , et al., Induction of antiphospholipid antibodies by immunization with synthetic viral and bacterial peptides. Lupus, 1999. 8(6): p. 449-55. [CrossRef]
- Schambeck, C.M. , et al., Venous thromboembolism and associated high plasma factor VIII levels: linked to cytomegalovirus infection? Thromb Haemost, 2000. 83(3): p. 510-1.
- Jenkins, R.E., B. S. Peters, and A.J. Pinching, Thromboembolic disease in AIDS is associated with cytomegalovirus disease. Aids, 1991. 5(12): p. 1540-2. [CrossRef]
- Agbanyo, F.R. and S. Wasi, Human cytomegalovirus interaction with platelets and adhesive glycoproteins: significance in viral pathogenesis. J Infect Dis, 1994. 170(5): p. 1120-7. [CrossRef]
- Pryzdial, E.L. and J.F. Wright, Prothrombinase assembly on an enveloped virus: evidence that the cytomegalovirus surface contains procoagulant phospholipid. Blood, 1994. 84(11): p. 3749-57. [CrossRef]
- Wang, F. , et al., Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front Aging Neurosci, 2018. 10: p. 376. [CrossRef]
- Saleem, M. , et al., Association Between Endothelial Function and Cognitive Performance in Patients With Coronary Artery Disease During Cardiac Rehabilitation. Psychosom Med, 2019. 81(2): p. 184-191. [CrossRef]
- Buie, J.J. , et al., Obesity-related cognitive impairment: The role of endothelial dysfunction. Neurobiol Dis, 2019. 132: p. 104580. [CrossRef]
- Riedel, B., K. Browne, and B. Silbert, Cerebral protection: inflammation, endothelial dysfunction, and postoperative cognitive dysfunction. Current Opinion in Anesthesiology, 2014. 27(1).
- Groeneveld, O.N. , et al., Oxidative stress and endothelial dysfunction are associated with reduced cognition in type 2 diabetes. Diab Vasc Dis Res, 2019. 16(6): p. 577-581. [CrossRef]
- Gozal, D. , et al., Neurocognitive and Endothelial Dysfunction in Children With Obstructive Sleep Apnea. Pediatrics, 2010. 126(5): p. e1161-e1167. [CrossRef]
- Heringa, S.M. , et al., Markers of low-grade inflammation and endothelial dysfunction are related to reduced information processing speed and executive functioning in an older population – the Hoorn Study. Psychoneuroendocrinology, 2014. 40: p. 108-118. [CrossRef]
- Yun, S.-M. , et al., Association of plasma endothelial lipase levels on cognitive impairment. BMC Psychiatry, 2019. 19(1): p. 187. [CrossRef]
- Martins-Filho, R.K. , et al., Biomarkers Related to Endothelial Dysfunction and Vascular Cognitive Impairment: A Systematic Review. Dementia and Geriatric Cognitive Disorders, 2020. 49(4): p. 365-374. [CrossRef]
- Øie, M.G. , et al., Subjective and objective cognitive function in adolescent with chronic fatigue following Epstein-Barr virus infection. Journal of Psychosomatic Research, 2022. 163: p. 111063.
- Shim, S.-M. , et al., Elevated Epstein-Barr Virus Antibody Level is Associated with Cognitive Decline in the Korean Elderly. Journal of Alzheimer's Disease, 2017. 55: p. 293-301. [CrossRef]
- Wang, Z. , et al., Human Cytomegalovirus Immediate Early Protein 2 Protein Causes Cognitive Disorder by Damaging Synaptic Plasticity in Human Cytomegalovirus-UL122-Tg Mice. Frontiers in Aging Neuroscience, 2021. 13. [CrossRef]
- Han, D. , et al., Human Cytomegalovirus IE2 Protein Disturbs Brain Development by the Dysregulation of Neural Stem Cell Maintenance and the Polarization of Migrating Neurons. J Virol, 2017. 91(17). [CrossRef]
- Tarchini, G. , Human Herpesviruses HHV-6A, HHV-6B &HHV-7: Diagnosis and Clinical Management. 3rd ed. Clinical Infectious Diseases, 2014. 60(3): p. 496-497. 2014. [Google Scholar]
- Huang, C. , et al., Association between human herpesvirus 6 (HHV-6) and cognitive function in the elderly population in Shenzhen, China. Aging Clinical and Experimental Research, 2022. 34(10): p. 2407-2415. [CrossRef]
- Tanaka, M. , et al., Fatigue-associated alterations of cognitive function and electroencephalographic power densities. PLoS One, 2012. 7(4): p. e34774. [CrossRef]
- Zerr, D.M. , et al., HHV-6 reactivation and its effect on delirium and cognitive functioning in hematopoietic cell transplantation recipients. Blood, 2011. 117(19): p. 5243-9. [CrossRef]
- Wirth, K.J., C. Scheibenbogen, and F. Paul, An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Journal of Translational Medicine, 2021. 19(1): p. 471. [CrossRef]
- Navone, S.E. , et al., Isolation and expansion of human and mouse brain microvascular endothelial cells. Nature Protocols, 2013. 8(9): p. 1680-1693. [CrossRef]
- Rosas-Hernandez, H. , et al., Isolation and Culture of Brain Microvascular Endothelial Cells for In Vitro Blood-Brain Barrier Studies. Methods Mol Biol, 2018. 1727: p. 315-331. [CrossRef]
- Bueno-Betí, C. , et al., An affordable method to obtain cultured endothelial cells from peripheral blood. Journal of Cellular and Molecular Medicine, 2013. 17(11): p. 1475-1483. [CrossRef]
- Okumura, N. , et al., Density-gradient centrifugation enables the purification of cultured corneal endothelial cells for cell therapy by eliminating senescent cells. Scientific Reports, 2015. 5(1): p. 15005. [CrossRef]
Figure 1.
An overview of the present paper. A) A delineation of our thought-approach and B) a chronological mind map (created on https://app.ayoa.com/mindmaps).
Figure 1.
An overview of the present paper. A) A delineation of our thought-approach and B) a chronological mind map (created on https://app.ayoa.com/mindmaps).
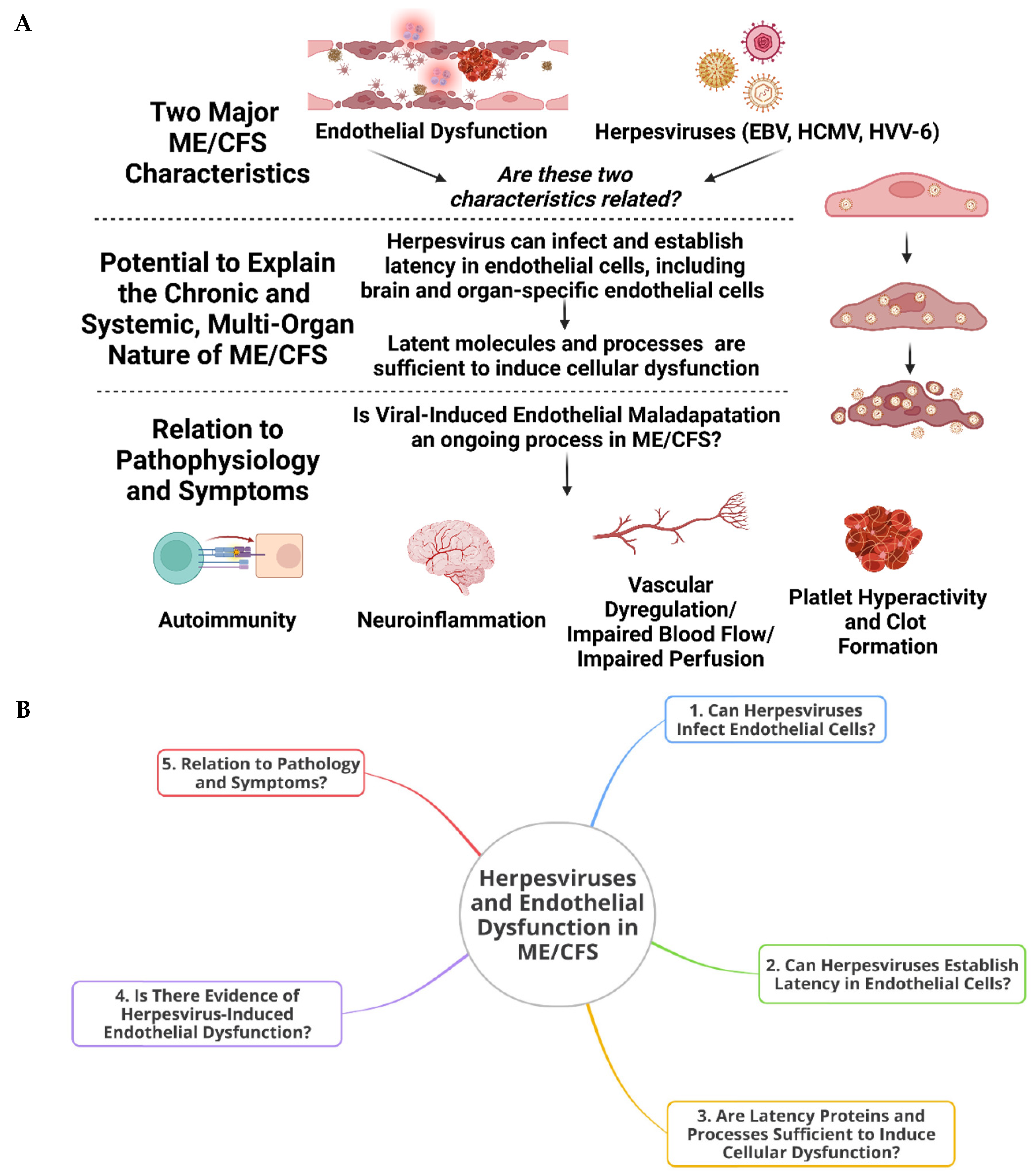
Figure 2.
Mechanisms by which HVV-8 latency and associated proteins cause endothelial dysfunction.
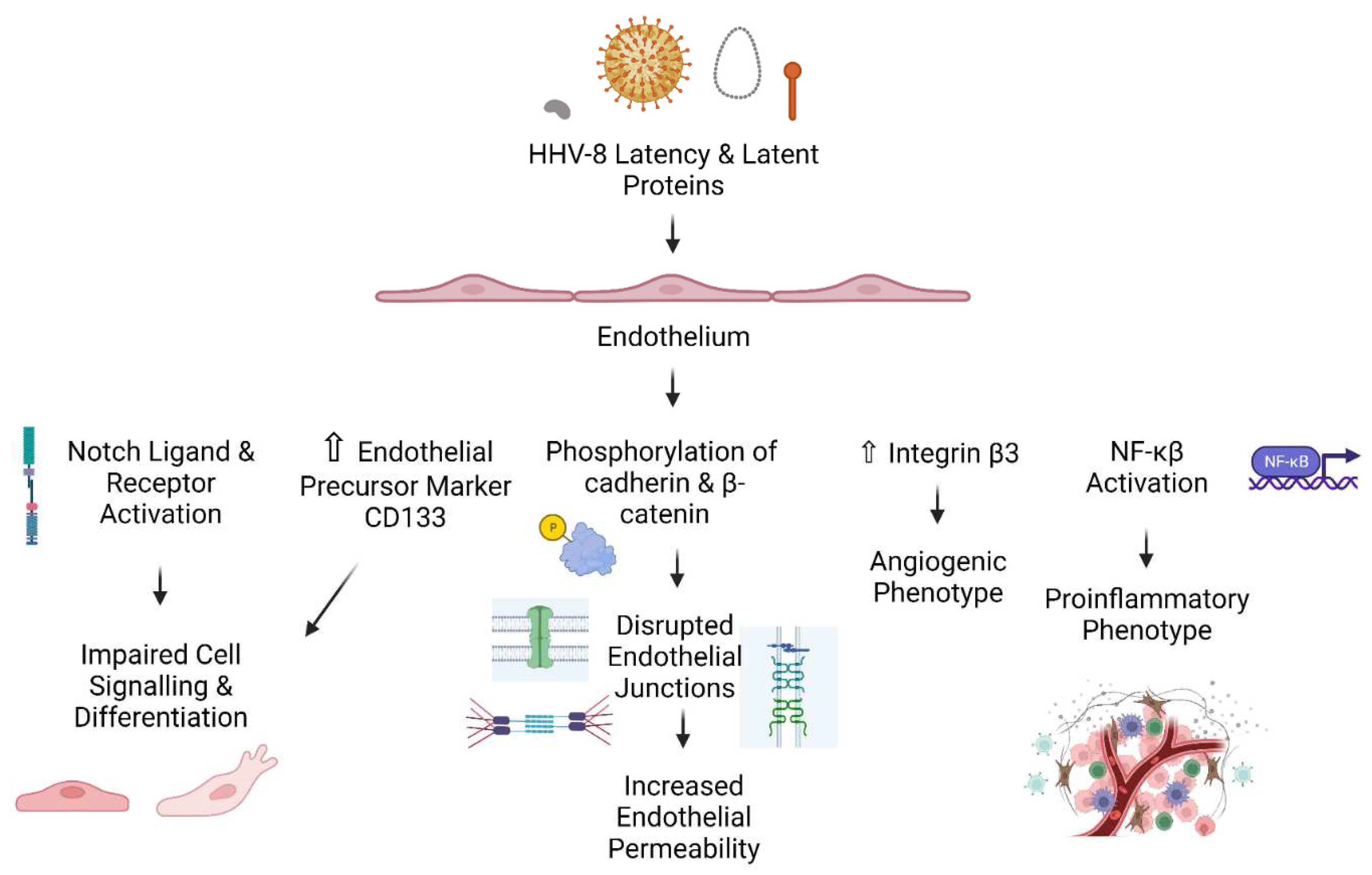
Figure 3.
Some mechanisms by which EBV latent proteins EBNA-1 and LMP-1 can induce cellular dysfunction in endothelial cells.
Figure 3.
Some mechanisms by which EBV latent proteins EBNA-1 and LMP-1 can induce cellular dysfunction in endothelial cells.
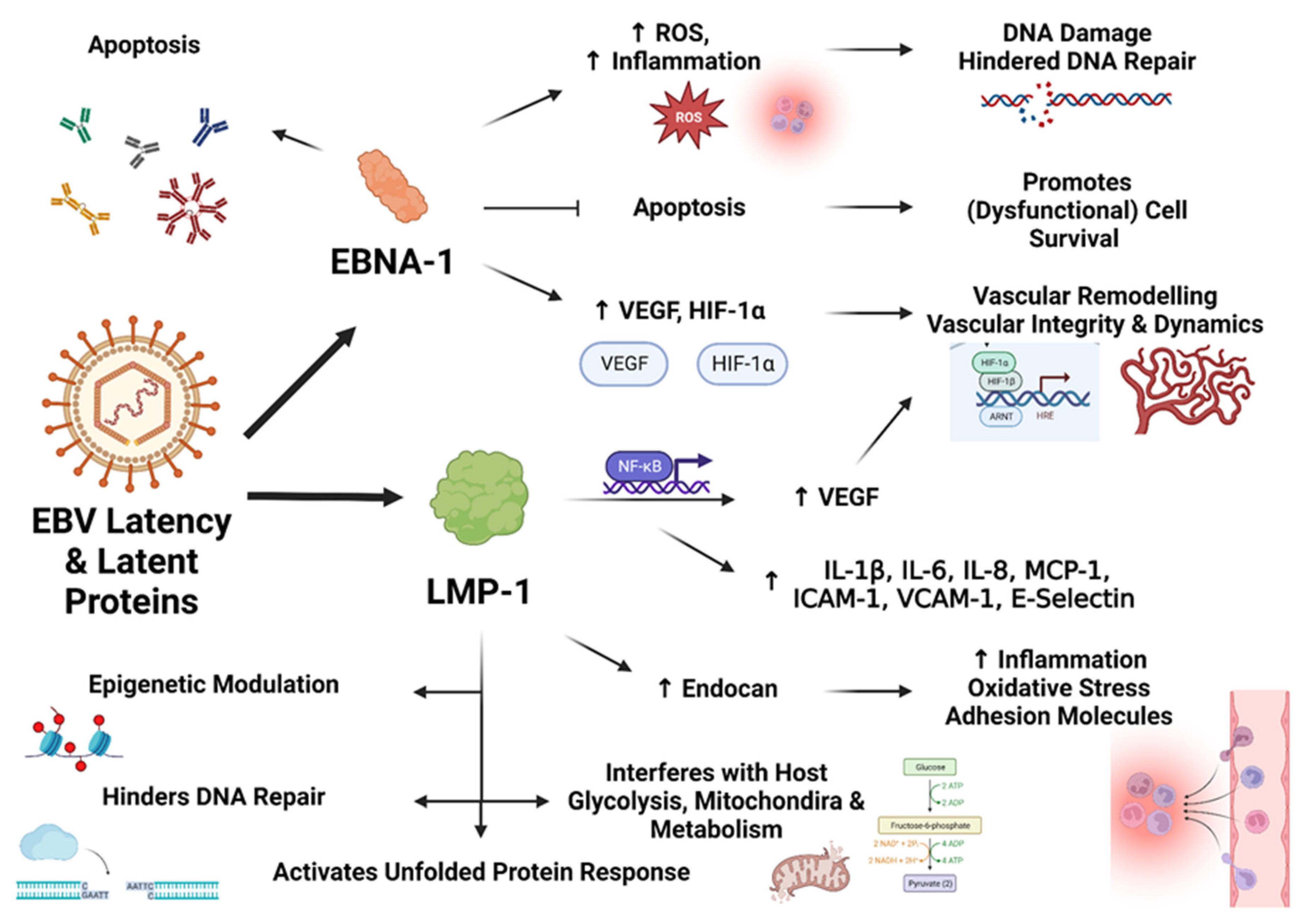
Figure 4.
Adopted figure from [50] showing that EBV load is inversely associated with hand skin perfusion (Open Access).
Figure 4.
Adopted figure from [50] showing that EBV load is inversely associated with hand skin perfusion (Open Access).
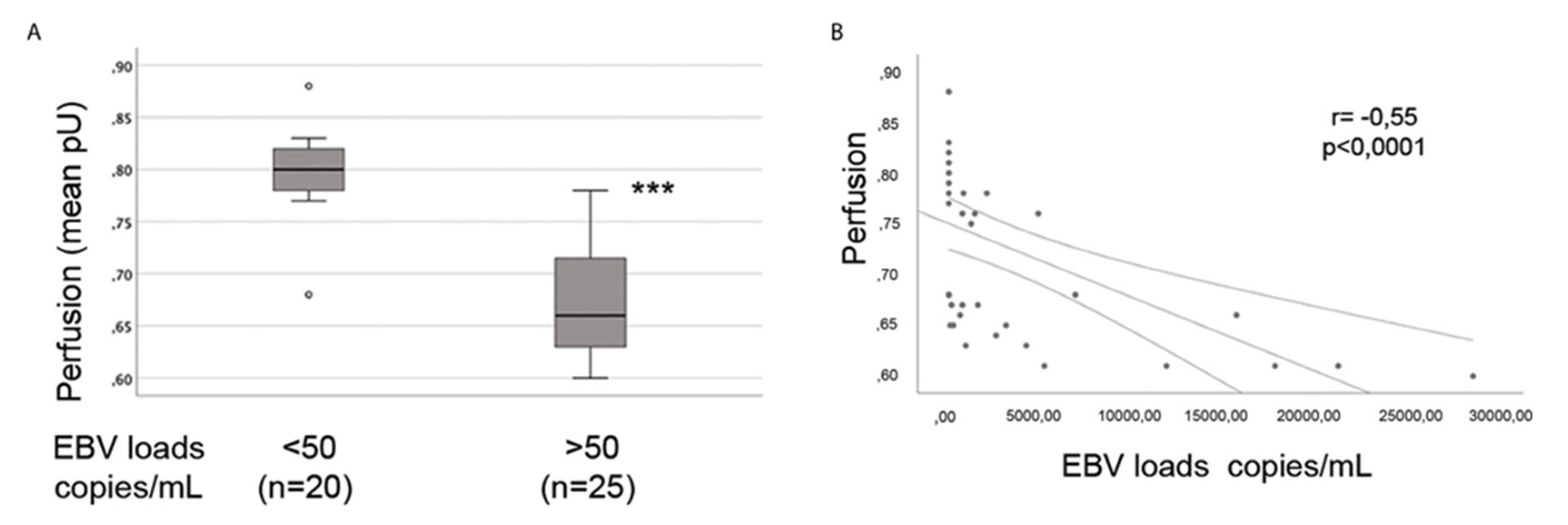

Table 1.
Links between herpesvirus infection and cellular dysfunction in ECs.
| Links Between Herpesvirus Infection and Endothelial Dysfunction | References |
|---|---|
| EBV | |
| ECs infected with EBV exhibit a proinflammatory phenotype, along with NF-κB and TLR9 activation, increased interferon, cytokine, and adhesion molecule expression, and increased clotting propensity | [50,113,147] |
| ECs increase expression of markers associated with vascular injury, such as endothelin-1, thrombospondin 1, and heparan sulfate proteoglycan 2 | [50] |
| Monocytes have the ability to transfer EBV infection to ECs | Monocytes have the ability to transfer EBV infection to ECs |
| Microvascular brain ECs infected by EBV exhibit a proinflammatory phenotype and lead to leukocyte recruitment | [49,148] |
| Upregulation of endothelial adhesion marker VCAM-1 upon infection | [149] |
| EBV-infected macrophages induce proinflammatory sequelae in ECs, and increase adhesion molecule expression | [150] |
| EBV dUTPase compromises blood-brain barrier integrity | [9] |
| EBV alters cholesterol, polysaccharide, nucleotides, nucleic acid and proline moieties in infected brain microvascular ECs | [147] |
| EBV-infected ECs of genital origin express LMP-1 on their membranes | [151] |
| Endothelial microenvironment is influenced by EBV infection | [152] |
| Extracellular vesicles from EBV-infected cells damage endothelial gap junctions, and prompt endothelial-to-mesenchymal transitions | [153] |
| Modulation of host autophagy in endothelial cells | [154] |
| Exosomes containing EBV-proteins can cross brain ECs and enter the central nervous system | [155] |
| EBV protein-containing exosomes can lead to long-term endothelial dysfunction | [156] |
| HCMV | |
| HCMV establishes latent infection in ECs | [68,69,157,158,159] |
| ECs of the microvasculature in the brain, lungs, heart, and gastrointestinal tract are target infection sites, but so are large vessel ECs | [14,160,161] |
| Reactivation causes endothelial dysfunction | [162] |
| HCMV can cause significant infection even when multiplicity of infection is low | [163] |
| MicroRNA (UL112) interferes with cell signalling pathways | [164] |
| HCMV infection is associated with endothelial inflammation and vascular disease, and the virus is localized in atherosclerotic plaques and non-plaque tissue surrounding lesions | [161,165,166,167,168] |
| HCMV increases atherosclerotic development in mice models with apolipoprotein E deficiency | [169] |
| HCMV induces the expression of leukocyte adhesion molecules on ECs and subsequent leukocyte activation and recruitment | [140,170,171,172,173,174] |
| Leukocytes undergoing transendothelial migration can be infected by infected ECs | [174] |
| In a patient with HCMV infection, ECs were found to be abnormal and containing viral inclusion bodies | [175] |
| The secretome of HCMV-infected fibroblasts contains over 1000 different proteins, most notably a profile that induces angiogenesis and wound healing in ECs | [176] |
| HCMV interferes with DNA protection mechanisms in ECs, specifically by interfering with the ataxia telangiectasia mutant pathway | [162] |
| EC autophagy is upregulated by HCMV | [173] |
| HCMV disrupts the mitochondrial transmembrane potential of endothelial mitochondria and leads to the release of cytochrome c and subsequent apoptosis | [177,178] |
| HHV-6 | |
| HHV-6 infection is associated with endothelial dysfunction and a greater extent of endothelial damage than HCMV | [66,179,180] |
| HHV-6 infects ECs but does not induce cytolytic effects, which led to the conclusion that ECs act as a reservoir for HHV-6 in vivo | [57] |
| HVV-6 is able to maintain a low-level of replication within ECs | [60,67] |
| An association between HHV-6 and endothelial dysfunction coupled to microcirculatory defects has been demonstrated | [181] |
| The induction of endothelial dysfunction by HHV-6 and subsequent influence on perfusion have been alluded to | [182] |
| HHV-6 antigens, DNA, and virus particles are found in ECs and associated vascular tissue from patients suffering from various cardiovascular diseases | [62,183,184,185,186,187] |
| Cardiac dysfunction, specifically reduced LVEF is associated with HHV-6 DNA persistence in endomyocardial biopsies, and is ameliorated when HHV-6 latency is resolved | [188] |
| Considered to be a major cause of viral myocarditis | [189] |
| HHV-6 also infects the CNS and ECs lining its vasculature | [190,191,192] |
| HVV-6 is implicated in neurological disease | [193,194,195,196,197,198] |
| Much like EBV, HHV-6 uses TLR9 to upregulate inflammation and promote lymphocyte filtration, as was revealed from a study where mice infected with HHV-6 subtypes resulted in CNS infection and viral persistence in brain tissue for up to 9 months | [199] |
| HHV-6 induces cellular inflammation and upregulates the expression of IL-8, RANTES, and monocyte chemoattractant protein-1 in ECs, even in a latent state, without viral DNA replication | [57,59,200] |
| It can also promote the reactivation of EBV | [201] |
| Lymphatic ECs also succumb to latent infection by HHV-6, where EC angiogenic and migratory properties are modulated | [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



