
Submitted:
22 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
Gliomas are central nervous system (CNS) derived tumors that originate from glial cells and affect six per 100,000 people in the United States. They are the most common form of CNS neoplasm and are highly diffusely infiltrative to surrounding brain tissue. Gliomas of diffusive nature are classified into different subcategories: astrocytomas, oligodendrogliomas, and ependymomas. Computed tomography (CT) and Magnetic Resonance Imaging (MRI) are used to determine tumor category and spread which may dictate treatment options. This paper focuses on emerging treatments of chemotherapy management with temozolomide and Avastin as well as emerging immune modulation treatment with vaccinations, manipulation of cellular checkpoints CTLA-4 and PD-1, co-receptor modulation of neuropilin-1, CAR-T cell therapy, and CRISPR-Cas9 gene editing.
The employment of TMZ and BEV has been supported in the literature, however, more research is necessitated to fully optimize regimens and to avoid immune- and toxicity-related effects of these drugs. Moreover, an even closer consideration is warranted for current immunotherapies as they are in their infancy compared to TMZ and BEV. Thus, although promising emerging treatment options, therapies involving cancer vaccines, ICIs, immune modulation of neuropilin-1, CAR-T, and CRISPR-Cas9 technology require further investigation into their long-term efficacy and utility in treating gliomas.
Keywords:
Glioma
; Immune Modulation
; Chemotherapy Management
; Temozolomide
; Avastin
Introduction
Gliomas are central nervous system (CNS) derived tumors that originate from glial cells and affect six per 100,000 people in the United States. Around 20,000 patients are diagnosed with gliomas every year in the United States, with 12,000 patients being diagnosed with glioblastomas [1]. Gliomas are the most common form of CNS neoplasm and are highly diffusely infiltrative to surrounding brain tissue. Gliomas of diffusive nature are classified into different subcategories: astrocytomas, oligodendrogliomas, and ependymomas. Pilocytic astrocytomas are the least malignant tumors while glioblastomas are the most malignant type [1].
Headaches are the most common symptom associated with gliomas and are thought to be caused by the growth of tumor cells. The tumor cells increase pressure in the vasculature and result in edema. Nausea, vomiting, and vision changes are other symptoms that can occur, with seizures being the second most common symptom experienced. In order to diagnose a glioma, imaging techniques are critical. Computed tomogram (CT) head can detect edema, asymmetry, and hemorrhage in the brain tissue. Magnetic resonance imaging (MRI) of the brain can be used to evaluate the grade of the tumor detected based on enhancement. Chest X-rays are also useful in determining tumor metastasis and play a vital role in determining treatment approaches.
Current treatment approaches include surgical resections, chemoradiation, antiepileptic medications, steroids, and deep venous thrombosis prophylaxis for symptom management [1]. This paper will focus on emerging treatments of chemotherapy management with temozolomide and Avastin as well as emerging immune modulation treatment with vaccinations, manipulation of cellular checkpoints CTLA-4 and PD-1, co-receptor modulation of neuropilin-1, CAR-T cell therapy, and CRISPR-Cas9 gene editing.
Prognostic Biomarkers for Treating Gliomas
Gliomas are tumors derived from neuroepithelial cells (e.g., glial cells) and cells with similar histologic characteristics which together comprise the most common primary tumors affecting the central nervous system [2]. Previously, these tumors were clumped into “low-grade” and “high-grade” by the World Health Organization (WHO) grading criterion, using primarily tumor histological and radiological findings [2,3]. Since the 2016 edition of the WHO classification system, the parameter for grading began integrating molecular change to guide patients and clinicians [3,4,5]. While new markers are continually being studied via immunohistochemistry and genomic sequencing, tailoring a multidisciplinary approach is essential to refining patients’ outcomes by tailoring the individual management of these heterogenous lesions [6].
Presently, gliomas are based on the 2021 WHO grading system which further evaluates gliomas for various biomarkers [5]. A glioma can be classified by histopathologic characteristics, such as cell morphology and glial fibrillary acidic protein (GFAP) staining, and nuclear atypia [5,6,7]. Molecular markers may be useful for certain subtypes, but not necessary to characterize all lesions [5]. Grading for pathologically equivocal lesions can be elucidated with molecular approaches, most commonly variants of isocitrate dehydrogenase (IDH), as well as the presence or absence of a 1p/19 chromosomal codeletion which may confer higher grading and worse prognoses, even independently of histological analysis [8,9]. In the case of larger glioblastomas, the methylation status of the promoter O6-methylguanine-DNA methyltransferase (MGMT) is also considered as it provides predictive value to patients’ prognoses and possible response to chemotherapeutic agents [10]. The MGMT biomarker is also better correlated with IDH-mutant glioblastomas, which may provide an opportunity for future therapeutic benefit [11].
Astrocytic and oligodendroglial tumors are collectively described as low-grade diffuse gliomas, largely on their histologic features of proliferation, cytologic patterns, and IDH mutation commonalities [2,3,5]. Low-grade gliomas include grades 1 and 2, while high-grade gliomas include grades 3 and 4. Features such as nuclear atypia and increased mitotic features are consistent with anaplastic (Grade 3) tumors, while vascularization and tumor necrosis are suggestive of glioblastomas (Grade 4) [5]. Additional neoplastic features correlate with an increase in histologic grading. The status of IDH-wildtype confers the status of lower grades 2 and 3, while those of IDH-mutant require further molecular and chemical assays to differentiate a glioblastoma versus a diffuse glioma, of which these are all grade 4 [8,9].
Currently, the standard management for gliomas follows their grading and includes maximal surgical resection of tumors with negative margins (when possible) followed by adjuvant series of chemotherapy and radiotherapy. Surgical intervention may serve for diagnostic and symptomatic relief but is often limited by the location and not curative for higher-grade gliomas [12]. Patients with a grade 2 or 3 glioma have been shown to have increased survival alkylation chemotherapeutics, such as the regimen of procarbazine, lomustine, and vincristine (PCV) in addition to their radiotherapy [12,13]. Temozolomide (TMZ) has been associated with improved results as adjuvant treatment in patients with 1p/19q codeletion anaplastic gliomas, and PCV has also been demonstrated to be effective as adjuvant therapy, especially in gliomas with 1p/19q codeletion mutation when compared to those without a mutation [14,15]. In general, TMZ is preferred by clinicians and patients since it is better tolerated, can be administered orally, and has more limited side effect profiles compared to PCV [15]. Additionally, some studies have shown a benefit of survival in patients with IDH-mutant grade 4 glioblastomas with TMZ relative to the IDH-wildtype variant, suggesting the potential of MGMT as a biomarker in glioma management [16]. Various guidelines currently recommend the use of TMZ over PCV since it is better tolerated due to its oral formulation as well as a smaller side effect profile [17].
Current research continues to discover the role of several novel markers and their potential benefits against gliomas. Chimeric antigen T-cell receptors (CAR-T) including the protein B7-H3, a cell product involved in lymphocyte activation, have revealed in preclinical findings of antitumor activity versus a member of the H3 family (e.g., B7-H3, CD276) that is highly expressed glioblastoma multiforme (e.g., grade 4 gliomas) and pediatric gliomas [18]. Another immune checkpoint involved in the cellular pathway of glioblastomas is the signal transducer and activator of the transcription-3 (STAT-3) signal transduction pathway. STAT-3 has been found in several studies to be abnormally overexpressed in glioblastoma tissue [19]. Other studies have shown that inhibitors of STAT3 induced apoptosis in gliomas [20,21]. Given its role in the dysregulation of proliferation in gliomas, STAT-3 serves as a promising therapeutic target for future studies.
As stated above, various immune checkpoint modulators play an active role in potential biomarker therapeutics for the management of gliomas. Programmed death-ligand 1 (PD-L1) is a molecule responsible for suppressing the activity of T-lymphocytes and mediates a tumor cell escape mechanism from immunosurveillance. A meta-analysis showed a correlation between decreased expression of PD-L1 and decreased survival from glioblastoma multiforme [22]. Several novel therapeutics currently undergoing clinical trials, and even more are currently being developed, with an emphasis on combining alkylating chemotherapy with novel immune regulatory markers involved in glioma cell signaling events. As MGMT is well described as a biomarker in various sensitivities to alkylating chemotherapeutics, it may promote effective treatment to various chemotherapeutics depending on the various biomarkers and sensitivities of different glioma subtypes [23].
Temozolomide
DNA alkalizing agents are one of the oldest and most prominent classes of drugs used in the treatment of cancer, especially brain tumors. They act by stopping DNA formation via the addition of alkyl groups to the specific nucleic acid bases. Temozolomide (TMZ) is a new drug that has shown positive results in the treatment of brain tumors as a chemotherapeutic alkalizing agent. However, even though it prolongs survival in patients, the progression and recurrence of the tumor are still observed [24]. Thus, to improve the anti-cancer properties of TMZ in treating gliomas, it is important to understand its mechanism of action.
TMZ is a second-generation imidazotetrazine prodrug that, under certain physiologic conditions, undergoes spontaneous hydrolyzation to its active form, MTIC (3-methyl-(triazine-1-yl) imidazole-4-carboxamide). Thus, little to no interaction with other drugs is observed due to TMZ not being metabolized in the liver. As for the mechanism of action, it works by most commonly methylating the N7 position of guanine, the O6 position of guanine, and the N3 position of adenine [24,25]. This methylation damages the DNA of tumor cells and aids in their apoptosis.
Figure 2.
The effect of TMZ on DNA in tumor cell lines.
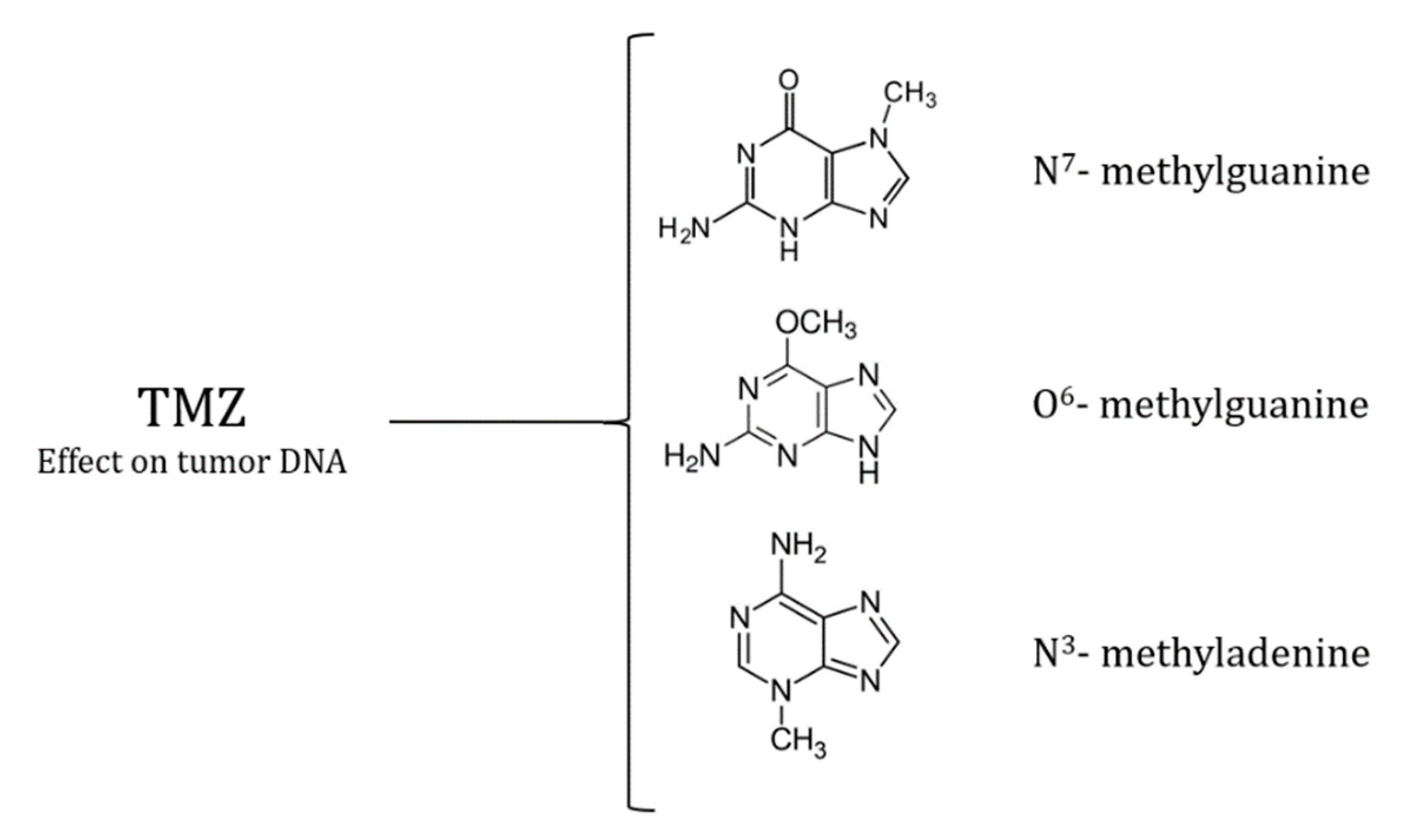
Moreover, these DNA methylations are observed to be effectively repaired by the DNA repair protein O6-alkylguanine alkyl transferase (AGT), which is encoded by the human gene O6-methylguanine-DNA methyltransferase (MGMT) [26,27]. This protein acts by the base excision repair (BER) pathway, which counteracts the damage induced by TMZ. Thus, glioma tumor cell lines that have lower levels of the gene encoding this protein are more sensitive to this drug, whereas the ones that have higher levels show resistive properties. Because of this reason, targeting the MGMT gene improves the efficacy of TMZ and improves its anti-tumor properties.
One of the ways to inhibit the function of AGT is by incorporating its inhibitor O6-benzylguanine (BG) in the treatment regimen of using TMZ for gliomas. The improved efficacy of this combination therapy is researched by many studies and is seen to be a promising way to treat resistant gliomas [28,29,30,31]. However, there is also clinical evidence suggesting systemic side effects of this combination therapy (hepatic toxicity, myelosuppression, pulmonary fibrosis, GI side effects) [32]. Thus, future research should explore the use of this drug combination by potentially limiting its delivery to just the site of interest and reducing its effects on healthy tissues [32,33].
Furthermore, in terms of using TMZ as a treatment regimen for gliomas, the protocol differs based on the type of glioma. The major diffused gliomas are classified by WHO based on the histological typing and grading, and the analyses of molecular markers [34]. For WHO grade II and III gliomas (mutant), TMZ chemotherapy is a standard adjuvant treatment post-surgery and radiotherapy [12,35]. For WHO grade IV glioblastoma (wild type), the Stupp protocol is seen to be beneficial [36].
In terms of Grade II gliomas, one study compared the use of TMZ and its effects on progression-free survival and overall survival in the patients [35]. The WHO-classified gliomas compared in this study were grade II astrocytoma (wild type), grade II astrocytoma (mutant type), and grade II oligodendroglioma. Moreover, the results of this study suggested that there was no significant difference found between the overall survival of patients with any type of grade II glioma. The progression-free survival, however, was significantly shorter in patients with grade II astrocytoma (wild type) compared to the other two [37,38,39,40,41].
Lastly, even though TMZ is widely used in the treatment of gliomas, there are certain prognostic factors established for this treatment regimen. This includes age, neurological status, the extent of tumor resection, IDH (isocitrate dehydrogenase) mutations, and inactivation of the MGMT region (via methylation) [42,43]. Now, despite the established effect of MGMT methylation on TMZ efficacy, the clinical use of this factor is very poor due to the lack of options for patients with unmethylated MGMT. One exception to this rule is, however, the younger patients with gliomas [44]. According to this study, the stratification of MGMT methylation status in younger patients showed a significantly longer overall survival than in the elderly population [44].
Avastin
There is mounting evidence that malignant gliomas show an exceptionally high degree of vascularization; therefore, a treatment targeting the tumor microenvironment's angiogenic factors has become the motivation for engineering pharmaceuticals like Bevacizumab (BEV) which is also known by its brand name “Avastin” [45,46,47]. In 2009, the FDA approved this antiangiogenic therapy for recurrent glioblastoma; previously, in 2004 it was the first-ever authorized agent to target tumor angiogenesis [48,49]. Currently, this treatment is approved for six types of cancer: metastatic colorectal cancer, recurrent non-small-cell lung cancer, metastatic renal cell carcinoma, metastatic cervical cancer, and our focus: recurrent glioblastoma, one type of glioma [50]. BEV is approved as a monotherapy option in patients with progressive and/or recurrent glioblastoma after previous therapy has been attempted [51].
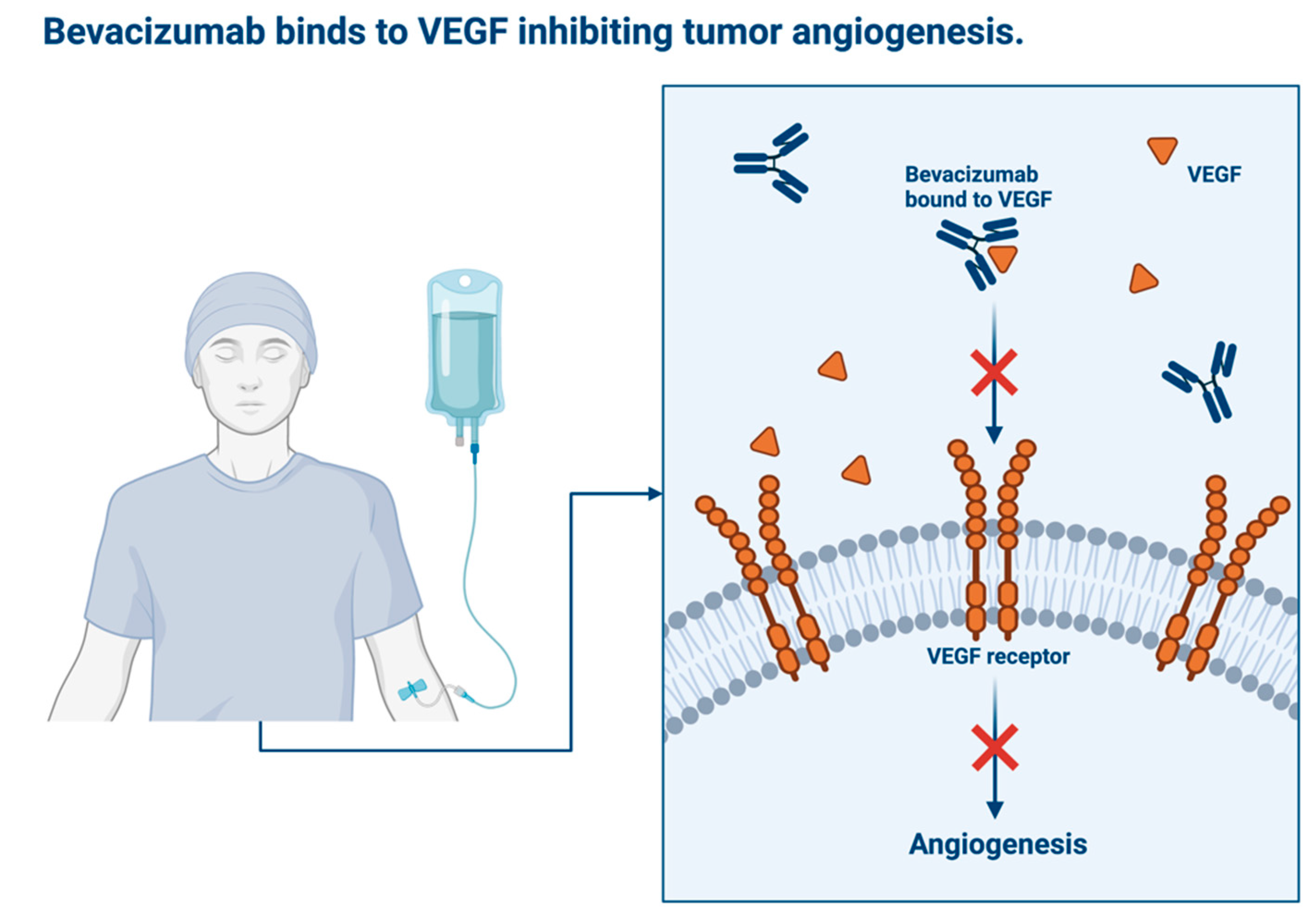
BEV is an angiogenesis inhibitor as it halts the growth of blood vessels; it is a humanized immunoglobulin G monoclonal antibody to vascular endothelial growth factor (VEGF) [47,50]. Malignant gliomas are known to express VEGF on cancer cell surfaces; also, greater expression of VEGF is associated with a poorer prognosis for patients [52,53,54]. VEGF is known to promote the tumor’s angiogenesis, vascular mimicry, and vasculogenesis [46]. In the body, BEV binds to VEGF with high specificity and inhibits VEGF's ability to bind to the VEGF receptors of vascular endothelial cells. This inhibits the usual downstream activity of VEGF thus reducing tumor blood supply and inhibiting further tumor vascularization [50,55]. Structural analysis revealed that Gly88 of human VEGF is required for BEV to bind [56]. Also, BEV does not initiate ligand structural changes but interrupts VEGF’s interaction with its receptors sterically [57]. In a mouse model of glioblastoma, when greater levels of VEGF were found in the serum, there was higher BEV treatment performance [58]. There is plenty of relevant clinical evidence that supports the use of BEV in recurrent glioblastoma [50].
Typically, BEV is administered intravenously every 2 weeks with a standard IV dosage of 10 mg/kg. This approved schedule was adopted in May 2009 [59]. The initial infusion usually lasts approximately 90 minutes to monitor for allergic reaction; however, subsequent infusions can range from 30 to 70 minutes [50]. One review found similar response and survival rates in 87 patients who were treated with 5 mg/kg rather than the usual 10 mg/kg [59]. However, the patients who received 5 mg/kg had much lower KPS scores, which is a measure of patient independence following treatment, and had more adverse events [59]. This medication is administered as long as it continues to demonstrate benefit to the patient. Common side effects of BEV treatment include hypertension, proteinuria, and fatigue [60]. However, it is important to note that hypertension during treatment is a possible biological marker of positive response to BEV therapy [50]. It’s thought that the associated hypertension is due to an increased vascular tone because VEGF-controlled vasodilation is inhibited by BEV; however, there are other theories about the mechanism of BEV-induced hypertension in patients [61]. Intracranial hemorrhage has also been observed after BEV treatment in high-grade glioma patients [62]. It’s thought that the potential for hemorrhage originates from BEV-induced disruption of the endothelial cell structure of the tumor vascular network; however, the observed rates of hemorrhage are considered nominal [51,62]. However, it is critical to acknowledge that patients who show signs of intracranial hemorrhage upon imaging and not candidates for BEV therapy [63].
BEV is not considered “chemotherapy” as it does not directly kill tumor cells; it can be used in combination with true chemotherapy regimens like lomustine, which hydrolyzes into reactive metabolites that destroy tumor nucleic acids [64]. One study found that patients suffering from recurrent glioblastoma saw higher overall survival rates when BEV was combined with lomustine rather than used as monotherapy [65]. A more recent study that included over 400 patients found that despite longer progression-free survival, treatment of glioblastoma with BEV and lomustine did not offer a justified survival advantage over lomustine alone [66]. When BEV was combined with irinotecan, a topoisomerase I inhibitor, recurrent glioblastoma patients showed greater progression-free survival, but lower rates of overall survival compared to patients who received BEV monotherapy [67,68]. A phase 2 clinical study concluded that combining BEV with carboplatin, a platinum-containing cytotoxic drug, resulted in too much toxicity to justify any clinical benefit [69]. BEV used in combination with cytotoxic treatments has also been shown to greatly reduce the size of glioma tumors and prevent the need for corticosteroids [70]. One 2017 meta-analysis, made up of four clinical trials and 607 patients, found that BEV combined with another chemotherapy treatment (irinotecan, carboplatin, and lomustine) significantly increased patients’ progression-free-survival rates compared to the use of BEV monotherapy or chemotherapy alone [71]. BEV treatment has also demonstrated its usefulness in reducing patients’ need for glucocorticoid supplementation to treat cerebral edema [72]. Currently, BEV monotherapy is considered a well-tolerated treatment option for recurrent glioblastoma patients [68]. The topic of BEV therapy combinations still requires exploration.
Unfortunately, the prolonged use of BEV can result in resistance to the medication. This resistance is thought to be the tumor’s activation of neovascularization pathways that are not yet targeted by anti-angiogenic treatments [46]. These vascularization pathways, like vascular mimicry, require more exploration to ensure recurrent glioma patients receive the best possible care [46].
Figure 3.
Mechanism of Bevacizumab to Inhibit Tumor Angiogenesis.
Emerging Immune Modulators
Within the central nervous system (CNS), microglia play an important immunologic function. These cells serve as an early line of defense for the brain and migrate to inflammatory regions of the CNS to mount an immune response [73]. They serve as phagocytes and antigen-presenting cells while also releasing chemokines and cytokines to promote the recruitment of other cells [74]. The blood-brain barrier poses a challenge for the use of cancer immunotherapy as it contains tight junctions which restrict the ability of drugs to pass through [75]. Additionally, the CNS has a lower number of T lymphocytes and lacks a lymphatic system [73].
High-grade gliomas are the most common primary tumors within the CNS [76]. Gliomas include anaplastic gliomas and glioblastomas [76]. Glioblastomas lead to a disorganized blood-brain barrier [77,78]. The tight junctions of the endothelial cells are broken down as tissue injury occurs, allowing the entry of leukocytes into the CNS [73]. Immune suppression occurs in patients with glioblastoma as the tumor microenvironment secretes immunosuppressive factors such as transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF) [79]. These factors prevent T cell proliferation and suppress their cytotoxic functions, while also preventing the maturation of dendritic cells [73,80,81]. In patients with glioblastoma, immunosuppression plays a key role in the progression of the tumor. Therefore, finding a method in which immunosuppression may be reversed offers a promising approach to improving patient outcomes and stopping tumor progression. Immunotherapy is a form of treatment that involves the activation of the host’s immune system to recognize and target cancer cells [82].
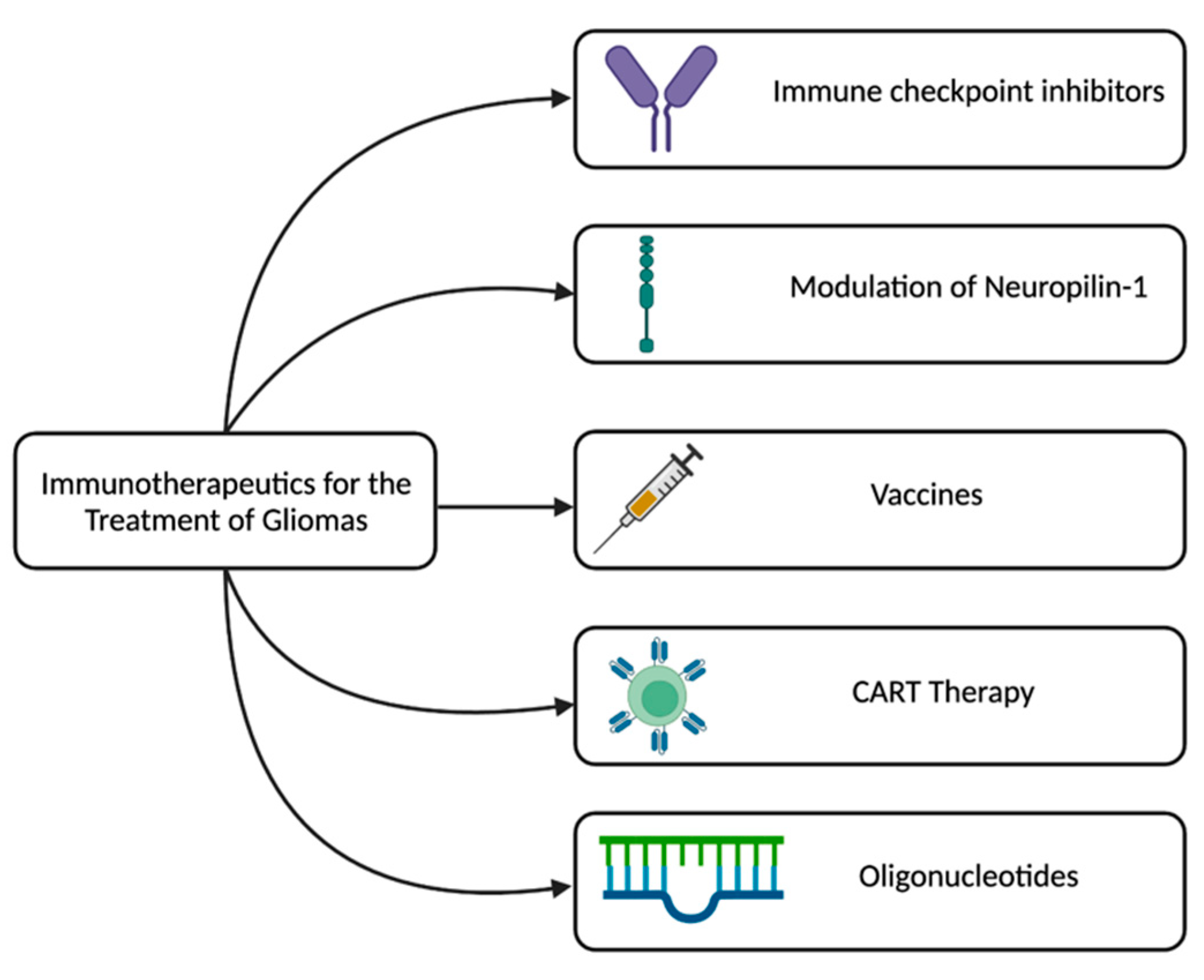
One form of immunotherapy includes the use of vaccines, which boost the patient’s immune system by exposure to an antigen. Vaccination is a form of active immunization and can be approached in two ways: cell-based therapy or peptide-based therapy [83]. Peptide-based therapy involves the injection of peptides as a vaccine to create an immune response. The selected peptides for vaccines are small and bind to MHC class I molecules, activating cytotoxic T cells [73]. Epidermal growth factor receptor (EGFR) III type mutant is the most common tumor-specific antigen in glioblastomas and is expressed in around 20-30% of tumors [84]. In the 1990s, the CDX-110 peptide vaccine was developed against tumor-specific antigens and although patients receiving the vaccine experienced a good humoral response, the vaccine did not significantly improve overall survival [85]. In cell-based immunotherapy, antigen-presenting cells, primarily dendritic cells, primed by the tumor are used to initiate an immune response [86]. Currently, the United States, Europe, and Japan are investigating the role of dendritic cell vaccines in the treatment of gliomas [87]. One phase II clinical trial found that the vaccine improved the survival of patients with glioblastoma but there haven’t been any phase III clinical trials and the cost of producing the vaccine is a limiting factor [84]. Current developments of dendritic cell vaccines involve pretreatment of vaccine sites with dendritic cells expressing cytomegalovirus phosphoprotein 65 RNA that leads to improvement of survival times and lymph node homing in mice models [88]. Although the development of vaccines for gliomas is limited, it offers a promising route for the management of gliomas. Further research is needed into optimizing a vaccine, but it offers a therapeutic approach with promising potential.
Immune checkpoints are used to control the movement through the cell cycle and affect the activation and inhibition of T cells [89]. In gliomas, CTLA-4 and PD-1 have been identified as immune checkpoint molecules [90]. PD-1 is expressed by T cells and interacts with PD-L1 on tumor cells. This interaction prevents the T cell from removing the tumor cell. As of 2014, the FDA has approved the use of immune checkpoint PD-1 targeting [82]. Mice models and human trials demonstrate that patients receiving anti-PD-1 therapy had an improved survival [91]. Ipilimumab was approved for use by the FDA in 2011 and is a monoclonal antibody that targets CTLA-4, a checkpoint molecule [82]. CTLA-4 acts to prevent T cells from eliminating other cells and to keep the immune system in check. However, this is disadvantageous in tumors. Additionally, more CTLA-4 expressed in gliomas was associated with a higher grader and severity, resulting in a poorer prognosis [92,93]. Ipilimumab works by preventing CTLA-4’s interaction with B7, allowing for T cell activation and proliferation [94]. In preclinical models, Ipilimumab has demonstrated a robust T-cell response, a decrease in tumor size, and increased survival [93,95,96]. One study investigated the addition of Ipilimumab with Nivolumab in treating glioblastomas in patients and found that patients were better able to tolerate Nivolumab alone rather than the combination [97]. Although Ipilimumab serves as a promising approach, further data is needed along with safety and dosing investigation.
Neuropilin-1 (Nrp1) is a co-receptor found on the surface of macrophages and microglia [98]. Nrp initiates angiogenesis in the tumor microenvironment [99]. Deletion of Nrp1 from myeloid cell types suppresses tumor growth by slowing tumor angiogenesis [99]. One study in mice removed Nrp1 and found that tumors in mice not expressing Nrp1 grew at a slower rate and demonstrated less vascularity [99]. Additionally, mice not expressing Nrp1 showed longer survival times [99]. Treatment with EG00229, a small molecule inhibitor of Nrp1’s b1 domain, yielded anti-tumorigenicity [99]. In humans, higher expression of Nrp1 is associated with a poorer prognosis of gliomas, therefore, research into how to target Nrp1 in gliomas offers a promising approach [100]. However, further research is needed in clinical trials.
Another type of immunotherapy used in the treatment of gliomas is chimeric antigen receptor (CAR) T-cell therapy [101]. CART therapy involves the extraction of a patient’s T cells and the engineering of them to express a receptor to an antigen [101]. The T cells are then inserted back into the patient. CART therapy offers an advantage as T cells can enter the CNS and eliminate tumor cells [102]. One common antigen targeted by CART therapy is EGFR, which is overexpressed in over 50% of gliomas [103]. However, the overall median survival following treatment was 6.9 months [104]. Although preclinical data shows promising results, clinical trials haven’t yielded the same results [101]. Antigen escape was a noted issue and current research is addressing this limitation by adding multivalent receptors to CAR T cells or through the use of combination therapy [101]. Further research is needed into the glioma microenvironment and the identification of glioma-specific antigens for CART therapy.
Gliomas are aggressive brain tumors with a high mortality rate. Gliomas rapidly progress and often develop resistance to therapies; therefore, the development of new therapeutics is crucial in glioma management. The development of oligonucleotides for the treatment of gliomas offers a promising approach as they’re able to penetrate the disrupted blood-brain barrier [105]. Oligonucleotides are short DNA or RNA-based synthetic polymers which affect the functions of nucleic acids [105]. Recent studies demonstrate that the use of oligonucleotides could activate gene transcription and provide an opportunity for genome-editing techniques [106,107]. Gene editing techniques offer a promising route through treating gliomas by deleting oncogenes, correcting mutations, and activating tumor suppressors [105]. Current research in mice models demonstrates efficacy and feasibility of using CRISPR-Cas9 gene editing in glioblastomas [105]. However, further research is needed to validate results along with clinical trials.
Figure 4.
Immunotherapy approaches for the treatment of gliomas.
Ongoing Clinical Trials
Treating gliomas, a complex and intricate process, involves considering tumor characteristics, patient factors, and preferences. Various literature explores ongoing clinical trials for glioma treatment, focusing on chemotherapy and immune modulators. More research is needed to refine these approaches for safety and effectiveness. Treatments fall into three categories: combination therapy with chemotherapy and immune checkpoint inhibitors, chemotherapy with adoptive cell therapy, or cancer vaccines. New diagnostic methods include combinations like Temozolomide and Nivolumab. Recurrent gliomas are treated with Atezolizumab and Bevacizumab or Temozolomide and Pembrolizumab. Trials evaluate standardization and synergistic effects of chemotherapy and immune checkpoint inhibitors, enhance the immune response through adoptive cell therapy, and stimulate targeted glioma cells with chemotherapies and cancer vaccines. Results suggest improved efficacy by leveraging chemotherapy's cytotoxic effects and immune modulators' properties, potentially reducing toxicity with lower doses. Personalized medicine, treatment resistance, and biomarkers are explored for tailored therapies. These treatments guide decisions and improve specificity in personalized therapies for patients.
Temozolomide (TMZ), an alkylating agent, is taken orally as a chemotherapy drug primarily used in specific brain tumors like gliomas. Molecularly, TMZ has a chemical structure consisting of a 5-carbon tetrazole ring with a methyl group (CH3) and a methyltriazenyl group (N3C-N=NC) [36,108,109]. This water-soluble prodrug of MTIC is optimally stable per a neutral pH condition and active at physiological pH conditions. Its development aimed to treat brain tumors and better the efficacy of penetration across the blood-brain barrier. This outcome usually leads to MTIC undergoing further degradation and forming reactive methyl diazonium ions that alkylate DNA, leading to DNA damage and ultimately tumor cell death, causing lymphopenia and affecting the function of immune cells, but it may also enhance antitumor immune responses when combined with certain immunotherapies. The timing and dose of temozolomide treatment are important factors in determining its effects on the immune system. Various factors such as tumor grade, molecular characteristics, patient age, and overall health are considered when determining the outcome of TMZ treatment. Being that this treatment is multifaceted, the treatment outcome of TMZ has been known to lead to a decrease in certain immune cell types, particularly CD4 T cells, and B cells, which can impact the immune response. This effect is further magnified when combined with radiation therapy [36,108,109]. Overall, the outcomes of temozolomide chemotherapy in gliomas involve a complex interplay between immune cell depletion, potential enhancement of antitumor activity, improved antigen presentation, and modulation of immunosuppressive cells. It highlights the need for further research to fully understand and optimize the immune-related effects of temozolomide treatment in glioma patients.
Bevacizumab, or Avastin, is a chemotherapy drug commonly used for treating gliomas. It is a monoclonal antibody that has been utilized in the treatment of various cancers. To fully comprehend the specific use of Avastin in chemotherapy and its outcomes, it is essential to consider its chemical properties, drug development, pharmacokinetics, and pharmacodynamics. Bevacizumab is a recombinant humanized monoclonal IgG1 antibody with a molecular weight of 149 kDa [110]. It was developed by Roche and Genentech with the aim of targeting angiogenesis, a process facilitated by a protein called VEGF. By inhibiting VEGF activity, Bevacizumab impedes the growth of blood vessels that supply tumors. The pharmacokinetic characteristics of Bevacizumab include intravenous administration and constant clearance rates at different doses. It is eliminated through proteolytic degradation. Pharmacodynamically, Bevacizumab disrupts the binding interactions of VEGF, leading to reduced angiogenesis and blood supply to tumors. Additionally, it normalizes tumor vasculature, facilitating the delivery of chemotherapy drugs to tumors [111]. Clinical trials have shown that Bevacizumab may offer modest benefits over other treatments for recurrent gliomas, particularly when combined with other agents like irinotecan. However, it is associated with potentially severe side effects such as gastrointestinal perforation, wound-healing complications, hemorrhage, and blood clots. The effectiveness of Bevacizumab in the initial treatment of glioblastoma is still under debate, and caution should be exercised when using it for recurrent cases. Further research is needed to determine the optimal approach, as different treatment options, including reoperation, alternating electric field therapy, chemotherapy, stereotactic radiotherapy, and combinations of these modalities, are being explored. While Bevacizumab and stereotactic radiotherapy or radiosurgery may offer some benefits for recurrent malignant gliomas, a consensus on the best approach has yet to be reached.
Glioma treatment, including immune therapies, is an active area of research with ongoing clinical trials. Immune checkpoint inhibitors (ICIs), immune modulation of Neuropilin-1, vaccines, and Chimeric Antigen Receptor T-cell (CAR-T) therapies show promise in various cancers. However, their efficacy in gliomas is still being investigated. ICIs like pembrolizumab and nivolumab have shown significant success in certain cancers but limited effectiveness in gliomas. Combination therapies with immunotherapies are being explored to enhance efficacy. Further clinical trials that aimed to evaluate the ICIs as monotherapy in gliomas have shown modest response rates, and combination therapies with immunotherapies have shown the potential to enhance efficacy [112]. Further research intends to navigate and explore predictive biomarkers that aid complementary selectivity for patients who are more likely to respond to checkpoint inhibitors. Immune modulation of Neuropilin-1, a protein involved in various cellular processes, including angiogenesis and immune regulation, and vaccine-based immunotherapies have shown potential in enhancing antitumor immune responses [113]. However, further research is needed to assess their efficacy and safety in gliomas. In gliomas, NRP-1 may assist in overcoming immunosuppressive mechanisms. At the same time, in vaccine-based immunotherapies, this enhancement is achieved by stimulating the immune system to recognize and attack tumor cells by presenting tumor-specific antigens [114]. Glioma treatment with Chimeric Antigen Receptor T-cell (CAR-T) involves the genetic modification of a patient's T cells to express a receptor that targets specific antigens present in tumor cells. CAR-T cell therapy has demonstrated remarkable success in hematological malignancies but faces challenges in solid tumors, including gliomas [114]. Ongoing studies explore CAR-T cell therapy targeting specific antigens in gliomas, but significant efficacy in solid tumors is yet to be established. Further investigation and optimization of these approaches are needed to improve treatment outcomes in gliomas.
Conclusions
In this discussion on the use of immune modulators and chemotherapeutics as viable options for the treatment of glioblastomas, this manuscript first delved into the nature of the malignancy as well as the role of prognostic biomarkers used prior to deploying said treatment. As previously mentioned, these biomarkers are crucial prognostic components that complement risk stratification, treatment monitoring, and disease progression. These biomarkers serve a dual responsibility of identifying characteristic markers expressed by a given neoplasm which allows focused targets for both immune modulation and chemotherapeutics. Thus, this discussion of biomarkers served as a segue into the utility of two prominent cancer therapies, namely temozolomide (TMZ) and bevacizumab (BEV). The first of these agents, TMZ, is a powerful DNA alkalizing agent that has served as a mainstay of glioma treatment. Nonetheless, due to limiting effects such as patient MGMT expression levels, TMZ administration is not foolproof. In contrast to the chemotherapeutic mechanisms of TMZ action, BEV, more well-known as Avastin, is a powerful antiangiogenic therapy for recurrent glioblastoma treatment. Although not considered chemotherapy, BEV has demonstrated some therapeutic benefit when coupled with true chemotherapy regimens and even other cytotoxic treatment options. Notably, however, there are numerous studies within the literature suggesting no justifiable survival advantage to BEV’s coadministration alongside traditional glioma treatment, and even toxic events across a few studies. Nonetheless, BEV monotherapy is considered well-tolerated for recurrent glioblastoma patients and harbors the unique ability to reduce cerebral edema in this group.
Considering the aforementioned shortcomings of modern chemotherapeutic and chemotherapeutic-like agents, the role of emerging immune modulators was thoroughly explored in the latter half of this discussion. Specifically, via the activation of the patient’s native immune system, immunotherapy has the potential to guide a mounted attack on “foreign” cancer cells. A few strategies discussed are the use of vaccinations to achieve the above-mentioned effect, the manipulation of cellular checkpoints such as CTLA-4 and PD-1, and co-receptor modulation of neuropilin-1 found on the surface of macrophages and microglia. Furthermore, more recent types of immunotherapies entail the treatment of gliomas with CAR-T cell therapy and prospectively CRISPR-Cas9 gene editing, with the latter tactic still in the early stages of product development. Finally, this discussion ends with an evaluation of ongoing trials in the context of chemotherapy treatment and immune therapies such as the ones detailed in the preceding sections. Overall, the employment of TMZ and BEV has been supported in the literature, however, more research is necessitated to fully optimize regimens and to avoid immune- and toxicity-related effects of these drugs. Moreover, an even closer consideration is warranted for current immunotherapies as they are in their infancy compared to TMZ and BEV. Thus, although promising emerging treatment options, therapies involving cancer vaccines, ICIs, immune modulation of neuropilin-1, CAR-T, and CRISPR-Cas9 technology require further investigation into their long-term efficacy and utility in treating gliomas.
References
- Zhang Y, Xie J, Han G, et al. [Detection and clinical significance of myeloid-derived suppressor cells in peripheral blood of patients with rectal carcinoma]. Zhonghua Wei Chang Wai Ke Za Zhi. 2017;20:798–802.
- Ostrom QT, Gittleman H, Truitt G, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018;20:iv1–iv86.
- Xu S, Tang L, Li X, et al. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020;476:1–12. [CrossRef]
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820. [CrossRef]
- Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23:1231–1251. [CrossRef]
- Velázquez Vega JE, Brat DJ, Ryken TC, et al. The role of neuropathology in the management of newly diagnosed glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2020;150:143–164.
- Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139:603–608.
- Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. [CrossRef]
- Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372:2499–2508.
- Watanabe T, Nakamura M, Kros JM, et al. Phenotype versus genotype correlation in oligodendrogliomas and low-grade diffuse astrocytomas. Acta Neuropathol. 2002;103:267–275. [CrossRef]
- Chai R, Li G, Liu Y, et al. Predictive value of MGMT promoter methylation on the survival of TMZ treated IDH-mutant glioblastoma. Cancer Biol Med. 2021;18:272–282. [CrossRef]
- Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18:170–186. [CrossRef]
- Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med. 2016;374:1344–1355. [CrossRef]
- van den Bent MJ, Tesileanu CMS, Wick W, et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053-22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2021;22:813–823.
- van den Bent MJ, Brandes AA, Taphoorn MJB, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31:344–350.
- Mohile NA, Messersmith H, Gatson NT, et al. Therapy for Diffuse Astrocytic and Oligodendroglial Tumors in Adults: ASCO-SNO Guideline. J Clin Oncol. 2022;40:403–426. [CrossRef]
- Brandes AA, Nicolardi L, Tosoni A, et al. Survival following adjuvant PCV or temozolomide for anaplastic astrocytoma. Neuro Oncol. 2006;8:253–260. [CrossRef]
- Majzner RG, Theruvath JL, Nellan A, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res. 2019;25:2560–2574. [CrossRef]
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res. 2008;6:675–684. [CrossRef]
- Rahaman SO, Harbor PC, Chernova O, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. [CrossRef]
- Iwamaru A, Szymanski S, Iwado E, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–2444. [CrossRef]
- Guo X, Zhang Y, Jiao H, et al. The prognostic significance of PD-L1 expression in patients with glioblastoma: A meta-analysis. Front Oncol. 2022;12:925560. [CrossRef]
- Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
- Barciszewska A-M, Gurda D, Głodowicz P, et al. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS One. 2015;10:e0136669. [CrossRef]
- Hombach-Klonisch S, Mehrpour M, Shojaei S, et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol Ther. 2018;184:13–41. [CrossRef]
- Spiro T, Liu L, Gerson S. New cytotoxic agents for the treatment of metastatic malignant melanoma: temozolomide and related alkylating agents in combination with guanine analogues to abrogate drug resistance. Forum (Genova). 2000;10:274–285.
- Jacinto FV, Esteller M. MGMT hypermethylation: a prognostic foe, a predictive friend. DNA Repair (Amst). 2007;6:1155–1160. [CrossRef]
- Kanzawa T, Bedwell J, Kondo Y, et al. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg. 2003;99:1047–1052. [CrossRef]
- Bobola MS, Silber JR, Ellenbogen RG, et al. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin Cancer Res. 2005;11:2747–2755.
- Qian L, Zheng J, Wang K, et al. Cationic core-shell nanoparticles with carmustine contained within O6-benzylguanine shell for glioma therapy. Biomaterials. 2013;34:8968–8978. [CrossRef]
- Stephen ZR, Gebhart RN, Jeon M, et al. pH-Sensitive O6-Benzylguanosine Polymer Modified Magnetic Nanoparticles for Treatment of Glioblastomas. Bioconjug Chem. 2017;28:194–202.
- Liang S, Xu H, Ye B-C. Membrane-Decorated Exosomes for Combination Drug Delivery and Improved Glioma Therapy. Langmuir. 2022;38:299–308. [CrossRef]
- Chu W, Houston ZH, Fletcher NL, et al. Development and Validation of a Targeted Treatment for Brain Tumors Using a Multi-Drug Loaded, Relapse-Resistant Polymeric Theranostic. Biomacromolecules. 2023;24:2674–2690. [CrossRef]
- Marquet G, Dameron O, Saikali S, et al. Grading glioma tumors using OWL-DL and NCI Thesaurus. AMIA Annu Symp Proc. 2007;2007:508–512.
- Ghaffari-Rafi A, Ghaffari-Rafi S, Leon-Rojas J. Role of Temozolomide Regimen on Survival Outcomes in Molecularly Stratified WHO Grade II Gliomas: A Systematic Review. Asian J Neurosurg. 2021;16:14–23. [CrossRef]
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. [CrossRef]
- Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17:1521–1532. [CrossRef]
- Wahl M, Phillips JJ, Molinaro AM, et al. Chemotherapy for adult low-grade gliomas: clinical outcomes by molecular subtype in a phase II study of adjuvant temozolomide. Neuro Oncol. 2017;19:242–251. [CrossRef]
- Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–1566. [CrossRef]
- Villani V, Merola R, Vidiri A, et al. Temozolomide low-dose chemotherapy in newly diagnosed low-grade gliomas: activity, safety, and long-term follow-up. Tumori. 2017;103:255–260. [CrossRef]
- Gao Y, Weenink B, van den Bent MJ, et al. Expression-based intrinsic glioma subtypes are prognostic in low-grade gliomas of the EORTC22033-26033 clinical trial. Eur J Cancer. 2018;94:168–178. [CrossRef]
- Fernandes C, Costa A, Osório L, et al. Current Standards of Care in Glioblastoma Therapy. In: De Vleeschouwer S, editor. Glioblastoma [Internet]. Brisbane (AU): Codon Publications; 2017 [cited 2023 Jun 25]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK469987/.
- Gorlia T, van den Bent MJ, Hegi ME, et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008;9:29–38.
- Brawanski KR, Sprung S, Freyschlag CF, et al. Influence of MMR, MGMT Promotor Methylation and Protein Expression on Overall and Progression-Free Survival in Primary Glioblastoma Patients Treated with Temozolomide. Int J Mol Sci. 2023;24:6184. [CrossRef]
- Stupp R, Pavlidis N, Jelic S, et al. ESMO Minimum Clinical Recommendations for diagnosis, treatment and follow-up of malignant glioma. Ann Oncol. 2005;16 Suppl 1:i64-65. [CrossRef]
- Angara K, Borin TF, Arbab AS. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl Oncol. 2017;10:650–660. [CrossRef]
- Wang H, Guo J, Wang T, et al. Efficacy and safety of bevacizumab in the treatment of adult gliomas: a systematic review and meta-analysis. BMJ Open. 2021;11:e048975. [CrossRef]
- Cohen MH, Shen YL, Keegan P, et al. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. [CrossRef]
- Ellis LM. Bevacizumab. Nat Rev Drug Discov. 2005;Suppl:S8-9.
- Ghiaseddin A, Peters KB. Use of bevacizumab in recurrent glioblastoma. CNS Oncol. 2015;4:157–169. [CrossRef]
- Gerriets V, Kasi A. Bevacizumab. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 Jun 25]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK482126/.
- Salmaggi A, Eoli M, Frigerio S, et al. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J Neurooncol. 2003;62:297–303. [CrossRef]
- Godard S, Getz G, Delorenzi M, et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 2003;63:6613–6625.
- Birner P, Piribauer M, Fischer I, et al. Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: evidence for distinct angiogenic subtypes. Brain Pathol. 2003;13:133–143. [CrossRef]
- Stefanik DF, Fellows WK, Rizkalla LR, et al. Monoclonal antibodies to vascular endothelial growth factor (VEGF) and the VEGF receptor, FLT-1, inhibit the growth of C6 glioma in a mouse xenograft. J Neurooncol. 2001;55:91–100. [CrossRef]
- Sullivan LA, Brekken RA. The VEGF family in cancer and antibody-based strategies for their inhibition. MAbs. 2010;2:165–175. [CrossRef]
- Muller YA, Chen Y, Christinger HW, et al. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998;6:1153–1167. [CrossRef]
- García-Romero N, Palacín-Aliana I, Madurga R, et al. Bevacizumab dose adjustment to improve clinical outcomes of glioblastoma. BMC Med. 2020;18:142. [CrossRef]
- Blumenthal DT, Mendel L, Bokstein F. The optimal regimen of bevacizumab for recurrent glioblastoma: does dose matter? J Neurooncol. 2016;127:493–502.
- Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. [CrossRef]
- Li M, Kroetz DL. Bevacizumab-induced hypertension: Clinical presentation and molecular understanding. Pharmacol Ther. 2018;182:152–160. [CrossRef]
- Lin X, Daras M, Pentsova E, et al. Bevacizumab in high-grade glioma patients following intraparenchymal hemorrhage. Neurooncol Pract. 2017;4:24–28. [CrossRef]
- Castro BA, Aghi MK. Bevacizumab for glioblastoma: current indications, surgical implications, and future directions. Neurosurg Focus. 2014;37:E9. [CrossRef]
- McBain C, Lawrie TA, Rogozińska E, et al. Treatment options for progression or recurrence of glioblastoma: a network meta-analysis. Cochrane Database Syst Rev. 2021;5:CD013579. [CrossRef]
- Taal W, Oosterkamp HM, Walenkamp AME, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014;15:943–953. [CrossRef]
- Wick W, Gorlia T, Bendszus M, et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N Engl J Med. 2017;377:1954–1963. [CrossRef]
- Vredenburgh JJ, Desjardins A, Reardon DA, et al. Experience with irinotecan for the treatment of malignant glioma. Neuro Oncol. 2009;11:80–91. [CrossRef]
- Szklener K, Mazurek M, Wieteska M, et al. New Directions in the Therapy of Glioblastoma. Cancers (Basel). 2022;14:5377. [CrossRef]
- Field KM, Simes J, Nowak AK, et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro Oncol. 2015;17:1504–1513. [CrossRef]
- Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787.
- Yang S-B, Gao K-D, Jiang T, et al. Bevacizumab combined with chemotherapy for glioblastoma: a meta-analysis of randomized controlled trials. Oncotarget. 2017;8:57337–57344. [CrossRef]
- Garcia J, Hurwitz HI, Sandler AB, et al. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017. [CrossRef]
- Thomas AA, Ernstoff MS, Fadul CE. Immunotherapy for the treatment of glioblastoma. Cancer J. 2012;18:59–68. [CrossRef]
- Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol. 2009;85:352–370. [CrossRef]
- Conrad CA. Chemotherapy for metastatic tumors to the central nervous system. Curr Oncol Rep. 2001;3:490–494. [CrossRef]
- Pellerino A, Franchino F, Soffietti R, et al. Overview on current treatment standards in high-grade gliomas. Q J Nucl Med Mol Imaging. 2018;62:225–238. [CrossRef]
- Vauleon E, Avril T, Collet B, et al. Overview of cellular immunotherapy for patients with glioblastoma. Clin Dev Immunol. 2010;2010:689171. [CrossRef]
- Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–1379. [CrossRef]
- Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011;13:3–13.
- Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech. 2001;52:401–410.
- Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol Today. 1991;12:370–374. [CrossRef]
- Ghouzlani A, Kandoussi S, Tall M, et al. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front Immunol. 2021;12:679425. [CrossRef]
- Yamanaka R. Cell- and peptide-based immunotherapeutic approaches for glioma. Trends Mol Med. 2008;14:228–235. [CrossRef]
- Zhao T, Li C, Ge H, et al. Glioblastoma vaccine tumor therapy research progress. Chin Neurosurg J. 2022;8:2. [CrossRef]
- Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–1385. [CrossRef]
- Van Gool S, Maes W, Ardon H, et al. Dendritic cell therapy of high-grade gliomas. Brain Pathol. 2009;19:694–712.
- Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. 2014;20:5620–5629. [CrossRef]
- Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–369. [CrossRef]
- Yeo AT, Charest A. Immune Checkpoint Blockade Biology in Mouse Models of Glioblastoma. J Cell Biochem. 2017;118:2516–2527. [CrossRef]
- 90. Kaminska B, Ciechomska IA, Cyranowski S. Autophagy in brain tumor immune evasion and responses to immunotherapy. Autophagy in Immune Response: Impact on Cancer Immunotherapy [Internet]. Elsevier; 2020 [cited 2023 Jun 25]. p. 29–52. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128196090000031.
- Goswami S, Walle T, Cornish AE, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26:39–46. [CrossRef]
- Peggs KS, Quezada SA, Korman AJ, et al. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18:206–213. [CrossRef]
- Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. [CrossRef]
- Brown NF, Ng SM, Brooks C, et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer. 2020;20:198. [CrossRef]
- Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res. 2016;4:124–135. [CrossRef]
- Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–5301. [CrossRef]
- Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20:674–686. [CrossRef]
- Miyauchi JT, Chen D, Choi M, et al. Ablation of Neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget. 2016;7:9801–9814. [CrossRef]
- Smith GT, Radin DP, Tsirka SE. From protein-protein interactions to immune modulation: Therapeutic prospects of targeting Neuropilin-1 in high-grade glioma. Front Immunol. 2022;13:958620. [CrossRef]
- Caponegro MD, Moffitt RA, Tsirka SE. Expression of neuropilin-1 is linked to glioma associated microglia and macrophages and correlates with unfavorable prognosis in high grade gliomas. Oncotarget. 2018;9:35655–35665. [CrossRef]
- Luksik AS, Yazigi E, Shah P, et al. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers (Basel). 2023;15:1414. [CrossRef]
- Bagley SJ, Desai AS, Linette GP, et al. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20:1429–1438. [CrossRef]
- Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res. 2005;11:1462–1466. [CrossRef]
- Goff SL, Morgan RA, Yang JC, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother. 2019;42:126–135. [CrossRef]
- Krichevsky AM, Uhlmann EJ. Oligonucleotide Therapeutics as a New Class of Drugs for Malignant Brain Tumors: Targeting mRNAs, Regulatory RNAs, Mutations, Combinations, and Beyond. Neurotherapeutics. 2019;16:319–347. [CrossRef]
- Yoon S, Rossi JJ. Therapeutic Potential of Small Activating RNAs (saRNAs) in Human Cancers. Curr Pharm Biotechnol. 2018;19:604–610. [CrossRef]
- Kelley ML, Strezoska Ž, He K, et al. Versatility of chemically synthesized guide RNAs for CRISPR-Cas9 genome editing. J Biotechnol. 2016;233:74–83. [CrossRef]
- Weller M, Cloughesy T, Perry JR, et al. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013;15:4–27.
- Brandes AA, Franceschi E, Tosoni A, et al. Temozolomide concomitant and adjuvant to radiotherapy in elderly patients with glioblastoma: correlation with MGMT promoter methylation status. Cancer. 2009;115:3512–3518.
- Kirkpatrick JP, Sampson JH. Recurrent malignant gliomas. Semin Radiat Oncol. 2014;24:289–298.
- Kim MM, Umemura Y, Leung D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. Cancer J. 2018;24:180–186.
- Thomas AA, Fisher JL, Hampton TH, et al. Immune modulation associated with vascular endothelial growth factor (VEGF) blockade in patients with glioblastoma. Cancer Immunol Immunother. 2017;66:379–389. [CrossRef]
- Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. [CrossRef]
- Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62:125–135. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



