
Submitted:
19 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
A reduction of melatonin function contributes to the acceleration of Alzheimer's disease (AD), and understanding molecular processes of melatonin-related signaling is critical for intervention of AD progression. Recently, we synthesized and tested a series of melatonin derivates with donepezil fragments in silico and in vitro. In this study, one of the most potent compounds, 3c, was studied in an in vivo rat model of pinealectomy (pin) and subsequent icvAβ1-42 infusion. Melatonin was used as a referent drug. The treatment with melatonin and 3c (10 mg/kg, i.p. for 14 days) exerted a beneficial effect on memory decline and concomitant rise of Aβ1-42 and pTAU in the hippocampus in the pin + icvAβ1-42 rats. Melatonin supplementation facilitated the non-amyloidogenic signaling via a non-receptor (histone deacetylase sirtuin 1, SIRT1) and receptor-related signaling (MT/ERK/CREB). The hybrid 3c derivate up-regulated the MT1A and MT2B receptors, pERK and pCREB. Our findings strongly support the hypothesis that melatonin-related derivates may become a promising drug candidate in AD therapy.
Keywords:
Alzheimer's disease
; Melatonin
; Melatonin analogue
; icvAβ1-42
; pTAU
; MT receptors
; ERK
; CREB
1. Introduction
Alzheimer’s disease (AD) is a progressive brain disorder and type of severe dementia that affects about 10 % of older people worldwide [1]. The accumulation of extracellular amyloid plaques, composed mainly of amyloid (A)β, and intracellular formation of hyperphosphorylated TAUprotein (pTAU) is considered the unique hallmark of AD pathogenesis. The disturbed balance between Aβ production and Aβ clearance with the generation of neurotoxic Aβ1-40 and Aβ1-42 fragments, leading to a surge in oxidative stress in the brain, is accepted as one of the hypotheses for the AD etiology. The concomitant behavioral impairments include impaired circadian rhythm, sleep disturbance, decreased cognitive function, and memory loss. Nowadays, the priority in the research on AD is focused on discovering the signaling pathways closely associated with predisposition to the development of AD pathogenesis. This issue is a critical step in finding novel targets for manipulation and, therefore, controlling the progression of this neurological disease. Although funding resources in the research of practical therapeutic approaches are numerous, there is still no approved drug possessing the three “golden” features needed to be introduced in the market, i.e., i) to be able to suppress the neurodegeneration and the progress of the disease; ii) to lack of side effect during chronic treatment and iii) to be at relatively low price.
Experimental and clinical findings suggest that circadian dysregulation of the sleep-wake cycle is part of the early symptoms of the diseases and predisposes to memory deficit and AD progress [2]. Treating disturbed sleep–wake cycle in the very beginning or before the expression of AD symptoms is suggested to lessen or even prevent pathogenesis development [3]. Thus, the orexin receptor antagonist Suvorexant, which the Food and Drug Administration recently approved in the USA, is a good sample for studying the efficacy and outcome of treatment strategy on impaired sleep-wake circadian pattern in early period or before to AD symptomatic [4]. The beneficial role of melatonin in the treatment of AD pathogenesis was also extensively studied [5]. The melatonin is released from the pineal gland during the dark phase when the inhibitory control from the suprachiasmatic nucleus is attenuated by light exposure. The hormone’s primary function is the regulatory control of the circadian rhythms in the body, including synchronization of the sleep-wake cycle and promotion of sleep. It is hypothesized that melatonin deficiency might be a critical trigger predisposing to AD pathogenesis [6]. Patients with AD are reported to have low levels of this hormone in cerebrospinal fluid [6,7,8]. The removal of the pineal gland in rodents is accepted as a relevant model to study the harmful consequences of melatonin deficiency that resemble the signaling dysfunction associated with neurodegenerative diseases, including AD [9]. Recently, we reported that simultaneous removal of the pineal gland and intracerebroventricular (icv) infusion of Aβ1-42 exacerbates behavioral responses and concomitant oxidative stress in the hippocampus and the frontal cortex [10]. Our research team synthesized and tested a series of new melatonin analogs and sulfonyl hydrazone compounds with a melatonin scaffold [11]. The docking analysis demonstrated a plausible mechanism of action of one of the most potent compounds, the 3c, associated with melatonin /MT/ receptors, and inhibition of acetylcholinesterase (AchE), and butyrylcholinesterase (BchE). In the present study, the most effective and potent compound, 3c, possessing low in vitro neurotoxicity, was selected and tested in a rat model of melatonin deficiency induced by pinealectomy (pin) and a subsequent icvAβ1-42 infusion. Melatonin was used as a referent drug. The underlying protective mechanism of the new compound was ascertained, including the possible signaling pathway closely related to the melatonin system and involved in the suppression of Aβ1-42 accumulation in the hippocampus. We hypothesized that this novel melatonin analog, 3c, behaves as a potent MT receptor agonist promoting the non-amyloidogenic pathway via facilitating the MT receptor–related ERK/CREB signaling process.
2. Results
2.1. The Circadian Pattern of Motor Activity and Effects of Melatonin and 3c Compound in a Rat Model of Pinealectomy and icvAβ1-42 Infusion
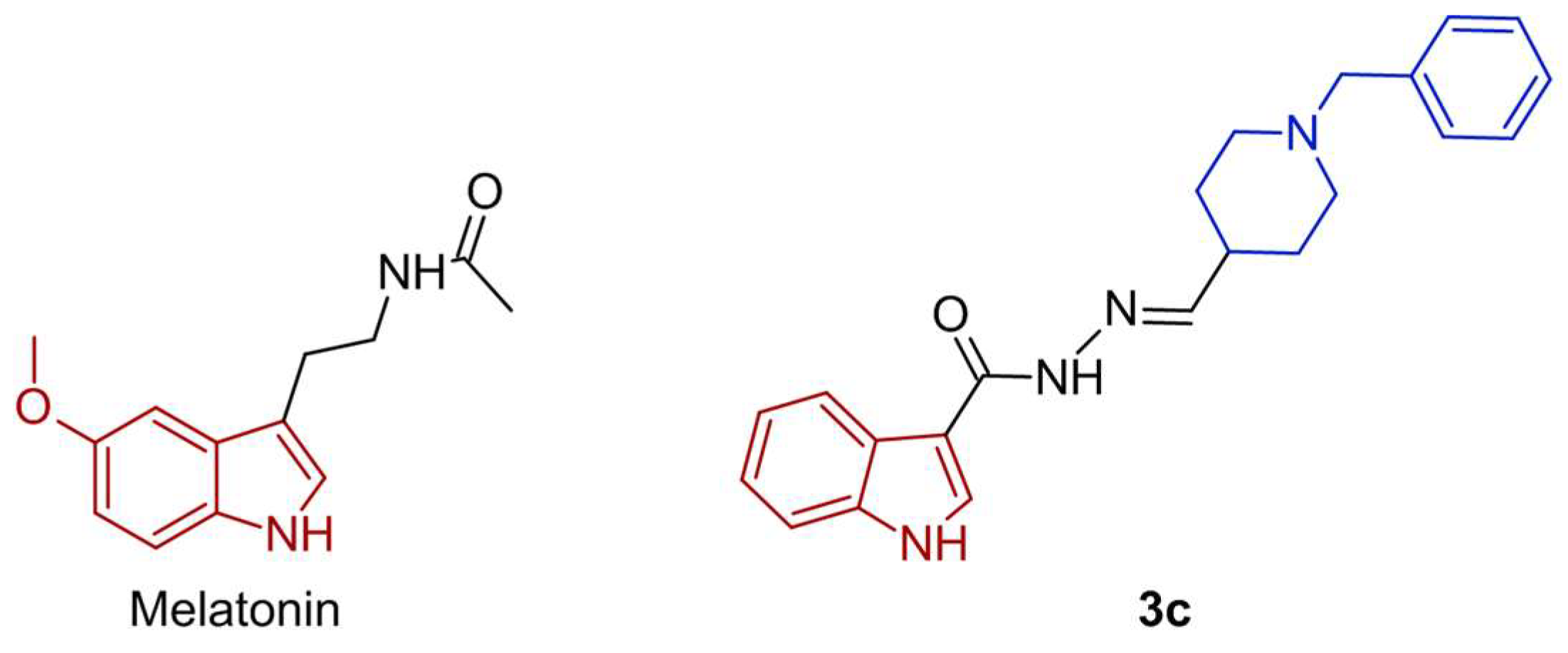
Figure 1.
The structures of the melatonin and the new compound 3c [11].
Figure 1.
The structures of the melatonin and the new compound 3c [11].
The 24-h registration of horizontal and vertical activity of the control group (sham-veh-veh group), model group (pin+Aβ1-42-veh), positive control (pin+Aβ1-42-mel) and treatment group (pin+Aβ1-42-3c) was conducted in the actimeter. All groups showed a circadian pattern of motor activity (p < 0.05 dark vs. light) that was also confirmed by the Cosinor analysis (Figure 2AB).
Further, the horizontal and vertical activity, total distance, and mean velocity measured in the light and the dark phase, respectively, were evaluated (Figure 3A-D). The daily pattern of activity was verified for all groups (p < 0.05, p < 0.001, dark vs. light phase). The model group was characterized by an elevated horizontal activity vs. control group (p = 0.03) as well as higher vertical activity vs. control group (p < 0.001) (Figure 3A,B). Both drugs tended to compensate the pin+Aβ1-42 –induced changes in horizontal activity (Figure 3A). At the same time only the melatonin treatment alleviated the enhanced effect of pin+Aβ1-42 on vertical activity (p < 0.001 vs. pin+Aβ1-42-veh group) (Figure 3B). There was no difference in total distance and velocity among groups (p > 0.05) (Figure 3C,D).
2.2. Effects of Melatonin and 3c Compound on Memory Impairment Induced by Pinealectomy and icvAβ1-42 Infusion
To evaluate the potency of the new hybrid substance 3c and melatonin, used as a reference to compensate for the pinealectomy-induced melatonin deficiency and the Aβ1-42-related memory decline, we conducted a battery of cognitive tests, including object recognition test (ORT), object location test (OLT) and Y-maze test.
2.2.1. Object Recognition Test
The ORT was used to assess cognitive capacity related to natural rodents’ curiosity to explore novel objects. The effects of melatonin and 3c in rats with pinealectomy and icvAβ1-42 infusion on short-term memory to recognize novelty was evaluated. The vehicle-treated pin+Aβ1-42 group significantly reduced the discrimination index (DI) for both time (p = 0.038 vs. sham-veh-veh group) and counts (p = 0.05 vs. sham-veh-veh group) (Figure 4A,B). While the referent drug significantly alleviated DI (count) vs. pin+Aβ1-42-veh group (p = 0.05), the sub-chronic treatment with melatonin or 3c showed a tendency to reverse the impaired memory recognition (p > 0.05 vs. sham-veh-veh group) and (Figure 4B).
2.2.2. Object Location Test
The OLT was used to evaluate the effect of melatonin and 3c on impaired short-term spatial memory due to pinealectomy and icvAβ1-42 infusion. For this purpose, the spontaneous response of rats to explore preferably the object relocated in an open field instead of a familiar object placed in the same position as in the sample phase (see Materials and methods) was detected. Like in the ORT, the pin+Aβ1-42-veh group had lower DI than the control group (p < 0.001), showing a disturbed short-term spatial memory (Figure 5). Although treatment with the hybrid compound 3c exhibited significantly reduced DI than the sham-veh-veh group (p = 0.024), both melatonin and 3c reversed the pin+Aβ1-42-induced memory impairment (p < 0.001 and p = 0.047, respectively).
2.2.3. Y-Maze Test
Spontaneous alternation behavior (SAB) was assessed in the Ist trial of a Y-maze test used to evaluate spatial memory. Lower SAB was detected in the pin-Aβ1-42-veh group as compared with the sham-veh-veh group (p < 0.001) (Figure 6A). Both the referent group melatonin and the hybrid 3c compound alleviated the impaired SAB (p = 0.02 and p < 0.001 vs. pin-Aβ1-42-veh group).
Short-term spatial memory was also assessed on the Y maze IInd trial. Like in the OLT, the model of melatonin deficit and subsequent icv Aβ1-42 infusion decreased DI (time) and DI (count) (p < 0.001 vs. control) (Figure 6B,C), respectively. The treatment with both the positive control melatonin and 3c compound reduced the pin+icvAβ1-42-induced impairment on the spatial memory (DI (time): p < 0.001, pin-Aβ1-42-mel group vs. pin-Aβ1-42-veh group; p = 0.036, pin-Aβ1-42-3c group vs. pin-Aβ1-42-veh group (Figure 6B); DI (count): p = 0.002, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel and pin-Aβ1-42-3c group, respectively, (Figure 6C). However, the treatment with 3c compound was unable to correct the pin-Aβ1-42-induced reduce of DI (time) (p < 0.001 vs. sham-veh-veh group) (Figure 6B).
2.3. The Expression of Markers of ADs and Effects of Melatonin and 3c Compound in a Rat Model of Pinealectomy and icvAβ1-42 Infusion
The effects of sub-chronic treatment with the positive control melatonin and the hybrid compound 3c on pathological markers closely associated with ADs, including the expression of Aβ1-42, pTAU protein and the level of AchE in the hippocampus were evaluated following induction of melatonin deficiency and concomitant icvAβ1-42 infusion a week after the procedure of removal of the pineal gland. The procedure of the hippocampus isolation was conducted 24 hours after the last memory test (see Figure with Experimental timeline).
The expression of Aβ1-42 and pTAU in the hippocampus was significantly elevated in the model group treated with a vehicle compared to the control group (p = 0.035 and p = 0.049, respectively) (Figure 7A,B). At the same time, there was no change in the level of AchE among the four groups (Figure 7C). The treatment with melatonin and compound 3c tended to reverse the model-related elevation of AD markers (p > 0.05 vs. control group).
2.4. Neuroprotective Effect of Melatonin and the Hybrid Compound 3c on Aβ1–42 Neurotoxicity via MT Receptors/ERK/CREB Signaling
Previous studies showed that melatonin exerted neuroprotection against Aβ neurotoxicity by directing APP metabolism to the non-amyloidogenic pathways. In the present study, we explored the possible involvement of this molecular mechanism in the beneficial effects on memory decline of melatonin and melatonin-related 3c compound in the pin+Aβ1-42 rat model. We measured the level of several signaling molecules associated with non-amyloidogenic pathway, including the expression of the two receptors MT1A and MT2B, respectively, sirtuin 1 (SIRT1), pERK1/2, and рCREB.
The effects of sub-chronic treatment with the positive control melatonin and the hybrid compound 3c on the signaling molecules associated with non-amyloidogenic pathway activated via MT1A/MT2B receptor subtypes in the hippocampus was assessed. The model of pinealectomy and subsequent icvAβ1-42-infusion down-regulated the two MT receptor subtypes in the hippocampus compared to the sham+veh-veh group (p < 0.05 and p = 0.003, respectively) (Figure 8A,B). The treatment with the positive control melatonin and 3c compound reversed the pin+icvAβ1-42-related diminished expression in the MT1A receptor subtype (p < 0.05 vs. control group), while only the novel melatonin-related hybrid up-regulated the pin-Aβ decrease in MT2B receptor subtype (p = 0.004 vs. pin-Aβ1-42-veh group) in the hippocampus. Interestingly, melatonin could not correct the pin-Aβ1-42-related down-regulation of the MT2B receptor subtype in the hippocampus (p = 0.027, vs. pin-Aβ1-42-veh group).
Further, the 3c compound reversed the pin-Aβ1-42-induced diminished expression of both the pERK1/2 (p = 0.002 vs. pin-Aβ1-42-veh group) the pCREB in the hippocampus (p < 0.05 vs. pin-Aβ1-42-veh group) (Figure 9A,B). At the same time, the positive effect of melatonin on these signaling molecules was even higher (p < 0.001 vs. pin-Aβ1-42-veh group, Figure 9A and p < 0.05 vs. sham-veh-veh and pin-Aβ1-42-veh group, Figure 9B, respectively). Similarly, the treatment with melatonin significantly elevated the SIRT1 in the hippocampus (p = 0.007 vs. sham-veh-veh group, p < 0.001 vs. pin-Aβ1-42-veh group) while the hybrid compound 3c was ineffective (p > 0.05 vs. pin-Aβ1-42-veh group; p = 0.002 vs. pin-Aβ1-42-mel group) (Figure 9C).
3. Discussion
Recently, our team designed, synthesized, and characterized a series of melatonin-based hybrid compounds possessing hydrazine scaffold to determine whether they have the potential for further evaluation in AD therapy [11]. Based on in silico and in vitro data, one of the two lead compounds, 3c, was chosen for further in vivo study in a rat model of pinealectomy and subsequent icvAβ1-42 infusion. Melatonin was used as a positive control. The potential protective activity of the hybrid 3c compound against neurotoxicity related to the Aβ protein and plausible underlying mechanism was explored. The hypothesis that melatonin and the compound 3c could stimulate non-amyloidogenic signaling in the hippocampus via activity on MT1A/MT2B receptors in the pin+icvAβ1-42 model was based on recently reported potency of the novel hybrid compound to bind to the two MT receptor subtypes by molecular docking analysis [11].
The major points directed to the close link between the melatoninergic system and AD pathogenesis are as follows: 1) impaired circadian rhythms (melatonin has a key role in the resynchronization of circadian rhythms); 2) impaired pro-oxidant/antioxidant balance (melatonin is a potent antioxidant and free radicals scavenger); 3) neuroinflammation (the hormone possess anti-inflammatory activity). As concerns the role of the melatonin system on the three main hallmarks of AD, i.e., formation of Aβ plaques, p-TAU, and cholinergic system dysfunction, a growing body of evidence suggests putative neuroprotective action of the hormone [12,13]. There is ongoing research to explore its therapeutic potential and the development of melatonin-related compounds for treating AD pathogenesis. However, the underlying mechanism associated with the beneficial effect of the melatonin system is still an area of ongoing research.
We reported earlier that simultaneous induction of melatonin deficiency by pinealectomy and AD-related pathogenesis by icv infusion of Aβ1-42 in rats provokes typical AD behavioral symptomatic alterations such as increased anxiety and cognitive disturbance [10]. Demir et al. (2017) and Zhu et al. (2004) reported on the exacerbated impact of melatonin deficiencies on AD-like alterations and memory decline [14,15].
The idea that cerebrospinal fluid (CSF) melatonin levels decrease in the preclinical stages of AD when patients show no cognitive impairment [8] suggests that melatonin deficiency might be a critical factor predisposing AD development. In the present study, we modified the reported earlier by our team model [10] to simulate the preclinical stage of AD with a drop of melatonin blood level induced by pinealectomy to assess the efficacy of advanced prophylactic treatment with the hormone and the novel melatonin hybrid compound 3c on AD pathogenesis induced by a subsequent icvAβ1-42-infusion. The long-term supplementation with a high dose of melatonin (50 mg/kg for 40 days) reversed behavioral impairments and concomitant elevation of oxidative stress in the frontal cortex and the hippocampus [10]. Melatonin may play a role in the modulation of spatial learning and memory, and its potential effects on these cognitive functions in AD models have been investigated in various studies [16,17,18,19]. The beneficial impact of the melatonin system on spatial memory might be associated with its potency to regulate impaired circadian rhythms, neuroprotection, antioxidant properties, and anti-inflammation. It is well known that the hippocampus is a brain region critical for spatial learning and memory. Melatonin receptors are present in the hippocampus [20], and thereby, the hormone might affect synaptic plasticity and processes closely related to memory formation in this region.
Moreover, the modulatory role of melatonin on acetylcholine [13], which neurotransmitter has a crucial role in cognition [21], is also an essential factor suggesting the beneficial effects of melatonin on spatial memory. Our results revealed that while the novel hybrid compound 3c is ineffective against impaired nonspatial cognitive performance, such as the ORT in a rat model of pin+icvAβ1-42, this melatonin analog possesses a comparable to the positive control melatonin potency to correct the impaired short-term and spatial hippocampus-dependent memory. The beneficial role of melatonin supplementation on spatial memory decline was demonstrated in various models of AD, including the hereditary form of AD, such as mouse transgenic and knockout mice [16,22,23], as well as sporadic form of AD, such as senescence-accelerated OXYS rats [18], streptozotosin- [24,25] and icvAβ1-42-induced models [26].
There is enough literature evidence to support the idea that the soluble Aβ oligomers and the total amount of Aβ, in particular, are responsible for progressive memory decline in AD [27,28], suggesting that these factors may be more critical than the formation of plaques in understanding the disease pathology. Both experimental reports from transgenic APP mice [29,30] and human brain tissue [27,28] demonstrated that cognitive changes appear before forming plaques, thereby supporting the presumption that soluble forms of Aβ may bring about cognitive impairment. Moreover, soluble Aβ oligomers, containing Aβ1–40 or Aβ1–42, detected in AD patients are assumed to contribute to the neurotoxicity associated with AD than Aβ deposits. Thereby, interventions focused on reducing or preventing the neurotoxicity associated with the soluble Aβ oligomers may be crucial for successive treatment of AD pathogenesis. To verify the model of melatonin deficiency induced by pinealectomy and subsequent icvAβ1–42 infusion, we first studied the expression of Aβ1–42, pTAU, and the level of AchE in the hippocampus. The detected increase of Aβ1–42 is in line with our previous report, where the pineal gland was removed simultaneously with the toxic oligomer infusion [10].
Furthermore, in the present study, the pTAU was also confirmed to be significantly elevated in the hippocampus. The lack of difference between the control and model group for this standard marker associated with the pathogenesis of AD as concerns the AChE, may be related to the fact that we measured the levels but not the enzyme activity, which is a limitation of the study and might be taken into consideration in further studies. We can also speculate that in our model, the detected enzyme levels in the hippocampus were conducted before the formation of β-amyloid plaques, where the increased activity of AChE was reported [31]. In addition, higher AChE activity in the vicinity of amyloid plaques was demonstrated to reinforce Aβ aggregation, making it more toxic than Aβ fibrils [32]. The pretreatment with both the positive control melatonin and the 3c compound reduced to control level the model-induced increase in the Aβ and pTAU expression in the hippocampus of pin+Aβ1-42 rats. Interestingly, both the Aβ1-40 and Aβ1-42 have a worsening impact on melatonin production and receptor signaling in cultured cells [33]. Therefore, melatonin deficiency might be associated not only with a removal of the pineal gland but also exacerbated hormonal production in other tissues in the pin+icvAβ1–42 rat model.
Further, the underlying molecular mechanism of melatonin and 3c compound was explored and discussed. Accumulated evidence supports the hypothesis that melatonin can trigger the non-amyloidogenic processing of APP while suppressing the amyloidogenic processing, thus preventing the formation of neurotoxic Aβ oligomers and further accumulation of Aβ into plaques [22]. The endogenous hormone stimulates the activity of alpha-secretases (ADAM10 and ADAM17) at the transcriptional level [12,34]. In contrast, melatonin suppresses amyloidogenic processing by downregulating beta-secretase (BACE1) (transcription, translation, and enzyme activity). Furthermore, diminished BACE1 activity keeps the cholinergic system intact and suppresses neuronal damage and memory decline in parallel with suppression of Aβ40/42 production [35,36].
The neuroprotective effects of melatonin might be exerted through both receptor-dependent and receptor-independent mechanisms [22]. The reduction in SIRT1 levels is negatively correlated with the duration of AD symptoms [37,38,39]. Melatonin promotes the expression of SIRT1 in primary neurons shortly after exposure for up to 24 hours via receptor-independent mode [40], suggesting that upregulation of SIRT1 may promote the expression of ADAM10 involved in non-amyloidogenic processing, thereby leading to reduced production of Aβ.
Melatonin can also exert its effects through MT1 and MT2 receptors, expressed jointly and individually in brain structures [20,41]. While the MT1 receptor is widely distributed in various tissues, the MT2 receptor is more restrictively distributed and is mainly found in the brain, including the hippocampus [20]. Melatonin, through its plasma receptors, induces ERK1/2 phosphorylation via distinct signaling pathways, activating transcription factors such as CREB [13]. Our results showed that while melatonin up-regulated the expression of SIRT1, the melatonin-related 3c compound was ineffective, suggesting that the hybrid compound could mimic the hormone effects mainly through MT receptor activation. Further, our results revealed that while melatonin and the 3c compound reversed the model-related down-regulation of MT1A receptors in the hippocampus, only the novel melatonin-like hybrid corrected the Aβ-induced reduced MT1B receptors. The positive impact of the two pretreatments (melatonin and 3c) on the pCREB expression suggests that the ERK1/2 / CREB signaling is the underlying mechanism involved in the protective effects of the two drugs against formation of Aβ via activation of MT1A receptors (melatonin and 3c) and MT2B (3c). However, the role of SIRT1 in the effects of melatonin that might be realized via non-receptor mode cannot be excluded.
4. Materials and Methods
The compound 3c were synthesized according to procedure described previously in the literature [11] and the structure were proved by 1H, 13C NMR spectra and HRMS.
4.1. Animals
Adult male Wistar rats (250-300 g bw), purchased by the vivarium of the Institute of Neurobiology, BAS, were acclimatized in standard conditions: plexiglas cages in groups of four; 12/12 cycled light/dark regime; average temperature at 22–23 oC; food and water ad libitum. The experimental design was prepared in full accordance with the European Communities Council Directive 2010/63/ E.U. and approved by the Bulgarian Food Safety Agency (research project: #347).
4.2. Experimental Design and Timetable
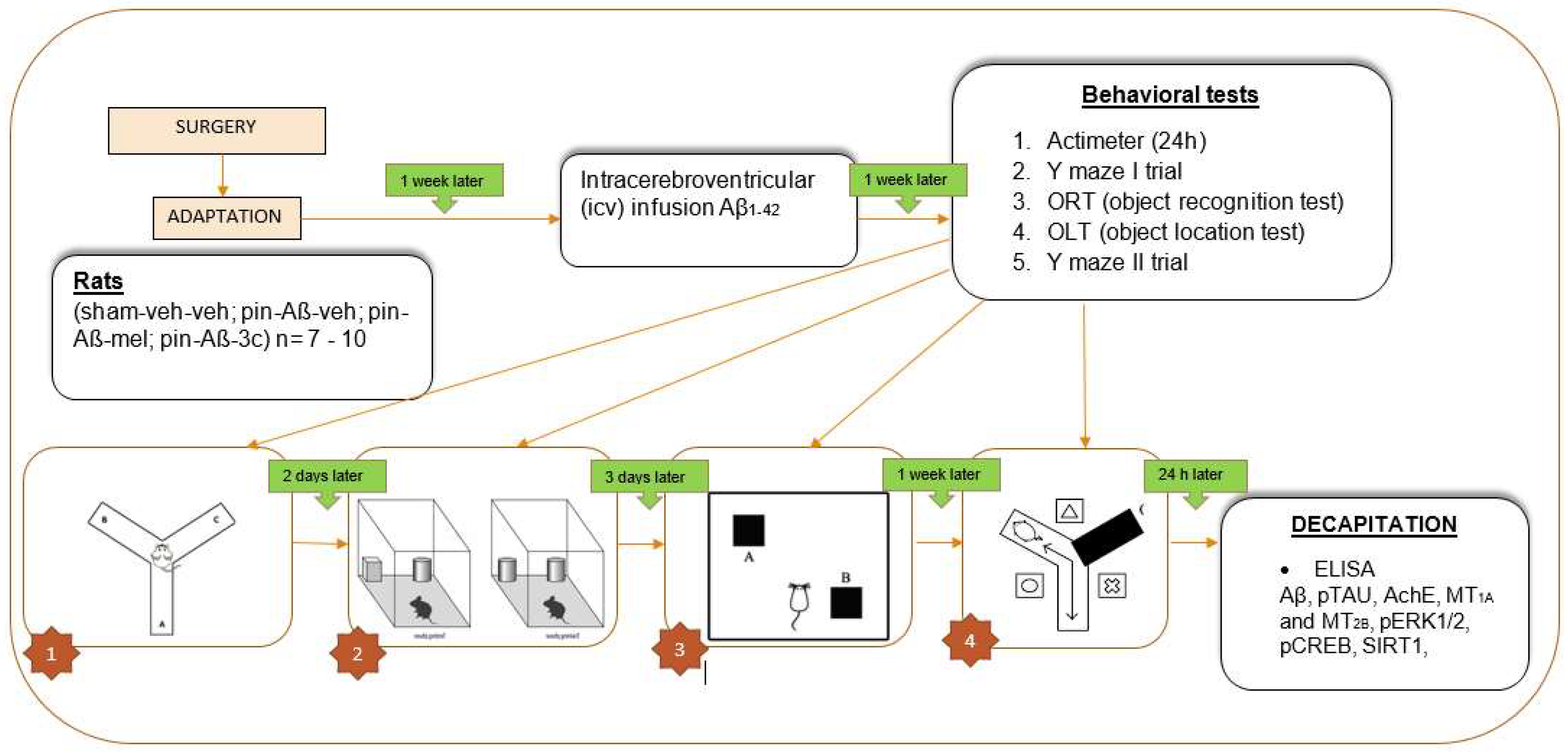
In Figure 10 outlines the timeline of experimental steps.
The rats were randomly assigned to four groups (n= 7 - 10 rats/group) as follows: Group 1 (sham-veh-veh; a control sham-operated rats, i.p injected after surgery with saline once a day for 14 days and icv infused with saline after a week), group 2 (pin-Aβ-veh; a model group with pinealectomy, i.p. injected after surgery with saline as controls and icv infused with Aβ1-42), group 3 (pin-Aβ-mel; a group with pinealectomy, i.p. injected after surgery with melatonin (10 mg/kg, i.p. for 14 days) and icv infused with Aβ1-42), group 4 (pin-Aβ-mel; a group with pinealectomy, i.p. injected after surgery with 3c compound(10 mg/kg, i.p. for 14 days) and icv infused with Aβ1-42),
4.2.1. Behavioral Test
Actimeter
Locomotion and diurnal rhythmic changes were tracked with an infrared actimeter (Bioseb, France) for 26 hours. The first two hours were counted as an adaptation period and were not included in the results. Each rat was tested in a sensor box (20 cm height, 45 cm width, and 45 cm length), measuring horizontal and vertical activity, distance and mean velocity.
Y-Maze Test
The Y-maze test comprised a setup featuring three steel arms positioned at 120° angles from each other. The working memory was evaluated in the Ist trial. Each rat was situated in the center of the apparatus and given 8 minutes for unrestricted exploration of the three arms. Entries into these arms were manually recorded by two individuals unaware of the schedule. The Spontaneous Alternation Behavior (SAB), based on visits to triads consisting of three arms, was calculated. SAB was determined using a formula that considered the number of arm entries and triads; alternation % = (Number of correct entries x 100) / (Total entries (N) – 2). In IInd trial, conducted at least 5 days later, a pretest was performed by closing off one arm and allowing the rat to explore only two arms for 10 minutes. After a 30-minute interval, the rat in the arm opposite to the initially explored two arms was placed. During this phase, the time spent in and the number of entries into both novel arms were measured.
Object Location Test (OLT)
The OLT involves several steps. During the adaptation phase, each animal was placed in a box (50 x 50 cm). The adaptation period was for 10 minutes, allowing the animals to acclimate to their environment without being scored. Twenty-four hours later, the animals were transferred to a dimly lit testing room 30 minutes before to the start of the test. Two identical objects were placed individually in the same box, positioned in opposite corners. The animals were given 5 minutes to explore and interact with the objects freely. After 60 minutes, the animals were subjected to the test phase. One of the objects was moved to the opposite corner, creating both a familiar and a novel location. The animals’ behavior during this 5-minute period was recorded. The duration (in seconds) of sniffing at the familiar and novel locations was recorded. The discrimination index was calculated using the formula: DI = (NO x 100) / (NO + FO). This index provided insights into the animals’ ability to distinguish between familiar and novel object locations. To maintain consistency and avoid any unwanted odors, the arms of the maze are wiped with vinegar or alcohol between each test.
Object Recognition Test (ORT)
The (ORT) was conducted following the procedures outlined in our previous study [42]. Briefly, after 24 hours of acclimation to an empty open field apparatus (50 × 50 × 50 cm), rats were placed within it alongside two identical plastic objects (referred to as “F”) for a 5-minute duration (Training phase). Following a sixty-minute interval, the rats were reintroduced into the same box for the Testing phase. In this phase, one of the objects was replaced with a novel object (referred to as “N”), which lasted 5 minutes. Throughout both the Training and Testing phases, meticulous observation was carried out, and the time spent and the number of times each object was explored through sniffing were recorded. Exploration time was quantified in seconds, and each object count (number of sniffing instances) was documented. The discrimination index was then calculated using the formula: (N) / (N + F).
4.2.2. Detection of Aβ, pTAU, AchE, pERK, pCREB and SIRT1 in the Homogenates from the Hippocampus
After the last memory test, the animals were euthanized by a guillotine, and the left and right hippocampi were carefully isolated, snap frozen in liquid nitrogen, and placed in the fridge until further analysis (8 samples/group). The hippocampal samples were weighted and homogenized with HEPES buffer (20 mM HEPES; 1 mM EGTA; 210 mM Mannithol ; 70 mM sucrose; pH 7,2) and protease inhibition cocktail (100mM PMSF, 100mM NaF, 35mM EDTA). The homogenates were centrifuged at 10 000x g for 5 min at 4oC. The supernatants were used for the determination of general protein via the Bradford method and afterwise several ELISA tests were performed, measuring Aβ, pTAU and AchE. The ELISA measurements were implemented according to the manufacturer’s guidance for the specific kits (Elabscience Rat Aβ1-42, E-EL-R1402; Elabscience Rat pMAPT/pTAU(phosphorylated microtubule-associated protein tau), E-EL-R1090; Elabscience Rat AChE(Acetylcholinesterase), E-EL-R0355).
Statistical analysis
The 24 circadian rhythms of motor activity in the actimeter was assessed by two-way repeated ANOVA. A two-way ANOVA was applied to determine the diurnal variations in horizontal, and vertical activity, distance, and mean velocity. Memory and biochemical data were evaluated by one-way ANOVA. Shapiro-Wilk test post hoc test was used in the case of homogenously distributed data, while Kruskal–Wallis on ranks followed by the Mann–Whitney U test was applied for non-parametric data. The significant level was set at p < 0.05.
5. Conclusions
Melatonin supplementation facilitated the non-amyloidogenic signaling via non-receptor (histone deacetylase sirtuin 1, SIRT1) and receptor MT1A-related signaling (ERK1/2 / CREB) in a rat model of pin + icvAβ1-42. The novel hybrid 3c analogue positively affected pin + icvAβ1-42-induced hippocampus-dependent spatial memory impairment through stimulation of MT1A/MT2B receptors that trigger ERK1/2 / CREB non-amyloidogenic signaling in rats. Melatonin supplementation or other interventions targeting the melatoninergic system may hold promise as potential strategies for managing aspects of AD. However, further research is needed to understand the underlying mechanisms fully and develop practical therapeutic approaches.
Author Contributions
Conceptualization, J.T. and V.A.; methodology, J.T., V.S., L.K. P.I., D.K.; validation, J.T.; formal analysis, D.K. and P.I.; investigation, P.I., D.K., J.T.,; resources, J.T.; data curation, P.I., D.K., J.T.; writing—original draft preparation, J.T. and D.K.; writing—review and editing, J.T.; project administration, J.T. and V.A.; funding acquisition, J.T. and V.A.; All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research was funded by the Bulgarian National Science Fund, grant number KP-06-N63/11; 14.12.2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595. [Google Scholar] [CrossRef]
- Benedict, C.; Byberg, L.; Cedernaes, J.; Hogenkamp, P.S.; Giedratis, V.; Kilander, L.; Lind, L.; Lannfelt, L.; Schiöth, H.B. Self-Reported Sleep Disturbance Is Associated with Alzheimer’s Disease Risk in Men. Alzheimers. Dement. 2015, 11, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, E.; Maestri, M.; Tognoni, G.; Fabbrini, M.; Nucciarone, B.; Manca, M.L.; Gori, S.; Iudice, A.; Murri, L. Daytime Sleepiness in Mild and Moderate Alzheimer’s Disease and Its Relationship with Cognitive Impairment. J. Sleep Res. 2005, 14, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Xiong, D.D.; Holtzman, D.M. Sleep, Circadian Rhythms, and the Pathogenesis of Alzheimer Disease. Exp. Mol. Med. 2015, 47, e148. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. A Role for the Regulation of the Melatonergic Pathways in Alzheimer’s Disease and Other Neurodegenerative and Psychiatric Conditions. Serotonin and Melatonin 2016, 443–466. [Google Scholar] [CrossRef]
- Claustrat, B.; Brun, J.; Borson-Chazot, F. Melatonin and Circadian Rhythms. Curr. Top. Med. Chem. 2002, 2. [Google Scholar] [CrossRef]
- Ohashi, Y.; Okamoto, N.; Uchida, K.; Iyo, M.; Mori, N.; Morita, Y. Daily Rhythm of Serum Melatonin Levels and Effect of Light Exposure in Patients with Dementia of the Alzheimer’s Type. Biol. Psychiatry 1999, 45, 1646–1652. [Google Scholar] [CrossRef]
- Zhou, J.N.; Liu, R.Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early Neuropathological Alzheimer’s Changes in Aged Individuals Are Accompanied by Decreased Cerebrospinal Fluid Melatonin Levels. J. Pineal Res. 2003, 35, 125–130. [Google Scholar] [CrossRef]
- Beriwal, N.; Namgyal, T.; Sangay, P.; Al Quraan, A.M. Role of Immune-Pineal Axis in Neurodegenerative Diseases, Unraveling Novel Hybrid Dark Hormone Therapies. Heliyon 2019, 5. [Google Scholar] [CrossRef]
- Tzoneva, R.; Georgieva, I.; Ivanova, N.; Uzunova, V.; Nenchovska, Z.; Apostolova, S.; Stoyanova, T.; Tchekalarova, J. The Role of Melatonin on Behavioral Changes and Concomitant Oxidative Stress in IcvAβ1-42 Rat Model with Pinealectomy. Int. J. Mol. Sci. 2021, 22, 12763. [Google Scholar] [CrossRef]
- Angelova, V.T.; Georgiev, B.; Pencheva, T.; Pajeva, I.; Rangelov, M.; Todorova, N.; Zheleva-Dimitrova, D.; Kalcheva-Yovkova, E.; Valkova, I. V.; Vassilev, N.; et al. Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease. Pharmaceuticals 2023, 16, 1194. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Htoo, H.H.; Wintachai, P.; Hernandez, J.F.; Dubois, C.; Postina, R.; Xu, H.; Checler, F.; Smith, D.R.; Govitrapong, P.; et al. Melatonin Stimulates the Nonamyloidogenic Processing of ΒAPP through the Positive Transcriptional Regulation of ADAM10 and ADAM17. J. Pineal Res. 2015, 58, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Govitrapong, P.; Boontem, P.; Reiter, R.J.; Satayavivad, J. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15. [Google Scholar] [CrossRef]
- Demir, M.; Yilmaz, U.; Colak, C.; Cigremis, Y.; Ozyalin, F.; Tekedereli, I.; Kilincli, A.; Sandal, S. The Effects of Lack of Melatonin in Experimental Rat Model of Alzheimer’s Disease: Relationship with FEZ1 Gene Expression. Med. Sci. | Int. Med. J. 2016, 6, 217–217. [Google Scholar] [CrossRef]
- Ling, Q.Z.; Shao, H.W.; Zhi, Q.L.; Dan, L.W.; Wang, J.Z. Effect of Inhibiting Melatonin Biosynthesis on Spatial Memory Retention and Tau Phosphorylation in Rat. J. Pineal Res. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Liu, Y.C.; Hsu, W.L.; Ma, Y.L.; Lee, E.H.Y. Melatonin Induction of APP Intracellular Domain 50 SUMOylation Alleviates AD through Enhanced Transcriptional Activation and Aβ Degradation. Mol. Ther. 2021, 29, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Jürgenson, M.; Zharkovskaja, T.; Noortoots, A.; Morozova, M.; Beniashvili, A.; Zapolski, M.; Zharkovsky, A. Effects of the Drug Combination Memantine and Melatonin on Impaired Memory and Brain Neuronal Deficits in an Amyloid-Predominant Mouse Model of Alzheimer’s Disease. J. Pharm. Pharmacol. 2019, 71, 1695–1705. [Google Scholar] [CrossRef]
- Rudnitskaya, E.A.; Muraleva, N.A.; Maksimova, K.Y.; Kiseleva, E.; Kolosova, N.G.; Stefanova, N.A. Melatonin Attenuates Memory Impairment, Amyloid-β Accumulation, and Neurodegeneration in a Rat Model of Sporadic Alzheimer’s Disease. J. Alzheimers. Dis. 2015, 47, 103–116. [Google Scholar] [CrossRef]
- Sun, C.; Qiu, X.; Wang, Y.; Liu, J.; Li, Q.; Jiang, H.; Li, S.; Song, C. Long-Term Oral Melatonin Alleviates Memory Deficits, Reduces Amyloid-β Deposition Associated with Downregulation of BACE1 and Mitophagy in APP/PS1 Transgenic Mice. Neurosci. Lett. 2020, 735. [Google Scholar] [CrossRef]
- Ng, K.Y.; Leong, M.K.; Liang, H.; Paxinos, G. Melatonin Receptors: Distribution in Mammalian Brain and Their Respective Putative Functions. Brain Struct. Funct. 2017, 222, 2921–2939. [Google Scholar] [CrossRef]
- Roy, J.; Tsui, K.C.; Ng, J.; Fung, M.L.; Lim, L.W. Regulation of Melatonin and Neurotransmission in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, Vol. 22, Page 6841 2021, 22, 6841. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin Regulates Aβ Production/Clearance Balance and Aβ Neurotoxicity: A Potential Therapeutic Molecule for Alzheimer’s Disease. Biomed. Pharmacother. 2020, 132. [Google Scholar] [CrossRef] [PubMed]
- Labban, S.; Alshehri, F.S.; Kurdi, M.; Alatawi, Y.; Alghamdi, B.S. Melatonin Improves Short-Term Spatial Memory in a Mouse Model of Alzheimer’s Disease. Degener. Neurol. Neuromuscul. Dis. 2021, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Kamsrijai, U.; Wongchitrat, P.; Nopparat, C.; Satayavivad, J.; Govitrapong, P. Melatonin Attenuates Streptozotocin-Induced Alzheimer-like Features in Hyperglycemic Rats. Neurochem. Int. 2020, 132. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.K.; Souza, L.C.; Azevedo, E.M.; Bail, E.L.; Zanata, S.M.; Andreatini, R.; Vital, M.A.B.F. Melatonin Reduces β-Amyloid Accumulation and Improves Short-Term Memory in Streptozotocin-Induced Sporadic Alzheimer’s Disease Model. IBRO Neurosci. Reports 2023, 14, 264. [Google Scholar] [CrossRef] [PubMed]
- Keymoradzadeh, A.; Komaki, A.R.; Bakhshi, A.; Faraji, N.; Golipoor, Z.; Shahshahani, P. The Effect of Different Doses of Melatonin on Learning and Memory Deficit in Alzheimer Model of Rats. Casp. J. Neurol. Sci. 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Ha, S.; Redmond, L. ERK Mediates Activity Dependent Neuronal Complexity via Sustained Activity and CREB-Mediated Signaling. Dev. Neurobiol. 2008, 68, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.; Castaño, E.; Kokjohn, T.A.; Kuo, Y.M.; Lyubchenko, Y.; Pinsky, D.; Connolly, E.S.; Esh, C.; Luehrs, D.C.; Blaine Stine, W.; et al. Physicochemical Characteristics of Soluble Oligomeric Abeta and Their Pathologic Role in Alzheimer’s Disease. Neurol. Res. 2005, 27, 869–881. [Google Scholar] [CrossRef]
- Holcomb, L.; Gordon, M.N.; Mcgowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-Type Phenotype in Transgenic Mice Carrying Both Mutant Amyloid Precursor Protein and Presenilin 1 Transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural Oligomers of the Amyloid-Beta Protein Specifically Disrupt Cognitive Function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Morán, M.A.; Mufson, E.J.; Gómez-Ramos, P. Colocalization of Cholinesterases with Beta Amyloid Protein in Aged and Alzheimer’s Brains. Acta Neuropathol. 1993, 85, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.E.; Chacón, M.A.; Dinamarca, M.C.; Cerpa, W.; Morgan, C.; Inestrosa, N.C. Acetylcholinesterase-Aβ Complexes Are More Toxic than Aβ Fibrils in Rat Hippocampus : Effect on Rat β-Amyloid Aggregation, Laminin Expression, Reactive Astrocytosis, and Neuronal Cell Loss. Am. J. Pathol. 2004, 164, 2163. [Google Scholar] [CrossRef] [PubMed]
- Cecon, E.; Chen, M.; Marçola, M.; Fernandes, P.A.C.; Jockers, R.; Markus, R.P. Amyloid β Peptide Directly Impairs Pineal Gland Melatonin Synthesis and Melatonin Receptor Signaling through the ERK Pathway. FASEB J. 2015, 29, 2566–2582. [Google Scholar] [CrossRef]
- Panmanee, J.; Nopparat, C.; Chavanich, N.; Shukla, M.; Mukda, S.; Song, W.; Vincent, B.; Govitrapong, P. Melatonin Regulates the Transcription of ΒAPP-Cleaving Secretases Mediated through Melatonin Receptors in Human Neuroblastoma SH-SY5Y Cells. J. Pineal Res. 2015, 59, 308–320. [Google Scholar] [CrossRef]
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer’s Disease. Neuron 2004, 41, 27–33. [Google Scholar] [CrossRef]
- Ohno, M.; Cole, S.L.; Yasvoina, M.; Zhao, J.; Citron, M.; Berry, R.; Disterhoft, J.F.; Vassar, R. BACE1 Gene Deletion Prevents Neuron Loss and Memory Deficits in 5XFAD APP/PS1 Transgenic Mice. Neurobiol. Dis. 2007, 26, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fivecoat, H.; Ho, L.; Pan, Y.; Ling, E.; Pasinetti, G.M. The Role of Sirt1: At the Crossroad between Promotion of Longevity and Protection against Alzheimer’s Disease Neuropathology. Biochim. Biophys. Acta 2010, 1804, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 Activator Resveratrol Protects SK-N-BE Cells from Oxidative Stress and against Toxicity Caused by Alpha-Synuclein or Amyloid-Beta (1-42) Peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 Activation as a Novel Mechanism Underlying the Prevention of Alzheimer Disease Amyloid Neuropathology by Calorie Restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef]
- Cristòfol, R.; Porquet, D.; Corpas, R.; Coto-Montes, A.; Serret, J.; Camins, A.; Pallàs, M.; Sanfeliu, C. Neurons from Senescence-Accelerated SAMP8 Mice Are Protected against Frailty by the Sirtuin 1 Promoting Agents Melatonin and Resveratrol. J. Pineal Res. 2012, 52, 271–281. [Google Scholar] [CrossRef]
- Lacoste, B.; Angeloni, D.; Dominguez-Lopez, S.; Calderoni, S.; Mauro, A.; Fraschini, F.; Descarries, L.; Gobbi, G. Anatomical and Cellular Localization of Melatonin MT1 and MT2 Receptors in the Adult Rat Brain. J. Pineal Res. 2015, 58, 397–417. [Google Scholar] [CrossRef]
- Shishmanova-Doseva, M.; Peychev, L.; Yoanidu, L.; Uzunova, Y.; Atanasova, M.; Georgieva, K.; Tchekalarova, J.; Shishmanova-Doseva, M.; Peychev, L.; Yoanidu, L.; et al. Anticonvulsant Effects of Topiramate and Lacosamide on Pilocarpine-Induced Status Epilepticus in Rats: A Role of Reactive Oxygen Species and Inflammation. Int. J. Mol. Sci. 2021, Vol. 22, Page 2264 2021, 22, 2264. [Google Scholar] [CrossRef]
Figure 2.
Effect of control (sham-veh-veh) group (n = 8), Aβ1-42-veh group (n = 7), Aβ1-42-mel group (n = 7), and Aβ1-42-3c group (n = ) on 24-h motor activity measured in the actimeter. Data are presented as mean ± SEM. Repeated two-way ANOVA demonstrated a main Time effect [F23,479 = 34,427, p < 0.001] as well as Group x Time interaction [F69,479 = 1,422, p = 0.026] for the horizontal activity. A main Time effect [F23,479 = 15,916, p < 0.001] was detected for the vertical activity. Post hoc test showed in all groups higher horizontal (Figure 1A) and vertical (Figure 1B) activity in the zeitgeber (ZT) 12-23 (the dark period) compared to the ZT 0-11 (the light period). Further, a significant difference in activity was detected at ZT8-ZT19 (pin+Aβ1-42-veh vs. sham-veh-veh group (p < 0.05) (Figure 1B). On the right of the figures are inserted Cosinor data.
Figure 2.
Effect of control (sham-veh-veh) group (n = 8), Aβ1-42-veh group (n = 7), Aβ1-42-mel group (n = 7), and Aβ1-42-3c group (n = ) on 24-h motor activity measured in the actimeter. Data are presented as mean ± SEM. Repeated two-way ANOVA demonstrated a main Time effect [F23,479 = 34,427, p < 0.001] as well as Group x Time interaction [F69,479 = 1,422, p = 0.026] for the horizontal activity. A main Time effect [F23,479 = 15,916, p < 0.001] was detected for the vertical activity. Post hoc test showed in all groups higher horizontal (Figure 1A) and vertical (Figure 1B) activity in the zeitgeber (ZT) 12-23 (the dark period) compared to the ZT 0-11 (the light period). Further, a significant difference in activity was detected at ZT8-ZT19 (pin+Aβ1-42-veh vs. sham-veh-veh group (p < 0.05) (Figure 1B). On the right of the figures are inserted Cosinor data.

Figure 3.
Effect of sham-veh-veh group (n = 8), Aβ1-42-veh group (n = 7), Aβ1-42-mel group (n = 7), and Aβ1-42-3c group (n = 7) on daily rhythm of 24-h registered horizontal activity (counts) (A), vertical activity (B), distance (C) and mean velocity (D) measured in the actimeter. Open and black rectangles present the light and the dark phases above the figures. Data are presented as mean ± SEM, n = 7-8. Two-way ANOVA demonstrated a main Phase effect for horizontal activity [F3,56 = 133,318, p < 0.001], a main Phase [F1,55 = 64.475, p < 0.001], Treatment [F3,56 = 6. 519, p = 0.001] as well as Phase x Treatment interaction [F3,56 = 6.273, p = 0.001] for vertical activity, a main Phase [F1,55 = 21.866, p < 0.001] and Phase x Treatment interaction [F3,56 = 18.262, p < 0.001] for distance, and a main Phase effect [F1,56 = 100.719, p < 0.001] for mean velocity. ###p < 0.001, #p < 0.05, dark vs. light phase (A-D); *p = 0.03, pin+Aβ1-42-veh group vs. sham-veh-veh (A); ***p < 0.001, pin+Aβ1-42-veh-mel group vs. pin+Aβ1-42-veh group (B).
Figure 3.
Effect of sham-veh-veh group (n = 8), Aβ1-42-veh group (n = 7), Aβ1-42-mel group (n = 7), and Aβ1-42-3c group (n = 7) on daily rhythm of 24-h registered horizontal activity (counts) (A), vertical activity (B), distance (C) and mean velocity (D) measured in the actimeter. Open and black rectangles present the light and the dark phases above the figures. Data are presented as mean ± SEM, n = 7-8. Two-way ANOVA demonstrated a main Phase effect for horizontal activity [F3,56 = 133,318, p < 0.001], a main Phase [F1,55 = 64.475, p < 0.001], Treatment [F3,56 = 6. 519, p = 0.001] as well as Phase x Treatment interaction [F3,56 = 6.273, p = 0.001] for vertical activity, a main Phase [F1,55 = 21.866, p < 0.001] and Phase x Treatment interaction [F3,56 = 18.262, p < 0.001] for distance, and a main Phase effect [F1,56 = 100.719, p < 0.001] for mean velocity. ###p < 0.001, #p < 0.05, dark vs. light phase (A-D); *p = 0.03, pin+Aβ1-42-veh group vs. sham-veh-veh (A); ***p < 0.001, pin+Aβ1-42-veh-mel group vs. pin+Aβ1-42-veh group (B).
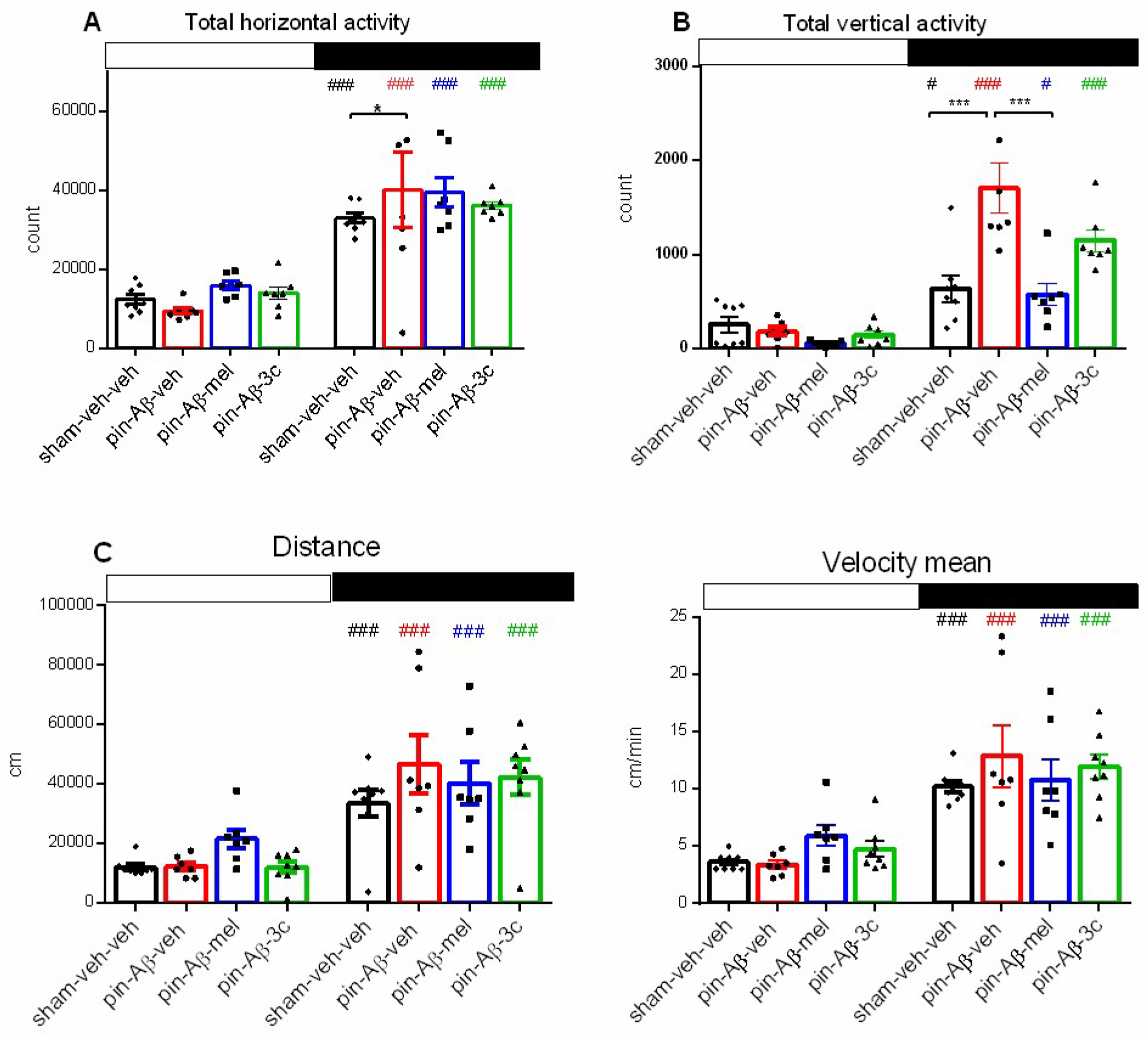
Figure 4.
Effect of control (sham-veh-veh) group, pin-Aβ1-42-veh group, pin-Aβ1-42-mel group, and pin-Aβ1-42-3c group on short-term recognition memory tested in the object recognition test. Data are presented as the mean ± SEM. The Kruskal-Wallis analysis revealed a main Group effect [H = 9.366, p = 0.025] for DI (sec) and [H = 21.933, p < 0.001] for DI (count); *p = 0.028, pin-Aβ1-42-veh group vs. sham-veh-veh group (A); *p = 0.05, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.05, pin-Aβ1-42-mel group vs. pin-Aβ1-42-veh group (B) (n = 8-10).
Figure 4.
Effect of control (sham-veh-veh) group, pin-Aβ1-42-veh group, pin-Aβ1-42-mel group, and pin-Aβ1-42-3c group on short-term recognition memory tested in the object recognition test. Data are presented as the mean ± SEM. The Kruskal-Wallis analysis revealed a main Group effect [H = 9.366, p = 0.025] for DI (sec) and [H = 21.933, p < 0.001] for DI (count); *p = 0.028, pin-Aβ1-42-veh group vs. sham-veh-veh group (A); *p = 0.05, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.05, pin-Aβ1-42-mel group vs. pin-Aβ1-42-veh group (B) (n = 8-10).
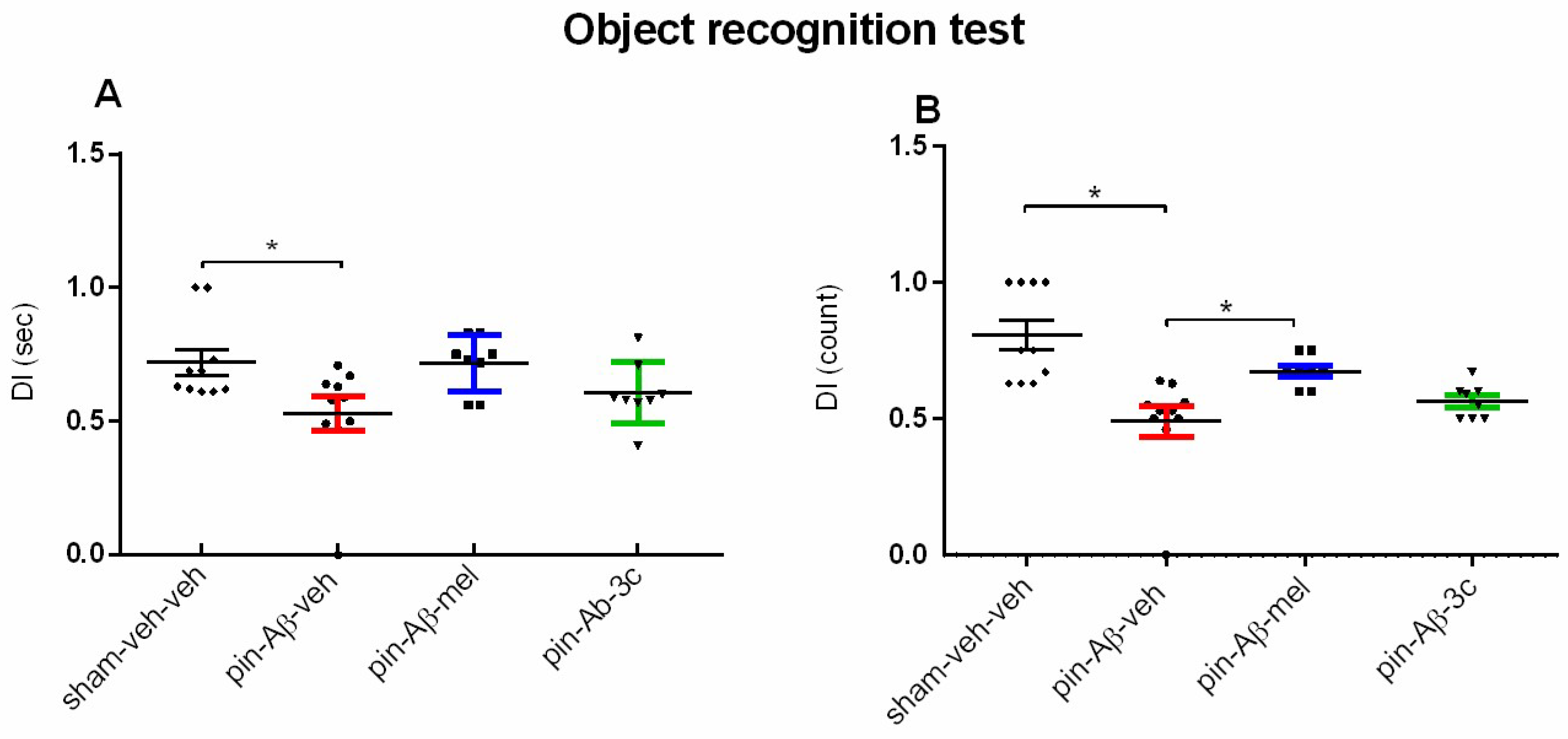
Figure 5.
Effect of control (sham-veh-veh) group, pin-Aβ1-42-veh group, pin-Aβ1-42-mel group, and pin-Aβ1-42-3c group on short-term spatial memory tested in the object location test. Data are presented as the mean ± SEM. The one-way ANOVA revealed a main Group effect [F3,35 = 11,367, p < 0.001]; ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; ***p < 0.001, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel; *p = 0.024, pin-Aβ1-42-3c vs. sham-veh-veh group; *p = 0.047, pin-Aβ1-42-3c vs. pin-Aβ1-42-veh group (n = 8 - 10).
Figure 5.
Effect of control (sham-veh-veh) group, pin-Aβ1-42-veh group, pin-Aβ1-42-mel group, and pin-Aβ1-42-3c group on short-term spatial memory tested in the object location test. Data are presented as the mean ± SEM. The one-way ANOVA revealed a main Group effect [F3,35 = 11,367, p < 0.001]; ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; ***p < 0.001, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel; *p = 0.024, pin-Aβ1-42-3c vs. sham-veh-veh group; *p = 0.047, pin-Aβ1-42-3c vs. pin-Aβ1-42-veh group (n = 8 - 10).
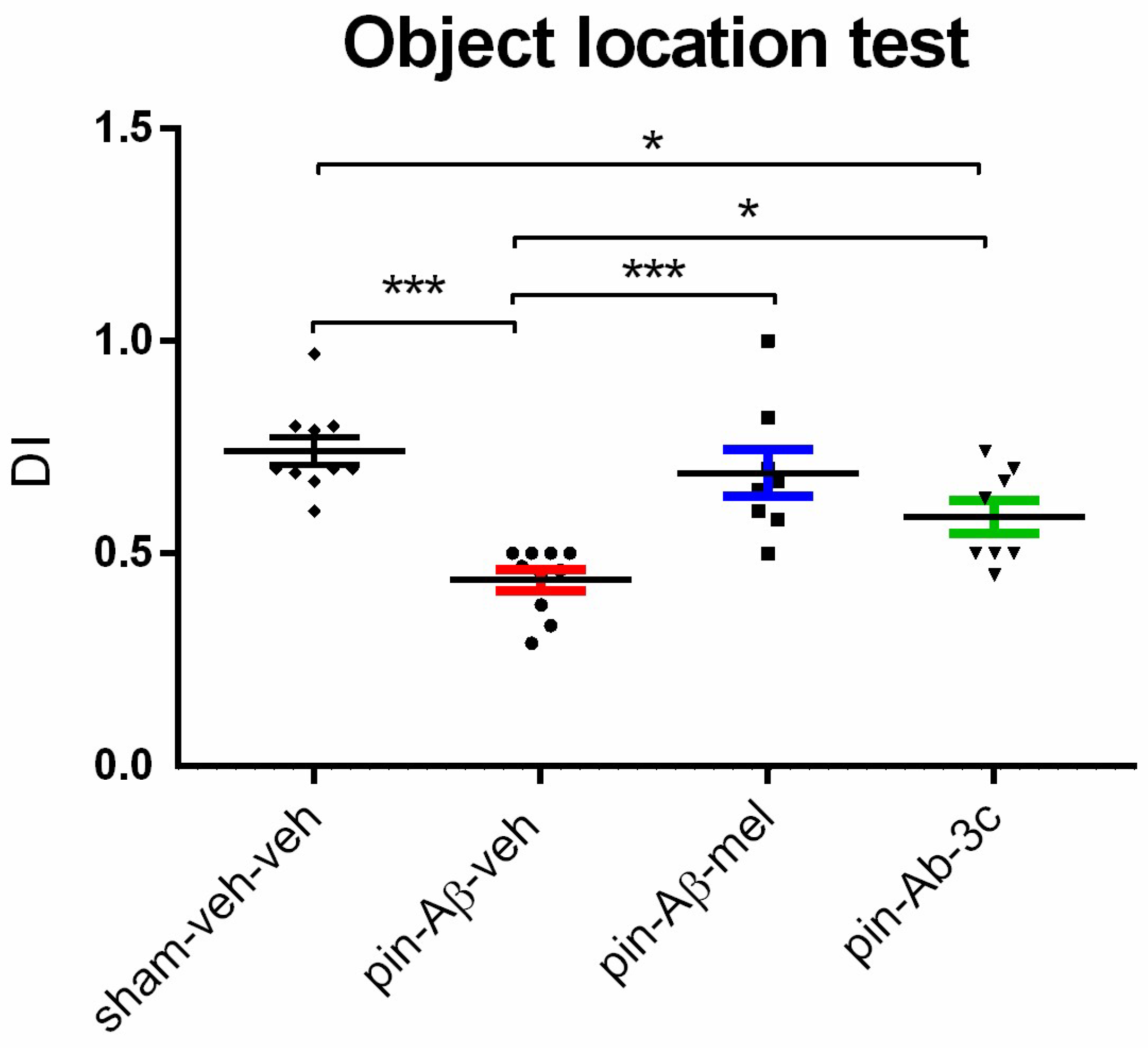
Figure 6.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on Spontaneous alternation behavior (SAB) (in percent) in the Ist trial (A), DI (sec) (B) and DI (count) (C), respectively, in the IInd trial in the Y-maze task. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,35 = 10,054, p < 0.001] for SAB; a main Group effect [F3,35 = 25,859, p < 0.001] for DI(time) and [H = 16,521, p < 0.001] for DI (counts) (Kruskal-Wallis test); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.02, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel group; ***p < 0.001 pin-Aβ1-42-veh group vs. pin-Aβ1-42-3c group (A); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; ***p < 0.01, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel group; ***p < 0.001 pin-Aβ1-42-3c group vs. sham-veh-veh group;*p = 0.036, pin-Aβ1-42-3c group vs. pin-Aβ1-42-veh group (B); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; **p = 0.002, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel and pin-Aβ1-42-3c group, respectively (C) (n = 8 - 10).
Figure 6.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on Spontaneous alternation behavior (SAB) (in percent) in the Ist trial (A), DI (sec) (B) and DI (count) (C), respectively, in the IInd trial in the Y-maze task. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,35 = 10,054, p < 0.001] for SAB; a main Group effect [F3,35 = 25,859, p < 0.001] for DI(time) and [H = 16,521, p < 0.001] for DI (counts) (Kruskal-Wallis test); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.02, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel group; ***p < 0.001 pin-Aβ1-42-veh group vs. pin-Aβ1-42-3c group (A); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; ***p < 0.01, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel group; ***p < 0.001 pin-Aβ1-42-3c group vs. sham-veh-veh group;*p = 0.036, pin-Aβ1-42-3c group vs. pin-Aβ1-42-veh group (B); ***p < 0.001, pin-Aβ1-42-veh group vs. sham-veh-veh group; **p = 0.002, pin-Aβ1-42-veh group vs. pin-Aβ1-42-mel and pin-Aβ1-42-3c group, respectively (C) (n = 8 - 10).
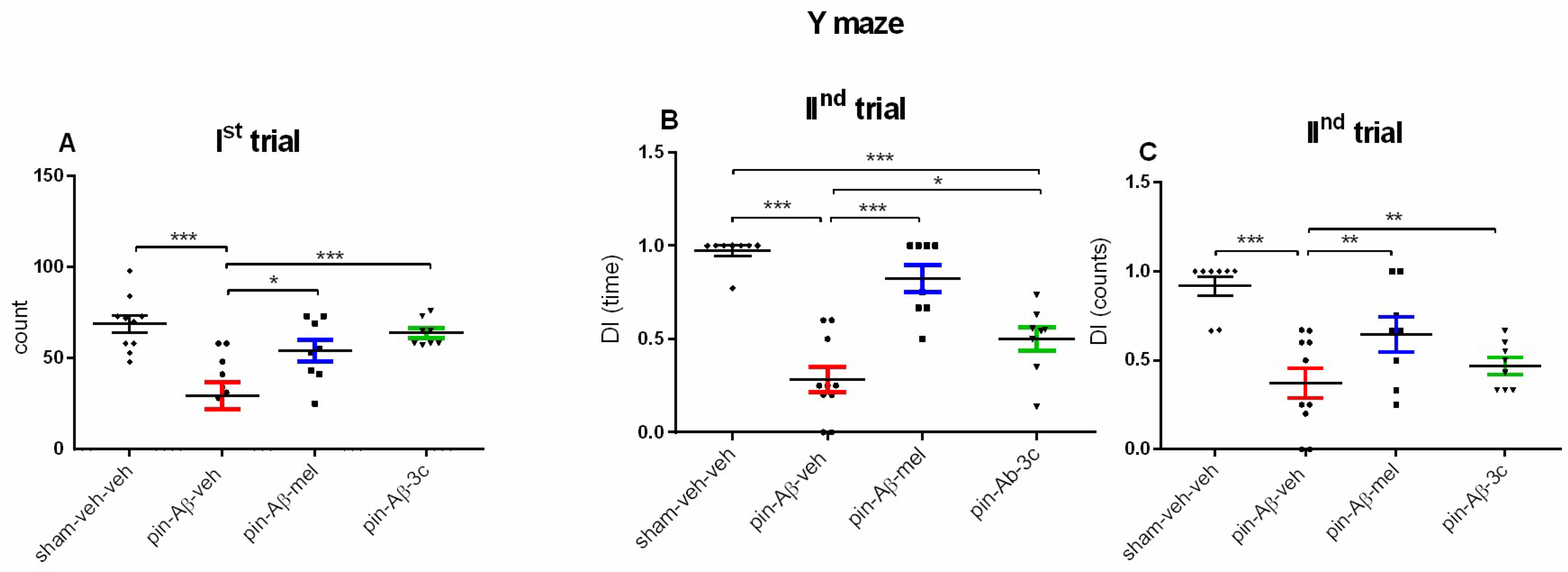
Figure 7.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on Aβ1-42 protein expression (A), p-TAU protein expression (B) and AchE level (C) in the hippocampus measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,31 = 3,125, p = 0.042] for Aβ1-42 protein, and [H = 8,936, p = 0.03] (Kruskal-Wallis test) for pTAU protein; *p = 0.035, pin-Aβ1-42-veh group vs. sham-veh-veh group (A); *p = 0.049, pin-Aβ1-42-veh group vs. sham-veh-veh group (B) (n = 8).
Figure 7.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on Aβ1-42 protein expression (A), p-TAU protein expression (B) and AchE level (C) in the hippocampus measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,31 = 3,125, p = 0.042] for Aβ1-42 protein, and [H = 8,936, p = 0.03] (Kruskal-Wallis test) for pTAU protein; *p = 0.035, pin-Aβ1-42-veh group vs. sham-veh-veh group (A); *p = 0.049, pin-Aβ1-42-veh group vs. sham-veh-veh group (B) (n = 8).
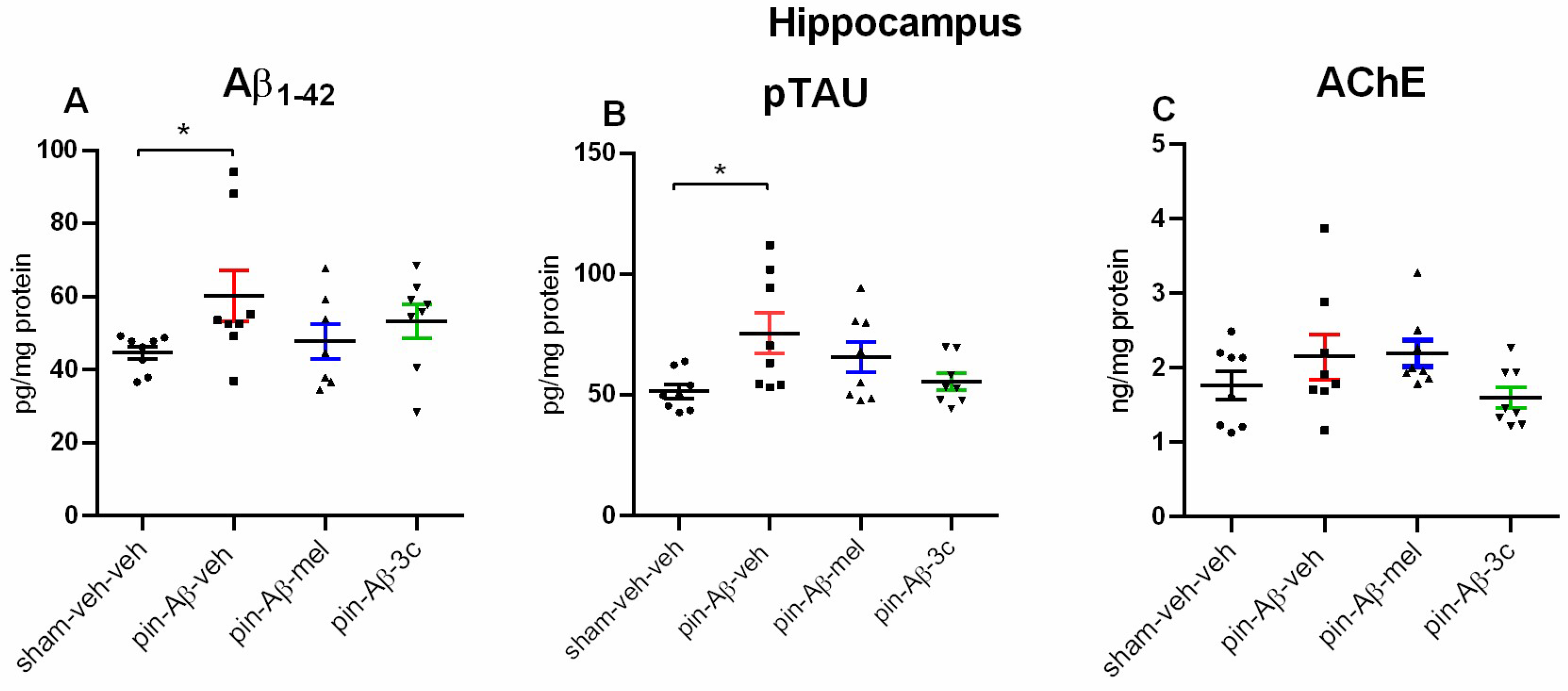
Figure 8.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on MT1A (A) and MT2B (B) receptor subtypes in the hippocampus, measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [H = 21,361, p < 0.001] (Kruskal-Wallis test) for MT1A and [F3,31 = 7,771, p < 0.001] (Shapiro-Wilk test) for MT2B receptor subtype; *p < 0.05, pin-Aβ1-42-veh group vs. sham-veh-veh group, pin-Aβ1-42-mel and pin-Aβ1-42-3c, respectively (A); **p = 0.003, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.027, pin-Aβ1-42-mel group vs. sham-veh-veh group; **p = 0.004, pin-Aβ1-42-3c group vs. pin-Aβ1-42-veh group (B) (n = 8).
Figure 8.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on MT1A (A) and MT2B (B) receptor subtypes in the hippocampus, measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [H = 21,361, p < 0.001] (Kruskal-Wallis test) for MT1A and [F3,31 = 7,771, p < 0.001] (Shapiro-Wilk test) for MT2B receptor subtype; *p < 0.05, pin-Aβ1-42-veh group vs. sham-veh-veh group, pin-Aβ1-42-mel and pin-Aβ1-42-3c, respectively (A); **p = 0.003, pin-Aβ1-42-veh group vs. sham-veh-veh group; *p = 0.027, pin-Aβ1-42-mel group vs. sham-veh-veh group; **p = 0.004, pin-Aβ1-42-3c group vs. pin-Aβ1-42-veh group (B) (n = 8).
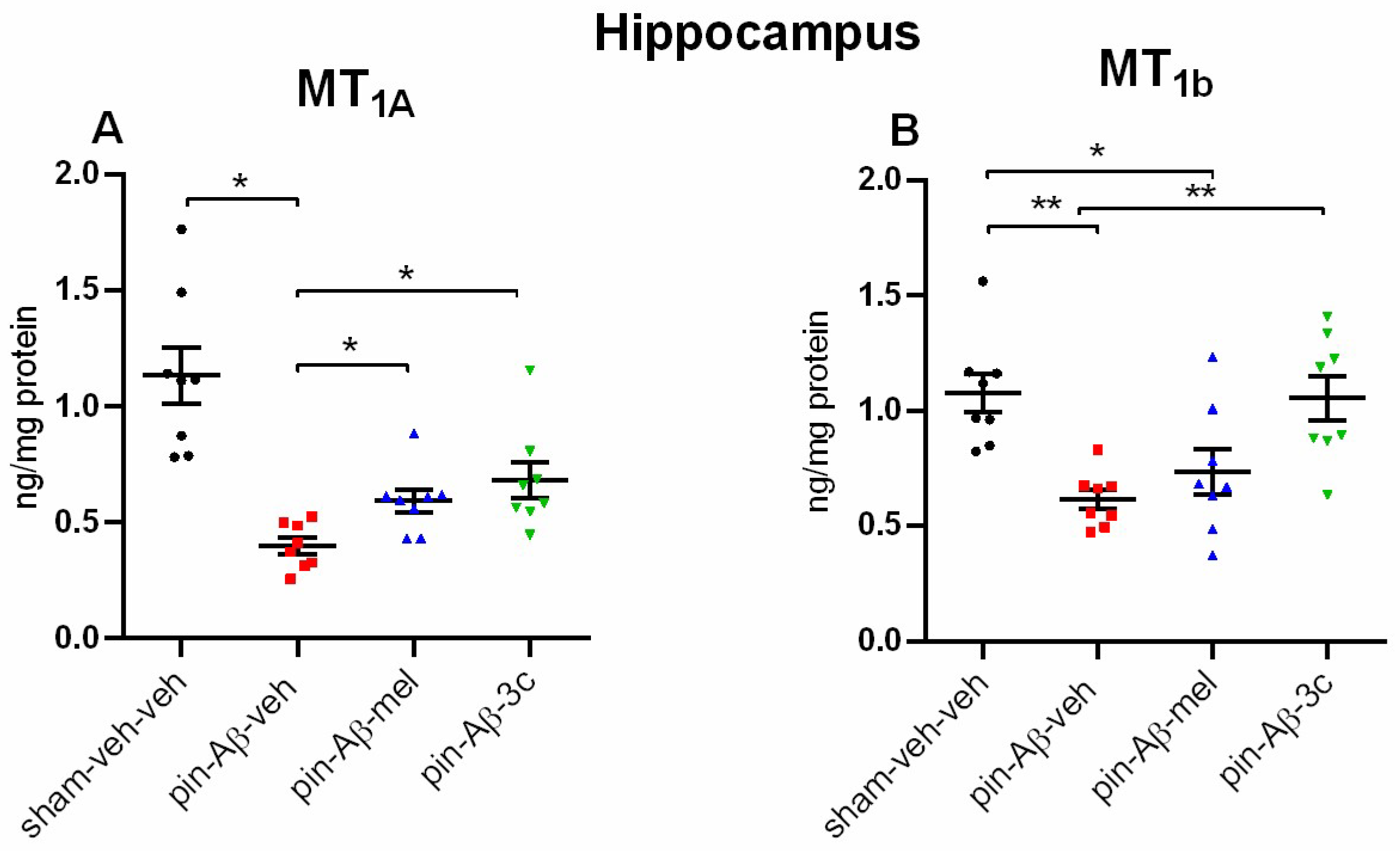
Figure 9.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on pERK 1/2 (A), pCREB (B), and SIRT1 (C) expression in the hippocampus, measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,31 = 10,069, p < 0.001] (Shapiro-Wilk) for pERK1/2; [H = 17,782, p < 0.001] (Kruskal-Wallis test) for pCREB, [F3,31 = 11,774, p < 0.001] (Shapiro-Wilk) for SIRT1;. ***p < 0.001, pin-Aβ1-42-mel group vs pin-Aβ1-42-veh group; **p = 0.002, pin-Aβ1-42-3c group vs pin-Aβ1-42-veh group (A); *p < 0.05, pin-Aβ1-42-mel group vs sham-veh-veh and pin-Aβ1-42-veh group; *p < 0.05, pin-Aβ1-42-3c group vs pin-Aβ1-42-veh group (B); ***p < 0.001, pin-Aβ1-42-mel group vs. pin-Aβ1-42-veh group; **p = 0.007, pin-Aβ1-42-mel group vs. sham-veh-veh group; **p = 0.002, pin-Aβ1-42-mel group vs. pin-Aβ1-42-3c group. .
Figure 9.
Effect of control (sham-veh-veh), pin-Aβ1-42-veh, pin-Aβ1-42-mel, and pin-Aβ1-42-3c on pERK 1/2 (A), pCREB (B), and SIRT1 (C) expression in the hippocampus, measured by the ELISA. Data are presented as the mean ± SEM. The one-way ANOVA demonstrated a main Group effect [F3,31 = 10,069, p < 0.001] (Shapiro-Wilk) for pERK1/2; [H = 17,782, p < 0.001] (Kruskal-Wallis test) for pCREB, [F3,31 = 11,774, p < 0.001] (Shapiro-Wilk) for SIRT1;. ***p < 0.001, pin-Aβ1-42-mel group vs pin-Aβ1-42-veh group; **p = 0.002, pin-Aβ1-42-3c group vs pin-Aβ1-42-veh group (A); *p < 0.05, pin-Aβ1-42-mel group vs sham-veh-veh and pin-Aβ1-42-veh group; *p < 0.05, pin-Aβ1-42-3c group vs pin-Aβ1-42-veh group (B); ***p < 0.001, pin-Aβ1-42-mel group vs. pin-Aβ1-42-veh group; **p = 0.007, pin-Aβ1-42-mel group vs. sham-veh-veh group; **p = 0.002, pin-Aβ1-42-mel group vs. pin-Aβ1-42-3c group. .
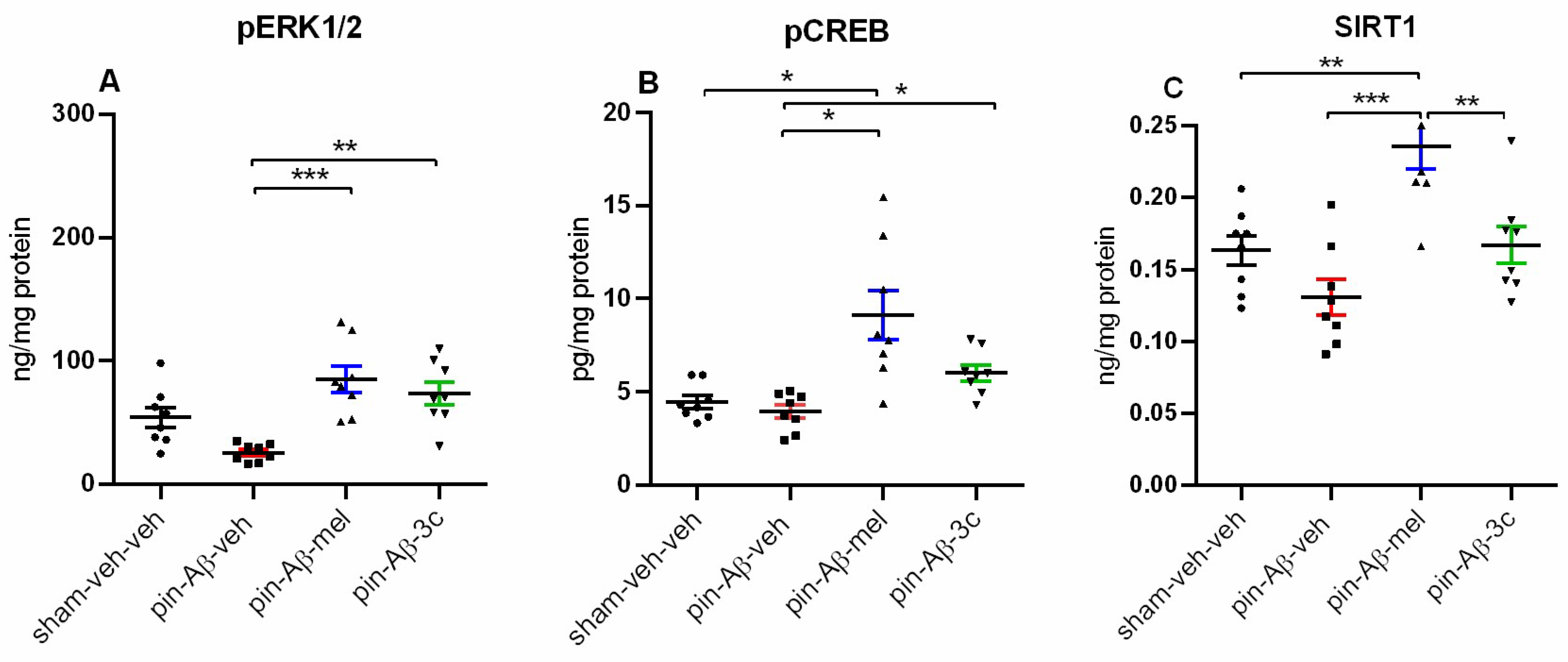
Figure 10.
Timeline of experimental steps.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



