
Submitted:
20 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
Ovarian cysts are linked to hormone imbalances and altered gene expressions, but the connection between cysts and ion channel expression is understudied. This study explored the role of TWIK-related acid-sensitive K+ (TASK) channels in bovine ovarian cyst formation. The ovarian follicles were split into small (5 to 10 mm in diameter) and large (> 25 mm in diameter) groups. Among the measured K+, Na+, and Cl- concentrations in follicular fluid (FF) obtained from small-sized follicles (SF) and large-sized follicles (LF), the K+ concentration was significantly lower in LFFF. Quantitative PCR, western blot, and immunocytochemistry data revealed that TASK-3 expression levels significantly decreased by approximately 50% in both LF and granulosa cells obtained from LF (LFGCs) compared to the corresponding controls. The TASK-3 protein was localized to the plasma membrane of GCs. The diameter of LFGCs was larger than that of SFGC. The cell swelling response to exposure to a hypotonic solution (200 mOsm/L) was highly reduced in TASK-3-overexpressing cells compared to vector-transfected cells. TASK-3-knockdown cells showed arrested growth. Senescence markers were detected in LFGCs and TASK-3-knockdown cells. These findings suggest that reduced TASK-3 expression in LF is associated with the inhibition of GC growth, leading to senescence and cyst formation.
Keywords:
granulosa cell
; ovarian cyst
; potassium ion
; TASK-3 channel
1. Introduction
Follicular cysts, a prevalent ovarian disorder, contribute to infertility in mammals. These cysts emerge when a follicle fails to undergo ovulation at its expected time due to various factors, such as genetics, imbalances in sex hormones and nutrition, aging, gene dysregulation, and stress. This leads to the cyst’s enlargement and accumulation of fluid [1,2]. Among these factors, an imbalanced distribution of sex hormones resulting from disruption in the hypothalamic-pituitary-ovarian (HPO) axis is widely acknowledged as the trigger for cyst formation [3]. However, beyond endocrine disturbances, alterations in intraovarian genes and proteins exert their influence on ovarian development and folliculogenesis, potentially contributing to the formation of cysts.
Cystic ovaries display distinctive gene and protein expression profiles in contrast to dominant or other follicles within a healthy ovary [3,4]. The shifts in gene expression within ovarian cells remain intricately connected to the etiology of cystic ovarian conditions [5,6]. In cattle, granulosa cells (GCs) obtained from persistent follicles show specific gene expression patterns [6]. These alterations in gene expression profiles within ovarian cells concurrently impact follicle diameter [7,8,9]. In cattle, ovaries harboring follicles exceeding a diameter of 25 mm in diameter are classified as cystic [10]. The precise causes and mechanisms underlying cyst formation are not yet fully elucidated.
GCs and follicle diameter are intricately linked, as GCs play a crucial role in orchestrating the growth and maturation of ovarian follicle, subsequently influencing their dimensions. By secreting an array of growth factors and signaling molecules, GCs facilitate the growth and differentiation of the oocyte, while also generating substantial levels of estrogen, a fundamental element in fostering follicular development. Anomalies in GC proliferation can precipitate the formation of cysts. Dysfunction or impairment of GCs can engender infertility, irregular menstrual cycles, and an array of other reproductive disorders. In cattle afflicted with cystic ovaries, the normal trajectory of folliculogenesis remains incomplete with abnormal GCs, culminating in the emergence of persistent cystic follicles [11,12].
Ion channels affect several cellular processes encompassing cell proliferation, apoptosis, senescence, migration, and volume regulation. The intricate dance of water and ions through ion channels located at the cell membrane plays a crucial role in regulating ovarian cellular activities, including the complex processes of folliculogenesis [13]. The interplay between large molecules and ions, secreted by oocytes or GCs, precipitates an osmotic pressure gradient that draws water into the antrum [14]. In the formation of the ovarian follicular antrum, cell volume regulation emerges as a cornerstone mechanism. Follicles undergo expansion until osmolality equilibrium is achieved both inside and outside the cells. This swelling, in turn, triggers the activation of Cl- or K+ channels. Notably, K+ and Cl− channels have emerged as pivotal agents orchestrating regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) processes [15].
In contrast to Cl- channels, there is a scarcity of reports regarding the involvement of K+ channels in the formation of ovarian cysts. A few K+ channels, KATP, KCa (IK, SK, BK), Kv4.2, and two-pore domain potassium (K2P) channels are detected in the ovary [16,17]. Among K+ channels, it is the K2P channels that distinctly orchestrate osmotic water movement through their adept control of the resting membrane potential and facilitation of K+ efflux in various cells, including mouse zygotes [18]. In the mammalian ovary and female germ cells, the TWIK-related acid-sensitive K+ channels (TASKs; TASK-1, TASK-3, and TASK-5), belong to the K2P channel family, are expressed [17,19,20]. Moreover, another member of the K2P channel family, TASK-2, is linked to apoptosis through volume regulation [21]. In breast cancer cells, downregulation of TASK-3 leads to heightened cell migration and invasion [22], while also inducing cellular senescence and inhibiting growth [23]. The modulation of TASK channels is expected to exert an influence on the formation of ovarian cysts by affecting cell volume and growth.
This study was conducted to investigate the potential influence of TASK channels on the formation of cystic follicles in bovine ovaries. Ion concentration and TASK channel expression level were compared in follicular fluid (FF) and GCs obtained from small-sized (5 to 10 mm in diameter) and large-sized (> 25 mm) follicles (SF and LF) in Korean cattle.
2. Results
2.1. Low K+ Concentration in Follicular Fluid Obtained from Large-Sized Follicles
Ovarian follicles were categorized based on their diameter, with sizes ranging from 5 to 10 mm (5~10 mm) or exceeding 25 mm (> 25 mm). The smaller follicles were referred to as small-sized follicles (SF), while the larger ones were referred to as large-sized follicles (LF). Comparing the follicular fluid (FF) obtained from LF (LFFF) to that obtained from SF (SFFF), significantly higher concentrations of estrogen (17β-estradiol, E2) and progesterone (P4) were observed in LFFF. In contrast, the concentration of testosterone (Te) decreased by approximately 63% in LFFF (Figure 1A; p < 0.05). Analysis of the concentrations of K+, Na+, and Cl- in SFFF revealed values of 10.4 ± 3.5 mEq/L, 138.0 ± 11.9 mEq/L, and 104.9 ± 7.0 mEq/L, respectively. The K+ concentration was significantly lower in LFFF (6.2 ± 0.8 mEq/L, Figure 1B; p < 0.05). There were no significant differences in Na+ and Cl- concentrations between SFFF and LFFF (p > 0.05). These results suggest that the regular movement of K+ is disrupted in the LF.
2.2. Downregulation of TASK Expression Level in GCs Obtained from LF
The TASK channels, known as one of the K+ transport systems in bovine ovarian follicles, were investigated in this study. Semi-quantitative PCR data showed a significant decrease in mRNA levels of TASK-1 and TASK-3 in both LF and GCs obtained from LF (LFGC) when compared to SF and SFGC (Figure 2A). Real-time PCR data further confirmed a significant reduction in the mRNA expression levels of TASK-1 and TASK-3 in LF and LFGC, compared to SF and SFGC (Figure 2B; p < 0.05, n = 4). In addition to mRNA levels, the protein level of TASK-3 was also found to be decreased in LFGC (Figure 2C; p < 0.05, n = 5). However, no significant change was observed in the protein level of TASK-1 between SFGC and LFGC (Figure 2C; p > 0.05, n=4). Immunofluorescence analysis revealed that both the intensity and the number of TASK-3-positive cells lower in LFGC compared to SFGC (Figure 2D). The changes observed in both the mRNA and protein levels of TASK-3 channels in LFGC were subsequently subjected to further analysis through immunostaining.
2.3. Localization of TASK-3 in Ovarian GCs
Two different immunohistochemical methods, fluorescence and 3, 3’-diaminobenzidine (DAB) staining, were employed to examine the expression pattern of TASK-3 in the ovary. The ovary, excluding follicles larger than 25 mm, was subjected to hematoxylin and eosin (H&E) staining and histological examination, revealing the presence of antrum, GC, and theca interna (TI) (Figure 3A). Immunofluorescence staining showed TASK-3 expression in both GC and TI (Figure 3B), while DAB staining highlighted predominant expression of TASK-3 in GC (Figure 3C), indicating that TASK-3 is expressed in GC. As shown in Figure 3D, fluorescence-based analysis revealed higher levels of TASK-3 expression in small-sized follicles compared to large-sized follicles.
2.4. Senescence Signals Increased in LFGC
Senescence markers, such as cell volume, concentrations of calcium, reactive oxygen species (ROS), and malondialdehyde (MDA), and SA-β-galactosidase signals, were analyzed in GCs. The diameter of LFGC (22.7±1.2 µm, n = 288) was larger than that of SFGC (16.6 ± 0.7 µm, n = 257) (Figure 4A), indicating that low expression of TASK-3 may induce cellular senescence and hinder water efflux. The majority (approximately 90%) of SFGC had a size of 15.38 µm. Measurement of calcium, ROS, and MDA concentrations in GCs revealed significantly higher levels in LFGC compared to SFGC (Figure 4B to 4D; p < 0.05, n = 6). Furthermore, the number of SA-β-galactosidase-positive (SA-β-gal+) cells was significantly higher in LFGC compared to SFGC (Figure 4E). In both SFGC and LFGC groups, TASK-3 knockdown resulted in an increase in the number of SA-β-gal+ cells. In addition, LFGC exhibited elevated expression levels of p53 and p21 compared to SFGC. Knockdown of TASK-3 in both groups also resulted in heightened expression of p53 and p21 (Figure 4F; p < 0.05, n = 4). As shown in Figure 4F, while the TASK-3 protein expression level saw a decline in the TASK-3 knockdown cells, the TASK-1 protein expression remained unchanged.
2.5. Hypotonic-Induced Swelling Reduced by TASK-3 Activation
To determine the role of TASK-3 in volume regulation, CHO cells transfected with TASK-3 were exposed to a hypotonic solution possessing an osmolarity of 200 mOsm. Introducing this hypotonic solution resulted in a boost of approximately 30% in TASK-3 currents (Figure 5A and 5B, n = 4). The cell’s response to the hypotonic solution was monitored by measuring the time course of basal fluorescence intensity and F0/Ft (proportional to cell volume) in cells loaded with calcein. A decrease in fluorescence intensity corresponds to an increase in cell volume. As cells encounter a hypotonic solution, they rapidly swell, leading to a decrease in the fluorescence intensity. However, cells overexpressing TASK-3 exhibited a negligible shift in fluorescence intensity in comparison to those transfected with the control vector. The overexpression of TASK-3 significantly curtails the cell’s swelling when subjected to the hypotonic solution (Figure 5C, p < 0.05, n = 4).
2.6. Senescence Markers Increased by TASK-3 Silencing
In CHO cells where TASK-3 was silenced, live (green) and dead (red) cell staining revealed that cell viability remained unchanged compared with cells transfected with scrambled siRNA (negative control siRNA, NCsiRNA) (Figure 6A). Notably, no dead cells were identified in TASK-3-silenced cells. The number of SA-β-gal+ cells was significantly greater in TASK-3 knockdown cells than in NCsiRNA cells (Figure 6B; p < 0.05, n = 4). Cells transfected with TASK-3 siRNA showed a low TASK-3 expression and a high p53 and p21 expression compared with NCsiRNA-transfected cells (Figure 6C and 6D, p < 0.05, n = 3).
3. Discussion
Ovaries harboring follicles larger than 25 mm in diameter are categorized as cystic in cattle [10]. The follicles utilized in this study, possessing diameters surpassing 25 mm, were characterized as follicular cysts based on histological features and hormone levels (E2/P4 ratio > 1 and low testosterone). A follicular cyst, typically a fluid-filled sac found on the ovarian surface, arises when a follicle grows without rupturing or releasing its oocyte due to hormonal signal dysfunction. This study focused on gene expression changes rather than hormonal variations, which have been established in numerous studies as contributors to follicular cyst development.
In the LFFF, the lower K+ concentration ([K+]) compared to SFFF enabled us to discern alterations in the expression of K+ channels. Consistent with our findings, the [K+] in FF declined as follicle size grew, despite the compared follicle sizes being slightly different [24]. Unlike K+, our findings indicated no notable difference in Na+ and Cl- concentrations between SF and LF. If the decrease in [K+] was due to dilution from fluid accumulation, we would have expected a concurrent reduction in in Na+ and Cl- concentrations. The isolated decrease in [K+] in LFFF is believed to result from the dysregulation of K+ channels or transporters. Regulating [K+] inside and outside the cell via K+ channels and transporters in the cell membrane is crucial for preserving the resting membrane potential and various cellular functions [25]. In general, when K+ carrier proteins function properly, the extracellular [K+] ([K+]e) remains tightly controlled within a specific range (typically 3.5-5.0 mmol/L). Changes in [K+] affect cellular processes, such as cell growth, apoptosis, and maintenance of cell volume [26,27]. A relatively high [K+] is sustained in FF.
An interesting finding emerged while investigating the significance of elevated [K+] levels in FF [28]. A past study demonstrated that the [K+] in in vivo FF closely resembled that in body fluid. However, FF obtained from ovaries at slaughterhouse exhibited a notable increase in [K+] compared to that in body fluid [28]. The underlying cause of this [K+] disparity between the two groups remains unresolved, possibly resulting from blockade of K+ transport and altered protein expression in ovaries obtained from slaughterhouse. The ovaries employed in this study were sourced from slaughterhouses and displayed a relatively elevated [K+] in comparison to body fluid. Further studies are essential to discern the underlying reasons for the augmented [K+] levels in ovarian FF obtained from the slaughterhouse.
Dysfunctional K+ channels in the reproductive system hinder the HPO axis, leading to reduced fertility [29]. Ovarian [K+] and K+ channels are pivotal in E2 and P4 synthesis and secretion [16,30], influencing follicular development. GCs release hormones, with a substantial portion of GC-produced estrogen entering the FF. Estrogen receptors alpha and beta (ERα and ERβ) function as hormone-responsive transcription factors, directly regulate the expression of their target genes. TASK-3 was one of the proteins regulated by E2. Utilizing the transcription element search system (TESS) software, it was revealed that TASK-3’s promoter region encompasses three predictive estrogen response elements. E2 treatment notably upregulated TASK-3 expression in GCs (data not shown). However, in LFGCs, ERα and ERβ expression was comparatively low when contrasted with SFGCs (data not shown). While E2 increases the expression level of TASK-3 in vitro, the limited ERs expression is projected to curtail its influence. Unraveling the intricate interplay among TASK-3, ERα, and mediating molecules necessitates further investigation. A reduced ER expression in LFGCs might be linked to the decreased expression of TASK-3.
Diminished TASK-3 expression in GCs is likely to lead to the inhibition of K+ efflux, impaired RVD, depolarized membrane potential, and cellular senescence. TASK-3 channel activation in GC cells drives water expulsion, with osmotic changes due to high molecular compounds in FF enhancing water movement to the FF [14,31]. Fluid outflow from GCs can boost FF volume, triggering ion channels in GCs to balance osmotic pressure. AQP4 might collaborate with TASK-3 for water and ion transport in GCs [32]. However, given the reduced levels of AQP4 in LFGCs [33], there may be limitations in water and ion transport.
Cellular senescence is a stable and ultimate growth arrest in which cells cannot proliferate despite optimal growth conditions and mitogenic stimuli [34]. The mechanism of ovarian senescence is shared between cattle and humans. An essential component of ovarian senescence involves the disruption of antioxidant signaling, specifically affecting early-stage oocytes and GCs [35]. Human GCs exhibit a down-regulation of antioxidative genes [35], which are closely linked to the decay of ovarian functionality. The TASK-3 channel functions as one of the mitochondrial channels that participate in protecting cells from damage [36]. Senescent cells exhibit distinct traits such as cellular enlargement, increased SA-β-gal activity, upregulated cell cycle regulators (p53 and p21), and an amplified senescence-associated secretory phenotype [34,37]. LFGCs that expressed lower levels of TASK-3 demonstrated an increased cell volume and elevated SA-β-gal activity, along with higher expressions of p53 and p21, when compared to SFGCs. This suggests that reduced TASK-3 expression could play a role in triggering senescence in GCs. The markers of senescence observed in LFGCs were similarly confirmed in cells where TASK-3 was silenced. Furthermore, LFGCs exhibited elevated levels of ROS and Ca2+, both of which have been linked to cellular senescence [38,39]. These factors contribute to a harmful microenvironment for the cells.
This study has several limitations. Firstly, the ovaries used for cellular isolation and gene analysis were obtained from a slaughterhouse. Consequently, we couldn’t determine the biological, genetic, and environmental factors of the donors that could potentially influence ovarian cyst development and TASK-3 expression levels. Additionally, while we observed differences in TASK-3 mRNA and protein levels between SF and LF, only TASK-1 mRNA varied between them. Generally, protein levels remain consistent across tissues [40], and although normally determined by mRNA levels, this isn’t always the case [41]. Given the changes at both the mRNA and protein levels, TASK-3 seemed more strongly linked to ovarian cysts than TASK-1, leading us to focus on it. Nonetheless, the role of TASK-1 deserves exploration in future studies, especially since mRNA and protein expression correlation can be low due to factors like post-transcriptional regulation [40]. Moreover, immunostaining did not provide a precise representation of TASK-3 expression levels in LF and SF, primarily because of the challenges in fixing the entire cystic follicles.
4. Materials and Methods
4.1. Sample Preparation
Bovine ovaries were obtained from a slaughterhouse and delivered to the laboratory within 2 h, preserved in phosphate-buffered saline (PBS) at 35~39°C containing 100 IU/ml of penicillin and 0.1 mg/ml streptomycin. Ovarian follicles were categorized based on their size: small (5 to 10 mm in diameter) and large (> 25 mm in diameter). Ovaries that had follicles exceeding 25 mm in diameter without a corpus luteum present in either the right or left ovary were identified as follicular cystic ovaries. The large follicles were isolated from follicular cystic ovaries. Follicles were prepared by cutting the perifollicular region with a razor blade. Experimental methods used in this study were partially modified from the procedures performed in the previous studies [42,43]. The 80 ovaries with large-sized follicles (LF) and 120 ovaries with only small-sized follicles (SF) were used in this study. Follicular fluid (FF) and granulosa cells (GCs) obtained from small-sized follicles (5 to 10 mm) of 5~10 ovaries were pooled and used for the experiments as a sample.
4.2. Isolation of Follicular Fluid and Granulosa Cells
The isolation of FF and GCs was performed as previously described [33]. Briefly, FF was carefully aspirated in follicles with a 10 mL syringe fitted with an 18- or 23-gauge needle. The fluid was centrifuged at 1,750 ×g for 10 min, and the supernatant and resultant pellets were used as FF and GCs, respectively. GCs were stained with trypan blue solution (0.4%, Thermo Fisher Scientific, Rockford, IL, USA) to analyze their number and size. GCs suspended in PBS were incubated with an equal volume of trypan blue reagent for 10 min at room temperature. The cells were washed twice with PBS and then observed under a BX-51 microscope (Olympus, Tokyo, Japan).
4.3. Measurement of 17β-Estradiol (E2), Progesterone (P4), and Testosterone
Hormone concentration was measured as detailed in references [42,43]. E2, P4, and testosterone levels in the FF were ascertained using the Dissociation Enhanced Lanthanide Fluorescence Immunoassay (DELFIA) system (PerkinElmer Life and Analytical Sciences, Wallac Oy, Turku, Finland). Following the manufacturer’s guidelines, each strip was first washed with DELFIA Platewash. Subsequently, 25 µL of either E2 standards or FF samples were dispensed into the strip wells. This was followed by the addition of 100 µL of the diluted E2 antiserum solution to each well, and the arrangement was then incubated for 30 min at room temperature with a slow shake. After this incubation, a solution of diluted E2 (50:1) in an amount of 100 µL was added to each well. The plate was then incubated for 2 h at room temperature with a slow shaking. After this incubation, each strip underwent six washes before an enhancement solution of 200 µL was introduced into every well. A subsequent gentle shake of the plate for 5 min was carried out. The fluorescence levels were recorded using a time-resolved fluorometer (Wallac 1420 VICTOR2TMD, PerkinElmer) within 1 h of this final shaking procedure. Both standards and FF samples were assessed in duplicate. While evaluating P4 and testosterone, the same protocol for E2 was applied, with the exception of a dilution stage that was conducted before the antiserum addition.
4.4. Measurement of Ion Concentrations
Potassium (K+), sodium (Na+), and chloride (Cl-) ion concentrations in the FF were determined with an automatic analyzer (DRI-CHEM 3500i, FUJIFILM, Tokyo, Japan) in line with the manufacturer’s guidelines. For quantitative analysis of the electrolytes in the FF, the potentiometric difference was assessed between two electrodes; one electrode was in contact with a reference liquid holding a known electrolyte concentration, while the other interfaced with the samples.
4.5. Reverse Transcriptase (RT)-Polymerase Chain Reaction (PCR) and Real-Time PCR
RT-PCR procedures were performed as previously described [42,43] utilizing specific primer pairs, detailed in Table 1. Bovine follicles and GCs were the source of the total RNA, which was isolated using the Trizol reagent (Invitrogen, CA, USA) following the manufacturer’s guidelines. The resultant total RNA (3 μg) underwent first-strand cDNA synthesis using oligo dT (SuperScript First-Strand Synthesis System, Invitrogen), which then served as a template for PCR amplification employing Taq polymerase (Takara Bio Inc, Otsu, Shiga, Japan). The synthesized first-strand cDNA’s concentration was determined with a spectrophotometer (NanoDrop® ND-1000, NanoDrop Technologies, Wilmington, DE, USA) and was subsequently employed as the PCR amplification template. The amplification process consisted of an initial denaturation at 94°C for 5 min, followed by 28 or 30 cycles at 94°C for 20 sec, 55°C for 20 sec, 72°C for 20 sec, and a 10 min extension at 72°C. Amplified products were run on 1.5% agarose gels with ethidium bromide. Resultant bands were extracted and underwent direct sequencing using an ABI PRISM® 3100-Avant Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
For real-time PCR, a combination of the ToprealTM qPCR 2X PreMIX kit (Enzynomics, Daejeon, South Korea), sapphire microplate 96-well, (Greiner bio-one, Kremsmünster, Austria), and the Light Cycler® 480 II System (Roche, Rotkreuz, Switzerland) was utilized. The PCR parameters included a denaturing cycle (95 °C for 10 min), 45 cycles of PCR (95 °C for 7 s, 56 °C for 7 s, and 72 °C for 10 s), a melting cycle (95 °C for 0 s and 65 °C for 60 s). Relative mRNA levels were determined using the 2-ΔΔCT method [44]. In both RT-PCR and real-time PCR analyses, target gene expression was standardized against the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level.
4.6. Western Blot Analysis
Western blotting was conducted based on established protocols [42,43]. GCs underwent homogenization in RIPA lysis buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with 20 mM Tris-HCl (pH 7.5), 150 mM NaCl/1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin). After incubation at 4°C for 30 min, the samples were centrifuged at 13,000 rpm (16,609 × g, Micro 17TR, Hanil, Korea) for 30 min at 4°C. The protein content of the supernatants was quantified by the Bradford protein assay (Bio-Rad, Hercules, CA, USA). The proteins (50-100 μg/lane) were then subjected to 10% SDS-polyacrylamide gels, followed by transfer to polyvinylidene fluoride (PVDF) membranes (0.45 μm, Millipore, Bedford, MA, USA) in TBS buffer solution containing 25 mM Tris-base, 190 mM glycine, and 20% methanol. Ponceau S staining verified effective transfer, and the destained blots were subsequently blocked. Primary antibodies specific to TASK-1 (1:1000 dilution, Alomone Labs, Jerusalem, Israel), TASK-3 (1:1000 dilution, Sigma, St Louis, MO, USA), p53 (1:200 dilution, Santa Cruz Biotechnology, Inc, Dallas, TX, USA), p21 (1:200 dilution, Santa Cruz Biotechnology), α-tubulin (1:5000 dilution, Sigma), and β-actin (1:5000 dilution, Sigma) were applied and left overnight at 4°C. Afterward, blots were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3000; Assay Designs, Ann Arbor, MI, USA) at room temperature for 1 h, and protein bands were visualized using the enhanced chemiluminescence (ECL Plus kit; ELPIS, Daejeon, Korea).
4.7. Hematoxylin-Eosin (H&E) Staining
H&E staining of ovaries was carried out based on the methods detailed in previous studies [42,43]. After a PBS (0.1 M, pH 7.0) wash, the ovaries were fixed in a 4% (w/v) paraformaldehyde solution, leading to the creation of 4 μm thick paraffin-embedded sections. For histological assessment of follicles, H&E staining was applied. The sections were laid on gelatin-coated slides and allowed to air dry before undergoing deparaffinization. After rinsing with tap water, they were submerged in hematoxylin for 5 min and then validated for thorough staining. A 3-min eosin stain followed. Thereafter, the sections were progressively dehydrated using a graded series of alcohols (70% to 100% ethanol, 3 min each), cleared in xylene, and finally mounted. Images of the stained sections were captured with a BX-51 microscope (Olympus, Tokyo, Japan) equipped with a high-resolution Camedia C-7070 camera (Olympus). For each sample, five sections underwent evaluation.
4.8. Immunostaining
Deparaffinized tissue sections were first washed in PBS, treated with 0.3% H2O2 for 30 min, and then rinsed again in PBS. To reduce non-specific IgG binding, sections were blocked using 1.5% normal goat serum in PBS at room temperature for 30 min. The sections were incubated overnight at 4°C in a humidified chamber with anti-TASK-3 primary antibody (1:200 dilution, Sigma). Following another PBS wash, sections were treated either with cyanine 3 (cy3) anti-rabbit secondary antibody (1:400 dilution, Abcam, Cambridge, UK), Alexa Fluor 488 anti-rabbit secondary antibody (1:400 dilution, Invitrogen), or biotin-conjugated secondary antibody (1:200 dilution, Sigma) diluted in 1.5% normal blocking serum at room temperature for 1 h, followed by three PBS washes. For visualization, immunofluorescence staining was counterstained with 4’,6-diamidino-2-phenylindole (DAPI). For the 3,3′-diaminobenzidine tetrahydrochloride (DAB) staining, sections exposed to biotin-conjugated secondary antibody were further treated with an avidin-biotin-peroxidase complex (ABC Elite kit; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. These samples were then washed with PBS and stained using DAB solution (Sigma) containing 0.03% H2O2 for 3 min. Hematoxylin was used for counterstaining. Images were captured using a confocal laser scanning microscope (Olympus) for fluorescence and a BX-51 microscope (Olympus) for DAB images.
For immunocytochemistry, the isolated GCs were cultured on a cover glass for 24 hours. The cells were permeabilized with 0.2% Triton X-100 for 10 min at room temperature and then washed three times in PBS. Blocking was performed using a buffer containing 2% normal goat serum in 0.1 M PBS for 1 h at room temperature. Cells were then incubated overnight at 4°C with rabbit polyclonal anti-TASK-3 primary antibody (1:100 dilution, Alomone Labs), followed by three additional PBS washes. Cells were next treated with Cy3-conjugated anti-rabbit IgG secondary antibody (1:100 dilution; Invitrogen) for 1 h in the dark, followed by another three washes. Nuclear staining was carried out using DAPI at a concentration of 0.1 μg/mL (Sigma). Finally, cells were wet-mounted using Permount mounting medium (Fisher Chemical, Geel, Belgium) and observed under an Olympus confocal laser scanning microscope.
4.9. Measurement of Free Radical Activity and Calcium and Malondialdehyde (MDA) Concentrations in CGs
The free radical activity, as well as calcium and MDA concentrations, were determined following the methodologies described in a previous study [45]. To evaluate free radical activity in cell lysates, the OxiselectTM In Vitro ROS/RNS assay kit (Cell Biolabs, San Diego, CA, USA) was utilized. The Calcium Detection Assay kit (Abcam) was used for measuring calcium concentration. MDA concentration was gauged using the OxiSelect™ TBARS assay kit (STA-330; Cell Biolabs).
4.10. Cellular Senescence Assay
Cellular senescence was evaluated using methods previously described [46] and following the manufacturer’s instruction (BioVision Inc, CA, USA). Briefly, bovine GCs at a density of 2 × 104 cells/mL and Chinese hamster ovary (CHO) cells at a density of 5 × 104 cells/mL were plated in a 24-well plate and incubated for 24 h at 37°C and 5% CO2. Subsequent to this incubation, the cells were rinsed twice with PBS and fixed with a fixative solution for 15 min at room temperature. After fixation, the cells were washed thrice with PBS. A staining solution mixture (comprising 470 µL of staining solution, 5 µL of staining supplement, and 25 µL of 20 mg/mL X-gal in DMF) was then added to each well, and the plate was incubated for an additional 48 h at 37°C. Images were captured from five distinct areas per dish using a microscope (Axiovert 40C, Zeiss, Jena, Germany), and cells were counted to determine the average stained cell number. The percentage of SA-β-gal-positive cells in each sample was calculated by taking the ratio of SA-β-gal-positive cells to the total cell count and then multiplying the result by 100.
4.11. Live/dead Cell Staining
Cell viability was assessed using calcein-AM (Thermo Fisher Scientific, Eugene, OR, USA) and propidium iodide (PI, Sigma) based on previously outlined methods [46]. Viable cells were stained green with calcein-AM, while dead cells with membrane damage were stained red using PI. CHO cells, post-transfection with siRNA at a density of 5 × 103 cells/100 μL, were cultured on a glass-bottomed culture dish (SPL, Pocheon, Korea) for 48 h. Post-culturing, cells were rinsed twice with free DMEM and subsequently stained with 2 μM calcein-AM and 3 μg/mL PI for 10 min at 37°C. After staining, cells were washed thrice and visualized using a confocal laser scanning microscope (Olympus) with filter sets tailored for fluorescein isothiocyanate (FITC) and Texas Red.
4.12. Recording of Whole-Cell Current
Whole-cell currents measurement were taken using an Axopatch 200 amplifier (Axon Instruments, Union City, CA, USA). The membrane potential was held at -80 mV, followed by a 1-sec depolarizing voltage pulse, varying between -120 and +60 mV. The patch pipettes exhibited a resistance ranging from 4 to 5 MΩ when filled with the pipette solution. The pipette solution comprised (in mM) 150 KCl, 1 MgCl2, 5 EGTA, and 10 HEPES. The bath solution contained (in mM) 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES. To achieve a pH of 7.3, adjustments were made using either HCl or NaOH (KOH). After the capacitative transients were cancelled, whole-cell currents were measured. The pCLAMP software (Version 8, Axon) was utilized for whole-cell current analysis. Recordings were conducted under ambient conditions.
4.13. Measurement of Cell Volume
Changes in the volume of individual cells were determined by monitoring shifts in the concentration of a trapped fluorescent dye inside the cell, as documented in prior research [47]. CHO cells, which had been transfected with DNA that encodes for rat TASK-3 (GenBank ID, AF192366) within the pcDNA3.1 vector, were cultured on 25-mm round coverslips. These cells were then treated with calcein-AM at a concentration of 5 μM for 5 min. Before initiating the experiment, they were exposed to an iso-osmotic solution for 30 min. All the experimental observations were made using a confocal laser imaging device (Olympus). The source of light for excitation was set at 488 nm, and emitted light was detected at wavelengths exceeding 515 nm. The results are depicted using the F0/Ft notation. In this context, F and t denotes fluorescence intensity and time, respectively. The value F0 is indicative of the fluorescence intensity when the cell is in an iso-osmotic solution and the time is set to zero. The ratio F0/Ft serves as an indicator of the cell volume.
4.14. Gene Silencing with Small Interfering RNA
Gene silencing assay was conducted according to established protocols [22]. CHO cells were transfected using either a scrambled siRNA as a negative control (NC, ON-TARGET Non-targeting Pool; Dharmacon, Lafayette, CO, USA) or TASK-3-specific ON-TARGETplus SMARTpool siRNA (Dharmacon). Transfections were performed in serum-free medium using the Magnetofection™ system (Chemcell GmbH, Berlin, Germany), in accordance with the manufacturer’s guidelines. For the transfection mixture, 75 nM of either NC siRNA and rat TASK-3 siRNA was combined with 1.0 μL of PolyMAG (Chemicell GmbH, Berlin, Germany). The mixture was incubated for 20 min at room temperature before being added to 500 μL of serum-free culture medium in individual wells of a 24-well plate. The plate was then placed on a MagnetoFACTOR plate 24 device and incubated for 30 min at room temperature. Following this, the culture medium was replaced with fresh serum-containing medium, and the cells were incubated for an additional 6 h at 37°C in a 95% air and 5% CO2. Subsequently, the medium was refreshed, and the cells were allowed to grow for an additional two days.
4.15. Data Analysis and Statistics
Images of agarose gels and Western blots were captured using a LAS-4000 luminescent image analyzer (Fujifilm Corp, Tokyo, Japan). Band intensities from gels and blotting membrane were quantified using Sigma Gel image analysis software (version 1.0, Jandel Scientific, CA, USA) or Quantity One software (version 4.6.3), which is compatible with a GS-800 calibrated densitometer (Bio-Rad, CA, USA). Data are presented as mean ± SD. Statistical significance was determined using Student’s t-test, with a p < 0.05 considered significant. All statistical analyses were conducted using OriginPro2020 software (OriginLab Corp., Northampton, MA, USA).
5. Conclusions
To our knowledge, this is the first study indicating a reduced TASK-3 channel gene in GCs from bovine follicular cystic ovaries. GCs with diminished TASK-3 expression displayed cellular senescence, a factor contributing to cyst formation. This decreased TASK-3 expression disrupts water and K+ movement in GCs. TASK-3 may be key to understanding bovine follicular cyst mechanisms. Our findings offer insight into follicular cyst pathophysiology and contribute to building a genetic understanding of its onset. Furthermore, these results may help understand human ovarian cysts because the female bovine reproductive tract shares many similarities with the human reproductive tract [48].
Author Contributions
Conceptualization, D.K. and C.-W.K.; methodology, E.-J.K., M.S.W., J.H.R. and D.K.; software, E.-J.K., M.S.W. and D.K.; validation, E.-J.K. and D.K.; formal analysis, E.-J.K., M.S.W., D.L.C., A.C.C, J.H.R. and D.K.; investigation, E.-J.K., C.-W.K. and D.K.; resources, I.-K.K., data curation, D.K.; writing—original draft preparation, C.W.K. and D.K.; writing—review and editing, D.K.; visualization, E.-J.K. and D.K.; supervision, D.K.L., S.-G.H., J.H. and D.K.; project administration, E.-J.K.; funding acquisition, C.W.K. and D.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (#2021R1I1A3044128 and #RS-2023-00219399).
Institutional Review Board Statement
The Korean legislature makes an exception to the ethical approval of experiments with bovine ovaries obtained from slaughterhouses. All experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee of Gyeongsang National University. This work was developed from Chang Woon Kim’s thesis for the degree of Ph.D. (Alteration of the gene expression patterns in follicular cyst of bovine ovary, 2016, Department of Medicine, Graduate School, Gyeongsang National University).
Informed Consent Statement
Not applicable
Data Availability Statement
The data related to this study are not currently stored in a publicly accessible repository. However, the authors are willing to provide the data upon reasonable request. Requests for access to the data can be directed to Prof. Dawon Kang (dawon@gnu.ac.kr).
Acknowledgments
We thank Eun-Sook Kim, a technician in our lab in 2006. She provided the results of the RT-PCR assay. We cannot contact her, so we acknowledge her in this section. We also thank Dr. Changyong Choe (cychi@korea.kr), who is in the National Institute of Animal Science, RDA, for providing bovine ovaries, the primary sample for the experiment.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analysis, or interpretation of the data; in the writing of the manuscript; or in the decision to publish the results.
References
- Peter, A.T. An update on cystic ovarian degeneration in cattle. Reprod Domest Anim 2004, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.A.; Garverick, H.A.; Keisler, D.H.; Xu, Z.Z.; Loos, K.; Youngquist, R.S.; Salfen, B.E. Characterization of ovarian follicular cysts and associated endocrine profiles in dairy cows. Biol Reprod 1995, 53, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.H.; Marelli, B.E.; Rey, F.; Amweg, A.N.; Diaz, P.U.; Stangaferro, M.L.; Salvetti, N.R. Molecular aspects of bovine cystic ovarian disease pathogenesis. Reproduction 2015, 149, R251–264. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, N.R.; Alfaro, N.S.; Velazquez, M.M.; Amweg, A.N.; Matiller, V.; Diaz, P.U.; Ortega, H.H. Alteration in localization of steroid hormone receptors and coregulatory proteins in follicles from cows with induced ovarian follicular cysts. Reproduction 2012, 144, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.M.; Colombero, M.; Amweg, A.N.; Huber, E.; Gareis, N.C.; Salvetti, N.R.; Ortega, H.H.; Rey, F. Involvement of PAPP-A and IGFR1 in Cystic Ovarian Disease in Cattle. Reprod Domest Anim 2015, 50, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Lingenfelter, B.M.; Dailey, R.A.; Inskeep, E.K.; Vernon, M.W.; Poole, D.H.; Rhinehart, J.D.; Yao, J. Microarray analysis of gene expression in granulosal cells from persistent follicles in cattle. Anim Reprod Sci 2008, 104, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Perks, C.M.; Denning-Kendall, P.A.; Gilmour, R.S.; Wathes, D.C. Localization of messenger ribonucleic acids for insulin-like growth factor I (IGF-I), IGF-II, and the type 1 IGF receptor in the ovine ovary throughout the estrous cycle. Endocrinology 1995, 136, 5266–5273. [Google Scholar] [CrossRef]
- Hastie, P.M.; Haresign, W. Modulating peripheral gonadotrophin levels affects follicular expression of mRNAs encoding insulin-like growth factors and receptors in sheep. Anim Reprod Sci 2008, 109, 110–123. [Google Scholar] [CrossRef]
- Beg, M.A.; Bergfelt, D.R.; Kot, K.; Wiltbank, M.C.; Ginther, O.J. Follicular-fluid factors and granulosa-cell gene expression associated with follicle deviation in cattle. Biol Reprod 2001, 64, 432–441. [Google Scholar] [CrossRef]
- Lapp, R.; Rottgen, V.; Viergutz, T.; Weitzel, J.M.; Vernunft, A. Induction of cystic ovarian follicles (COFs) in cattle by using an intrafollicular injection of indomethacin. J Reprod Dev 2020, 66, 181–188. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Yu, D.; Chen, J.; Xing, C.; Li, J.; Li, J.; Cai, Y. The effects of mouse ovarian granulosa cell function and related gene expression by suppressing BMP/Smad signaling pathway. Animal cells and systems 2018, 22, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Dentis, J.L.; Schreiber, N.B.; Burress, A.M.; Spicer, L.J. Effects of angiogenin on granulosa and theca cell function in cattle. Animal: an international journal of animal bioscience 2017, 11, 811–819. [Google Scholar] [CrossRef] [PubMed]
- McConnell, N.A.; Yunus, R.S.; Gross, S.A.; Bost, K.L.; Clemens, M.G.; Hughes, F.M. Water permeability of an ovarian antral follicle is predominantly transcellular and mediated by aquaporins. Endocrinology 2002, 143, 2905–2912. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.J.; Irving-Rodgers, H.F. Formation of the ovarian follicular antrum and follicular fluid. Biol Reprod 2010, 82, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Maeno, E.; Shimizu, T.; Dezaki, K.; Wang, J.; Morishima, S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 2001, 532, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Song, K.S.; Xu, B.; Wang, T. Role of potassium channels in female reproductive system. Obstetrics & gynecology science 2020, 63, 565–576. [Google Scholar] [CrossRef]
- Hur, C.G.; Choe, C.; Kim, G.T.; Cho, S.K.; Park, J.Y.; Hong, S.G.; Han, J.; Kang, D. Expression and localization of two-pore domain K(+) channels in bovine germ cells. Reproduction 2009, 137, 237–244. [Google Scholar] [CrossRef]
- Trimarchi, J.R.; Liu, L.; Smith, P.J.; Keefe, D.L. Apoptosis recruits two-pore domain potassium channels used for homeostatic volume regulation. Am J Physiol Cell Physiol 2002, 282, C588–594. [Google Scholar] [CrossRef]
- Bai, X.; Lacey, H.A.; Greenwood, S.L.; Baker, P.N.; Turner, M.A.; Sibley, C.P.; Fyfe, G.K. TASK channel expression in human placenta and cytotrophoblast cells. J Soc Gynecol Investig 2006, 13, 30–39. [Google Scholar] [CrossRef]
- Hur, C.G.; Kim, E.J.; Cho, S.K.; Cho, Y.W.; Yoon, S.Y.; Tak, H.M.; Kim, C.W.; Choe, C.; Han, J.; Kang, D. K+ efflux through two-pore domain K+ channels is required for mouse embryonic development. Reproduction 2012. [Google Scholar] [CrossRef]
- Niemeyer, M.I.; Cid, L.P.; Barros, L.F.; Sepulveda, F.V. Modulation of the two-pore domain acid-sensitive K+ channel TASK-2 (KCNK5) by changes in cell volume. J Biol Chem 2001, 276, 43166–43174. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.W.; Park, H.S.; Kim, E.J.; Cho, Y.W.; Kim, G.T.; Mun, Y.J.; Choi, E.J.; Lee, J.S.; Han, J.; Kang, D. Reduction of breast cancer cell migration via up-regulation of TASK-3 two-pore domain K+ channel. Acta Physiol (Oxf) 2012, 204, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, R.; Valenzuela, C.; Concha, G.; Brown, N.; Zuniga, L. TASK-3 Downregulation Triggers Cellular Senescence and Growth Inhibition in Breast Cancer Cell Lines. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Galeska, E.; Wrzecinska, M.; Kowalczyk, A.; Araujo, J.P. Reproductive Consequences of Electrolyte Disturbances in Domestic Animals. Biology 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Gumz, M.L.; Rabinowitz, L.; Wingo, C.S. An Integrated View of Potassium Homeostasis. N Engl J Med 2015, 373, 1787–1788. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Foller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J Membr Biol 2005, 205, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Nielsen, B. Comparative physiology of cellular ion and volume regulation. J Exp Zool 1975, 194, 207–219. [Google Scholar] [CrossRef]
- Knudsen, J.F.; Litkowski, L.J.; Wilson, T.L.; Guthrie, H.D.; Batta, S.K. follicular fluid electrolytes and osmolality in cyclic pigs. J Reprod Fertil 1979, 57, 419–422. [Google Scholar] [CrossRef]
- Fatima, S.S.; Rehman, R.; Martins, R.S.; Alam, F.; Ashraf, M. Single nucleotide polymorphisms in Renalase and KCNQ1 genes and female infertility: A cross-sectional study in Pakistan. Andrologia 2019, 51, e13434. [Google Scholar] [CrossRef]
- Liu, J.; Li, Q.; Ren, J.; Liang, X.; Zhang, Q.; Han, Y. Association of Sex with Serum Potassium, Sodium, and Calcium Disorders after Hypertensive Intracerebral Hemorrhage. World neurosurgery 2020, 141, e367–e373. [Google Scholar] [CrossRef]
- Clarke, H.G.; Hope, S.A.; Byers, S.; Rodgers, R.J. Formation of ovarian follicular fluid may be due to the osmotic potential of large glycosaminoglycans and proteoglycans. Reproduction 2006, 132, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.L.; Ding, J.H.; Fan, Y.; Zhang, J.; Gao, L.; Hu, G. Aquaporin 4 regulates the effects of ovarian hormones on monoamine neurotransmission. Biochem Biophys Res Commun 2007, 353, 457–462. [Google Scholar] [CrossRef]
- Kim, C.-W.; Choi, E.-J.; Kim, E.-J.; Siregar, A.S.; Han, J.; Kang, D. Aquaporin 4 expression is downregulated in large bovine ovarian follicles. Journal of Animal Reproduction and Biotechnology (KSARB) 2020, 35, 315–322. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Tesarik, J.; Galan-Lazaro, M.; Mendoza-Tesarik, R. Ovarian Aging: Molecular Mechanisms and Medical Management. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Jarmuszkiewicz, W.; Kunz, W.S. Mitochondrial potassium channels. IUBMB Life 2009, 61, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Sakemura, R.; Kumagai, A.; Sumikawa, E.; Fujii, M.; Ayusawa, D. Nuclear swelling occurs during premature senescence mediated by MAP kinases in normal human fibroblasts. Bioscience, biotechnology, and biochemistry 2008, 72, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca(2+) in aging and age-related diseases. Redox biology 2020, 36, 101678. [Google Scholar] [CrossRef]
- Belrose, J.C.; Xie, Y.F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Molecular brain 2012, 5, 11. [Google Scholar] [CrossRef]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC genomics 2017, 18, 305. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Choe, C.; Cho, Y.W.; Kim, C.W.; Son, D.S.; Han, J.; Kang, D. Identification of differentially expressed genes in bovine follicular cystic ovaries. Korean J Physiol Pharmacol 2010, 14, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Siregar, A.S.; Nyiramana, M.M.; Kim, E.-J.; Shin, E.-J.; Kim, C.-W.; Lee, D.; Hong, S.-G.; Han, J.; Kang, D. TRPV1 Is Associated with Testicular Apoptosis in Mice. J Anim Reprod Biotechnol 2019, 34, 7. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Woo, M.S.; Cao, D.L.; Kim, E.J.; Jeong, Y.Y.; Koh, E.H.; Cho, K.M.; Kang, S.S.; Kang, D. Fermented and Aged Ginseng Sprouts (Panax ginseng) and Their Main Component, Compound K, Alleviate Asthma Parameters in a Mouse Model of Allergic Asthma through Suppression of Inflammation, Apoptosis, ER Stress, and Ferroptosis. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Nyiramana, M.M.; Cho, S.B.; Kim, E.J.; Kim, M.J.; Ryu, J.H.; Nam, H.J.; Kim, N.G.; Park, S.H.; Choi, Y.J.; Kang, S.S.; et al. Sea Hare Hydrolysate-Induced Reduction of Human Non-Small Cell Lung Cancer Cell Growth through Regulation of Macrophage Polarization and Non-Apoptotic Regulated Cell Death Pathways. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, M.I.; Cid, L.P.; Sepulveda, F.V. K+ conductance activated during regulatory volume decrease. The channels in Ehrlich cells and their possible molecular counterpart. Comp Biochem Physiol A Mol Integr Physiol 2001, 130, 565–575. [Google Scholar] [CrossRef]
- Way, A.L. Isolation and culture of bovine oviductal epithelial cells for use in the anatomy and physiology laboratory and undergraduate research. Adv Physiol Educ 2006, 30, 237–241. [Google Scholar] [CrossRef]
Figure 1.
Hormone and ion concentrations in bovine ovarian follicular fluid obtained from small-sized and large-sized follicles. (A) The concentrations of 17β-estradiol (E2), progesterone (P4), and testosterone (Te). (B) The concentrations of K+, Na+, and Cl-. Each bar represents the mean ± SD of five different samples. *Significant difference from the corresponding control value (p < 0.05). SFFF and LFFF denote follicular fluid obtained from small-sized (5 to 10 mm) and large-sized (> 25 mm) follicles, respectively.
Figure 1.
Hormone and ion concentrations in bovine ovarian follicular fluid obtained from small-sized and large-sized follicles. (A) The concentrations of 17β-estradiol (E2), progesterone (P4), and testosterone (Te). (B) The concentrations of K+, Na+, and Cl-. Each bar represents the mean ± SD of five different samples. *Significant difference from the corresponding control value (p < 0.05). SFFF and LFFF denote follicular fluid obtained from small-sized (5 to 10 mm) and large-sized (> 25 mm) follicles, respectively.
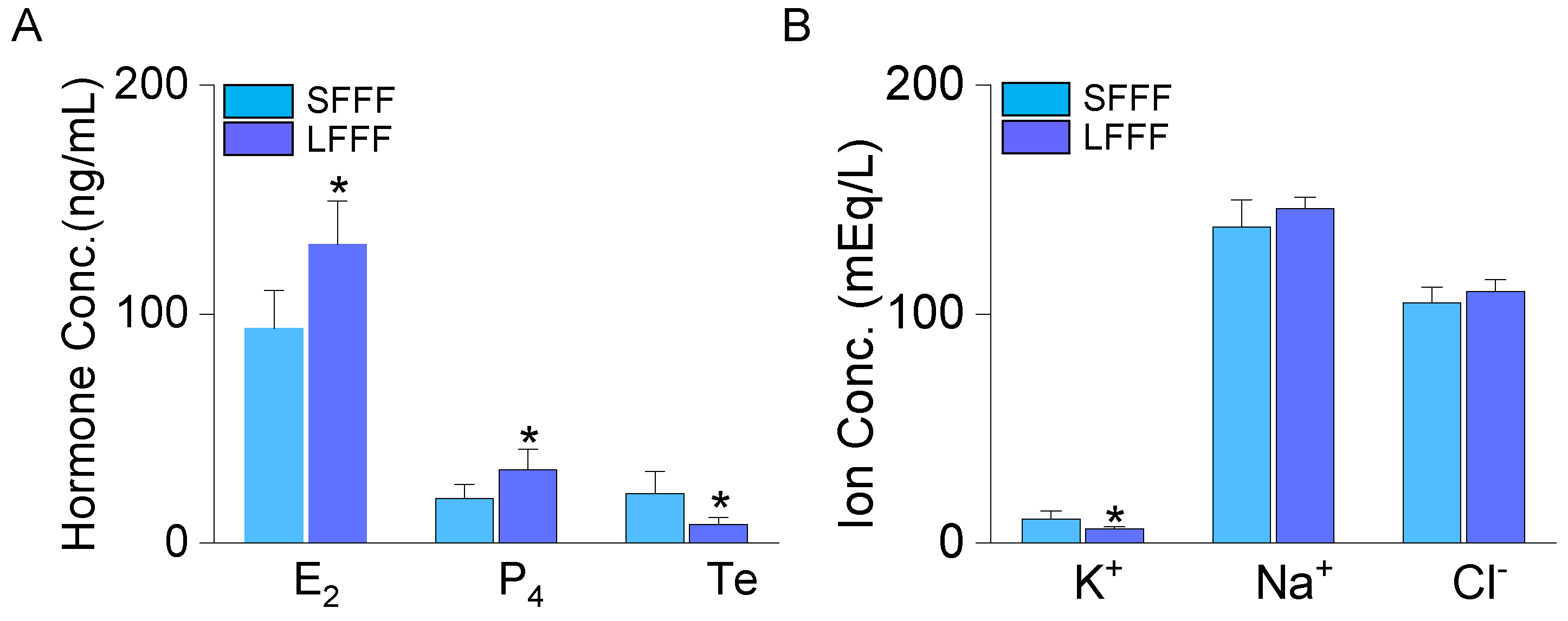
Figure 2.
Down-regulation of TASK-3 expression level in the bovine ovarian follicles and granulosa cells. (A) TASK-1 and TASK-3 mRNA expression in follicles (SF and LF) and GCs (SFGC and LFGC). TASK-1 (287 bp) and TASK-3 (433 bp). (B) Real-time PCR data for TASK-1 and TASK-3 in follicles and GCs. The expression levels were normalized to GAPDH. (C) Western blot analysis for TASK-1 and TASK-3 in SFGC and LFGC. The protein expression levels were normalized to α-tubulin. (D) TASK-3 immunofluorescence intensity in GCs. The isolated GCs were cultured on a glass coverslip and subjected to immunostaining to detect the expression of TASK-3 within the cells. TASK-3 was immunostained with an anti-TASK-3 antibody and cyanine (Cy3)-conjugated anti-rabbit IgG (red). The 4’,6’-diamidino-2-phenylindole (DAPI) was used for nuclear staining (blue). Each bar represents the mean ± SD of four different samples (see results). *Significant difference from the corresponding control value (p < 0.05). Scale bar, 50 μm.
Figure 2.
Down-regulation of TASK-3 expression level in the bovine ovarian follicles and granulosa cells. (A) TASK-1 and TASK-3 mRNA expression in follicles (SF and LF) and GCs (SFGC and LFGC). TASK-1 (287 bp) and TASK-3 (433 bp). (B) Real-time PCR data for TASK-1 and TASK-3 in follicles and GCs. The expression levels were normalized to GAPDH. (C) Western blot analysis for TASK-1 and TASK-3 in SFGC and LFGC. The protein expression levels were normalized to α-tubulin. (D) TASK-3 immunofluorescence intensity in GCs. The isolated GCs were cultured on a glass coverslip and subjected to immunostaining to detect the expression of TASK-3 within the cells. TASK-3 was immunostained with an anti-TASK-3 antibody and cyanine (Cy3)-conjugated anti-rabbit IgG (red). The 4’,6’-diamidino-2-phenylindole (DAPI) was used for nuclear staining (blue). Each bar represents the mean ± SD of four different samples (see results). *Significant difference from the corresponding control value (p < 0.05). Scale bar, 50 μm.
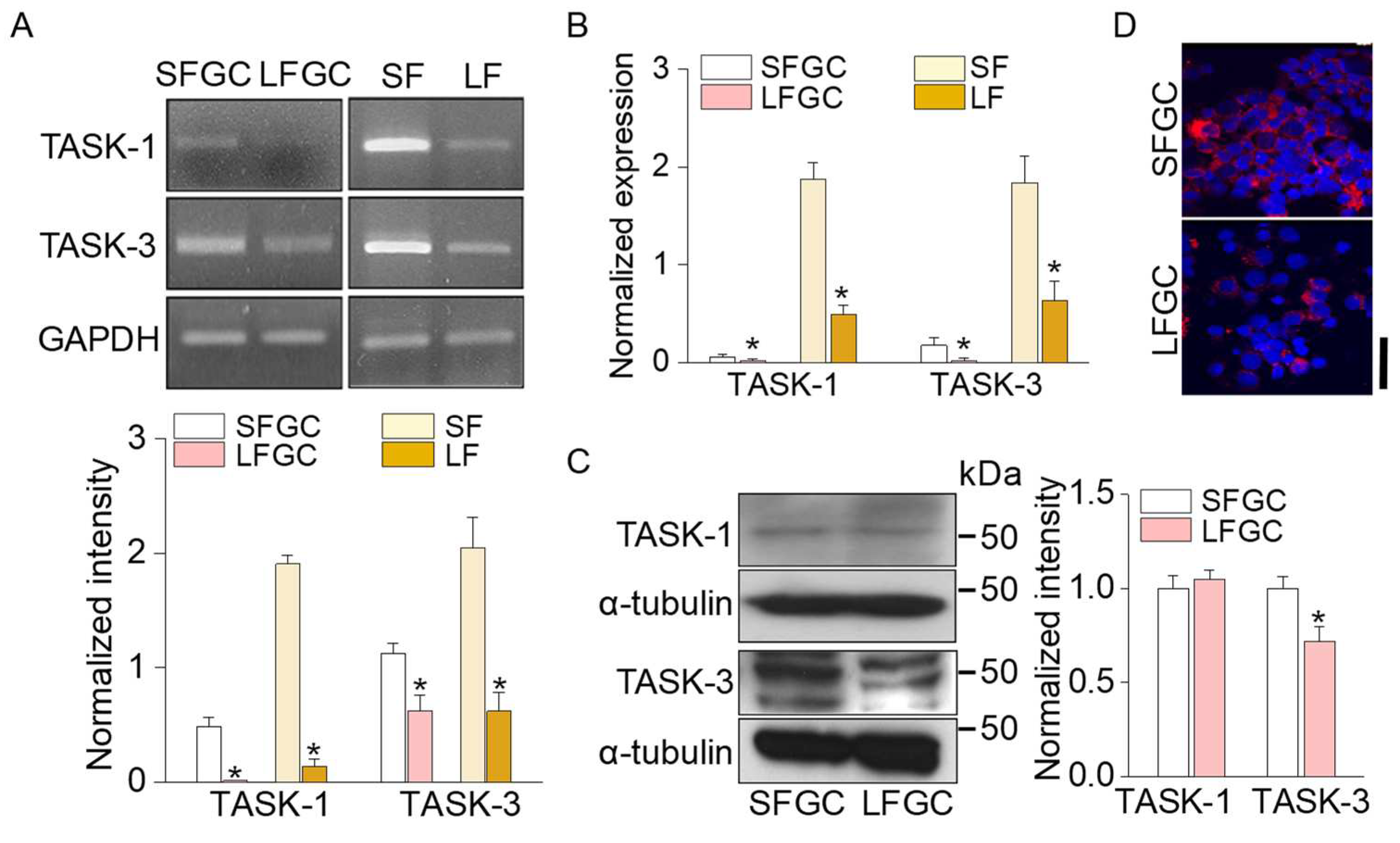
Figure 3.
Expression of TASK-3 in granulosa cells. (A) H&E staining of a bovine ovary tissue section. GC and TI represent granulosa cells and theca interna, respectively. (B) Expression of TASK-3 in both GC and TI. Fluorescent images labeled with anti-TASK-3 antibody and cyanine (Cy3)-conjugated anti-rabbit IgG (orange/red). 4’,6’-diamidino-2-phenylindole (DAPI) for nuclear staining (blue). NC represents negative control omitting anti-TASK-3 antibody. (C) TASK-3 expression in GC detected by DAB staining. (D) The immunofluorescence intensity of TASK-3 detected in follicles of different sizes. Green and blue indicate TASK-3 expression and nucleus stained with Alexa Fluor 488 and DAPI, respectively. The center and right panels display magnified views of the left image (indicated by the same colored boxes). The left and center images exhibit H&E staining, while the right side features the TASK-3 fluorescence image. Scale bars, 50 μm.
Figure 3.
Expression of TASK-3 in granulosa cells. (A) H&E staining of a bovine ovary tissue section. GC and TI represent granulosa cells and theca interna, respectively. (B) Expression of TASK-3 in both GC and TI. Fluorescent images labeled with anti-TASK-3 antibody and cyanine (Cy3)-conjugated anti-rabbit IgG (orange/red). 4’,6’-diamidino-2-phenylindole (DAPI) for nuclear staining (blue). NC represents negative control omitting anti-TASK-3 antibody. (C) TASK-3 expression in GC detected by DAB staining. (D) The immunofluorescence intensity of TASK-3 detected in follicles of different sizes. Green and blue indicate TASK-3 expression and nucleus stained with Alexa Fluor 488 and DAPI, respectively. The center and right panels display magnified views of the left image (indicated by the same colored boxes). The left and center images exhibit H&E staining, while the right side features the TASK-3 fluorescence image. Scale bars, 50 μm.
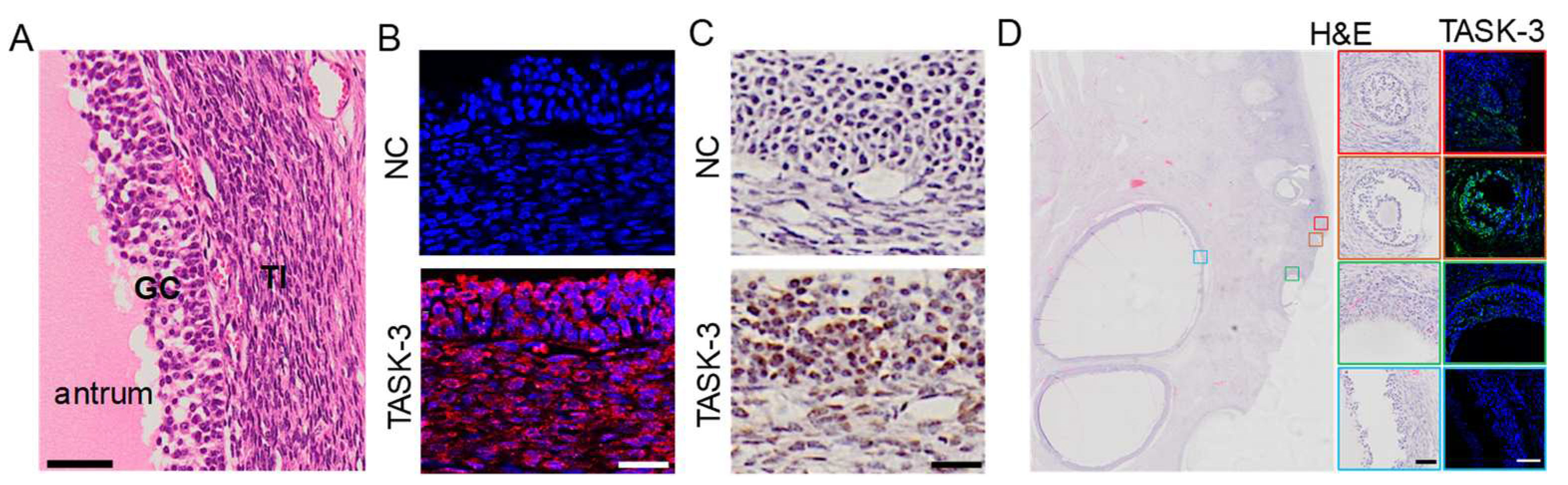
Figure 4.
Upregulation of senescence signals in LFGC. (A) Trypan blue-stained cells. The size of the cells was measured with a software ruler. (B) Calcium concentration in GCs. (C) ROS levels in GCs. RFU represents a relative fluorescence unit. (D) MDA concentration in GCs. The label referenced in D was also common to B and C. (E) Representative images stained with SA-β-gal. Images depict cells stained with SA-β-gal, which appear in a dark cyan color when stained. (F) Detection of p53 and p21 proteins. The expression levels were normalized to β-actin. Each bar represents the mean ± SD of four to six samples (see results). *Significant difference from the corresponding control value (p < 0.05). Scale bars, 100 μm.
Figure 4.
Upregulation of senescence signals in LFGC. (A) Trypan blue-stained cells. The size of the cells was measured with a software ruler. (B) Calcium concentration in GCs. (C) ROS levels in GCs. RFU represents a relative fluorescence unit. (D) MDA concentration in GCs. The label referenced in D was also common to B and C. (E) Representative images stained with SA-β-gal. Images depict cells stained with SA-β-gal, which appear in a dark cyan color when stained. (F) Detection of p53 and p21 proteins. The expression levels were normalized to β-actin. Each bar represents the mean ± SD of four to six samples (see results). *Significant difference from the corresponding control value (p < 0.05). Scale bars, 100 μm.
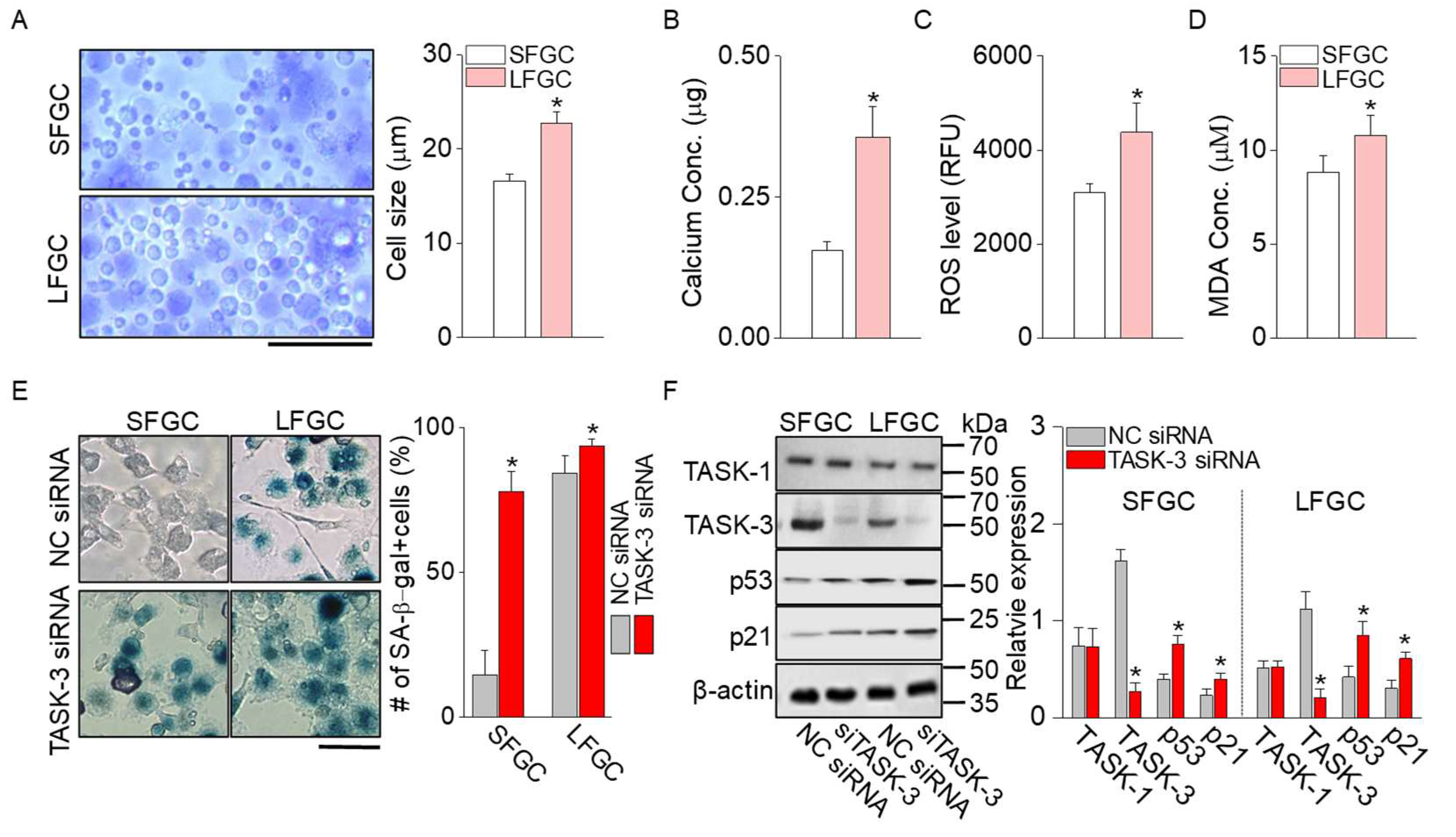
Figure 5.
Volume regulation by TASK-3 channel. (A) Activation of the TASK-3 channel by hypotonic solution. The whole-cell current of TASK-3 was responsive to the tonicity shift in the extracellular solution from 300 mOsm to 200 mOsm. (B) Summary of changes in TASK-3 currents upon applying a hypotonic solution. (C) Volume changes in response to a hypotonic solution. The volume changes were monitored by examining the time course of relative fluorescence. Bar graphs showed fluorescence intensity after a 3-minute exposure to the hypotonic solution. Each bar represents the mean ± SD of four different samples. *Significant difference from the corresponding control value (p < 0.05). F0 represents fluorescence intensity in iso-osmotic solution at time = 0. The ratio F0/Ft is proportional to cell volume.
Figure 5.
Volume regulation by TASK-3 channel. (A) Activation of the TASK-3 channel by hypotonic solution. The whole-cell current of TASK-3 was responsive to the tonicity shift in the extracellular solution from 300 mOsm to 200 mOsm. (B) Summary of changes in TASK-3 currents upon applying a hypotonic solution. (C) Volume changes in response to a hypotonic solution. The volume changes were monitored by examining the time course of relative fluorescence. Bar graphs showed fluorescence intensity after a 3-minute exposure to the hypotonic solution. Each bar represents the mean ± SD of four different samples. *Significant difference from the corresponding control value (p < 0.05). F0 represents fluorescence intensity in iso-osmotic solution at time = 0. The ratio F0/Ft is proportional to cell volume.
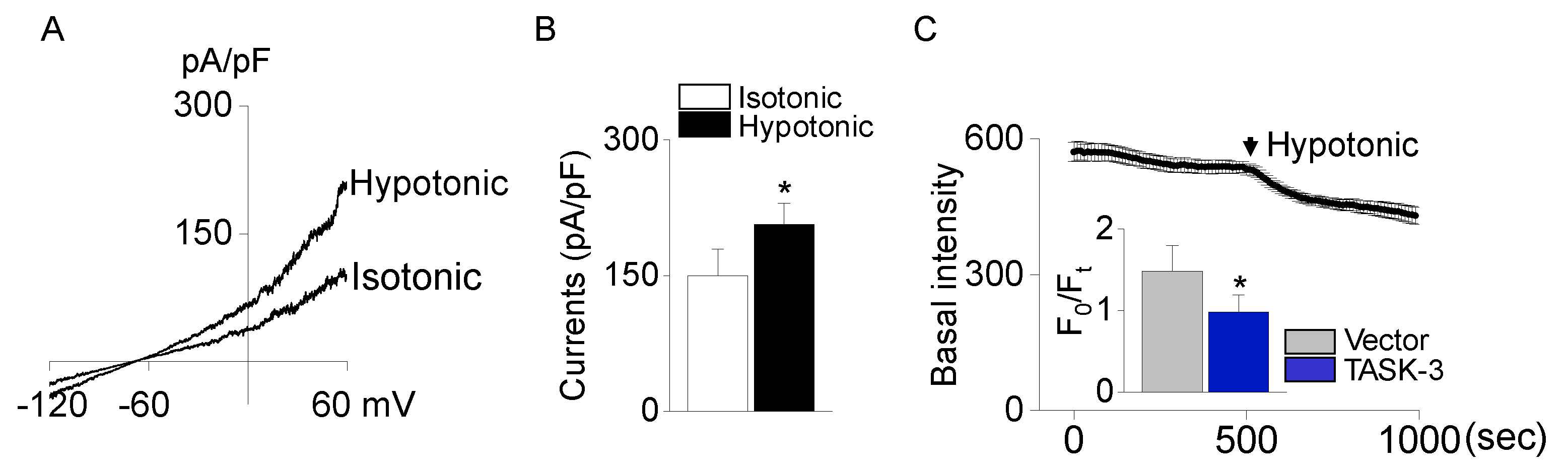
Figure 6.
Senescence triggered by TASK-3 suppression. (A) Live and dead cell staining. Red signals (PI-positive cells) were not detected in both NCsiRNA- and TASK-3 siRNA-transfected cells. (B) Increase in the number of SA-β-gal-stained cells in TASK-3 siRNA-transfected cells. (C) Upregulation of p53 and p21 protein in TASK-3 siRNA-transfected cells. (D) Comparative expression of p53 and p21 in TASK-3 modulated cells. The expression levels were normalized to β-actin. Each bar represents the mean ± SD. *Significant difference from the corresponding control value (p < 0.05). Scale bars, 100 μm.
Figure 6.
Senescence triggered by TASK-3 suppression. (A) Live and dead cell staining. Red signals (PI-positive cells) were not detected in both NCsiRNA- and TASK-3 siRNA-transfected cells. (B) Increase in the number of SA-β-gal-stained cells in TASK-3 siRNA-transfected cells. (C) Upregulation of p53 and p21 protein in TASK-3 siRNA-transfected cells. (D) Comparative expression of p53 and p21 in TASK-3 modulated cells. The expression levels were normalized to β-actin. Each bar represents the mean ± SD. *Significant difference from the corresponding control value (p < 0.05). Scale bars, 100 μm.
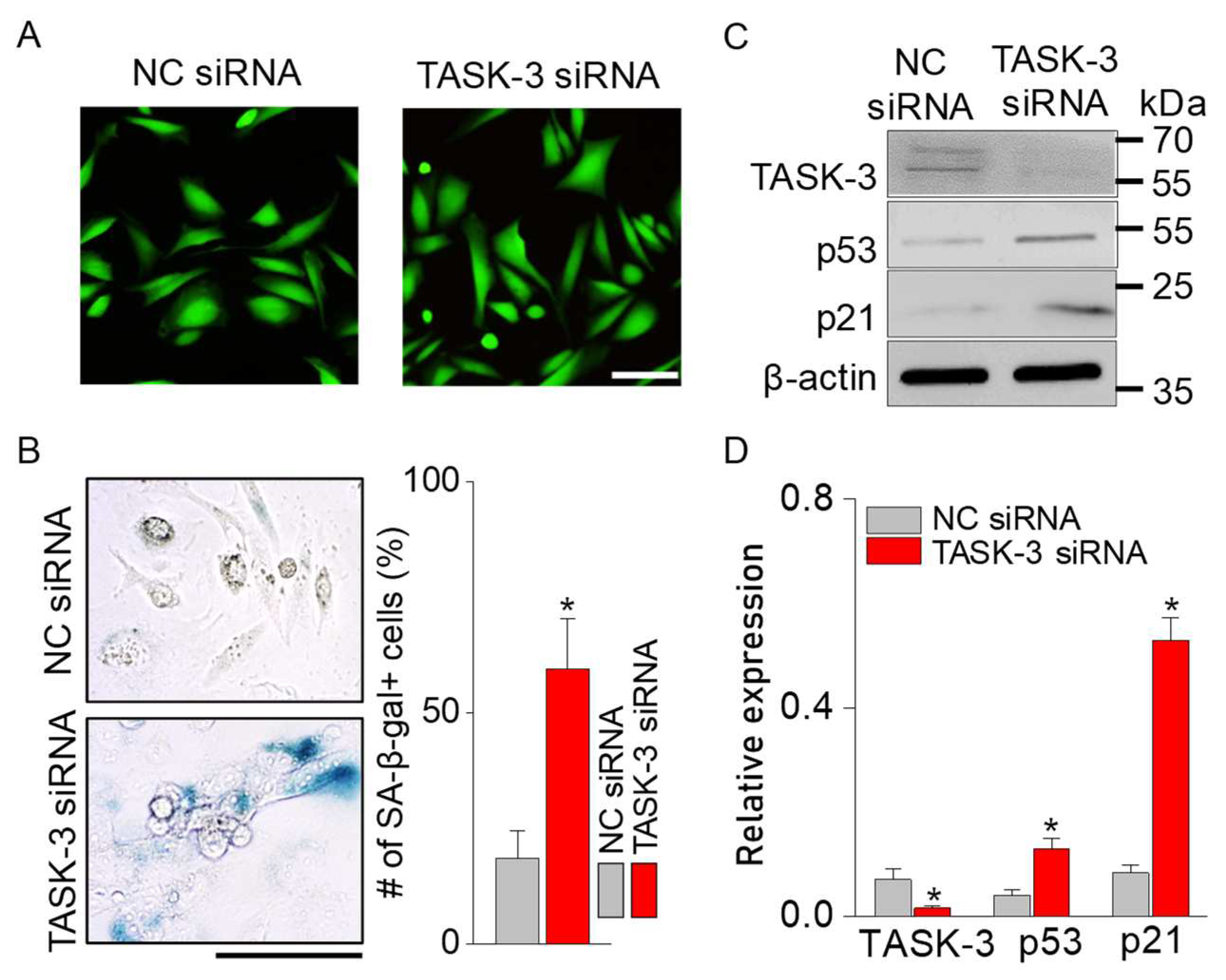

Table 1.
Primer sequences for PCR in bovine samples.
| Gene Name (channel name) |
Species | GenBank Accession Number |
Primer Sequences (5′–3′) | Application | Expected size (bp) |
|---|---|---|---|---|---|
|
Kcnk3 (TASK-1) |
Bovine | XM_597401 | F: CAGGCCTACTACTACTGCT R: GGCCCGTGAGGATGTAGA |
qRT-PCR | 133 |
| F: ACACCTTCGTGAAGTACCTG R: GGATGTAGACGAAGCTGAAG |
RT-PCR | 287 | |||
|
Kcnk9 (TASK-3) |
Bovine | XM_588194 | F: CTACTACTGCTTCATCACGTTG R: CCCACCAGGATATACATAAAGCTA |
qRT-PCR | 123 |
| F: CTACGTGGCCTTTAGCTTTA R: GTCGGTAAAGCTGTGTAACC |
RT-PCR | 433 | |||
| GAPDH | Bovine | NM_001034034 | F: ATGGTCTACATGTTCCAG R: AAGATGGTGATGGCCTTT |
qRT-PCR | 104 |
| F: CAGCGACACTCACTCTTCTAC R: GGAAGTCAGGAGATTCTCAGT |
RT-PCR | 250 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



