
Submitted:
20 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is one of the most widespread neurodegenerative diseases affecting people averaging over 65 years. It manifests with severe cognitive damage, loss of memory, impairment in performing activities, ventricular expansion and final dementia. AD is associated with the deposit of amyloid β protein (Aβ) plaques, intracellular neurofibrillary tangles (NFTs), progressive inflammation and impairment of synaptic transmission and of mitochondrial function. Due to the poor diagnostic tools for the early stages of the disease, proteomics biomarkers have gained a paramount role because they can monitor the prodromal variations of the disease-linked molecular changes. Further, proteome biomarkers can help the follow up of AD progression over time and aid in setting personalized medical care, before the catastrophic consequences of dementia occurs. Research has focused on the identification of proteome biomarkers in cerebrospinal fluid (CSF) and plasma, which are discussed in the present review, but also in other matrices, as saliva and urine, revealing the high potentiality of proteomics approach and, at the same time, the difficulty to identify, for the different stages of the disease, sensitive and specific biomarkers clinically available.
Keywords:
Alzheimer’s disease
; biomarker
; proteomics
; cerebrospinal fluid
; Aβ protein
; neurofibril- 14 lary tangles
; plasma
1. Introduction
Neurological degenerative diseases affect a huge number of people each year worldwide and Alzheimer’s disease (AD) is the most growing one. The World Alzheimer Report 2023 foretells an increase of AD patients from 55 to 130 million in the next 25 years with a significant increase in poor and medium richness countries [1]. AD is the main form of dementia accounting for about 60% of all cases clinically diagnosed and it has a typical onset after the age of 65. It is a severely invalidating disease characterized by brain changes beginning years or even decades before clinical symptoms, such as memory loss and confusion, occur [2]. It is characterized by spread death of neuronal cells which is responsible for cognitive impairment, memory loss, decline of daily habits and visuospatial abilities [3,4]. Once AD is diagnosed, the disease shows irreversible progression with huge brain damage, reduction of grey matter in the hippocampus and enthorinal cortex which constitute the long-term memory system [5]. As a consequence, it is a pivotal concern to find valid molecular tools in screening programs to detect people at early stage of AD, long before the appearance of clinical symptoms, to identify the disease with no doubt and to define the possible progression rate. Further, the discovery of molecular targets would help in setting proper medical treatments before the brain’s degeneration, which characterizes AD, is complete.
Specific molecules, referred to as biomarkers, work as indicators of pathological states and can now be reliably measured in patient fluid samples, to suggest the presence or absence of the disease or the likelihood to later develop AD. To be a valid diagnostic tool a biomarker must be specific, sensitive and able to univocally define the progressive AD stages. Further, a validated biomarker should distinguish AD dementia from prodromal mild cognitive impairment (MCI), which in turn can progress to AD and from other types of dementia, such as frontotemporal lobe dementia (FTLD) or Lewy body dementia (LBD) [6]. A good panel of biomarkers should be able to correlate with the severity progression rate supporting both the evaluation of the patient condition and the discovery of possible and specific drug medical targets. Proteins are very sensitive to diagnose a specific disease and, for this reason, the most promising approach for the identification of AD biomarkers is proteomics. AD proteomics biomarkers are proteins derived from brain homogenate [7,8], cerebrospinal fluid (CSF) [9,10,11,12,13,14], blood [12,15,16], but even saliva [17] and urine [18], which can help to follow the staging of the disease from presymptomatic conditions [19], to MCI [20] and more severe grades up to AD dementia [21] (see Figure 1).
The implementation of proteomics techniques and bioinformatic tools in biomarker research has gained a huge consensus because it allows large scale detection and identification of proteins rising the probability to detect novel biomarkers [24,25,26,27]. Proteomic studies benefit of multi approaches ranging from two-dimensional electrophoresis (2DE), mass spectrometry (MS) and protein microarrays [14].
The clinical diagnosis of AD is based on loss of memory and general cognitive decline, but the underlying pathological mechanisms which occur far ahead are extremely complex and difficult to identify because of confusing factors such as age, gender and genetic tendency. The first AD event which takes place is the aggregation process of amyloid protein (A) causing phosphorylation of tau protein (p-tau) and its aggregation in intraneuronal neurofibrillary tangles (NFTs) [28,29].
From then on, many studies concluded that aggregation of A and the accumulation of NFTs were the main players in AD onset [30]. As a consequence, the patient, over time, develops at first short-term memory problems, within a few years, followed by severe cognitive impairments, loss of anterograde and retrograde memory and of the normal behavioral functions [31].
The amyloid plaques are formed by aggregates of A protein with 40-42/43 residues, coming from the amyloid precursor protein (APP) cleaved by two different convertase enzymes [32]. For these reasons A42 or the ratio A42/A40, together with total tau (t-tau) and p-tau [33], the dominant proteins of the NFTs, still represent the main indicators of the disease and they are the worldwide recognized criteria for AD diagnosis [34,35].
Proteomics biomarkers have brought a great advance to in vivo diagnosis of AD, uncovering the major proteins involved in the disease pathogenesis. The important development of MS together with improved analysis and dedicated software has gained a deep insight into AD mechanisms. Proteomics has been a step forward in AD study, because the proteins are the effective players of the organism biological functions and, despite the fact that the human genome is known [36], many processes take place between gene transcription, protein translation and post translational modifications. To fully understand the events which underline complex and unknown pathologies as AD, it is pivotal to know the role of the involved proteins. Genetic analysis can estimate the risk of disease occurrence as reported for subjects with apolipoprotein E (ApoE) allele 4 [37,38], but it can not be used to ascertain the disease and its severity grade. For this reason, ApoE 4 is not considered a real biomarker for AD [39]. On the contrary, proteomics enables to better define the final product of a gene, how it works in the protein networks and its qualitative and quantitative modifications following pathological processes. While the genome is quite constant for each individual, proteome is a highly dynamic entity, variable in different cellular types. It exhibits strong expression dependence on time and environmental conditions, making it a suitable monitor of different conditions in health and illness. A protein, as final outcome of a gene, is reminiscent of the function much more of the gene itself, making the study of the dynamic proteome the best tool for AD knowledge. Bioinformatics and data analysis, now strengthen by machine-learning and artificial intelligence are key points in AD proteomics [40,41]. One important strength is that proteomics can allow the quantification of thousands of proteins, detect post translational modifications, such as phosphorylation, methylation, oxidation [42] which are known to play a pivotal role in AD [43], gaining important insights into the molecular mechanisms responsible for AD pathogenesis [44,45,46].
In addition to A plaque and NFTs formation, other changes take place in AD development affecting many structures of the central nervous system (CNS) which result highly compromised. First degenerative events occur in the entorhinal cortex and hippocampus, which shows a significant volume contraction. Later on ventricular enlargement and general loss of cortical matter influence a wide range of physiological functions [47,48,49], affecting language, reasoning and social behavior. Synaptic neurotransmission [50,51] and mitochondrial metabolism impairments [52,53] are described as well. Amyloid plaques disrupt postsynaptic structures impairing synaptic transmission [54]. Alterations in mitochondrial morphology, distribution, increased oxidative stress and enzymatic deregulation are all reported in AD [55]. So, although A42, or, better, the ratio A42/A40 and p-tau [10,56] mostly correlate with cognitive defects and neurotoxicity, respectively, the critical point is that they are unable to give information concerning the wide heterogeneal aspects of the disease.
Modification of cellular properties and even altered behavior of astrocytes, microglia and vasculature [57] are key points in the onset and progression of AD, beyond the amyloid cascade hypothesis [58] and must be fully understood to better know the disease. Since the detection of A deposits and NFTs, different antibodies have been tested for immunotherapy treatment of AD, but the percentage of success has been quite low, with very limited potential treatment [59]. More recently two promising molecules showed clinical results in patients with mild form of AD. Donanemab is an antibody targeting modified form of A[60] which can improve performing activities in the patient. Similarly, Lecanemab binds A protofibrils slowing down the cognitive impairment [61]. Unfortunately, both the antibodies display ancillary negative effects and the good medical treatment is still lacking.
In this view, the main goal in AD handling is to define novel early biomarkers and, at the same time, possible targets for the setting of new adjuvant pharmaceuticals. This could open to a better handling of the patients giving them optimist chances to face the disease and to find the correct medical approaches before reaching the devastating condition of dementia. Proteomics techniques have been searching for AD biomarkers in different body compartments being the CSF and the blood the most investigated. CFS is the best sample because it flows directly in the CNS where AD takes the lead and has the higher concentrations of neurodegenerative biomarkers. The negative point for its use is that the sample extraction with the lumbar puncture is quite invasive and, for this reason, blood could be preferred. Blood is of easier access and availability and CNS biomarkers can be found as a consequence of modification of the blood-brain barrier [62] and can be used for AD proteome analysis too [63]. In 2014 the National Institutes on Aging initiated a multiapproach program pointing at the discovery of new biomarkers [64]. The current status of this program relies on the possibility of a high throughput quantification and comparison of thousands of proteins derived from samples of healthy and AD individuals. MS improvement allowed high sensitivity and specificity in the detection of very low protein amounts derived both from CSF and blood. The refined technique has brought to the definition of the underlying molecular pathways of AD at different stages [25].
The review will focus on the proteomics biomarker landscape for AD in CSF and blood considering some of the mostly investigated pathological pathways which characterize this widespread disease.
2. Proteomics research for AD biomarkers
Proteomics based biomarker research
AD protein biomarkers are a promising class of molecules which provide insights into AD molecular mechanisms and a great utility for the diagnosis, prognosis and therapeutic assessments of the disease [65,66,67]. When the disease starts, many proteins change their expression profile, which can be detected over time, enlightening basic differences between healthy and AD individuals [68]. Proteomics allows the profiling of different sample matrices, from brain tissue [7] to biofluids [69], resulting in the identification of several biomarker candidates.
One of the easier approaches is the 2DE-based proteomics coupled with MS analysis, which allows great sensitivity and the identification of thousands of proteins [12,70]. MS is a powerful technique which unveils molecular mechanisms of biological processes which take place in disease progression. There are two different approaches for the identification and characterization of proteins retrieved from either cells, tissues or biofluids as CSF, blood, saliva or urine. The first is a bottom-up approach which uses a proteolytic digestion of the extracted proteins before MS analysis. The main limitation is the low percentage coverage of the original protein and, in this case, a significant amount of information can be lost. The second approach is a top-down workflow which is a good technique to identify and characterize whole proteins from complex biological mixtures and their possible post translational modifications, thanks to the possibility of having access to the complete sequence of the protein [71].
Concerning AD, the first step was the identification of different A peptides and modified tau proteins [72,73]. Later on, with the informatic supply of the human protein database, it has been easier to face with the AD proteome and sub-proteome studies, where sub-proteome defines a more restricted analysis in the region of interest. For example, the sub-proteome of amyloid plaques, which has been referred to as amyloidome, has evidenced about 900 proteins which result differently expressed in rapidly progressive AD and sporadic AD [74]. Out of more than 4000 proteins of amyloidome, 40 proteins are mostly present in the plaque region [75]. Sub-proteome analysis has been conducted in the synapse environment, looking at receptors, membrane and scaffold proteins. Brain derived samples allowed the identification of more than 5000 proteins in synaptic sub-proteome [76]. On the complex, MS techniques have allowed the identification of thousands of proteins involved in AD and playing a role in multiple molecular processes underlying synaptic activity [77], mitochondrial function [78], metabolism and glia. Asymptomatic and symptomatic stages of AD are discriminated by the different expression of proteins involved in glial biology [79]. Many proteomic studies report proteome changes in single brain regions [79,80,81]. The ability to uncover proteins which are expressed at different levels between AD patients and normal individuals, starting from the early pathological modifications, is a promising tool for AD diagnosis and prognosis.
Proteomics and Aβ cascade hypothesis
AD is a complex, multifactorial disease which is characterized by many factors taking part in its onset [82]. The main theory is the amyloid cascade, proposed in 1991 by Hardy and Higgins [83], which considers the accumulation of A plaques in the brain parenchyma as the central starting point for every form of AD. A plaques were identified in AD patients since 1984 [84]. A huge amount of evidence accumulated through the years led to the cloning of APP [85], whose mutations are linked to the familiar disease development [86]. One of the main consequences of plaque deposition is a widespread synaptic damage in the brain through the aggregationn of p-tau [87].
A protein comes from the amyloidogenic pathway of APP, cleaved by the enzymes - and -secretase which give rise, in normal conditions to soluble fragments. Due to the cleavage at different sites, -secretase produces different A peptides with A40 and A42 being the most abundant [88]. Mutated APP and/or secretase genes lead to the formation of insoluble A fragments, mainly A42, which are very active in plaque formation [89]. This is the leading event which triggers the CNS degeneration observed in AD that can begin up to twenty years before clinical symptoms [90]. The different fragments of A, soluble or insoluble, produce synaptotoxic effects with alteration of neurotransmitter systems, cytoskeleton damage and compromised synaptic plasticity up to neuronal death [91,92].
At the beginning, the deposit of the plaques is counteracted by microglia action, which contributes to the maintenance of synaptic contacts by preventing synaptical degradation through a phagocytose activity [87], but as soon as the A load increases, it becomes responsible for the formation of NFTs and neuronal impairment both in the familial AD form, characterized by gene mutations involved in the amyloid pathway [93] and in sporadic AD, whose etiology is still quite unknown [94]. The A plaques spread across the brain causing synaptic damage, p-tau formation, disassembling of cytoskeleton and tangle structures evolving in neuronal loss and disassembling of neuronal networks [95]. The evolution of these changes underlies the progressive patient decline.
Strictly connected to plaques deposition, many other events occur such as deregulation of calcium homeostasis and consequent toxicity [96], inflammatory responses [97] and mitochondrial dysfunction [53]. The mitochondrial damage, which is one of the hallmarks of AD, is responsible for the leakage of reactive oxygen species [98] and disruption of glutamatergic synaptic plasticity because of damage of N-methyl-D-aspartate (NMDA) and -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [99].
More recent studies raise the possibility of other causative processes in AD onset as neuroinflammation mechanisms [100], oxidative stress [101], autophagy mechanisms [102] suggesting that the real culprit for AD onset must still be unmasked [103]. Despite this last consideration, the identification of A fragments, especially the decrease of the fragment A42, aggregating faster than A40, and the increase of p-tau proteins, which are considered the major determinants of brain damage, have been the first established biomarkers [104,105] and are currently the only molecular markers to confirm AD diagnosis used in clinical practice, sided by neuroimaging techniques which detect aggregates of A within the brain [106].
Although proteomic studies were formerly conducted on brain tissue homogenates, revealing a notable number of differently expressed proteins along with the AD progression [107], the availability of early and reliable biofluid diagnostic biomarkers allows AD detection and searching of preventive measures to avoid neuronal damage. Unlike other diseases, as cancer, it is quite hard to take a sample of the brain from the living patient and, consequently, a biofluid biomarker can provide evidence for the pathological changes in the CNS. The identification of biomarkers is thus, achievable faster by using fluid samples respect to tissue extracts. Fluid biomarkers are less invasive, offer a major feasibility to obtain samples from the patients over time, do not expose the patient to radiation and can give a quantification to follow up the disease progression [108]. The most studied fluid biomarkers are available in CSF and in the blood, but AD biomarkers are reported in saliva [109] and ocular fluids, where aggregates of A have been found [110].
Cerebrospinal (CSF) fluid biomarkers
The protein changes which characterize the neurodegenerative process of AD are best seen in CSF and, although blood is easier to collect for proteomics sample extractions, the CSF is the best biofluid indicator for AD proteome biomarkers, because it reflects the main neuropathological processes which take place in the CNS [111]. CSF is an ultra-filtrate of plasma circulating in the ventricular system of the brain and, thus, it is in contact with brain tissue, collecting the proteins of the extracellular space. The APP protein and secretase enzymes originate A fragments which are responsible for the early stages of AD [112]. A fragments include two main isoforms, the soluble A40 fragment and the insoluble, hydrophobic A42 one which appears in higher percentages in AD patients and is the main component of the AD plaques [113,114]. It aggregates with higher probability, reducing its concentration in CSF, probably because of its sequestration in A plaques [115]. It is a strong predictor of progression to MCI or AD in patients with subjective complaints [116]. It must be stated that A42 results reduced even in other central pathologies as bacterial meningitis [117] and CSF A42 is also dependent on the total A peptides of the brain [118]. For these reasons, more than the evaluation of A42 alone, the ratio A42/A40 has been proposed as CSF predictive biomarker of AD [119], gaining better prediction accuracy and better discrimination between AD and non-AD dementia [118,119,120,121]. It is detected early in the disease onset, before the cognitive impairment occur [122]. On the contrary, total A proteins do not represent a biomarker for AD diagnosis having similar concentration in healthy and AD individuals [123].
A plaques initiate the misfolding of tau proteins [124] spreading through the whole brain and causing neuronal degeneration and brain matter loss followed by cognitive impairment. At the present time no valuable medical treatment is available for the disease, the real progression still remains unclear and much research work is in progress to find out other proteins involved [66]. A panel of different variable biomarkers could, indeed, help on the establishment of protocols aimed at early AD identification and at evaluation of the disease progression rate.
Soluble and insoluble A fragments, isolated from the plaques, were identified, at first, through liquid chromatography (LC)-electrospray ionization (ESI)-MS [125] and matrix-assisted laser desorption ionization (MALDI)-time of flight (TOF)-MS [126], but, until 1992, they were the only well known AD-linked proteins because of the limited availability of protein databases. It was only after the Human Genoma Project was completed in 2003, that protein databases were available, providing the fundamentals for full proteome analysis. Afterwards A peptides have been analyzed with multiple MS-based techniques and then they have been developed into targeted assays for quantitation [127].
t-tau increases in AD probably because of neuron loss [128], but it is correlated also to altered levels of CSF proteins involved in neuronal plasticity and blood–brain barrier dysfunction [129]. p-tau accumulation has been investigated with MS [130] and is mostly linked to the formation of NFTs. It shows multiple isoforms as p-tau181, now considered a validated marker for AD and often used in routine biochemical assessments. [131,132], p-tau231, p-tau217 which all increase in AD patients. The different phosphorylated or dephosphorylated status accounts for detachment of p-tau from microtubules. It accumulates leading to NFTs formation, one of the characteristic hallmark of AD. As a consequence the destabilization of cytoskeleton system, the block of axonal transport path, strongly impairing the synaptic activity, alteration of the neuronal communication and disruption of synaptic plasticity take place [133]. At the present time A, t-tau and p-tau are considered the gold standard method to establish reliable AD diagnosis [12] and for clinical application in MCI patients to predict the progression to AD [134].
CSF A only or combined with p-tau is considered a molecular cue which can distinguish, with high sensitivity, between AD and healthy subjects [135]. Abnormal increase of t-tau and p-tau, combined with low A42 are strong biomarkers of the AD-associated pathological changes in the brain and predict AD features with high accuracy [105]. The analysis of CSF has reported a correlation with amyloid plaques in cortical brain biopsies between low CSF A42 and high CSF t-tau and p-tau levels, respectively [136] assessing a strong relation between AD damages and proteins of the CSF [137]. The reduction of A42 reflects the increased aggregation into plaques [138] and correlates with amyloid plaque load in post-mortem studies [105].
Although A and tau proteins are still considered reliable AD biomarkers and have high specificity, these cause only a fraction of the biological changes that take place in AD, though involved in metabolism alterations, oxidative processes, vascular effects and inflammatory responses [97,139,140,141]. Many proteins have been identified in the plaques, correlated to different forms of AD [74]. Different molecules which play important roles in the disease have been proposed as biomarker candidates to be possibly inserted in etiologic pathways beyond A cascade. Several studies have focused on putative biomarker including inflammation- and oxidative stress-related proteins which are considered, together with the A hypothesis, sided mechanisms of AD onset.
In the last years the marked immune response of the brain has suggested an immune etiology of AD [97] with astrocytes and microglia, which are part of the CNS immune response, taking a pivotal role in AD’s development [142], exacerbating the mechanisms of A and tau symptoms [143,144]. Microglia cells at first degrade A plaques, but the excessive immune response drives to microgliosis with the over production of cytokines, free radicals and subsequent neuronal damage [145]. On the other hand, the oxidative stress etiology has been reported since almost three decades and reviewed recently [146,147,148]. Inflammation and oxidative biomarkers are thus considered potential AD biomarkers [14,149,150,151,152].
The CSF AD-linked protein neurofilament light (NFl) is a non-specific biomarker of axonal degeneration. Indeed, NFl increases in AD and in other neurodegenerative disorders any time that brain cells are damaged [153]. NFl cannot be used for diagnosis of AD, but, in a person who has already been diagnosed, NFl results potentially useful as powerful monitor for prediction of disease progression. The ratio NFl/A42 is reported as one of the best predictor of brain atrophy and cognitive scores [154].
AD is a complex pathology and the discovery of valuable biomarkers indexing an early diagnosis, the disease severity, the progression rate and an effective treatment is still a continuous need for AD research framework, thus, many efforts are addressed to identify novel protein biomarkers reflecting these points [155]. The proteomics approach has been and still is a valid method to discover novel biomarkers [150,156,157]. MS based proteomics of the CSF allows the analysis of thousands of proteins, growing the possibility to identify novel biomarkers of the disease and a selective and precise quantification providing protein identity and abundance [115,158,159,160].
One of the major problems encountered in protein biomarker research is the identification of low-abundance proteins, which can be masked by high-abundance ones. For this reason new technological improvements are aimed to the enrichment of the sample to better find out the proteins of interest [161,162,163]. Indeed, the involvement of tau in AD progression was known since many years, but the ability to its quantification depended on the depletion technique of the most abundant proteins [150]. With better technological improvements, the MS-based research for novel AD biomarkers has gained multiple results and different conceivable biomarkers have been proposed.
A high-performance CSF proteome study has been done on 200 participants of three different cohorts, whose A and p-tau values were known aiming at the classification in AD and non-AD individuals. Notable proteome changes were detected for 40 up- and down-regulated proteins as the astrocyte-derived YKL-40/chitinase-3-like protein 1, involved in neurodegenerative processes and the fatty acid binding protein 3 (FABP3), a small protein expressed in neurons, astrocytes and brain endothelial cells [150]. YKL-40, which reflects astrocyte activation, was tested as putative AD biomarker in line with previous studies considering it as possible indicator for disease progression from mild condition to AD dementia [164]. Since YKL-40 is related to inflammatory response and astrocyte-mediated neuroinflammatory conditions, the detection of high concentrations in CSF could suggest medical treatments targeting inflammation mechanisms [165]. Further, the progression of clinical symptoms and cortical atrophy are closely associated with increases of YKL-40 levels [166].
The increasing interest for the immune response led to find out in AD CSF higher concentrations for the soluble triggering receptor of myeloid cells 2 (sTREM2), a possible AD biomarker candidate which results correlated with t-tau and p-tau181 [167].
Increased levels of osteopontin, an inflammatory marker which could potentiate the AD immune response[168], interleukine-10 [169], macrophage migration inhibitory factor [170] and monocyte chemoattractant protein 1 [171], have all been found changed in their expression level in AD vs non-AD patients, but these proteins need more investigation to be really confirmed as AD biomarkers.
High-resolution MS and tandem mass-tags-based multiplexing combined with immunodepletion technique on 5 control and 5 AD patient samples, identified 139 out of 2327 differentially expressed proteins, including t-tau, neuronal pentraxin-2 (NPTX2), glial fibrillary acidic protein (GFAP) and neuronal cell adhesion molecule-1 (NCAM1). Glucose metabolism-associated proteins were higher in AD CSF, probably released into CSF from brain tissue, according to previous reports [24]. NPTX2 is a modulator of synaptic activity which is known to facilitate excitatory synapse formation, contributing to brain plasticity, learning and memory [172]. It is down-regulated in AD patients, representing an important factor for cognitive dysfunction and disease progression [173], able to predict memory loss and brain atrophy [174].
A MS-based shotgun proteomics study reports the association between 790 proteins of the CSF proteome with the core markers of AD, A42 and p-tau. Positive correlation of four proteins (cannabinoid receptor 1, neuroendocrine convertase 2, NPTX2 and somatostatin) with A42 were detected. The endocannabinoid system was already reported as potential target of medical treatment in AD [175]. 50 proteins were found to be associated with tau and 46 with p-tau, of which 41 were in common providing new insights in CSF proteome alterations related to the disease. Strong associations with the core AD A and/or tau were reported also for proteins implicated in energy metabolism, synaptic activity, nitric oxide production. Several proteins involved in synaptic activity are altered in AD CSF. Presynaptic synaptosomal-associated protein 25 (SNAP-25) has been shown to have higher concentrations in CSF of AD early stage. Synaptotagmin-1 (SYT-1) shows increased values in MCI patients progressing to AD and, for this reason, it could be considered a marker of progressive cognitive decline [176]. Neurogranin and neuromodulin, were positively correlated both with tau and p-tau [177]. High CSF concentration of neurogranin, a protein having a key role in synaptic plasticity, reflects synaptic loss in AD patients and may be a valid indicator of future cognitive decline linked to dendritic instability and degeneration [178]. Neuromodulin is a presynaptic protein involved in synaptic plasticity as well, which results down-regulated in AD patients providing indication for impaired cognitive abilities.
Wang and colleagues [179] report an integrated ultra-deep proteome analysis in cortex, CSF and serum revealing 37 proteins as potential AD markers. The methodological approach of integrating multiple proteomes and the MS-techniques combined with a systems biology view, gained the interesting result of 59% of these proteins involved in mitochondrial dysfunction. The result was in line with the hypothesis of important mitochondrial changes as putative causative agents of AD [53,180]. Consistently with these results, decreased levels of mitochondrial thioredoxin-dependent peroxide reductase (PRDX3), a protective antioxidant enzyme, were observed in AD CSF sample. This suggest an impairment of mitochondrial function and, since mitochondria are the main source of reactive oxygen species, an imbalance in redox equilibrium [181]. Ubiquitin C-terminal hydrolase L1 (UCHL1) involved in degradation of misfolded or damaged proteins, FABP3, involved in lipid metabolism are shown to have good diagnostic value of AD [182,183]. Pyruvate kinase M (PKM) results increased in AD and it has been considered as putative biomarker for either glucose metabolism or neurodegeneration with higher levels due to release in CSF following cell death. The alteration of glucose metabolism could be an early sign of the disease even if the evaluation of other glycolytic enzymes in AD e non-AD individuals must be taken into account [184].
Immunodepletion was also used in a multiplex tandem mass tag labeling study which revealed 225 down-regulated and 303 up-regulated out of 2875 profiled proteins in CSF samples from 20 controls and 20 AD patients. The proteins, including tau, NPTX2, NCAM1 were collected in five different panels involved in the deregulation of synaptic activity, vascular function and coagulation, cellular structure and myelination, inflammatory and metabolic pathways [185]. The meta-analysis of six CSF datasets derived from previous studies [24,150,179,185] provided a panel of 5939 proteins. To improve the choice of high specific AD markers the datasets were integrated with brain proteome, leading to evidence 65 up-regulated proteins and 44 down-regulated ones [25].
A multiplex proteomics study on AD considered analog CSF and blood biomarkers in different cohorts. The authors identified alterations of proteins involved in inflammatory response, apoptosis and other biological processes as possible links with A and tau. Several chemokines, interleukins and immune markers result changed in AD patients and are proposed as markers of disease diagnosis. Caspase 8, involved in synaptic plasticity, amyloid processing and microglial pro-inflammatory activation, shows increase in CSF and blood of AD patients [186]. Its inhibition could be a speculative therapeutic strategy for Alzheimer patients to help neuronal survival. The same authors report a down-regulation for the junctional adhesion molecule B (JAM-B) protein which participates in synaptic adhesion and could be associated to cognitive abilities, up-regulation of matrix metalloproteinase-9/10 (MMP9, MMP10) correlating with cognitive abilities and metalloproteinase-linked proteins which can play a determinant role in direct degradation of A deposits [187].
Mannosylated-glycan transferrin (Man-Tf) is a post-translationally modified transferrin isoform produced from cortical neurons, which is increased in AD CSF samples, probably following oxidative stress of endoplasmic reticulum, as detected through ultra LC-MS study. It displays high correlation with p-tau, consistently with the observation of hippocampal neurons co-stained for both the proteins, leading to the proposal that combined p-tau and Man-Tf could be a biomarker for MCI and AD [188]. AD is thus associated with strong CSF biomarkers as low levels of A42 and/or A42/A40 ratio and high levels of p-tau and t-tau. On the other hand, the huge diversity of differentially expressed proteins encountered in multiple proteomics study confirms the complexity of AD etiology and pathology and its multifaceted aspects. As a consequence other novel biomarkers are looked for in a continuous effort to ameliorate AD diagnosis and prognosis (see Figure 2).
Plasma biomarkers
There are two valid reasons to investigate the availability of AD biomarkers in plasma. The first one if that blood samples are obtained with minimally invasive procedures, preventing the patient from lumbar puncture. The second is that the plasma biomarkers can be measured at relatively low cost if a standard measurement system will be reached.
Indeed, many laboratories around the world base clinical diagnosis on plasma sample, as for C-reactive protein level for coronary disease [189]. However, the blood represents a very complex matrix which poses many difficulties for proteomics biomarker detection and performing MS analysis is a really hard matter. The first of all is the abundance of plasma proteins such as albumin which has a concentration of 10 orders of magnitude higher compared to the rarest proteins [190] and it accounts for 50% of the most abundant. There are about 22 proteins which account for 99% of plasmatic protein weight. The detectability of low abundance proteins can be managed through depletion of high abundance ones, enriching the sample, although one potential risk is the loss of the low-weight proteins which bind to albumin [191].
AD is characterized by damage of the brain-blood barrier with increasing permeability of the vascular endothelium [62,192] and this allows the detection of AD protein-based biomarkers in the blood flow, though with lower concentrations respect to CSF biomarkers, as reported for the core AD protein A [121] and t-tau which results 100 times lower respect to CSF [193]. A further problem is that a protein can be produced only in the CNS or even in peripheral tissues/organs and, in this latter case, its differential expression could be due to systemic effects without any link with AD. This effect would make really hard to uncover AD-dependent mechanisms.
Earlier studies on AD plasma biomarkers were directed to molecules known to be related with AD etiology, namely APP, A and p-tau [194]. A fragments are much more difficult to be evaluated because they stick to plasma proteins and, in part, are produced by platelets causing a sort of disturbance in the concentration measure [195]. One of the classical CSF biomarkers, A42/A40 ratio shows a similar decrease in plasma of AD patients. Most of the studies report a reduction of the ratio in MCI and AD patients, progressive MCI subjects and individuals at risk of developing MCI/AD, but, in a few cases, the data were partially contradictory because of the ratio increase linked to a major risk of AD development as well [191].
Immunoprecipitation coupled with MS allowed since 2018 a good evaluation of the ratio A42/A40, despite the fact that the ratio decrease averages 50% in CSF and only 10-15% in the blood [196]. A decrease of A42/A40 in plasma is now recognized as a detector of early AD stages with high accuracy [197].
Following plasma A, MS assays allowed detection of p-tau in plasma samples [198] and it is recognized as specific AD biomarker for the early stage of the disease [199]. The biomarker value compared to CSF p-tau one was evidenced by very recent works dealing with different phosphorylation states of the protein [200]. In pre-symptomatic individuals plasma p-tau increases over ten years before symptoms onset [201]. It associates with rapid cognitive impairment and hippocampal atrophy [202,203] and it discriminates between AD individuals and controls better than other plasma biomarkers such as A42/A40 ratio and NFl [204]. Different p-tau fragments can be detected in plasma as putative biomarkers and show optimal diagnostic accuracies. Plasma p-tau181, p-tau231 and p-tau217, phosphorylated on threonine residues, are excellent monitor for AD symptomatic stage, increasing with the disease severity and show an excellent association with amyloid and tau pathologies [131]. Plasma p-tau181 is higher in AD patients compared to control individuals and well correlates with CSF p-tau181. It increases with disease progression and is a good marker for AD dementia compared to non-AD dementia [205]. MS analysis of different p-tau forms, namely p-tau181, p-tau217 and p-tau205, has focused on the utility of these proteins to define AD progression over time [198]. Recent data indicate that elevated plasma p-tau181 is associated with future deposition of A plaques in different brain regions, suggesting its use as potential biomarker for amyloid deposits [206]. p-tau217 shows many fold increase in symptomatic AD patients and it is able to separate between AD and non-AD samples [200].
NFl increases in the AD prodromal stage [207], but it is associated with different neurodegenerative diseases making NFl alone a less specific AD diagnosis biomarker [208].
GFAP, released from astrocytes, shows increased concentration in CSF of patients affected by different neurodegenerative disorders, including AD [209]. It is proposed as plasma biomarker correlated to worse outcomes of AD [210]. Further, GFAP shows negative correlation with A42/A40 and, despite the fact GFAP is not specific for AD, it should be inserted in the blood biomarker panel as useful indicator of astroglia activation [211]. At the present state of art A42/A40, p-tau, NFl and GFAP are the most reliable blood-based biomarkers recognized.
A meta-analysis approach [151] presents highly reproducible AD plasma-based biomarker candidates. Beyond several proteins of inflammatory and oxidative processes, the authors retrieved six putative and highly reproducible biomarkers replicated in different independent cohorts. Alpha-2-macroglobulin (2M) results significantly higher in AD patients. It is linked to p-tau of CSF [212] and it correlates with cognitive decline, compared to healthy individuals. 2M is connected to vascular dysfunction and to up- or down-regulated proteins of the complement cascade [7,213]. Its role in the inhibition of coagulation could delay the repair of endothelial cells of the blood-brain barrier allowing the entrance of pro-inflammatory molecules in the brain [214]. It appears stage-dependent since it results down-regulated in pre-symptomatic subjects. Apolipoprotein A1 (ApoA-1) is down-regulated in plasma, similarly to the decrease of CSF ApoA-1 of AD patients, probably due to the binding to A[215]. Afamin is involved in antioxidant mechanisms since it transports vitamin E and it results down-regulated in AD patients whose brain would be more vulnerable to oxidative stress. Fibrinogen--chain results up-regulated and can be connected to increased vascular damage as already proposed for fibrinogen detected in CSF sample [216] and fibrinogen isoforms detected in plasma with a two-dimensional differential in gel electrophoresis combined with MALDI TOF/TOF-MS [151]. Pancreatic polypeptide (PP) is up-regulated in plasma samples from five different independent cohorts, but a clear role in AD pathophysiology is still missed. Insulin like growth factor binding protein-2 (IGFBP2) participates in energy production in neurons and it is supposed interacting with tau and A in CSF worsening cognitive abilities [217]. According to the authors view all these proteins would be implicated in systemic inflammatory response which, in turn, could trigger A aggregation and tau phosphorylation in the CNS, brain inflammation and oxidative damage.
In 2008 a 5-protein biomarker panel has been proposed as signature for AD diagnosis. The panel includes the S100 calcium-binding protein A9 (S100A9), directly connected to AD [218,219], alpha-globulin 1, endothelial cell-adhesion molecule, CD84 and CD226. On the complex the 5-protein panel is able to diagnose AD and even differentiate AD from other neurological disease [15], suggesting that multi protein panel approach could be a potent tool to face this invalidating disease over time continuum.
A recent investigation combines a multiple reaction monitoring (MRM)-MS approach with machine learning, leading to the quantification of 125 plasma proteins and the prediction of damage progression. Afamin, ApoE, biotinidase and paraoxonase/arylesterase show a significant decrease and they are sided by other proteins which include previously reported blood-based biomarker candidates [220].
The studies of AD plasma biomarkers have led to a high number of proteins differentially expressed compared with healthy individuals [151,221]. This number is still growing due to the improvements of the technical skills. Nevertheless, much work still remains to be done to ascertain the specificity and sensitivity of blood-based protein biomarkers, in order to have reliable diagnosis and to improve therapeutic cares.
3. Conclusions
Molecular biomarkers are molecules which can be used to discriminate between healthy and illness conditions. In general, such molecules can have diagnostic value in pathology detection, a staging value for the disease progression, a prognostic value to predict the final outcome and also are able to monitor the clinical response. Protein biomarkers have received large consensus thanks to the employment of proteomics techniques, even because proteins are very sensitive and can be detected in tiny amount of sample to allow early diagnosis. Further, proteomics can recognize protein posttranslational modifications which potentially assume pivotal role in disease onset and progression [44]. In the recent years proteomics applied to AD research has widened the expectations on early diagnosis and prognosis. AD is the most worldwide spread neurodegenerative disease, affecting preferentially people over 65 years of age with gradual loss of independence, memory and cognitive abilities. Up to now the most common diagnostic methods include magnetic resonance imaging (MRI), or positron emission tomography (PET) together with collected CSF analysis to measure the proteins mostly associated with AD. Much work has been done for the discovery of proteome-based biomarkers in biofluid, setting important milestones in AD knowledge. The former CSF biomarkers investigated were A fragments, t-tau and p-tau proteins which are associated with the great pathological variations inside the brain. A42, A42/A40 and p-tau show significant differences between AD and non-AD patients, assuming a relevant diagnostic value. The ratio A42/A40 is probably the most reliable parameter discriminating between patients with AD and without AD [121,222,223]. The combination of all these biomarkers constitutes a signature of AD to detect the disease at early stages. In addition to these established CSF biomarkers, other candidates have been proposed to improve AD follow up. NFl is a biomarker for neuronal injury and, although it is not specific for AD, it can add information on the disease progression over time. Similarly, YKL-40 reflects glia alteration, NPTX2 correlates with cognitive decline with neurogranin and neuromodulin as well. Unfortunately, at the present time most of the novel biomarkers identified are available as a research tool and not for real clinical practice.
To allow an easier approach with the patient, other biofluid matrices have been studied. Blood protein biomarkers could increase the application in clinical practice. Blood presents some advantages because it is accessible and prevents patient from lumbar puncture. The proved dysfunction of the blood brain barrier can allow the detection of AD proteins in the blood flow. However, the reported concentrations for A40, A42 and tau are much lower and, most of all, blood is a very complex matrix and it could render really difficult to detect the proteins involved in AD etiology and time course. The high quantity of antibodies can give false results [224] and protein biomarkers are subjected to proteolytic degradation in liver or can be metabolized and cleaved from the blood flow [225]. In conclusion, the use of AD blood biomarkers is still under progress and need more time to have clinical validated molecules with diagnostic and prognostic value. In the next time proteome-based biomarkers, with improved MS techniques and together with artificial intelligence (AI) tools should aim to the setting of protein platforms, including more and less specific AD markers, which could escape the present available data analysis. These platforms would be able to give a very early diagnosis, a real-time information on disease progression and trustable indicators for medical treatment. AI approach will be of great benefit to integrate multiple data sources, even with little significance, put together a huge amount of experimental data compiling a biomarker panel for different biological materials, in order to disclose full knowledge of one of the most impairing neurodegenerative diseases.
Author Contributions
Conceptualization, original draft prepration, review and editing, Valeria Magnelli.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 2M | Alpha2 Macroglobulin |
| A | Amyloid beta protein |
| AI | Artificial intelligence |
| AMPA | -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ApoA-1 | Apolipoprotein 1 |
| ApoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| ESI | Electrospray ionization |
| FABP3 | Fatty acid binding protein 3 |
| FTLD | Frontotemporal lobe dementia |
| GFAP | Glial fibrillary acidic protein |
| IGFBP2 | Insulin like growth factor binding protein 2 |
| JAM-B | Junctional adhesion molecule B |
| LC | Liquid chromatography |
| LBD | Lewy body dementia |
| MALDI | Matrix assisted laser desorption ionization |
| Man-Tf | Mannosylated glycan transferrin |
| MMP9 | Metallo proteinase 9 |
| MMP10 | Metalloproteinase 10 |
| MRI | Magnetic resonance imaging |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry |
| NCAM1 | Neuronal cell adhesion molecule 1 |
| NFl | Neurofilament light |
| NFTs | Neurofibrillary tangles |
| NMDA | N-methyl-D-aspartate |
| NPTX2 | Neuropentraxin 2 |
| p-tau | Hyperphosphorylated tau protein |
| PET | Positron emission tomography |
| PKM | Protein kinase M |
| PRDX3 | Mitochondrial thioredoxin-dependent peroxide reductase |
| SNAP-25 | Synaptosomal-associated protein 25 |
| sTREM2 | Soluble triggering receptor of myeloid cells 2 |
| SYT-1 | Synaptotagmin-1 |
| S100A9 | S100 calcium binding protein A9 |
| t-tau | Total tau protein |
| TOF | Time-of-flight |
| UCHL1 | Ubiquitin C-terminal hydrolase L1 |
| YKL-40 | Astrocyte-derived chitinase-3-like protein 1 |
References
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. The Lancet Neurology 2016, 15, 455–532. [Google Scholar] [CrossRef] [PubMed]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in brain function occur years before the onset of cognitive impairment. Journal of Neuroscience 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2010 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2010, 6, 158–194. [Google Scholar] [CrossRef] [PubMed]
- Dubbelman, M.A.; Jutten, R.J.; Tomaszewski Farias, S.E.; Amariglio, R.E.; Buckley, R.F.; Visser, P.J.; Rentz, D.M.; Johnson, K.A.; Properzi, M.J.; Schultz, A.; et al. Decline in cognitively complex everyday activities accelerates along the Alzheimer’s disease continuum. Alzheimer’s Research & Therapy 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Matsuda, H.; Tabira, T.; Asada, T.; Uno, M. Changes in Brain Morphology in Alzheimer Disease and Normal Aging: Is Alzheimer Disease an Exaggerated Aging Process? American Journal of Neuroradiology 2001, 22, 1680–1685. [Google Scholar] [PubMed]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends in Biotechnology 2011, 29, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xia, W. Proteomic profiling of plasma and brain tissue from Alzheimer’s disease patients reveals candidate network of plasma biomarkers. Journal of Alzheimer’s Disease 2020, 76, 349–368. [Google Scholar] [CrossRef]
- Askenazi, M.; Kavanagh, T.; Pires, G.; Ueberheide, B.; Wisniewski, T.; Drummond, E. Compilation of reported protein changes in the brain in Alzheimer’s disease. Nature Communications 2023, 14, 4466. [Google Scholar] [CrossRef]
- Blennow, K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef]
- Anoop, A.; Singh, P.K.; Jacob, R.S.; Maji, S.K.; et al. CSF biomarkers for Alzheimer’s disease diagnosis. International journal of Alzheimer’s disease 2010, 2010. [Google Scholar] [CrossRef]
- McGrowder, D.A.; Miller, F.; Vaz, K.; Nwokocha, C.; Wilson-Clarke, C.; Anderson-Cross, M.; Brown, J.; Anderson-Jackson, L.; Williams, L.; Latore, L.; et al. Cerebrospinal fluid biomarkers of Alzheimer’s disease: current evidence and future perspectives. Brain Sciences 2021, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Korecka, M.; Shaw, L.M. Mass spectrometry-based methods for robust measurement of Alzheimer’s disease biomarkers in biological fluids. Journal of neurochemistry 2021, 159, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Gunes, S.; Aizawa, Y.; Sugashi, T.; Sugimoto, M.; Rodrigues, P.P. Biomarkers for Alzheimer’s disease in the current state: A narrative review. International journal of molecular sciences 2022, 23, 4962. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Dhariwal, R.; Patil, N.; Ojha, S.; Tendulkar, R.; Tendulkar, M.; Dhanda, P.S.; Yadav, A.; Kaushik, P. Unveiling the Molecular Footprint: Proteome-Based Biomarkers for Alzheimer’s Disease. Proteomes 2023, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Mubeen, S.; Khan, A.; Ibrahim, S.; Meer, B. Plasma biomarkers: potent screeners of Alzheimer’s disease. American Journal of Alzheimer’s Disease & Other Dementias® 2019, 34, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Buckley, N.J.; Bos, I.; Engelborghs, S.; Sleegers, K.; Frisoni, G.B.; Wallin, A.; Lléo, A.; Popp, J.; Martinez-Lage, P.; et al. Plasma proteomic biomarkers relating to Alzheimer’s disease: a meta-analysis based on our own studies. Frontiers in aging neuroscience 2021, 13, 712545. [Google Scholar] [CrossRef]
- Ashton, N.J.; Ide, M.; Zetterberg, H.; Blennow, K. Salivary biomarkers for Alzheimer’s disease and related disorders. Neurology and Therapy 2019, 8, 83–94. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Zhu, J.; Guan, Y.; Xie, F.; Cai, X.; Deng, J.; Wei, Y.; He, R.; Fang, Z.; et al. Systematic evaluation of urinary formic acid as a new potential biomarker for Alzheimer’s disease. Frontiers in Aging Neuroscience 2022, 14, 1364. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Sikkes, S.A.; Van Den Hout, A.; Handels, R.; Bos, I.; Van Der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s & Dementia 2019, 15, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.O.; Aakre, J.A.; Kremers, W.K.; Vassilaki, M.; Knopman, D.S.; Mielke, M.M.; Alhurani, R.; Geda, Y.E.; Machulda, M.M.; Coloma, P.; et al. Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population-based setting. JAMA neurology 2018, 75, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Sathe, G.; Na, C.H.; Renuse, S.; Madugundu, A.K.; Albert, M.; Moghekar, A.; Pandey, A. Quantitative proteomic profiling of cerebrospinal fluid to identify candidate biomarkers for Alzheimer’s disease. PROTEOMICS–Clinical Applications 2019, 13, 1800105. [Google Scholar] [CrossRef]
- Bai, B.; Vanderwall, D.; Li, Y.; Wang, X.; Poudel, S.; Wang, H.; Dey, K.K.; Chen, P.C.; Yang, K.; Peng, J. Proteomic landscape of Alzheimer’s Disease: novel insights into pathogenesis and biomarker discovery. Molecular neurodegeneration 2021, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Dalal, V.; Dhankhar, P.; Biswas, S. Proteomics as a Potential Tool for Biomarker Discovery. In High Altitude Sickness–Solutions from Genomics, Proteomics and Antioxidant Interventions; Springer, 2022; pp. 119–141. [CrossRef]
- Awasthi, S.; Spellman, D.S.; Hatcher, N.G. Proteomic Discovery and Validation of Novel Fluid Biomarkers for Improved Patient Selection and Prediction of Clinical Outcomes in Alzheimer’s Disease Patient Cohorts. Proteomes 2022, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues in clinical neuroscience 2003, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; De Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. The Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Zvěřová, M. Clinical aspects of Alzheimer’s disease. Clinical biochemistry 2019, 72, 3–6. [Google Scholar] [CrossRef]
- O’brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annual review of neuroscience 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Blennow, K.; Hanse, E. Amyloid β and APP as biomarkers for Alzheimer’s disease. Experimental gerontology 2010, 45, 23–29. [Google Scholar] [CrossRef]
- Bouwman, F.H.; Frisoni, G.B.; Johnson, S.C.; Chen, X.; Engelborghs, S.; Ikeuchi, T.; Paquet, C.; Ritchie, C.; Bozeat, S.; Quevenco, F.C.; et al. Clinical application of CSF biomarkers for Alzheimer’s disease: From rationale to ratios. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2022, 14, e12314. [Google Scholar] [CrossRef]
- Mahaman, Y.A.R.; Embaye, K.S.; Huang, F.; Li, L.; Zhu, F.; Wang, J.Z.; Liu, R.; Feng, J.; Wang, X. Biomarkers used in Alzheimer’s disease diagnosis, treatment, and prevention. Ageing research reviews 2022, 74, 101544. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Leoni, V. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer’s disease. Clinical chemistry and laboratory medicine 2011, 49, 375–383. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature Reviews Neurology 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Jack Jr, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Provenzano, F.A.; Small, S.A.; Initiative, A.D.N. A deep learning MRI approach outperforms other biomarkers of prodromal Alzheimer’s disease. Alzheimer’s Research & Therapy. [CrossRef]
- Saleem, T.J.; Zahra, S.R.; Wu, F.; Alwakeel, A.; Alwakeel, M.; Jeribi, F.; Hijji, M. Deep Learning-Based Diagnosis of Alzheimer’s Disease. Journal of Personalized Medicine 2022, 12, 815. [Google Scholar] [CrossRef]
- Ren, R.J.; Dammer, E.B.; Wang, G.; Seyfried, N.T.; Levey, A.I. Proteomics of protein post-translational modifications implicated in neurodegeneration. Translational neurodegeneration 2014, 3, 1–13. [Google Scholar] [CrossRef]
- Sathe, G.; Na, C.H.; Renuse, S.; Madugundu, A.; Albert, M.; Moghekar, A.; Pandey, A. Phosphotyrosine profiling of human cerebrospinal fluid. Clinical proteomics 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.R.; Bach, S.B.; Perry, G. Analysis of post-translational modifications in Alzheimer’s disease by mass spectrometry. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2019, 1865, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Kayed, R.; Abate, G.; Uberti, D.; Kinnon, P.; Piccirella, S. Post-translational Modifications of the p53 Protein and the Impact in Alzheimer’s Disease: A Review of the Literature. Frontiers in Aging Neuroscience 2022, 14, 835288. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.; Jacquemin, C.; Villain, N.; Fenaille, F.; Lamari, F.; Becher, F. Mass spectrometry for neurobiomarker discovery: The relevance of post-translational modifications. Cells 2022, 11, 1279. [Google Scholar] [CrossRef] [PubMed]
- Mouton, P.R.; Martin, L.J.; Calhoun, M.E.; Dal Forno, G.; Price, D.L. Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s dementia. Neurobiology of aging 1998, 19, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Nestor, S.M.; Rupsingh, R.; Borrie, M.; Smith, M.; Accomazzi, V.; Wells, J.L.; Fogarty, J.; Bartha, R.; Initiative, A.D.N. Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain 2008, 131, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: a review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Beata, B.K.; Wojciech, J.; Johannes, K.; Piotr, L.; Barbara, M. Alzheimer’s disease—biochemical and psychological background for diagnosis and treatment. International Journal of Molecular Sciences 2023, 24, 1059. [Google Scholar] [CrossRef] [PubMed]
- Meftah, S.; Gan, J. Alzheimer’s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Frontiers in Synaptic Neuroscience 2023, 15, 1129036. [Google Scholar] [CrossRef]
- Tang, J.; Oliveros, A.; Jang, M.H. Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. International neurourology journal 2019, 23, S5. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Molecular Neurodegeneration 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sabatini, B.L.; Südhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harbor perspectives in biology 2012, 4, a005777. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial dysfunction in Alzheimer’s disease: opportunities for drug development. Current Neuropharmacology 2022, 20, 675. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Ashton, N.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; Zetterberg, H.; et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho-tau as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. European journal of nuclear medicine and molecular imaging 2021, 48, 2121–2139. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Biological psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. New England Journal of Medicine 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. New England Journal of Medicine 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiology of disease 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Hye, A.; Lynham, S.; Thambisetty, M.; Causevic, M.; Campbell, J.; Byers, H.; Hooper, C.; Rijsdijk, F.; Tabrizi, S.; Banner, S.; et al. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 2006, 129, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Hodes, R.J.; Buckholtz, N. Accelerating medicines partnership: Alzheimer’s disease (AMP-AD) knowledge portal aids Alzheimer’s drug discovery through open data sharing. Expert opinion on therapeutic targets 2016, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Hernández, F.; Avila, J. Protein biomarkers for the diagnosis of Alzheimer’s disease at different stages of neurodegeneration. International Journal of Molecular Sciences 2020, 21, 6749. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Sharma, A.; Kumar, D.; Asthana, M.K.; Lalhlenmawia, H.; Kumar, A.; Bhattacharyya, S.; Kumar, D. Promising protein biomarkers in the early diagnosis of Alzheimer’s disease. Metabolic Brain Disease 2022, 37, 1727–1744. [Google Scholar] [CrossRef] [PubMed]
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvänen, S.; Schiöth, H.B. Advances in the development of new biomarkers for Alzheimer’s disease. Translational Neurodegeneration 2022, 11, 1–24. [Google Scholar] [CrossRef]
- Haytural, H.; Benfeitas, R.; Schedin-Weiss, S.; Bereczki, E.; Rezeli, M.; Unwin, R.D.; Wang, X.; Dammer, E.B.; Johnson, E.C.; Seyfried, N.T.; et al. Insights into the changes in the proteome of Alzheimer disease elucidated by a meta-analysis. Scientific data 2021, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Biofluid-based biomarkers for Alzheimer’s disease–related pathologies: An update and synthesis of the literature. Alzheimer’s & Dementia 2022, 18, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Penque, D. Two-dimensional gel electrophoresis and mass spectrometry for biomarker discovery. Proteomics–Clinical Applications 2009, 3, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Catherman, A.D.; Skinner, O.S.; Kelleher, N.L. Top down proteomics: facts and perspectives. Biochemical and biophysical research communications 2014, 445, 683–693. [Google Scholar] [CrossRef]
- Mori, H.; Takio, K.; Ogawara, M.; Selkoe, D. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. Journal of Biological Chemistry 1992, 267, 17082–17086. [Google Scholar] [CrossRef]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. Journal of Biological Chemistry 1992, 267, 17047–17054. [Google Scholar] [CrossRef]
- Drummond, E.; Nayak, S.; Faustin, A.; Pires, G.; Hickman, R.A.; Askenazi, M.; Cohen, M.; Haldiman, T.; Kim, C.; Han, X.; et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta neuropathologica 2017, 133, 933–954. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Ge, W.; Ma, C. Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimer’s & Dementia 2019, 15, 429–440. [Google Scholar] [CrossRef]
- Hesse, R.; Hurtado, M.L.; Jackson, R.J.; Eaton, S.L.; Herrmann, A.G.; Colom-Cadena, M.; Tzioras, M.; King, D.; Rose, J.; Tulloch, J.; et al. Comparative profiling of the synaptic proteome from Alzheimer’s disease patients with focus on the APOE genotype. Acta neuropathologica communications 2019, 7, 1–18. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Nouwens, A.S.; Dodd, P.R.; Etheridge, N. The synaptic proteome in Alzheimer’s disease. Alzheimer’s & Dementia 2013, 9, 499–511. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Molecular brain 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Johnson, E.C.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature medicine 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.H.; Hortobágyi, T.; Winblad, B.; Ballard, C.; Lehtiö, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 2020, 105, 975–991. [Google Scholar] [CrossRef]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. The nature and pathogenesis of the amyloid deposits in Alzheimer’s disease. In Amyloidosis; Springer, 1986; pp. 227–242. [CrossRef]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Edwards, F.A. A unifying hypothesis for Alzheimer’s disease: from plaques to neurodegeneration. Trends in neurosciences 2019, 42, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.w. The γ-secretase complex: from structure to function. Frontiers in cellular neuroscience 2014, 8, 427. [Google Scholar] [CrossRef]
- Di Carlo, M.; Giacomazza, D.; San Biagio, P. Alzheimer’s disease: biological aspects, therapeutic perspectives and diagnostic tools. Journal of Physics: Condensed Matter 2012, 24, 244102. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. The Lancet Neurology 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Frontiers in cellular neuroscience 2015, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Hyman, B.T.; Spires-Jones, T.L. Alzheimer’s disease: synapses gone cold. Molecular neurodegeneration 2011, 6, 1–9. [Google Scholar] [CrossRef]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-β peptide. Journal of Biological Chemistry 2017, 292, 3172–3185. [Google Scholar] [CrossRef]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β modification: a key to the sporadic Alzheimer’s disease? Frontiers in genetics 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 2021, 144, 310–324. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aß to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, X.; Ma, L.; Wei, W.; Li, Z.; Chang, S.; Wen, J.; Sun, J.; Li, H. Role of Aβ in Alzheimer’s-related synaptic dysfunction. Frontiers in Cell and Developmental Biology 2022, 10, 964075. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Frontiers in pharmacology 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomedical reports 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L. Targeting autophagy for the treatment of Alzheimer’s disease: challenges and opportunities. Frontiers in molecular neuroscience 2019, 12, 203. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Current neuropharmacology 2017, 15, 926–935. [Google Scholar] [CrossRef]
- Hampel, H.; Goernitz, A.; Buerger, K. Advances in the development of biomarkers for Alzheimer’s disease: from CSF total tau and Aβ1–42 proteins to phosphorylated tau protein. Brain research bulletin 2003, 61, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Archives of neurology 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Shigemoto, Y.; Sato, N. Neuroimaging of Alzheimer’s disease: focus on amyloid and tau PET. Japanese journal of radiology 2019, 37, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, N.T.; Dammer, E.B.; Swarup, V.; Nandakumar, D.; Duong, D.M.; Yin, L.; Deng, Q.; Nguyen, T.; Hales, C.M.; Wingo, T.; et al. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell systems 2017, 4, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Experimental & molecular medicine 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Shi, J.; Arnold, L.; McGeer, P. [P2–255]: SALIVARY AMYLOID-BETA PROTEIN LEVELS CAN DIAGNOSE ALZHEIMER DISEASE AND PREDICT ITS FUTURE ONSET. Alzheimer’s & Dementia 2017, 13, P710–P711. [Google Scholar] [CrossRef]
- Hussain, A.; Sheikh, Z.; Subramanian, M. The Eye as a Diagnostic Tool for Alzheimer’s Disease. Life 2023, 13, 726. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Sauer, M.; Visser, P.J.; Tijms, B.M.; Vorontsov, E.; Blennow, K.; Zetterberg, H.; Gobom, J. Optimized sample preparation and data analysis for TMT proteomic analysis of cerebrospinal fluid applied to the identification of Alzheimer’s disease biomarkers. Clinical Proteomics 2022, 19, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gholami, A. Alzheimer’s disease: The role of proteins in formation, mechanisms, and new therapeutic approaches. Neuroscience Letters, 1375. [Google Scholar] [CrossRef]
- Lewczuk, P.; Esselmann, H.; Meyer, M.; Wollscheid, V.; Neumann, M.; Otto, M.; Maler, J.M.; Rüther, E.; Kornhuber, J.; Wiltfang, J. The amyloid-β (Aβ) peptide pattern in cerebrospinal fluid in Alzheimer’s disease: evidence of a novel carboxyterminally elongated Aβ peptide. Rapid Communications in Mass Spectrometry 2003, 17, 1291–1296. [Google Scholar] [CrossRef]
- Spies, P.E.; Verbeek, M.M.; van Groen, T.; Claassen, J.A. Reviewing reasons for the decreased CSF Abeta42 concentration in Alzheimer disease 2012. [CrossRef]
- Begcevic, I.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Goldhardt, O.; Grimmer, T.; Magdolen, V.; Batruch, I.; Diamandis, E.P. Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. Journal of Proteomics 2018, 182, 12–20. [Google Scholar] [CrossRef]
- van Harten, A.C.; Visser, P.J.; Pijnenburg, Y.A.; Teunissen, C.E.; Blankenstein, M.A.; Scheltens, P.; van der Flier, W.M. Cerebrospinal fluid Aβ42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimer’s & dementia 2013, 9, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, M.; Gisslén, M.; Vanmechelen, E.; Blennow, K. Low cerebrospinal fluid β-amyloid 42 in patients with acute bacterial meningitis and normalization after treatment. Neuroscience letters 2001, 314, 33–36. [Google Scholar] [CrossRef]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. The world journal of biological psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Andreasson, U.; Londos, E.; Minthon, L.; Blennow, K. Prediction of Alzheimer’s disease using the CSF Aβ42/Aβ40 ratio in patients with mild cognitive impairment. Dementia and geriatric cognitive disorders 2007, 23, 316–320. [Google Scholar] [CrossRef]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Annals of clinical and translational neurology 2016, 3, 154–165. [Google Scholar] [CrossRef]
- Lewczuk, P.; Esselmann, H.; Otto, M.; Maler, J.M.; Henkel, A.W.; Henkel, M.K.; Eikenberg, O.; Antz, C.; Krause, W.R.; Reulbach, U.; et al. Neurochemical diagnosis of Alzheimer’s dementia by CSF Aβ42, Aβ42/Aβ40 ratio and total tau. Neurobiology of aging 2004, 25, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Fandos, N.; Pérez-Grijalba, V.; Pesini, P.; Olmos, S.; Bossa, M.; Villemagne, V.L.; Doecke, J.; Fowler, C.; Masters, C.L.; Sarasa, M.; et al. Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2017, 8, 179–187. [Google Scholar] [CrossRef]
- Hansson, O.; Zetterberg, H.; Vanmechelen, E.; Vanderstichele, H.; Andreasson, U.; Londos, E.; Wallin, A.; Minthon, L.; Blennow, K. Evaluation of plasma Aβ40 and Aβ42 as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiology of aging 2010, 31, 357–367. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Mori, H.; Takio, K.; Ogawara, M.; Selkoe, D.J. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. Journal of Biological Chemistry 1992, 267, 17082–17086. [Google Scholar] [CrossRef]
- Wang, R.; Sweeney, D.; Gandy, S.E.; Sisodia, S.S. The Profile of Soluble Amyloid β Protein in Cultured Cell Media: detection and quantification of amyloid β protein and variants by immunoprecipitation-mass spectrometry. Journal of Biological Chemistry 1996, 271, 31894–31902. [Google Scholar] [CrossRef] [PubMed]
- Oe, T.; Ackermann, B.L.; Inoue, K.; Berna, M.J.; Garner, C.O.; Gelfanova, V.; Dean, R.A.; Siemers, E.R.; Holtzman, D.M.; Farlow, M.R.; et al. Quantitative analysis of amyloid β peptides in cerebrospinal fluid of Alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass spectrometry. Rapid Communications in Mass Spectrometry: An International Journal Devoted to the Rapid Dissemination of Up-to-the-Minute Research in Mass Spectrometry 2006, 20, 3723–3735. [Google Scholar] [CrossRef] [PubMed]
- Bos, I.; Vos, S.; Verhey, F.; Scheltens, P.; Teunissen, C.; Engelborghs, S.; Sleegers, K.; Frisoni, G.; Blin, O.; Richardson, J.C.; et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer’s disease spectrum. Alzheimer’s & Dementia 2019, 15, 644–654. [Google Scholar] [CrossRef]
- Visser, P.J.; Reus, L.M.; Gobom, J.; Jansen, I.; Dicks, E.; Van der Lee, S.J.; Tsolaki, M.; Verhey, F.R.; Popp, J.; Martinez-Lage, P.; et al. Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer’s disease. Molecular neurodegeneration 2022, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Gabelle, A.; Hirtz, C.; Fenaille, F.; Sergeant, N.; Schraen-Maschke, S.; Vialaret, J.; Buée, L.; Junot, C.; Becher, F.; et al. Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with lewy bodies. Journal of Alzheimer’s Disease 2016, 51, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ortiz, F.; Kac, P.R.; Brum, W.S.; Zetterberg, H.; Blennow, K.; Karikari, T.K. Plasma phospho-tau in Alzheimer’s disease: towards diagnostic and therapeutic trial applications. Molecular neurodegeneration 2023, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Benedet, A.L.; Pascoal, T.A.; Karikari, T.K.; Lantero-Rodriguez, J.; Brum, W.S.; Mathotaarachchi, S.; Therriault, J.; Savard, M.; Chamoun, M.; et al. Cerebrospinal fluid p-tau231 as an early indicator of emerging pathology in Alzheimer’s disease. EBioMedicine 2022, 76. [Google Scholar] [CrossRef] [PubMed]
- Metaxas, A.; Kempf, S.J. Neurofibrillary tangles in Alzheimer’s disease: elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural regeneration research 2016, 11, 1579. [Google Scholar] [CrossRef] [PubMed]
- Herukka, S.K.; Simonsen, A.H.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S.; et al. Recommendations for cerebrospinal fluid Alzheimer’s disease biomarkers in the diagnostic evaluation of mild cognitive impairment. Alzheimer’s & Dementia 2017, 13, 285–295. [Google Scholar] [CrossRef]
- Pottiez, G.; Yang, L.; Stewart, T.; Song, N.; Aro, P.; Galasko, D.R.; Quinn, J.F.; Peskind, E.R.; Shi, M.; Zhang, J. Mass-spectrometry-based method to quantify in parallel tau and amyloid β 1–42 in CSF for the diagnosis of Alzheimer’s disease. Journal of proteome research 2017, 16, 1228–1238. [Google Scholar] [CrossRef]
- Seppälä, T.; Nerg, O.; Koivisto, A.; Rummukainen, J.; Puli, L.; Zetterberg, H.; Pyykkö, O.; Helisalmi, S.; Alafuzoff, I.; Hiltunen, M.; et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 2012, 78, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nature Reviews Neurology 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Strozyk, D.; Blennow, K.; White, L.; Launer, L. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Ardanaz, C.G.; Ramírez, M.J.; Solas, M. Brain metabolic alterations in Alzheimer’s disease. International Journal of Molecular Sciences 2022, 23, 3785. [Google Scholar] [CrossRef] [PubMed]
- Rummel, N.G.; Butterfield, D.A. Altered metabolism in Alzheimer disease brain: Role of oxidative stress. Antioxidants & Redox Signaling 2022, 36, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.V.; Proza, J.F.; Sohrabji, F.; Lawler, J.M. Vascular and metabolic dysfunction in Alzheimer’s disease: a review. Experimental biology and medicine 2011, 236, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunology and cell biology 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. The international journal of biochemistry & cell biology 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for neuroinflammation in Alzheimer’s disease. Progress in Neurology and Psychiatry 2016, 20, 25–31. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. Journal of Neuroscience 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radical Biology and Medicine 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends in pharmacological sciences 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative stress and beta amyloid in Alzheimer’s disease. Which comes first: The chicken or the egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.; Montine, T.J. Biomarkers of oxidative damage and inflammation in Alzheimer’s disease. Biomarkers in medicine 2010, 4, 27–36. [Google Scholar] [CrossRef]
- Bader, J.M.; Geyer, P.E.; Müller, J.B.; Strauss, M.T.; Koch, M.; Leypoldt, F.; Koertvelyessy, P.; Bittner, D.; Schipke, C.G.; Incesoy, E.I.; et al. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Molecular systems biology 2020, 16, e9356. [Google Scholar] [CrossRef]
- Rehiman, S.H.; Lim, S.M.; Neoh, C.F.; Majeed, A.B.A.; Chin, A.V.; Tan, M.P.; Kamaruzzaman, S.B.; Ramasamy, K. Proteomics as a reliable approach for discovery of blood-based Alzheimer’s disease biomarkers: A systematic review and meta-analysis. Ageing research reviews 2020, 60, 101066. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of oxidative damage in alzheimer’s disease and neurodegeneration: From pathogenic mechanisms to biomarker discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Mielke, M.M.; Syrjanen, J.A.; Blennow, K.; Zetterberg, H.; Vemuri, P.; Skoog, I.; Machulda, M.M.; Kremers, W.K.; Knopman, D.S.; Jack Jr, C.; et al. Plasma and CSF neurofilament light: relation to longitudinal neuroimaging and cognitive measures. Neurology 2019, 93, e252–e260. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, K.; Gupta, V.B.; Villemagne, V.L.; Eratne, D.; Graham, P.L.; Fowler, C.; Bourgeat, P.; Li, Q.X.; Collins, S.; Bush, A.I.; et al. Cerebrospinal fluid neurofilament light concentration predicts brain atrophy and cognition in Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2020, 12, e12005. [Google Scholar] [CrossRef] [PubMed]
- Khoonsari, P.E.; Häggmark, A.; Lönnberg, M.; Mikus, M.; Kilander, L.; Lannfelt, L.; Bergquist, J.; Ingelsson, M.; Nilsson, P.; Kultima, K.; et al. Analysis of the cerebrospinal fluid proteome in Alzheimer’s disease. PloS one 2016, 11, e0150672. [Google Scholar] [CrossRef]
- Davidsson, P.; Sjögren, M. The use of proteomics in biomarker discovery in neurodegenerative diseases. Disease markers.
- Ho, L.; Sharma, N.; Blackman, L.; Festa, E.; Reddy, G.; Pasinetti, G.M. From proteomics to biomarker discovery in Alzheimer’s disease. Brain research reviews 2005, 48, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Cilento, E.M.; Jin, L.; Stewart, T.; Shi, M.; Sheng, L.; Zhang, J. Mass spectrometry: A platform for biomarker discovery and validation for Alzheimer’s and Parkinson’s diseases. Journal of neurochemistry 2019, 151, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Haque, R.U.; Dammer, E.B.; Duong, D.M.; Ping, L.; Johnson, E.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Targeted mass spectrometry to quantify brain-derived cerebrospinal fluid biomarkers in Alzheimer’s disease. Clinical proteomics 2020, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.M.; Dammer, E.B.; Ping, L.; Duong, D.M.; Modeste, E.; Carter, E.K.; Johnson, E.C.; Levey, A.I.; Lah, J.J.; Roberts, B.R.; et al. Quantitative Mass Spectrometry Analysis of Cerebrospinal Fluid Protein Biomarkers in Alzheimer’s Disease. Scientific Data 2023, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brandi, J.; Noberini, R.; Bonaldi, T.; Cecconi, D. Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. Journal of Chromatography A, 4633. [Google Scholar]
- Sarihan, M.; Albayrak, M.G.B.; Kasap, M.; Akpinar, G.; Kocyigit, E. An experimental workflow for enrichment of low abundant proteins from human serum for the discovery of serum biomarkers. Journal of biological methods 2023, 10. [Google Scholar] [CrossRef]
- Cellar, N.A.; Karnoup, A.S.; Albers, D.R.; Langhorst, M.L.; Young, S.A. Immunodepletion of high abundance proteins coupled on-line with reversed-phase liquid chromatography: a two-dimensional LC sample enrichment and fractionation technique for mammalian proteomics. Journal of Chromatography B 2009, 877, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Baldacci, F.; Lista, S.; Cavedo, E.; Bonuccelli, U.; Hampel, H. Diagnostic function of the neuroinflammatory biomarker YKL-40 in Alzheimer’s disease and other neurodegenerative diseases. Expert review of proteomics 2017, 14, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Mavroudis, I.; Chowdhury, R.; Petridis, F.; Karantali, E.; Chatzikonstantinou, S.; Balmus, I.M.; Luca, I.S.; Ciobica, A.; Kazis, D. YKL-40 as a Potential Biomarker for the Differential Diagnosis of Alzheimer’s Disease. Medicina 2021, 58, 60. [Google Scholar] [CrossRef]
- Gispert, J.D.; Monté, G.C.; Falcon, C.; Tucholka, A.; Rojas, S.; Sánchez-Valle, R.; Antonell, A.; Lladó, A.; Rami, L.; Molinuevo, J.L. CSF YKL-40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiology of aging 2016, 38, 47–55. [Google Scholar] [CrossRef]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Molecular neurodegeneration 2016, 11, 1–7. [Google Scholar] [CrossRef]
- Sun, Y.; Yin, X.S.; Guo, H.; Han, R.K.; He, R.D.; Chi, L.J.; et al. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediators of inflammation 2013, 2013. [Google Scholar] [CrossRef]
- Gangishetti, U.; Christina Howell, J.; Perrin, R.J.; Louneva, N.; Watts, K.D.; Kollhoff, A.; Grossman, M.; Wolk, D.A.; Shaw, L.M.; Morris, J.C.; et al. Non-beta-amyloid/tau cerebrospinal fluid markers inform staging and progression in Alzheimer’s disease. Alzheimer’s research & therapy 2018, 10, 1–10. [Google Scholar] [CrossRef]
- Khan, W.; Aguilar, C.; Kiddle, S.J.; Doyle, O.; Thambisetty, M.; Muehlboeck, S.; Sattlecker, M.; Newhouse, S.; Lovestone, S.; Dobson, R.; et al. A subset of cerebrospinal fluid proteins from a multi-analyte panel associated with brain atrophy, disease classification and prediction in Alzheimer’s disease. PloS one 2015, 10, e0134368. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. Journal of neuroinflammation 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Elbaz, I.; Lerer-Goldshtein, T.; Okamoto, H.; Appelbaum, L. Reduced synaptic density and deficient locomotor response in neuronal activity-regulated pentraxin 2a mutant zebrafish. The FASEB Journal 2015, 29, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.F.; Xu, D.; Craig, M.T.; Pelkey, K.A.; Chien, C.C.; Shi, Y.; Zhang, J.; Resnick, S.; Pletnikova, O.; Salmon, D.; et al. NPTX2 and cognitive dysfunction in Alzheimer’s Disease. Elife 2017, 6, e23798. [Google Scholar] [CrossRef]
- Swanson, A.; Willette, A.; Initiative, A.D.N.; et al. Neuronal Pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer’s disease spectrum. Brain, behavior, and immunity 2016, 58, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Stumm, C.; Hiebel, C.; Hanstein, R.; Purrio, M.; Nagel, H.; Conrad, A.; Lutz, B.; Behl, C.; Clement, A.B. Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiology of aging 2013, 34, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Camporesi, E.; Nilsson, J.; Brinkmalm, A.; Becker, B.; Ashton, N.J.; Blennow, K.; Zetterberg, H. Fluid biomarkers for synaptic dysfunction and loss. Biomarker insights 2020, 15, 1177271920950319. [Google Scholar] [CrossRef]
- Dayon, L.; Núñez Galindo, A.; Wojcik, J.; Cominetti, O.; Corthésy, J.; Oikonomidi, A.; Henry, H.; Kussmann, M.; Migliavacca, E.; Severin, I.; et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimer’s research & therapy 2018, 10, 1–12. [Google Scholar] [CrossRef]
- Blennow, K. A review of fluid biomarkers for Alzheimer’s disease: moving from CSF to blood. Neurology and therapy 2017, 6, 15–24. [Google Scholar] [CrossRef]
- Wang, H.; Dey, K.K.; Chen, P.C.; Li, Y.; Niu, M.; Cho, J.H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Molecular neurodegeneration 2020, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimer’s & Dementia 2023, 19, 333–342. [Google Scholar] [CrossRef]
- Liu, P.; Li, L.; He, F.; Meng, F.; Liu, X.; Su, Y.; Su, X.; Luo, B.; Peng, G. Identification of Candidate Biomarkers of Alzheimer’s Disease via Multiplex Cerebrospinal Fluid and Serum Proteomics. International Journal of Molecular Sciences 2023, 24, 14225. [Google Scholar] [CrossRef] [PubMed]
- Öhrfelt, A.; Johansson, P.; Wallin, A.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Svensson, J. Increased cerebrospinal fluid levels of ubiquitin carboxyl-terminal hydrolase L1 in patients with Alzheimer’s disease. Dementia and geriatric cognitive disorders extra 2016, 6, 283–294. [Google Scholar] [CrossRef]
- Chiasserini, D.; Parnetti, L.; Andreasson, U.; Zetterberg, H.; Giannandrea, D.; Calabresi, P.; Blennow, K. CSF levels of heart fatty acid binding protein are altered during early phases of Alzheimer’s disease. Journal of Alzheimer’s Disease 2010, 22, 1281–1288. [Google Scholar] [CrossRef]
- Paciotti, S.; Wojdała, A.L.; Bellomo, G.; Toja, A.; Chipi, E.; Piersma, S.R.; Pham, T.V.; Gaetani, L.; Jimenez, C.R.; Parnetti, L.; et al. Potential diagnostic value of CSF metabolism-related proteins across the Alzheimer’s disease continuum. Alzheimer’s Research & Therapy 2023, 15, 124. [Google Scholar] [CrossRef]
- Higginbotham, L.; Ping, L.; Dammer, E.B.; Duong, D.M.; Zhou, M.; Gearing, M.; Hurst, C.; Glass, J.D.; Factor, S.A.; Johnson, E.C.; et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science advances 2020, 6, eaaz9360. [Google Scholar] [CrossRef] [PubMed]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta neuropathologica communications 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Miners, J.S.; Baig, S.; Palmer, J.; Palmer, L.E.; Kehoe, P.G.; Love, S. SYMPOSIUM: Clearance of Aβ from the Brain in Alzheimer’s Disease: Aβ-Degrading Enzymes in Alzheimer’s Disease. Brain pathology 2008, 18, 240–252. [Google Scholar] [CrossRef]
- Hoshi, K.; Ito, H.; Abe, E.; Fuwa, T.J.; Kanno, M.; Murakami, Y.; Abe, M.; Murakami, T.; Yoshihara, A.; Ugawa, Y.; et al. Transferrin biosynthesized in the brain is a novel biomarker for Alzheimer’s disease. Metabolites 2021, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clinical chemistry 2010, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: history, character, and diagnostic prospects. Molecular & cellular proteomics 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.L.; Westwood, S.; Lovestone, S. Blood-based proteomic biomarkers of Alzheimer’s disease pathology. Frontiers in neurology 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews Neurology 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Budelier, M.M.; Bateman, R.J. Biomarkers of Alzheimer disease. The journal of applied laboratory medicine 2020, 5, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Hampel, H.; Buerger, K. Biological marker candidates of Alzheimer’s disease in blood, plasma, and serum. CNS neuroscience & therapeutics 2009, 15, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Vigo-Pelfrey, C.; Teplow, D.B.; Miller, C.; Schenk, D.; Johnston, J.; Winblad, B.; Venizelos, N.; Lannfelt, L.; Selkoe, D.J. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proceedings of the National Academy of Sciences 1994, 91, 11993–11997. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Perez-Grijalba, V.; Romero, J.; Pesini, P.; Sarasa, L.; Monleon, I.; San-Jose, I.; Arbizu, J.; Martinez-Lage, P.; Munuera, J.; Ruiz, A.; et al. Plasma Aβ42/40 ratio detects early stages of Alzheimer’s disease and correlates with CSF and neuroimaging biomarkers in the AB255 study. The Journal of Prevention of Alzheimer’s Disease 2019, 6, 34–41. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. Journal of Experimental Medicine 2020, 217, e20200861. [Google Scholar] [CrossRef] [PubMed]
- Chhatwal, J.P.; Schultz, A.P.; Dang, Y.; Ostaszewski, B.; Liu, L.; Yang, H.S.; Johnson, K.A.; Sperling, R.A.; Selkoe, D.J. Plasma N-terminal tau fragment levels predict future cognitive decline and neurodegeneration in healthy elderly individuals. Nature communications 2020, 11, 6024. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. Jama 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, A.; Karikari, T.K.; Poole, T.; Ashton, N.J.; Lantero Rodriguez, J.; Khatun, A.; Swift, I.; Heslegrave, A.J.; Abel, E.; Chung, E.; et al. Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: a longitudinal cohort study. Molecular psychiatry 2021, 26, 5967–5976. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, D.S.; Ashton, N.J.; Blennow, K.; Zetterberg, H.; Simrén, J.; Lantero-Rodriguez, J.; Karikari, T.K.; Hiniker, A.; Rissman, R.A.; Salmon, D.P.; et al. Plasma biomarkers for Alzheimer’s disease in relation to neuropathology and cognitive change. Acta neuropathologica 2022, 143, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Tissot, C.; L. Benedet, A.; Therriault, J.; Pascoal, T.A.; Lussier, F.Z.; Saha-Chaudhuri, P.; Chamoun, M.; Savard, M.; Mathotaarachchi, S.S.; Bezgin, G.; et al. Plasma pTau181 predicts cortical brain atrophy in aging and Alzheimer’s disease. Alzheimer’s Research & Therapy 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Simrén, J.; Leuzy, A.; Karikari, T.K.; Hye, A.; Benedet, A.L.; Lantero-Rodriguez, J.; Mattsson-Carlgren, N.; Schöll, M.; Mecocci, P.; Vellas, B.; et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimer’s & Dementia 2021, 17, 1145–1156. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nature medicine 2020, 26, 379–386. [Google Scholar] [CrossRef]
- McGrath, E.R.; Beiser, A.S.; O’Donnell, A.; Yang, Q.; Ghosh, S.; Gonzales, M.M.; Himali, J.J.; Satizabal, C.L.; Johnson, K.A.; Tracy, R.P.; et al. Blood phosphorylated tau 181 as a biomarker for amyloid burden on brain PET in cognitively healthy adults. Journal of Alzheimer’s Disease 2022, 87, 1517–1526. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. The Lancet Neurology 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nature Reviews Neurology 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Kamada, M.; Kawamura, Y.; Terao, C.; Shimoda, F.; Tomita, N.; Arai, H.; Furukawa, K. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. Journal of neurochemistry 2016, 136, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.B.; Aggarwal, N.T.; McAninch, E.A.; Weuve, J.; Barnes, L.L.; Wilson, R.S.; DeCarli, C.; Evans, D.A. Remote blood biomarkers of longitudinal cognitive outcomes in a population study. Annals of neurology 2020, 88, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Cicognola, C.; Janelidze, S.; Hertze, J.; Zetterberg, H.; Blennow, K.; Mattsson-Carlgren, N.; Hansson, O. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer’s research & therapy 2021, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Maxwell, S.; Bena, J.; Bekris, L.M.; Rao, S.M.; Chance, M.; Lamb, B.T.; Leverenz, J.B.; Initiative, A.D.N. Key inflammatory pathway activations in the MCI stage of Alzheimer’s disease. Annals of Clinical and Translational Neurology 2019, 6, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Meri, S. Yin and Yang: complement activation and regulation in Alzheimer’s disease. Progress in neurobiology 2003, 70, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Suzuki, H.; Meno, K.; Liu, S.; Korenaga, T.; Uchida, K. Identification of Plasma Proteins as Biomarkers for Mild Cognitive Impairment and Alzheimer’s Disease Using Liquid Chromatography–Tandem Mass Spectrometry. International Journal of Molecular Sciences 2023, 24, 13064. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Wang, L.; Prabakaran, S.; Wengenroth, M.; Lockstone, H.; Koethe, D.; Gerth, C.; Gross, S.; Schreiber, D.; Lilley, K.; et al. Independent protein-profiling studies show a decrease in apolipoprotein A1 levels in schizophrenia CSF, brain and peripheral tissues. Molecular psychiatry 2008, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Namkoong, H.; Kim, H.K.; Kim, S.; Hwang, D.W.; Na, H.R.; Ha, S.A.; Kim, J.R.; Kim, J.W. Fibrinogen gamma-A chain precursor in CSF: a candidate biomarker for Alzheimer’s disease. BMC neurology 2007, 7, 1–6. [Google Scholar] [CrossRef]
- Lane, E.M.; Hohman, T.J.; Jefferson, A.L.; Initiative, A.D.N. Insulin-like growth factor binding protein-2 interactions with Alzheimer’s disease biomarkers. Brain imaging and behavior 2017, 11, 1779–1786. [Google Scholar] [CrossRef]
- Zhao, L.; Jia, X.; Shankar, S.; Olofsson, A.; Brännström, T.; Mu, Y.; Gräslund, A.; Morozova-Roche, L.A.; Jarvet, J.; Wang, C.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade 2013. [CrossRef]
- Wang, C.; Iashchishyn, I.A.; Pansieri, J.; Nyström, S.; Klementieva, O.; Kara, J.; Horvath, I.; Moskalenko, R.; Rofougaran, R.; Gouras, G.; et al. S100A9-driven amyloid-neuroinflammatory cascade in traumatic brain injury as a precursor state for Alzheimer’s disease. Scientific Reports 2018, 8, 12836. [Google Scholar] [CrossRef] [PubMed]
- Kononikhin, A.S.; Zakharova, N.V.; Semenov, S.D.; Bugrova, A.E.; Brzhozovskiy, A.G.; Indeykina, M.I.; Fedorova, Y.B.; Kolykhalov, I.V.; Strelnikova, P.A.; Ikonnikova, A.Y.; et al. Prognosis of Alzheimer’s disease using quantitative mass spectrometry of human blood plasma proteins and machine learning. International Journal of Molecular Sciences 2022, 23, 7907. [Google Scholar] [CrossRef] [PubMed]
- Kiddle, S.J.; Sattlecker, M.; Proitsi, P.; Simmons, A.; Westman, E.; Bazenet, C.; Nelson, S.K.; Williams, S.; Hodges, A.; Johnston, C.; et al. Candidate blood proteome markers of Alzheimer’s disease onset and progression: a systematic review and replication study. Journal of Alzheimer’s Disease 2014, 38, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Oudart, J.B.; Djerada, Z.; Nonnonhou, V.; Badr, S.; Bertholon, L.A.; Dammak, A.; Jaidi, Y.; Novella, J.L.; Pallet, N.; Gillery, P.; et al. Incremental Value of CSF Biomarkers in Clinically Diagnosed AD and Non-AD Dementia. Frontiers in Neurology 2020, 11, 560. [Google Scholar] [CrossRef] [PubMed]
- Peña-Bautista, C.; Álvarez-Sánchez, L.; Pascual, R.; Moreno, M.J.; Baquero, M.; Cháfer-Pericás, C. Clinical usefulness of cerebrospinal fluid biomarkers in Alzheimer’s disease. European Journal of Clinical Investigation 2023, 53, e13910. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Blennow, K. From cerebrospinal fluid to blood: the third wave of fluid biomarkers for Alzheimer’s disease. Journal of Alzheimer’s Disease 2018, 64, S271–S279. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nature Reviews Neurology 2018, 14, 639–652. [Google Scholar] [CrossRef]
Figure 1.
Progression from presymptomatic Alzheimer’s disease (AD) to severe AD condition. The percentage of subjects progressing from presymptomatic AD to mild cognitive impairment (MCI) ranges between 20 and 30%[22]. The percentage of subjects progressing from MCI to severe AD is higher, ranging between 43% up to 70% [23].
Figure 1.
Progression from presymptomatic Alzheimer’s disease (AD) to severe AD condition. The percentage of subjects progressing from presymptomatic AD to mild cognitive impairment (MCI) ranges between 20 and 30%[22]. The percentage of subjects progressing from MCI to severe AD is higher, ranging between 43% up to 70% [23].
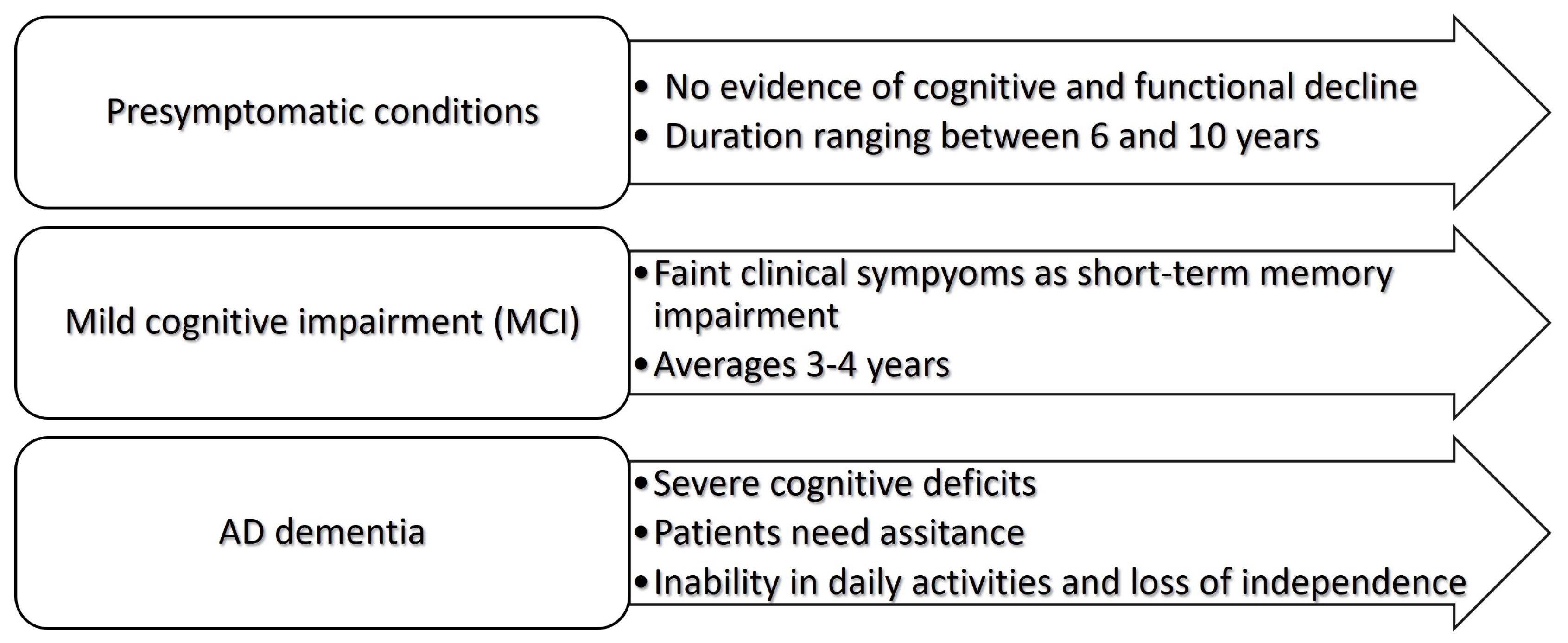
Figure 2.
Well-established (left table) and novel candidates (right table) for AD protein biomarkers in cerebrospinal fluid (CSF). Amyloid protein (A); total-tau (t-tau); hyperphosphorylated tau (p-tau); neurofilament light (NFl); astrocyte-derived chitinase-3-like protein 1 (YKL-40); neuropentraxin 2 (NPTX2); synaptotagmin-1 (SYT-1); synaptosomal-associated protein 25 (SNAP-25); soluble triggering receptor of myeloid cells 2 (sTREM2).
Figure 2.
Well-established (left table) and novel candidates (right table) for AD protein biomarkers in cerebrospinal fluid (CSF). Amyloid protein (A); total-tau (t-tau); hyperphosphorylated tau (p-tau); neurofilament light (NFl); astrocyte-derived chitinase-3-like protein 1 (YKL-40); neuropentraxin 2 (NPTX2); synaptotagmin-1 (SYT-1); synaptosomal-associated protein 25 (SNAP-25); soluble triggering receptor of myeloid cells 2 (sTREM2).
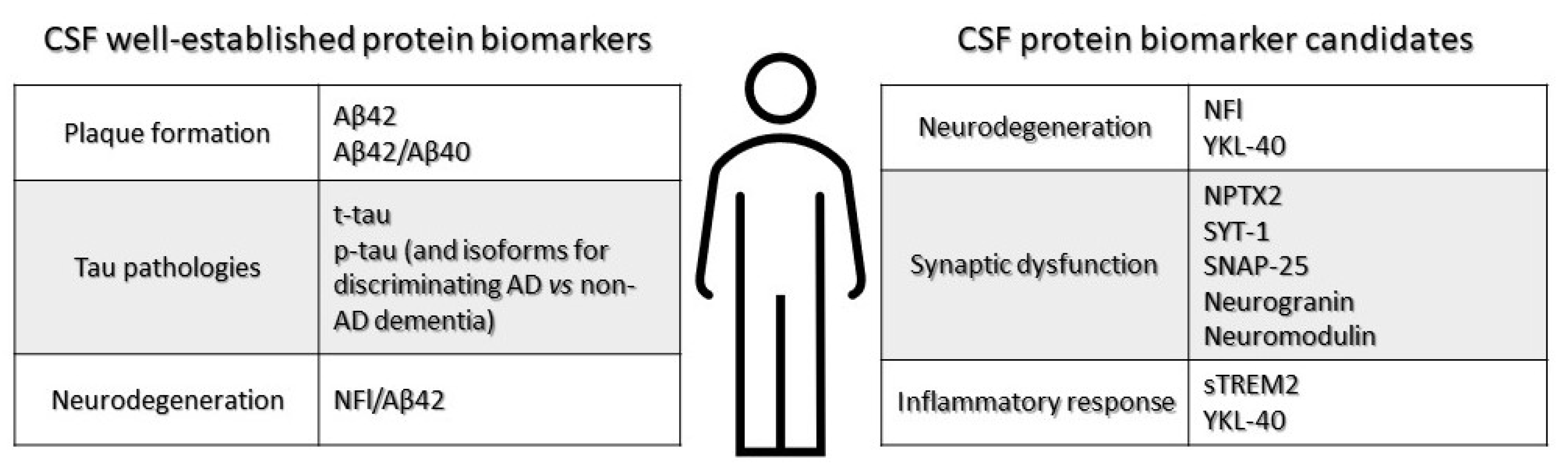
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



