Submitted:
19 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Purpose of Review Multipotent Mesenchymal stromal cells (MSCs) have recently risen to prominence in regenerative medicine for their powerful intrinsic properties of self-regeneration, immunomodulation, and multi-potency. They exhibited an excellent safety profile in early-phase clinical trials. Nevertheless, MSCs-based therapy suffers reduced efficacy in randomized clinical trials due to a poor understanding of their proper delivery modalities. Here, we intend to explore the current application of MSCs after tissue injuries. We specifically focus on tissue source, delivery routes, metabolic fitness, and cell dose. We further discuss potential approaches to optimize in vivo engraftment.
Recent findings MSCs therapeutic effect mediates by paracrine and contact factors, which are cells' intrinsic physiological processes. The cell culture expansion process does not destroy MSCs' intrinsic properties. In vivo persistence of administered MSCs determines their therapeutic potency. Cell viability, fitness, immune match, and delivery route meticulously regulate their in vivo persistency. Different genetic or chemical modification strategies are currently applied to extend their in vivo lifespan and boost their pharmaceutical effect.
Summary Improving the in vivo persistence of implanted MSCs could facilitate its clinical translation. This review highlights the pros and cons of different MSCs’ delivery strategies used in clinical or preclinical studies, emphasizing various modification approaches. These can promote prolonged graft cell survival.
Keywords:
Mesenchymal stromal cells
; route of delivery
; dose
; Immunogenicity
; Metabolic fitness
; Potency assay
Introduction
Mesenchymal stromal cells (MSCs) show great promise as a biological therapeutic for numerous inflammatory and autoimmune ailments owing to their unique immunoregulatory and regenerative properties (1). Culture-adopted MSCs secrete a wide range of small molecules, peptides, chemokines, cytokines, and morphogens, collectively named secretomes. Host immune cells reciprocally interact with secretomes to modulate the intensity of immune response at the tissue injury site (2). Besides the immunomodulatory and regenerative capacities, the unique tumor-homing property makes them an ideal candidate for carrying anti-cancer agents, for which they can participate in the resistance to various anti-cancer drugs (Table 1).
MSCs were first clinically explored as a cellular therapeutic in 1995 by Hillard Lazarus to accelerate hematopoietic recovery in human subjects (3). Since then, multiple clinical and preclinical studies have diligently tried to establish MSCs as a biopharmaceutical for several diseases. Despite the advancements of MSCs research over time, most of these MSC-based trials are still in early phase I or II, whereas only a few portions of them (less than 50) are in phase III, and currently, only nine products (approximately 1%) have been permitted worldwide for market authorization (4). To date, the European Medicines Agency approved only one MSCs-based product as a living cell pharmaceutical against perianal fistulas, and the US Food and Drug Administration has not yet approved any MSCs-related product (5).
Despite the failure of clinical trials to meet primary efficacy endpoints, numerous preclinical animal tests identified the promising outcome of MSCs adoptive transfer in several mouse disease models. Cell drug deployment variables in human clinical trials are the primary hurdle that impedes the direct translation of MSCs’ effect on murine outcomes to the clinical situation (6). Though it is not decisive that MSCs engraftment and differentiation convey their in vivo paracrine support, recent studies evidenced the correlation between MSCs in vivo persistence and clinical potency (5, 7). Parameters that control the stable in vivo engraftment of implanted MSCs are the route of delivery, immune compatibility, dosing, and fitness of culture-adapted MSCs (5, 8). Hence the primary target of future MSCs-based research should be on exploring engineering or priming approaches to modulate cell drug deployment strategy for inducing graft cell survival. In this review, we reappraise different MSCs’ delivery strategies so far applied in clinical and preclinical situations and discuss some future directions for the clinical translation of this therapeutic intervention to promote MSCs’ clinical application.
- A.
- Cell processing and mode of delivery: current status and translational hurdles
To develop MSCs based therapy in human clinical trials, it is pivotal to understand the feasibility and safety of their current therapeutic modality. A complete understanding of cell manufacturing variables like processing, fitness, and functionality along with non-manufacturing variables like handling at the point of Care, route of delivery, dosing of MSCs will aid in the appropriate and safe use of the cells for clinical therapy.
- MSCs’ tissue source
Mesenchymal stromal cells (MSCs) can be isolated from multiple organ and tissue sources such as bone marrow, umbilical cord, adipose tissue, placenta, peripheral blood, periodontal ligament, amniotic fluid (15). Of these, bone marrow-derived MSCs are the most regularly tested tissue source in clinical trials. MSCs isolated from different tissue sources have been well characterized by the presence of common surface antigen such as expression of CD105, CD73, and CD90, and lack of CD45, CD34, CD14 or CD11b, CD79α or CD19 and HLA-DR (16). MSCs derived from distinct sources possess similar multipotential differentiation capacities (including osteoblasts, chondrocytes, and adipocytes). However, their differentiation efficiency varies during specific lineage differentiation for tissue regeneration (17). MSCs' differentiation potential is regulated by their tissue origin, through epigenetic regulations such as DNA methylation of important transcription factors. During lineage-specific differentiation studies, it appears that adult stem cells do not differentiate with the same efficacy, as the tissue-specific stem cells are generally more effective when differentiating toward their origin tissues. For instance, bone marrow-derived stem cells (BMSCs) express a higher magnitude of osteogenic genes, while adipose-derived stem cells (ADSCs) have significant upregulation of adipogenic genes after in vitro induction (18). Therefore, in a successful clinical trial suitable tissue source selection is imperative to produce clinical-grade MSCs (Figure 1).
- 2.
- MSCs’ immunogenicity
Off-the-shelf, cryobanked allogeneic MSCs propose significant logistic and cost-reductive advantages in clinical arrangement compared to their autologous counterpart. However, to reach the efficacy endpoint, major pre-clinical mouse data support the use of syngeneic MSCs(5).
Since MSCs express low levels of MHC class I molecules on their surface and lack the expression of MHC class II, previously MSCs were considered “immunoprivileged”(19). Moreover, in early studies, researchers identified that allogeneic MSCs significantly delay the proliferation of MHC-mismatched lymphocytes in in vitro mixed leukocyte reactions (MLR)(20). Therefore, in clinical trials, mass-produced MSCs from a few donors were extensively used to treat allogeneic unrelated recipients for a range of diseases, without concern for immune rejection. However, recent studies showed the antibody production against and immune rejection of allogeneic donor MSCs(21, 22), raising the question of employing MSCs as the universal donor. Using a mouse model of Colitis, current findings identified that the administration of both allogeneic and syngeneic MSCs can improve the disease following the first course of treatment. However, like syngeneic MSCs, allogenic MSCs are impotent to maintain sustained responsiveness in relapsing colitis(5). Indeed, allogeneic MSCs protect themselves from immune detection, in some cases, where they exert their therapeutic activity through a brief “hit and run” mechanism. This is possible in some cases where persistence is dispensable to exert its therapeutic effect(19). Collective evidence indicates that allogeneic MSCs are the only reasonable deployment strategy for use in acute tissue injury syndromes such as stroke, sepsis, or myocardial infarction, where the delays in manufacturing autologous MSCs would forfeit their efficiency in affecting outcomes. Nonetheless, in chronic degenerative disorders where repeated long-term infusions are necessary to maintain MSCs therapeutic benefit, there would be a bias in favor of the syngeneic (autologous) MSCs(5). Despite the immunogenic nature of allo-MSCs, clinical studies often rely of cryopreserved allogenic cells because of the donor related issues (age or disease state of patient) and in the need of low-cost immediate care(23). Hence cell modification approaches to avoid allo-rejection and alleviate transplantation shock would be beneficial to improve MSCs therapeutic utility in clinical trials (Figure 1).
- 3.
- MSCs administration routes:
MSCs delivery route is one of the critical parameters that can dictate an effective and safe therapeutic outcome(6). Though plenty of pre-clinical and clinical trials are attempting to optimize specific routes of delivery, presently there is no consensus on the optimal MSCs delivery method in human clinical trials. Clinical trials frequently select the cost-effective and convenient intravenous transfusion method, though their therapeutic outcome shows variable effectiveness(5). Depending on the treatment requirement, MSCs can be implanted through local and systemic administration.
a) Systemic administration: Intravenous (IV) and intra-arterial injection are the major approaches for systemic MSCs’ delivery, where IV delivery is considered the conventional and safe approach in human clinical trials. According to a recent survey of human MSC clinical trials, >40% of studies implant a median of 100 million MSCs IV for a wide range of clinical disorders(5). Nevertheless, a plurality of reports identified that the IV-transfused MSCs are trapped in lung, which are cleared shortly afterward. Since the paracrine functions support MSCs’ therapeutic activity, reduced lifespan of grafted cells debilitates their therapeutic activity. A recent mouse preclinical study identified that repeated IV delivery of maximally tolerated dose (50 million cells/kg body weight) of fit MSCs failed to affect colitis clinical outcomes(5). On the contrary, a research group identified the beneficial effect of intravenously transfused mouse MSCs in reduction of lethal sepsis. They explained this event by the process of efferocytosis, where more than half of lung-trapped MSCs is rapidly phagocytosed by lung-resident tissue macrophages. Production of interleukin-10 (IL-10) by phenotypically altered macrophages reduce tissue inflammation(24). A similar study identified the role of IP/IV delivered apoptotic MSCs in improvement of mouse GVHD outcomes, which is mediated by the interaction with host phagocytic cells and a secondary efferocytotic response(25). Repeated IV administration of adipose derived MSCs into diabetic rat, reduced kidney damage and induced the secretion of glial cell-derived neurotrophic factor to recover podocyte(26).
Compare to intravenous, intra-arterial (IA) approach is more efficient as it reduces cell-trapping in the lungs and induces engrafted cell migration at the target tissue injury site(27). Intriguingly, few studies identified adverse effect associated with IA delivery of MSCs. Though IA injection is preferable route for efficient cell targeting to the brain, recent survey identified probable risk of cerebral infarcts associated with IA delivery of MSCs for stroke. Hence, for optimal use of IA approach, cell size, cell dose and infusion speed must be carefully considered(28).
Though intra-peritoneal (IP) or subcutaneous (SC) delivery is not frequently used in clinical trial like IV route, preclinical studies identified their therapeutic utility in various disease recovery. IP delivered MSCs-condition media in a rat model of vaginal distention injury, recovered urethral sphincter function through increasing leak point pressure(29). A recent study identified that maximum tolerated IV bolus of MSCs fail to improve toxic colitis in mice, whereas SC or IP delivered MSCs show significant effect on colitis clinical and pathologic endpoints(5).
b) Local administration
Local administration of MSCs is intended to increase the engraftment of therapeutic cells at the target sites for immediate generation of local action or differentiation into the functional cells. Topical approach of MSCs delivery has identified as least invasive method and is efficacious in case of wound healing, and skin graft survival in burn or diabetic-related wounds, and also to repair injury in solid organs and their related tissues, such as heart, brain, spinal tissue, liver etc(30).
Intra-muscular infusion (IM), like IV delivery route, is also considered as the minimally invasive and simple route of MSCs delivery. Additionally, IM route promotes prolonged survival of implanted MSCs compare to other conventional routes, which is supportive of improved therapeutic outcomes(31). In the study by Mao et al. IM transfusion of human umbilical cord MSCs in a rat model of dilated cardiomyopathy led to improved cardiac function(32). In a separate study by Wang et al., the combination of small gap neurorrhaphy and bone marrow derived MSCs transfusion through IM injection in a rat model of peripheral nerve injury showed significant improvement of peripheral nerve regeneration and recovery of nerve function(33). Though MSCs local injection can promote improved therapeutic outcome, sometimes it causes traumatic event and its invasion may cause massive bleeding and secondary damage.
Collective evidences indicate that each delivery approach has its own advantages and restrictions. Hence route of cell delivery should be selected according to the study’s aim, size of the target organ, and animal models to be used.
- 4.
- Optimal Timing/ dose/ fitness of MSCs
a) Time: The optimal cell delivery timing is imperative in maximizing MSCs clinical effect, as it regulates the survival time of engrafted cells. Comparative study is needed to optimize the time of MSCs delivery at the three phases of the tissue or organ healing process: i.e., injury phase (hours), repair phase (days), and remodeling phase (weeks) before any successful clinical trial(34). The cytotoxic environment at the acute injury phase of myocardial infarction, diminish the functional property of transplanted MSCs(35). Whereas, stem cell delivery at the repair phase i.e., 4 to 7 days after acute myocardial infarction, showed maximal therapeutic benefits(36). MSCs delivery within two hours and one week of thoracic irradiation resulted in beneficial outcomes in the treatment of Radiation-induced lung fibrosis (RILF) through anti-inflammation in the pneumonitic phase and anti-fibrosis during the whole lung injury period(37).
b) Dose: Number of delivered MSCs is an essential factor that can be modified to achieve optimal therapeutic benefit. The appropriate number of transplanted MSCs depends upon type of organ, tissue, and animal species. For instance, large number of cells in a single treatment may not always bias to positive therapeutic outcome. A very high dose of intravenously transplanted cells may cause blockages in the capillaries of the lungs, results in poor therapeutic effect(38). In clinical trial optimal dose range identified for treating spinal injury patient is, 0.5 x 106 - 5 x 106 MSCs/kg body weight of the recipient(39). Also, in some cases, repeated long-term infusion at certain intervals seems to stimulate the positive outcome. Although intravenous transfusion of MSCs is the safest mode, the major disadvantage is the limitation of maximum amount of delivered cells, which differs with the rodent model. In rodent, the optimal cell concentration is 50M/Kg for intravenous transplantation, whereas in case of human the optimal dose is 1-2M/Kg body weight. This species-specific variation of MSCs dosing, would prognosticate a negative bias in outcomes for humans if the functioning biological mechanisms are comparable between species and are dose-dependent(6).
c) Fitness: Various preclinical mechanistic studies established that the MSCs exert a local and systemic immunosuppressive effect via the release of multiple paracrine soluble factors. Since metabolic fitness is essential for paracrine functionality, the major animal studies utilize culture rescued, log phase MSCs with optimal metabolic fitness and high replication capacity. However, human clinical trials often use pre-banked cryopreserved allogeneic MSCs thawed immediately and infused at the point of care. Importantly, in the first 24 h following recovery from cryostorage, MSCs express different cell injury markers. Previous studies identified that immediate post-thawed MSCs show defective immune functionality(1), increased susceptibility to lysis by host T cells, and short-term in vivo persistence(40, 41). In a murine model of Colitis, short in vivo persistence of post-thawed MSCs led to impaired cell-dependent functionality(5). Besides, human clinical trials regularly use an industrial-scale expansion of MSCs. Emerging studies identified that prolonged culture expansion of MSCs leads to replicative exhaustion/senescence and impairment of immunosuppressive ability(42). In supporting this notion, researchers identified that the late passage MSCs are clinically less effective in mitigating GVHD than early passage cells(43, 44). Hence collective evidence indicates that metabolic fitness is one of the primary factors responsible for the disparity in interspecies outcomes. The development of strategies to reduce cellular senescence or rescue MSCs from cryoinjury would improve MSCs clinical application.
- B.
- Improvement of MSCs application in tissue injury
MSCs therapeutic potential is attributed to their capacity to undergo lineage-specific differentiation, immune modulatory and regenerative response. After in vivo transplantation, MSCs confront adverse microenvironment of injured tissue, such as oxidative stress, chronic inflammation, extracellular matrix degradation, which promotes apoptosis or rejection of graft MSCs. Genetic modification or preconditioning approaches have been extensively studied to improve MSCs in vivo persistence and therapeutic utility.
1. Improvement of tissue homing and adhesion property: One of the key features of adoptively transferred MSCs is their ability to home at tissue injury site in response to chemokine gradient. In this multistep process, MSCs surface chemokine receptor, CXCR4 or CXCR7, serves as specific receptor for stromal cell-derived factor 1 (SDF-1), which is one of the most powerful chemokines involved in cellular migratory processes(45). It has been identified that the overexpression of CXCR4/CXCR7 in the adipose tissue-derived MSCs, endorses their paracrine, proliferative and migratory abilities, which has further potent therapeutic impact in liver regeneration and rat renal transplantation(46, 47). Optimum adhesion of the transplanted MSCs is the primary contributor to cell engraftment and tissue/organ regeneration, where detachment causes anoikis. Cellular adhesion is associated with the function of integrins, which regulates cell extracellular matrix (ECM) and cell–cell adhesion mechanisms via the binding of ECM with adhesion molecules(48). Integrin-linked kinase (ILK) overexpression in MSCs induced cell survival and hence improved myocardial damage recovery, when transplanted into an ischemic myocardium model. ILK-overexpression also leads to rapid angiogenesis through AKT and mTOR signaling pathways in an infarcted myocardium(49). Since in in vivo condition, MSCs thrive to low oxygen tension environment, one classical method of enhancing the migratory, proliferative and therapeutic functionality of MSCs is hypoxic preconditioning. Studies identified that, in murine hind-limb ischemia model, hypoxia preconditioned (2% O2) MSCs show better proliferation and migration effect through elevated expression of heat shock protein, 78-kD glucose-regulated protein (GRP78)(50).
2. Immunosuppressive property: Different approaches like cytokine pre-licensing or genetic modification, have been identified to amplify the immunosuppressive effect of MSCs. Priming MSCs with TLR3 ligand Poly (I:C), enhanced the therapeutic activity of MSCs in TNBS induced colitis, through the production of immunosuppressive molecule PGE2(51). Administration of (TNFα/IL1β)-prelicensed MSCs in rat cornea transplant model, promoted immune-regulatory CD11b+B220+ monocyte/macrophage population and significantly increased Treg cells in the lungs and spleen(52). In a study by François et al. it was identified that the simultaneous treatment of MSCs with TNF-α and IFN-γ leads to increased production of IDO, which contributes in the differentiation of CD14+ monocytes into IL-10 producing anti-inflammatory CD206+ M2 macrophages(53). MSCs preconditioned with IL-1β produce enhanced level of immunomodulatory cytokines such as TNF-α, IL-6, IL-8 and IL-23A and chemokines such as CCL5, CCL20, CXCL1, CXCL3, CXCL5, CXCL6, CXCL10, and CXCL11. Additionally, IL1β prelicensing improved MSCs ability to recruit neutrophils, monocytes, lymphocytes, and eosinophils in vitro(54).
3. Cellular senescence and graft survival: After in vivo transplantation, the harsh microenvironment of host tissue often leads to premature death or cellular senescence of MSCs, followed by reduced MSCs' functions. Graft survival can be improved by genetically modifying the cells with overexpressing Integrin-linked kinase (ILK) in MSCs via activation of AKT and mTOR signaling pathways in an infarcted myocardium(49). MSCs overexpressing Hepatocyte growth factor (HGF) or Hypoxia-inducible factor 1α (HIF1A), also reported to have an anti-apoptotic effect and hence high therapeutic impact in several disease recovery(55),(56, 57). Prevention of senescence is another MSCs modification approach that can arrest irreversible cellular proliferation and improve MSCs' therapeutic functionality. Intriguingly, overexpressing human Oct4 and Sox2 into adipose MSCs enhances cellular stemness and proliferation capacity(58). In a separate study, telomerase reverse transcriptase (TERT) transfection was identified as an effective way to delay MSCs senescence by inducing higher proliferative and cell cycle-related gene expression factors(59), and also improve neural and osteogenic lineages proliferation(60, 61). IFN-γ stimulation is also identified as a cell apoptosis-prevention approach, which protects MSCs from NK cell-mediated cytotoxicity in vitro(62).
4. Strategies to prolong allo-MSCs’ persistence: Since preclinical studies identified a direct correlation between MSCs persistence and therapeutic efficacy(5), approaches that prevent allo-rejection and extend MSCs persistence would be a demanding tool in clinical research. Modifying host or transplanted MSCs is the possible way to avoid Allo-MSCs rejection. Administering specific doses of immunosuppressive drugs with allo-MSCs, is the most well-studied approach to prevent graft rejection. In a cardiac allograft mouse model, combinative therapy of low-dose rapamycin with MHC-mismatched allo-MSCs was found to be effective in the long-term persistence of MSCs and tolerance (100 days) of a heart graft of MSC donor origin(63). Strategically suppressing the MHC class I surface expression using viral immunoevasins is the other method of immunogenicity reduction and boosting MSCs in vivo persistence(64). Deletion of MHC class II surface expression was advantageous for allo-MSCs persistence. Though MSCs express low MHC II surface antigen, in vitro or in vivo differentiation of MSCs triggered increased MHC II expression, inducing a transition from immune privileged to immunogenic phenotype. Researchers developed immunoprivileged MSCs by knocking out Class II transactivator (CIITA), a known regulator of MHC II expression. CIITA knock-out MSCs did not rejectafter transplantation into the myocardium, followed by myocardial repair(65).
5. Importance of in vitro potency assay: Advanced clinical trial mandates MSCs’ identity and potency assay to measure their therapeutic utility. This functionality test ensures the quality or effectiveness of each of the MSCs manufactured lots, which are diverse at the level of cell source and manufacturing process(6). MSCs secrete a plurality of immunomodulatory and regenerative molecules in response to in vivo environmental cues. Hence, the major challenge of developing the assay matrix approach is identifying the combination of factors critical for MSCs’ regenerative functionality in humans. According to the International Society for Cell Therapy (ISCT), the assay matrix approach should capture the aggregate of effector pathways significant to MSCs immunomodulation, regeneration, and homing properties(66). The regenerative functionality of donor MSCs partially depends upon host immune cells-mediated licensing process. Hence, investigation of the intercation between donor MSCs and recipient leukocytes, may serve as a surrogate measure of potency. Obeying the declaration of ISCT, recently an assay matrix approach has been developed, which able to test two assay systems to quantify the potency of human MSCs: MSCs secretome analysis and a quantitative RNA-based array for specific genes responsible for immunomodulatory and homing properties of MSCs(67).
Although plenty of studies are involved in developing effective MSCs’ potency assay, more effort is needed to formulate approaches that can define and simplify the multifunctional or matrix responses of MSCs and serve as a platform for robust potency analysis.
Conclusions
A successful MSCs-based clinical trial relies on MSCs’ intrinsic functionality, such as MSCs’ homing property, effective differentiation, proliferation, and application of paracrine or endocrine influences. Tissue source, metabolic fitness, dosages, and delivery route are the focal regulatory factors that maintain MSCs intrinsic functionality.
For an effective and safe MSCs-based clinical trial, it is pivotal to understand the molecular mechanism of the cross-talk between host immune cells and the MSCs-derived secretomes. The technical knowledge derived from the successful MSC-based ADMIRE CD clinical trial will increase MSCs' clinical utility. However, more studies are needed to gain an in-depth understanding of mechanism of action and the biological properties of MSCs, collected by different manufacturing processes to enhance their therapeutic potency. Knowledge of proper selection of optimal cell delivery route, timing, and dosing is also pivotal to improve MSCs survival and overall function. Additionally, for further progression, methods that combine optimal delivery, dosages, preconditioning, and tracking should be explored in varying inflammatory disease models that currently lack effective treatment.
Acknowledgments
Laboratory space and institutional support from Dr. Geetanjali Sachdeva and Dr. Deepak Modi. A funding from DBT/Wellcome Trust India Alliance Early career fellowship to J.G.
References
- Francois, M.; et al. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-gamma licensing. Cytotherapy 2012, 14, 147–152. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef]
- Lazarus, H.M.; E Haynesworth, S.; Gerson, S.L.; Rosenthal, N.S.; I Caplan, A. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995, 16, 557–64. [Google Scholar] [PubMed]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Challenges in Clinical Development of Mesenchymal Stromal/Stem Cells: Concise Review. STEM CELLS Transl. Med. 2019, 8, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Giri, J.; Galipeau, J. Mesenchymal stromal cell therapeutic potency is dependent upon viability, route of delivery, and immune match. Blood Adv. 2020, 4, 1987–1997. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensébé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef]
- Galipeau, J., et al. (2021) Mesenchymal stromal cell variables influencing clinical potency: the impact of viability, fitness, route of administration and host predisposition. Cytotherapy. [CrossRef]
- Galipeau, J.; Krampera, M.; Leblanc, K.; Nolta, J.A.; Phinney, D.G.; Shi, Y.; Tarte, K.; Viswanathan, S.; Martin, I. Mesenchymal stromal cell variables influencing clinical potency: the impact of viability, fitness, route of administration and host predisposition. Cytotherapy 2021, 23, 368–372. [Google Scholar] [CrossRef]
- Hosseini-Asl, S.-K.; Mehrabani, D.; Karimi-Busheri, F. Therapeutic Effect of Mesenchymal Stem Cells in Ulcerative Colitis: A Review on Achievements and Challenges. J. Clin. Med. 2023, 9, 3922. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.M.; et al. Multiple infusions of mesenchymal stromal cells induce sustained remission in children with steroid-refractory, grade III-IV acute graft-versus-host disease. Br. J. Haematol. 2013, 163, 501–509. [Google Scholar] [CrossRef]
- Kurtzberg, J.; et al. Allogeneic human mesenchymal stem cell therapy (remestemcel-L, Prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2014, 20, 229–235. [Google Scholar] [CrossRef]
- Schlosser, S.; Dennler, C.; Schweizer, R.; Eberli, D.; Stein, J.V.; Enzmann, V.; Giovanoli, P.; Erni, D.; Plock, J.A. Paracrine effects of mesenchymal stem cells enhance vascular regeneration in ischemic murine skin. Microvasc. Res. 2012, 83, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, R.; Kamat, P.; Schweizer, D.; Dennler, C.; Zhang, S.; Schnider, J.T.; Salemi, S.; Giovanoli, P.; Eberli, D.; Enzmann, V.; et al. Bone marrow-derived mesenchymal stromal cells improve vascular regeneration and reduce leukocyte-endothelium activation in critical ischemic murine skin in a dose-dependent manner. Cytotherapy 2014, 16, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, H.; Zeng, X.; Guo, W.; Jin, Y.; Wang, S.; Tian, R.; Han, Y.; Guo, L.; Han, J.; et al. Efficient lung cancer-targeted drug delivery via a nanoparticle/MSC system. Acta Pharm. Sin. B 2019, 9, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells - current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Vallone, V.B.; et al. Mesenchymal stem cells and their use in therapy: what has been achieved? Differ. ; Res. Biol. Divers. 2013, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Szala, S.; Wisniewska, E.; Czapla, J. [Mesenchymal stromal cells]. Postep. Hig. I Med. Dosw. 2014, 68:1287-1298. [CrossRef]
- Xie, L.; Zhang, N.; Marsano, A.; Vunjak-Novakovic, G.; Zhang, Y.; Lopez, M.J. In Vitro Mesenchymal Trilineage Differentiation and Extracellular Matrix Production by Adipose and Bone Marrow Derived Adult Equine Multipotent Stromal Cells on a Collagen Scaffold. Stem Cell Rev. Rep. 2013, 9, 858–872. [Google Scholar] [CrossRef] [PubMed]
- A Ankrum, J.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringden, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- Schu, S.; Nosov, M.; O'Flynn, L.; Shaw, G.; Treacy, O.; Barry, F.; Murphy, M.; O'Brien, T.; Ritter, T. Immunogenicity of allogeneic mesenchymal stem cells. J. Cell. Mol. Med. 2012, 16, 2094–2103. [Google Scholar] [CrossRef]
- Zangi, L.; Margalit, R.; Reich-Zeliger, S.; Bachar-Lustig, E.; Beilhack, A.; Negrin, R.; Reisner, Y. Direct Imaging of Immune Rejection and Memory Induction by Allogeneic Mesenchymal Stromal Cells. STEM CELLS 2009, 27, 2865–2874. [Google Scholar] [CrossRef]
- Berglund, A.K.; Fortier, L.A.; Antczak, D.F.; Schnabel, L.V. Immunoprivileged no more: measuring the immunogenicity of allogeneic adult mesenchymal stem cells. Stem Cell Res. Ther. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Németh, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2–dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.S.; Bertolino, G.M.; Giacomini, C.; Bornhäuser, M.; Dazzi, F.; Galleu, A. Mesenchymal Stromal Cells for Graft Versus Host Disease: Mechanism-Based Biomarkers. Front. Immunol. 2020, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Aigner, L. Adult mesenchymal stem cell therapy for myelin repair in Multiple Sclerosis. Biol. Res. 2012, 45, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Mangera, A.; Chapple, C.R. Tissue engineering in urethral reconstruction—an update. Asian J. Androl. 2013, 15, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Caplan, H.; Olson, S.D.; Kumar, A.; George, M.; Prabhakara, K.S.; Wenzel, P.; Bedi, S.; Toledano-Furman, N.E.; Triolo, F.; Kamhieh-Milz, J.; et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenges to Clinical Application. Front. Immunol. 2019, 10, 1645. [Google Scholar] [CrossRef]
- Cai, J.; Yu, X.; Xu, R.; Fang, Y.; Qian, X.; Liu, S.; Teng, J.; Ding, X. Maximum Efficacy of Mesenchymal Stem Cells in Rat Model of Renal Ischemia-Reperfusion Injury: Renal Artery Administration with Optimal Numbers. PLOS ONE 2014, 9, e92347. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Zhang, X.; Liu, Y.; Chen, J.; Hu, B.; Song, J.; Zhang, Y. Strategies to Optimize Adult Stem Cell Therapy for Tissue Regeneration. Int. J. Mol. Sci. 2016, 17, 982. [Google Scholar] [CrossRef]
- Braid, L.R.; Wood, C.A.; Wiese, D.M.; Ford, B.N. Intramuscular administration potentiates extended dwell time of mesenchymal stromal cells compared to other routes. Cytotherapy 2018, 20, 232–244. [Google Scholar] [CrossRef]
- Mao, C.; Hou, X.; Wang, B.; Chi, J.; Jiang, Y.; Zhang, C.; Li, Z. Intramuscular injection of human umbilical cord-derived mesenchymal stem cells improves cardiac function in dilated cardiomyopathy rats. Stem Cell Res. Ther. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Y.; Zhao, J.; Jiang, B. Intramuscular injection of bone marrow mesenchymal stem cells with small gap neurorrhaphy for peripheral nerve repair. Neurosci. Lett. 2015, 585, 119–125. [Google Scholar] [CrossRef]
- Jarvinen, T.; Jarvinen, M.; Kalimo, H. Regeneration of injured skeletal muscle after the injury. Muscle Ligaments Tendons J. 2013, 03, 337–45. [Google Scholar] [CrossRef]
- Deten, A.; Volz, H.C.; Briest, W.; Zimmer, H.-G. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef]
- Hampel, C.; Wienhold, D.; Benken, N.; Eggersmann, C.; Thüroff, J. Definition of overactive bladder and epidemiology of urinary incontinence. Urology 1997, 50, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, X.; Ren, L. Optimization of the adipose-derived mesenchymal stem cell delivery time for radiation-induced lung fibrosis treatment in rats. Sci. Rep. 2019, 9, 5589. [Google Scholar] [CrossRef] [PubMed]
- van Koppen, A. , et al. Healthy bone marrow cells reduce progression of kidney failure better than CKD bone marrow cells in rats with established chronic kidney disease. Cell Transplant. 2012, 21, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.R.; Pollock, K.; Hubel, A.; McKenna, D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion 2014, 54, 1418–1437. [Google Scholar] [CrossRef]
- Chinnadurai, R.; et al. Cryopreserved Mesenchymal Stromal Cells Are Susceptible to T-Cell Mediated Apoptosis Which Is Partly Rescued by IFNgamma Licensing. Stem Cells 2016, 34, 2429–2442. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; et al. Cryopreserved or Fresh Mesenchymal Stromal Cells: Only a Matter of Taste or Key to Unleash the Full Clinical Potential of MSC Therapy? Adv. Exp. Med. Biol. 2016, 951, 77–98. [Google Scholar] [PubMed]
- Sensebé, L.; Tarte, K.; Galipeau, J.; Krampera, M.; Martin, I.; Phinney, D.G.; Shi, Y. Limited Acquisition of Chromosomal Aberrations in Human Adult Mesenchymal Stromal Cells. Cell Stem Cell 2012, 10, 9–10. [Google Scholar] [CrossRef]
- Galipeau, J. The mesenchymal stromal cells dilemma--does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 2013, 15, 2–8. [Google Scholar] [CrossRef]
- von Bahr, L.; et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol. Blood Marrow Transplant. : J. Am. Soc. Blood Marrow Transplant. 2012, 18, 557–564. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, A.; Tao, C.; Li, X.; Jin, P. The role of SDF-1-CXCR4/CXCR7 axis in biological behaviors of adipose tissue-derived mesenchymal stem cells in vitro. Biochem. Biophys. Res. Commun. 2013, 441, 675–680. [Google Scholar] [CrossRef]
- Du, Z.; et al. Mesenchymal stem cells overexpressing C-X-C chemokine receptor type 4 improve early liver regeneration of small-for-size liver grafts. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2013, 19, 215–225. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, G.; Wang, F.; Liu, H.; Liu, L.; Han, Y.; Zhang, J.; Yuan, J. Protective Effects of Mesenchymal Stem Cells with CXCR4 Up-Regulation in a Rat Renal Transplantation Model. PLOS ONE 2013, 8, e82949. [Google Scholar] [CrossRef]
- Lee, S.; Choi, E.; Cha, M.-J.; Hwang, K.-C. Cell Adhesion and Long-Term Survival of Transplanted Mesenchymal Stem Cells: A Prerequisite for Cell Therapy. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Song, S.-W.; Chang, W.; Song, B.-W.; Song, H.; Lim, S.; Kim, H.-J.; Cha, M.-J.; Choi, E.; Im, S.-H.; Chang, B.-C.; et al. Integrin-Linked Kinase Is Required in Hypoxic Mesenchymal Stem Cells for Strengthening Cell Adhesion to Ischemic Myocardium. STEM CELLS 2009, 27, 1358–1365. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. Hypoxic Preconditioning Promotes the Bioactivities of Mesenchymal Stem Cells via the HIF-1α-GRP78-Akt Axis. Int. J. Mol. Sci. 2017, 18, 1320. [Google Scholar] [CrossRef]
- Qiu, Y.; Guo, J.; Mao, R.; Chao, K.; Chen, B.-L.; He, Y.; Zeng, Z.-R.; Zhang, S.-H.; Chen, M.-H. TLR3 preconditioning enhances the therapeutic efficacy of umbilical cord mesenchymal stem cells in TNBS-induced colitis via the TLR3-Jagged-1-Notch-1 pathway. Mucosal Immunol. 2017, 10, 727–742. [Google Scholar] [CrossRef]
- Murphy, N.; et al. TNF-alpha/IL-1beta-licensed mesenchymal stromal cells promote corneal allograft survival via myeloid cell-mediated induction of Foxp3(+) regulatory T cells in the lung. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9404–9421. [Google Scholar]
- Francois, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. : J. Am. Soc. Gene Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef]
- Carrero, R.; et al. IL1beta induces mesenchymal stem cells migration and leucocyte chemotaxis through NF-kappaB. Stem Cell Rev. Rep. 2012, 8, 905–916. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, X.; Zhang, Y.; Liang, X.; Ding, Y.; Xu, Y.; Fang, Z.; Zhang, F. Enhanced cell survival and paracrine effects of mesenchymal stem cells overexpressing hepatocyte growth factor promote cardioprotection in myocardial infarction. Exp. Cell Res. 2016, 344, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Li, F.; Fang, J.; Xu, L.; Sun, C.; Han, J.; Hua, T.; Zhang, Z.; Feng, Z.; Jiang, X. Hypoxia inducible factor 1α promotes survival of mesenchymal stem cells under hypoxia. Am. J. Transl. Res. 2017, 9, 1521–1529. [Google Scholar]
- Lv, B.; Hua, T.; Li, F.; Han, J.; Fang, J.; Xu, L.; Sun, C.; Zhang, Z.; Feng, Z.; Jiang, X. Hypoxia-inducible factor 1 α protects mesenchymal stem cells against oxygen-glucose deprivation-induced injury via autophagy induction and PI3K/AKT/mTOR signaling pathway. Am. J. Transl. Res. 2017, 9, 2492–2499. [Google Scholar] [PubMed]
- Han, S.M. , et al. Enhanced proliferation and differentiation of Oct4- and Sox2-overexpressing human adipose tissue mesenchymal stem cells. Exp. Mol. Med. 2014, 46, e101. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Choi, J.-H.; Jung, J.; Kim, J.K.; Lee, S.S.; Kim, G.J. Changes in PTTG1 by human TERT gene expression modulate the self-renewal of placenta-derived mesenchymal stem cells. Cell Tissue Res. 2014, 357, 145–157. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Chen, C.-L.; Liu, H.-C.; Lee, Y.-T.; Wang, H.-W.; Hou, L.-T.; Hung, S.-C. Overexpression of hTERT increases stem-like properties and decreases spontaneous differentiation in human mesenchymal stem cell lines. J. Biomed. Sci. 2010, 17, 64–64. [Google Scholar] [CrossRef]
- Saeed, H.; Qiu, W.; Li, C.; Flyvbjerg, A.; Abdallah, B.M.; Kassem, M. Telomerase activity promotes osteoblast differentiation by modulating IGF-signaling pathway. Biogerontology 2015, 16, 733–745. [Google Scholar] [CrossRef]
- Noone, C.; Kihm, A.; English, K.; O'Dea, S.; Mahon, B.P. IFN-gamma stimulated human umbilical-tissue-derived cells potently suppress NK activation and resist NK-mediated cytotoxicity in vitro. Stem cells and development 2013, 22, 3003–3014. [Google Scholar] [CrossRef]
- Ge, W. , et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am. J. Transplant. : Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2009, 9, 1760–1772. [Google Scholar] [CrossRef]
- de la Garza-Rodea, A.S. , et al. Exploitation of herpesvirus immune evasion strategies to modify the immunogenicity of human mesenchymal stem cell transplants. PloS one 2011, 6, e14493. [Google Scholar] [CrossRef]
- Huang, X.P. , et al. Class II transactivator knockdown limits major histocompatibility complex II expression, diminishes immune rejection, and improves survival of allogeneic bone marrow stem cells in the infarcted heart. FASEB J. : Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 3069–3082. [Google Scholar] [CrossRef]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Rajan, D.; Qayed, M.; Arafat, D.; Garcia, M.; Liu, Y.; Kugathasan, S.; Anderson, L.J.; Gibson, G.; Galipeau, J. Potency Analysis of Mesenchymal Stromal Cells Using a Combinatorial Assay Matrix Approach. Cell Rep. 2018, 22, 2504–2517. [Google Scholar] [CrossRef]
Figure 1.
MSCs therapeutic efficiency depends upon donor source (1) and stromal tissue source variability (2), and Cell manufacturing process (3). Immune matching, metabolic fitness and route of delivery regulate MSCs persistence inside recipient (5). Before in vivo administration, chemical licensing/ genetic modification (4) is the key strategy to improve graft cell survival (5b).
Figure 1.
MSCs therapeutic efficiency depends upon donor source (1) and stromal tissue source variability (2), and Cell manufacturing process (3). Immune matching, metabolic fitness and route of delivery regulate MSCs persistence inside recipient (5). Before in vivo administration, chemical licensing/ genetic modification (4) is the key strategy to improve graft cell survival (5b).
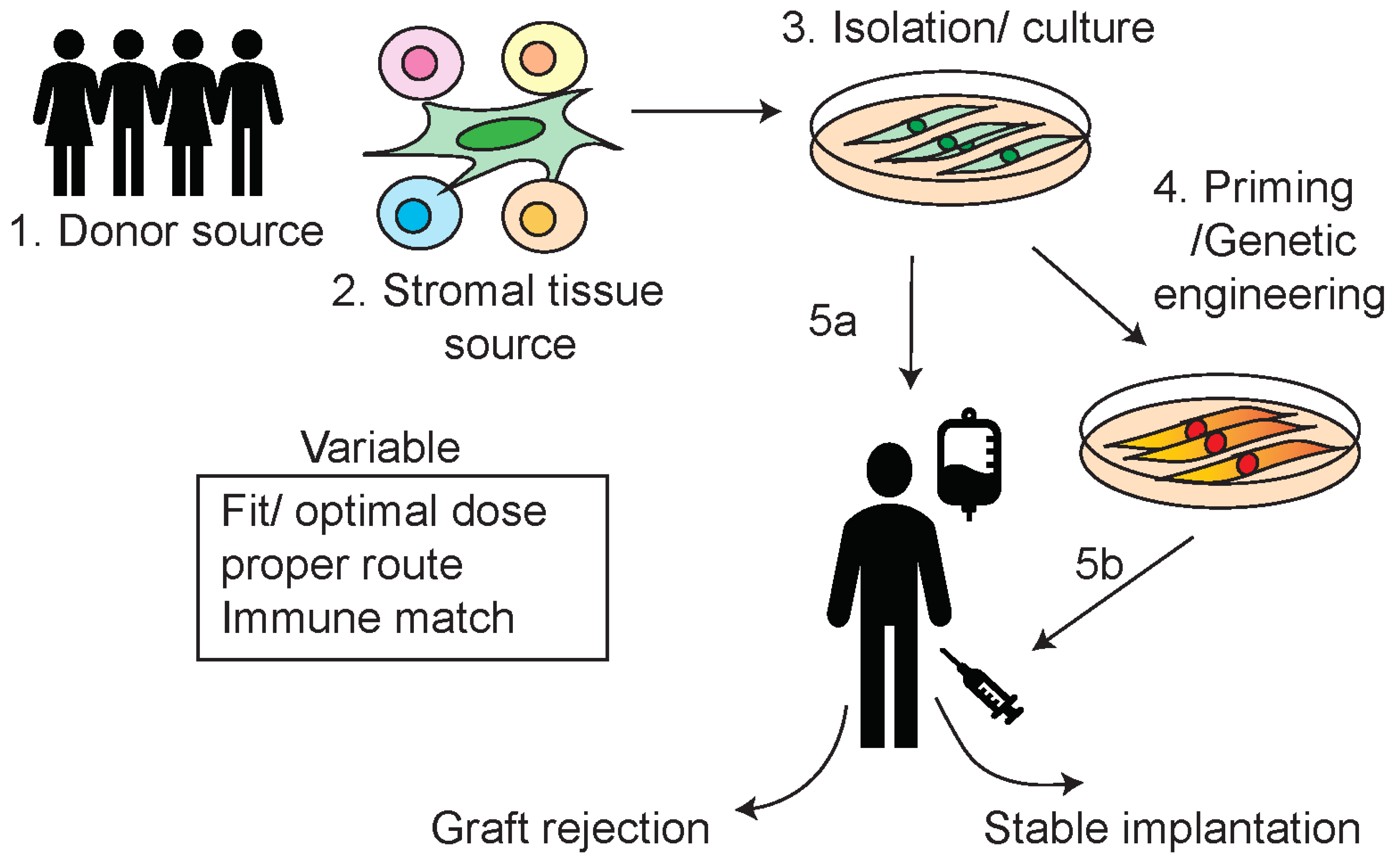

Table 1.
MSCs' role in regenerative medicine.
| MSCs Functionality | Therapeutic Outcome |
|---|---|
| Immunomodulatory | reduce tissue inflammation during colitis (9) Resolving acute steroid-refractory graft vs. host disease (10, 11) |
| Angiogenic/vascular regeneration & and tissue healing | vascular regeneration in ischemic murine skin (12) Angiogenesis in myocardial infarction (13) |
| Anti-cancer drug delivery agent | MSCs as a drug carrier to deliver chemotherapeutic agent Docetaxel (DTX) to lung tumors (14) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



