Submitted:
22 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
With the first case of human immunodeficiency virus (HIV) infection confirmed in US in 1981, the efforts of discovering anti-HIV therapeutics have been continued ever since. Ten years later, the first HIV drug zidovudine (AZT) was developed to inhibit HIV reverse transcriptase. At meantime, scientists were enlightened to discover new drugs of different targets acting on HIV integrase, protease, and host receptors. The advent of combination antiretroviral therapy (cART) is completely a game changer, with high efficiency in suppressing viremia for people with HIV (PWH) by controlling the viral load below the detectable level. On the bright side, the ART treatment has made HIV a chronic infection rather than a fatal disease. However, it cannot eradicate integrated HIV DNA from the host cells, thus the latent viral reservoir has become a lifelong threaten. In this review, we first discuss the scientific history of conventional HIV drug discovery, with more and more anti-HIV agents have been developed to solve drug resistant issue and relieve the side effect. As a complementary therapy, advanced gene editing technologies have been applied to excise HIV provirus from host genome. Within four decades, novel research conducted on HIV treatment and their contributions to eliminate HIV have been altogether summarized in our review.
Keywords:
HIV
; cART
; eene editing
; CRISPR
1. Introduction
It has been four decades since human immunodeficiency virus (HIV) was identified as the pathogen which caused the disease of acquired immunodeficiency syndrome (AIDS) [1,2]. Compared to the number of antiviral drugs developed against different viral infection, the discovery of HIV drugs has achieved great success with over thirty drugs approved by Food and Drug Administration (FDA) until present [3]. Based on their molecular mechanism and targets to each step of viral lifecycle, these drugs can be classified into six different groups: (1) coreceptor inhibitors (CRIs) and (2) fusion inhibitors (FIs) targeting viral entry; (3) nucleoside reverse transcriptase inhibitors (NRTIs) and (4) non–nucleoside reverse transcriptase inhibitors (NNRTIs) targeting reverse transcription; (5) integrase strand transfer inhibitors (InSTIs) targeting viral integration; (6) protease inhibitors (PIs) targeting viral maturation [4,5]. From the history of anti-HIV drug discovery, the first generation of drugs are all reverse transcriptase inhibitors including the first HIV drug zidovudine (3'-azido-3'-deoxythymidine, or AZT) approved in 1987 [6,7]. However, the AIDS lethality has not decreased until the advent of antiretroviral therapy (ART) after the development of reverse transcriptase inhibitors and protease inhibitors in the middle 1990s.
In general, ART is the combination of three or more drugs designed against at least two steps in the lifecycle of virus. People with HIV (PWH) should be treated with ART as soon as possible, usually the initial ART combination contains InSTIs, such as first generation InSTIs raltegravir (RAL), elvitegravir (EVG), and second generation InSTIs dolutegravir (DLG) and bictegravir (BIC), to specifically block the viral DNA integrated into the host genome [8,9]. With the clinical application of ART, HIV viral replication was strongly suppressed and the plasma viral load of PWH was significantly reduced to the undetectable level [10,11]. Although not universally appreciated, the statement of viral load undetectable equals untransmittable (Undetectable = Untransmittable or U = U), relying on the science-based evidence [12,13]. There are some clinical studies have demonstrated no transmissions happened to their partners during the condomless intercourse, no matter heterosexual acts or men who have sex with men (MSM), when the undetectable viral load maintained for more than six months by ART [14,15,16]. Eventually, ART development has successfully changed AIDS from a fatal disease into a controlled viral infection with the restored immune function [17,18] and with the mortality rates of PWH close to general mortality rates [19,20,21].
Despite current ART can durably suppress HIV viremia, the viral load will get rebound within several weeks of ART interruption even in patients with very small HIV reservoir and minimal on-going viral transcription [22,23]. The lifelong treatment of ART for PWH has brought new challenges into our attention, such as side effects [24], drug resistant [25,26] and unable to eradicate the integrated virus [27,28]. The persistence of HIV viral reservoir integrated into the host cells is always the threaten for PLWH, just like a ticking time bomb. Therefore, continuous efforts are demanded to discover novel drugs against different targets with less toxicity and develop alternative therapeutics including the gene editing technologies application into eliminating viral reservoirs for HIV functional cure.
In this review, we provide an overview of HIV-1 life cycle and summarize the most updated anti-HIV drugs that US Food and Drug Administration (FDA) approved, regarding with the steps during the viral replication. In addition, we describe the most advanced gene editing tools based on the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated nuclease 9 (Cas9) system as a complementary strategy for completely HIV cure. Along with it, the effective in vivo delivery system for programmable elements will be briefly discussed at last.
2. HIV life cycle and correlated target for drug discovery
2.1. Overview of the life cycle of HIV-1
The HIV-1 life cycle can be viewed as a series of steps processed in order, with some events may happen simultaneously. The first step is the attachment of viral particle to host cell membrane, by a specific interaction between HIV glycoprotein gp120 and the host cell receptor CD4 that expressed on the cell surface [29]. Under the observation of variable tropism displayed by HIV infected CD4+ cells, two coreceptors including α-chemokine receptor CXCR4 and β-chemokine receptor CCR5 have been identified to be critical for T cell tropic (T-tropic) and macrophage tropic (M-tropic) respectively [30,31]. After receptor binding, the membrane fusion takes place under a sequence of conformational change of glycoprotein gp41. The exposed fusion peptide inserts into the target cell membrane, at the meantime, the N-terminal heptad repeat (NHR) and the C-terminal heptad repeat (CHR) form the coiled-coil six helix bundle (6HB) which bends HIV viral membrane closer to host cell membrane that forces the membrane fusion [32]. As a result, viral RNA is released into the host cell cytoplasm.
Once viral RNA releasing into the cell cytoplasm, it starts reverse transcription to generate viral DNA using the viral genome encoded enzyme reverse transcriptase (RT), also known as RNA-dependent DNA polymerase [33]. The synthesized DNA associates with several viral and cellular proteins, forming a large nucleoprotein complex named preintegration complex (PIC) [34]. The viral DNA as part of PIC, will be tranported into cell nucleus by passing through the nuclear pore on the nucleus membrane. The viral genome encoded enzyme integrase (IN), within part of the PIC, presents in nucleus to catalyze viral DNA integration into host cell chromosome. The integrated viral DNA, referred to as the “provirus”, is essential for HIV viral replicaiton. It serves as the template for viral RNA synthesis, the transcribed mRNA is then transported to the cytoplasm and start translation for making viral proteins. The viral envelop glycoprotein (Env) and the precursor of Gag protein, Gag-pol polyprotein are synthesized in the endoplasmic reticulum (ER) and transported to the plasma membrane at where viral particle assembly takes place [35]. The N-terminal myristoylated matrix (MA) domain of Gag is observed by structure with exposure a patch of highly basic residues, these positively charged residues could interact with negatively charged acidic phospholipids on the plasma membrane to stablize binding [35]. During the process of viral protein transportation to the membrane, the Gag precursor recruits two copies of single-stranded viral RNA interacts with Gag-pol precursor and assembles as viral particles [36]. The assembled Gag protein complex induces membrane budding, with the viral Env protein incorporated into the particles [37]. During or immediately after the budding, viral protease (PR) cleaves Gag and Gag-pol polyprotein precursors to the mature Gag and Pol proteins [38]. The viral particles pinch off from the plasma membrane and are capable of initiating a new round of infection.
2.2. Viral entry inhibitors
It is efficient to design drugs targeting viral entry as it could block viral infection at the first step. Up to date, there are three entry inhibitors that approved by FDA listed in Table 1 [39]. Enfuvirtide, also known as T20 or ENF (trade name as Fuzeon), is the first HIV entry inhibitor approved in 2003 [40]. Enfuvirtide is a short peptide derived from HIV gp41 amino acids 127-162, it binds to viral native NHR to prevent the formation of 6HB fusion core so that inhibit membrane fusion [41]. However, enfuvirtide is clinically treated in high doses due to its relatively low antiviral activity and short half-life in vivo [42]. Another drawback of enfuvirtide is that it easily induces drug resistance because of the viral mutations detected in NHR [43]. The second entry inhibitor is maraviroc (also known as MVC, trade name as Selzentry), which approved in 2007 as a potent antiviral drug for M-tropic (R5) strains of HIV-1 [44]. Maraviroc is a CCR5 antagonist that inhibits the binding of chemokine ligand to CCR5 receptor at a low nanomolar range and blocks downstream CCR5-signalling [45]. Maraviroc is a small molecule with distribution throughout the body, it can penetrate the blood–brain barrier and can be detected in cerebrospinal fluid (CSF) [46], seminal plasma (SP) [47] and cervicovaginal fluids (CF) [48]. In clinic, the resistance to maraviroc appears when virus switches coreceptor from CCR5 to CXCR4, or to a dual tropism under the pressure of drug treatment [49]. The last entry inhibitor is ibalizumab-uiyk (also known as IBA, trade name as Trogarzo) that FDA approved in 2018, as HIV treatment for adult patients that other drugs have not worked [50,51]. Ibalizumab-uiyk is the only monoclonal antibody (MAb) developed for HIV treatment with intravenous (i.v.) injection over decades. Ibalizumab-uiyk is a recombinant humanized IgG4 MAb that derived from a parent mouse MAb. It binds to the interface between extracellular domains 1 and 2 of human CD4 receptor that induces steric hindrance to prevent HIV gp120 binding to CD4 molecule [52]. Ibalizumab-uiyk has its unique advantages for HIV therapy, including low toxicity, ability to restore CD4 T cells, minimal potential for resistance. It is an important addition to manage HIV infection in adults who are failing with the current treatment.
2.3. Reverse transcriptase inhibitors
The current FDA approved anti-HIV drugs for post entry inhibition are targeting viral genome encoded enzyme RT, IN and PR (Table 1). In fact, more than half of the approved drugs are RT inhibitors. These small molecules can block the viral RNA conversion into DNA which will be integrated into the host genome later. The first effective anti-HIV drug that FDA approved in 1987 was a RT inhibitor zidovudine, also known as azidothymidine (AZT) [7]. Approved RT inhibitors can be grouped into two classes: nucleoside reverse transcriptase inhibitors (NRTIs) and non–nucleoside reverse transcriptase inhibitors (NNRTIs). NRTIs are deoxynucleoside triphosphate analogs, but lack a free 3′-hydroxyl group. Once NRTIs are incorporated into the nascent viral DNA, further viral DNA synthesis catalyzed by RT enzyme is effectively terminated [53]. Within this class, tenofovir, also known as tenofovir disoproxil fumarate (TDF), is part of recommended initial ART regimes in clinic [54]. These recommended regimes usually have a high rate of viral suppression, minimal toxicity and less likely to involve drug resistance. NNRTIs are small hydrophobic compounds that binding to an allosteric site located approximately 10 Å away from the RT catalytic active site, to inhibit DNA polymerization [55]. Until present, there are six HIV-1 NNRTIs approved by FDA in a timeline order as : nevirapine (NVP), delavirdine (DLV), efavirenz (EFV), etravirine (ETR), rilpivirine (RPV), doravirine (DOR). NNRTIs are essential component in ART and currently under extensive development. Compare to NRTIs, NNRTIs seem to block viral RNA reverse transcription at initial steps [56]. However, the emearging drug-resistance of NNRTIs makes it failure in beating the growing number of mutated HIV-1 variants. For example, efavirenz, nevirapine and delavirdine could effectively inhibit the wild-type HIV-1 in clinic, but they are less effective against RT mutant HIV-1, such as clinical strains containing K103N and Y181C mutations [57].
2.4. Protease inhibitors
It has been studied that proteolytic cleavage of Gag and Gag-pol polyprotein precursors by HIV PR is required for viral infectivity. The budded uncleaved virions have lost the infection cability [58]. The critical of enzyme PR in the maturation process of virus makes it an attractive target for anti-HIV drugs consequently. So far, there are ten FDA approved protease inhibitors (PIs) including: saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, lopinavir, fosamprenavir, atazanavir, tipranavir, and darunavir (Table 1). The first one, saquinavir (SQV), approved in 1995, which marked the beginning of combination antiretroviral therapy for HIV patients. The clinical data showed that ART with saquinavir and RT inhibitor zalcitabine significantly extended the patient lifespan, compared with zalcitabine alone [59]. Protease inhibitor thus becomes one of the most important regimens in the combination therapy. Unfortunately, most of the PIs are associated with side effects in long-term treatment, such as the increased risk of cardiovascular and cerebrovascular diseases as well as dyslipidemia, diabetes [60,61,62]. There was no significance regarding of adverse effects when comparing PI as a monotherapy compound with the ART combination of PIs and RT inhibitors, indicating that PIs were in major responsibility for the side effects [63].
It is possible to optimize the chemical struture of PI molecule to improve the clinical benefits. The successful attempts are darunavir and lopinavir, which were modified from amprenavir and ritonavir, respectively. However, the further modification of lopinavir was failed, none of the analogues compounds has shown better than original lopinavir [64]. Other attempts have been tried to optimize PIs to avoid side effects. For exmaple, GS-8374 modified from a scaffold of TMC-126 (darunavir analog), has a favorable resistance profile against a spectrum of patient-derived HIV-1 variants highly resistant to multiple PIs [65]. In addition, this new inhibitor GS-8374 neither affects insulin-stimulated glucose uptake in adipocytes in culture nor acutely alters peripheral glucose disposal in a rodent model system as preclinical evaluation, which is similar to atazanavir but unlike ritonavir and lopinavir [66].
2.5. Integrase inhibitors
Like all retroviruses, HIV-1 integrates its viral DNA which generated from viral RNA reverse transcription into the host chromosome. Integration provides a favorable environment for virus long term persistence. The process of viral integration is mediated by viral genome encoded enzyme IN, which is a specialized DNA recombinase. IN is an attractive drug target because it is essential for infective virions production, and there is no mammalian homologue of IN [67]. However, the progress for drug discovery against IN was delayed for more than 10 years than anti-HIV drug targeting RT and PR. It is mainly because a lack of good lead compounds obtained from in vitro screening and there were no reliable assays to evaluate integrase inhibition. Until now, there are five compounds approved by FDA (Table 1), including the first IN inhibitor raltegravir that got approval in 2007, and elvitegravir, dolutegravir, bictegravir, cabotegravir.
After synthesis of the viral DNA, IN assembles at the ends of it by binding to the HIV-1 LTR region, in consequence of DNA-IN binding complex formation. Then IN catalyze to remove two terminal nucleotides at each end of LTRs to produce a new 3’ hydroxyl ends, as refer to 3’ processing [68]. At the second catalytic step, IN is responsible to transfer viral DNA to the human chromosomal DNA, namely strand transfer or transesterification. IN binds to the host chromosomal DNA and mediates a concerted nucleophilic attack by the 3’ hydroxyl residues of the viral DNA on phosphodiester bridges located in the target DNA. Then the processed 3’ hydroxyl ends of viral DNA are ligated to the 5’-O-phosphate ends of the host DNA, irreversibly binding the viral DNA to the target DNA [69]. Last step of integration is the gap between virus and host DNA filled by host repair machinery. The current approved InSTIs were developed to primarily target the strand transfer step of HIV-1 integration, thus become the only class that interacts with two essential elements of the virus within the DNA-IN complex [70]. InSTIs are generally well tolerated by PWH, but emerging data suggest that some InSTIs contribute to weight gain. Because of its high efficacy and less side effects, InSTIs are recommended as an initiating ART component [71].
3. CRISPR/Cas9 based gene editing in HIV treatment
3.1. Gene editing targets host cell
In March 2008 at the Conference on retroviruses and opportunistic infections, the Berlin patient was declared as the first person “cured of HIV” who remained free of HIV for more than 13 years after ART interruption. With the success of stem cells transplantation from a healthy donor with homozygous CCR5△32 mutation, people start to see the silver lining behind the cloud. After the Berlin patient, there are London patient, New York patient, City of Hope patient, and Düsseldorf patient, who are cured of HIV from haematopoietic stem cell (HSC) transplantation [72]. The lesson we learnt is that 32-base-pair deletion of coreceptor CCR5 renders host cells resistant to R5-tropic HIV-1 variants infection [73]. Therefore, intensive work on disruption CCR5 expression by gene editing tools is currently being conducted. The rapidly updated CRISPR/Cas9 technology has boosted the research for curative HIV therapy, such as the recently developed base editing. Base editing uses programmable DNA-binding proteins to directly convert C⋅G to T⋅A base pairs (cytosine base editing, CBE) [74], or convert C⋅G to T⋅A base pairs (adenine base editing, ABE) [75]. Base editors modify targeted nucleotides with a single base change in the genome (Figure 1), no introduction of double-strand breaks (DSBs) while CRISPR/Cas9 does.
Xu et al. established a HSC transplantation model with CRISPR/Cas9 confered CCR5-ablated human CD34+ hematopoietic stem/progenitor cells (HSPCs) [76]. HSPCs reconstituted in mice for over 1 year and achieved robust CCR5 disruption, which mediated an HIV-1 resistance effect in vivo. Liu et al. designed two different guide RNA (gRNA) combinations targeting both CXCR4 and CCR5, in a single vector [77]. The simultaneous genome editing of HIV-1 coreceptors CXCR4 and CCR5 in primary CD4+ T cells with CRISPR/Cas9 system, protects modified cells from X4-tropic or R5-tropic HIV-1 viral infection. Knipping et al. applied base editors to simultaneously disrupt both co-receptors in primary human CD4+ T cells, it prevents transduction with R5-tropic and X4-tropic viral vectors [78]. Except targeting coreceptors, Chinnapaiyan et al. used CRISPR/Cas9 to knockdown cellular co-factor cyclin T1 which is crucial for HIV transcription, and demonstrated cyclin T1 inhibition mediated HIV silencing [79]. Overall, the research of CCR5 gene modification is dominant rather than other host cellular target.
3.2. Gene editing targets HIV genome
Although the current HIV-1 treatment including ART and broadly neutralizing antibody could suppress plasma viral load below the detectable level, they could not eliminate the integrated provirus. Virus will get rebound within few weeks after ART withdrawal. This challenge can be overcome with gene editing on provial DNA in CD4+ T cells. Ebina et al. designed gRNA to target HIV-1 long terminal repeat (LTR), in specific location of TAR sequence of the R region and NF-κB binding sequence in the U3 region, respectively. This LTR-targeted CRISPR/Cas9 system can disrupt HIV-1 provirus and also excise provirus from the cellular genome [80]. Kaminski et al. employed CRISPR/Cas9 editing system to precisely remove the integrated copies of the proviral DNA fragment from latently infected human CD4+ T cells, by targeting the highly conserved sequence of LTR U3 region among all HIV-1 viral isolates [81]. Liao et al. adapted the CRISPR/Cas9 system to disrupt latently integrated viral genome and provide long-term defense against new viral infection. They screened multiple potential gRNA target sites in the HIV-1 genome, including the structural (gag and env), enzymatic (pol) and accessory genes (vif and rev), as well as LTRs. By using a multiplexed CRISPR/Cas9 system with gRNAs targeting LTR sequences (especially the R region), they achieved an elevated level of disruption and excision of pre-integrated proviral genome [82]. Zhu et al. tested 10 sites in HIV-1 DNA by CRISPR/Cas9, and revealed a highly efficient target site within the second exon of Rev that could inactivate provirus efficiently by significantly reduction of HIV-1 gene expression and virus production [83]. In general, among different sites targeted on viral genome, targeting HIV-1 LTR has a strong impact on viral disruption because LTR serves as a critical element for viral transcription. And especially targeting the LTR-R region, which contains the TAR sequence that is highly conserved of all HIV-1 subtypes.
In addition, there are combinational targets on both host cells and viral genome. To improve the efficiency of viral elimination, Dash et al. developed dual CRISPR therapies targeting both proviral DNA and CCR5 [84]. There are two CRISPR reagents, including one set designed to target LTR and Gag to excise HIV-1 LTR-Gag region from latent proviral DNA intagrated cells, and the other set designed to target host coreceptor CCR5. The viral outgrowth assay (VOA) demonstrated that no progeny virus was recovered from plasma and tissues from CRISPR-treated virus-free mice.
3.3. Clinical trials of gene editing applied in HIV treatment
On December 8, 2023, the U.S. FDA approved two milestone treatments, Casgevy and Lyfgenia, for sickle cell disease (SCD) in patients 12 years and older [85]. Casgevy is the first FDA-approved therapy utilizing CRISPR/Cas9, which greatly inspired the development of HIV treatment in the field of gene therapy. To date, EBT-101 is the only CRISPR/Cas9 based gene therapy for HIV treatment in clinical trial and now is in the status of recruiting (NCT05144386). EBT-101 is comprised of an all-in-one CRISPR/Cas9 system that expressing dual gRNAs targeting viral LTRs and the Gag gene, thereby generating three possible deletions: 5′LTR to Gag, Gag to 3′LTR, and 5′LTR to 3′LTR [86]. The dual gRNAs excise large sections of proviral DNA (Figure 2), eliminating viral escape and reproduction.
Besides, there are zinc finger nucleases (ZFNs) enabled CCR5 gene editing for HIV treatment in clinical trial. A phase I study of autologous T cells genetically modified of CCR5 by ZFNs was completed (NCT00842634). It demonstrated the removal of CCR5 protein on the T cells is permanent in the follow-up study up to 6 years. Meanwhile, another phase I (NCT01044654) and phase 1/2 (NCT01252641) clinical trials regarding dose escalation study and single dose infusion of autologous T cells genetically modified of CCR5 by ZFNs in HIV infected patients was completed almost the same time. It both approves the safety and feasibility of ZFN-CCR5-modified CD4+ T cells re-infused back to humans as HIV therapy. Futher, the phase II clinical trial randomly comparing the effect of infusing expanded autologous CD4+ T cells with or without CCR5 modification ex vivo by ZFNs among HIV-infected patients is ongoing (NCT03666871). For stem cell gene modification, there is a phase I pilot study to evaluate the feasibility, safety and engraftment of ZFN-CCR5-modified CD34+ HSPCs in R5 tropic HIV-1 infected patients (NCT02500849).
4. Conclusions and future perspectives
The discovery of anti-HIV drug is arguably among the most successful treatment for any human disease, regarding the number of available anti-HIV agents that developed for four decades since the first HIV-1 viral infection case was confirmed in 1981. More than 30 antiretroviral drugs have been approved, and combination therapy is demonstrated with high efficiency and controllable toxicity for PWH. However, the lifelong treatment of ART and acquired drug resistance remain as major hurdle to treatment. Continuous efforts are demanded to develop new compounds and new drug combinations to achieve therapy success. Although these ART drugs are highly potent to suppress viremia, they are unable to eradicate the integrated viral DNA which will be potentially reactivated at certain condition. With the development of gene editing tools, such as ZFNs, CRISPR/Cas9, transcription activator-like effectors (TALENS) ect. More and more research conducted on provirus elimination using these new technologies. Especially for recent years, base editing and prime editing derived from CRISPR/Cas9 system are much safer because no double-strand breaks introduced in the targeted DNA. With efficient design of target sites, low off-target effects, these gene modification tools can be applied quickly in various research fields including anti-HIV therapy. It is convenient in application since for different target, it only needs to design different gRNA. And the flexibility is that different gRNAs can be combined in one vector for better knock-out efficiency. Such as the current ongoing clinical trial of EBT-101, it is a combination of gRNAs targeting HIV-1 LTR and Gag genes.
The CRISPR/Cas9 system is indeed a promising tool applied for HIV treatment, but the obstacle is low delivery efficiency in vivo. The delivery components can be plasmid, ribonucleoprotein (RNP) and mRNA. The delivery vehicle can be viral vector-based delivery, lipid nanoparticle and microinjection. Adeno-associated virus (AAV) vector is an efficient and widely used delivery agent, which remains the only gene therapy vector that FDA approved for human disease. Viral vector delivery for gene editing element also made remarkable progress for clinical HIV treatment. In specific, EBT-101 adapts AAV9 for CRISPR/Cas9 delivery. Other clinical trials of CCR5 gene modified T cells and stem cells are transduced with ZFNs expressed in adenoviral vector. It is obvious that lacking specific targeting CD4 T cells is the most significant challenge for CRISPR/Cas9 therapeutics in removing HIV reservoir in clinic. It is urgent to develop methods for specifically targeting T cells. Hamann et al. modified the AAV2 capsid proteins, with a set of novel nanobodies with high affinity for the human CD4 receptor. This CD4 antibody modified capsid was demonstrated to improve targeting efficiency of human primary CD4+ T cells in vitro [87]. It provides a promising strategy for changing AAV tropism to particularly targeting specific cell. Another limitation is the packaging capacity of AAV vector, which is less than 5 kb. The large size of SpCas9 (~4.1 kb) decreases the efficiency of delivery, so it seems impossible to package the base editor and prime editor in AAV. Therefore, Levy et al. came up with a dual AAVs system for the delivery of split base editors [88]. Another way to solve the issue is to optimize SpCas9 protein into a truncated variant or use smaller SaCas9. While it is a long way to go, but the combination of antiretroviral drug and the gene editing technology will lead to the promising progress in HIV-1 therapeutics.
Author Contributions
Y.S. took care of conceptualization; writing the original draft; review and editing. L.W. took care of supervision; resources; conceptualization; writing the original draft; review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent Detection and Isolation of Cytopathic Retroviruses (HTLV-III) from Patients with AIDS and at Risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int J Biol Macromol 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 2012, 2, a007161. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res 2013, 98, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, R.; Klecker, R.W.; Weinhold, K.J.; Markham, P.D.; Lyerly, H.K.; Durack, D.T.; Gelmann, E.; Lehrman, S.N.; Blum, R.M.; Barry, D.W.; et al. Administration of 3'-azido-3'-deoxythymidine, an inhibitor of HTLV-III/LAV replication, to patients with AIDS or AIDS-related complex. Lancet 1986, 1, 575–580. [Google Scholar] [CrossRef] [PubMed]
- McLeod, G.X.; Hammer, S.M. Zidovudine: five years later. Ann Intern Med 1992, 117, 487–501. [Google Scholar] [CrossRef]
- Zhao, A.V.; Crutchley, R.D.; Guduru, R.C.; Ton, K.; Lam, T.; Min, A.C. A clinical review of HIV integrase strand transfer inhibitors (INSTIs) for the prevention and treatment of HIV-1 infection. Retrovirology 2022, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Mbhele, N.; Chimukangara, B.; Gordon, M. HIV-1 integrase strand transfer inhibitors: a review of current drugs, recent advances and drug resistance. Int J Antimicrob Agents 2021, 57, 106343. [Google Scholar] [CrossRef] [PubMed]
- Lecher, S.L.; Fonjungo, P.; Ellenberger, D.; Toure, C.A.; Alemnji, G.; Bowen, N.; Basiye, F.; Beukes, A.; Carmona, S.; de Klerk, M.; et al. HIV Viral Load Monitoring Among Patients Receiving Antiretroviral Therapy - Eight Sub-Saharan Africa Countries, 2013-2018. MMWR Morb Mortal Wkly Rep 2021, 70, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Han, W.M.; Law, M.G.; Egger, M.; Wools-Kaloustian, K.; Moore, R.; McGowan, C.; Kumarasamy, N.; Desmonde, S.; Edmonds, A.; Davies, M.A.; et al. Global estimates of viral suppression in children and adolescents and adults on antiretroviral therapy adjusted for missing viral load measurements: a multiregional, retrospective cohort study in 31 countries. Lancet HIV 2021, 8, e766–e775. [Google Scholar] [CrossRef]
- Eisinger, R.W.; Dieffenbach, C.W.; Fauci, A.S. HIV Viral Load and Transmissibility of HIV Infection: Undetectable Equals Untransmittable. JAMA 2019, 321, 451–452. [Google Scholar] [CrossRef]
- LeMessurier, J.; Traversy, G.; Varsaneux, O.; Weekes, M.; Avey, M.T.; Niragira, O.; Gervais, R.; Guyatt, G.; Rodin, R. Risk of sexual transmission of human immunodeficiency virus with antiretroviral therapy, suppressed viral load and condom use: a systematic review. CMAJ 2018, 190, E1350–E1360. [Google Scholar] [CrossRef]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011, 365, 493–505. [Google Scholar] [CrossRef]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H. , et al. Antiretroviral Therapy for the Prevention of HIV-1 Transmission. N Engl J Med 2016, 375, 830–839. [Google Scholar] [CrossRef]
- Mujugira, A.; Celum, C.; Coombs, R.W.; Campbell, J.D.; Ndase, P.; Ronald, A.; Were, E.; Bukusi, E.A.; Mugo, N.; Kiarie, J. , et al. HIV Transmission Risk Persists During the First 6 Months of Antiretroviral Therapy. J Acquir Immune Defic Syndr 2016, 72, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Hou, Y.; Zhao, Y.; Dou, Z.; Ma, Y.; Zhang, D.; Wu, Y.; Zhao, D.; Liu, Z. , et al. Immune restoration in HIV-1-infected patients after 12 years of antiretroviral therapy: a real-world observational study. Emerg Microbes Infect 2020, 9, 2550–2561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.X.; Jiao, Y.M.; Zhang, C.; Song, J.W.; Fan, X.; Xu, R.N.; Huang, H.H.; Zhang, J.Y.; Wang, L.F.; Zhou, C.B. , et al. HIV Reservoir Decay and CD4 Recovery Associated With High CD8 Counts in Immune Restored Patients on Long-Term ART. Front Immunol 2020, 11, 1541. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Cole, S.R.; Breger, T.L.; Rudolph, J.E.; Filiatreau, L.M.; Buchacz, K.; Humes, E.; Rebeiro, P.F.; D'Souza, G.; Gill, M.J. , et al. Mortality Among Persons Entering HIV Care Compared With the General U.S. Population : An Observational Study. Ann Intern Med 2021, 174, 1197–1206. [Google Scholar] [CrossRef]
- Fontela, C.; Aguinaga, A.; Moreno-Iribas, C.; Reparaz, J.; Rivero, M.; Gracia, M.; Floristan, Y.; Fresan, U.; Miguel, R.S.; Ezpeleta, C. , et al. Trends and causes of mortality in a population-based cohort of HIV-infected adults in Spain: comparison with the general population. Sci Rep 2020, 10, 8922. [Google Scholar] [CrossRef] [PubMed]
- Reis, K.G.; Desderius, B.; Kingery, J.; Kirabo, A.; Makubi, A.; Myalla, C.; Lee, M.H.; Kapiga, S.; Peck, R.N. Blood pressure, T cells, and mortality in people with HIV in Tanzania during the first 2 years of antiretroviral therapy. J Clin Hypertens (Greenwich) 2020, 22, 1554–1562. [Google Scholar] [CrossRef]
- Pannus, P.; Rutsaert, S.; De Wit, S.; Allard, S.D.; Vanham, G.; Cole, B.; Nescoi, C.; Aerts, J.; De Spiegelaere, W.; Tsoumanis, A. , et al. Rapid viral rebound after analytical treatment interruption in patients with very small HIV reservoir and minimal on-going viral transcription. J Int AIDS Soc 2020, 23, e25453. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Aga, E.; Bosch, R.J.; Pilkinton, M.; Kroon, E.; MacLaren, L.; Keefer, M.; Fox, L.; Barr, L.; Acosta, E. , et al. Time to Viral Rebound After Interruption of Modern Antiretroviral Therapies. Clin Infect Dis 2022, 74, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Kolakowska, A.; Maresca, A.F.; Collins, I.J.; Cailhol, J. Update on Adverse Effects of HIV Integrase Inhibitors. Curr Treat Options Infect Dis 2019, 11, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Liu, K.; Liu, H.; Hu, Y.; Zhang, Z.; Qin, J.; Xu, Q.; Peng, K.; Jin, X.; Wang, J.H. , et al. Corrigendum to "Trend of HIV-1 drug resistance in China: A systematic review and meta-analysis of data accumulated over 17 years (2001-2017)" [EClinicalMedicine 18 (2020) 100238]. EClinicalMedicine 2021, 33, 100696. [Google Scholar] [CrossRef] [PubMed]
- Chimukangara, B.; Lessells, R.J.; Rhee, S.Y.; Giandhari, J.; Kharsany, A.B.M.; Naidoo, K.; Lewis, L.; Cawood, C.; Khanyile, D.; Ayalew, K.A. , et al. Trends in Pretreatment HIV-1 Drug Resistance in Antiretroviral Therapy-naive Adults in South Africa, 2000-2016: A Pooled Sequence Analysis. EClinicalMedicine 2019, 9, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L. , et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat Commun 2019, 10, 3193. [Google Scholar] [CrossRef] [PubMed]
- Bandera, A.; Gori, A.; Clerici, M.; Sironi, M. Phylogenies in ART: HIV reservoirs, HIV latency and drug resistance. Curr Opin Pharmacol 2019, 48, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Geleziunas, R.; Wainberg, M.A. The human immunodeficiency virus type 1 (HIV-1) CD4 receptor and its central role in promotion of HIV-1 infection. Microbiol Rev 1995, 59, 63–93. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- 33. Telesnitsky, A. and S.P. Goff, Reverse Transcriptase and the Generation of Retroviral DNA, in Retroviruses, J.M. Coffin, S.H. Hughes, and H.E. Varmus, Editors. 1997: Cold Spring Harbor (NY).
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat Rev Microbiol 2017, 15, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.E.; Saad, J.S. The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Rein, A. RNA Packaging in HIV. Trends Microbiol 2019, 27, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr Opin Virol 2019, 36, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.V. Enfuvirtide, a new drug for HIV infection. Lancet 2003, 361, 1577–1578. [Google Scholar] [CrossRef] [PubMed]
- He, Y. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr Pharm Des 2013, 19, 1800–1809. [Google Scholar] [CrossRef]
- Patel, I.H.; Zhang, X.; Nieforth, K.; Salgo, M.; Buss, N. Pharmacokinetics, pharmacodynamics and drug interaction potential of enfuvirtide. Clin Pharmacokinet 2005, 44, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O'Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr. , et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C. , et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J. , et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Tiraboschi, J.M.; Niubo, J.; Curto, J.; Podzamczer, D. Maraviroc concentrations in cerebrospinal fluid in HIV-infected patients. J Acquir Immune Defic Syndr 2010, 55, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Tiraboschi, J.M.; Niubo, J.; Curto, J.; Podzamczer, D. Maraviroc concentrations in seminal plasma in HIV-infected patients. J Acquir Immune Defic Syndr 2010, 55, e35–36. [Google Scholar] [CrossRef]
- Dumond, J.B.; Patterson, K.B.; Pecha, A.L.; Werner, R.E.; Andrews, E.; Damle, B.; Tressler, R.; Worsley, J.; Kashuba, A.D. Maraviroc concentrates in the cervicovaginal fluid and vaginal tissue of HIV-negative women. J Acquir Immune Defic Syndr 2009, 51, 546–553. [Google Scholar] [CrossRef]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B. , et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N Engl J Med 2008, 359, 1442–1455. [Google Scholar] [CrossRef]
- Beccari, M.V.; Mogle, B.T.; Sidman, E.F.; Mastro, K.A.; Asiago-Reddy, E.; Kufel, W.D. Ibalizumab, a Novel Monoclonal Antibody for the Management of Multidrug-Resistant HIV-1 Infection. Antimicrob Agents Chemother 2019, 63. [Google Scholar] [CrossRef]
- Markham, A. Ibalizumab: First Global Approval. Drugs 2018, 78, 781–785. [Google Scholar] [CrossRef]
- Pace, C.S.; Fordyce, M.W.; Franco, D.; Kao, C.Y.; Seaman, M.S.; Ho, D.D. Anti-CD4 monoclonal antibody ibalizumab exhibits breadth and potency against HIV-1, with natural resistance mediated by the loss of a V5 glycan in envelope. J Acquir Immune Defic Syndr 2013, 62, 1–9. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Arion, D.; Parniak, M.A. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs). Cell Mol Life Sci 2000, 57, 1408–1422. [Google Scholar] [CrossRef]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C. , et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef]
- Tronchet, J.M.; Seman, M. Nonnucleoside inhibitors of HIV-1 reverse transcriptase: from the biology of reverse transcription to molecular design. Curr Top Med Chem 2003, 3, 1496–1511. [Google Scholar] [CrossRef]
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res 1998, 38, 153–179. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci U S A 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Faulds, D. Saquinavir. A review of its pharmacology and clinical potential in the management of HIV infection. Drugs 1996, 52, 93–112. [Google Scholar] [CrossRef]
- Bozzette, S.A.; Ake, C.F.; Tam, H.K.; Chang, S.W.; Louis, T.A. Cardiovascular and cerebrovascular events in patients treated for human immunodeficiency virus infection. N Engl J Med 2003, 348, 702–710. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Moorman, A.C.; Williamson, J.M.; Tong, T.C.; Ward, D.J.; Wood, K.C.; Greenberg, A.E.; Janssen, R.S.; investigators, H.I.V.O.S. Protease inhibitors and cardiovascular outcomes in patients with HIV-1. Lancet 2002, 360, 1747–1748. [Google Scholar] [CrossRef] [PubMed]
- Alvi, R.M.; Neilan, A.M.; Tariq, N.; Awadalla, M.; Afshar, M.; Banerji, D.; Rokicki, A.; Mulligan, C.; Triant, V.A.; Zanni, M.V. , et al. Protease Inhibitors and Cardiovascular Outcomes in Patients With HIV and Heart Failure. J Am Coll Cardiol 2018, 72, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Paton, N.I.; Kityo, C.; Hoppe, A.; Reid, A.; Kambugu, A.; Lugemwa, A.; van Oosterhout, J.J.; Kiconco, M.; Siika, A.; Mwebaze, R. , et al. Assessment of second-line antiretroviral regimens for HIV therapy in Africa. N Engl J Med 2014, 371, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Sham, H.L.; Zhao, C.; Li, L.; Betebenner, D.A.; Saldivar, A.; Vasavanonda, S.; Kempf, D.J.; Plattner, J.J.; Norbeck, D.W. Novel lopinavir analogues incorporating non-Aromatic P-1 side chains--synthesis and structure--activity relationships. Bioorg Med Chem Lett 2002, 12, 3101–3103. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, C.; Stray, K.; Tsai, L.; Williams, M.; Yang, Z.Y.; Cannizzaro, C.; Leavitt, S.A.; Liu, X.; Wang, K.; Murray, B.P. , et al. In vitro characterization of GS-8374, a novel phosphonate-containing inhibitor of HIV-1 protease with a favorable resistance profile. Antimicrob Agents Chemother 2011, 55, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Hruz, P.W.; Yan, Q.; Tsai, L.; Koster, J.; Xu, L.; Cihlar, T.; Callebaut, C. GS-8374, a novel HIV protease inhibitor, does not alter glucose homeostasis in cultured adipocytes or in a healthy-rodent model system. Antimicrob Agents Chemother 2011, 55, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- LaFemina, R.L.; Schneider, C.L.; Robbins, H.L.; Callahan, P.L.; LeGrow, K.; Roth, E.; Schleif, W.A.; Emini, E.A. Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cells. J Virol 1992, 66, 7414–7419. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.L.; Felock, P.J.; Hastings, J.C.; Blau, C.U.; Hazuda, D.J. The role of manganese in promoting multimerization and assembly of human immunodeficiency virus type 1 integrase as a catalytically active complex on immobilized long terminal repeat substrates. J Virol 1996, 70, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J. , et al. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc Natl Acad Sci U S A 2002, 99, 6661–6666. [Google Scholar] [CrossRef]
- Scarsi, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649–1676. [Google Scholar] [CrossRef]
- Payra, S.; Manjhi, P.K.; Singh, S.; Kumar, R.; Singh, S.K.; Kumar, A.; Maharshi, V. HIV cure: Are we going to make history? HIV Med 2023. [Google Scholar] [CrossRef]
- Allers, K.; Schneider, T. CCR5Delta32 mutation and HIV infection: basis for curative HIV therapy. Curr Opin Virol 2015, 14, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W. , et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol Ther 2017, 25, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S. , et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Knipping, F.; Newby, G.A.; Eide, C.R.; McElroy, A.N.; Nielsen, S.C.; Smith, K.; Fang, Y.; Cornu, T.I.; Costa, C.; Gutierrez-Guerrero, A. , et al. Disruption of HIV-1 co-receptors CCR5 and CXCR4 in primary human T cells and hematopoietic stem and progenitor cells using base editing. Mol Ther 2022, 30, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Chinnapaiyan, S.; Santiago, M.J.; Panda, K.; Rahman, M.S.; Alluin, J.; Rossi, J.; Unwalla, H.J. A conditional RNA Pol II mono-promoter drives HIV-inducible, CRISPR-mediated cyclin T1 suppression and HIV inhibition. Mol Ther Nucleic Acids 2023, 32, 553–565. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 2013, 3, 2510. [Google Scholar] [CrossRef]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep 2016, 6, 22555. [Google Scholar] [CrossRef]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J. , et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun 2015, 6, 6413. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Chen, C.; Kaminski, R.; Su, H.; Mancuso, P.; Sillman, B.; Zhang, C.; Liao, S.; Sravanam, S.; Liu, H. , et al. CRISPR editing of CCR5 and HIV-1 facilitates viral elimination in antiretroviral drug-suppressed virus-infected humanized mice. Proc Natl Acad Sci U S A 2023, 120, e2217887120. [Google Scholar] [CrossRef] [PubMed]
- https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease.
- Burdo, T.H.; Chen, C.; Kaminski, R.; Sariyer, I.K.; Mancuso, P.; Donadoni, M.; Smith, M.D.; Sariyer, R.; Caocci, M.; Liao, S., et al.; et al. Preclinical safety and biodistribution of CRISPR targeting SIV in non-human primates. Gene Ther 2023. [Google Scholar] [CrossRef]
- Hamann, M.V.; Beschorner, N.; Vu, X.K.; Hauber, I.; Lange, U.C.; Traenkle, B.; Kaiser, P.D.; Foth, D.; Schneider, C.; Buning, H. , et al. Improved targeting of human CD4+ T cells by nanobody-modified AAV2 gene therapy vectors. PLoS One 2021, 16, e0261269. [Google Scholar] [CrossRef]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng 2020, 4, 97–110. [Google Scholar] [CrossRef]
Figure 1.
Mechanism schematic of cytidine base editor (CBE) and adenine base editor (ABE) system. (A) CBE contains a Cas9 nickase fused to a deaminase and two UGI (uracil glycosylase inhibitor). C is changed to U under deamination, then change to T with host DNA repair. As a result, CBE converts C/G base pair into T/A base pair. (B) ABE are composed of a Cas9 nickase fused to deaminase that change A to I, then I to G. As a result, ABE converts A/T base pair into G/C base pair. .
Figure 1.
Mechanism schematic of cytidine base editor (CBE) and adenine base editor (ABE) system. (A) CBE contains a Cas9 nickase fused to a deaminase and two UGI (uracil glycosylase inhibitor). C is changed to U under deamination, then change to T with host DNA repair. As a result, CBE converts C/G base pair into T/A base pair. (B) ABE are composed of a Cas9 nickase fused to deaminase that change A to I, then I to G. As a result, ABE converts A/T base pair into G/C base pair. .
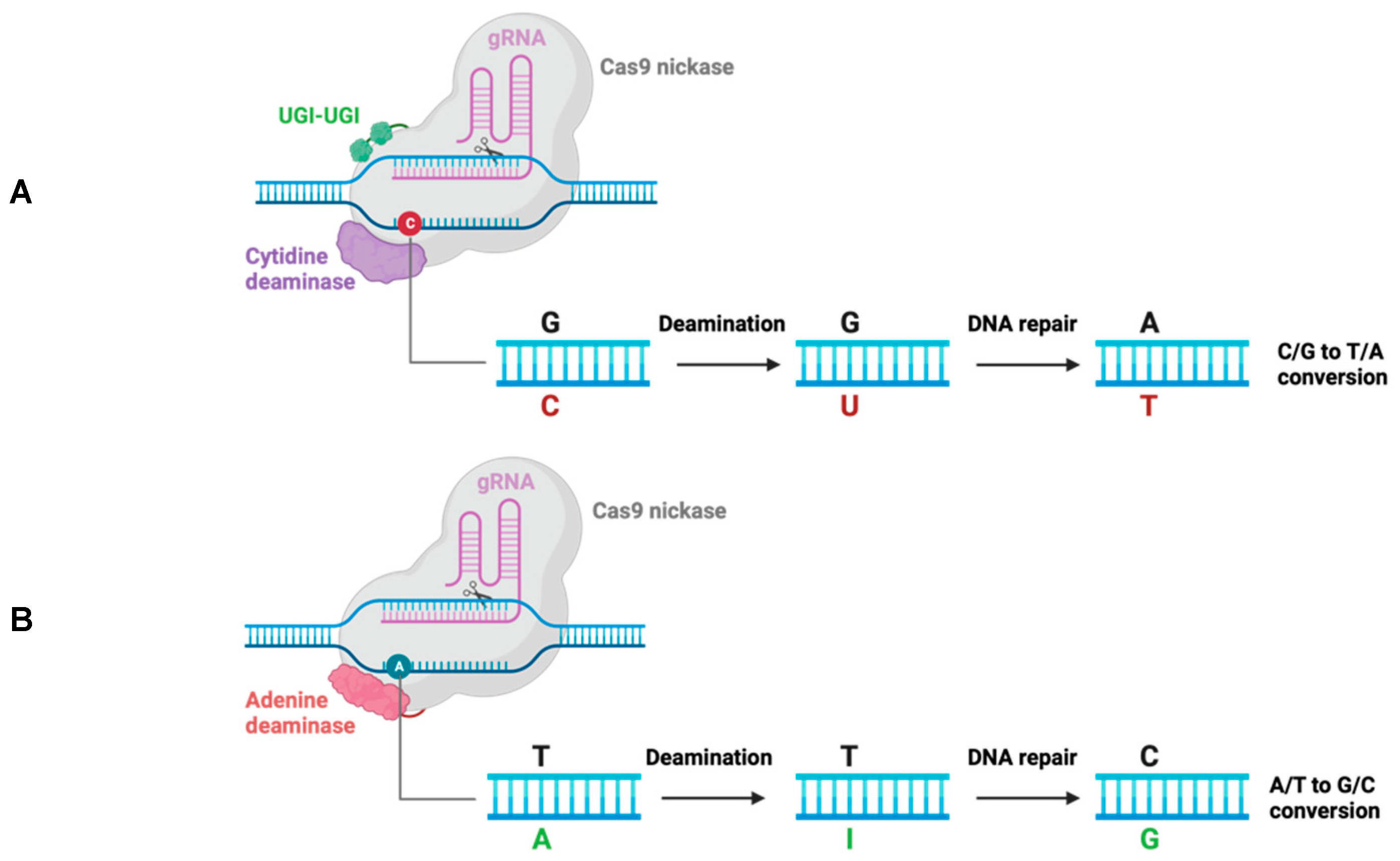
Figure 2.
Schematic of CRISPR/Cas9 system applied for HIV provirus excision. Two gRNAs were designed to target HIV LTR (5’ and 3’) and Gag at specific site. The induced double strand breaks are repaired by non-homologous end joining, which will induce deletion of fragment from 5’LTR to Gag (A), or Gag to 3’LTR (B), or 5’LTR to 3’LTR (C).
Figure 2.
Schematic of CRISPR/Cas9 system applied for HIV provirus excision. Two gRNAs were designed to target HIV LTR (5’ and 3’) and Gag at specific site. The induced double strand breaks are repaired by non-homologous end joining, which will induce deletion of fragment from 5’LTR to Gag (A), or Gag to 3’LTR (B), or 5’LTR to 3’LTR (C).
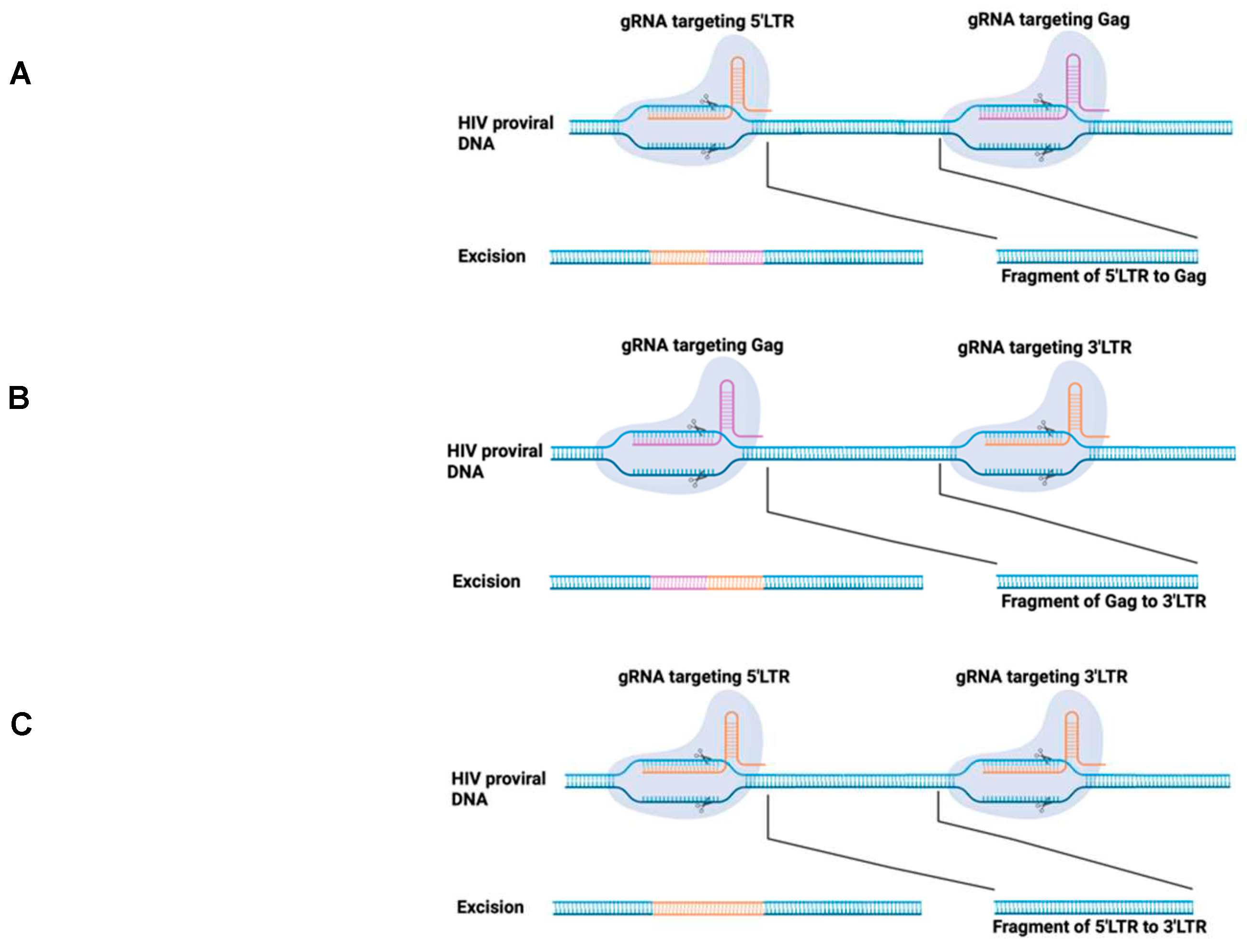

Table 1.
List of FDA approved HIV drugs by targets.
| Year | Trade name | Generic name | Molecule type | Target | Manufacturer | Class |
|---|---|---|---|---|---|---|
| 2003 | Fuzeon | Enfuvirtide (T20) | Peptide | GP41 | Trimeris/Roche | FI |
| 2007 | Selzentry | Maraviroc (MVC) | Small molecule | CCR5 | Pfizer | CRI |
| 2018 | Trogarzo | Ibalizumab-uiyk (IBA) | monoclonal antibody | CD4 | TaiMed Biologics | RI |
| 1987 | Retrovir | Zidovudine (AZT) | Small molecule | RT | GlaxoSmithKline | NRTI |
| 1991 | Videx | Didanosine (ddI) | Small molecule | RT | Bristol-Myers Squibb | NRTI |
| 1992 | Hivid | Zalcitabine (ddC) | Small molecule | RT | Roche | NRTI |
| 1994 | Zerit | Stavudine (d4T) | Small molecule | RT | Bristol-Myers Squibb | NRTI |
| 1995 | Epivir | Lamivudine (3TC) | Small molecule | RT | GlaxoSmithKline | NRTI |
| 2003 | Fuzeon | Enfuvirtide (T20) | Peptide | GP41 | Trimeris/Roche | FI |
| 2007 | Selzentry | Maraviroc (MVC) | Small molecule | CCR5 | Pfizer | CRI |
| 2018 | Trogarzo | Ibalizumab-uiyk (IBA) | monoclonal antibody | CD4 | TaiMed Biologics | RI |
| 1996 | Viramune | Nevirapine (NVP) | Small molecule | RT | Boehringer Ingelheim | NNRTI |
| 1997 | Rescriptor | Delavirdine (DLV) | Small molecule | RT | ViiV Healthcare | NNRTI |
| 1998 | Sustiva | Efavirenz (EFV) | Small molecule | RT | DuPont Pharmaceuticals | NNRTI |
| 1998 | Ziagen | Abacavir (ABC) | Small molecule | RT | ViiV Healthcare | NRTI |
| 2001 | Viread | Tenofovir disoproxil (TDF) | Small molecule | RT | Gilead | NRTI |
| 2003 | Emtriva | Emtricitabine (FTC) | Small molecule | RT | Gilead | NRTI |
| 2008 | Intelence | Etravirine (ETR) | Small molecule | RT | Johnson & Johnson | NNRTI |
| 2011 | Edurant | Rilpivirine (RPV) | Small molecule | RT | Tibotec | NNRTI |
| 2018 | Pifeltro | Doravirine (DOR) | Small molecule | RT | Merck | NNRTI |
| 1995 | Invirase | Saquinavir (SQV) | Small molecule | PR | Roche | PI |
| 1996 | Crixivan | Indinavir sulfate (IDV) | Small molecule | PR | Merck | PI |
| 1996 | Norvir | Ritonavir (RTV) | Small molecule | PR | Abbott | PI |
| 1997 | Viracept | Nelfinavir (NFV) | Small molecule | PR | Agouron Pharmaceuticals | PI |
| 1999 | Agenerase | Amprenavir (APV) | Small molecule | PR | GlaxoSmithKline | PI |
| 2000 | Kaletra | Lopinavir (LPV) | Small molecule | PR | Abbott | PI |
| 2003 | Lexiva | Fosamprenavir (FPV) | Small molecule | PR | GlaxoSmithKline | PI |
| 2003 | Reyataz | Atazanavir (ATV) | Small molecule | PR | Bristol-Myers Squibb | PI |
| 2005 | Aptivus | Tipranavir (TPV) | Small molecule | PR | Boehringer Ingelheim | PI |
| 2006 | Prezista | Darunavir (DRV) | Small molecule | PR | Tibotec | PI |
| 2007 | Isentress | Raltegravir (RAL) | Small molecule | IN | Merck | InSTI |
| 2013 | Tivicay | Dolutegravir (DTG) | Small molecule | IN | ViiV Healthcare | InSTI |
| 2014 | Vitekta | Elvitegravir (EVG) | Small molecule | IN | Gilead | InSTI |
| 2018 | Biktarvy | Bictegravir (BIC) | Small molecule | IN | Gilead | InSTI |
| 2021 | Vocabria | Cabotegravir (CAB) | Small molecule | IN | ViiV Healthcare | InSTI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



