
Submitted:
23 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Inhibitors of the factor FXI represent a new class of anticoagulant agents that are facing the clinical approval for the treatment of acute coronary syndrome (ACS), venous thromboembolism (VTE), and stroke prevention of atrial fibrillation (AF). These new inhibitors include chemical small molecules (asundexian and milvexian), monoclonal antibodies (abelacimab, osocimab, and xisomab), and antisense oligonucleotides (IONIS-FXIRX and fesomersen), and thus with very peculiar and different pharmacokinetic and pharmacodynamic properties. Besides their clinical efficacy and safety, based on their pharmacological heterogeneity, the use of these drugs in patients with comorbidities may undergo to drug-drug interactions (DDI) with other concomitant therapies. Although only few clinical evidence are available, it is possible to predict clinical relevant DDI by taking into consideration their pharmacokinetic properties, such as the CYP450-dependent metabolism, the interaction with drug transporters and/or the route of elimination. These characteristics may be useful to differentiate their use with the direct oral anticoagulant (DOAC) anti -FXa (rivaroxaban, apixaban, edoxaban) and thrombin (dabigatran), whose pharmacokinetics are strongly dependent from P-gp inhibitors/inducers. In the present review, we summarize the current clinical evidence on DDI of new anti FXI with CYP450/P-gp inhibitors and inducers and indicate potential differences with DOAC anti FXa.
Keywords:
DOAC
; FXIa
; mAbs
; ASO
; drug-drug interactions
1. Introduction
Direct oral anticoagulants (DOACs) represent a valid alternative to warfarin and, more in general, to vitamin K antagonists, as standard of care anticoagulation therapy. The therapeutic class of DOACs include one thrombin (Factor II activated, FIIa) inhibitor, dabigatran, and three FX inhibitors, rivaroxaban, apixaban and edoxaban. DOACs are indicated for prevention of stroke in non-valvular atrial fibrillation (NVAF) patients, venous thromboembolism (VTE), and for treatment of acute coronary syndrome (ACS). Since the first approval in 2010, DOACs have shown in randomized, placebo controlled, clinical trials the same efficacy than warfarin in preventing thrombotic events but with approximately 50% lower risk of intracranial bleeding events [1]. This advantageous safety profile is partially limited by a 25% increased risk of gastrointestinal bleeding risk compared to warfarin [1]. The administration of DOACs is also more convenient, than heparins and warfarin, for their used at fixed doses without the routine coagulation monitoring. Despite their improved safety profile and compliance, DOACs still have limitations, including the high risk for gastrointestinal bleeding and relevant drug-drug interactions (DDI) with other therapies [2].
In search of a new anticoagulant agents with a better clinical profile, the inhibition of FXI has been considered very attractive in consideration to the reduced risk of thrombosis with minor bleeding tendency of subjects carrying a genetic defect of this contact pathway coagulation factor [3,4].
In the new era of pharmacology, it is imperative to use different strategies for a proper inhibition of a particular molecular target, i.e., FXI. Currently, there are three classes of anti FXI under clinical development, small molecules (asundexian and milvexian), monoclonal antibodies (abelacimab, osocimab, xisomab), and antisense oligonucleotides (ASO) IONIS-FXIRX, and IONIS-FXI-LRX (Figure 1).
Promising results of phase 2 trials in patients undergoing major orthopedic surgery, and in those with end-stage renal disease (ESRD), NVAF and ACS have led to large phase 3 trials that are currently ongoing. Among these, the OCEANIC-AF phase III study has been stopped early due to inferior efficacy of asundexian versus apixaban for preventing stroke in patients with NVAF. Since the available safety data are consistent with previously reported safety profiles of asundexian, the drug is still under investigation in the OCEANIC-STROKE phase III study as planned [5].
The fully human monoclonal antibody (mAb) abelacimab binds and inhibit both FXI and FXIa [6], while osocimab blocks specifically FXI in the active configuration (FXIa) [7]. Differently, xisomab prevents FXI activation by FXIIa, thus even though it binds FXI functions as a FXIIa inhibitor. These drugs are administered by i.v or s.c injection and have a very long half-life time of elimination (25-40 days) (Figure 1).
Asundexian and milvexian are chemically synthesized molecule, which potently inhibit the active form of FXI (FXIa) in a reversible manner. Asundexian show a relatively longer half-life time than milvexian (15 h vs 10 h). Accordingly, asundexian is administered orally once a day while milvexian can be used also with twice daily posology [7].
Different from small molecule drugs or biologics which bind and affect target proteins, ASOs are short chemically modified oligonucleotides that bind to complementary RNA in cells and modulate RNA function to produce a pharmacological effect [8] (Figure 1). IONIS-FXIRX (BAY2306001) is an ASO that inhibits the synthesis of coagulation factor XI (FXI), while FXI-LICA (BAY2976217, IONIS-FXI-LRX, fesomersen) shares the same RNA sequence as IONIS-FXIRX but contains a GalNAc-conjugation that facilitates asialoglycoprotein receptor (ASGPR)-mediated uptake into hepatocytes. The ASO are administered subcutaneously and showed a half-life time of elimination of approximately 1-2 weeks [9]. The GalNAc-conjugation, presents in the FXI-LICA, has a strong influence on the pharmacokinetic and pharmacodynamic allowing lower and less frequent dosing (once-monthly vs. once-weekly) to yield comparable pharmacological activity as unconjugated ASOs [10] (Figure 1).
2. Pharmacological considerations of drug-drug interaction with mAbs and ASO
To predict the possible drug interactions of mAbs and ASO with small chemical drugs it is mandatory to know their metabolism pathways and elimination processes. On the other hands the absorption phase can be excluded from the prediction of possible drug interactions since both types of drugs are administered subcutaneously.
Abelacimab, osocimab, and xisomab, like other mAbs are eliminated by means of catabolic processes. mAbs are catabolized into peptides and amino acids primarily by the reticulum endothelial system (RES), which consists of phagocytic cells, such as macrophages and monocytes [11]. For this reason, their elimination is mainly independent from the liver and kidney function. A second relevant route of elimination is target-mediated, consisting in the internalization within the target-expressing cells, and consequent intracellular catabolism within the lysosomes. However, this mechanism of elimination can be excluded for mAbs targeted to soluble factors, such as the anti FXIa. For the same reason the elimination of mAbs anti FXIa can be predicted to be constant, independently from the dose. However, some exceptions have been described, with mAbs that, even if they are targeted to soluble antigens, may undergo to dose-dependent clearance. The soluble mAb-antigen complex can be recognized by the Fcγ receptors express on monocytes and macrophages, which subsequently triggers its internalization and catabolism [12,13].
Finally, it must be considered that the administration of therapeutic mAbs, can lead to the generation of autoantibodies by immune system. The antibody response still represents a clinically relevant event that may interfere with the pharmacological activity of mAbs and contribute to their clearance [14,15]. The activation of the antibody response depends on numerous factors including the state of the patient’s immune system, the co- administration of other drugs, the structure and the anti-body composition (glycosylation), but more importantly on the similarity with the endogenous IgG [16]. Usually, fully human antibodies are less immunogenic than humanized one, difference that had determined, for instance, the failure of the clinical development of the anti PCSK9 antibody bococizumab [17].
After a single dose i.v. and multiple dose s.c. of abelacimab no anti-drug antibodies (ADA) have been detected, confirming the non-immunogenicity of this fully-human mAb [18]. Conversely, after single i.v. or s.c. doses in healthy volunteers of fully human mAb osocimab, ADA formation was confirmed in six volunteers out of 56 included in the study [19]. In phase 1, first in human study, xisomab, a humanized anti-FXI antibody, did not show immunogenic reactivity with no subjects with anti-drug antibodies detected [20]. The apparent higher immunogenicity of osocimab compared to xisomab, may have a potential impact on its clearance that can be reduced in patients treated with immunosuppressive drugs, such as methotrexate, azathioprine or mercaptopurine, as observed with infliximab and adalimumab [21,22].
The absorption of ASO and GalNAc-conjugated ASOs is rapid following subcutaneous injection with a time to maximum concentration (Tmax) between 1-4 h [10]. GalNAc-conjugated ASOs are highly bound to plasma proteins, the extent of protein binding is mostly greater than 98%. The GalNAc conjugation showed good stability in blood after absorption, although metabolites with 1, 2, and/or 3 GalNAc sugar deletions or unconjugated ASO can be detected. The total exposure of each of its metabolites accounted for less than 13% of the total full length oligonucleotide [23]. The AUC for unconjugated ASO accounted for ≤5% of the total full-length oligonucleotides within the first 24-h post dose. After 24 h, the unconjugated ASO became the major circulating species in plasma [10]. GalNAc-conjugated ASOs have a much higher plasma clearance than unconjugated ASOs due to a more efficient uptake to hepatocytes (over 5-fold) [10]. GalNAc-conjugated plasma clearance is driven by tissue distribution that occurs rapidly and extensively mainly to liver and kidney.
The three GalNAc sugars are cleaved off one after another by lysosomal N-acetyl-β-glucosaminidase, then, the lysosomal DNase II (deoxyribonuclease II) remove the linker from the ASO. This hydrolysis
creates an active phosphate species which is rapidly deactivated either by alkaline phosphatase or being esterified with one of the exposed hydroxyl groups to form cyclized metabolites. Upon further oxidation, these linker metabolites are eliminated as acids by biliary excretion. The phosphate removed linkers are also metabolized by O- or N-dealkylation and eliminated by renal excretion [24].
Similar to unconjugated ASOs, renal excretion of GalNAc-conjugated ASOs is limited due to their highly bound to proteins. No protein binding related DDIs would be expected for GalNAc-conjugated ASOs with small drug molecules. On the other hand, DDI may occur when two or multiple ASOs are co-administered. Studies in animals have shown that an excipient ASO may enhance the pharmacological activity of another ASO, probably via protein binding displacement in the intracellular space [25,26]. However, the clinical relevance of these findings remains to be elucidated.
The major species excreted in urine is the unconjugated ASO, which represents only 1% of the total dose administered, together with various short nucleotide metabolites, as observed for unconjugated ASOs dosed alone [27]. After subcutaneous administration, 25.7% of the radioactive GalNAc is recovered in urine and 71.7% in the feces [28].
In vitro studies demonstrated that GalNAc-conjugated ASOs are not inhibitor nor inducer of major cytochrome P450 isoforms (e.g. CYP1A2, CYP2B6, and CYP3A4) and are not substrates or inhibitors of uptake or efflux membrane transporters (e.g. OATP, OAT, MDR1, etc.) [29,30]. Thus, ASO and GalNAc-conjugated ASOs are predicted to have no DDI with small drug molecules and to be safely administered in patients with CKD, and ESRD [9,31].
Thus, the clearance of mAbs and ASO does not involve CYP450 activity or interaction with cell membrane transporters, such as P-glycoprotein (P-gp), therefore their pharmacokinetics interactions with small molecule drugs can be excluded. It remains to be established the possible pharmacodynamic interaction with drugs affecting the coagulation cascade or the platelet activities. Although the inhibitors of FXIa are considered safe in terms of risk of major bleeding [7,32,33], it cannot be excluded a clinically relevant interaction with antiplatelets, aspirin, or NSAIDs.
3. DOAC and small drug molecules anti FXIa: potential differences on DDIs
Differently from mAbs, small molecules can be cleared by means of CYP450 enzyme activities and by binding to drug transporters, such as P-gp, OATPs, OATs, and OCTs. Preclinical results indicate that milvexian is metabolized by CYP3A4/5 and it is a substrate of P-gp, but not OATP [34]. The renal excretion ranged from 6.9% to 17.8% of the administered dose and it is lower in the fasted state (6.9%-12.3%) than in the fed state (16.3%-17.8%). This difference is more likely due to the higher oral bioavailability in the presence of food [35]. The total exposures to milvexian, after a single oral 60 mg dose, were 41% and 54% greater in participants with eGFR values of 30 and 15 mL/min/1.73 m2, respectively, than in subjects with normal renal function. The median time to maximum concentration (Tmax) was similar for the three groups (4.5-5.0 h) and the half-life increased for participants with moderate (18.0 h) or severe (17.7 h) renal impairment compared with those with normal renal function (13.8 h) [36]. Thus, according to the low renal excretion, a single dose of milvexian 60 mg was safe and well tolerated in participants with moderate or severe renal impairment [36].
Asundexian was shown to be predominantly excreted via feces and to a lower extent with urine (80.3% and 20.3%, respectively), primarily either as a product of amide hydrolysis (~47%) or as unchanged drug (~37%), with oxidative biotransformation playing a minor role (~13%) [37]. More recently, in vitro analysis determined that asundexian is a P-gp substrate and it is predominantly metabolized via carboxylesterase 1 and, to a lesser extent, via CYP3A4 [38]. Asundexian did not affect CYP1A2, CYP2A6, CYP2B6, CYP2C19, CYP2E1, CYP2J2, and CYP3A4 while a weak inhibitory potential on CYP2C8, CYP2C9, CYP1A1, and CYP2D6 was observed [39]. Thus, both milvexian and asundexian are both P-gp substrate as the currently approved DOACs (dabigatran, edoxaban, apixaban, and rivaroxaban) (Table 1), therefore strong inhibitors of this drug transporter can increase absorption and exposure of these anticoagulants and the risk of bleeding [2].
On the other hand, an induction of P-gp can reduce their absorption and expose patients to thrombosis. The most likely interaction mechanism for all DOACs consists of significant gastrointestinal re-secretion over a P-gp transporter after absorption in the gut and in their renal clearance [40]. The intensity of the inhibition or induction of P-gp transporters can predict the entity of the change in drug exposure (Table 2).
Dabigatran and edoxaban seem to be less influenced by CYP450 inhibitors and inducers, since their metabolism does not involve this enzymatic pathway [41]. On the contrary, rivaroxaban, and to a lower extent apixaban, are metabolized by CYP3A4, thus opening to possible an additional mechanism of DDI beyond the modulation of P-gp function (Table 1 and 2).
Individual variability of DOAC plasma concentrations may significantly impact the interaction with P-gp inhibitors or inducers. Thus, dabigatran, that shows a low oral bioavailability, and rivaroxaban, whose gastrointestinal absorption is strongly increased by the presence of food [2], may be more exposed to clinically significant DDI.
The anti FXIa are also substrate for P-gp and, thus, can undergo to similar DDI of DOACs [2]. In addition, both milvexian and asundexian are metabolized by CYP3A4, and thus can interact with drugs that influence this metabolic pathway (inducers or inhibitors) (Table 1 and 2). The gastrointestinal absorption of both drugs is partially influenced by the presence of food, with a higher bioavailability for milvexian and lower for asundexian. However, these effects appear to have a lower impact compared to rivaroxaban, whose absorption is strongly reduced under fasting condition.
Another level of differentiation between the DOAC and the new oral anti FXIa is the renal excretion. Milvexian and asundexian are marginally eliminated by the kidney and thus, can be potentially administered in patients with moderate or severe renal impairment (Table 1), as documented by the results of a clinical trial [36].
4. Clinical evidence of DDI with milvexian and asundexian
The potential effect of asundexian to inhibit and/or induce CYP3A4 over time was investigated in a nonrandomized, open-label study. The AUC and maximum plasma concentration (Cmax) of midazolam, CYP3A4 substrate, and its metabolites α-hydroxymidazolam, were determined before and after the coadministration of midazolam with a single or multiple doses of asundexian [43].
The time to reach the Cmax (Tmax) of midazolam and α-hydroxymidazolam were rapidly reached (0.5-0.75 h) and were not affected by the co-administration of asundexian. Further, AUC, Cmax and the half-life time (T1/2) of midazolam and were shown to be similar across the treatment groups (Table 3). Coadministration of asundexian 25 mg for one day resulted in an 4.2% and 4.4% increase in the AUC of midazolam and α-hydroxymidazolam, respectively. Similar effect was observed after the coadministration of 75 mg of asundexian. The AUC ratios of α-hydroxymidazolam to midazolam were comparable before and after the administration of asundexian at 25 mg and 75 mg. Following repeated dosing with asundexian 25 mg once daily for 9 consecutive days, it has been observed an increase of AUCs for midazolam and α-hydroxymidazolam by 6.3% and 6.0%, respectively. A slightly higher effect was observed with 75 mg of asundexian with an increase in the AUC for midazolam of 17% and a decrease in the AUC for α-hydroxymidazolam of 7.2%. Based on these results, asundexian appeared to have no relevant effect on the AUC of midazolam and α-hydroxymidazolam either after 1 day or 10 days of treatment (Table 3). The 90% confidence interval (CI) of the AUC ratio for both midazolam and α-hydroxymidazolam were within the 0.80 to 1.25 values, except for the upper 90% CI for AUC ratio for midazolam on day 10 in the 75 mg group, which was numerically only marginally above the 1.25 boundary [43]. Taken together, this trial demonstrated that asundexian 25 mg or 75 mg showed no indication of any clinically relevant inhibition and/or induction of CYP3A4 in healthy volunteers.
A second phase 1 clinical trial was performed to investigate the pharmacokinetics of asundexian upon co-administration with the two P-gp inhibitors itraconazole and verapamil with strong and moderate inhibitory capacity towards CYP3A4, respectively (Table 2) or the moderate CYP3A4 inhibitor fluconazole.
Itraconazole increased mean asundexian AUC by 103% (ratio: 2.0; 90% CI: 1.9-2.2) with a prolongation of the t1/2 from 16.2 to 28.9 hours (Table 4). No effect on Cmax of asundexian was observed in response to itraconazole administration (2.6%; ratio: 1.025, 90% CI: 0.95–1.10). A less potent CYP3A4 inhibitor verapamil increased asundexian exposure (AUC) of 75.6% (ratio: 1.75, 90% CI: 1.66-1.85). Also in this case, the effect on Cmax was less pronounced (Table 4). Terminal t1/2 increased from 13.9 to 22.6 hours compared with asundexian alone. Co-administration with fluconazole had a less pronounced effect on asundexian AUC and Cmax, with a 16.8% and 3.3% increased, respectively. Terminal half-life was comparable between treatments (15.5 h [with fluconazole] vs 13.9 h [asundexian alone]).
The CYP3A4 inducer carbamazepine reduced the concentrations of asundexian accelerating the elimination rate. Median tmax was slightly shorter, at 2.0 h with carbamazepine compared with 3.0 h for asundexian alone. Mean exposure (AUC) of asundexian decreased by 44.4% (ratio: 0.56, 90% CI: 0.50-0.61) with concomitant administration of carbamazepine, whereas Cmax was less affected (-18.2%) (Table 4).
Importantly, these studies observed a close correlation between the plasma concentration of asundexian and activated partial thromboplastin time (aPTT) or FXIa activity, indicating a direct inhibition of the coagulation cascade. Accordingly, the aPTT and FXIa activity returned to baseline levels after 48 h, while both parameters were still affected in the presence of itraconazole (1.17 and 0.37, respectively), suggesting a longer duration of action of asundexian [38]. Indeed, the actual return to baseline was achieved at 96 h post-dose when asundexian was co-administered with itraconazole. Co-treatment with verapamil or fluconazole yielded intermediate results according to their lower capacity to inhibit CYP3A4 and P-gp. On the contrary, the induction of CYP3A4 and P-gp by carbamazepine determined a fastest return to baseline for both pharmacodynamic markers (aPTT and FXIa) with values approaching baseline at 24 h post-dose [38].
Taken together, these results indicate a possible DDI of asundexian with drugs having a combined inhibitory effect on both P-gp and CYP3A4 (itraconazole and verapamil). However, no effect was observed in response to moderate CYP3A inhibition (fluconazole). A lack of efficacy of asundexian can also be predicted in the presence of strong P-gp and CYP3A4 inducers, i.e. carbamazepine.
Asundexian is excreted, as parental drug, via feces (28%) and urine (9%) [37], thus the increased asundexian exposure observed with P-gp inhibitors on top of CYP3A4 inhibition (itraconazole, verapamil) indicates a relevant contribution of this transporter to active renal and non-renal secretion. The observed high oral bioavailability of asundexian [44] indicates a saturated gastrointestinal P-gp efflux and negligible first-pass metabolism. Thus, the effect of itraconazole and verapamil on asundexian exposure may be related to a decrease in systemic CYP3A4-mediated biotransformation together with renal/non-renal P-gp driven excretion. In line with this hypothesis, asundexian renal clearance increased and decreased after the administration with P-gp and CYP3A4 inducers and inhibitors, respectively, but was not affected by CYP3A4 inhibition alone as observed with fluconazole (Table 4).
Taken together, asundexian exposure is mainly driven by P-gp inhibition and reduced excretion as unchanged drug via feces and urine. Nevertheless, the strongest DDI was observed with a combined P-gp and strong CYP3A4 inhibition, suggesting the contribution of both pathways on asundexian disposition.
Similar DDI clinical studies have been performed with milvexian, where the effect of rifampicin (CYP3A4 and P-gp inducer) and two CYP450 inhibitors itraconazole (strong) and diltiazem (moderate) on the pharmacokinetic of milvexian were investigated [45,46].
These studies were carried out considering the preclinical data demonstrating that milvexian is a substrate of CYP3A4/5 and P-gp [34]. Single dose of rifampicin does not affect the exposure of milvexian, however, following repeated doses the t1/2 was shorter (9h vs. 13 h), Tmax values similar and a significant decreased of both Cmax, and AUC was observed with their values equal to 22% and 15%, respectively, compared to milvexian alone [46] (Table 5).
It has been established that acute administration of rifampin results in potent inhibition of hepatic OATP1B uptake transporters, thus, the fact that a single dose of rifampicin did not affect milvexian exposure indicated that this drug is not an OATP1B substrate, as observed in vitro [34]. On the contrary, after multiple doses of rifampicin a strong induction of CYP3A4 and P-gp can occur. Under this treatment, a significant reduction in Cmax of milvexian was observed, indicative of a high level of induction of CYP3A and P-gp in the gut, leading to a limited systemic absorption of milvexian. Consistent with the lower plasma concentration of milvexian following repeated doses of rifampin, the activity of two pharmacodynamic biomarkers, aPTT or FXI, were minimally affected compared to when milvexian was administered alone [46].
An open-label, non-randomized, two-period crossover study conducted in healthy participants assessed the effects of co-administration with strong and moderate CYP3A inhibitors itraconazole and diltiazem, respectively, on the pharmacokinetic and pharmacodynamic properties of milvexian [45]. Considering the different inhibitory potency towards CYP3A4, the AUC of milvexian increased by 3.8-fold and by 38% with co-administered with itraconazole, and diltiazem, respectively. Cmax was 28% higher in the presence of itraconazole and by 9.6% with diltiazem, compared with milvexian alone (Table 5). Finally, the t1/2 values were approximately 1.5- and 1.1-fold longer with itraconazole and diltiazem co-administration, respectively, while Tmax values were similar in the two treatment arms [45]. As expected, aPTT was prolonged by milvexian administration either alone or when co-administered with itraconazole or diltiazem, with maximal mean aPTT prolongation occurring near Cmax. As observed with asundexian, the magnitude of aPTT prolongation was directly dependent by milvexian plasma concentration, and thus a longer effect on aPTT was seen in the presence of itraconazole [45]. These results confirmed that milvexian is a substrate for CYP3A4 and P-gp. However, the impact of a strong CYP3A4 inhibitor was below 5-fold by the observed increases in milvexian exposure, and the impact of a moderate CYP3A4 inhibitor was less than 2.5-fold. This suggests that milvexian would not be considered a sensitive CYP3A4 substrate (i.e. AUC ≥ 5-fold) [47].
5. Conclusions
Considering the different pharmacokinetic characteristics of the three types of FXIa inhibitors, it appears clear that both monoclonal antibodies and ASO clearance is not affected by kidney and liver impairment and the do not undergo to significant DDI with conventional small molecule drugs. Thus, their use appears to be less problematic in older and multimorbidity patients, although their very long half lifetime (one month and 2-4 weeks, respectively) may represent a critical issue in case of side effects, such as major bleeding. It remains, indeed, to be established the pharmacodynamic interaction with drugs that increase the risk of bleeding, such as antiplatelets, selective serotonin reuptake inhibitors (SSRI), tyrosine kinase inhibitors, bevacizumab and alemtuzumab [2].
On the contrary, the oral small drug molecule anti FXIa (milvexian and asundexian) may face similar DDI issues observed with the DOAC (dabigatran, apixaban, edoxaban, and rivaroxaban). For both drugs we have results of dedicated clinical trial assessing their potential DDI with both inducers and inhibitors of CYP3A4 and P-gp. Although preclinical studies suggested a possible interaction dependent from CYP3A4 pathway, milvexian and asundexian demonstrated to undergo to clinically relevant interaction with P-gp inhibitors and inducers, while their pharmacokinetics seem to be less influenced by modulators of CYP3A4. However, a potential advantage compared to the DOAC, is represented by their very limited renal extraction, and thus may be administered also in patients with CKD.
Author Contributions
Writing—original draft preparation, N.F.; writing—review and editing, E.C.; A.C.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Ferri, N.; Colombo, E.; Tenconi, M.; Baldessin, L.; Corsini, A. Drug-Drug Interactions of Direct Oral Anticoagulants (DOACs): From Pharmacological to Clinical Practice. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Chan, N.C.; Weitz, J.I. New Therapeutic Targets for the Prevention and Treatment of Venous Thromboembolism With a Focus on Factor XI Inhibitors. Arterioscler Thromb Vasc Biol 2023, 43, 1755–1763. [Google Scholar] [CrossRef]
- Duga, S.; Salomon, O. Congenital factor XI deficiency: an update. Seminars in thrombosis and hemostasis 2013, 39, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.bayer.com/media/en-us/oceanic-af-study-stopped-early-due-to-lack-of-efficacy/.
- Koch, A.W.; Schiering, N.; Melkko, S.; Ewert, S.; Salter, J.; Zhang, Y.; McCormack, P.; Yu, J.; Huang, X.; Chiu, Y.H.; et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood 2019, 133, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Simioni, P.; Prandoni, P.; Ferri, N. Clinical Pharmacology of Factor XI Inhibitors: New Therapeutic Approaches for Prevention of Venous and Arterial Thrombotic Disorders. Journal of clinical medicine 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.; Bethune, C.; Smyth, A.; Tyrwhitt, J.; Jung, S.W.; Yu, R.Z.; Wang, Y.; Geary, R.S.; Weitz, J.; Bhanot, S.; et al. Phase 2 Study of the Factor XI Antisense Inhibitor IONIS-FXIRx in Patients With ESRD. Kidney Int Rep 2022, 7, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, R.Z.; Henry, S.; Geary, R.S. Pharmacokinetics and Clinical Pharmacology Considerations of GalNAc(3)-Conjugated Antisense Oligonucleotides. Expert opinion on drug metabolism & toxicology 2019, 15, 475–485. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clinical pharmacokinetics 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.A.; Tseng, C.M.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Marathe, A.; Peterson, M.C.; Mager, D.E. Integrated cellular bone homeostasis model for denosumab pharmacodynamics in multiple myeloma patients. The Journal of pharmacology and experimental therapeutics 2008, 326, 555–562. [Google Scholar] [CrossRef]
- Ridker, P.M.; Tardif, J.C.; Amarenco, P.; Duggan, W.; Glynn, R.J.; Jukema, J.W.; Kastelein, J.J.P.; Kim, A.M.; Koenig, W.; Nissen, S.; et al. Lipid-Reduction Variability and Antidrug-Antibody Formation with Bococizumab. N Engl J Med 2017, 376, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Roskos, L.K.; Kellermann, S.; Foon, K.A. Human Antiglobulin Responses. Measuring Immunity. Basic Biology and Clinical Assessment 2005, Chapter 13, 172–186. [Google Scholar]
- Clark, M. Antibody humanization: a case of the ’Emperor’s new clothes’? Immunol Today 2000, 21, 397–402. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Bococizumab for the treatment of hypercholesterolaemia. Expert opinion on biological therapy 2017, 17, 909–910. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.A.; Freedholm, D.; Widener, N.; Wang, X.; Simard, E.; Cullen, C.; Al-Saady, N.M.; Lepor, N.E.; Coulter, S.; Lovern, M.; et al. Pharmacokinetics and pharmacodynamics of Abelacimab (MAA868), a novel dual inhibitor of Factor XI and Factor XIa. Journal of thrombosis and haemostasis : JTH 2022, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, B.; Thomas, D.; Schwers, S.; Wiegmann, S.; Prange, W.; Yassen, A.; Boxnick, S. First randomized evaluation of safety, pharmacodynamics, and pharmacokinetics of BAY 1831865, an antibody targeting coagulation factor XI and factor XIa, in healthy men. Journal of thrombosis and haemostasis : JTH 2022, 20, 1684–1695. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Verbout, N.G.; Wallisch, M.; Hagen, M.W.; Shatzel, J.J.; Olson, S.R.; Puy, C.; Hinds, M.T.; McCarty, O.J.T.; Gailani, D.; et al. Contact Activation Inhibitor and Factor XI Antibody, AB023, Produces Safe, Dose-Dependent Anticoagulation in a Phase 1 First-In-Human Trial. Arteriosclerosis, thrombosis, and vascular biology 2019, 39, 799–809. [Google Scholar] [CrossRef]
- Maini, R.N.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Davis, D.; Macfarlane, J.D.; Antoni, C.; Leeb, B.; Elliott, M.J.; Woody, J.N.; et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis and rheumatism 1998, 41, 1552–1563. [Google Scholar] [CrossRef]
- Seitz, K.; Zhou, H. Pharmacokinetic drug-drug interaction potentials for therapeutic monoclonal antibodies: reality check. Journal of clinical pharmacology 2007, 47, 1104–1118. [Google Scholar] [CrossRef]
- Gunawan, P.I.; Idarto, A.; Saharso, D. Acanthamoeba Infection in a Drowning Child. Ethiop J Health Sci 2016, 26, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Graham, M.J.; Post, N.; Riney, S.; Zanardi, T.; Hall, S.; Burkey, J.; Shemesh, C.S.; Prakash, T.P.; Seth, P.P.; et al. Disposition and Pharmacology of a GalNAc3-conjugated ASO Targeting Human Lipoprotein (a) in Mice. Mol Ther Nucleic Acids 2016, 5, e317. [Google Scholar] [CrossRef]
- Donner, A.J.; Wancewicz, E.V.; Murray, H.M.; Greenlee, S.; Post, N.; Bell, M.; Lima, W.F.; Swayze, E.E.; Seth, P.P. Co-Administration of an Excipient Oligonucleotide Helps Delineate Pathways of Productive and Nonproductive Uptake of Phosphorothioate Antisense Oligonucleotides in the Liver. Nucleic Acid Ther 2017, 27, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Wancewicz, E.; Matson, J.; Pearce, M.; Siwkowski, A.; Swayze, E.; Bennett, F. Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2’-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem Pharmacol 2009, 78, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Yu, R.Z.; Watanabe, T.; Henry, S.P.; Hardee, G.E.; Chappell, A.; Matson, J.; Sasmor, H.; Cummins, L.; Levin, A.A. Pharmacokinetics of a tumor necrosis factor-alpha phosphorothioate 2’-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug metabolism and disposition: the biological fate of chemicals 2003, 31, 1419–1428. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Yu, R.Z.; Gaus, H.J.; Greenlee, S.; Post, N.; Schmidt, K.; Migawa, M.T.; Seth, P.P.; Zanardi, T.A.; Prakash, T.P.; et al. Elucidation of the Biotransformation Pathways of a Galnac3-conjugated Antisense Oligonucleotide in Rats and Monkeys. Mol Ther Nucleic Acids 2016, 5, e319. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Yu, R.Z.; Warren, M.S.; Liu, M.; Jahic, M.; Nichols, B.; Post, N.; Lin, S.; Norris, D.A.; Hurh, E.; et al. Assessment of the Drug Interaction Potential of Unconjugated and GalNAc(3)-Conjugated 2’-MOE-ASOs. Mol Ther Nucleic Acids 2017, 9, 34–47. [Google Scholar] [CrossRef]
- Yu, R.Z.; Warren, M.S.; Watanabe, T.; Nichols, B.; Jahic, M.; Huang, J.; Burkey, J.; Geary, R.S.; Henry, S.P.; Wang, Y. Lack of Interactions Between an Antisense Oligonucleotide with 2’-O-(2-Methoxyethyl) Modifications and Major Drug Transporters. Nucleic Acid Ther 2016, 26, 111–117. [Google Scholar] [CrossRef]
- Willmann, S.; Marostica, E.; Snelder, N.; Solms, A.; Jensen, M.; Lobmeyer, M.; Lensing, A.W.A.; Bethune, C.; Morgan, E.; Yu, R.Z.; et al. PK/PD modeling of FXI antisense oligonucleotides to bridge the dose-FXI activity relation from healthy volunteers to end-stage renal disease patients. CPT Pharmacometrics Syst Pharmacol 2021, 10, 890–901. [Google Scholar] [CrossRef]
- Verhamme, P.; Yi, B.A.; Segers, A.; Salter, J.; Bloomfield, D.; Buller, H.R.; Raskob, G.E.; Weitz, J.I.; Investigators, A.-T. Abelacimab for Prevention of Venous Thromboembolism. The New England journal of medicine 2021, 385, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Bauersachs, R.; Becker, B.; Berkowitz, S.D.; Freitas, M.C.S.; Lassen, M.R.; Metzig, C.; Raskob, G.E. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. Jama 2020, 323, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Dilger, A.K.; Pabbisetty, K.B.; Corte, J.R.; De Lucca, I.; Fang, T.; Yang, W.; Pinto, D.J.P.; Wang, Y.; Zhu, Y.; Mathur, A.; et al. Discovery of Milvexian, a High-Affinity, Orally Bioavailable Inhibitor of Factor XIa in Clinical Studies for Antithrombotic Therapy. Journal of medicinal chemistry 2022, 65, 1770–1785. [Google Scholar] [CrossRef]
- Perera, V.; Wang, Z.; Luettgen, J.; Li, D.; DeSouza, M.; Cerra, M.; Seiffert, D. First-in-human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin Transl Sci 2022, 15, 330–342. [Google Scholar] [CrossRef]
- Perera, V.; Abelian, G.; Li, D.; Wang, Z.; Zhang, L.; Lubin, S.; Bello, A.; Murthy, B. Single-Dose Pharmacokinetics of Milvexian in Participants with Normal Renal Function and Participants with Moderate or Severe Renal Impairment. Clinical pharmacokinetics 2022, 61, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Piel, I.; Engelen, A.; Lang, D.; Schulz, S.I.; Gerisch, M.; Brase, C.; Janssen, W.; Fiebig, L.; Heitmeier, S.; Kanefendt, F. Metabolism and Disposition of the Novel Oral Factor XIa Inhibitor Asundexian in Rats and in Humans. European journal of drug metabolism and pharmacokinetics 2023, 48, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Kanefendt, F.; Brase, C.; Jungmann, N.; Fricke, R.; Engelen, A.; Schmitz, S. Pharmacokinetics of asundexian with combined CYP3A and P-gp inhibitors and an inducer: Target in vitro and in vivo studies. British journal of clinical pharmacology 2023. [Google Scholar] [CrossRef]
- Roehrig, S.; Ackerstaff, J.; Jimenez Nunez, E.; Teller, H.; Ellerbrock, P.; Meier, K.; Heitmeier, S.; Tersteegen, A.; Stampfuss, J.; Lang, D.; et al. Design and Preclinical Characterization Program toward Asundexian (BAY 2433334), an Oral Factor XIa Inhibitor for the Prevention and Treatment of Thromboembolic Disorders. Journal of medicinal chemistry 2023, 66, 12203–12224. [Google Scholar] [CrossRef]
- Gnoth, M.J.; Buetehorn, U.; Muenster, U.; Schwarz, T.; Sandmann, S. In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. The Journal of pharmacology and experimental therapeutics 2011, 338, 372–380. [Google Scholar] [CrossRef]
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology 2021, 23, 1612–1676. [Google Scholar] [CrossRef]
- Corsini, A.; Ferri, N.; Proietti, M.; Boriani, G. Edoxaban and the Issue of Drug-Drug Interactions: From Pharmacology to Clinical Practice. Drugs 2020, 80, 1065–1083. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Heckmann, M.; Distler, J.; Koechel, A.; Schwers, S.; Kanefendt, F. Pharmacokinetics, pharmacodynamics and safety of BAY 2433334, a novel activated factor XI inhibitor, in healthy volunteers: A randomized phase 1 multiple-dose study. British journal of clinical pharmacology 2022, 88, 3447–3462. [Google Scholar] [CrossRef] [PubMed]
- Kanefendt, F.; Brase, C.; Unger, S.; Kubitza, D. Effects of Tablet Formulation, Food, or Gastric pH on the Bioavailability of Asundexian. Clinical pharmacology in drug development 2023, 12, 219–230. [Google Scholar] [CrossRef]
- Perera, V.; Wang, Z.; Lubin, S.; Christopher, L.J.; Chen, W.; Xu, S.; Seiffert, D.; DeSouza, M.; Murthy, B. Effects of Itraconazole and Diltiazem on the Pharmacokinetics and Pharmacodynamics of Milvexian, A Factor XIa Inhibitor. Cardiol Ther 2022, 11, 407–419. [Google Scholar] [CrossRef]
- Perera, V.; Wang, Z.; Lubin, S.; Christopher, L.J.; Chen, W.; Xu, S.; Seiffert, D.; DeSouza, M.; Murthy, B. Effects of rifampin on the pharmacokinetics and pharmacodynamics of milvexian, a potent, selective, oral small molecule factor XIa inhibitor. Sci Rep 2022, 12, 22239. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Guidance for industry. Drug interaction studies—design, data analysis, implications for dosing, and labeling instructions. 2018. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf (accessed on 6 June 2018).
Figure 1.
Pharmacological characteristics of anti FXIa under development.
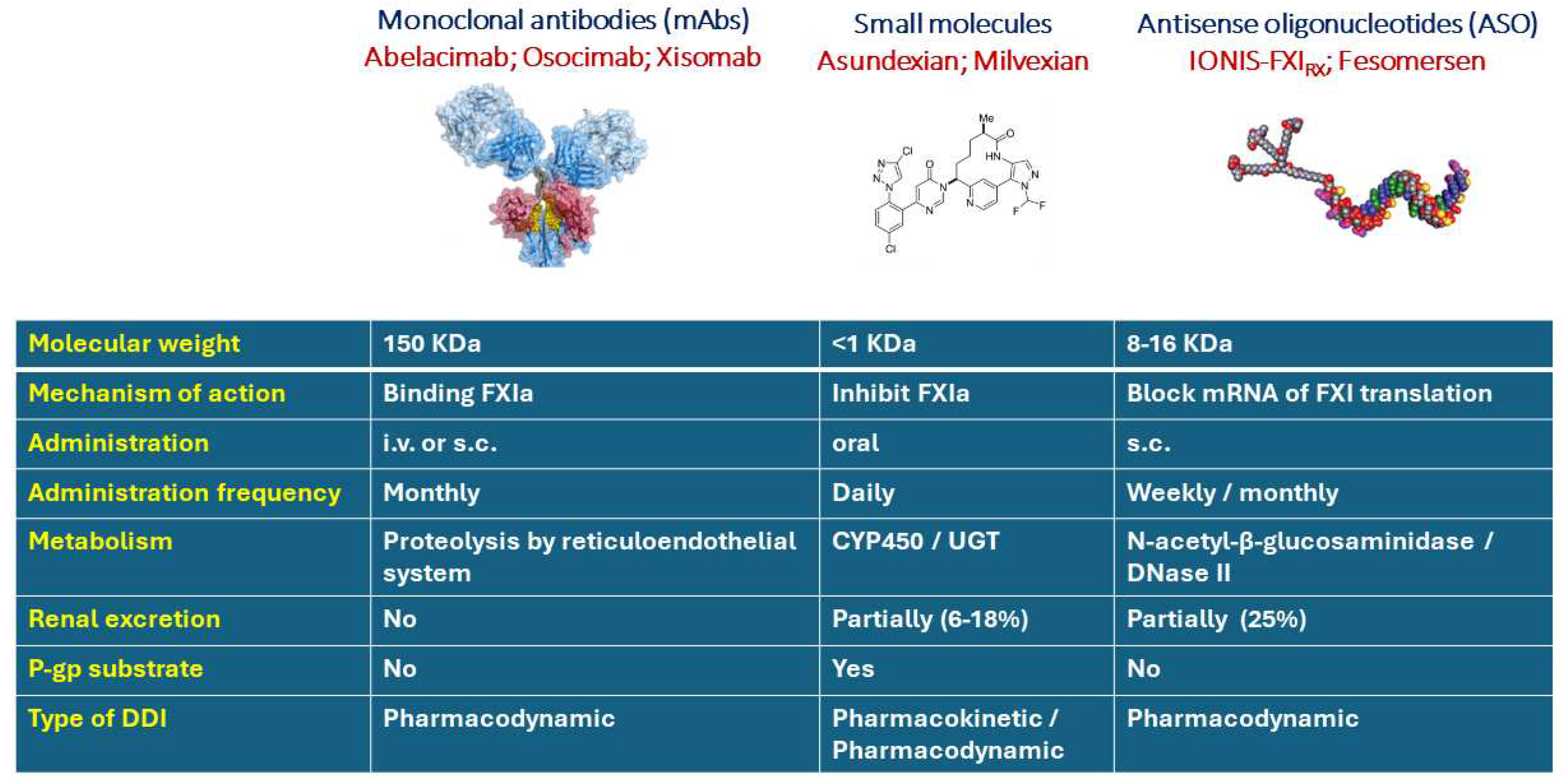

Table 1.
pharmacokinetic and pharmacodynamic characteristics of oral anti thrombin, FXa and FXIa.
| Drug | Dabigatran | Rivaroxaban | Apixaban | Edoxaban | Milvexian | Asundexian |
|---|---|---|---|---|---|---|
| Target | Thrombin | FXa | FXa | FXa | FXIa | FXIa |
| Ki (nmol/L) | 4.5 | 0.4 | 0.08 | 0.56 | 0.11 | 1.00 |
| Bioavailability | 6.5% | 80% | 50% | 60% | ND | 94% |
| Effect of food | Prolonged but not reduced | Increased (20 mg) |
None | None | Partially increased | Partially reduced |
| Vd | 60-70 L | 50 L | 21 L | >300L | 347L | NA |
| Proteins bound | 35% | >90% | 87% | 40-59% | 92% | 94% |
| Prodrug | Yes | No | No | No | No | No |
| Tmax (h) | 1-3 | 2-4 | 3-4 | 2 | 3-4 | 2.5-3 |
| Half lifetime (h) | 12-17 | 5-9 (sani) | 8-15 | 8-11 | 9-10 | 14-17 |
| Metabolism (CYP) | Conjugation | 3A4 (18%), 2J2, Independent from CYP |
CYP3A4 (25%), CYP1A2, CYP2J2, CYP2C8, CYP2C9, CYP2C19 | 3A4 (<4%) | 3A4 | 3A4 Modest 2C8, 2C9, 1A1 and 2D6 inhibitor Modest CYP3A4 inducer |
| Substrate P-gp | Yes (only prodrug) | Yes | Yes | Yes | Yes | Yes |
| Substrate of other transporters | NA | BCRP/ABCG2 | BCRP/ABCG2 | NA | No OATP | NA |
| Renal excretion | 80% | 65% | 27% | 35% | 7-18% | 6% |
| Posology | BID | OD | BID | OD | BID/OD | BID/OD |
BID: bis in die; OP: once daily; BCRP: breast cancer resistance protein; ABCG: ATP Binding Cassette Subfamily G Member; OATP: Organic Anion Transporting Polypeptides; NA: not available.
Table 2.
Inducers and inhibitors of CYP3A and P-gp. Modified from Corsini et al [42].
Table 2.
Inducers and inhibitors of CYP3A and P-gp. Modified from Corsini et al [42].
| P-gp Inhibitor | Non-P-gp inhibitor | P-gp Inducer | |
|---|---|---|---|
| Strong CYP3A inhibitor | itraconazole, ketoconazole, clarithromycin, lopinavir, indinavir, ritonavir, telaprevir | voriconazole, fluconazole | |
| Moderate CYP3A inhibitor | erythromycin, verapamil, diltiazem, dronedarone | not identified | doxorubicin |
| Weak CYP3A inhibitor | lapatinib, quinidine, cyclosporine, felodipine, azithromycin, ranolazine, ticagrelor, chloroquine, hydroxychloroquine | cimetidine | vinblastine |
| CYP3A Inducers | carbamazepine, phenytoin, phenobarbital, rifampin, dexamethasone, tocilizumab, St. John’s Wort |
Table 3.
AUC of midazolam and α-hydroxymidazolam following treatment with asundexian.
| Treatment | Analyte | AUC ratio day 1/day 1 (90% CI) |
AUC ratio day 10/day 1 (90% CI) |
|---|---|---|---|
| Asundexian 25 mg OD + midazolam | Midazolam | 1.04 (0.97-1.12) | 1.06 (0.99-1.14) |
| α-Hydroxymidazolam | 1.07 (0.97-1.17) | 1.06 (0.96-1.16) | |
| Asundexian 75 mg OD + midazolam | Midazolam | 1.04 (0.98-1.12) | 1.17 (1.10-1.26) * |
| α-Hydroxymidazolam | 0.99 (0.92-1.06) | 0.99 (0.92-1.06) |
Modified from Kubitza et al, 2022 [43]. *Ratio above 1.25.
Table 4.
Pharmacokinetic parameters of asundexian in the presence or absence of itraconazole, verapamil, fluconazole, or carbamazepine.
Table 4.
Pharmacokinetic parameters of asundexian in the presence or absence of itraconazole, verapamil, fluconazole, or carbamazepine.
| Parameter | asundexian | asundexian + Itraconazole | Ratio asundexian +Itraconazole / asundexian | ||
| AUC μg h L−1 | 6920 | 14000 | 2.02 | ||
| Cmax μg L−1 | 377 | 387 | 1.03 | ||
| tmax | 3.48 | 2.49 | 0.71 | ||
| t1/2 | 16.2 | 28.9 | 1.78 | ||
| CL/F | 3.61 | 1.78 | 0.49 | ||
| CLR | 0.307 | 0.180 | 0.59 | ||
| Parameter | asundexian | asundexian + verapamil | Ratio asundexian+ verapamil / asundexian | asundexian + fluconazole | Ratio asundexian+ fluconazole / asundexian |
| AUC μg h L−1 | 6360 | 11200 | 1.76 | 7430 | 1.168 |
| Cmax μg L−1 | 347 | 396 | 1.14 | 359 | 1.035 |
| tmax | 3.47 | 3.00 | 0.87 | 3.45 | 0.994 |
| t1/2 | 21.8 | 22.6 | 1.04 | 15.5 | 0.711 |
| CL/F | 3.93 | 2.24 | 0.57 | 3.37 | 0.858 |
| CLR | 0.297 | 0.215 | 0.72 | 0.313 | 1.054 |
| Parameter | asundexian | asundexian + carbamazepine | Ratio asundexian+ carbamazepine / asundexian | ||
| AUC μg h L−1 | 11500 | 6380 | 0.55 | ||
| Cmax μg L−1 | 623 | 509 | 0.82 | ||
| tmax | 2.00 | 3.00 | 1.50 | ||
| t1/2 | 14.4 | 11.0 | 0.76 | ||
| CL/F | 4.36 | 7.84 | 1.80 | ||
| CLR | 0.265 | 0.377 | 1.42 | ||
Asundexian was administered at 25 mg oral single dose alone or in combination with itraconazole and verapamil, while for the study with carbamazepine was used 50 mg oral dose. Modified from Kanefendt F. et al, 2023 [38].
Table 5.
Pharmacokinetic parameters of milvexian in the presence or absence of rifampicin, itraconazole or diltiazem.
Table 5.
Pharmacokinetic parameters of milvexian in the presence or absence of rifampicin, itraconazole or diltiazem.
| Parameter | milvexian |
milvexian following repeated doses of rifampicin |
Ratio milvexian+ rifampicin / milvexian |
| Cmax, ng/mL | 599 | 132 | 0.22 |
| AUC, ng·h/mL | 6153 | 923 | 0.15 |
| Tmax, h | 3.5 | 4.0 | 1.14 |
| T1/2, h | 13.21 | 8.85 | 0.67 |
| Parameter | milvexian | milvexian / itraconazole | Ratio milvexian+ itraconazole / milvexian |
| Cmax, ng/mL | 229 | 293 | 1.28 |
| AUC, ng·h/mL | 2144 | 5342 | 2.49 |
| Tmax, h | 3.0 | 4.0 | 1.33 |
| T1/2, h | 11.6 | 17.1 | 1.47 |
| Parameter | milvexian | milvexian / diltiazem | Ratio milvexian+ diltiazem / milvexian |
| Cmax, ng/mL | 248 | 272 | 1.10 |
| AUC, ng·h/mL | 2220 | 3059 | 1.38 |
| Tmax, h | 3.0 | 4.0 | 1.33 |
| T1/2, h | 12.3 | 13.6 | 1.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



