
Submitted:
24 January 2024
Posted:
24 January 2024
You are already at the latest version
Abstract
Vitamin D has important anti-inflammatory, anti-microbial properties and plays a central role in host immune response. Due to the crucial role of the kidneys in the metabolism of vitamin D, patients with chronic kidney disease (CKD), are prone to vitamin D deficiency. The resultant reduction in the production of calcitriol, the activated form of vitamin D in patients with CKD is responsible for exacerbating the existing renal impairment and periodontal inflammation. Recent evidence suggests a bidirectional, causal relationship between periodontitis and renal functional status. Both conditions have shared pathophysiologic mechanisms including oxidative stress, increases in the systemic inflammatory burden and impaired host response. This review explores the association between vitamin D, CKD and periodontitis. The review summarises the current evidence base for the classical and non-classical vitamin D metabolic pathways, the biological mechanisms linking vitamin D deficiency, CKD and periodontitis, as well as the bidirectional relationship between the two chronic inflammatory conditions. Finally, the paper explores the impact of vitamin D deficiency on CKD, periodontitis, and related co-morbidities.
Keywords:
Vitamin D
; Chronic kidney disease
; periodontitis
1. Introduction
Vitamin D deficiency is associated with various noncommunicable diseases, among which are chronic kidney disease (CKD) and periodontal disease. As well as its fundamental roles in calcium and phosphate homeostasis, vitamin D possesses important antibacterial, anti-inflammatory and host modulatory effects [1], which exert “renoprotective” and “perio-protective” effects.
CKD is a global health concern affecting 5-10% of the world population [2]. According to Kidney Disease Improving Global Outcomes, CKD is diagnosed on the basis of an estimated glomerular filtration rate (eGFR) and values less than 60ml/min/1.73m2 have been identified as the threshold for CKD [3,4], together with abnormalities of renal structure or function, present for more than 3 months with implications for health [4] There are 5 stages in CKD, the higher the staging the lower the eGFR [4].
Periodontitis is a chronic multifactorial inflammatory disease with an overall prevalence of 11.2% and is the 6th most prevalent disease worldwide [5]. It is associated with dysbiosis of the oral flora, characterised by the progressive destruction of the periodontium, with loss of clinical attachment, loss of alveolar bone, presence of periodontal pockets and gingival bleeding [6]. Periodontitis is a serious public health issue, as it can cause not only local symptoms, but it can also have a negative impact on the individual’s general health, contributing to the development, and to the worsening, of chronic non-communicable degenerative diseases, such as chronic kidney disease (CKD) [7].
Periodontal disease and CKD have shared pathophysiological mechanisms, namely increased inflammatory state, impaired immune response, and oxidative stress [8]. Therefore, the occurrence of both conditions is likely to result in an amplification of adverse outcomes [9]. A recent large cohort study suggested a bidirectional, causal relationship between periodontal inflammation and renal function [10], such that a 10% increase in periodontal inflamed surface area (PISA) led to 3.0% decrease in eGFR and a 10% decrease in eGFR led to a 25.0% increase in PISA [10].
This review will discuss 1) the vitamin D metabolic pathway and how this is altered in CKD patients 2) the bidirectional relationship between periodontal inflammation and renal impairment; 3) the biological mechanisms linking vitamin D deficiency, CKD, and periodontitis, and 4) the impact of Vitamin D deficiency on systemic inflammation and co-morbidities associated with CKD and periodontitis.
2. Vitamin D Metabolic Pathways
Vitamin D is a fat-soluble hormone that can be obtained from two main sources. Firstly, dietary sources such as fatty fish and mushrooms. It is found in the form of Ergocalciferol (D2) from plant sources or Cholecalciferol (D3) from animal sources [11]. Secondly, through the action of sunlight ultraviolet rays on skin in the form of 7-Dehydrocholesterol which is converted into previtamin Cholecalciferol (D3) [12]. Due to the relatively small proportion of vitamin D from the diet, dermal synthesis accounts for 90% of vitamin D provision [13].
2.1. Classical Pathway
The classical pathway for activation of vitamin D involves two stages of hydroxylation. Firstly, the Vitamin D2 and D3 precursors are transported to the liver by Vitamin D binding protein (DBP) 14. Precursors D2 and D3 are then converted into inactive 25-hydroxvitamin D (25(OH)D) by hydroxylation at the C25 position by 25-hydroxylase, coded by cytochrome P2R1 (CYP2R1)15. 25(OH)D acts as the main circulating and storage form of vitamin D in the body. 25(OH)D is then circulated in blood as 25(OH)-DBP complex and undergoes glomerular filtration and uptake into the kidney proximal tubule cell by the receptor megalin. 25(OH)D-DBP then undergoes 1-α-hydroxylation by the Cytochrome P450 Family 27 Subfamily B Member 1 gene (CYP27B1) to its most activated state, 1,25-dihydroxyvitamin D (1,25(OH)2D) also known as calcitriol 15. This is illustrated in (Figure 1).
Calcitriol has wide ranging physiological and pharmacological effects [1]. Calcitriol is responsible for increasing intestinal calcium and phosphate absorption when serum calcium and phosphate levels are low. It also increases phosphorus resorption from bone and is involved in the production of antimicrobial peptides, epithelial defence mechanisms, host modulatory effects, maintenance of the renin-angiotensin system, inhibition of host tumour cells and suppression of parathyroid hormone (PTH) release1.Calcitriol exerts these affects by binding to intracellular vitamin D receptors (VDRs), which are steroid hormone nuclear receptors and function as transcription factors [16].
In health, the activation of CYP27B gene is regulated by PTH, phosphorus, calcium, and fibroblast growth factor-23 (FGF-23) levels, and subsequently calcitriol levels [17]. Increased PTH levels combined with decreased phosphorus and calcium levels activate CYP27B gene and lead to increased calcitriol levels [13]. Meanwhile, increased FGF-23 levels inhibit CYP27B gene and decrease calcitriol levels [18].
2.2. Non-Classical Pathway
Aside from the classical metabolism of vitamin D and its role in calcium and phosphate homeostasis, a non-classical pathway of calcitriol synthesis appears to be present in various tissues, both including and peripheral to the kidneys. Additionally, 1--hydroxylase (which is primarily expressed in the kidneys) may also be expressed in extrarenal cells and tissues [19]. Thus, the extra-renal production of calcitriol primarily functions as an autocrine or paracrine factor at extra-renal sites and thus play a role in non-classical actions of vitamin D [20,21].
Central to the non-classical function is the regulation of the renin-angiotensin- aldosterone system (RAAS). Calcitriol is regarded as a negative endocrine regulator of the renin gene, thereby inhibiting RAAS and preserving renal function. RAAS stimulates the production of renin, which cleaves angiotensin into angiotensin I, which is then processed into angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II binds to the type 1 angiotensin II receptor (AT1R) to produce various delirious effects for renal and cardiovascular tissues, including hypertension [22,23]. This is described in more detail in (Figure 2).
There is also evidence that VDR activation by calcitriol may also downregulate other RAS components aside from renin, including the Ang II type one receptor, renin receptor and transforming growth factor-beta [37].This can aid in the reduction of renal blood pressure and fibrogenesis. VDR activation by calcitriol could also have anti-inflammatory effects, via suppression of nuclear factor-kB (NF-kB) activation. It also appears that VDR can form complexes with various transcription factors and engage in crosstalk with a wide range of cellular signals [38], thus illustrating the depth of the relationship between VDR activation and the RAAS components.
The suppression of the NF-kB pathway by calcitriol has extra-renal consequences too. Its suppression of the pathway is twofold: calcitriol suppresses NF-kB nuclear migration and phosphorylation, and it downregulates IkB phosphorylation (a protein involved in NF-kB signalling) by suppressing ROS activity [39]. The NF-kB pathway promotes pro-inflammatory cytokine expression, and thus a reduction in NF-kB activity results in a reduction in inflammatory markers. Thus, the suppression of NF-kB pathway in turn can prevent insulin resistance [40], have neuroprotective effects against ischaemic strokes [39], and protect against other inflammatory disorders. Finally, it is worthy to note that both the inhibitions of the RAAS and NF-kB pathway are responsible for the “reno-protective” effects of vitamin D [41].
3. Serum 25(OH)D Thresholds
Serum 25-(OH)vitamin D level is the ideal indicator of deficiency. However, there is a lack of agreement in the defining serum concentrations associated with deficiency and adequacy [1]. Most guidelines currently define vitamin D sufficiency as any value above 50 nmol/L (20ng/ml) [42,43]. However, various studies have indicated that optimal serum levels should be anywhere between 100 to 200 nmol/L (40-80ng/ml) [44,45], and that a concentration below 50nmol/L (20ng/ml) is considered vitamin D deficient [46].
4. Biological Mechanisms Linking Vitamin D Deficiency, CKD and Periodontitis
Vitamin D plays a central role in host immune response and possesses important anti-inflammatory, anti-microbial and host modulatory properties [1]. Due to the wide- ranging physiological and pharmacological role of calcitriol [1], vitamin D deficiency is responsible for the exacerbation of renal impairment and progression of periodontitis. CKD is characterised by altered vitamin metabolism, as well as elevations in PTH and FGF23. Additionally, oxidative stress which promotes inflammation and impaired host response provide the pathophysiological mechanisms for disease progression in both CKD and periodontitis. Finally, vitamin D binding protein (DBP) polymorphisms which are associated with bioavailable 25(OH)D, is linked to severity and progression of CKD and periodontitis.
4.1. Altered Vitamin D Pathways in CKD
The kidney plays a central role in vitamin D metabolism and regulation of its circulating levels. Vitamin D deficiency has been identified more than 80% of patients with CKD [47]. The trend is that the deficiency worsens with progressive renal impairment, ultimately the onset of hyperparathyroidism. There are several mechanisms responsible for the reduced production of calcitriol: 1) in CKD, there is an overall reduction in renal mass, limiting the 1-α-hydroxylase available for production of calcitriol [38]; 2) reduced eGFR also limits the conversion of 25(OH)D to 1-α-hydroxylase, further reducing the production of the calcitriol [20]; 3) reduced renal megalin receptors in CKD will lead to reduced uptake of 25(OH)D and therefore reduced production of calcitriol; 4) the elevation of FGF23 in CKD, inhibits the CYP27B gene [48] [49], which also reduces 1-α-hydroxylase activity [50,51]; 5) hyperphosphatemia due to impaired renal phosphate excretion in CKD also contributes to reduced 1-α-hydroxylase activity [52]. Thus, the downregulation of 1-α-hydroxylase activity reduces the overall production of calcitriol from the kidneys.
While the inverse relationship between serum 25(OH)D, and renal function has traditionally been explained by alterations in the classical vitamin D metabolism (section 2.1), it is clear that non-classical functions of calcitriol also play a role (section 2.2). A deficiency in vitamin D and thus a reduction in calcitriol promotes RAS activity, and the sequential activation of angiotensin II could raise blood pressure and damage the renal microvasculature. Aside from its direct action on RAS, vitamin D promotes insulin resistance via a wide range of molecular mechanisms involved in glucose homeostasis and immune modulation [53]. Therefore, a deficiency in vitamin D might lead to diabetes, and diabetic nephropathy has been shown to activate RAS [54]. Indeed, intrarenal angiotensin II levels may be up to a hundred times higher in diabetic patients [54],and diabetic nephropathy remains a leading cause of CKD [55]. Locally synthesised angiotensin II is also detrimental to the cardiovascular system, and cardiovascular disease is responsible for much of the mortality of CKD [56]. In these ways, a deficiency in vitamin D can lead to profound consequences for renal, endocrine and cardiovascular function, all of which exacerbate the development and severity of CKD.
4.2. Elevations of PTH and FGF23 Levels in CKD
Vitamin D levels are also regulated by PTH and FGF23. Increases in serum levels of FGF23 and PTH are responsible for vitamin D deficiency (Figure 1). PTH increases renal calcium reabsorption, excretion of phosphorus, and stimulates calcitriol synthesis [57,58].
FGF23 is a phosphaturic hormone secreted by osteoblasts and osteocytes which is strongly associated with inflammation. The elevation of FGF23, which is to offset phosphorus retention in CKD, inhibits the renal expression of 1-a-hydroxylase [59],and reducing the production of calcitriol. Thus, serum FGF23 increase with decline in eGFR and increased phosphate level. This results in downstream reduction of 1,25(OH)D concentrations and onset of secondary hyperparathyroidism, SHPT [59,60] ,due to elevated PTH but low or normal calcium levels. SHPT ultimately progresses to tertiary hyperparathyroidism (THPT), where both PTH and calcium levels are elevated [61]. As a result, vitamin D deficiency (<20 ng/mL) and insufficiency (20–29 ng/mL) are common in individuals with CKD [62].
4.3. Oxidative Stress
Oxidative stress promotes inflammation by stimulating the release of pro-inflammatory medicators (NF-kB related cascade) [63]. A recent animal study showed that renal tissue damage is linked to oxidative stress following periodontitis [64]. This concept was reinforced in a recent longitudinal study that pointed to oxidative stress, rather than inflammatory load, as the biological basis for the bidirectional relationship between oxidative stress and periodontitis [65]. Thus, oxidative stress is responsible for progressive renal impairment [66], tissue damage in periodontitis [66]; as well as systemic implications for the development of atherosclerosis [67], and cardiovascular disease [68]. Interestingly, oxidative stress is considered a non-traditional risk factor for all-cause mortality [69,70].
4.4. Impaired Host Response
The mechanisms linking vitamin D status, CKD and periodontitis are related to the biological functions of calcitriol, which possesses various immunomodulatory properties that affect both the innate and adaptive immune system. Calcitriol downregulates the expression of MHC class II (and co-stimulatory molecules) on antigen-presenting dendritic cells (Figure 3). It also suppresses pro-cytokine production – whilst stimulating anti-inflammatory cytokine production – and both these mechanisms suppress subsequent T-cell activation and differentiation [71,72].
Vitamin D exercises various effects on macrophages and their monocyte precursors, which are involved in phagocytosis and cytokine production (Figure 3). Upon pathogen recognition and antigen presentation, monocytes and macrophages upregulate VDR expression and metabolise vitamin D into calcitriol. Calcitriol activates intracellular VDRs to promote the production of antibiotic peptides including -defensins and cathelicidins [76,81,82]. Calcitriol also epigenetically regulates the immunological memory and differentiation of monocytes and macrophages (Figure 3) [83].
Calcitriol also suppresses the activity of T and B-lymphocytes. It reduces differentiation of T-helper cells into the Th1-type and pro-inflammatory cytokine generation, in favour of Th2-type differentiation and the production of anti-inflammatory cytokines [78]. It also suppresses Th17-type differentiation and its inflammatory cytokines [79]. Calcitriol also suppresses naïve B-cell differentiation and maturation into memory and plasma cells [80].
Overall, a lack of calcitriol promotes chronic inflammation through the inhibition of the aforementioned mechanistic pathways. Therefore, a vitamin D deficiency enhances the risk of both CKD and periodontitis through impaired host response.
4.5. DBP Genetic Polymorphisms and Bioavailable 25(OH)D
DBP serves to transport vitamin D and its metabolites such as 25(OH)D in the blood to specific target tissues where vitamin D will exert its biological effects. Only the bioavailable 25(OH)D, rather than the total serum 25(OH)D that is associated with serum calcium, and plasma PTH concentrations in patients on haemodialysis [84]. Bioavailable forms of vitamin D include the free fraction (<1% of total 25(OH)D) and the fractions bound to albumin or lipoprotein 10-15% of total 25(OH)D.
DBP genetic polymorphism rs7041 and rs4588 result in different phenotypes of the DBP that have varying binding affinities to 25(OH)D [82]. These polymorphisms have been associated with differences in bioavailable levels of vitamin D and have been implicated in elevated risk of CKD [85] as well as periodontitis [86].
Additionally, DBP polymorphisms potentially explain racial predilection for a particular DBP phenotype [87,88], as well as differential responses to vitamin D supplementation in patients with low serum total 25(OH)D concentrations [89]. Thus, DBP polymorphisms can impact the relationship between with serum total 25(OH)D concentrations and clinical outcomes and the effect of vitamin D supplementation [90].
Therefore, it may be inappropriate to assess the vitamin D status of individual patients by serum total 25(OH)D concentrations alone. Rather, the DBP phenotype (affinity) should be taken into account, due to the likely implications in the clinical prognosis of CKD and periodontitis, as well as responses to vitamin D supplementation.
5. Impact of CKD on Periodontal Inflammation
Longitudinal studies have demonstrated an association between CKD and progression of periodontal disease [91]. CKD is associated with a two-fold increase in the prevalence of periodontitis [9]. A recent meta-analysis also suggested that individuals with CKD presents with higher mean PPD, and CAL, compared to healthy subjects without CKD. The difference in PPD and CAL between CKD and healthy subjects was 0.25mm and 0.041mm respectively [92].
5.1. CKD- Mineral and Bone Disorder and Impact on Periodontitis
Homeostatic imbalances associated with CKD leads to changes in the regulation of calcium, phosphorous, PTH, and fibroblast growth factor 23 (FGF23) levels, resulting in increased bone demineralisation, often referred to as CKD-mineral and bone disorder (CKD-MBD) [93]. In failing kidneys, there is a significant reduction in the hydroxylation of inactive vitamin D (25-hydroxvitamin D) [25(OH)D] into active calcitriol (1,25(OH)2D) by 1-α-hydroxylase enzyme [1] . Hence, reduced calcitriol levels combined with reduced renal phosphate excretion, leads to systemic hypocalcaemia and hyperphosphataemia, and secondary hyperparathyroidism. Eventually, secondary hyperparathyroidism progresses to tertiary hyperparathyroidism and the development of hyperparathyroid bone disease, that is characterised by high bone turnover, thinned cortical bone, and increased abnormal trabecular bone [93].
CKD-MBD can exacerbate periodontitis by accelerating alveolar bone loss [94]. This concept was reinforced in an animal study where mice with chronic uraemia and hyperparathyroidism showed significantly reduced cortical alveolar bone compared to healthy controls, and that a further decrease in bone levels was seen after a high phosphate diet was given to increase PTH hormone serum levels [95].
6. Impact of Periodontal Inflammation on Renal Function
Large epidemiologic surveys such NHANES III [96] have demonstrated that periodontitis was predictive for the occurrence of CKD [64,73,97]. In particular, periodontitis has been identified as a non-traditional risk factor for eGFR decline [98], and a contributor of oxidative stress [65]. Furthermore, P. gingivalis lipopolysaccharide (LPS) exposure resulted in the elevation of FGF23 in the kidneys [99]. FGF23 has been identified as a risk factor for cardiovascular mortality in CKD patients [67,100,101].
Recently, it was shown that presence of periopathogenic bacteria, resulted in an elevation of tumour necrosis factor- alpha (TNFα), that was predictive of the severity of renal impairment reflected by eGFR, and periodontal clinical parameters such as plaque index (PI), gingival index (GI), probing pocket depths (PPD) and clinical attachment loss (CAL) [102]. A meta-analysis suggested that periodontitis significantly increased the risk of all-cause mortality in CKD [103]. However, this result was refuted by a large database study from Taiwan – where it was concluded that periodontitis was not a predictor for long-term mortality or morbidity in patients with advanced CKD [104]. Therefore, future well-designed prospective studies are needed to verify these findings.
Further evidence supporting the impact of periodontal inflammation on renal function comes from improved renal outcomes following non-surgical periodontal treatment. Recent systematic reviews and meta-analyses which have only included a limited number of studies suggested that periodontal treatment improved renal function in CKD patients [105,106]. This is also demonstrated by two case series studies, which showed improved eGFR and creatinine, as well as periodontal clinical outcomes at 3-6 months after non-surgical periodontal therapy [107]. The biological plausibility underlying the favourable outcomes relates to the shared pathophysiologic mechanisms between CKD and periodontitis including oxidative stress, and impaired host response (section 4). Therefore, these findings reinforce the impact of periodontal inflammation on progression of renal failure.
7. Impact of Vitamin D Deficiency on CKD
Vitamin D deficiency is associated with a higher risk of mortality, secondary and tertiary hyperparathyroidism [108]. Due to impaired renal function, the eGFR is reduced, and therefore a decline in the conversion of 25(OH)D to calcitriol, the active form of vitamin D. The latter reduces intestinal calcium absorption and, together with phosphate retention, contributes to the onset of secondary and tertiary hyperparathyroidism.
CKD Patients who are vitamin D deficient have high mortality rates [109] and increased cardiovascular risk [110]. In addition, the elevation of FGF23, which is linked with vitamin D deficiency, is associated with progression of CKD towards end stage renal disease (ESRD), occurrence of cardiovascular (CVS) events and increased mortality rates in patients with CKD [100,101].
Serum total 25(OH)D concentrations was predictive of renal outcomes such as doubling of serum creatinine in ESRD, and associated with disease progression and morality [55,111],. A meta-analysis of prospective studies demonstrated an increase relationship between all- cause mortality in patients with CKD and serum total 25(OH)D concentrations [112]. Conversely, A recent systematic review and meta-analysis concluded that Higher levels of serum 25(OH)D were associated with lower risk of all-cause mortality [113].
Therefore, to ensure that patients with CKD avoid vitamin D deficiency and prevent complications such as secondary and tertiary hyperparathyroidism and other co-morbidities [62,114,115], the Kidney Disease Outcomes Quality Initiative (KDOQI) and Kidney Disease Improving Global Outcomes (KDIGO) group have suggested the use of vitamin D supplementation. Therefore, to ensure that patients with CKD avoid vitamin D deficiency and prevent complications such as secondary and tertiary hyperparathyroidism and other co-morbidities [59,114,115], the Kidney Disease Outcomes Quality Initiative (KDOQI) and Kidney Disease Improving Global Outcomes (KDIGO) group have suggested the use of vitamin D supplementation.
7.1. Vitamin D Supplementation in CKD Patients
Vitamin D supplementation is associated with reduced risk of all-cause mortality [116] and cardiovascular mortality in patients in CKD, including those with ESRD [78,115]. In particular, therapies with calcitriol and analogues are associated with reduced mortality in CKD patients, particularly those suffering from SHPT [117].
CKD patients are deficient in vitamin D, even in the early stages of disease [118]. Vitamin D plays a vital role not only in mineral homeostasis, but also in systemic health. As such, it is advised that vitamin D supplementation in CKD patients begins as soon as possible, to ensure a pool of vitamin D can be turned into calcitriol.
During the early stages of CKD, where there is still evidence of residual renal function, supplementation can be achieved CKD patients with oral forms of inactive vitamin D3 or D2. Vitamin D3 (cholecalciferol) is the natural form synthesised in the dermis, whilst vitamin D2 (ergocalciferol) is a synthetic product made using fungi [119]. Another reason is that vitamin D2 is associated with higher catabolic processes and therefore the overall improvement in serum vitamin D levels is not as sustainable as that seen with D3. Therefore, vitamin D3 is superior to vitamin D2 in raising total 25(OH)D and thus ideally used for supplementation [120].
It is important to appreciate that renal production of calcitriol becomes suppressed during Stage 3 of CKD, with the significant loss of renal 1--hydroxylase. Therefore, calcitriol replacement should begin during Stage 3 to avoid the development of secondary hyperparathyroidism and other associated co-morbidities [44]. This is particularly necessary since vitamin D is rarely present in foods, and sun exposure is commonly insufficient in the general population [45]. Therefore, the generally accepted strategies involve starting vitamin D supplementation (ideally with D3) immediately after diagnosis and beginning calcitriol therapy only once the CKD has reached Stage 3.
8. Impact of Vitamin D Deficiency on Periodontitis
The relationship between vitamin D and periodontitis was recently reviewed [1]. In general, an inverse association exists between serum 25(OH)D and periodontal disease inflammation. Most of the studies supporting this association were cross-sectional or case-control studies. One of these was the NHANES III study which showed that low serum vitamin D was associated with periodontal inflammation [121]. Furthermore, it was demonstrated in an RCT that administration of vitamin D (700IU/day) and calcium (500mg/day) significantly reduced tooth loss in older patients during 3 years of observation [122]. A more recent RCT showed that vitamin D supplementation resulted in significant but modest improvement in periodontal outcomes [123].
Interestingly, it was shown in a case-control study that patients with CKD and periodontitis showed lower serum levels of vitamin D compared to control patients without periodontitis [124]. In other words, vitamin D deficiency was more severe in patients with CKD and periodontitis than patients with CKD only [124]. This is plausible, given the shared pathophysiologic mechanisms between CKD and periodontitis, including the elevation of pro-inflammatory cytokines, impaired host response cytokines, and an increase in oxidative stress.
9. Impact of Vitamin D Deficiency on Co-Morbidities Associated to CKD and Periodontitis
Vitamin D has wide-ranging roles in promoting inflammation and reducing the risk of various co-morbidities. Therefore, aside from exacerbating CKD and periodontitis, a deficiency in vitamin D can also negatively and independently affect systemic health. The impact of vitamin D deficiency on systemic health will be specifically discussed here for cardiovascular disease, diabetes mellitus, autoimmune disease. However, as both CKD and periodontitis are directly and independently associated with a myriad of co-morbidities [86,125,126,127], it can be challenging to establish whether it is the contribution from vitamin D deficiency or the cause and effect of CKD and periodontitis in driving the systemic inflammatory burden.
9.1. Cardiovascular Disease
A severe deficiency in vitamin D is positively correlated with a higher risk of cardiovascular disease (CVD) [128]. This is partly due to the reduction of cardiovascular risk factors by vitamin D, but also due to the direct effects of vitamin D on vascular tissues and cardiomyocytes, which express VDRs that respond to calcitriol [129]. Calcitriol has various positive effects on the vascular wall, namely reduced thrombogenicity and vasoconstriction, the inhibition of atherogenesis and the promotion of endothelial repair [130]. These effects protect against atherosclerosis and hypertensive damage.
In cardiomyocytes, calcitriol regulates intracellular calcium metabolism [131]. Calcitriol binds to VDRs on the cell-surface and in the cytoplasm. Membrane-bound receptors activate adenylate cyclase, which increases cytoplasmic calcium via downstream pathways. Cytosolic receptors complex with retinoid-X receptors, migrate to the nucleus and upregulate synthesis of the calcium-binding protein cholecalcin.
Interestingly, cardiovascular synthesis of calcitriol from its vitamin D precursor can be regulated by PTH, as is known to be the case in renal tissues [132]. Calcitriol is a known inhibitor of PTH action [133]. In this way, a deficiency in serum vitamin D (and therefore calcitriol) can lead to secondary and tertiary hyperparathyroidism, which has its own consequences for cardiovascular health. These include an increase in oxidative stress, RAAS, thrombogenicity and foam cell formation, which can all lead to cardiovascular disease [129]. Given the fact that nutritional rickets, hypocalcaemia and SHPT have all been associated with heart failure [134], this PTH-mediated pathway could explain the negative cardiovascular effects of vitamin D deficiency, aside from VDR activation.
Finally, CVD and associated mortality are significant concerns for patients with CKD [135]. Studies have also shown that periodontitis is a major contributor to the development of atherosclerosis by potentiating endothelial dysfunction, inflammation, and the advancement of atherosclerotic plaque [126]. As such periodontitis is considered to be independently linked to cardiovascular morbidity in patients with CKD [127].
9.2. Diabetes Mellitus
Diabetes mellitus is largely characterised by insulin resistance, where systemic inflammation plays a key role [136]. Diabetic nephropathy is responsible for almost 50% of ESRD cases [137]. Serum 25(OH)D levels of patients with type II diabetic nephropathy has been linked to renal disease progression [138]. In patients with diabetes, periodontitis is also independently associated with the progression of renal disease [139], while individuals suffering with diabetic nephropathy have a higher risk of periodontitis resulting in missing teeth compared to those patients without CKD [140].
Calcitriol protects tissues against inflammatory damage, by suppressing the systemic production of pro-inflammatory cytokines whilst encouraging the release of anti-inflammatory cytokines [141], as well as playing a protective role against insulin resistance by modulating pancreatic β cell activity [142]. Therefore, vitamin D deficiency could lead to insulin resistance and potentiate the development of diabetes mellitus.
Calcitriol also maintains insulin secretion by -cells, which, when reduced can lead to the development of DM. It modulates calcium-mediated exocytosis, which is necessary for the secretion of insulin vesicles [143]. During the progression of insulin resistance, the -cells secrete more insulin in response and this hyperactivity results in β cell dysfunction and eventually apoptosis [144]. Calcitriol also controls intracellular reactive oxygen species (ROS) levels, by promoting the expression of cellular antioxidants, maintaining mitochondrial function [145], maintaining redox homeostasis [146] and decreasing nitrogen oxide production [147]. Vitamin D also regulates target cell response to insulin, by promoting insulin receptor expression [148]. Vitamin D deficiency can lead to secondary and tertiary hyperparathyroidism, which are also linked to glucose intolerance and insulin resistance [149]. In these ways, calcitriol protects the tissues against insulin resistance.
9.3. Autoimmune Disease
Vitamin D deficiency is correlated with the development of various autoimmune diseases [152]. Rheumatoid arthritis (RA) is an autoimmune disease of the joints, characterised by chronic synovial inflammation. Various studies found a negative correlation between serum vitamin D levels and disease severity [152,153], though this could be blamed – at least in part – on the associated lack of mobility and sunlight exposure in RA patients [154]. Even so, a decrease in vitamin D is associated with increased TNF- and interleukin-6 (IL-6) secretion by inflammatory cells, and suppression of endothelial function [155]. The pharmacological use of calcitriol in RA patients has been shown to inhibit pro-inflammatory cytokines and matrix metallopeptidase production in synoviocytes [156], reducing the recruitment of inflammatory cells to the site and consequent joint destruction. In these ways, vitamin D deficiency could contribute to the onset or progression of rheumatoid arthritis.
Systemic lupus erythematosus (SLE) is an autoimmune disease characterised by systemic inflammation and the presence of immune complexes. SLE patients tend to have lower serum vitamin D levels, and there is some evidence of a relationship between vitamin D levels and disease severity [156,157,158]. The main reason for this association is due to the role of calcitriol, which suppresses T lymphocytes and antinuclear antibody production by B lymphocytes and thus dampens the formation of immune complexes [159]. However, the depth of the relationship between SLE and vitamin D is still poorly understood.
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system and is more common in high-latitude regions where sunlight exposure and consequent vitamin D synthesis is limited [160]. It is known that reduced serum vitamin D levels are negatively correlated with disease severity in MS [161]. Calcitriol inhibits the differentiation and proliferation of type 1 T helper (Th1) cells by promoting the production of pro-inflammatory cytokines, such as IL-10 and transforming growth factor-beta (TGF-) over anti-inflammatory cytokines (such as IL-12 and TNF-) in dendritic cells [72,162,163]. The anti-inflammatory effects of calcitriol on dendritic cells in the CNS can suppress the onset or exacerbation of MS, thus increasing the risk of MS without vitamin D and calcitriol.
10. Conclusion
A wealth of evidence supports the association between Vitamin D deficiency and chronic inflammatory conditions such as chronic kidney disease and periodontitis. Both conditions share common pathophysiologic mechanisms including insufficient inflammation, impaired host response and oxidative stress. The kidneys play a crucial role in the metabolism of vitamin D. Thus, altered vitamin D metabolic pathways and elevations of PTH and FGF23 are key biochemical observations in CKD. Emerging evidence also support to the bidirectional relationship between renal impairment and periodontal inflammation. Vitamin D plays a significant role in host immune response and possesses important anti-inflammatory, anti-microbial and host modulatory properties. Vitamin D status is also associated with renal outcomes and clinical outcomes related to periodontitis. However, as many of studies were based on large observational studies, further prospective randomised controlled trials are needed to provide deeper insights into this relationship.
Acknowledgments
This work was supported in part by grant received from Royal College of Surgeons Edinburgh.
Conflicts of Interest
There are no conflicts of interest to declare.
References
- Lu EMC. The role of vitamin D in periodontal health and disease. J Periodontal Res. 2023;58(2):213-224. [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl (2011). 2017;7(1):1-59. [CrossRef]
- Schaeffner ES, Ebert N, Delanaye P, et al. Two novel equations to estimate kidney function in persons aged 70 years or older. Ann Intern Med. 2012;157(7):471-481. [CrossRef]
- Stevens PE, Levin A, Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825-830. [CrossRef]
- Tonetti MS, Jepsen S, Jin L, Otomo-Corgel J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J Clin Periodontol. 2017;44(5):456-462. [CrossRef]
- Papapanou PN, Sanz M, Buduneli N, et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. 2018;89 Suppl 1:S173-S182. [CrossRef]
- Parsegian K, Randall D, Curtis M, Ioannidou E. Association between periodontitis and chronic kidney disease. Periodontol 2000. 2022;89(1):114-124. [CrossRef]
- Tonetti MS, Van Dyke TE, working group 1 of the joint EFP/AAP workshop. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Periodontol. 2013;84(4 Suppl):S24-9. [CrossRef]
- Deschamps-Lenhardt S, Martin-Cabezas R, Hannedouche T, Huck O. Association between periodontitis and chronic kidney disease: Systematic review and meta-analysis. Oral Dis. 2019;25(2):385-402. [CrossRef]
- Sharma P, Fenton A, Dias IHK, et al. Oxidative stress links periodontal inflammation and renal function. J Clin Periodontol. 2021;48(3):357-367. [CrossRef]
- Benedik E. Sources of vitamin D for humans. International Journal for Vitamin and Nutrition Research. 2022;92(2):118-125. [CrossRef]
- Lehmann B, Genehr T, Knuschke P, Pietzsch J, Meurer M. UVB-Induced Conversion of 7-Dehydrocholesterol to 1α,25-Dihydroxyvitamin D3 in an In Vitro Human Skin Equivalent Model. Journal of Investigative Dermatology. 2001;117(5):1179-1185. [CrossRef]
- Chang SW, Lee HC. Vitamin D and health - The missing vitamin in humans. Pediatr Neonatol. 2019;60(3):237-244. [CrossRef]
- Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(3):319-329. [CrossRef]
- Christakos S, Ajibade D V, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: Metabolism I. SYNTHESIS OF 1,25(OH) 2 D 3 FROM VITAMIN D 3. [CrossRef]
- Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173-179. [CrossRef]
- Silver J, Naveh-Many T. FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat Rev Nephrol. 2013;9(11):641-649. [CrossRef]
- Nakashima A, Yokoyama K, Yokoo T, Urashima M. Akio Nakashima, Keitaro Yokoyama, Takashi Yokoo, Mitsuyoshi Urashima. World J Diabetes. 2016;7(5):89-100. [CrossRef]
- Hewison M. Vitamin D and the immune system: new perspectives on an old theme. [CrossRef]
- Dusso AS. Kidney disease and vitamin D levels: 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and VDR activation. Kidney Int Suppl (2011). 2011;1(4):136-141. [CrossRef]
- Adams JS, Hewison M. Update in Vitamin D. J Clin Endocrinol Metab. 2010;95(2):471-478. [CrossRef]
- Maranduca M, Clim A, Pinzariu A, et al. Role of arterial hypertension and angiotensin II in chronic kidney disease (Review). Exp Ther Med. 2023;25(4):153. [CrossRef]
- Paz Ocaranza M, Riquelme JA, García L, et al. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17(2):116-129. [CrossRef]
- Ferrario CM. Role of angiotensin II in cardiovascular disease - Therapeutic implications of more than a century of research. JRAAS - Journal of the Renin-Angiotensin-Aldosterone System. 2006;7(1). [CrossRef]
- Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98(3). [CrossRef]
- Huang BS, Chen A, Ahmad M, Wang HW, Leenen FHH. Mineralocorticoid and AT1 receptors in the paraventricular nucleus contribute to sympathetic hyperactivity and cardiac dysfunction in rats post myocardial infarct. Journal of Physiology. 2014;592(15). [CrossRef]
- Iyer SN, Lu D, Katovich MJ, Raizada MK. Chronic control of high blood pressure in the spontaneously hypertensive rat by delivery of angiotensin type 1 receptor antisense. Proc Natl Acad Sci U S A. 1996;93(18). [CrossRef]
- Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RAK, Thaiss F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors. Kidney Int. 2002;61(6). [CrossRef]
- Schieffer B, Wirger A, Meybrunn M, et al. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation. 1994;89(5). [CrossRef]
- Aguilera G. Role of angiotensin II receptor subtypes on the regulation of aldosterone secretion in the adrenal glomerulosa zone in the rat. Mol Cell Endocrinol. 1992;90(1). [CrossRef]
- Qadri F, Culman J, Veltmar A, Maas K, Rascher W, Unger T. Angiotensin II-induced vasopressin release is mediated through alpha-1 adrenoceptors and angiotensin II AT1 receptors in the supraoptic nucleus. Journal of Pharmacology and Experimental Therapeutics. 1993;267(2).
- Sadoshima JI, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts critical role of the AT1 receptor subtype. Circ Res. 1993;73(3). [CrossRef]
- Viswanathan M, Strömberg C, Seltzer A, Saavedra JM. Balloon angioplasty enhances the expression of angiotensin II AT1 receptors in neointima of rat aorta. Journal of Clinical Investigation. 1992;90(5). [CrossRef]
- Jara ZP, Icimoto MY, Yokota R, et al. Tonin overexpression in mice diminishes sympathetic autonomic modulation and alters angiotensin type 1 receptor response. Front Med (Lausanne). 2019;6(JAN). [CrossRef]
- Kramár EA, Krishnan R, Harding JW, Wright JW. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul Pept. 1998;74(2-3). [CrossRef]
- Williams S, Malatesta K, Norris K. Vitamin D and Chronic Kidney Disease.
- Freundlich M, Quiroz Y, Zhang Z, et al. Suppression of renin–angiotensin gene expression in the kidney by paricalcitol. Kidney Int. 2008;74(11):1394-1402. [CrossRef]
- Andress DL. Vitamin D in chronic kidney disease: A systemic role for selective vitamin D receptor activation. Kidney Int. 2006;69(1):33-43. [CrossRef]
- Tajalli-Nezhad S, Karimian · Mohammad, Cordian Beyer ·, et al. The regulatory role of Toll-like receptors after ischemic stroke: neurosteroids as TLR modulators with the focus on TLR2/4. Cellular and Molecular Life Sciences. 2019;76:523-537. [CrossRef]
- Wamberg L, Kampmann U, Stødkilde-Jørgensen H, Rejnmark L, Pedersen SB, Richelsen B. Effects of vitamin D supplementation on body fat accumulation, inflammation, and metabolic risk factors in obese adults with low vitamin D levels - results from a randomized trial. Eur J Intern Med. 2013;24(7):644-649. [CrossRef]
- Li YC. Renoprotective effects of vitamin D analogs. Kidney Int. 2010;78(2):134-139. [CrossRef]
- Francis R, Aspray T, Fraser W, et al. Vitamin D and Bone Health : A Practical Clinical Guideline for Patient Management. National Osteoporosis Society. 2018;2.
- Compston J, Cooper A, Cooper C, et al. Vitamin D and Health - Scientific Advisory Committee on Nutrition. Osteoporosis International. 2016;27(4).
- Jones G. Expanding role for vitamin D in chronic kidney disease: importance of blood 25-OH-D levels and extra-renal 1alpha-hydroxylase in the classical and nonclassical actions of 1alpha,25-dihydroxyvitamin D(3). Semin Dial. 2007;20(4):316-324. [CrossRef]
- Heaney RP. Vitamin D in Health and Disease. Clinical Journal of the American Society of Nephrology. 2008;3(5):1535-1541. [CrossRef]
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78. [CrossRef]
- Filipov JJ, Zlatkov BK, Dimitrov EP, Svinarov D. Relationship between vitamin D status and immunosuppressive therapy in kidney transplant recipients. Biotechnol Biotechnol Equip. 2015;29(2):331-335. [CrossRef]
- Shimada S, Hirose T, Takahashi C, et al. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci Rep. 2018;8(1):16808. [CrossRef]
- Faul C. FGF23 effects on the heart—levels, time, source, and context matter. Kidney Int. 2018;94(1):7-11. [CrossRef]
- Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. Journal of Clinical Investigation. 2004;113(4):561-568. [CrossRef]
- Perwad F, Azam N, Zhang MYH, Yamashita T, Tenenhouse HS, Portale AA. Dietary and Serum Phosphorus Regulate Fibroblast Growth Factor 23 Expression and 1,25-Dihydroxyvitamin D Metabolism in Mice. Endocrinology. 2005;146(12):5358-5364. [CrossRef]
- Usatii M, Rousseau L, Demers C, et al. Parathyroid hormone fragments inhibit active hormone and hypocalcemia-induced 1,25(OH)2D synthesis. Kidney Int. 2007;72(11):1330-1335. [CrossRef]
- Szymczak-Pajor I, Drzewoski J, Śliwińska A. The Molecular Mechanisms by Which Vitamin D Prevents Insulin Resistance and Associated Disorders. Int J Mol Sci. 2020;21(18):6644. [CrossRef]
- Carey RM, Siragy HM. Newly Recognized Components of the Renin-Angiotensin System: Potential Roles in Cardiovascular and Renal Regulation. Endocr Rev. 2003;24(3):261-271. [CrossRef]
- Li P, He L, Sha Y, Luan Q. Relationship of Metabolic Syndrome to Chronic Periodontitis. J Periodontol. 2009;80(4):541-549. [CrossRef]
- Mehrotra R, Kermah DA, Salusky IB, et al. Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int. 2009;76(9):977-983. [CrossRef]
- Kestenbaum B, Belozeroff V. Mineral metabolism disturbances in patients with chronic kidney disease. Eur J Clin Invest. 2007;37(8):607-622. [CrossRef]
- Ravani P, Malberti F, Tripepi G, et al. Vitamin D levels and patient outcome in chronic kidney disease. Kidney Int. 2009;75(1):88-95. [CrossRef]
- Ho BB, Bergwitz C. FGF23 signalling and physiology. J Mol Endocrinol. 2021;66(2):R23-R32. [CrossRef]
- Wahl P, Wolf M. FGF23 in chronic kidney disease. Adv Exp Med Biol. 2012;728:107-125. [CrossRef]
- Jamal SA, Miller PD. Secondary and tertiary hyperparathyroidism. J Clin Densitom. 2013;16(1):64-68. [CrossRef]
- Al-Aly Z, Qazi RA, González EA, Zeringue A, Martin KJ. Changes in serum 25-hydroxyvitamin D and plasma intact PTH levels following treatment with ergocalciferol in patients with CKD. Am J Kidney Dis. 2007;50(1):59-68. [CrossRef]
- Rapa SF, Di Iorio BR, Campiglia P, Heidland A, Marzocco S. Inflammation and Oxidative Stress in Chronic Kidney Disease—Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int J Mol Sci. 2019;21(1):263. [CrossRef]
- França LFC, Vasconcelos ACCG, da Silva FRP, et al. Periodontitis changes renal structures by oxidative stress and lipid peroxidation. J Clin Periodontol. 2017;44(6):568-576. [CrossRef]
- Sharma P, Fenton A, Dias IHK, et al. Oxidative stress links periodontal inflammation and renal function. J Clin Periodontol. 2021;48(3):357-367. [CrossRef]
- Gyurászová M, Gurecká R, Bábíčková J, Tóthová Ľ. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxid Med Cell Longev. 2020;2020:5478708. [CrossRef]
- Yang X, Li Y, Li Y, et al. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front Physiol. 2017;8:600. [CrossRef]
- Hertiš Petek T, Petek T, Močnik M, Marčun Varda N. Systemic Inflammation, Oxidative Stress and Cardiovascular Health in Children and Adolescents: A Systematic Review. Antioxidants (Basel). 2022;11(5). [CrossRef]
- Annuk M, Zilmer M, Lind L, Linde T, Fellström B. Oxidative Stress and Endothelial Function in Chronic Renal Failure. Journal of the American Society of Nephrology. 2001;12(12):2747-2752. [CrossRef]
- Locatelli F, Canaud B, Eckardt KU, Stenvinkel P, Wanner C, Zoccali C. Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrology Dialysis Transplantation. 2003;18(7):1272-1280. [CrossRef]
- Berer A, Stöckl J, Majdic O, et al. 1,25-Dihydroxyvitamin D3 inhibits dendritic cell differentiation and maturation in vitro. Exp Hematol. 2000;28(5):575-583. [CrossRef]
- Penna G, Adorini L. 1α,25-Dihydroxyvitamin D3 Inhibits Differentiation, Maturation, Activation, and Survival of Dendritic Cells Leading to Impaired Alloreactive T Cell Activation. The Journal of Immunology. 2000;164(5):2405-2411. [CrossRef]
- Ao T, Kikuta J, Ishii M. The effects of vitamin D on immune system and inflammatory diseases. Biomolecules. 2021;11(11). [CrossRef]
- Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D 3 . The FASEB Journal. 2005;19(9). [CrossRef]
- Yuk JM, Shin DM, Lee HM, et al. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host Microbe. 2009;6(3). [CrossRef]
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770-1773. [CrossRef]
- Carlberg C. Vitamin D Signaling in the Context of Innate Immunity: Focus on Human Monocytes. Front Immunol. 2019;10. [CrossRef]
- Skrobot A, Demkow U, Wachowska M. Immunomodulatory Role of Vitamin D: A Review. In: ; 2018:13-23. [CrossRef]
- Ikeda U, Wakita D, Ohkuri T, et al. 1α,25-Dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol Lett. 2010;134(1):7-16. [CrossRef]
- Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory Effects of 1,25-Dihydroxyvitamin D3 on Human B Cell Differentiation. The Journal of Immunology. 2007;179(3). [CrossRef]
- Wang TT, Nestel FP, Bourdeau V, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173(5):2909-2912. [CrossRef]
- Diamond G, Beckloff N, Ryan LK. Host defense peptides in the oral cavity and the lung: similarities and differences. J Dent Res. 2008;87(10):915-927. [CrossRef]
- Carlberg C. Vitamin D: A Micronutrient Regulating Genes. Curr Pharm Des. 2019;25(15):1740-1746. [CrossRef]
- Bhan I, Powe CE, Berg AH, et al. Bioavailable vitamin D is more tightly linked to mineral metabolism than total vitamin D in incident hemodialysis patients. Kidney Int. 2012;82(1). [CrossRef]
- Denburg MR, Bhan I. Vitamin D-Binding Protein in Health and Chronic Kidney Disease. Semin Dial. 2015;28(6). [CrossRef]
- Dhaif YG, Garcia-Sanchez R, Albuquerque R, Lu E. The association between Vitamin D binding protein levels and periodontal status: a systematic review. J Periodontal Res. Published online December 2023.
- Powe CE, Evans MK, Wenger J, et al. Vitamin D–Binding Protein and Vitamin D Status of Black Americans and White Americans. New England Journal of Medicine. 2013;369(21). [CrossRef]
- Robinson-Cohen C, Hoofnagle AN, Ix JH, et al. Racial differences in the association of serum 25-hydroxyvitamin D concentration with coronary heart disease events. JAMA. 2013;310(2). [CrossRef]
- Parikh A, Chase HS, Vernocchi L, Stern L. Vitamin D resistance in chronic kidney disease (CKD). BMC Nephrol. 2014;15(1). [CrossRef]
- Chun RF, Lauridsen AL, Suon L, et al. Vitamin D-binding protein directs monocyte responses to 25-hydroxy- and 1,25-dihydroxyvitamin D. Journal of Clinical Endocrinology and Metabolism. 2010;95(7). [CrossRef]
- Taylor GW, Sato M, Minagawa K, Yoshihara A, Iwasaki M, Ansai T. Effect of chronic kidney disease on progression of clinical attachment loss in older adults: A 4-year cohort study. J Periodontol. 2019;90(8). [CrossRef]
- Serni L, Caroti L, Barbato L, et al. Association between chronic kidney disease and periodontitis. A systematic review and metanalysis. Oral Dis. 2023;29(1). [CrossRef]
- Cannata-Andía JB, Martín-Carro B, Martín-Vírgala J, et al. Chronic Kidney Disease-Mineral and Bone Disorders: Pathogenesis and Management. Calcif Tissue Int. 2020;108:410-422. [CrossRef]
- Costacurta M, Basilicata M, Marrone G, et al. The Impact of Chronic Kidney Disease on Nutritional Status and Its Possible Relation with Oral Diseases. Nutrients. 2022;14(10). [CrossRef]
- Allen MR, Chen NX, Ii VHG, Moe SM. E-Mail Adverse Mandibular Bone Effects Associated with Kidney Disease Are Only Partially Corrected with Bisphosphonate and/or Calcium Treatment. Published online 2013. [CrossRef]
- Fisher MA, Taylor GW, West BT, McCarthy ET. Bidirectional relationship between chronic kidney and periodontal disease: A study using structural equation modeling. Kidney Int. 2011;79(3). [CrossRef]
- Lertpimonchai A, Rattanasiri S, Tamsailom S, et al. Periodontitis as the risk factor of chronic kidney disease: Mediation analysis. J Clin Periodontol. 2019;46(6). [CrossRef]
- Grubbs V, Vittinghoff E, Taylor G, et al. The association of periodontal disease with kidney function decline: A longitudinal retrospective analysis of the MrOS dental study. Nephrology Dialysis Transplantation. 2016;31(3). [CrossRef]
- Kajiwara K, Sawa Y, Fujita T, Tamaoki S. Immunohistochemical study for the expression of leukocyte adhesion molecules, and FGF23 and ACE2 in P. gingivalis LPS-induced diabetic nephropathy. BMC Nephrol. 2021;22(1). [CrossRef]
- Nakano C, Hamano T, Fujii N, et al. Combined use of vitamin D status and FGF23 for risk stratification of renal outcome. Clinical Journal of the American Society of Nephrology. 2012;7(5). [CrossRef]
- Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82(7). [CrossRef]
- Mahendra J, Palathingal P, Mahendra L, et al. Impact of Red Complex Bacteria and TNF-α Levels on the Diabetic and Renal Status of Chronic Kidney Disease Patients in the Presence and Absence of Periodontitis. Biology (Basel). 2022;11(3). [CrossRef]
- Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: A meta-analysis of cohort studies. BMC Nephrol. 2017;18(1). [CrossRef]
- Tai YH, Chen JT, Kuo HC, et al. Periodontal disease and risk of mortality and kidney function decline in advanced chronic kidney disease: a nationwide population-based cohort study. Clin Oral Investig. 2021;25(11). [CrossRef]
- Delbove T, Gueyffier F, Juillard L, et al. Effect of periodontal treatment on the glomerular filtration rate, reduction of inflammatory markers and mortality in patients with chronic kidney disease: A systematic review. PLoS One. 2021;16(1):e0245619. [CrossRef]
- da Silva TA, Abreu LG, Esteves Lima RP. A meta-analysis on the effect of periodontal treatment on the glomerular filtration rate of chronic kidney disease individuals: A systematic review and meta-analysis was conducted to assess the impact of the periodontal treatment on the glomerular filtration rate of individuals with chronic kidney disease. Spec Care Dentist. 2021;41(6):670-678. [CrossRef]
- Almeida S, Figueredo CM, Lemos C, Bregman R, Fischer RG. Periodontal treatment in patients with chronic kidney disease: a pilot study. J Periodontal Res. 2017;52(2):262-267. [CrossRef]
- Jean G, Terrat JC, Vanel T, et al. Evidence for persistent vitamin D 1-alpha-hydroxylation in hemodialysis patients: Evolution of serum 1,25-dihydroxycholecalciferol after 6 months of 25-hydroxycholecalciferol treatment. Nephron Clin Pract. 2008;110(1). [CrossRef]
- Walker JP, Hiramoto JS, Gasper WJ, et al. Vitamin D deficiency is associated with mortality and adverse vascular access outcomes in patients with end-stage renal disease. In: Journal of Vascular Surgery. Vol 60. ; 2014. [CrossRef]
- Lopez AG, Kerlan V, Desailloud R. Non-classical effects of vitamin D: Non-bone effects of vitamin D. Ann Endocrinol (Paris). 2021;82(1):43-51. [CrossRef]
- Melamed ML, Astor B, Michos ED, Hostetter TH, Powe NR, Muntner P. 25-Hydroxyvitamin D levels, race, and the progression of kidney disease. Journal of the American Society of Nephrology. 2009;20(12). [CrossRef]
- Pilz S, Tomaschitz A, März W, et al. Vitamin D, cardiovascular disease and mortality. Clin Endocrinol (Oxf). 2011;75(5). [CrossRef]
- Jayedi A, Soltani S, Shab-Bidar S. Vitamin D status and all-cause mortality in patients with chronic kidney disease: A systematic review and dose-response meta-analysis. Journal of Clinical Endocrinology and Metabolism. 2017;102(7). [CrossRef]
- DeVille J, Thorp ML, Tobin L, Gray E, Johnson ES, Smith DH. Effect of ergocalciferol supplementation on serum parathyroid hormone and serum 25-hydroxyvitamin D in chronic kidney disease. Nephrology. 2006;11(6). [CrossRef]
- Ikizler TA, Cuppari L. The 2020 Updated KDOQI Clinical Practice Guidelines for Nutrition in Chronic Kidney Disease. Blood Purif. 2021;50(4-5). [CrossRef]
- Chowdhury R, Kunutsor S, Vitezova A, et al. Vitamin D and risk of cause specific death: Systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ (Online). 2014;348. [CrossRef]
- Duranton F, Rodriguez-Ortiz ME, Duny Y, Rodriguez M, Daurès JP, Argilés A. Vitamin D treatment and mortality in chronic kidney disease: A systematic review and meta-analysis. Am J Nephrol. 2013;37(3). [CrossRef]
- Mehrotra R, Kermah DA, Salusky IB, et al. Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int. 2009;76(9):977-983. [CrossRef]
- Cheng S, Coyne D. Vitamin D and outcomes in chronic kidney disease. Curr Opin Nephrol Hypertens. 2007;16(2). [CrossRef]
- Armas LAG, Hollis BW, Heaney RP. Vitamin D 2 Is Much Less Effective than Vitamin D 3 in Humans. J Clin Endocrinol Metab. 2004;89(11):5387-5391. [CrossRef]
- Dietrich T, Joshipura KJ, Dawson-Hughes B, Bischoff-Ferrari HA. Association between serum concentrations of 25-hydroxyvitamin D 3 and periodontal disease in the US population. American Journal of Clinical Nutrition. 2004;80(1). [CrossRef]
- Krall EA, Wehler C, Garcia RI, Harris SS, Dawson-Hughes B. Calcium and vitamin D supplements reduce tooth loss in the elderly. American Journal of Medicine. 2001;111(6). [CrossRef]
- Gao W, Tang H, Wang D, Zhou X, Song Y, Wang Z. Effect of short-term vitamin D supplementation after nonsurgical periodontal treatment: A randomized, double-masked, placebo-controlled clinical trial. J Periodontal Res. 2020;55(3). [CrossRef]
- Bastos J do A, Andrade LCF de, Ferreira AP, et al. Serum levels of vitamin D and chronic periodontitis in patients with chronic kidney disease. J Bras Nefrol. 2013;35(1):20-26. [CrossRef]
- MacRae C, Mercer SW, Guthrie B, Henderson D. Comorbidity in chronic kidney disease: A large cross-sectional study of prevalence in Scottish primary care. British Journal of General Practice. 2021;71(704). [CrossRef]
- Dietrich T, Sharma P, Walter C, Weston P, Beck J. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. J Clin Periodontol. 2013;40(SUPPL. 14). [CrossRef]
- Hajishengallis G. Interconnection of periodontal disease and comorbidities: Evidence, mechanisms, and implications. Periodontol 2000. 2022;89(1). [CrossRef]
- Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117(4). [CrossRef]
- Zittermann A, Trummer C, Theiler-schwetz V, Lerchbaum E, März W, Pilz S. Vitamin d and cardiovascular disease: An updated narrative review. Int J Mol Sci. 2021;22(6). [CrossRef]
- Chen S, Glenn DJ, Ni W, et al. Expression of the vitamin D receptor is increased in the hypertrophic heart. Hypertension. 2008;52(6). [CrossRef]
- Zittermann A, Schulze Schleithoff S, Tenderich G, Berthold HK, Körfer R, Stehle P. Low vitamin D status: A contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol. 2003;41(1). [CrossRef]
- Somjen D, Weisman Y, Kohen F, et al. 25-Hydroxyvitamin D3-1α-hydroxylase is expressed in human vascular smooth muscle cells and is upregulated by parathyroid hormone and estrogenic compounds. Circulation. 2005;111(13). [CrossRef]
- Friedlaender MM, Kornberg Z, Wald H, Popovtzer MM. Renal effect of vitamin D metabolites: evidence for the essential role of the 25(OH) group. American Journal of Physiology-Renal Physiology. 1983;244(6):F674-F678. [CrossRef]
- Elidrissy ATH, Munawarah M, Alharbi KM. Hypocalcemic rachitic cardiomyopathy in infants. J Saudi Heart Assoc. 2013;25(1). [CrossRef]
- Subbiah AK, Chhabra YK, Mahajan S. Cardiovascular disease in patients with chronic kidney disease: A neglected subgroup. Heart Asia. 2016;8(2). [CrossRef]
- Kolb H, Mandrup-Poulsen T. An immune origin of type 2 diabetes? Diabetologia. 2005;48(6). [CrossRef]
- Tuttle KR, Bakris GL, Bilous RW, et al. Diabetic kidney disease: A report from an ADA consensus conference. Diabetes Care. 2014;37(10). [CrossRef]
- Fernández-Juárez G, Luño J, Barrio V, et al. 25 (OH) vitamin D levels and renal disease progression in patients with type 2 diabetic nephropathy and blockade of the renin-angiotensin system. Clinical Journal of the American Society of Nephrology. 2013;8(11). [CrossRef]
- Shultis WA, Weil EJ, Looker HC, et al. Effect of periodontitis on overt nephropathy and end-stage renal disease in type 2 diabetes. Diabetes Care. 2007;30(2). [CrossRef]
- Naruishi K, Oishi K, Inagaki Y, et al. Association between periodontal condition and kidney dysfunction in Japanese adults: A cross-sectional study. Clin Exp Dent Res. 2016;2(3). [CrossRef]
- Andrukhov O, Andrukhova O, Hulan U, Tang Y, Bantleon HP, Rausch-Fan X. Both 25-hydroxyvitamin-D3 and 1,25-dihydroxyvitamin- D3 reduces inflammatory response in human periodontal ligament cells. PLoS One. 2014;9(2). [CrossRef]
- Lips P, Eekhoff M, van Schoor N, et al. Vitamin D and type 2 diabetes. Journal of Steroid Biochemistry and Molecular Biology. 2017;173. [CrossRef]
- Muñoz-Garach A, García-Fontana B, Muñoz-Torres M. Vitamin D status, calcium intake and risk of developing type 2 diabetes: An unresolved issue. Nutrients. 2019;11(3). [CrossRef]
- Berridge MJ. Vitamin D deficiency and diabetes. Biochemical Journal. 2017;474(8). [CrossRef]
- Berridge MJ. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016;371(1700). [CrossRef]
- Jain SK, Micinski D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem Biophys Res Commun. 2013;437(1). [CrossRef]
- Tagliaferri S, Porri D, De Giuseppe R, Manuelli M, Alessio F, Cena H. The controversial role of Vitamin D as an antioxidant: results from randomised controlled trials. Nutr Res Rev. 2019;32(1). [CrossRef]
- Park S, Kim DS, Kang S. Vitamin D deficiency impairs glucose-stimulated insulin secretion and increases insulin resistance by reducing PPAR-γ expression in nonobese Type 2 diabetic rats. Journal of Nutritional Biochemistry. 2016;27. [CrossRef]
- Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Endocrinol Metab Clin North Am. 2010;39(2). [CrossRef]
- Kang S, Tsai LT, Zhou Y, et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat Cell Biol. 2015;17(1). [CrossRef]
- Ong LTC, Booth DR, Parnell GP. Vitamin D and its Effects on DNA Methylation in Development, Aging, and Disease. Mol Nutr Food Res. 2020;64(23). [CrossRef]
- Murdaca G, Tonacci A, Negrini S, et al. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun Rev. 2019;18(9). [CrossRef]
- Mouterde G, Gamon E, Rincheval N, et al. Association Between Vitamin D Deficiency and Disease Activity, Disability, and Radiographic Progression in Early Rheumatoid Arthritis: The ESPOIR Cohort. J Rheumatol. 2020;47(11):1624-1628. [CrossRef]
- Rossini M, Maddali Bongi S, La Montagna G, et al. Vitamin D deficiency in rheumatoid arthritis: Prevalence, determinants and associations with disease activity and disability. Arthritis Res Ther. 2010;12(6). [CrossRef]
- Caraba A, Crişan V, Romoşan I, Mozoş I, Murariu M. Vitamin D status, disease activity, and endothelial dysfunction in early rheumatoid arthritis patients. Dis Markers. 2017;2017. [CrossRef]
- Dankers W, Dankers W, Davelaar N, et al. THU0047 1,25(OH)2d3 and dexamethasone additively suppress synovial fibroblast activation by ccr6+ th memory cells and enhance the effect of tnf-alpha blockade. In: ; 2018. [CrossRef]
- Mok CC. Systemic lupus erythematosus: What should family physicians know in 2018? Hong Kong Medical Journal. 2018;24(5). [CrossRef]
- Schoindre Y, Jallouli M, Tanguy ML, et al. Lower Vitamin D levels are associated with higher systemic lupus erythematosus activity, but not predictive of disease flare-up. Lupus Sci Med. 2014;1(1). [CrossRef]
- Linker-Israeli M, Elstner E, Klinenberg JR, Wallace DJ, Koeffler HP. Vitamin D3 and its synthetic analogs inhibit the spontaneous in vitro immunoglobulin production by SLE-derived PBMC. Clinical Immunology. 2001;99(1). [CrossRef]
- Sellner J, Kraus J, Awad A, Milo R, Hemmer B, Stüve O. The increasing incidence and prevalence of female multiple sclerosis-A critical analysis of potential environmental factors. Autoimmun Rev. 2011;10(8). [CrossRef]
- Hiremath GS, Cettomai D, Baynes M, et al. Vitamin D status and effect of low-dose cholecalciferol and high-dose ergocalciferol supplementation in multiple sclerosis. Multiple Sclerosis. 2009;15(6). [CrossRef]
- Lemire JM, Ince A, Takashima M. 1,25-dihydroxyvitamin d3 attenuates of expression of experimental murine lupus of MRL/1 mice. Autoimmunity. 1992;12(2). [CrossRef]
- Mattner F, Smiroldo S, Galbiati F, et al. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D3. Eur J Immunol. 2000;30(2). [CrossRef]
- Fichna M, Zurawek M, Januszkiewicz-Lewandowska D, et al. Association of the CYP27B1 C(- 1260)A polymorphism with autoimmune Addison’s disease. Experimental and Clinical Endocrinology and Diabetes. 2010;118(8). [CrossRef]
- Yazici D, Yavuz D, Tarcin O, Sancak S, Deyneli O, Akalin S. Vitamin D receptor gene ApaI, TaqI, FokI and BsmI polymorphisms in a group of Turkish patients with Hashimoto’s thyroiditis. In: Minerva Endocrinologica. Vol 38. ; 2013.
- Zhang J, Li W, Liu J, et al. Polymorphisms in the vitamin D receptor gene and type 1 diabetes mellitus risk: An update by meta-analysis. Mol Cell Endocrinol. 2012;355(1). [CrossRef]
Figure 1.
| Vitamin D classical activation pathway in the human body. Sources of Vitamin D such as UV rays and diet, deliver vitamin D in their precursor forms D2 and D3 [11]. These precursors then bind to the Vitamin D binding protein (DBP) at site of synthesis and form the DBP-D protein complex. Vitamin D is then carried in DBP-D complex through the blood plasma to the liver. Precursors D2 and D3 are hydroxylated into inactive 25-hydroxvitamin D [25(OH)D] by 25-hydroxylase, coded by cytochrome P2R1 (CYP2R1) gene [14]. 25(OH)D uptake into kidney from blood and activated by 1-α-hydroxylation by CYP27B gene to 1,25-dihydroxyvitamin D (1,25(OH)2D) activated state. Determinants of CYP27B gene activity include increased parathyroid hormone (PTH), decreased phosphorous and calcium activating CYP27B; and increased fibroblast growth factor-23 (FGF-23) inhibiting its activity [15].
Figure 1.
| Vitamin D classical activation pathway in the human body. Sources of Vitamin D such as UV rays and diet, deliver vitamin D in their precursor forms D2 and D3 [11]. These precursors then bind to the Vitamin D binding protein (DBP) at site of synthesis and form the DBP-D protein complex. Vitamin D is then carried in DBP-D complex through the blood plasma to the liver. Precursors D2 and D3 are hydroxylated into inactive 25-hydroxvitamin D [25(OH)D] by 25-hydroxylase, coded by cytochrome P2R1 (CYP2R1) gene [14]. 25(OH)D uptake into kidney from blood and activated by 1-α-hydroxylation by CYP27B gene to 1,25-dihydroxyvitamin D (1,25(OH)2D) activated state. Determinants of CYP27B gene activity include increased parathyroid hormone (PTH), decreased phosphorous and calcium activating CYP27B; and increased fibroblast growth factor-23 (FGF-23) inhibiting its activity [15].
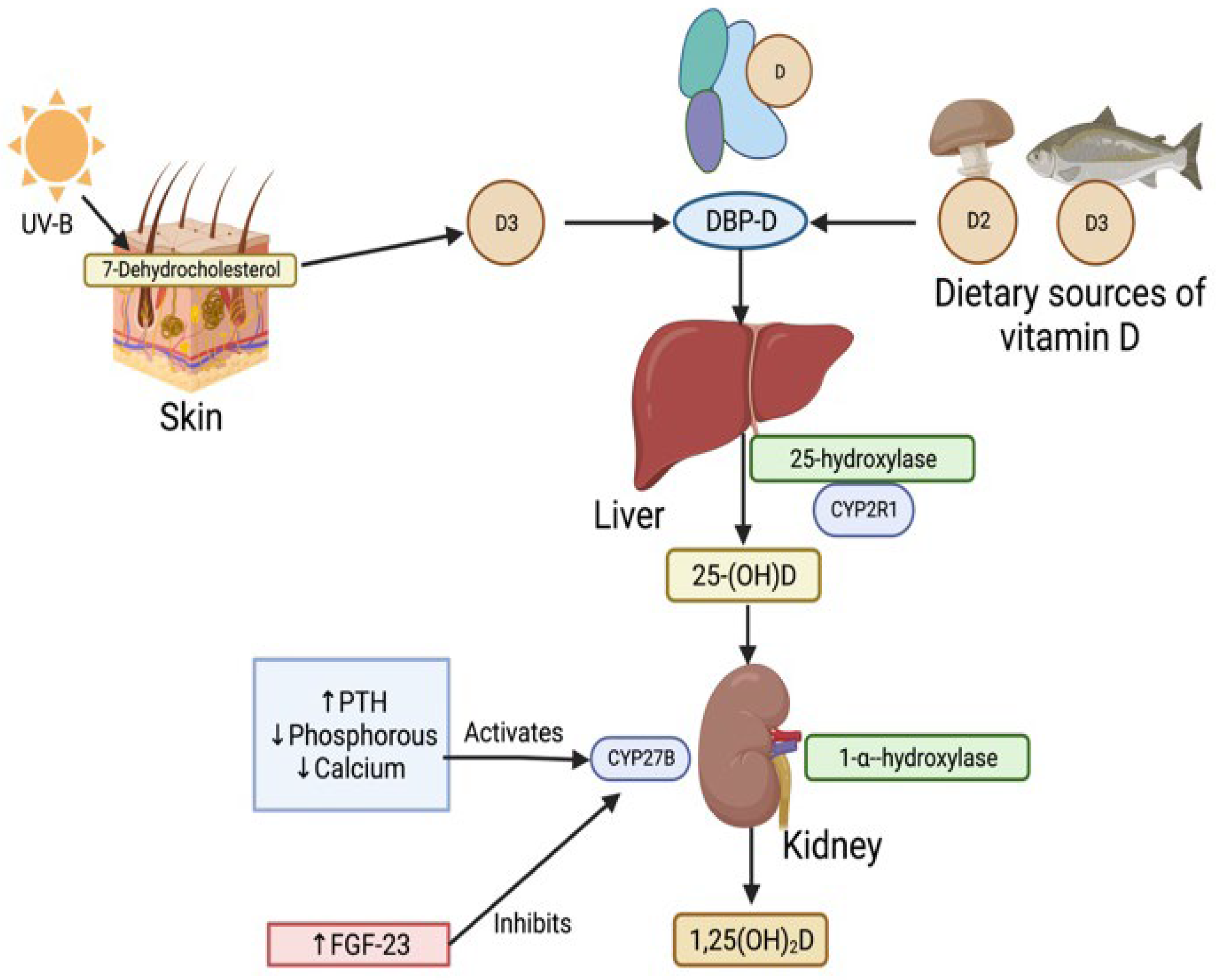
Figure 2.
– Vitamin D deficiency leads to the progression of chronic kidney disease (CKD) and cardiovascular disease (CVD). In health, vitamin D binds to vitamin D receptors (VDRs) and increases intracellular calcium in the juxtaglomerular apparatus, suppressing the renin-angiotensin-aldosterone system (RAAS) and the secretion of renin. Renin cleaves angiotensinogen into angiotensin I, which is then converted into angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin-II can produce more downstream angiotensin sub-types [23,24]. Angiotensin-II binds to type 1 angiotensin-II receptor (AT1R) [25], increasing sympathetic tone [26], blood pressure [22,27], inflammation [28], fibrosis [29], aldosterone production [30], anti-diuretic hormone (ADH) production [31] and cardiac hypertrophy [32]. Aside from the vascular damage caused, AT1R activation increases vascular smooth muscle cell dedifferentiation, leading to atherosclerosis [33]. AT1R activation also decreases parasympathetic tone [34] and nitric oxide production [35], contributing to hypertension. These effects culminate as renal and cardiovascular damage [22,23,36]. Vitamin D works to protect against this damage by suppressing RAAS, but this su.ppression is reversed during CKD.
Figure 2.
– Vitamin D deficiency leads to the progression of chronic kidney disease (CKD) and cardiovascular disease (CVD). In health, vitamin D binds to vitamin D receptors (VDRs) and increases intracellular calcium in the juxtaglomerular apparatus, suppressing the renin-angiotensin-aldosterone system (RAAS) and the secretion of renin. Renin cleaves angiotensinogen into angiotensin I, which is then converted into angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin-II can produce more downstream angiotensin sub-types [23,24]. Angiotensin-II binds to type 1 angiotensin-II receptor (AT1R) [25], increasing sympathetic tone [26], blood pressure [22,27], inflammation [28], fibrosis [29], aldosterone production [30], anti-diuretic hormone (ADH) production [31] and cardiac hypertrophy [32]. Aside from the vascular damage caused, AT1R activation increases vascular smooth muscle cell dedifferentiation, leading to atherosclerosis [33]. AT1R activation also decreases parasympathetic tone [34] and nitric oxide production [35], contributing to hypertension. These effects culminate as renal and cardiovascular damage [22,23,36]. Vitamin D works to protect against this damage by suppressing RAAS, but this su.ppression is reversed during CKD.
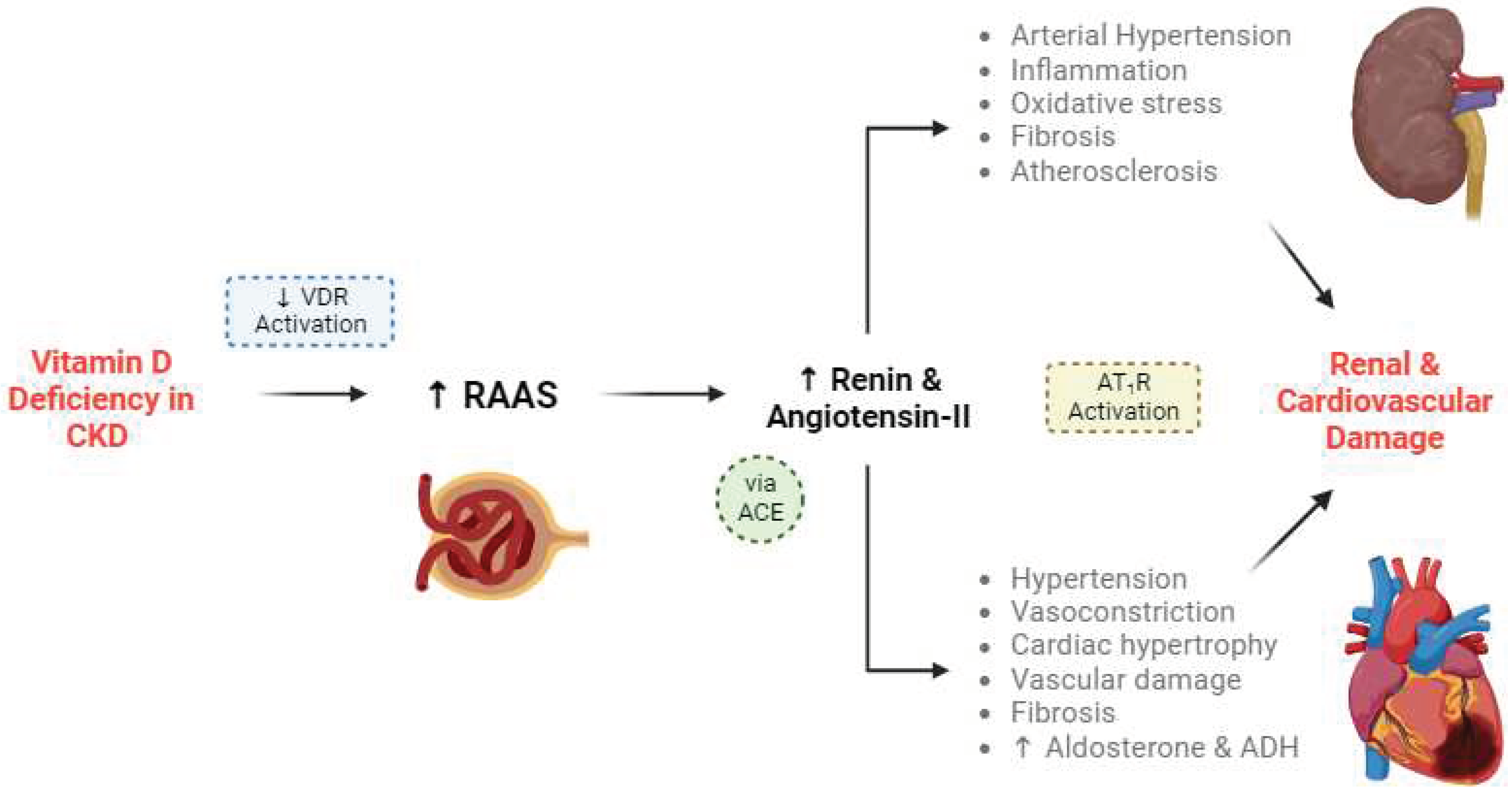
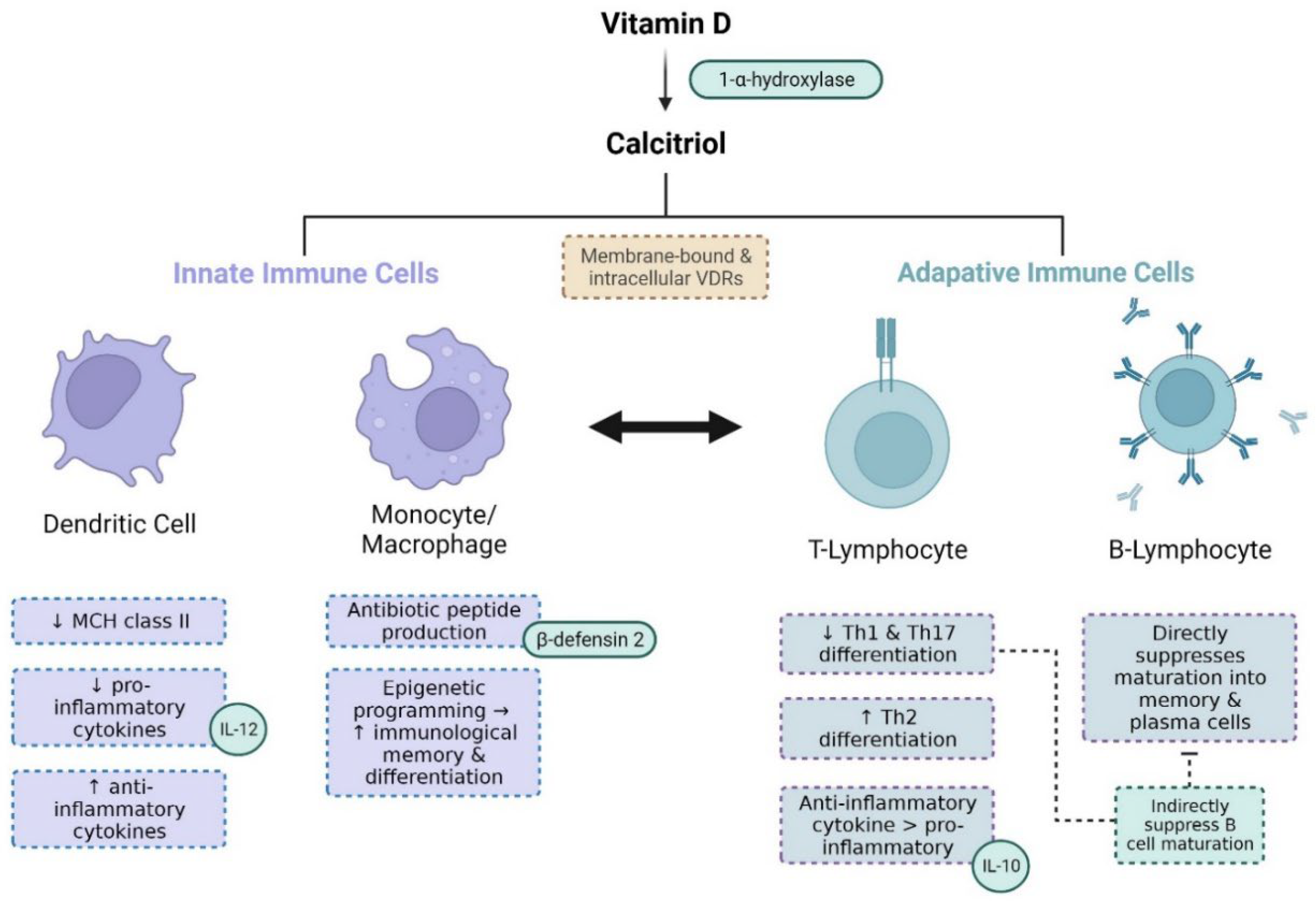
Figure 3.
– Vitamin D modulates the functions of various immune cells. 25-hydroxyvitamin D (vitamin D) is metabolised into its active form 1,25-dihydroxyvitamin D (calcitriol) by CYP27BQ (1--hydroxylase) [1]. Calcitriol binds to vitamin D receptors (VDRs) both on the cell-surface (membrane-bound VDRs) and in the cytoplasm (intracellular VDRs) of immune cells [73]. Calcitriol downregulates the production of MHC class II in dendritic cells, which is necessary for antigen recognition and dendritic cell activation. Activated dendritic cells stimulate T-lymphocyte (T-cell) activity, so the suppression of dendritic cells leads to reduced T-cell function [74,75]. Dendritic cells contribute to innate immunity, whilst T-cells contribute to adaptive immunity, so in this way, innate and adaptive immunity are linked. Calcitriol also suppresses the production of pro-inflammatory cytokines like interleukin-2 (IL-12) in dendritic cells, whilst stimulating the production of anti-inflammatory cytokines. Calcitriol binds to intracellular VDRs in macrophages and their monocyte precursors, forming heterodimers with retinoid-X receptor [33]. This stimulates the production of cell membrane-destroying antibiotic peptides, including β-defensins [70,76],. Calcitriol also epigenetically regulates the immunological memory and differentiation of macrophages and monocytes [77]. Calcitriol reduces the differentiation of T-helper cells into types Th1 and Th17 and their production of pro-inflammatory cytokines [78,79], whilst stimulating differentiation into the Th2-type and favouring their production of anti-inflammatory cytokines like interleukin 10 (IL-10). T-cell-derived pro-inflammatory cytokines are important for B-lymphocyte (B-cell) differentiation, and so the suppression of T-cell pro-inflammatory activity also suppresses B-cell activity. Calcitriol also directly suppresses naïve B-cell differentiation and maturation into memory and plasma cells [80].
Figure 3.
– Vitamin D modulates the functions of various immune cells. 25-hydroxyvitamin D (vitamin D) is metabolised into its active form 1,25-dihydroxyvitamin D (calcitriol) by CYP27BQ (1--hydroxylase) [1]. Calcitriol binds to vitamin D receptors (VDRs) both on the cell-surface (membrane-bound VDRs) and in the cytoplasm (intracellular VDRs) of immune cells [73]. Calcitriol downregulates the production of MHC class II in dendritic cells, which is necessary for antigen recognition and dendritic cell activation. Activated dendritic cells stimulate T-lymphocyte (T-cell) activity, so the suppression of dendritic cells leads to reduced T-cell function [74,75]. Dendritic cells contribute to innate immunity, whilst T-cells contribute to adaptive immunity, so in this way, innate and adaptive immunity are linked. Calcitriol also suppresses the production of pro-inflammatory cytokines like interleukin-2 (IL-12) in dendritic cells, whilst stimulating the production of anti-inflammatory cytokines. Calcitriol binds to intracellular VDRs in macrophages and their monocyte precursors, forming heterodimers with retinoid-X receptor [33]. This stimulates the production of cell membrane-destroying antibiotic peptides, including β-defensins [70,76],. Calcitriol also epigenetically regulates the immunological memory and differentiation of macrophages and monocytes [77]. Calcitriol reduces the differentiation of T-helper cells into types Th1 and Th17 and their production of pro-inflammatory cytokines [78,79], whilst stimulating differentiation into the Th2-type and favouring their production of anti-inflammatory cytokines like interleukin 10 (IL-10). T-cell-derived pro-inflammatory cytokines are important for B-lymphocyte (B-cell) differentiation, and so the suppression of T-cell pro-inflammatory activity also suppresses B-cell activity. Calcitriol also directly suppresses naïve B-cell differentiation and maturation into memory and plasma cells [80].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



