
Submitted:
20 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
This review article focuses on the upstream pertinent pathophysiology leading to neurodegenerative disease. Specifically, the nexus appears to be blood brain barrier (BBB) leakiness resulting in a two-prong inflammatory disease spectrum damaging the microvasculature and corrupted protein synthesis and degradation with accumulating misfolded toxic proteins. The suboptimal results of removing misfolded proteins means a new approach to disease in the preclinical state is required aimed at other targets.Validated noninvasive imaging and serologic biomarkers of early preclinical disease implemented in the high-risk patient cohort along with periodic surveillance once effective treatments are developed will be required.This review discusses the physiology and pathophysiology of the BBB, new MRI imaging techniques identifying the leak and altered fluid dynamic effects in the preclinical state. The risk factors for disease development, preventative measures, and potential treatment targets are also discussed.
Keywords:
Blood brain barrier
; Microvascular pathology
; Neurodegenerative Inflammatory changes
; ASL MRI
; DCE MRI
; Serologic markers
Introduction
Throughout the past few years, Pharmacologic focus on removal of β amyloid has proven disappointing [1,2] . Their accumulation occurs later and perhaps reaches a non-recoverable point in the neurodegenerative process. The early inflammatory phase begins years or decades prior to their accumulation in association with microvascular disease [3,4,5,6,7]. It has become clear that the nexus of this complex disease is blood brain barrier dysfunction (BBB) [3,6,7,8]. The scope of this chapter includes discussion of the normal physiology of the BBB, mechanisms of damage, the physiologic consequences of dysfunction and methods of early detection of vascular injury. What is known of the normal restorative function after BBB damage will be discussed in the context of apparent decline with aging. This will precede discussion of the pathophysiology of BBB damage, and its perpetuation and acceleration by accumulating toxic protein end products (β Amyloid and hp Tau) from corrupted synthesis and degradation. Evidence based discussion of prevention and focus on methods of noninvasive early detection and probable future therapeutic research directions follow.
Here-to-fore therapeutic efforts at mitigating Alzheimer disease have focused on late disease stage removal of toxic protein end products (β Amyloid) with limited effect and great expense[1,2]. It is clear from these clinical trials however that early intervention is necessary for success, perhaps in the preclinical state [9]. Further a change in therapeutic target(s) addressing the upstream nexus (BBB dysfunction) leading to defined pathologic and clinical damage may suppress disease development. We will address what is known of early BBB pathophysiology, the multiplicity of potential triggers, and consequent altered fluid dynamics. The details of the various signaling pathways are not included for simplicity but several excellent review articles have been referenced for those interested [6,8,11].
Leveraging the early perfusion dynamic alterations, preclinical disease may be identified using invasive and noninvasive techniques to serve initially as an outcome measure of efficacy for new therapeutic endeavors [12,13,14,15]. They potentially will serve as both a diagnostic and surveillance tool to identify and assess outcomes of novel early disease phase treatments before cognitive decline develops.
Normal BBB Physiology and regulation
The brain is a complex highly metabolically active organ requiring a steady flow of nutrients, oxygen and selected substrates and waste removal for maximum efficient function. This requires highly regulated and unfettered rapid access to the above depending on moment to moment need at the same time excluding potential damaging substances. The capillary vasculature provides both functions with unique highly specialized endothelial cells which channel in nutrients passively, and actively via ionic, nonionic, and small molecule transporters as well as specialized vesicular clathrin pathways for selected larger molecules [3,8,11,16,18,19] Figure 1a. Together with pericytes signaling, tight junctions (TJs) are formed between endothelial cells which exclude potentially harmful molecular patterns, so called damage associated molecular patterns (DAMPS) from unwanted but innately produced substances and pathogen associated molecular patterns (PAMPS) from foreign substances from infectious agents. It is the pericytes via communication with glial cells of the neurovascular unit (NVU) to regulate uptake via BBB transport mechanisms of needed substrate. Waste byproducts, CO2 and water accumulating within the neuropil from oxidative metabolism are dealt with by both venous transport (≈60%) and ≈40% exits via an ingenious use of paravascular space between the basement membranes of the pericytes and astrocyte end feet dubbed the glymphatic space [17]. Counterintuitively, its flow is retrograde through the arterial wall intermixing ultimately with CSF and reabsorbed through the arachnoid granulations, cribriform plate, dural meningeal lymphatics and then ultimately draining through the deep cervical lymph nodes. A minor component leaves via sleeves of exiting spinal nerve roots [20].
Adapted from Galea. The blood brain barrier in systemic infection and inflammation. Cellular&Molecular Immunology(2021)18:2489–2501; https://doi.org/10.1038/s41423-021-00757-x
Blood Brain Barrier Cellular makeup and their function
The complex nature of the brain BBB requires interaction among all elements of the neurovascular unit (NVU) [5,8,9,16]. The high metabolic needs of the brain (8% of body mass but consumes 25% of produced energy) translates into the need for steady inflow of nutrients and outflow of waste for optimal function. The system is highly dynamic in nature adapting to rapid changes in metabolic requirements by expressing appropriate transporters and allowing for the selective ingress and egress of ions, lipids, and solutes as well as transcytosis of selective larger molecules( Figure 2). O2 and CO2 move passively through the barrier. Maintenance of the tight junctions is also responsive to metabolic needs and highly selective at the same time excluding potential toxins [21,22]. These functions are controlled by a choreography of signaling amongst all the NVU players (capillary endothelium, astrocytes, pericytes, and associated basement membranes). The exclusive nature of what passes through the barrier is controlled by layered protection. Starting from the intravascular space, negatively charged glycocalyx expressed by endothelial cell repels entry of unwelcome plasma associated proteins. The endothelial cells themselves express selective transporters and transcytosis pathways and the tight junctions between cells expressed via direction from pericytes/astrocytes. Pericytes and astrocyte end feet and their basement membranes provide yet the final barrier [22,23] (Figure 1b).
Auto regulation of blood inflow to each region of the brain is controlled by smooth muscle cells (SMCs) in the precapillary arterioles and post capillary venules with pericytes lining the capillaries [16,24]. SMCs at the arterial-capillary and capillary-vein junctions have cytoplasmic actin which allows for fine tuning of pre and postcapillary flow thereby maximizing oxygen and nutrient extraction [16,24]. Local energy requirements controlling blood flow is signaled by astrocytes to pericytes locally which in turn optimize vascular tone [25]. The autoregulatory vascular tone is balanced between local energy requirements and the mean arterial pressure (MAP). The vascular tone regulation is dependent on the rapidity of MAP changes and is more effective with MAP elevations than decreases. The dogma of stable CA over a wide range of MAP has been effectively disputed [26]. This latter issue is tangential to our discussion yet is critical in the clinical considerations of blood pressure management and its effect on cerebral perfusion. We will now address the function of the cellular components of the NVU in homeostasis and in disease.
Capillary Endothelial cells
The capillary endothelial cells (EC) within the central nervous system take on enhanced roles in homeostasis providing gatekeeper function for entry of nutrients and egress of waste [28,29]. The increased functionality brain ECs relative to peripheral ECs requires additional energy needs thus more mitochondria are present in the former [8,9,11]. The restrictive tight junctions consisting of intercellular protein strands are expressed by ECs [8,9,11]. Pericytes regulate expression of these tight junctions and are in effect transducers of signals from the glial and neuronal NVU components [8,11]. The endothelial cells are responsible for the passive ingress of oxygen, egress of CO2, lipid, and soluble molecules under four hundred Daltons or eight or less carbon bonds [8,11]. The active transport of ionic and nonionic (glucose, amino acids) small molecules and the transcytosis of selected larger molecules occurs via clathrin vesicular pathways [18] Figure 1a.
The basement membrane matrix (BMM) of the capillaries is expressed on the abluminal side of and by EC s and pericytes. A second contiguous outer layer is expressed by astrocyte end feet. The BMM connects the endothelial cells firmly to the NVU [8,11]. Further the BMM component, and integrin protein ligands, are a signal pathway from astrocyte to endothelial cell [30,31]. This intimate pathway signals the release of growth factors and receptors, influences cell survival, migration, and polarity [30,31].
On the luminal side of endothelial cells glycocalyx is secreted consisting of proteoglycans and heparan sulfate which are negatively charged thus repelling negatively charged plasma as well as blood cells from entry into the neuropil [32]. Further, heparan sulfates inhibit intravascular coagulation. In the presence of hypertension, the glycocalyx transduce sheer force energy, signaling the nitrous oxide synthetase to produce vasodilator nitrous oxide[32]. The subsequent effect is vasodilatation but in addition ROS species are generated causing upregulation of metalloprotease enzymes with deleterious effects on tight junctions resulting in leaks [32]. The glycocalyx thus adds the first layer of protection of the BBB and its integrity effects the BBB stability [32].
Pericytes
Capillary endothelial cells are encased by pericytes which regulate them. Pericytes express PDGF receptors with the cells migrating embiologically to endothelial cells which express PDGF [7,8]. The two cell types become intimately connected via a ‘peg and socket’ arrangement which allows for ease of signaling between them [11]. The ratio of endothelial cells to pericytes in the CNS is 1:1 demonstrating the importance of the relationship for continued maintenance of the TJ and expression of transporters and transcytosis [7,8,11]. Their basement membrane and the BM of the surrounding astrocyte end feet serve as the conduit defined as the glymphatic space which is thought to manage ≈40% of NVU metabolic waste egress[]. In addition, they secrete important protein components incorporated in the tight junctions [11]. Pericytes also are necessary for aquaporin 4 channel expression in the astrocyte end feet tethered together by laminin proteins [33]. Further pericytes express a variety of ionic and solute transporters but their exact role is unknown [7,8,11]. Of importance though is their expression of Lipoprotein receptor LRP1 which facilitates uptake and clearance of β amyloid. Unlike APOE 2 > 3 isoforms which bind preferentially to LRP1 preventing upregulation of CypA and upregulation of MMP-9, which if activated, degrades BBB tight junctions, APOE 4 does not [8,11]. The latter isoform allows for accumulation of intracellular β amyloid in pericytes which is toxic and results in their loss disrupting the tight junctions (TJ) [8,34]. With damage to the BBB, pericytes are an early casualty which causes loss of polarization of the aquaporin channels (return to the soma from end feet). The loss of pericytes, and Aquaporin channels in Astrocyte foot processes plus leaking TJs halts the glymphatic clearance of waste pathway [7,8]. With the aging process, there is loss of capillary pericyte: the mechanism is unknown [35,36,37].
Tight junctions
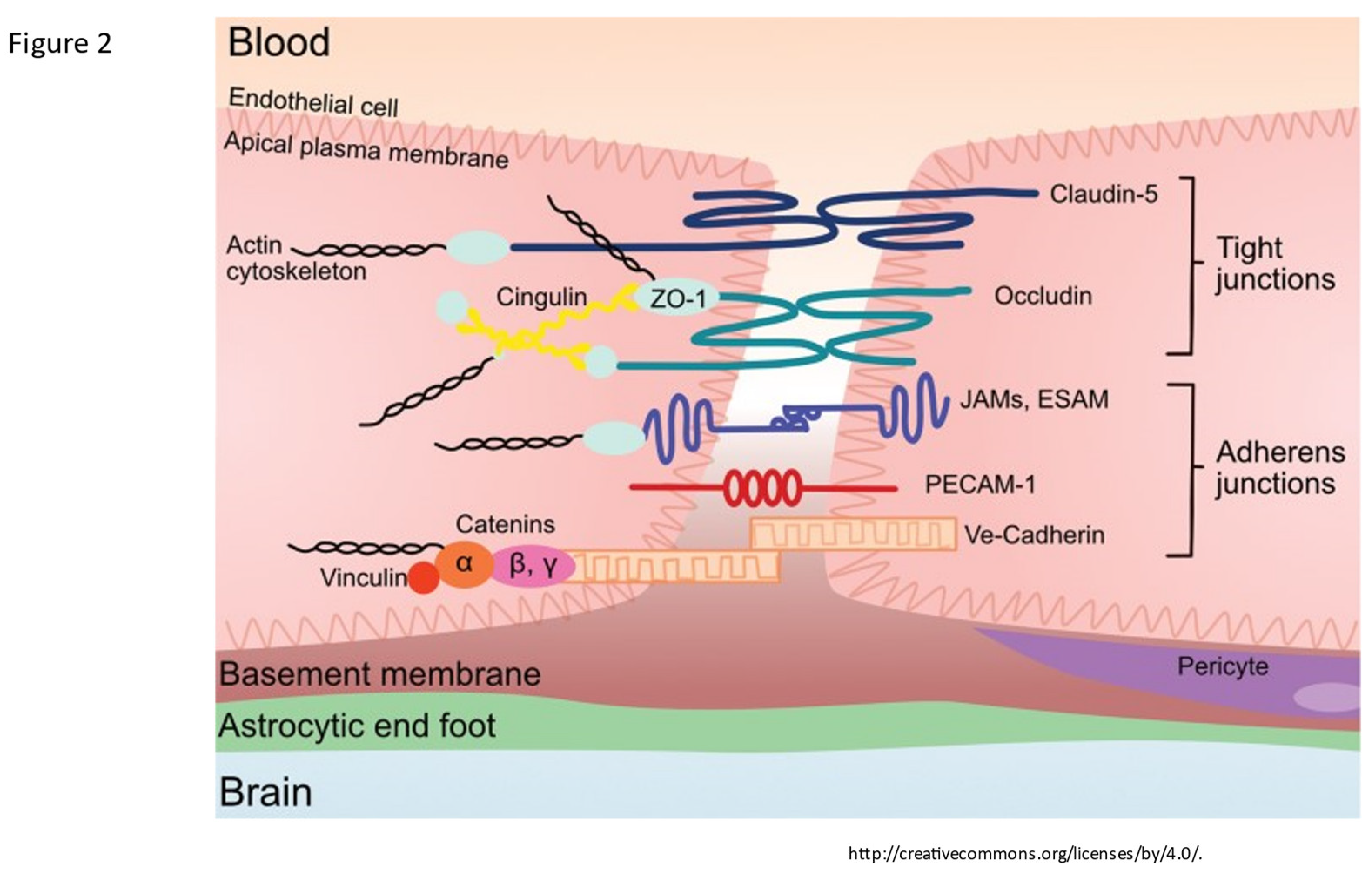
The endothelial cells express cellular adhesion molecules and junctional proteins between endothelial cells which function as a fine mesh and electrical barrier and effectively reduces entry of unwanted small molecules and cells [21,22]. Starting on the luminal side of the endothelial cell junctions, Cadherin, PECAM 1 and other junctional proteins limit ingress of leukocytes and allow for inter-endothelial cell signaling [21,22]. Transcellular junctional proteins of importance expressed by endothelial cells include transmembrane protein claudin 5 and occludins anchored to endothelial myosin filaments by intracellular Zona occludins which restrict paracellular ingress of ions and solutes [21,22]]. The endothelial adhesion protein expression is upregulated by pericytes via signaling from angiopoietin release thus inducing tight junction formation [8]. Astrocytes also express junctional proteins CX30/43 further strengthening the cell-to-cell adhesion [8]. Release of pro-maintenance retinoic acid, (SHH and angiopoetin-1 enhances endothelial BBB maintenance [23]. In the presence of inflammation, microglia convert to proinflammatory Type 1 cells which express C3a which alters the phenotypic expression of endothelial cells thereby retracting the tight junctions (see below) [8,35,36,37]. Figure 2
Figure 2.
The tight junction proteins include claudin-5, occludin, and zonula occludins (ZO-1,2,3). Claudin-5 and occludin are both transmembrane proteins while the zonula occludens are intracellular proteins. The adherens junctions include transcellular components, JAMs, ESAM, PECAM-1, and Ve-cadeherin. The cytoplasmic catenins form a complex with Ve-cadeherin. Actin cytoskeleton helps to anchor the junctional proteins in endothelial cells. Knox, Emily G., et al. "The blood-brain barrier in aging and neurodegeneration." Molecular psychiatry 27.6 (2022): 2659-2673.
Figure 2.
The tight junction proteins include claudin-5, occludin, and zonula occludins (ZO-1,2,3). Claudin-5 and occludin are both transmembrane proteins while the zonula occludens are intracellular proteins. The adherens junctions include transcellular components, JAMs, ESAM, PECAM-1, and Ve-cadeherin. The cytoplasmic catenins form a complex with Ve-cadeherin. Actin cytoskeleton helps to anchor the junctional proteins in endothelial cells. Knox, Emily G., et al. "The blood-brain barrier in aging and neurodegeneration." Molecular psychiatry 27.6 (2022): 2659-2673.
Astrocytes
Astrocytes also contribute to the BBB via their contribution in forming the outer basement membrane layer of glia limitans by secretion of basement membrane proteins. This double layer of basement membrane serves as a second waste egress or glymphatic pathway [38]. The astrocyte end feet then surround the basement membrane and is thus involved in the ingress and egress of solutes, ions, lipids, waste [38,39]. By monitoring and regulating neuronal metabolic activity, astrocytes signal the ECs required transporter expression for importation of needed substrates from the blood [8,38,39]. Water balance is also regulated via the end foot AQ 4 channels.
Microglia
The anti-inflammatory M2 microglia are homeostatic and protective of the BBB by providing anti-inflammatory cytokines, glucocorticoids, and matrix proteins . Their conversion to proinflammatory cell (type 1) when stimulated by either the ROS cascade or vascular or intrinsic borne cytokines upregulates expression of inflammatory chemokines, cytokines and complement with release of the C3a fragment and subsequent phenotypic conversion of endothelial cells [40]. They convert ECs from homeostatic phenotype to an in effect immune attractant cell. As part of their conversion, the TJs are retracted thus allowing inflammatory cells and substrates to enter the interstitium [41]. The effect of complement activation within the CNS is a clear contributor to accelerating continued BBB dysfunction and leak.
BBB dysfunction
This extraordinarily complex barrier requires the full unfettered interaction of all the above-mentioned elements for maximal protection and maintenance of brain function. The barrier likewise must have sustained resilience to constantly maintain function against the constant daily barrage of endogenous and exogenous insults. At the same time with overwhelming infection or injury the system must allow access of inflammatory cells and solute [34]. The balance in youth is disrupted by traumatic brain injury (TBI), inflammation or infection but with resolution there is spontaneous repair [42]. This balance begins to fail with the aging process allowing a slow leak and a diminished repair capacity [36]. The pathways of restoration following low level sustained or more significant injury are incompletely understood and clearly wane in advanced age [36,37]. Identified elements include age related low-level inflammation or TBI with reduction in pericytes and upregulation of in situ inflammation resulting in leaky capillaries and microvascular anatomic disruption. Additionally impaired glycocalyx protection and upregulation and circulation of inflammatory proteins systemically and within the neuropil enhance the loss of BBB integrity [43,44,45]. There is consequent loss of pericyte homeostatic signaling results in reduced endothelial expression of junctional proteins and increased leakiness [36,37]. This provides a pathway for indiscriminate entry into the interstitium of inflammatory cells , and normally restricted substances (fibrin, albumin, thrombin, hemoglobin, hemosiderin, immunoglobulins, free iron, plasmin) [8,36,37]. Further there is loss of basement membrane integrity with consequent dysfunction of glymphatic flow, and reduced communication with the astrocyte-pericyte-endothelial signaling impairing proper absorption of required solutes, ions lipids, and proteins [44,45,46]. Post-endothelial injury, these cells employ Caveolae transcytosis pathway in lieu of the homeostatic Cathrin pathway, the former being less discriminant in transporting appropriate proteins and lipids into the interstitium thus adding more potential toxins [8,11]. Finally, astrocytes morph into proinflammatory phenotype and pile on with expression of Vascular endothelial growth factor (VEGF), matrix metalloprotease enzymes (MMPs) and endothelin which add to loss of BBB integrity and normal NVU signaling [46].
In youth, the WNT/βCatenin signaling pathway from astrocytes to endothelial structures enhances expression and restoration of TJ proteins, restores transporter expression, and restores the homeostatic use of the clathrin transcytosis pathway [27]. The WNT/ βCatenin pathway is enhanced by endothelial, pericyte or astrocyte Netrin activated endothelial Unc5b receptors [27]. Whether this is the only general restoration path restoring BBB integrity for all varieties of insults causing loss of BBB integrity is unknown.
In broad strokes the main culprits affecting BBB integrity are blunt trauma, inflammation, viral or bacterial infection and vascular injury. The causes are not mutually exclusive but are often co-occurring. The BBB targets of each of these insults varies, and as such the initial downstream damage whether it is vascular, metabolic or both may vary. We will explore each of the main culprits individually. Figure 3
Head trauma causes biphasic injury with the initial blunt force trauma initially disrupting normal BBB maintenance by its affect on the cellular and membranous components of the NVU[42]. Specifically, loss of pericytes and their resulting effect on loss of tight junction integrity and altered NVU signaling cause cerebral edema, and leakage of solute and cellular components [42]. The second later phase is inflammatory which yields a double hit to the BBB. Influx of cytokines, inflammatory cells, and DAMPS from innate immune system and PAMPS from fragments of viral, bacterial, or fungal elements add additional secondary BBB injury [42,47]. Both mechanisms alter the normal perfusion dynamics. In a recent study we conducted evaluating college athletes post-mild acute traumatic brain injury using ASL MRI technique (discussed below), we were able to identify reduced capillary mean transit time (cMTT)/ glymphatic clearance rate acutely and demonstrated return to normal clearance at recovery [48]. Thus, a restoration pathway exists in youth at least with minor TBI but becomes dysfunctional with repetitive trauma leading to CTE and in ”normal” aging adults [31,33]. With future clearer understanding of the repair mechanisms in youth, restorative treatments could potentially be developed and applied to advanced age-related BBB leakiness.
With age, low grade inflammatory insults via leaked in and subsequent glial upregulation of inflammatory interleukin cytokines IL1b, IL6, IL-12, IL-23, TNF-α), monocyte/lymphocyte attracting chemokines CCL2-CCL4 , CCL7, leukocyte attracting CXCL10. Microglial cells convert to Type 1 with expression of complement components C1q, C3a with inflammation or presence of PAMPS from prior/ dormant viral encephalitis (HSV-1 , HHV6, HHV7, EB virus), or chronic infection (periodontal disease, chlamydia, fungi) [49,50,51,52,53,54,55,56].
Additionally, diabetes, obesity, autoimmune inflammatory processes, whole brain radiation treatments, tobacco abuse, and trauma impacts the BBB TJs [32,42,47] . The expression of C3a fragment of complement and upregulate the endothelial C3a receptor which causes profound phenotypic endothelial cellular changes [54,56]. The endothelial cells transform into immune permissive/attractant cells with retraction of the TJ proteins via cytoskeletal actin contraction causing leaks as well as expression of VCAM1 attracting CD8 cells to the cell surface and entry into the interstitium with inflammation [54]. Here it should be noted that there is a substantial contribution to the upregulation of interstitial inflammation by complement prevalent in microglia and astrocytes [54,56]. If low level leakiness is sustained the resulting altered metabolic processes longer term cause corruption of protein synthesis and degradation with accumulating misfolded toxic proteins, and microvasculopathy in the case of neurodegenerative diseases [30,31,32,33,34,46]. The rapidity of development and severity of misfolded protein accumulation is enhanced by well know genetic genotypes most commonly APOE 4 carriers [3,4,5,8,9]. Figure 4
The enterobiome is also a factor increasingly being investigated in development of neurodegenerative disease with clear involvement in Parkinson’s disease [60,61]. Enterobiome production of α synuclein protein with ascent and brain stem incorporation via Vagal afferents is well documented [60]. Bacterial derived Lipopolysaccharide (LPS) and High mobility group box 1 (HMGB1) a nonhistone nucleoprotein which normally has intracellular maintenance function, once excreted becomes a potent proinflammatory substance once extracellular [62]. LPS is a classic PAMP and HMGB1 a classic DAMP, with resultant upregulation of inflammation and have been shown to induce BBB leak [49,62].
Vascular insult related to sustained hypertension causes vascular shear stress, damage to glycocalyx resulting in reduced heparin sulfate and proteoglycan content with loss of antithrombotic and anti-inflammatory effects. The damage that ensues is produced by accumulating reactive oxygen species with resultant upregulation of metalloproteases and damage to the BBB as well as upregulation of inflammatory cytokines [33,34,35,36,37,63]. If severe or sustained each of these injuries cause structural alterations to the microvasculature with thinning , elongation, and increased tortuosity of the affected vessels due to upregulation of metalloprotease enzymes related to increased oxidative stress and inflammatory changes [63]. The normal regional perfusion of the brain region is thus altered such that there is reduced blood flow, prolonged mean capillary transit time and glymphatic flow impairment [64]. These same vascular changes develop in both head injury and chronic inflammatory related BBB leak in absence of normal repair signaling/or pathways [12,65,66]. Figure 5
String vessels (black) with no blood cells form as endothelial cells undergo apoptosis. Neutrophils (blue dots) block a small portion (∼2%) of capillaries, reducing blood flow in the network. Penetrating vessels have a tortuous shape, with a tendency to decrease in diameter with aging in AD. (Dorr, Adrienne, et al. "Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease." Brain 135.10 (2012): 3039-3050.)
Histopathology shows vascular abnormalities such as tortuous arterioles and string vessels (arrows) in the AD brain. Celloidin sections were stained for collagen [215]. c Scanning electron microscopy of corrosion casts of the cerebral vasculature of an arcAbeta mouse revealed degenerated vessels (arrow). In addition, stenosis and bulging of the vessel wall are observed both on arteries and arterioles (asterisks). Scale bar 50μm. Modified from [31]. d Table with effects of changes in vessel morphology on biomechanical properties. This article is licensed under the Creative Commons Attribution-Noncommercial-No Derivatives 4.0 International License (CC BY-NC-ND). Klohs, Jan. "An integrated view on vascular dysfunction in Alzheimer’s disease." Neurodegenerative Diseases 19.3-4 (2020): 109-127.
The ”normal” aging process is associated with loss of pericytes cause unknown with low level leakiness of the BBB [12,65,66]. Is their loss the consequence or cause of BBB leak? There is also thickening of the endothelial basement membrane and alteration in constituent proteins along with loss of pericytes and presence of reactive astrocytes and microglia reducing neurovascular coupling [30,31,32,33,34,35]. The BBB leakiness goes undetected for years until there is mild loss of cognitive abilities [58,66]. Progression to more severe dementia is not a certainty with progression over 10 years in about 40% of patients. Modifiable risk factors for developing cognitive impairment include uncontrolled hypertension, obesity, poorly controlled diabetes, TBI, tobacco and excess alcohol use are well recognized and listed with level of evidence in the combined WHO and 2020 Lancet Commission Report [67,68] Table 1.
Insulin resistance has also been identified, but whether it is a cause or result of endothelial cells dysfunction with decreased glut 1 transporter expression is unclear [8]. Those carrying 1 copy of APOE 4 have a 40% greater chance of developing AD dementia progression and 2 copies 10 times increased risk of early age or late age AD development and is currently non-modifiable [67,68]. In addition, the potential exists to therapeutically target the dysfunctional BBB by inhibiting the microglial and astrocyte conversion to their proinflammatory phenotype, inhibit the astrocyte secreted inflammatory species and ROS activated metalloprotease enzyme systems , and promote expression of the cadherin proteins at the BBB [69]. Management of the early neurodegenerative disease development will require repair of the blood brain barrier dysfunction. Before we tackle this, more robust delineation of BBB repair mechanisms, operational in youth, is required which can potentially be upregulated in age related leakiness.
Direct intraarterial vascular accumulation of βamyloid and resulting pro-hemorrhage and vasoocclusive effect has been addressed elsewhere and will not be addressed here [70,71].
This mounting evidence for BBB dysfunction being the nexus of neurodegenerative disease development can no longer be ignored. The co-occurrence of microvascular disease in addition to the interstitial metabolic inflammatory changes in most patients suggests they are 2 prongs of the same disease process and likely develop concurrently [41,43]. The accelerant factor of the ongoing accumulation of toxic misfolded proteins results in progressive expansion of the BBB dysfunction and ultimately loss of neurons and supporting structures with the well-known cognitive impairments resulting [46,58]. The pattern of involvement typically begins in the highly metabolically active hippocampal structures but with pertinent exceptions such as Frontal temporal, Parietal, and rarely Occipital lobe presentations clinically [35].
The cooccurrence of both “small vessel disease” (by MRI) in the absence of large vessel atherosclerosis and associated neurodegeneration with accumulation of βamyloid hpTau. as seen almost universally in neurodegenerative disease strongly suggests the two processes are a spectrum of the same disease process [33,34]. The nexus is the loss of stability/integrity of the BBB Figure 4. If this is so, then the physiologic consequences on perfusion must be measurable in the early preclinical stages of disease. This produces clinical challenges for early recognition, surveillance, and a major change in therapeutic focus to addressing potential means of restoring BBB integrity. The next section will discuss the early BBB leak and potential noninvasive means of detection. Without valid safe methods of BBB leak identification outcomes from future early intervention trials is impossible in a reasonable time frame, as cognitive decline may take years to develop.
Altered Fluid Dynamic
Hemodynamic flow is altered regionally post BBB leak [72,73]. The mechanism is related to the microvascular/glymphatic flow anatomic changes alluded to above. There is reduced blood volume in the affected areas but in the absence of concomitant large or medium size arterial disease the mean arterial transit time (aMTT) remains normal [57,64]. Just as a leaky garden hose reduces transit time of water out its end when compared to an intact one, so does a leaky BBB alter regional mean capillary transit times (cMTT) [57,64]. In addition, the disruption in the BBB reduces both signaling from astrocytes to endothelial cells of its metabolic needs and thus reduced expression of required transporters and vesicular pathways [57,64]. This cascade is the result of glycocalyx and pericyte loss, basement membrane damage, and metabolic disruption caused by extravascular leak of restricted substances [32]. The basement membrane damage shuts down the associated glymphatic system due to retraction of astrocyte aquaporin channels from end feet to soma. Put simply, with BBB dysfunction reduced volume of fluid is slow to arrive and cannot get out. The pathologic sequalae allows for persistent smoldering of inflammatory processes that down stream over time results in well recognized misfolded protein accumulations associated with neurodegenerative disease [3,4,5,6,7,8,33].
Early Identification of BBB Dysfunction
Redirecting clinical research efforts to remedy the preclinical BBB dysfunction will require validated outcome measures to assess efficacy. A combination of imaging techniques addressing the leak or resulting physiologic consequences coupled with serologic as opposed to CSF evidence of brain injury would be ideal. Paradigms testing cognitive dysfunction/function are abnormal late in the disease course at a time when treatment is probably futile. So, in order to demonstrate positive effect of future early treatments, outcome measures addressing the effects of BBB leak must be validated. To identify early BBB disruption, an imaging study should either survey the vascular leak or reflect the altered physiology present. The leak in can be directly imaged using dynamic contrast enhancement (DCE) MRI imaging. BBB leak has been identified in aging adults without cognitive impairment localized initially in the hippocampal region by DCE-MRI [35,66,74]. This technique measures gadolinium contrast extravasation into the brain parenchyma relative to the arterial concentration in specific brain regions. Most notably the Hippocampal region and sub regions within were found to demonstrate quantifiable contrast leak [35]. Figure 6 This technique uses the Patlak analysis to identify subtle leak of contrast. The technique requires gadolinium contrast injection with scan times currently of 25-30 minutes. Faster scan methods are under development. Limitations for this technique are long scan times per sequence and required gadolinium making multiple follow up studies problematic.
Another approach for early identification of BBB leak in the preclinical and early clinical state of disease leverages the regional hemodynamic changes resulting from disruption in normal microvascular anatomy and leaky vessels [46,64,66]. The disruption in neurodegenerative disease causes reduced cerebral vascular flow volume but does not result in changes in the arterial mean transit time (aMTT) [64,65,66]. The mean capillary mean transit time (cMTT) however is prolonged and glymphatic outflow reduced. Noninvasive 3D ASL MRI indirectly assesses clearance of labeled protons in the late stage of perfusion by assessing perfusion signal at multiple time points post labeling (PLD) and, using linear analysis, measures the slope of clearance [75]. This method identifies perfusion alterations resulting from BBB leak, before the accumulation of β Amyloid or hpTau. In our effort to develop a usable MRI technique for identifying perfusion changes in AD, three specific goals had to be met. The first was availability of the technique to any community hospital with a 3T MRI scanner. The second was it had to be time efficient and noninvasive since multiple scans would likely be required over time, hence a non-contrast study was imperative. The third was cost efficiency. The medical system here or elsewhere cannot absorb high volume high-cost diagnostic studies in a pervasive disease of this sort.
This technique uses 3D arterial spin labeling capturing the signal averages at multiple time points late in the perfusion cycle. The technique subtracts background signal leaving residual labeled proton signal from perfusion. The paradigm records residual signal after time delays post labeling beginning at 2800 ms through 4000 ms at 200ms intervals (7 data points) [75]. By doing so we can legitimately use linear analysis for “glymphatic clearance rate” as the correlation with the T1 decay times of the constituent components in the late phase of perfusion have over as 96% correlation coefficient [75]. Signal averages in 6 regions of interest , bitemporal, bifrontal, and biparietal are investigated using standard volume and anatomic locations [75]. Figure 7
The resultant signal averages are then easily transferred to a spread sheet for graphical analysis the slope being the glymphatic clearance rate. This technique fulfills our major requirements in that it is non-invasive, time efficient (about 2 minutes per ASL sequence and with T2 flair and susceptibility imaging 20-minute total scan time) and cost efficient (about $300 Medicare reimbursement Quote from Medicare website). In a small case series report (COVID interrupted), we were able to demonstrate progression in one subject with MCI to precursor MRI changes of more advanced dementia prior to significant MMSE changes, a second patient with stable MRI changes and no cognitive decline and a third patient with typical AD dementia and associated ASL MRI changes [76]. The technique is still in the development stage but will be enhanced with the general release of 3D ASL acquiring multiple PLD determinations following a single spin labeling. Also, the segmentation programs should speed up the cumbersome data analysis by ROI. The limitations of ASL is that low level of signal obtained in the late phases of perfusion. By using multiple PLDs and large ROI’s, artifact is reduced.
It is important to note that both directly imaging BBB leak of contrast and the determination of clearance by ASL are not specific for neurodegenerative disease, but can be found in acute head injury, CNS infection, brain tumor, acute stroke or in other words, in any condition which alters the integrity of the BBB. That said given the clinical suspicion of early disease or high-risk profile for developing neurodegenerative disease, surveillance in the future when effective means of aborting the BBB damage is available, will be necessary.
Serologic markers of NVU injury would enhance the sensitivity of the testing. GFAP is a sensitive measure of β Amyloid accumulation and thus helpful in identifying mid stage disease [14,15]. Small acidic calcium-binding protein S100β is a sensitive but nonspecific marker of BBB leak [14]. The 2 markers may serve to corroborate imaging findings in the early phase of disease (S100β). GFAP concentration if elevated would suggest disease progression along with imaging study changes. Hence the concomitant use would provide a convenient and sensitive noninvasive means of diagnosis and surveillance of initial stages of neurodegenerative disease.
From a clinician/patient standpoint, sensitive early detection using noninvasive MRI technology with a serologic marker of brain injury to establish the preclinical diagnosis would be a welcome advance particularly when effective early treatment exists.
Current methods of risk reduction
There is unambiguous evidence that smoking cessation, successfully managing diabetes, hyperlipidemia, hypertension, and obesity risk of late life dementia is reduced [77]. Likewise treating hearing impairment in middle age, reduction in alcohol use to no more than 21 U of alcohol (1unit =10cc alcohol) [68]. Education level less than high school (12 years formal education) is associated with higher dementia risk[]. Increasing aerobic exercise to 45-60 minutes per session at any frequency reduced the incidence of dementia [67,68]. Food supplements are of no value in preventing dementia , but a healthy diet (Mediterranean diet) was indirectly beneficial by reducing cardiovascular risk. Social isolation and depression are also risk factors, with reduced (<4hrs/night) or excessive sleep (>10 hrs sleep) an additional risk. Table 1 Formal WHO 9-point risk factors with added 3 additional risk factors of excessive alcohol consumption, TBI and possibly air pollution added by the Lancet study group. A phase 1 trial using Senolytic therapy has been put forward as a potential treatment in alleviating cognitive deficits in AD [78,79].
To that end, early identification of patients at risk related to chronic poorly controlled diabetes, hypertension, chronic infection, identifiable enterobiome factors, genetic predisposition, multiple head traumas will require surveillance for BBB leak at an early age and on a periodic basis. Once treatment/s are initiated then continued surveillance will be required to assure reversal/ cessation of BBB leakiness and cognitive change. Figure 8 Cheap, noninvasive, time efficient and validated diagnostic testing will be mandatory. In short, once early successful treatment is available, a major shift in clinical thinking must take place specifically, we will need to find it before it finds us!
Figure 8 Flow diagram of potential future approach to preclinical diagnosis and surveillance of high-risk individuals for developing AD once effective treatment paradigms are available.
Conclusion- where do we go from here?
An innovative approach to early diagnosis and preemptive treatment of neurodegenerative disease based on recognition and mitigation of blood brain barrier dysfunction, the nexus of these diseases must be addressed. AD is highly likely a spectrum disease causing both microvasculopathy and altered protein synthesis and degradation with accumulating toxic misfolded proteins. The latter events accelerate the BBB dysfunction producing a vicious cycle with loss of neural components and ultimately cognitive decline. The disappointing effect of removing accumulating misfolded proteins (β amyloid) demonstrates the need to intervene early but address a different target, specifically repairing the BBB leakiness.
Step 1: For now, reducing the peripheral inflammatory load (multiple sources) and control of vascular/glycocalyx shear forces from uncontrolled hypertension are essential thereby reducing the burden on the age-related leaking BBB. Middle age treatment of hypertension, obesity, type 2 diabetes, and depression, smoking cessation, avoidance of high-risk TBI activities and reduced alcohol use should be carefully explained to patients in terms of reducing risk of long-term cognitive loss. Social interaction, exercise, hearing aids when required, and cognitive stimulation are all positive influences on maintaining intellectual function.
Step 2 is leveraging the body’s natural BBB repair mechanism that is slowly lost with the aging process. To move in this direction will require firmer knowledge of the repair signaling pathways that are present in youth but begin to slowly fail with age. Once this is clarified, a target/s for therapeutic intervention can be exploited. Validated, non-invasive, economically sound MRI/serologic methods will need to be in place to both make pre-clinical diagnosis and judge efficacy of future treatment outcomes.
Funding
No external grant funding was provided outside of intramural publication funding by LUCOM. The author has no conflicts of interest.
Acknowledgements
I would like to thank Riley Lutz for his time and attention adapting Figure s 1, 2, 4, and 5. I would also like to thank Dr. Carl Hoegerl for reviewing this manuscript. His suggestions were extremely helpful.
References
- Van Dyck, Christopher H., et al. "Lecanemab in early Alzheimer’s disease." New England Journal of Medicine 388.1 (2023): 9-21. [CrossRef]
- Bateman, Randall J., et al. "Two Phase 3 Trials of Gantenerumab in Early Alzheimer’s Disease." New England Journal of Medicine 389.20 (2023): 1862-1876. [CrossRef]
- Huang, Zhangsen, et al. "Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease." Frontiers in neuroendocrinology 59 (2020): 100857. [CrossRef]
- Barisano, Giuseppe, et al. "Blood–brain barrier link to human cognitive impairment and Alzheimer’s disease." Nature cardiovascular research 1.2 (2022): 108-115.
- Hussain, Basharat, Cheng Fang, and Junlei Chang. "Blood–brain barrier breakdown: an emerging biomarker of cognitive impairment in normal aging and dementia." Frontiers in neuroscience 15 (2021): 688090. [CrossRef]
- Nehra, Geetika, Bjoern Bauer, and Anika MS Hartz. "Blood-brain barrier leakage in Alzheimer’s disease: From discovery to clinical relevance." Pharmacology & Therapeutics 234 (2022): 108119. [CrossRef]
- Sweeney, Melanie D., et al. "The role of brain vasculature in neurodegenerative disorders." Nature neuroscience 21.10 (2018): 1318-1331. [CrossRef]
- Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Bloodbrain.
- barrier: from physiology to disease and back. Physiol Rev.2019;99:21–78. [CrossRef]
- Andjelkovic, Anuska V., et al. "Blood-brain barrier dysfunction in normal aging and neurodegeneration: mechanisms, impact, and treatments." Stroke 54.3 (2023): 661-672. [CrossRef]
- Selkoe, Dennis J. "Treatments for Alzheimer's disease emerge." Science 373.6555 (2021): 624-626. [CrossRef]
- Castro Dias, Mariana, et al. "Structure and junctional complexes of endothelial, epithelial and glial brain barriers." International journal of molecular sciences 20.21 (2019): 5372. [CrossRef]
- Zhang, Qianqian, et al. "Altered regional cerebral blood flow and brain function across the Alzheimer's disease spectrum: a potential biomarker." Frontiers in Aging Neuroscience 13 (2021): 630382. [CrossRef]
- Thrippleton, Michael J., et al. "Quantifying blood-brain barrier leakage in small vessel disease: review and consensus recommendations." Alzheimer's & Dementia 15.6 (2019): 840-858. [CrossRef]
- Sun, Huixin, et al. "Methods used for the measurement of blood-brain barrier integrity." Metabolic brain disease 36 (2021): 723-735. [CrossRef]
- Pereira, Joana B., et al. "Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease." Brain 144.11 (2021): 3505-3516. [CrossRef]
- Kadry, Hossam, Behnam Noorani, and Luca Cucullo. "A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity." Fluids and Barriers of the CNS 17.1 (2020): 1-24.
- Zhao, Lucy, et al. "Physiology of glymphatic solute transport and waste clearance from the brain." Physiology 37.6 (2022): 349-362. [CrossRef]
- Preston, Jane E., N. Joan Abbott, and David J. Begley. "Transcytosis of macromolecules at the blood–brain barrier." Advances in pharmacology 71 (2014): 147-163.
- van Leeuwen, E., Hampton, M. B., and Smyth, L. C. D. (2020). Redox signaling and regulation of the blood-brain barrier. Int. J. Biochem. Cell Biol. 125:105794. [CrossRef]
- Proulx, Steven T. "Cerebrospinal fluid outflow: a review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics." Cellular and Molecular Life Sciences 78.6 (2021): 2429-2457.
- Lochhead, Jeffrey J., et al. "Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders." Frontiers in physiology 11 (2020): 914. [CrossRef]
- Hudson, Natalie, and Matthew Campbell. "Tight junctions of the neurovascular unit." Frontiers in Molecular Neuroscience 14 (2021): 752781. [CrossRef]
- Engelhardt, Britta, and Stefan Liebner. "Novel insights into the development and maintenance of the blood–brain barrier." Cell and tissue research 355.3 (2014): 687-699. [CrossRef]
- Iadecola, Costantino. "The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease." Neuron 96.1 (2017): 17-42. [CrossRef]
- Bhowmick, Saurav, et al. "Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury." Experimental neurology 317 (2019): 260-270. [CrossRef]
- Brassard, Patrice, et al. "Losing the dogmatic view of cerebral autoregulation." Physiological reports 9.15 (2021): e14982. [CrossRef]
- Boyé, Kevin, et al. "Endothelial Unc5B controls blood-brain barrier integrity." Nature Communications 13.1 (2022): 1169. [CrossRef]
- Santisteban, Monica M., et al. "Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension." Hypertension 76.3 (2020): 795-807. [CrossRef]
- Roudnicky, Filip, et al. "Inducers of the endothelial cell barrier identified through chemogenomic screening in genome-edited hPSC-endothelial cells." Proceedings of the National Academy of Sciences 117.33 (2020): 19854-19865. [CrossRef]
- Profaci, Caterina P., et al. "The blood–brain barrier in health and disease: Important unanswered questions." Journal of Experimental Medicine 217.4 (2020). [CrossRef]
- Knox, Emily G., et al. "The blood-brain barrier in aging and neurodegeneration." Molecular psychiatry 27.6 (2022): 2659-2673. [CrossRef]
- Yang, Rui, et al. "The Role of Heparin and Glycocalyx in Blood–Brain Barrier Dysfunction." Frontiers in Immunology 12 (2021): 754141. [CrossRef]
- Lendahl, Urban, Per Nilsson, and Christer Betsholtz. "Emerging links between cerebrovascular and neurodegenerative diseases—a special role for pericytes." EMBO reports 20.11 (2019): e48070. [CrossRef]
- Searson, Peter Charles, et al. "The influence of physiological and pathological perturbations on blood-brain barrier function." Frontiers in Neuroscience 17 (2023): 1289894. [CrossRef]
- Montagne, Axel, et al. "Blood-brain barrier breakdown in the aging human hippocampus." Neuron 85.2 (2015): 296-302. [CrossRef]
- Banks WA, Reed MJ, Logsdon AF, Rhea EM, Erickson MA. Healthy aging and the blood–brain barrier. Nat Aging. 2021;1:243–54.
- Verheggen, Inge CM, et al. "Increase in blood–brain barrier leakage in healthy, older adults." Geroscience 42 (2020): 1183-1193.
- Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., … Kipnis, J. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature. [CrossRef]
- Mestre, Humberto, Yuki Mori, and Maiken Nedergaard. "The brain’s glymphatic system: current controversies." Trends in neurosciences 43.7 (2020): 458-466.
- Ronaldson, Patrick T., and Thomas P. Davis. "Regulation of blood–brain barrier integrity by microglia in health and disease: a therapeutic opportunity." Journal of Cerebral Blood Flow & Metabolism 40.1_suppl (2020): S6-S24. [CrossRef]
- Iturria-Medina, Yasser, et al. "Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis." Nature communications 7.1 (2016): 11934. [CrossRef]
- Cash, Alison, and Michelle H. Theus. "Mechanisms of blood–brain barrier dysfunction in traumatic brain injury." International journal of molecular sciences 21.9 (2020): 3344. [CrossRef]
- Furman, David, et al. "Chronic inflammation in the etiology of disease across the life span." Nature medicine 25.12 (2019): 1822-1832. [CrossRef]
- Katsi, Vasiliki, et al. "Blood–brain barrier dysfunction: the undervalued frontier of hypertension." Journal of Human Hypertension 34.10 (2020): 682-691. [CrossRef]
- Lucas, Samuel JE, et al. "Influence of changes in blood pressure on cerebral perfusion and oxygenation." Hypertension 55.3 (2010): 698-705. [CrossRef]
- Klohs, Jan. "An integrated view on vascular dysfunction in Alzheimer’s disease." Neurodegenerative Diseases 19.3-4 (2020): 109-127. [CrossRef]
- Vázquez-Rosa, Edwin, et al. "P7C3-A20 treatment one year after TBI in mice repairs the blood–brain barrier, arrests chronic neurodegeneration, and restores cognition." Proceedings of the National Academy of Sciences 117.44 (2020): 27667-27675. [CrossRef]
- Kang, Jubin, et al. "Delay in Clearance of Labeled Protons in Patients with Acute Head Trauma Utilizing MRI 3D TGSE PASL MRI (Arterial Spin Labeling)(P8-1.007)." (2023). [CrossRef]
- Vigasova, Dana, et al. "Multi-pathogen infections and Alzheimer’s disease." Microbial Cell Factories 20.1 (2021): 1-13.
- Itzhaki, Ruth F., et al. "Do infections have a role in the pathogenesis of Alzheimer disease?." Nature Reviews Neurology 16.4 (2020): 193-197. [CrossRef]
- Jacob, Alexander, and Jessy John Alexander. "Complement and blood–brain barrier integrity." Molecular immunology 61.2 (2014): 149-152. [CrossRef]
- Li, Zhilian, et al. "Correlation of serum complement factor 5a level with inflammatory response and cognitive function in patients with Alzheimer’s disease of different severity." BMC neurology 23.1 (2023): 319.
- Eadon, M.T., Jacob, A., Cunningham, P.N., Quigg, R.J., Garcia, J.G., Alexander, J.J. Transcriptional profiling reveals that C5a alters miRNA in brain endothelial cells. Immunology. 2014 May 6. (epub ahead of print). [CrossRef]
- Propson, N. E., Roy, E. R., Litvinchuk, A., Köhl, J., & Zheng, H. (2021). Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. The Journal of Clinical Investigation, 131(1). [CrossRef]
- Shah, Akash, Uday Kishore, and Abhishek Shastri. "Complement system in Alzheimer’s disease." International Journal of Molecular Sciences 22.24 (2021): 13647. [CrossRef]
- Krance, Saffire H., et al. "The complement cascade in Alzheimer’s disease: a systematic review and meta-analysis." Molecular Psychiatry 26.10 (2021): 5532-5541. [CrossRef]
- Elschot, Elles P., et al. "A comprehensive view on MRI techniques for imaging blood-brain barrier integrity." Investigative Radiology 56.1 (2021): 10-19. [CrossRef]
- Nation, Daniel A., et al. "Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction." Nature medicine 25.2 (2019): 270-276. [CrossRef]
- Chang, Junlei, et al. "Gpr124 is essential for blood–brain barrier integrity in central nervous system disease." Nature medicine 23.4 (2017): 450-460. [CrossRef]
- Travagli, R. Alberto, Kirsteen N. Browning, and Michael Camilleri. "Parkinson disease and the gut: new insights into pathogenesis and clinical relevance." Nature Reviews Gastroenterology & Hepatology 17.11 (2020): 673-685. [CrossRef]
- Liu, Shan, et al. "Microbiota-gut-brain axis and Alzheimer’s disease: implications of the blood-brain barrier as an intervention target." Mechanisms of Ageing and Development 199 (2021): 111560. [CrossRef]
- Ahumada-Castro, Ulises, et al. "Keeping zombies alive: The ER-mitochondria Ca2+ transfer in cellular senescence." Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1868.11 (2021): 119099. [CrossRef]
- Takata, Fuyuko, et al. "Blood-brain barrier dysfunction amplifies the development of neuroinflammation: understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction." Frontiers in cellular neuroscience 15 (2021): 661838. [CrossRef]
- Yoshiura, Takashi, et al. "Simultaneous measurement of arterial transit time, arterial blood volume, and cerebral blood flow using arterial spin-labeling in patients with Alzheimer disease." American Journal of Neuroradiology 30.7 (2009): 1388-1393. [CrossRef]
- Binnewijzend, Maja AA, et al. "Cerebral perfusion in the predementia stages of Alzheimer’s disease." European radiology 26 (2016): 506-514. [CrossRef]
- Zheng, Weimin, et al. "Disrupted regional cerebral blood flow, functional activity and connectivity in Alzheimer’s disease: a combined ASL perfusion and resting state fMRI study." Frontiers in Neuroscience 13 (2019): 738. [CrossRef]
- Chowdhary, Neerja, et al. "Reducing the risk of cognitive decline and dementia: WHO recommendations." Frontiers in neurology 12 (2022): 765584. [CrossRef]
- Livingston, Gill, et al. "Dementia prevention, intervention, and care: 2020 report of the Lancet Commission." The Lancet 396.10248 (2020): 413-446. [CrossRef]
- Archie, Sabrina Rahman, Abdullah Al Shoyaib, and Luca Cucullo. "Blood-brain barrier dysfunction in CNS disorders and putative therapeutic targets: an overview." Pharmaceutics 13.11 (2021): 1779. [CrossRef]
- Greenberg, Steven M., et al. "Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways." Nature Reviews Neurology 16.1 (2020): 30-42.
- Jäkel, Lieke, et al. "Prevalence of cerebral amyloid angiopathy: a systematic review and meta-analysis." Alzheimer's & Dementia 18.1 (2022): 10-28. [CrossRef]
- Fazlollahi, Amir, et al. "Increased cerebral blood flow with increased amyloid burden in the preclinical phase of alzheimer's disease." Journal of Magnetic Resonance Imaging 51.2 (2020): 505-513. [CrossRef]
- Van De Haar, Harm J., et al. "Blood-brain barrier leakage in patients with early Alzheimer disease." Radiology 281.2 (2016): 527-535. [CrossRef]
- Montagne, Axel, et al. "Imaging subtle leaks in the blood–brain barrier in the aging human brain: potential pitfalls, challenges, and possible solutions." GeroScience 44.3 (2022): 1339-1351.
- Joseph, C. R., Benhatzel, C. M., Stern, L. J., Hopper, O. M., & Lockwood, M. D. (2020). Pilot study utilizing MRI 3D TGSE PASL (arterial spin labeling) differentiating clearance rates of labeled protons in the CNS of patients with early Alzheimer disease from normal subjects. Magnetic Resonance Materials in Physics, Biology and Medicine, 33(4), 559–568. [CrossRef]
- Joseph, Charles R., et al. "Utilizing Reduced Labeled Proton Clearance to Identify Preclinical Alzheimer Disease with 3D ASL MRI." Case Reports in Neurology 15.1 (2023): 177. [CrossRef]
- Mahapatra, Manoj K., Muthukumar Karuppasamy, and Biswa M. Sahoo. "Therapeutic potential of semaglutide, a newer GLP-1 receptor agonist, in abating obesity, non-alcoholic steatohepatitis and neurodegenerative diseases: a narrative review." Pharmaceutical Research 39.6 (2022): 1233-1248. [CrossRef]
- Gonzales, Mitzi M., et al. "Senolytic therapy to modulate the progression of Alzheimer’s disease (SToMP-AD): a pilot clinical trial." The journal of prevention of Alzheimer's disease (2022): 1-8.
- Zhang, P., Kishimoto, Y., Grammatikakis, I. et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22, 719–728 (2019). [CrossRef]
Figure 1.
a Compendium of normal transport mechanisms through the BBB endothelium with NVU cellular relationships. The neurovascular unit/blood–brain barrier (NVU/BBB) is composed of specialized endothelial cells and support cells, including pericytes and astrocytes. The cross-sectional view illustrates that the majority of the abluminal surface of the endothelial cell is covered by pericytes and astrocytic foot processes. Paracellular transport across the BBB/NVU is restricted by tight junction proteins, and even small, lipophilic molecules. Facilitated active transport, receptor-mediated transport, and ion transporters allow the brain to be supplied with nutrients while maintaining strict homeostasis. Adapted from Griffith, Jessica I., et al. "Addressing BBB heterogeneity: a new paradigm for drug delivery to brain tumors." Pharmaceutics 12.12 (2020): 1205. b Normal trans axial anatomy of the NVU/vascular (capillary) BBB. From the inside out the layers that form the blood–brain barrier include the endothelial expressed glycocalyx, specialized endothelial cells, double layer endothelial and astrocyte expressed basement membrane with glymphatic space sandwiched between the layers, pericytes, and astrocytes.
Figure 1.
a Compendium of normal transport mechanisms through the BBB endothelium with NVU cellular relationships. The neurovascular unit/blood–brain barrier (NVU/BBB) is composed of specialized endothelial cells and support cells, including pericytes and astrocytes. The cross-sectional view illustrates that the majority of the abluminal surface of the endothelial cell is covered by pericytes and astrocytic foot processes. Paracellular transport across the BBB/NVU is restricted by tight junction proteins, and even small, lipophilic molecules. Facilitated active transport, receptor-mediated transport, and ion transporters allow the brain to be supplied with nutrients while maintaining strict homeostasis. Adapted from Griffith, Jessica I., et al. "Addressing BBB heterogeneity: a new paradigm for drug delivery to brain tumors." Pharmaceutics 12.12 (2020): 1205. b Normal trans axial anatomy of the NVU/vascular (capillary) BBB. From the inside out the layers that form the blood–brain barrier include the endothelial expressed glycocalyx, specialized endothelial cells, double layer endothelial and astrocyte expressed basement membrane with glymphatic space sandwiched between the layers, pericytes, and astrocytes.
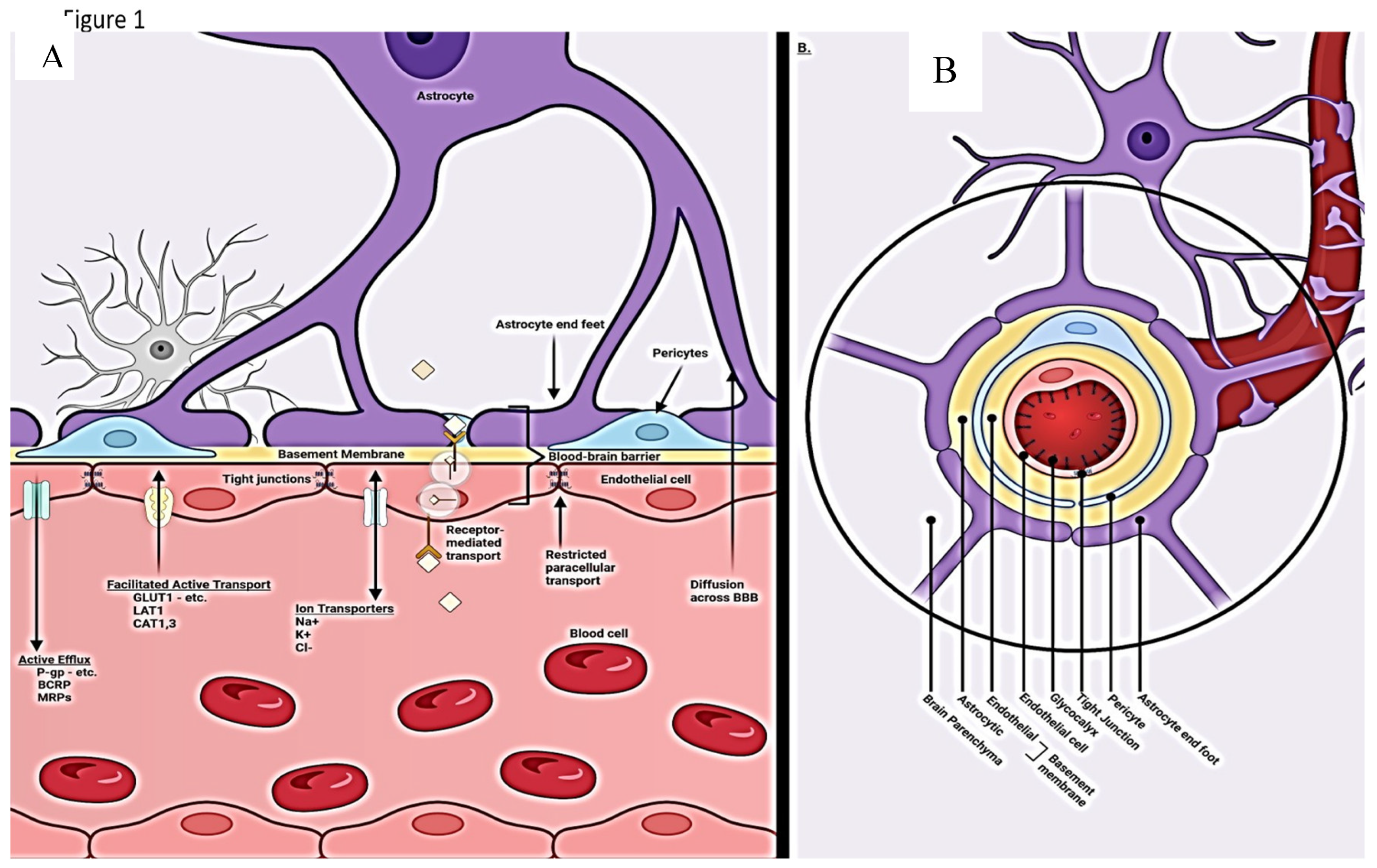
Figure 3.
Flow chart of developing BBB dysfunction in youth with robust repair potential post injury and loss of repairability with advanced age and consequent worsening BBB damage and leak. The delayed result of the latter is 2-fold: microvascular damage and production and accumulation of misfolded proteins. The accelerant effect of accumulating β Amyloid and hpTau on further BBB disruption causes a viscous cycle of more widespread BBB dysfunction leading to more widespread vascular damage and inflammatory infiltration with ultimate loss of neural elements and cognitive function decline.
Figure 3.
Flow chart of developing BBB dysfunction in youth with robust repair potential post injury and loss of repairability with advanced age and consequent worsening BBB damage and leak. The delayed result of the latter is 2-fold: microvascular damage and production and accumulation of misfolded proteins. The accelerant effect of accumulating β Amyloid and hpTau on further BBB disruption causes a viscous cycle of more widespread BBB dysfunction leading to more widespread vascular damage and inflammatory infiltration with ultimate loss of neural elements and cognitive function decline.
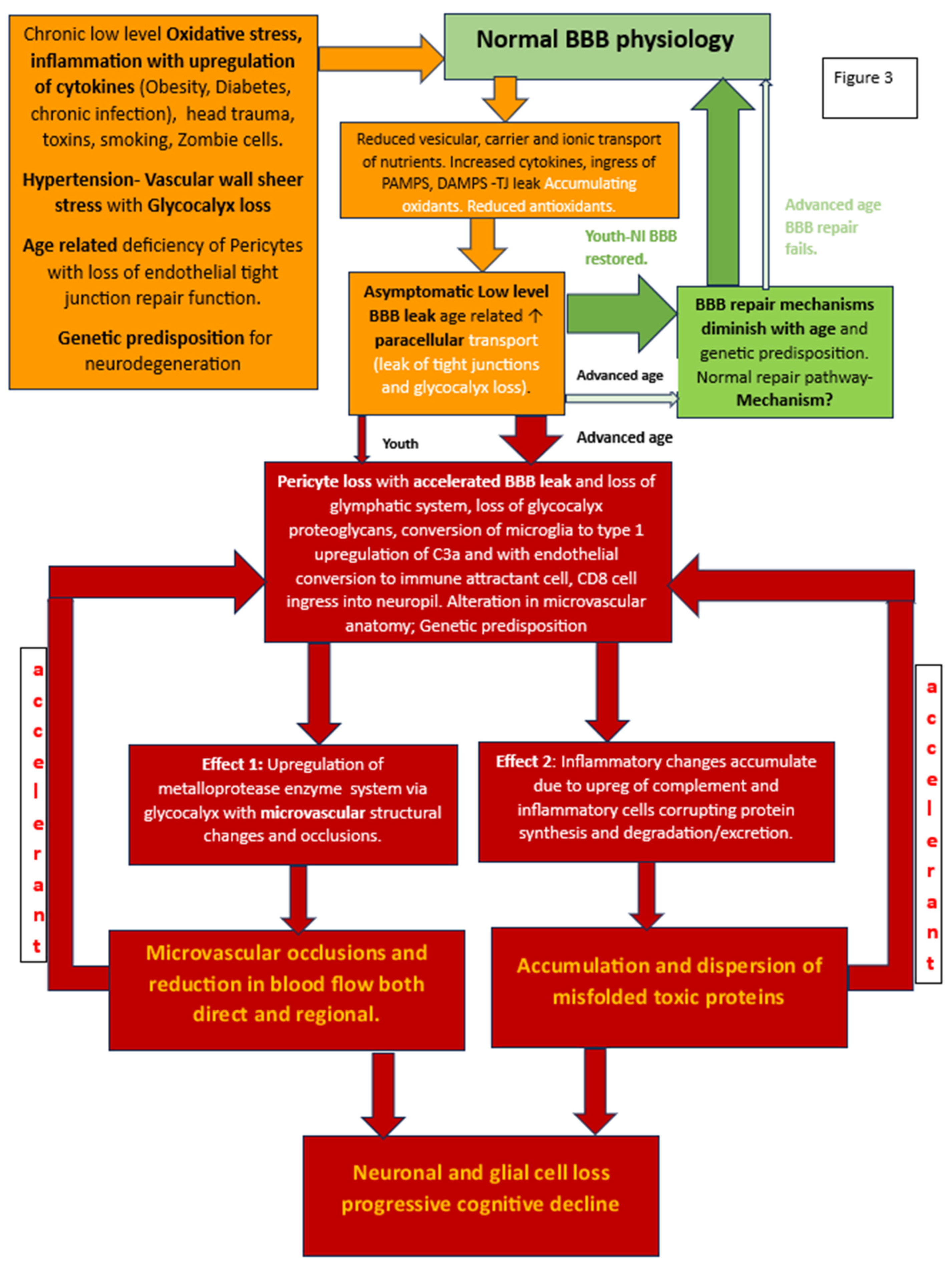
Figure 4.
Schematic illustration summarizing the effects of brain damage on BBB integrity. The production and activation of MMPs, VEGFs, and ETs are upregulated in various brain cells following brain damage. These factors can then impair the viability and integrity of the BBB by negatively impacting the tight junctions between adjacent endothelial cells.
Figure 4.
Schematic illustration summarizing the effects of brain damage on BBB integrity. The production and activation of MMPs, VEGFs, and ETs are upregulated in various brain cells following brain damage. These factors can then impair the viability and integrity of the BBB by negatively impacting the tight junctions between adjacent endothelial cells.
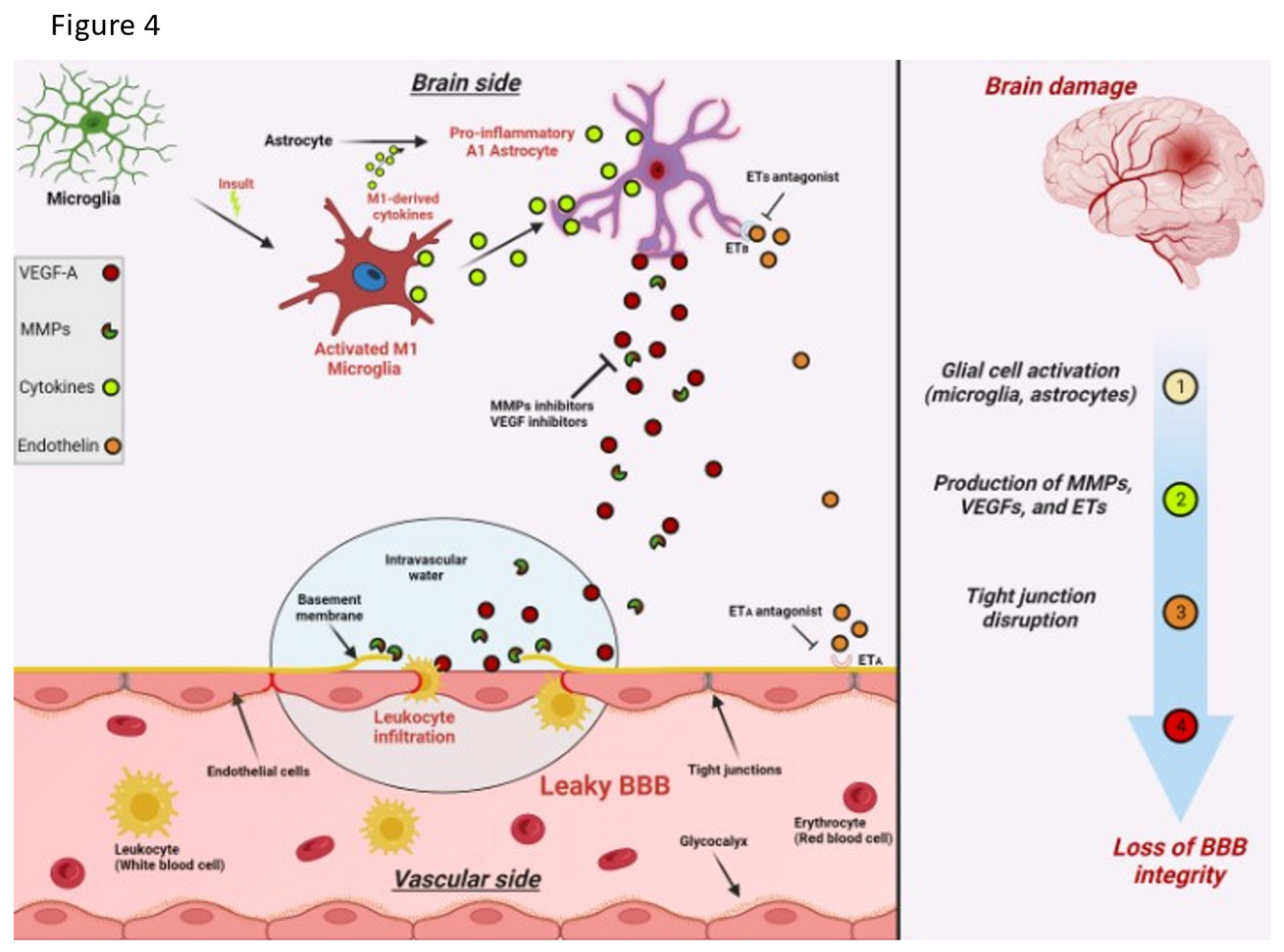
Figure 5.
Diagram of healthy and Alzheimer’s brain vasculature. (A) Diagram of healthy brain vasculature. Penetrating arteries (red) and veins (blue) are connected via a capillary mesh (gray). Steinman, Joe, Hong-Shuo Sun, and Zhong-Ping Feng. "Microvascular alterations in Alzheimer's disease." Frontiers in Cellular Neuroscience 14 (2021): 618986. (B) Diagram of Alzheimer’s brain vasculature. Deposition of amyloid (yellow dots) in the vascular bed causes vascular dysfunction and loss in AD, with proliferation of vessels around missing vasculature (Meyer, Eric P., et al. "Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease." Proceedings of the national academy of sciences 105.9 (2008): 3587-3592.).
Figure 5.
Diagram of healthy and Alzheimer’s brain vasculature. (A) Diagram of healthy brain vasculature. Penetrating arteries (red) and veins (blue) are connected via a capillary mesh (gray). Steinman, Joe, Hong-Shuo Sun, and Zhong-Ping Feng. "Microvascular alterations in Alzheimer's disease." Frontiers in Cellular Neuroscience 14 (2021): 618986. (B) Diagram of Alzheimer’s brain vasculature. Deposition of amyloid (yellow dots) in the vascular bed causes vascular dysfunction and loss in AD, with proliferation of vessels around missing vasculature (Meyer, Eric P., et al. "Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease." Proceedings of the national academy of sciences 105.9 (2008): 3587-3592.).
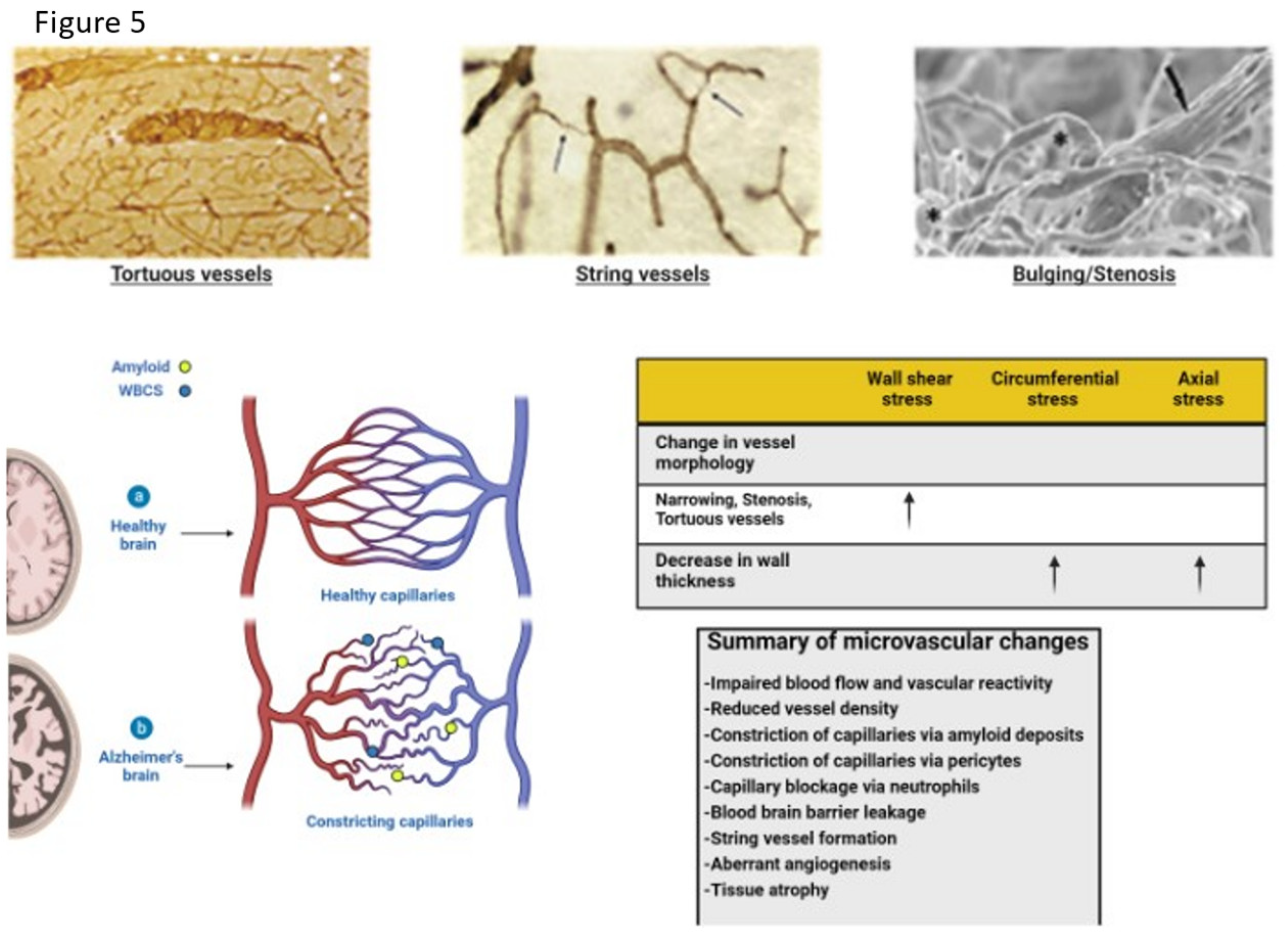
Figure 6.
DCE Magnetic resonance brain image of an Alzheimer’s patient with color-coded (Red Yellow) blood-brain barrier leakage. Note the greater extent of leakage in AD. Adapted from Evidence of BBB damage in the AD brain: (a) extensive leakage of gadobutrol (an MRI contrasting agent) through a damaged BBB in brains of patients with early signs of AD; (b) less extensive leakage of the agent in brains of normal patients. Used with permission of The Radiological Society of North America, from van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Hofman PAM, Verhey FRJ, Backes WH, Blood-brain barrier leakage in patients with early Alzheimer disease, Radiology (2016) 281, 527-535. Available via license: CC BY 4.0.
Figure 6.
DCE Magnetic resonance brain image of an Alzheimer’s patient with color-coded (Red Yellow) blood-brain barrier leakage. Note the greater extent of leakage in AD. Adapted from Evidence of BBB damage in the AD brain: (a) extensive leakage of gadobutrol (an MRI contrasting agent) through a damaged BBB in brains of patients with early signs of AD; (b) less extensive leakage of the agent in brains of normal patients. Used with permission of The Radiological Society of North America, from van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Hofman PAM, Verhey FRJ, Backes WH, Blood-brain barrier leakage in patients with early Alzheimer disease, Radiology (2016) 281, 527-535. Available via license: CC BY 4.0.
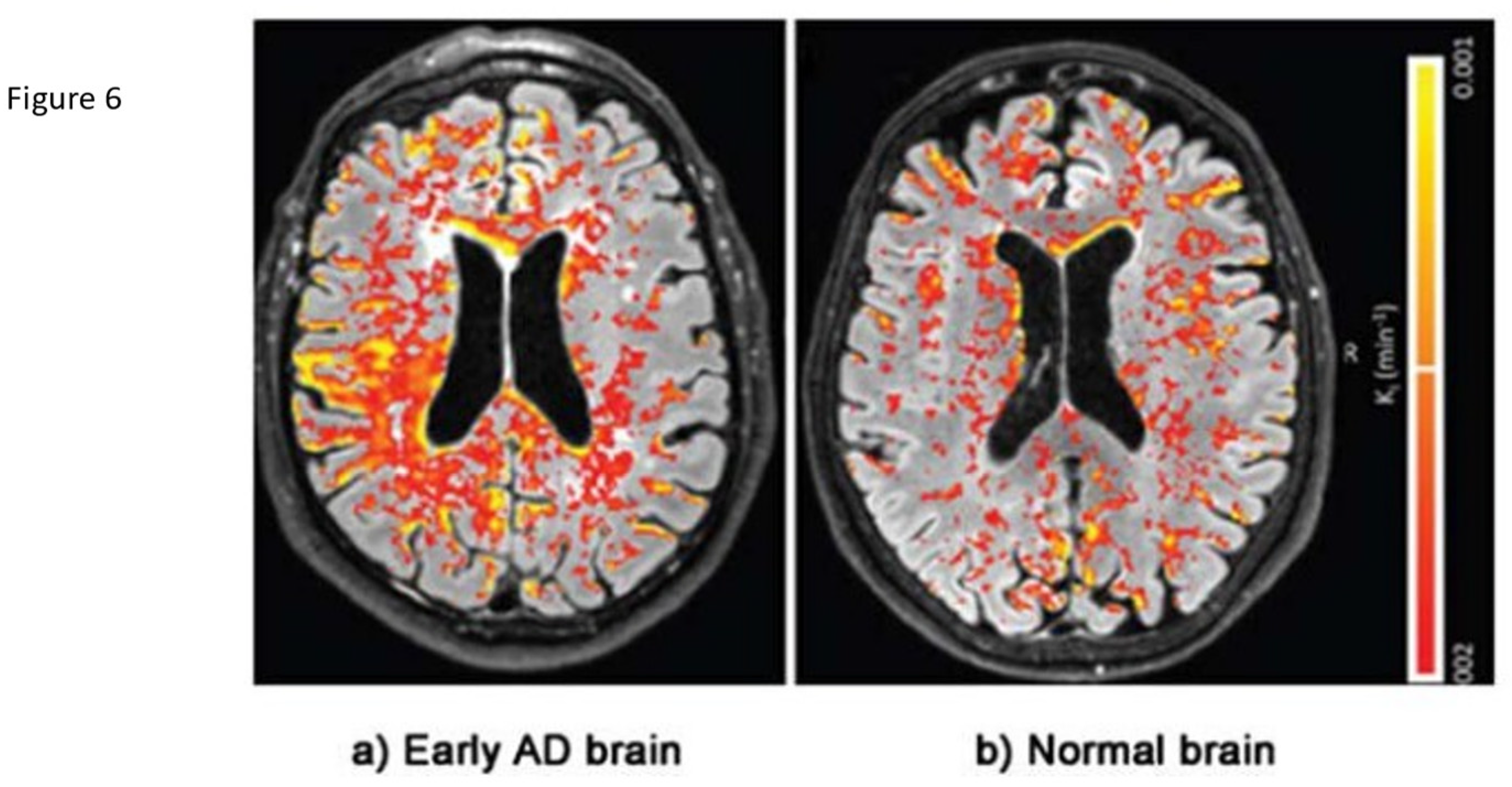
Figure 7.
3D ASL glymphatic clearance rate in MCI and AD. The upper 3 images denote the region of interest and size recorded for each region held constant bilaterally and across all subjects. The 4 mm slice angle and level for each region was also held constant. The lower images demonstrate the linear analysis of the 7 data points.
Figure 7.
3D ASL glymphatic clearance rate in MCI and AD. The upper 3 images denote the region of interest and size recorded for each region held constant bilaterally and across all subjects. The 4 mm slice angle and level for each region was also held constant. The lower images demonstrate the linear analysis of the 7 data points.
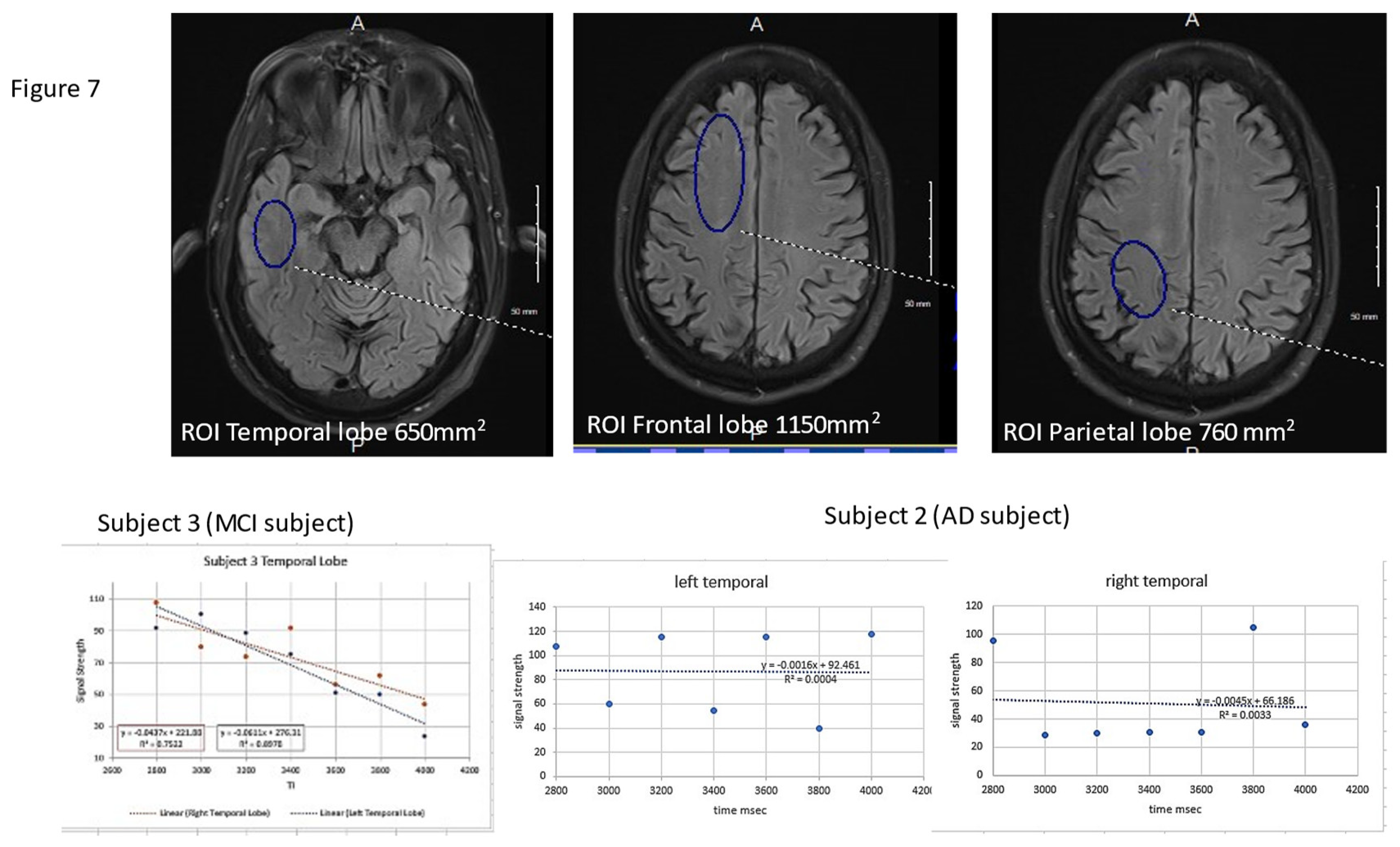
Figure 8.
Flow diagram of potential future approach to preclinical diagnosis and surveillance of high-risk individuals for developing AD once effective treatment paradigms are available.
Figure 8.
Flow diagram of potential future approach to preclinical diagnosis and surveillance of high-risk individuals for developing AD once effective treatment paradigms are available.
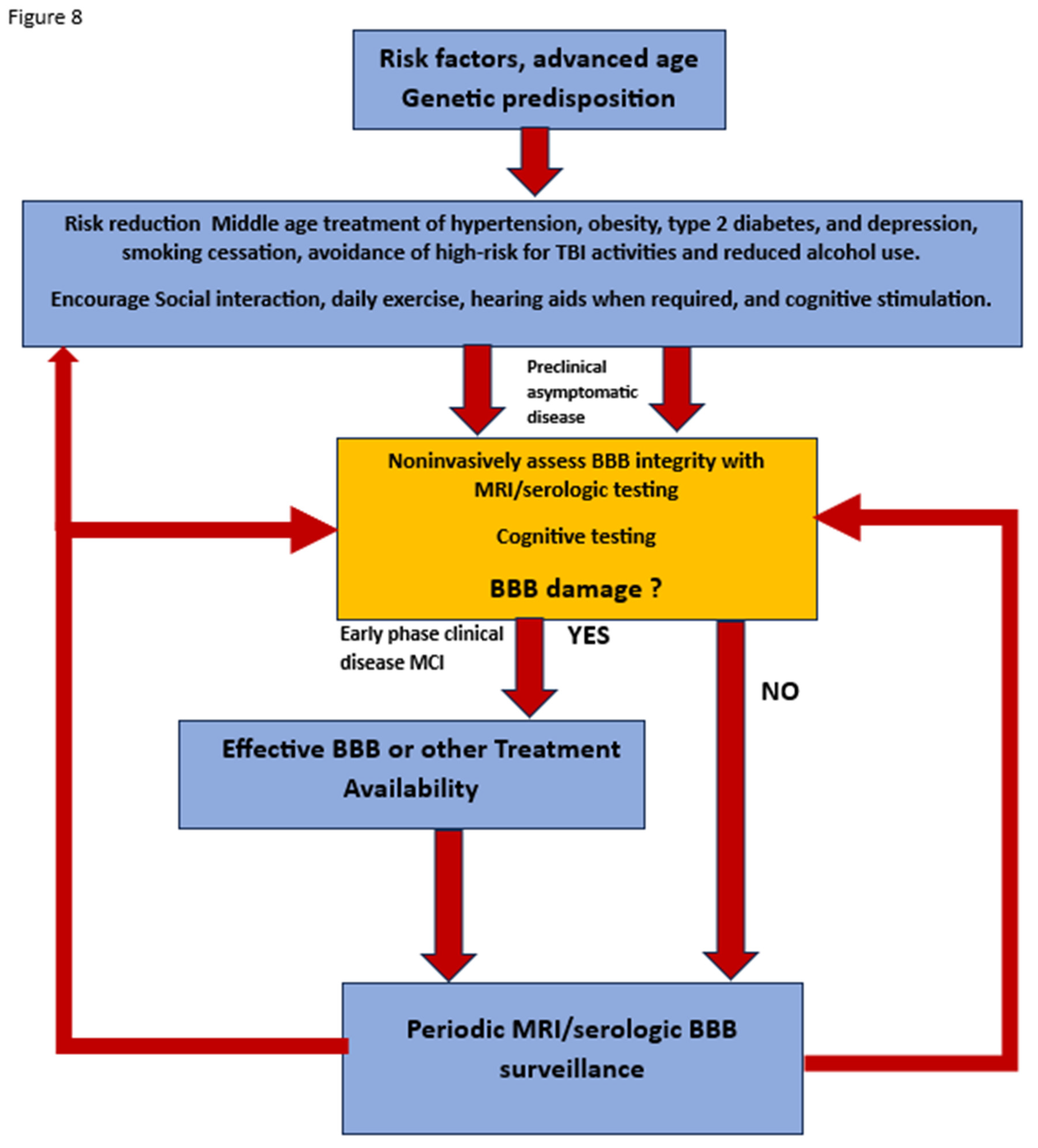

Table 1.
WHO recommendations for dementia prevention with quality and strength of medical evidence determinations. Chowdhary N, Barbui C, Anstey KJ, Kivipelto M, Barbera M, Peters R, Zheng L, Kulmala J, Stephen R, Ferri CP, Joanette Y, Wang H, Comas-Herrera A, Alessi C, Suharya (Dy) K, Mwangi KJ, Petersen RC, Motala AA, Mendis S, Prabhakaran D, Bibi Mia Sorefan A, Dias A, Gouider R, Shahar S, Ashby-Mitchell K, Prince M, and Dua T (2022) Reducing the Risk of Cognitive Decline and Dementia: WHO Recommendations. Front. Neurol. 12:765584. doi: 10.3389/fneur.2021.765584.
Table 1.
WHO recommendations for dementia prevention with quality and strength of medical evidence determinations. Chowdhary N, Barbui C, Anstey KJ, Kivipelto M, Barbera M, Peters R, Zheng L, Kulmala J, Stephen R, Ferri CP, Joanette Y, Wang H, Comas-Herrera A, Alessi C, Suharya (Dy) K, Mwangi KJ, Petersen RC, Motala AA, Mendis S, Prabhakaran D, Bibi Mia Sorefan A, Dias A, Gouider R, Shahar S, Ashby-Mitchell K, Prince M, and Dua T (2022) Reducing the Risk of Cognitive Decline and Dementia: WHO Recommendations. Front. Neurol. 12:765584. doi: 10.3389/fneur.2021.765584.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



