
Submitted:
24 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
Parkinson’s Disease (PD) is a debilitating neurodegenerative disease, causing loss of motor function, and in some instances, cognitive decline and dementia, in those affected. The quality of life can be improved, and disease progression delayed through early interventions, however, current methods of confirming a PD diagnosis are extremely invasive. This prevents their use as a screening tool for the early onset stages of PD. We propose a surface imprinted polymer (SIP) electroimpedance spectroscopy (EIS) biosensor for detecting α-Synuclein (αSyn) and its aggregates, a biomarker that appears in saliva and blood during the early stages of PD as the blood brain barrier degrades. The surface imprinted polymer stamp is fabricated by low temperature melt stamping polycaprolactone (PCL) on interdigitated EIS electrodes. The result is a low-cost, small footprint biosensor that is highly suitable for non-invasive monitoring of the disease biomarker. The sensors were tested with αSyn dilutions in deionized water, and in constant ionic concentration matrix solutions with decreasing concentrations of αSyn to remove the background effects of concentration. The device response confirmed the specificity of these devices to the target protein of monomeric αSyn. The sensor limit of detection was measured to be 5 pg/L and its linear detection range was 5 pg/L – 5 µg/L. This covers the physiological range of αSyn in saliva and makes this a highly promising method of quantifying αSyn monomers for PD patients in the future. The SIP surface was regenerated, and the sensor reused to demonstrate its capability for repeat sensing as a potential continuous monitoring tool for the disease biomarker.
Keywords:
surface imprinted polymers
; electroimpedance spectroscopy
; label-free biosensors
; parkinson’s disease
; α-synuclein
Introduction
Neurological disorders are the leading cause of disability in the world, affecting 15% of people, with neurodegenerative diseases such as Parkinson’s disease (PD) and Alzheimer’s disease currently accounting for 31-36% of neurological disorders [1]. The prevalence of neurodegenerative disease is rising [2], yet, despite the rapidly aging population, there is limited access to neurological healthcare and accessible diagnostic tests [3]. At present, neurodegenerative diseases are mainly diagnosed by neurological and physical exams [4], however, observable symptoms occur years or even decades after the onset of disease pathology. In order to detect neurodegenerative diseases in their earliest state, early identification of pathological biomarkers could potentially be a powerful tool.
α-Synuclein (αSyn) is a neural protein with remarkable conformational plasticity in its physiological form, fulfilling multiple roles in the body [5], [6]. However, when misfolded and/or phosphorylated, αSyn becomes pathological and aggregates into fibrils leading to synucleinopathies such as PD [7]. Aggregation of αSyn and subsequent neurodegeneration of midbrain dopaminergic neurons produces the loss of motor symptoms used for initial diagnosis of PD [8]. Pathological αSyn misfolding and aggregation precede clinical symptom manifestation by several years. Once PD is suspected, the diagnosis can be confirmed using cerebral spinal fluid (CSF) seeding activity testing which measures the rate at which αSyn forms toxic aggregates [9]. Unfortunately, this test cannot be used as a screening tool for early detection of PD as it is highly invasive, requires specialized laboratory setup, and can take from 5 to 13 days [10]. There are a few promising examples of biosensing platforms suitable for less invasive, less cumbersome, and hence more accessible αSyn quantification, such as our organic electrolyte gated FET aptasensor platform and Adam et al’s electrochemical biosensor [11], [12] to list a few. These emerging biosensors rely on a bioreceptor molecule, either an aptamer or an antibody, adding unique complexities to sensor shelf-life and usability as a continuous monitoring device.
As an alternative to electrochemical and electrolyte gated biosensors, electroimpedance spectroscopy (EIS) sensors, transduce sample target biomarker binding by measuring the change of reactance and resistance as a function of angular frequency [13]. EIS is capable of rapid, non-destructive, label-free characterization and without current production to perform measurements [14]. EIS is highly sensitive to near surface effects, making it ideal for affinity biosensors, with a simple electrode design requiring only interdigitated structures of counter and reference electrodes. Conventional EIS biosensors rely on changes resulting from enzymatic reactions facilitated by gold nanoparticles, or selective binding action in presence of a bioreceptor such as an antibody or aptamer (short oligomer DNA chains). Karaboğa et al produced an electrochemical EIS – Gold nanoparticle-Polyglutamic acid biosensor (ECB) for αSyn with a linear range of 4-200 pg/mL in blood, a limit of detection (LOD) of 1.35pg/mL and recovery rates of 96.81-102.65% [15]. Their early results were promising; however, ECB EIS face significant challenges with sensitivity to surface variations and complex device architecture.
EIS combined with affinity-based recognition is a facile, rapid, and exceptionally durable platform for biosensing [16,17]. Synthetic methods of selectively binding target molecules focus on highly repeatable, selective, and cost-effective recognition processes. Conventional immunoassays, the gold standard of biomolecule quantification, rely on the selective binding of immunoglobulins (antibodies; Ig) [16]. These biologically sourced materials are highly sensitive to environment and fabrication processes, which makes integrating them into commercial biosensors challenging. In contrast, synthetic ‘antibody mimics’ such as surface imprinted polymers are extremely simple to fabricate, low cost, and with good chemical and thermal resistance and rejuvenation abilities.
Surface Imprinted Polymers (SIP) are polymers imprinted with a biomolecule of interest to form three-dimensional stereo cavities that bind the target biomolecule with high specificity. Molecular imprinting, and stamp imprinting are the most commonly reported methods of fabricating SIPS [17]. In molecular imprinting a monomer is polymerized, or a polymer is crosslinked around a biomarker target. Yang et al produced a P-glycoprotein SIP with an LoD of 22 fg/L, however, a key challenge with this approach is the complexity of cross linking on surface [18]. Polymerizing and crosslinking reagents can interfere with the biomarker structure, whilst milder processes such UV cross linkable materials are often water soluble. [14]. Stamp imprinting avoids the negative impacts of crosslinking to targets by using deposited polymer layers [17]. Werner et al compared two methods of surface imprinting polymers, polymerization and Escherichia coli cell stamp imprinting [19]. They demonstrated through Atomic Force Microscopy (AFM) that both methods produce smooth surfaces and the presence of stereo cavities for detection [19]. Pressing the biomarker target into a polymer surface forms specific cavities as small as ions, and as large as cells [20].
In this work we report a highly specific EIS biosensor combined with a SIP nanomaterial as bioreceptor, for simple and rapid quantification of αSyn. The SIP was prepared using stamp imprinted Polycaprolactone (PCL). PCL is a low temperature solution processable, biocompatible, biodegradable polymer, with a dielectric constant of 3.2 [21], [22]. PCL melts at 60⁰C, low enough to minimally affect lyophilized proteins and it does not dissolve in water or swell (less than 0.25 % swelling over 10 hours [23]), making it robust. In our previous work demonstrating a proof-of-concept PCL SIP EIS biosensor, we implemented a thermally pressed PCL SIP layer over interdigitated electrodes (IDEs) on a passivated silicon substrate. The stamp used for imprinting consisted of αSyn on polydimethylsiloxane (PDMS) [24]. We were able to demonstrate concentration dependent EIS behavior, but with significant challenges. The first was the fabrication process relied on thermal pressing, leading to a thick, non-uniform PCL SIP layer ranging between 10µm-200µm. The high thicknesses contributed to the low resolution between concentrations. The PDMS αSyn stamp had highly variable material density due to the hydrophobic nature of the polymer, and the adhesion to PCL led to damage and low device success rate. We have greatly improved the device structure and fabrication process to produce a much more sensitive and robust biosensor. We used a solution processed PCL to control the layer thickness and a novel PVA stamp to improve the biomarker distribution. The resultant biosensor can detect low levels of αSyn in tested solutions. In order to minimize concentration dependent signal, we tested the sensitivity of the device with solutions of 1 µg/mL, with a varying ratio of αSyn and a homologous control biomolecule ß-synuclein (ßSyn). ßSyn was selected as a control material as it is a synuclein neural protein that has a similar primary structure to αSyn with slight structural differences (αSyn has 140 amino acids whereas ßSyn has 137) [25]. αSyn is more prone to agglomeration due to its different charge distribution and shape. Testing of αSyn and ßSyn combinations showed the PCL SIP device has a linear range of 5 pg/L to 500 ng/L. With an integrated PCL microfluidic channel, the linear range is observed to be extended over 5 pg/L to 5 µg/L. Overall, we have produced a novel SIP EIS biosensor with a facile, scalable fabrication process leveraging low temperature processing.
MATERIALS AND METHODS
2.1. Device Fabrication
Stamp: 400 nm of polyvinyl alcohol (PVA) is static deposited spin coated from a 10 wt.% solution of PVA in chloroform onto a glass slide. The stamp was prepared by dropping 50µL of 1 mg/mL αSyn in deionized water (DI) onto a 0.5cm2 area and dried at room temperature for 2 hours (Figure 1a).
PCL SIP: Kapton substrates (500 EN, Dow Chemicals, USA) are patterned with 100 nm of Aluminum (Al) and 100nm of chromium (Cr) using standard lift-off photolithography techniques. The interdigitated electrodes (IDE) with an area of 25mm2, with 20 fingers and a measured spacing of 102 µm and width of 153 µm (Figure 1b). A 600 nm thick layer of PCL is deposited on the IDEs by dynamic deposition spin coating (at 6000 rpm). The stamp is placed in contact with the PCL surface, heated to 60⁰C and pressed using a 200g weight for 2 minutes. The structure is then removed from the heat and allowed to cool. The PCL surface and stamp are submerged in water allowing the PVA stamp polymer to dissolve, releasing the EIS device without damage. The SIP EIS biosensors are washed with 0.5 mM ascorbic acid and deionized water (DI) to ensure removal of the stamp biomolecules prior to testing.
PCL microfluidic channel: A microfluidic channel was made by melting PCL into a mold (Figure 1c). The well depth was 2 mm deep, and luer lock tubing was melted into the microfluidic channel for simple sample loading and waste removal. The microfluidic channel was then adhered to the PCL microfluidic channel using chloroform as a solvent. The PCL of the microfluidic channel and the surface forms an excellent seal after the chloroform off gases, leaving an integrated sample handling method.
2.2. Sample Preparation
αSyn in DI test samples were created by serial dilutions of from 10 mg/L of dried αSyn material in DI. 10-fold serial dilutions were produced from 10 mg/L to 100 ag/L. Constant ionic concentration solutions were created using αSyn and ßSyn in varying ratios to produce solutions of serially decreasing αSyn solutions, but with a constant total ionic concentration. The concentrations of αSyn in these solutions was 10-fold dilutions from 50 µg/L down to 100 ag/L.
αSyn monomer and ßSyn were supplied by the LADDER group in Chemistry Department, Carleton University. To avoid unwanted aggregation of the material, all materials were stored at -20⁰C when not in use, and vortexed prior to use.
2.3. Testing Processes
Impedance analysis is performed using an Agilent 4294A impedance analyzer (Fig 1d). The impedance magnitude and phase angle are collected during a logarithmic frequency sweep from 40 Hz to 100 MHz with an amplitude of 500mV. During testing, 10µL of sample is incubated on the surface for 1 minute prior to testing. Each data collection was repeated three times. The surface is then rinsed with DI, followed by 0.5 mM ascorbic acid, and a final DI rinse and N2 drying to ensure all material is removed from the surface between tests. The device is then ready for the next test.
The impedance magnitudes were converted to the real (Z’) and imaginary (Z”) components and plotted as a Nyquist plot for parameter extraction during sensor optimization. Experimental data was analyzed using MATLAB.
2.4. Atomic Force Microscopy of Soft-Printed SIP Surface
SEM was performed with a Tescan Vega-II XMU VPSEM. Figure 2a shows the SEM topography of a 238.1 µm by 238.1 µm scan of a SIP on EIS electrode post testing and regeneration. The scale-like appearance of the PCL is a factor of the heat-melt process involved in the stamping process. The important factor here is the size of the crystals formed. Without the presence of a stamp, we observe crystals on the scale of 100 µm – mm. In the presence of the stamp, we observe significantly smaller crystals (scale of 2-5 µm) formed by the stamp protein acting as nucleation points. Clear cavities are observed well distributed across the surface. These are the binding sites of the PCL SIP. Based on the partial specific volume, calculating the approximate volume occupied by a protein of mass M (kDa) is volume (nm3) =1.212*M, giving a volume of 17.5nm3, so assuming a globular protein the diameter should be 2.78nm for αSyn monomers [26]. We expect the surface cavities to be in this range for single αSyn monomers, which we further examined using atomic force microscopy.
Atomic Force measurements took place in air using a Veeco Dimension 3100 AFM in tapping mode with a silicon probe tip. Nanoscale AFM lateral resolution is dependent on tip sharpness and profile, lateral feature size is inflated for adjacent particles or rough surfaces. z dimension deflection is a reliable indicator of feature size. Figure 2b shows the AFM topography of a 1 µm by 1 µm scan of a PCL SIP post testing and regeneration. The largest surface cavities have depths of 9.2 ± 5.5 nm, with the smallest cavities down to a few nm. This indicates that there is some agglomeration of αSyn. Figure 2c shows the AFM topography of a 1 µm by 1 µm scan of an αSyn Stamp with lyophilized αSyn on the surface. The z dimension sizes of molecules were between 3 - 9.2 nm. Material size variation is observed on the slides. This confirms that there is some anticipated agglomeration. Thus, the imprints on the PCL SIP shown in figure 2b were consistent with the size of lyophilized material on the surface of the stamp. We observed surface cavities consistent with effective stamping of the PCL surface.
RESULTS AND DISCUSSION
3.1. Impedance Spectroscopy Data Analysis
The SIP EIS biosensor impedance response for five separate devices was repeat tested n=3 times with each test solution. Given that the EIS biosensor tests electrolytes, it is suitable to analyze the impedance response data with a Randles-Ershler equivalent circuit model (Figure 3a) [27]. The expected graph shape from the Randles-Ershler Nyquist plot is shown in Figure 3b. This is a basic model that is applied to both faradaic and non-faradaic EIS biosensors. Faradaic biosensors are defined as having a redox species that generates charge. Non-faradaic biosensors do not rely on charge generation and are generally label free. It is important to note though that there is not necessarily a direct correspondence between circuit elements and underlying physical processes; for example, the simplified Randles-Ershler circuit model lumps the entirety of the sensing mechanism processes into a single element CG.
There are 4 main parameters, RS or solution resistance, CG or geometric capacitance, ZW the Warburg element and RCT, the charge transfer resistance. The prevalence of the elements is dictated by the device architecture and materials. Solution resistance (RS) is dependent on the finite conductance of ions in bulk solution. Therefore, it is affected by concentration, but not by binding processes. The Warburg impedance, ZW is usually physically insignificant in non-faradaic biosensors, as it is a delay arising from the diffusion of electroactive species to the electrode. Thereby, it only has an appreciable effect at low frequencies, and is affected by convection. The ideal Warburg element has a phase shift of -45⁰. RCT captures two effects, the energy barrier to redox species (caused by electrostatic repulsion or steric hinderance) and the overpotential. In non-faradaic EIS biosensors, it also models the leakage current from imperfect insulator dielectrics.
CG is the capacitance between the electrodes and the electrolyte solution. It can be modelled as a series of capacitances including surface insulators, double layer capacitances and surface modifications. The electric double layer is created by the alignment of charged materials in solution to electrodes of opposite charge. Thus, electric fields in ionic solutions decay exponentially because the alignment of ions negates the effective field. The length of this decay is called the Debye length and is proportional to the square root of ion concentrations. Another contributor to the CG is the adsorbed molecules on the surface. In the absence of charge production, CG is the dominant capacitance term. The CG also contains a constant phase element that dominates at low frequency that can account for the complex double layer capacitance of the remaining fluid on surface, adsorbed molecules, and porous surface structures.
In the ideal situation of non-faradaic biosensors, RCT would be theoretically infinite as no charge would be crossing the perfect insulator. However, due to the polarizability of polymers and confirmational changes of materials RCT is finite. Under these conditions, the imaginary portion of the impedance is inversely proportional to the electrical double layer capacitance [28]. This creates the incomplete semicircular shape with the slow transition to the linear behavior even in non-faradaic biosensors. This deviation from the ideal can be attributed to surface non-uniformity, roughness, and potentially porosity. These kinds of surface effects can create sub-microscopic areas each with a unique resistance-capacitance contribution to the overall behavior. Parasitic impedances and frequency dispersion—transformation of dielectric response from one mode of polarization to another—are usually described in the ZW [29]. As the sensing mechanism is from the change in near surface effects and particularly the double layer capacitance in the geometric capacitance, repeatable CG extraction is imperative. Using the Randles equivalent circuit model and parameter extraction methods has been established in literature for approximating the non-faradaic biosensors.
The x intercepts of the semicircle represent the contact resistance (Rs) and surface resistance (Rct). Eqns. (2) are used to extract the CG and Zw, with the tail slope used to determine α, the phase change of the constant phase element. The peak frequency value is used to determine CG, the geometric capacitance. MATLAB is used to identify the high corner frequency of the Nyquist plot and calculate the RS and RCT. Real data is shown in Figure 3c for device 5 tested with a 100 pg/L αSyn constant ionic concentration solution.
Figure 3d shows unfiltered data for dilutions of αSynuclein in DI, showing non-ideal Randles-Ershler impedance output curve for non-faradaic electrolytes under AC. The data trend shows a smaller semicircular curve and lower maximum real and imaginary capacitances for increasing concentration. There are two distinct behaviors that contribute to the shapes of the graph: increasing concentration and increasing binding. As RS is a factor of solution concentration, it will decrease with increasing concentration of charged materials. With the increasing concentration, there is an increasing contribution to CG from an increasing double layer capacitance, with decreasing capacitance from an increase in binding to the surface. There are multiple explanations for the decrease in capacitance with increasing binding. It could be that the presence of proteins changes the conductivity in the near surface region, the binding interrupts the formation of the EDL, or it could change the surface energy of the insulator [30]. The effect can be seen in the decreasing size of the semicircular portions of the Nyquist plots, and the increasing impedance with increasing concentration. In order to observe the effects of binding alone, the ionic concentration of solutions was kept constant.
3.2. Characterizing Sensor Performance in
Figure 4a shows real, unfiltered Nyquist plots showing the concentration dependent change in CG for our PCL SIP EIS biosensors tested in a constant ionic concentration environment with varying concentrations of αSyn. The solutions all have a total synuclein protein concentration of 100 µg/L, but with a decreasing ratio of αSyn to ßSyn. The purpose of testing only in a constant ionic concentration environment is that these devices do have a non-specific response to electrolyte concentration. ßSyn is a homologous protein to αSyn that is structurally different, making it ideal as a control. As the ionic concentration remains constant, the capacitance change will be from increasing binding.
Plotting the Nyquist data clearly shows that not only is there a change in peak frequency (a dependent variable of CG) but the RS is clearly increasing with concentration. This is a good indicator that there are indeed changes occurring at the surface of the biosensor. As these devices are tested in an aqueous environment under an applied bias, the electrolyte forms an electric double layer. The freely moving materials in the electrolyte align themselves to the surface. The effective thicknesses of these layers are on the angstrom to nm level. As the non-Faradaic EIS biosensors do not have any charge transfer the biosensing mechanism is due to changes in the electric double layer capacitance. With αSyn binding into the stereo cavities, the development of the EDL is interrupted which decreases the capacitance. The impact of binding is great because the nm and angstrom scale thicknesses of the EDL layers make the EIS SIP sensitive to near surface interactions.
The concentration to percent change in geometric capacitance is shown in Figure 4b. The data was averaged across the 5 different sensor devices, with a 95% confidence interval. Parameters were extracted from the plots using the Randles Erschler equivalent circuit model and fitted to a four-parameter logistical curve. Data is normalized using the 500 fg/L limit of detection test completed on each device prior to experimental data collection to allow for comparison between tested SIP EIS devices. LoD was determined by linear fitting using the standard method using LoD = 3.3(Sy/S) where Sy is the standard deviation of the sensor response (Sy) extracted using linearly fitted data and S, is the slope of the sensor calibration curve. In a simplistic estimation, concentrations that deviate by more than three standard deviations are considered outside of the linear detectable range. The biosensor has a linear range of 5 pg/L to 5 µg/L, with a LoD of 5 pg/L. The clear concentration-dependent geometric capacitance shows that the simplistic soft-printed SIP fabrication process is sufficient for quantifying minute changes to αSyn monomer concentration. Re-usability of our devices was investigated further by repeatedly testing the baseline geometric capacitance where we found a low standard variation of 7.2 % (n=12), indicating that the dilute acid wash and DI wash are effective in removing bound targets from the template between tests.
3.3. Preliminary Data from Microfluidic Channel SIP EIS Biosensor
The final test was to create an EIS SIP biosensor device with the sensing area enclosed within a PCL microfluidic channel, with luer lock interconnects for efficient sample handling. The test volume of the PCL microfluidic channel device was kept at 100 µL. Repeating the same constant ionic concentration testing as with the previous device (shown in Figure 4) produced the data shown in Figure 5a,b.
The device was tested in the linear range established with the open face biosensor. The EIS SIPs showed the same linear behavior and range enclosed in a microfluidic channel as when tested on an open surface, a highly desirable outcome (Figure 5b). It also highlights a source of work for the future, as enclosing the sensor in the microfluidic channel affects the change in geometric capacitance. The percent change in CG has decreased over the same linear range when the microfluidic channel was enclosed. Another significant change is the Nyquist plot shape. The absence of the low frequency linear range can be explained by the enclosed channel minimizing dielectrophoretic droplet spreading. Investigating the bulk effects from the microfluidic channel will be the next stage of device development.
The SIP EIS is therefore a promising biosensor device for detection of biomolecules and the device fabrication process is simple and facile, making it ideal for large-scale manufacturing and rapid prototyping. Not only are our biosensors capable of detecting αSyn in the dilute levels present in saliva, but the nature of non-invasive testing makes the devices desirable [31]. Saliva has fewer risk factors than serum or cerebral spinal fluid and can be repeatedly sampled [32]. Furthermore, the sensor can be easily interfaced with off-the shelf portable EIS readers making it point-of-care ready. We have previously reported a PCB-integrated EIS sensor for portable and quantitative analysis of 8-Isoprostane in exhaled breadth [14]. Integrating the proposed SIP EIS in a similar way will be adopted in the next phase of platform testing and validation.
Future work will focus on improving the repeatability of stamp production to reduce device variability. The binding between SIP and target biomolecule is dependent only on steric forces, which in a complex media such as whole blood, serum or interstitial fluid biological samples would be greatly influenced by high energy media components.
Conclusions
αSynuclein is a key biomarker for Parkinson’s Disease, which presently lacks a non-invasive and accessible method of clinical diagnostics. We have demonstrated the first PCL-based surface imprinted polymer EIS biosensor for αSyn, fabricated using solution processable, low-temperature soft imprinting process. Benefits of our EIS biosensor are scalable printing process, environmental stability of the PCL-based SIP bioreceptor surface, a large linear detection range of 5 pg/L – 5 µg/L which covers the physiological concentration range of αSyn in saliva samples. The sensor LoD was measured to be 5 pg/L which is comparable to that of αSyn biosensors that rely on more expensive and less scalable bioreceptors such as antibody and aptamer. The regenerative capabilities of the PCL SIP surface make this device suitable for rapid and repeated testing of the biomarker. The biosensor testing using constant ionic concentration solutions of αSyn and ßSyn, a comparable synuclein protein, demonstrated no concentration dependent behavior for ßSyn, confirming the specificity of these biosensor towards the target protein, i.e., monomeric αSyn protein. These outcomes make the PCL-based SIP EIS biosensor a highly promising method of quantifying pathogenic forms of αSyn monomers in clinical biofluid samples such as saliva and serum in future applications.
Author Contributions
Conceptualization, R.P. and R.M.; methodology, R.P. and R.M.; formal analysis, R.M.; Sensor fabrication and data collection, R.M and R.A; writing—original draft preparation, R.M.; writing—review and editing, R.P.; supervision, R.P.; funding acquisition, R.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Sciences and Engineering Research Council of Canada, Discovery Grant.
Acknowledgments
The authors acknowledge Dr. Maria DeRosa and Dr. Matthew Holahan for providing αSyn and ßSyn materials for device testing. The authors also thank Rob VanDusen and Angela Mccormick of Carleton Microfab and Dr. Peter Gordon of Carleton Nanofab for their assistance with device fabrication and PCL-SIP surface characterization.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- E. Beghi et al. ‘Articles The burden of neurological diseases in Europe: an analysis for the Global Burden of Disease Study 2017’, Lancet Public Health, vol. 5, pp. 551–67, 2020, Accessed: Nov. 17, 2023. [Online]. Available: www.thelancet.com/.
- ‘Global action plan on the public health response to dementia’, Geneva, 2017. Accessed: Nov. 17, 2023. [Online]. Available: http://apps.who.int/bookorders.
- E. R. Dorsey, A. M. Glidden, M. R. Holloway, G. L. Birbeck, and L. H. Schwamm. Teleneurology and mobile technologies: the future of neurological care. Nature Reviews Neurology 2018 14:5. vol. 14, no. 5, pp. 285–297, Apr. 2018. [CrossRef]
- D. Grimes et al. Canadian guideline for Parkinson disease. CMAJ. vol. 191, no. 36, pp. E989–E1004, Sep. 2019. [CrossRef]
- H. K. Chung, H.-A. Ho, D. Pérez-Acuña, and S.-J. Lee. Modeling α-Synuclein Propagation with Preformed Fibril Injections. J Mov Disord. vol. 12, no. 3, p. 139, Sep. 2019. [CrossRef]
- L. Suescún, E. Sanchez, M. Gómez, F. L. Garcia-Arias, and V. M. Núñez Zarantes. Protein. In Human Nutrition, 2020th ed.; Cámara de Comercio de Bogotá. Editorial Kimpres Ltda, 2011, p. 53. Accessed: Nov. 17, 2023. [Online]. Available: http://pressbooks.oer.hawaii.edu/humannutrition/.
- R. M. Meade, D. P. Fairlie, and J. M. Mason. Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Molecular Neurodegeneration. 2019 14:1, vol. 14, no. 1, pp. 1–14, Jul. 2019. [CrossRef]
- Z. A. Sorrentino and B. I. Giasson. The emerging role of α-synuclein truncation in aggregation and disease. Journal of Biological Chemistry. vol. 295, no. 30, pp. 10224–10244, Jul. 2020.. [CrossRef]
- M. J. Russo et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun. vol. 9, no. 1, pp. 1–13, Dec. 2021.. [CrossRef]
- C. D. Orrù et al. A rapid α-synuclein seed assay of Parkinson’s disease CSF panel shows high diagnostic accuracy. Ann Clin Transl Neurol. vol. 8, no. 2, p. 374, Feb. 2021. [CrossRef]
- R. Massey, E. McConnell, D. Chan, M. Holahan, M. DeRosa, and R. Prakash. Non-invasive Monitoring of Alpha-Synuclein in Saliva for Parkinson’s Disease using Organic Electrolyte Gated FET Aptasensor. ACS Sens. vol. 8, no. 8, pp. 3116–3126, Jul. 2023. [CrossRef]
- H. Adam, S. C. B. Gopinath, M. K. M. Arshad, N. A. Parmin, and U. Hashim. Distinguishing normal and aggregated alpha-synuclein interaction on gold nanorod incorporated zinc oxide nanocomposite by electrochemical technique. Int J Biol Macromol. vol. 171, pp. 217–224, Feb. 2021.. [CrossRef]
- K. Smolinska-Kempisty, A. Guerreiro, F. Canfarotta, C. Cáceres, M. J. Whitcombe, and S. Piletsky. A comparison of the performance of molecularly imprinted polymer nanoparticles for small molecule targets and antibodies in the ELISA format. Scientific Reports 2016 6:. vol. 6, no. 1, pp. 1–7, Nov. 2016. [CrossRef]
- R. S. Massey, B. Gamero, and R. Prakash. A System-on-Board Integrated Multi-analyte PoC Biosensor for Combined Analysis of Saliva and Exhaled Breath. Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, EMBS. pp. 904–909, 2022. [CrossRef]
- M. N. S. Karaboǧa and M. K. Sezgintürk. Cerebrospinal fluid levels of alpha-synuclein measured using a poly-glutamic acid-modified gold nanoparticle-doped disposable neuro-biosensor system. Analyst. vol. 144, no. 2, pp. 611–621, Jan. 2019. [CrossRef]
- M. G. Sande, J. L. Rodrigues, D. Ferreira, C. J. Silva, and L. R. Rodrigues. Novel Biorecognition Elements against Pathogens in the Design of State-of-the-Art Diagnostics. Biosensors 2021, Vol. 11, Page 418. vol. 11, no. 11, p. 418, Oct. 2021. [CrossRef]
- C. Unger and P. A. Lieberzeit. Molecularly imprinted thin film surfaces in sensing: Chances and challenges. React Funct Polym. vol. 161, p. 104855, Apr. 2021.. [CrossRef]
- H. Yang et al. A molecularly imprinted electrochemical sensor based on surface imprinted polymerization and boric acid affinity for selective and sensitive detection of P-glycoproteins. Anal Chim Acta. vol. 1207, p. 339797, May 2022. [CrossRef]
- M. Werner, M. S. Glück, B. Bräuer, A. Bismarck, and P. A. Lieberzeit. Investigations on sub-structures within cavities of surface imprinted polymers using AFM and PF-QNM. Soft Matter. vol. 18, no. 11, pp. 2245–2251, Mar. 2022. [CrossRef]
- Z. El-Schich et al. Molecularly imprinted polymers in biological applications. Biotechniques. vol. 69, no. 6, pp. 407–420, Dec. 2020.. [CrossRef]
- S. M. Aguilar, J. D. Shea, M. A. Al-Joumayly, B. D. Van Veen, N. Behdad, and S. C. Hagness. Dielectric Characterization of PCL-Based Thermoplastic Materials for Microwave Diagnostic and Therapeutic Applications. IEEE Trans Biomed Eng. vol. 59, no. 3, p. 627, Mar. 2012. [CrossRef]
- C. J. Luo, E. Stride, and M. Edirisinghe. Mapping the influence of solubility and dielectric constant on electrospinning polycaprolactone solutions. Macromolecules. vol. 45, no. 11, pp. 4669–4680, Jun. 2012. [CrossRef]
- N. Malik, S. Shrivastava, and S. B. Ghosh. Moisture Absorption Behaviour of Biopolymer Polycapralactone (PCL) / Organo Modified Montmorillonite Clay (OMMT) biocomposite films’. IOP Conf Ser Mater Sci Eng. vol. 346, no. 1, May 2018.. [CrossRef]
- R. S. Massey, Y. H. Li, and R. Prakash. Surface Imprinted Electroimpedance Biosensor for Detecting α-Synuclein for Parkinson’s Disease. 2023 IEEE Sensors Applications Symposium (SAS). Ottawa, ON, Canada, 2023, pp. 1-4. [CrossRef]
- L. Barba et al. Alpha and Beta Synucleins: From Pathophysiology to Clinical Application as Biomarkers. Movement Disorders. vol. 37, no. 4, pp. 669–683, Apr. 2022. [CrossRef]
- M. S. Chae, J. H. Park, H. W. Son, K. S. Hwang, and T. G. Kim. IGZO-based electrolyte-gated field-effect transistor for in situ biological sensing platform. Sens Actuators B Chem. vol. 262, pp. 876–883, Jun. 2018. [CrossRef]
- H. S. Magar, R. Y. A. Hassan, and A. Mulchandani. Electrochemical Impedance Spectroscopy (EIS): Principles, Construction, and Biosensing Applications. Sensors 2021, Vol. 21, Page 6578. vol. 21, no. 19, p. 6578, Oct. 2021. [CrossRef]
- 28. A. Sanjeev Tanak, B. Jagannath, Y. Tamrakar, S. Muthukumar, and S. Prasad. Non-faradaic electrochemical impedimetric profiling of procalcitonin and C-reactive protein as a dual marker biosensor for early sepsis detection. Anal Chim Acta X. vol. 3, p. 100029, 2019. [CrossRef]
- 29. S. Cruz-Manzo and P. Greenwood. An impedance model based on a transmission line circuit and a frequency dispersion Warburg component for the study of EIS in Li-ion batteries. Journal of Electroanalytical Chemistry. vol. 871, p. 114305, Aug. 2020. [CrossRef]
- 30. J. S. Daniels and N. Pourmand. Label-Free Impedance Biosensors: Opportunities and Challenges. Electroanalysis. vol. 19, no. 12, pp. 1239–1257, Jun. 2007. [CrossRef]
- 31. M. Kang et al. An electrochemical sensor based on rhodamine B hydrazide-immobilized graphene oxide for highly sensitive and selective detection of Cu(II) †. New J. Chem. vol. 39, p. 3137, 2015. [CrossRef]
- M. Gröschl. Saliva: a reliable sample matrix in bioanalytics. Bioanalysis. vol. 9, no. 8, pp. 655–668, May 2017. [CrossRef]
Figure 1.
(a) aSyn Stamp fabrication process (b) SIP fabrication on IDEs (c) microfluidic channel addition method (d)Sample testing and regeneration process (e) Photograph showing PCL SIP DUT (e) EIS IDES prior to PCL deposition (f) Microfluidic channel SIP EIS DUT.
Figure 1.
(a) aSyn Stamp fabrication process (b) SIP fabrication on IDEs (c) microfluidic channel addition method (d)Sample testing and regeneration process (e) Photograph showing PCL SIP DUT (e) EIS IDES prior to PCL deposition (f) Microfluidic channel SIP EIS DUT.
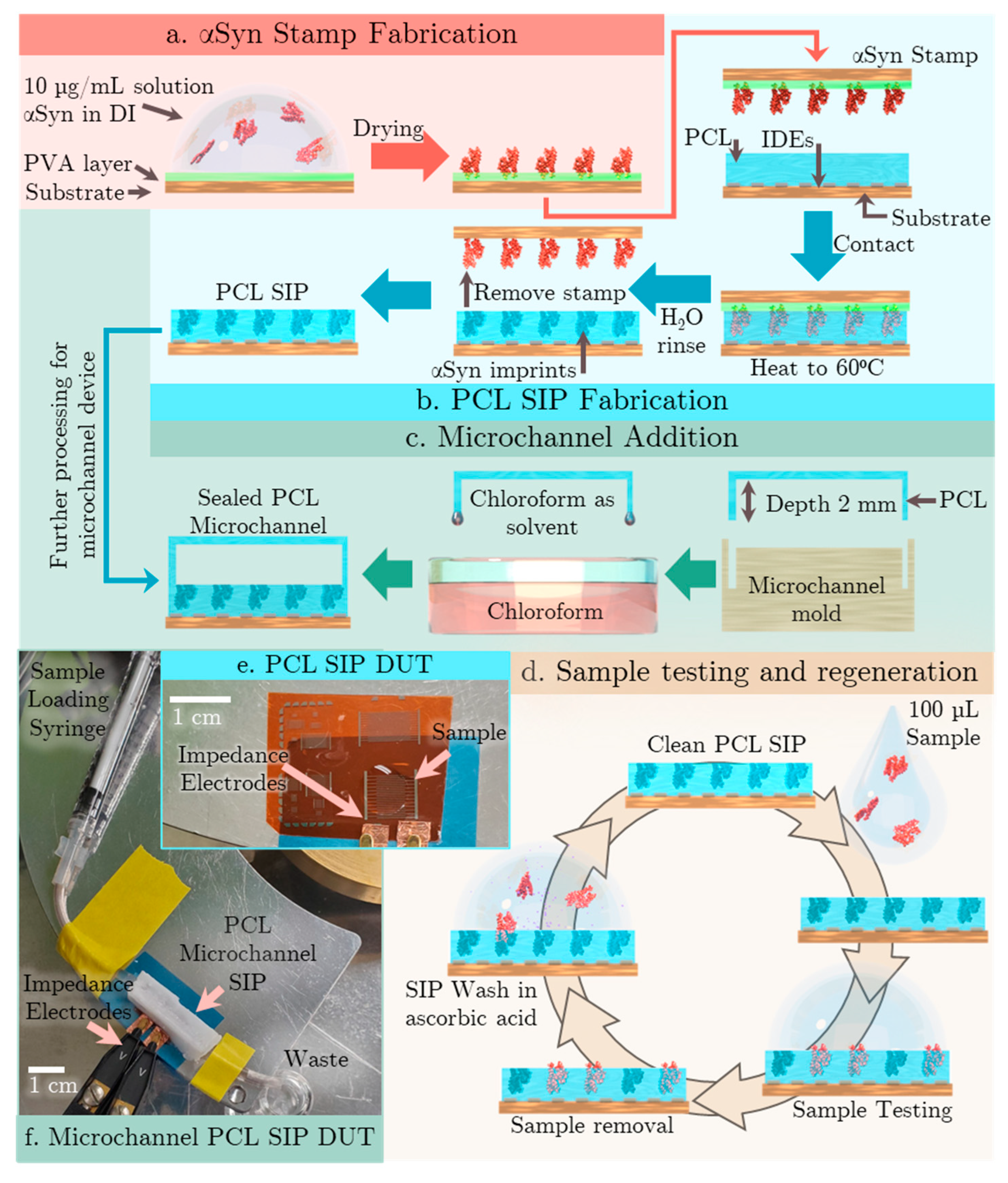
Figure 2.
(a) SEM image of PCL SIP on IDE electrode (b) AFM image of PCL SIP post testing and regeneration (c) AFM image of αSyn stamp showing variable sizes of lyophilized material.
Figure 2.
(a) SEM image of PCL SIP on IDE electrode (b) AFM image of PCL SIP post testing and regeneration (c) AFM image of αSyn stamp showing variable sizes of lyophilized material.

Figure 3.
(a) Equivalent Circuit Model (b) Equivalent circuit data shape and extraction parameters (c) Real sensor data example, and extraction locations for Randles-Ershler behaviour (d) Real data for concentrations of αSyn in water compared to equivalent circuit model data (d) EIS response of real data for dilutions series of αSyn in DI.
Figure 3.
(a) Equivalent Circuit Model (b) Equivalent circuit data shape and extraction parameters (c) Real sensor data example, and extraction locations for Randles-Ershler behaviour (d) Real data for concentrations of αSyn in water compared to equivalent circuit model data (d) EIS response of real data for dilutions series of αSyn in DI.
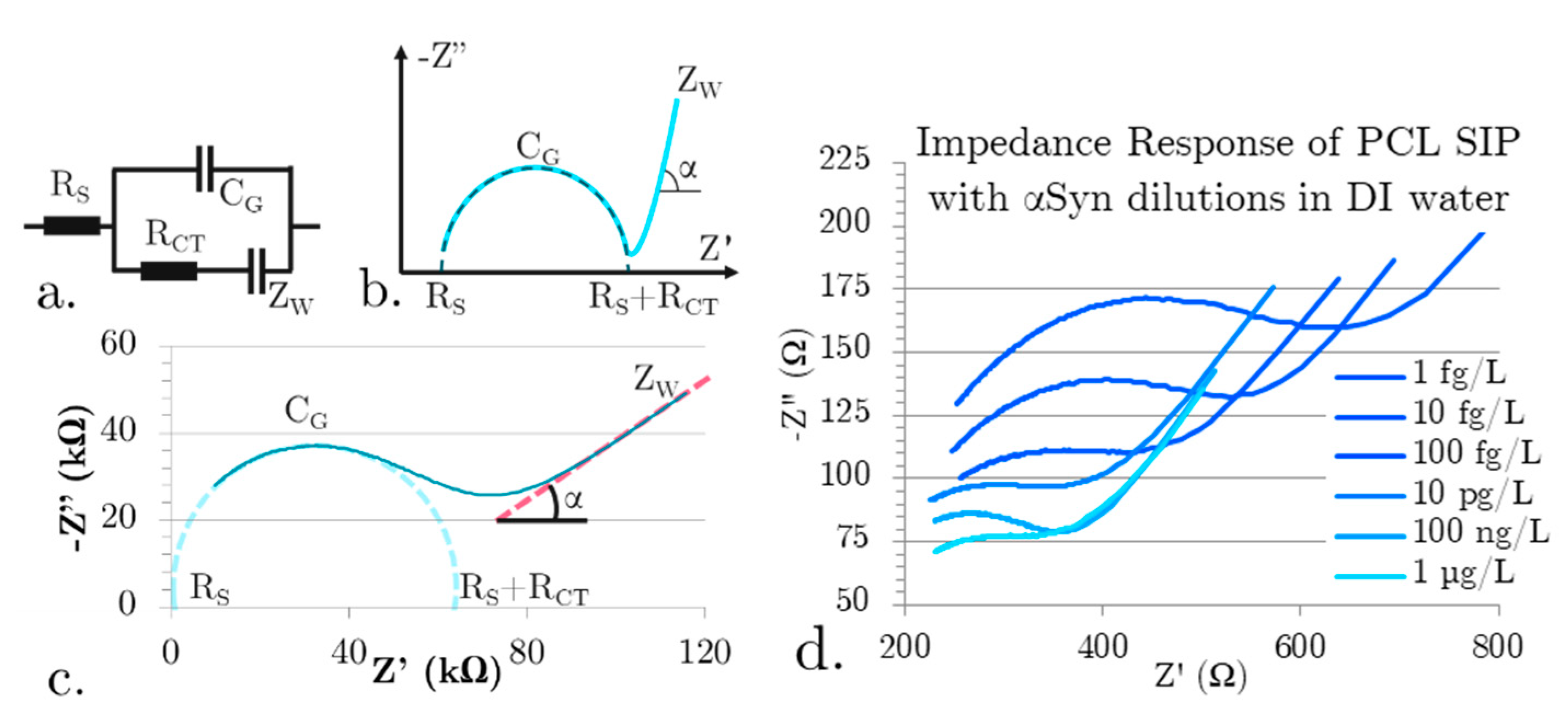
Figure 4.
(a) Raw impedance data for the total synuclein protein concentration of 100 µg/L with decreasing ratios of αSyn to ßSyn (b) αSyn concentration dependence in constant ionic concentration solutions.
Figure 4.
(a) Raw impedance data for the total synuclein protein concentration of 100 µg/L with decreasing ratios of αSyn to ßSyn (b) αSyn concentration dependence in constant ionic concentration solutions.
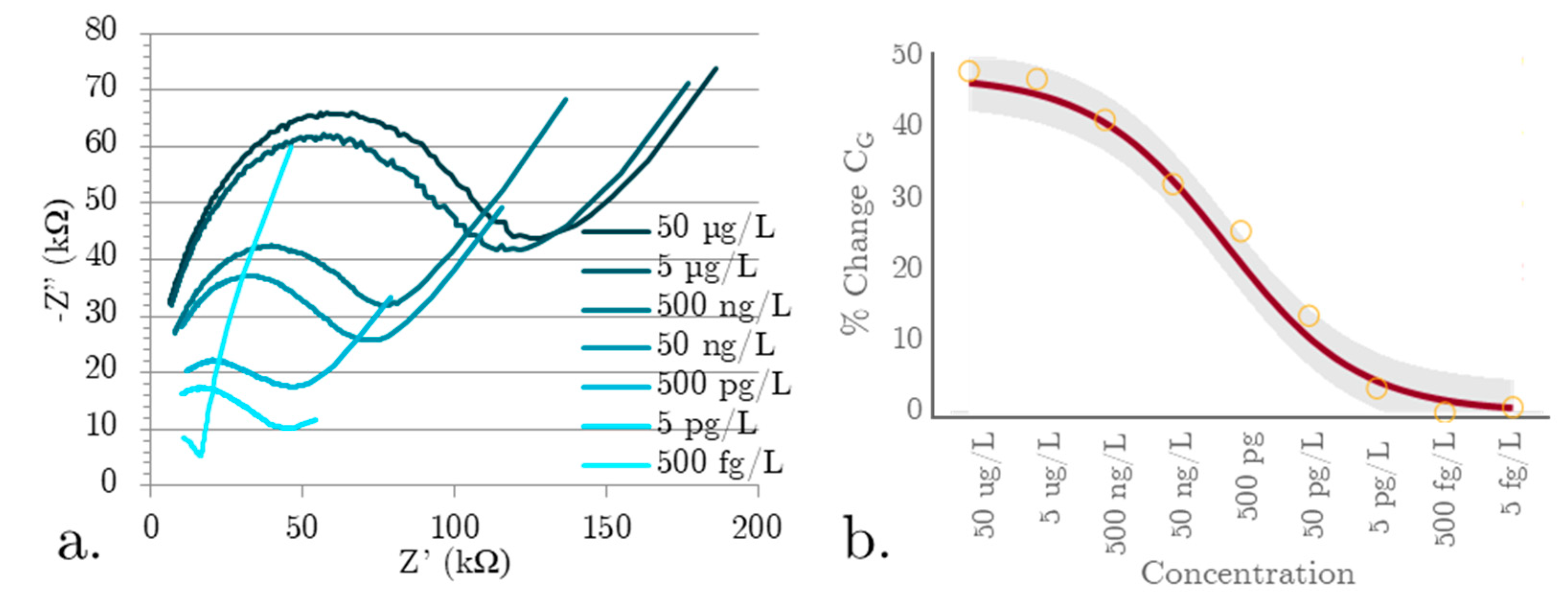
Figure 5.
a) Raw impedance data for series dilutions of αSyn in the total synuclein protein concentration in a 100 µg/L solution, b) linear fit graph showing the demonstrable linear range response to αSyn for the microfluidic channel integrated SIP EIS biosensor.
Figure 5.
a) Raw impedance data for series dilutions of αSyn in the total synuclein protein concentration in a 100 µg/L solution, b) linear fit graph showing the demonstrable linear range response to αSyn for the microfluidic channel integrated SIP EIS biosensor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



