
Submitted:
25 January 2024
Posted:
26 January 2024
You are already at the latest version
Abstract
The aberrant glycosylation is a hallmark of cancer progression and chemoresistance. It is also an immune therapeutic target for various cancers. Tunicamycin (TM) is one of the potent nucleo-side antibiotics and an inhibitor of aberrant glycosylation in various cancer cells, including breast cancer, gastric cancer, and pancreatic cancer, parallel with inhibition of cancer cell growth and progression of tumors. Thus, TM can be considered a potent antitumor drug in various cancers and may promote chemosensitivity. Mechanistically, TM impedes the role of the UDP-HexNAc enzyme in the biosynthesis of oligosaccharides, specialized macromolecules instrumental in N-linked glycosylation. Further, like chemotherapies such as doxorubicin (DOX), 5'fluorouracil, etoposide, and Cisplatin, TM induces the unfolded protein response (UPR) by blocking aberrant glycosylation. Despite TM's antitumor effectiveness, its lack of cell-type specific cytotoxicity impedes its anticancer efficacy. Thus, various nanoencapsulation techniques and materials have been considered for use in experiments to reduce TM's cytotoxicity and improve efficacy as a targeted therapy. The current review is a profound audit of the benefits and pitfalls of TM in various cancers, focusing on breast, colon, and pancreatic cancers. Additional progressive studies have also discussed the use of TM in immune checkpoints and other unique pathways. Cytotoxicity and other possibly adverse effects of TM are highlighted based on the data from in vitro and in vivo assays. In addition, the recent advances in nano-based drug delivery systems regarding TM have been emphasized. However, the potential of this nucleoside inhibitor re-quires thorough investigation and research to determine its likeliness as a viable chemotherapeutic.
Keywords:
Tunicamycin
; Nanoparticle
; Immunotherapy
; Glycosylation
; Drug resistance
; Multidrug therapy
; cancer
1. Introduction
Protein glycosylation is a unique physiological and pathophysiological process that attaches glycans (carbohydrate-based polymers) to the protein backbone via N-linked glycosylation (at the asparagines or glutamines carboxamide side chain) or O-linked glycosylation (at the hydroxyl group of serine/threonine side chains) [1,2]. Glycosylation is the most frequent post-translational modification of a protein [3] and the key molecular event associated with innate and adaptive immune systems [4]. It is involved in cell membrane formation, folding and unfolding, and protein stability [5]. Thus, the functional utilities of protein glycosylation in health and diseases are growing interest in biomedical research [4,6].
The aberrant glycosylation with unique structures, known as tumor-associated carbohydrate antigens (TACAs) [7,8,9], plays a vital role in malignant transformation, cancer progression, and metastasis [10,11]. Aberrant Glycosylation, particularly that associated with enzymes, is useful as a biomarker for cancer cells. The enzymes that cause aberrant glycosylation express cancer-specific changes, especially over-expression of certain enzymes in cellular biosynthesis. For example, in the proteomics study, analysis of the glycans and glycoproteins in cancer and normal cells can yield personalized diagnosis and treatments, partly because cancer cells' metabolism differs from normal cells. By continuing to study the cancer-modified glycans, these biomarkers can better be used to detect and diagnose specific types of cancer [12]. The spreading of primary cancer cells to distant organs is coupled with alterations and degradation of the extracellular matrix (ECM), impacting cellular interactions and structural changes of surface proteins by glycosylation [4,5,6,13,14,15,16]. Thus, glycosylation is now reconsidered in the era of cancer-targeted therapy [17]. Thereby, particular emphasis on glycosylation inhibitors is given for the rational development of immunologically targeted cancer therapies.
Tunicamycin (TM), a mixture of homologous nucleoside antibiotic inhibitors of N-linked glycosylation, may inhibit drug resistance in multidrug therapy. This drug impedes the transfer of UDP-N-acetylglucosamine (GlcNAc) to dolichol phosphate in the endoplasmic reticulum (ER) of eukaryotic cells, thus disrupting protein maturation by blocking oligosaccharides biosynthesis [18]. Oligosaccharides are instrumental in N-linked glycosylation, linking to nitrogen atoms from asparagine residue. The formulation and structure of proteins depend on this natural process. The inhibition of this process results in unfolded protein response and cellular stress, therefore resulting in cell death. However, as effective as it is, this stalwart offensive of canceling N-linked glycosylation is not cell-type specific. TM in cancer treatment could prove hazardous, as residual cytotoxicity on surrounding normal tissue could worsen the state of the disease. Nanocarriers, a prospective asset in the pharmaceutical world, may present a solution to hinder this drug's cytotoxicity.
2. Origin and Basic Biology of Tunicamycin
Tunicamycin (TM) is a nucleoside antibiotic consisting of tunicamine, uracil, N-acetylglucosamine, and fatty acyl side chain (Figure 1). Streptomyces lysosuperificus, classified by Tamura et al. in 1970, is the sole producer of this antibiotic [19]. This bacterium was cultivated in a fluid of 2% glucose, 2% gluten meal, 2% pharma media, 3% K2HPO4, 1.5% KH2PO4, and 0.3% CaCO3. After 30 hours in the culture broth, TM was detected as a product of fermentation of streptomyces lysosuperificus and accumulated over 100 hours. Newcastle disease virus was used as a subject to test the antibiotic activity. TM exhibits antibacterial activity due to its inhibition of phospho-MurNAc-pentapeptide translocase (MraY), an enzyme essential in bacterial cell wall synthesis [20]. MraY is classified as a translocase that helps in cell wall biosynthesis utilizing UDP-MurNAc, a pentapeptide necessary in peptidoglycan synthesis. TM inhibits MraY similarly to how it blocks dolichol phosphate in N-linked glycosylation. In a study conducted by Duskin et al., three variants of 3T3 fibroblasts were treated with TM: normal 3T3 cells, SV40-transformed 3T3 cells, and polyomavirus-transformed 3T3 cells [21]. TM was administered to variants in cell plates and displayed high cytotoxic effects on all 3T3 cell types, differing in some factors. After 2-4 days, the cytotoxicity of TM altered the morphology of their regular epithelioid shapes into long, pointed structures. These changes were present when TM was present in all cell plates. However, when TM was removed, any physiological changes soon regressed into a normal state, proving that TM lingers around any targeted subject and applies cytotoxicity until removed from a subject.
3. How does Tunicamycin work?
A multifaceted mode of action of TM has been reported as a therapeutic drug. In any cancer treatment, the development of drug resistance seems to be almost imminent, and this limiting factor prevents achieving cures in patients with cancers [22]. The aberrant glycosylation is one of the leading factors in developing drug resistance phenotypes in cancer cells [23]. Some oncoproteins and oncogenes promote aggressive phenotypes such as proliferation and drug resistance via N-linked glycosylation [24,25]. TM inhibits N-linked glycosylation and other N-glycans via inducing ER stress and hindering the development of invasive behavior of cancer cells. These include cancer stemness and drug resistance [26]. However, it is uncertain if TM could regulate the mutant effect of proto-oncogenes associated with the above pathological events. Mechanistically, TM can enter eukaryotes via the protein transporter major facilitator domain containing 2A (MFSD2A) and permeates through the endoplasmic reticulum (ER) membrane, causing stress [27,28]. TM induces ER stress through the unfolded protein response (UPR), promoting TRAIL-induced apoptosis [29,30]. The whole genome microarray analysis found that under sustained ER stress by TM, endothelial nitric oxide synthase (eNOS) upregulated in prostate cancer cells that activated mTORC1 through eNOS-RagC pathway and promote p62-reactive oxygen species (ROS) dependent mitochondrial apoptosis [30]. The UPR work through three receptors are located on the ER membrane. These include Protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme (IRE1), and activating transcription factor 6 (ATF6) [31,32,33,34,35]. These receptors are usually found inactive in scenarios where UPR is nonexistent due to its regulation by glucose-reacting protein 78 (GRP78). The induction of TM inhibits N-acetyl glucosamine-phosphate transmission to UDP-dolichol phosphate, resulting in unfolded proteins. GRP78 then activates the three UPR mediating receptors. The cytosolic portion of ATF6 then enters the nucleus and encodes a homologous C/EBP protein (CHOP) [36]. Overexpression of this CHOP protein leads to autophagy, which inevitably leads to apoptosis (Figure 2).
4. Tunicamycin in Cancer Therapy
Despite having some issues, TM's anticancer properties, which are mediated through UPR induction due to N-linked glycosylation in eukaryotes, are well established [37,38,39]. The following sections briefly and categorically highlight TM's impact on various cancers. The in vitro and animal studies found that the most targeted tumors for Tunicamycin are the breast, liver, colon, lungs, and pancreas, depicted in Figure 3.
Breast cancer- Breast cancers (BC) with metastasis are a global threat to female health as treatment options are still limited and weakly effective. Systemic drug therapy is still the first-line option for metastatic breast cancer treatment with surgery and radiation therapy. These include hormone therapy, chemotherapy, targeted therapy, and immunotherapy. However, the current systemic drug therapies can most likely not remove all cancer cells. Thus, new drugs and therapeutic options are urgently needed. With several preclinical drugs, TM could be a potential drug for treating BC with metastasis, as preclinical studies showed TM blocks BC growth and metastasis through the regulation of the Akt/NF-kB-signaling pathway [40] and enhance trastuzumab antitumor activity on BC [41], suggesting a promising approach of combination therapy of TM and trastuzumab to improve trastuzumab's clinical efficiency [41]. The role of Wnt/β-captain in TM-induced ER-stressed mediated apoptosis in triple-negative breast cancer (TNBC) cells has been documented, providing additional TM mechanisms [42].
Cancer stem cells (CSCs) play a significant role in tumor growth and drug resistance [43]. They are characterized to have the cell surface phenotypes CD44+/CD24- [43]. About 1-4% of BC cells carry CD44+/CD24-, which is considered highly aggressive [43]. TM-induced ER stress reduces the growth and invasion of CD44+/CD24- cells, indicating TM therapy is an exciting approach to target breast cancer stem cells [44].
Colon Cancer- Colon cancer, which is also called colorectal cancer (CRC), is one of the most common digestive tract tumors and the leading cause of cancer-related death in the elderly population in the United States and globally [45]. However, early screening can find non-cancerous abnormal growths in the colon, called polyps, which can be removed (polypectomy) and prevent cancerous growth in the colon. In the advanced stage, chemotherapy is the only option before or after radiation. Targeted and immunotherapy is also considered for advanced colon cancer.
The studies suggest TN could be an anti-CRC drug as TN treatment efficiently inhibits CRC cell growth and aggressive behavior by downregulating vimentin, FIB, and Ecacollagen type I and Slug expression levels and tumor-bearing mice's survival [46]. Further, TN has been found to enhance TRAIL-induced apoptosis in CRC cells by JNK-CHOP-mediated DR5 upregulation and inhibiting the EGFR pathway [47]. TN reprogramed cell-cell adhesion in undifferentiated CRC cells by inducing E-cadherin [48]. Interestingly, separate indirect studies claimed TM is a promoting factor of CRC [49,50].
Lung Cancer- Lung cancer is the most common cancer-related death in the human population and is highly correlated with cigarette smoking [51]. The non-small cell lung cancer (NSCLC) is the primary subtype of lung cancer and accounts for about eighty percent of the new cases of lung cancer [51,52]. The therapeutic options and efficacies of NSCLC are limited due to drug resistance. Thus, the 5-year survival rate of NSCLC remains at 15% [51,52]. In lung cancer, the therapeutic roles of TN are well documented. The studies showed that TM enhances the anticancer effects of Cisplatin on lung cancer growth via deglycosylation of PTX3 through the AKT/NF-κB signaling pathway [52]. Furthermore, TM promotes anti-immunity in a lung cancer mouse model with combination therapy [53].
Pancreatic ductal adenocarcinoma (PDAC)- Among pancreatic cancers, PDAC is the deadliest cancer in the pancreas and constitutes 90% of pancreatic cancer. PDAC represents the 7th most common cause of cancer-related death globally and has an overall 5-year survival rate not exceeding 9% [54,55]. It is anticipated that PDAC could be the second most common cause of cancer-related death in the USA by 2040 [54,56]. Based on genomic alterations, PDAC can be classified into four groups. These include stable genomes (< 50 percent structural variants per genome), scattered genomes (50-200 structural variants per genome), locally rear-ranged genomes (>200 structural variants clustered on less than three chromosomes), and unstable genomes (>200 structural variants distributed in the genome) [57,58]. This genome classification provides precision oncology knowledge and has clinical potential for PDAC patients [57].
Despite having a limited effect or no outcome of the therapy due to quick development resistance to drugs, the first-line treatments of PDAC are still FOLFIRINOX (combination of 5FU, Leucovirin, Oxaliplatin, and Irinotecan), Gemcitabine alone or in combination with nab-Paclitaxel [54,59]. The therapeutic limitations of PDAC show the urgency of finding new therapeutic options [54,59]. Mutant K-RAS in the ductal epithelial cells is the driving force of the initiation and progression of PDAC [57,60,61,62,63,64], which can be promoted by multiple factors, including CCN-family proteins [65,66]. Several preclinical studies are actively involved in discovering small molecules to target K-RAS mutants [67].
Mutant K-RAS plays a critical role in the immunosuppression of PDAC microenvironment [68,69,70]. Thus, immunologically, mutant K-RAS has an equally important target [71,72,73]. Despite having mixed and descriptive information on T-helper 2 (Th2) cells and associated cytokines in cancer, recent studies found that Th2 cells may be crucial regulators of colon and pancreas cancer progression by affecting immune cell composition and type II immune responses. In murine allograft models of colon and pancreatic cancers, the adoptive transfer of Th2 cells into tumor-bearing mice significantly reduces tumor growth by inducing cytotoxicity via eosinophils and macrophages' innate immune response. The studies also showed that IL-5 protects against tumor growth by recruiting and activating eosinophils [74]. Although glycosylation plays a vital role in regulating the above proteins, the impact of TM is underdetermined.
Overexpression of eukaryotic translational initiation factors in various cancers, including in PDAC, is a common alteration. The studies showed that one of the factors, eIF4E, is overexpressed in poorly differentiated and metastatic PDAC [54,75]. eIF4E phosphorylation (p-eIF4E) at serine209 by MNK1/2 serine/threonine kinases promotes its transformation activity [76,77]. Thus, upon mutant K-RAS activation in PDAC, eIF4E activated and promoted tumor invasion and metastasis. Studies have shown that TM treatment can inactivate eIF4E [78]. Based on these studies, we can deduce that TM treatment can be an ideal therapeutic approach for PDAC.
5. Tunicamycin in Immunotherapy
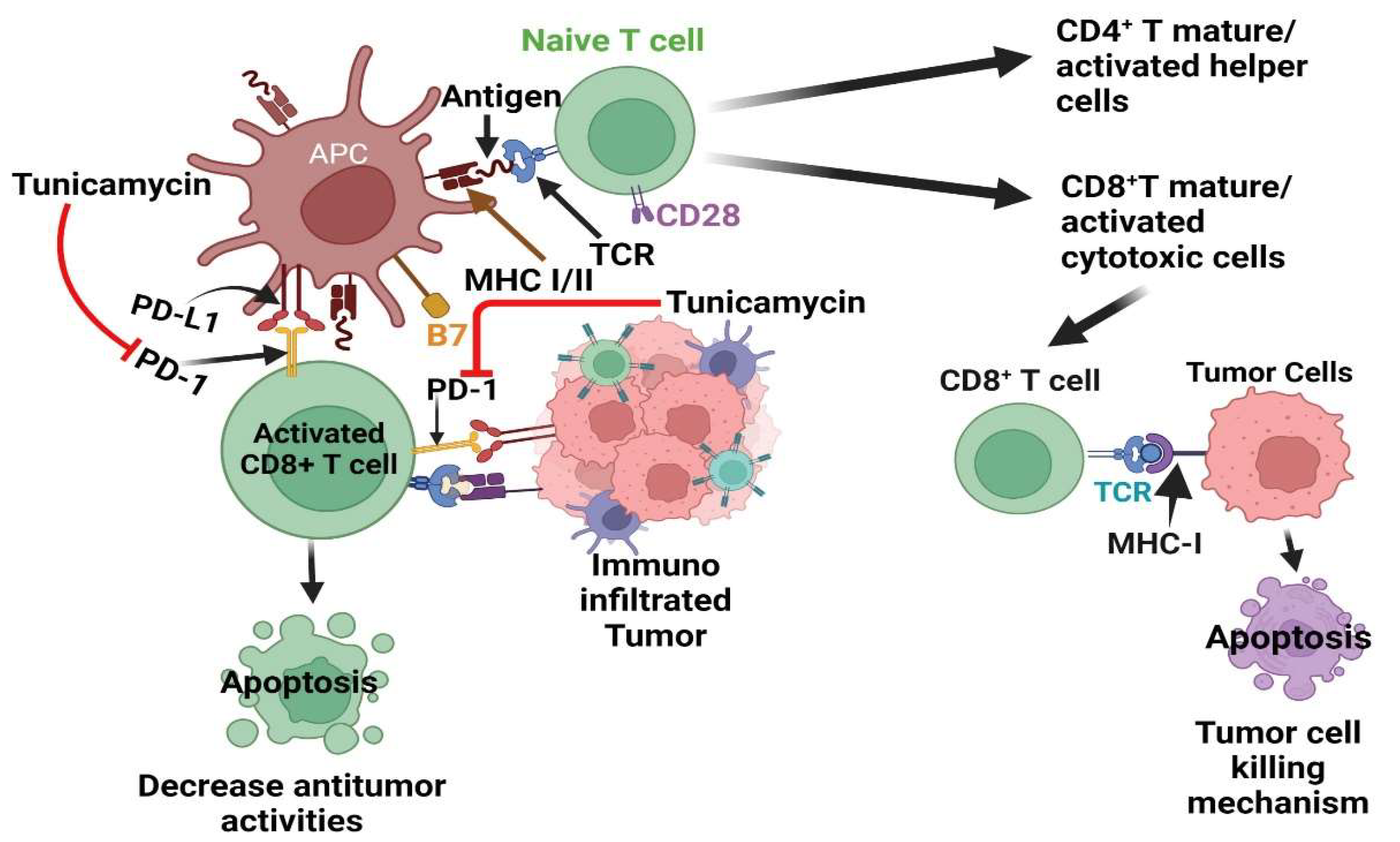
Tumor initiation and progression are linked with altered or deluded immunity [79] or reprogramming of immune cells within the tumor microenvironment [80,81]. The communication between tumor cells and the immune system is dynamic, reminiscent of a balance of immunity factors [79,80]. Thus, immunooncology is now at the forefront of basic, preclinical, and clinical research and has recently been harnessed as one of the pillars of cancer therapy [79,82,83,84,85,86]. The central principle of cancer immunotherapy is eliminating cancer cells by host cytotoxic immune CD8+ T cells [79,84,85,86,87]. However, this does not always happen in the tumor ecosystem, as tumor cells manipulate the system, cause tumor-specific CD8+ T cell dysfunction via antigen-derive differentiation program [88], or activate various immune checkpoint suppressive mechanisms. These include regulatory T (Treg) cells, over-expression of program death-1 (PD-1) receptor and its ligand PD-L1, CTLA-4, and others [83,84,89,90], which are briefly depicted in Figure 4.
In many different malignancies, immune-targeted therapy primarily focuses on the T-cell base [79], as the T-cells have the potency to suppress tumor growth clinically [79,87,91,92]. Despite the clinical successes of immune checkpoint inhibitors, the mechanisms underlying inhibitors' upregulation in cancer cells and infiltrating T cells have only begun to be understood. The emerging findings showed that Indoleamine 2,3-dioxygenase 1 (IDO1) is overproduced in highly tumorigenic self-renewing repopulating cancer cells [93]. IDO1 catalyzes the committing and rate-limiting step of the kynurenine (KYN) metabolic pathway, leading to abundant KYN release [93,94]. KYN is then absorbed in CD8+ T cells, activating the aryl hydrocarbon receptor (AhR) that causes PD-1 overexpression [93]. This sequential event adopts a tumor immunological microenvironment that is defective in recognizing and killing cancer cells [94].
The PD-L1 regulation in cancer cells is mediated by multiple pathways depending on the cancer type. These pathways are genomic alterations, epigenetic modifications, transcriptional and post-transcriptional regulation, post-translation modifications, and exosomal transport [95,96,97].
In the tumor microenvironment, the PD-1 receptor is highly N-glycosylated in T cells to maintain PD-1 stability and interaction with the PD-L1 ligand expressed in tumor cells to dampen the activity of T-cell receptor (TCR)/CD28) signals [98]. The studies showed several N-glycosylated inhibitors, including TM treatment deglycosylated PD-1 [98,99] (Figure 4). Thus, TM could be considered an ideal therapy to target PD-1 alone or in combination of other PD-1 inhibitors.
Like PD-1, PD-L1 is also highly N-glycosylated to maintain its stability and binding affinity with PD-1 in various cancers and eradicate Triple-negative Breast cancer (TNBC) by targeting glycosylated PD-L1 [100]. Thus, studies suggest targeting PD-L1 glycosylation could be an ideal therapeutic option by rationale combination of cancer immunotherapies [101,102,103,104].
Interleukins (ILs) are secreted cytokines that play vital roles in the biology of cancers and immune functions [105]. Although some ILs (i.e., IL-6, IL-10, and IL-17) boost tumor growth, some ILs may promote the immune system's antitumor response. These include IL-2, IL-12 and IL-5 [105,106]. Previous studies have shown that glycosylation plays a role in IL-12 biogenesis, but the loss of glycosylation has little or no effect on IL-12 and IL-23 secretion, heterodimerization, and biological activities [107]. IL-5 is produced by both hematopoietic and non-hematopoietic cells [108]. T-cell-derived IL-5 controls eosinophil production and their activation via JAK-STAT and RAS/Raf-ERK signaling pathways [108]. Recent studies have shown that IL-5-producing CD4-positive T cells and eosinophils collectively enhance response to immune checkpoint blockade (ICB) in breast cancer. Further, it has also been reported that ICB increased IL-5 production by CD4 T-cells elevated eosinophil production from bone marrow and infiltration that eventually enhanced ICB efficacy [106,109]. T-cell-derived IL-5 exhibits heterogeneous glycosylation with four N-glycosylation sites in the extracellular regions, and removal of N- or O-linked glycosylation on IL-5 enhances the potency of the cytokine [110], without having effect on lymphocyte proliferation in a lymphocyte proliferation assay following the treatment of glycosylation inhibitor tunicamycin (TM) [108,111]. Previously, however, it has been shown that in humans, IL-5 acts only on eosinophils and basophils for their growth, maturation and activation as these cells express IL-5 receptors [112,113,114]. Thus, the results of these studies suggest that inhibition of glycosylation in IL-5 by TM could be effective only on eosinophil production, maturation and their activation. However, further studies are warranted.
In pancreatic cancer progression, IL-5/IL-5Rα roles are possibly a double-edged sword, with both pro- and antitumor activities being discovered recently [74,115,116,117]. However, the mechanistic roles of IL-5 are still unclear. Studies have shown that TM can inhibit endoplasmic-reticulum (ER)-stress-mediated autophagy and promote the effectiveness of chemotherapies on pancreatic cancer cells [118]. Further, studies showed ER-stressed inhibitors reduce IL-5 production in the Th-2 cells (naïve CD4+ T cells) [119], which inhibit colon and pancreatic cancer growth by promoting antitumorigenic responses from macrophages and eosinophils [74]. However, the effect of ER-stressed inhibitors, including TM, on IL-5 in pancreatic cancer cells and localized eosinophils is still uncertain.
In conclusion, TM shows promise in drug-induced immunotherapy when combined with other PD-1 and PD-L1 inhibitors, such as monoclonal antibodies or various chemotherapeutics (Figure 4).
6. The Pitfalls of the Use of Tunicamycin
As much as TM promises to be an efficient drug in chemo-and immunotherapy, evidence of its cytotoxicity proves that it affects all cells, whether healthy or cancerous. Thus, TM could prove consequential if TM's residual cytotoxicity affects surrounding tissues around a tumor, possibly weakening body systems depending on where TM is inducted. Direct administration of TM in murine models of leukemia resulted in potent liver toxicity. Unfortunately, as much as TM is adequate in meeting the thresholds of chemotherapy, TM's cytotoxicity is not cell-type specific. This makes TM a liability in its free form. However, nanoparticle encapsulation technology can bypass this problem. Nanoscale particles, particularly those engineered with cell-specific ligands, can recognize specific cell types. As such, encapsulating TM inside a nanoparticle can rescue healthy cells from non-specific effects of TM. Further, chemical conjugation of TM to an antibody or to a macromolecule like PEG, can result in the formation of antibody-drug conjugates (ADCs) or polymer-drug conjugates (PDCs) respectively. Both variants can be used to augment cellular accumulation of TM inside cancer cells. Collectively, nontechnology can also provide exciting prospects that may improve TM's implications with cytotoxicity and efficiency [120,121,122,123].
7. Outlook
Tunicamycin shows promise if it is to be proven applicable in targeted therapy; however, in vivo, experiments and assays may prove the need for more research and trials in mice models before it can be approved. For such a cytotoxic drug with an effect as such, many precautions need to be taken when it comes to nanoparticle delivery. Glycotherapy modeled after a TM-focused approach can suppress the invasion of cancer cells that spread beyond the tumor mass. The UPR holds many risks as TM could have a toxic buildup in surrounding healthy tissue. It should be recalled that TM's cytotoxicity is not cell-type specific. However, the morphological effects of TM can be reversed by taking TM out of the equation, as shown earlier. The antitumorigenic effects of TM can be used in next-generation cancer therapy. Many things can go wrong as the carrier does not reach the predetermined deposition site. This method should be perfected as, with many nanocarriers, toxic buildup in the organs usually leads to more complications and may result in a weaker prognosis.
Author Contributions
Conceptualization, SB and SKB; validation, MQ, SU, and DJM; investigation, SB, AAA, and SKB; resources, XX; data curation, SB, AAA, and SKB; writing—SB and AAA; writing (editing and reviewing)—AAA, SB, AK, PG, MQ and SKB; visualization, SB, AAA, and SKB; supervision, SB and SKB; project administration, SKB and SB; funding acquisition, SB, MQ, and SKB. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by VA Merit Review grants (SKB and SB) VA Research Career Award Fund (SKB), and this research was supported by NIH grant number 1P20 GM109024 from the National Institute of General Medical Sciences (NIGMS) (MQ).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Dr. Inamul Haque and Lauren Birkeness for their editorial help. Graphical abstracts and figures were created on Biorender.com.
Conflicts of Interest
"The authors declare no conflicts of interest.".
References
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22(6):736-56. Epub 20111218. [CrossRef]
- Clausen H, Bennett EP. A family of UDP-GalNAc: polypeptide N-acetylgalactosaminyl-transferases control the initiation of mucin-type O-linked glycosylation. Glycobiology. 1996;6(6):635-46. [CrossRef] [PubMed]
- Varki A. Biological roles of glycans. Glycobiology. 2017;27(1):3-49. Epub 20160824. [CrossRef]
- Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291(5512):2370-6. [CrossRef] [PubMed]
- Schoberer J, Shin YJ, Vavra U, Veit C, Strasser R. Analysis of Protein Glycosylation in the ER. Methods Mol Biol. 2018;1691:205-22. [CrossRef]
- Takahashi K, Raska M, Stuchlova Horynova M, Hall SD, Poulsen K, Kilian M, et al. Enzymatic sialylation of IgA1 O-glycans: implications for studies of IgA nephropathy. PLoS One. 2014;9(2):e99026. Epub 20140611. [CrossRef]
- Hakomori S. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anticancer vaccines. Adv Exp Med Biol. 2001;491:369-402. [CrossRef] [PubMed]
- Matsumoto Y, Ju T. Aberrant Glycosylation as Immune Therapeutic Targets for Solid Tumors. Cancers (Basel). 2023;15(14). Epub 20230708. [CrossRef]
- Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, Mechref Y. Glycosylation Changes in Brain Cancer. ACS Chem Neurosci. 2018;9(1):51-72. Epub 20171107. [CrossRef]
- Hauselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol. 2014;4:28. Epub 20140213. [CrossRef]
- Munkley J, Elliott DJ. Hallmarks of glycosylation in cancer. Oncotarget. 2016;7(23):35478-89. [CrossRef]
- Meany DL, Chan DW. Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin Proteomics. 2011;8(1):7. Epub 20110603. [CrossRef]
- Arnal-Estape A, Nguyen DX. Sweets for a bitter end: lung cancer cell-surface protein glycosylation mediates metastatic colonization. Cancer Discov. 2015;5(2):109-11. [CrossRef]
- Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21(3):505-15. [CrossRef] [PubMed]
- MacDougall JR, Matrisian LM. Contributions of tumor and stromal matrix metalloproteinases to tumor progression, invasion and metastasis. Cancer Metastasis Rev. 1995;14(4):351-62. [CrossRef] [PubMed]
- Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10(6):415-33. [CrossRef] [PubMed]
- Mereiter S, Balmana M, Campos D, Gomes J, Reis CA. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell. 2019;36(1):6-16. [CrossRef] [PubMed]
- Wu J, Chen S, Liu H, Zhang Z, Ni Z, Chen J, et al. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J Exp Clin Cancer Res. 2018;37(1):272. Epub 20181109. [CrossRef]
- Akira Takatsuki KA, Gakuzo Tamura. Tunicamycin, A New Antibiotic, I Isolation And Characterization of Tunicamycin. The Journal of Antibiotics. 1970;24(4):215-23. [CrossRef]
- Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K, Kaeser B, Ichikawa S, et al. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol. 2018;25(3):217-24. Epub 2018/02/21. [CrossRef]
- Duksin D, Bornstein P. Changes in surface properties of normal and transformed cells caused by tunicamycin, an inhibitor of protein glycosylation. Proc Natl Acad Sci U S A. 1977;74(8):3433-7. Epub 1977/08/01. [CrossRef]
- Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299-309. Epub 20191113. [CrossRef]
- Arey BJ. The Role of Glycosylation in Receptor Signaling IntechOpen. 2012. Epub 09/26/2012. [CrossRef]
- Seales EC, Jurado GA, Singhal A, Bellis SL. Ras oncogene directs expression of a differentially sialylated, functionally altered beta1 integrin. Oncogene. 2003;22(46):7137-45. [CrossRef] [PubMed]
- Very N, Lefebvre T, El Yazidi-Belkoura I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget. 2018;9(1):1380-402. Epub 20171103. [CrossRef]
- Wang Y, Zhang L, He Z, Deng J, Zhang Z, Liu L, et al. Tunicamycin induces ER stress and inhibits tumorigenesis of head and neck cancer cells by inhibiting N-glycosylation. Am J Transl Res. 2020;12(2):541-50. Epub 2020/03/21.
- Bassik MC, Kampmann M. Knocking out the door to tunicamycin entry. Proc Natl Acad Sci U S A. 2011;108(29):11731-2. Epub 20110706. [CrossRef]
- Reiling JH, Clish CB, Carette JE, Varadarajan M, Brummelkamp TR, Sabatini DM. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc Natl Acad Sci U S A. 2011;108(29):11756-65. Epub 20110615. [CrossRef]
- de-Freitas-Junior JC, Bastos LG, Freire-Neto CA, Rocher BD, Abdelhay ES, Morgado-Diaz JA. N-glycan biosynthesis inhibitors induce in vitro anticancer activity in colorectal cancer cells. J Cell Biochem. 2012;113(9):2957-66. [CrossRef] [PubMed]
- Guha P, Kaptan E, Gade P, Kalvakolanu DV, Ahmed H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget. 2017;8(40):68191-207. Epub 20170715. [CrossRef]
- Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22(16):R622-6. [CrossRef] [PubMed]
- Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108(12):2777-93. Epub 20110809. [CrossRef]
- Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3(6):411-21. [CrossRef] [PubMed]
- Read A, Schroder M. The Unfolded Protein Response: An Overview. Biology (Basel). 2021;10(5). Epub 20210429. [CrossRef]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110(10):1383-8. [CrossRef]
- Lei Y, Wang S, Ren B, Wang J, Chen J, Lu J, et al. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS One. 2017;12(8):e0183680. Epub 20170825. [CrossRef]
- Hasegawa A, Osuga Y, Hirota Y, Hamasaki K, Kodama A, Harada M, et al. Tunicamycin enhances the apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand in endometriotic stromal cells. Hum Reprod. 2009;24(2):408-14. Epub 20081101. [CrossRef] [PubMed]
- Delom F, Emadali A, Cocolakis E, Lebrun JJ, Nantel A, Chevet E. Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ. 2007;14(3):586-96. Epub 20060721. [CrossRef] [PubMed]
- Banerjee A, Lang JY, Hung MC, Sengupta K, Banerjee SK, Baksi K, et al. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem. 2011;286(33):29127-38. Epub 20110615. [CrossRef]
- Wang X, Xiong W, Tang Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase B/nuclear factor-kappaB signaling pathway. Oncol Lett. 2018;15(4):4137-42. Epub 20180126. [CrossRef]
- Han X, Zhang X, Li H, Huang S, Zhang S, Wang F, et al. Tunicamycin enhances the antitumor activity of trastuzumab on breast cancer in vitro and in vivo. Oncotarget. 2015;6(36):38912-25. [CrossRef]
- You Z, He L, Yan N. Tunicamycin-Induced Endoplasmic Reticulum Stress Promotes Breast Cancer Cell MDA-MB-231 Apoptosis through Inhibiting Wnt/beta-Catenin Signaling Pathway. J Healthc Eng. 2021;2021:6394514. Epub 20210715. [CrossRef]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983-8. Epub 20030310. [CrossRef]
- Nami B, Donmez H, Kocak N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24- phenotype breast cancer stem cells. Exp Toxicol Pathol. 2016;68(7):419-26. Epub 20160624. [CrossRef] [PubMed]
- Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73(3):233-54. Epub 20230301. [CrossRef] [PubMed]
- You S, Li W, Guan Y. Tunicamycin inhibits colon carcinoma growth and aggressiveness via modulation of the ERK-JNK-mediated AKT/mTOR signaling pathway. Mol Med Rep. 2018;17(3):4203-12. Epub 20180117. [CrossRef]
- Guo X, Meng Y, Sheng X, Guan Y, Zhang F, Han Z, et al. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs. 2017;28(1):66-74. [CrossRef] [PubMed]
- de Freitas Junior JC, Silva Bdu R, de Souza WF, de Araujo WM, Abdelhay ES, Morgado-Diaz JA. Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell-cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother Pharmacol. 2011;68(1):227-38. Epub 20101007. [CrossRef] [PubMed]
- Chern YJ, Wong JCT, Cheng GSW, Yu A, Yin Y, Schaeffer DF, et al. The interaction between SPARC and GRP78 interferes with ER stress signaling and potentiates apoptosis via PERK/eIF2alpha and IRE1alpha/XBP-1 in colorectal cancer. Cell Death Dis. 2019;10(7):504. Epub 20190626. [CrossRef]
- Liu CY, Hsu CC, Huang TT, Lee CH, Chen JL, Yang SH, et al. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol Oncol. 2018;12(10):1706-17. Epub 20180820. [CrossRef]
- Massion PP, Carbone DP. The molecular basis of lung cancer: molecular abnormalities and therapeutic implications. Respir Res. 2003;4(1):12. Epub 20031007. [CrossRef]
- Ahmmed B, Khan MN, Nisar MA, Kampo S, Zheng Q, Li Y, et al. Tunicamycin enhances the suppressive effects of Cisplatin on lung cancer growth through PTX3 glycosylation via AKT/NF-kappaB signaling pathway. Int J Oncol. 2019;54(2):431-42. Epub 20181128. [CrossRef]
- Liu L, Li S, Qu Y, Bai H, Pan X, Wang J, et al. Ablation of ERO1A induces lethal endoplasmic reticulum stress responses and immunogenic cell death to activate anti-tumor immunity. Cell Rep Med. 2023;4(10):101206. Epub 20230927. [CrossRef]
- Shin S, Solorzano J, Liauzun M, Pyronnet S, Bousquet C, Martineau Y. Translational alterations in pancreatic cancer: a central role for the integrated stress response. NAR Cancer. 2022;4(4):zcac031. Epub 20221028. [CrossRef]
- Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017;18(7). Epub 2017/06/24. [CrossRef]
- Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw Open. 2021;4(4):e214708. Epub 20210401. [CrossRef]
- Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16(4):207-20. Epub 2019/02/06. [CrossRef] [PubMed]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495-501. Epub 2015/02/27. [CrossRef]
- Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. Epub 20160421. [CrossRef] [PubMed]
- Banerjee SK, Maity G, Haque I, Ghosh A, Sarkar S, Gupta V, et al. Human pancreatic cancer progression: an anarchy among CCN-siblings. J Cell Commun Signal. 2016;10(3):207-16. Epub 2016/08/20. [CrossRef]
- Basu SK, Lee S, Salotti J, Basu S, Sakchaisri K, Xiao Z, et al. Oncogenic RAS-Induced Perinuclear Signaling Complexes Requiring KSR1 Regulate Signal Transmission to Downstream Targets. Cancer Res. 2018;78(4):891-908. Epub 2017/12/21. [CrossRef]
- Banerjee SK, Makdisi WF, Weston AP, Campbell DR. A two-step enriched-nested PCR technique enhances sensitivity for detection of codon 12 K-ras mutations in pancreatic adenocarcinoma. Pancreas. 1997;15(1):16-24. Epub 1997/07/01. [CrossRef] [PubMed]
- Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63(9):2005-9. [PubMed]
- di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220-9. Epub 2013/04/30. [CrossRef]
- Birkeness LB, Banerjee S, Quadir M, Banerjee SK. The role of CCNs in controlling cellular communication in the tumor microenvironment. J Cell Commun Signal. 2022. Epub 2022/06/09. [CrossRef] [PubMed]
- Dhar G, Mehta S, Banerjee S, Gardner A, McCarty BM, Mathur SC, et al. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: a potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 2007;254(1):63-70. Epub 2007/03/27. [CrossRef] [PubMed]
- Bannoura SF, Khan HY, Azmi AS. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front Oncol. 2022;12:1013902. Epub 2022/12/20. [CrossRef]
- Hafezi S, Saber-Ayad M, Abdel-Rahman WM. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. Int J Mol Sci. 2021;22(19). Epub 20210923. [CrossRef]
- Dias Carvalho P, Guimaraes CF, Cardoso AP, Mendonca S, Costa AM, Oliveira MJ, et al. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018;78(1):7-14. Epub 20171220. [CrossRef] [PubMed]
- Hamarsheh S, Gross O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun. 2020;11(1):5439. Epub 20201028. [CrossRef]
- Cheng NC, Vonderheide RH. Immune vulnerabilities of mutant KRAS in pancreatic cancer. Trends Cancer. 2023;9(11):928-36. Epub 20230729. [CrossRef]
- Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell. 2018;33(4):563-9. [CrossRef]
- Thind K, Padrnos LJ, Ramanathan RK, Borad MJ. Immunotherapy in pancreatic cancer treatment: a new frontier. Therap Adv Gastroenterol. 2017;10(1):168-94. Epub 20161017. [CrossRef]
- Jacenik D, Karagiannidis I, Beswick EJ. Th2 cells inhibit growth of colon and pancreas cancers by promoting anti-tumorigenic responses from macrophages and eosinophils. Br J Cancer. 2023;128(2):387-97. Epub 20221114. [CrossRef]
- Baylot V, Andrieu C, Katsogiannou M, Taieb D, Garcia S, Giusiano S, et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011;2(10):e221. Epub 20111020. [CrossRef]
- Topisirovic I, Ruiz-Gutierrez M, Borden KL. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res. 2004;64(23):8639-42. [CrossRef] [PubMed]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21(24):3232-7. Epub 20071130. [CrossRef]
- Xing Y, Ge Y, Liu C, Zhang X, Jiang J, Wei Y. ER stress inducer tunicamycin suppresses the self-renewal of glioma-initiating cell partly through inhibiting Sox2 translation. Oncotarget. 2016;7(24):36395-406. [CrossRef]
- Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell. 2016;164(6):1233-47. [CrossRef]
- Ng MSF, Kwok I, Tan L, Shi C, Cerezo-Wallis D, Tan Y, et al. Deterministic reprogramming of neutrophils within tumors. Science. 2024;383(6679):eadf6493. Epub 20240112. [CrossRef] [PubMed]
- Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front Immunol. 2018;9:1977. Epub 20180831. [CrossRef]
- Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447-62. [CrossRef] [PubMed]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44(2):343-54. Epub 20160209. [CrossRef]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56-61. [CrossRef] [PubMed]
- Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, et al. Immune Checkpoint Inhibitors in Cancer Therapy. Curr Oncol. 2022;29(5):3044-60. Epub 20220424. [CrossRef]
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450-61. Epub 20150406. [CrossRef]
- Raskov H, Orhan A, Christensen JP, Gogenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. 2021;124(2):359-67. Epub 20200915. [CrossRef]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 2016;45(2):389-401. Epub 20160809. [CrossRef]
- Jiang Y, Chen M, Nie H, Yuan Y. PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother. 2019;15(5):1111-22. Epub 20190319. [CrossRef]
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651-68. Epub 20200520. [CrossRef]
- Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33(17):1974-82. Epub 20150120. [CrossRef]
- Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23. Epub 20150616. [CrossRef]
- Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell. 2018;33(3):480-94 e7. [CrossRef] [PubMed]
- Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol Sci. 2018;39(3):307-25. Epub 20171215. [CrossRef] [PubMed]
- Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14(1):10. Epub 20210107. [CrossRef]
- Escors D, Gato-Canas M, Zuazo M, Arasanz H, Garcia-Granda MJ, Vera R, et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct Target Ther. 2018;3:26. Epub 20180928. [CrossRef]
- Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell. 2019;76(3):359-70. Epub 20191024. [CrossRef]
- Sun L, Li CW, Chung EM, Yang R, Kim YS, Park AH, et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020;80(11):2298-310. Epub 20200310. [CrossRef]
- Salatino M, Girotti MR, Rabinovich GA. Glycans Pave the Way for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell. 2018;33(2):155-7. [CrossRef] [PubMed]
- Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell. 2018;33(2):187-201 e10. [CrossRef]
- Wang YN, Lee HH, Hsu JL, Yu D, Hung MC. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J Biomed Sci. 2020;27(1):77. Epub 20200703. [CrossRef]
- Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell. 2019;36(2):168-78 e4. Epub 20190718. [CrossRef]
- Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. Epub 20160830. [CrossRef]
- Yaghoubi N, Soltani A, Ghazvini K, Hassanian SM, Hashemy SI. PD-1/ PD-L1 blockade as a novel treatment for colorectal cancer. Biomed Pharmacother. 2019;110:312-8. Epub 20181203. [CrossRef] [PubMed]
- Samadi M, Kamrani A, Nasiri H, Shomali N, Heris JA, Shahabi P, et al. Cancer immunotherapy focusing on the role of interleukins: A comprehensive and updated study. Pathol Res Pract. 2023;249:154732. Epub 20230801. [CrossRef] [PubMed]
- Blomberg OS, Spagnuolo L, Garner H, Voorwerk L, Isaeva OI, van Dyk E, et al. IL-5-producing CD4(+) T cells and eosinophils cooperate to enhance response to immune checkpoint blockade in breast cancer. Cancer Cell. 2023;41(1):106-23 e10. Epub 20221215. [CrossRef] [PubMed]
- Bohnacker S, Hildenbrand K, Aschenbrenner I, Muller SI, Bieren JE, Feige MJ. Influence of glycosylation on IL-12 family cytokine biogenesis and function. Mol Immunol. 2020;126:120-8. Epub 20200818. [CrossRef] [PubMed]
- Takatsu K. Interleukin-5 and IL-5 receptor in health and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(8):463-85. [CrossRef]
- Grisaru-Tal S, Munitz A. T cell-eosinophil crosstalk-A new road for effective immune checkpoint blockade in breast cancer? Cancer Cell. 2023;41(1):9-11. Epub 20221215. [CrossRef] [PubMed]
- Kodama S, Tsujimoto M, Tsuruoka N, Sugo T, Endo T, Kobata A. Role of sugar chains in the in-vitro activity of recombinant human interleukin 5. Eur J Biochem. 1993;211(3):903-8. [CrossRef] [PubMed]
- Kunimoto DY, Allison KC, Watson C, Fuerst T, Armstrong GD, Paul W, et al. High-level production of murine interleukin-5 (IL-5) utilizing recombinant baculovirus expression. Purification of the rIL-5 and its use in assessing the biologic role of IL-5 glycosylation. Cytokine. 1991;3(3):224-30. [CrossRef] [PubMed]
- Greenfeder S, Umland SP, Cuss FM, Chapman RW, Egan RW. Th2 cytokines and asthma. The role of interleukin-5 in allergic eosinophilic disease. Respir Res. 2001;2(2):71-9. Epub 20010308. [CrossRef]
- Hirai K, Yamaguchi M, Misaki Y, Takaishi T, Ohta K, Morita Y, et al. Enhancement of human basophil histamine release by interleukin 5. J Exp Med. 1990;172(5):1525-8. [CrossRef]
- Resnick MB, Weller PF. Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol. 1993;8(4):349-55. [CrossRef] [PubMed]
- Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. 2022;40(2):153-67 e11. Epub 20220203. [CrossRef]
- Davis BP, Rothenberg ME. Eosinophils and cancer. Cancer Immunol Res. 2014;2(1):1-8. [CrossRef] [PubMed]
- Gitto SB, Beardsley JM, Nakkina SP, Oyer JL, Cline KA, Litherland SA, et al. Identification of a novel IL-5 signaling pathway in chronic pancreatitis and crosstalk with pancreatic tumor cells. Cell Commun Signal. 2020;18(1):95. Epub 20200617. [CrossRef]
- Thakur PC, Miller-Ocuin JL, Nguyen K, Matsuda R, Singhi AD, Zeh HJ, et al. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J Transl Med. 2018;16(1):190. Epub 20180709. [CrossRef]
- Li A, Song NJ, Riesenberg BP, Li Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front Immunol. 2019;10:3154. Epub 20200211. [CrossRef]
- Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1(1):10-29. Epub 20160603. [CrossRef]
- Chen Z, Wang Z, Gu Z. Bioinspired and Biomimetic Nanomedicines. Acc Chem Res. 2019;52(5):1255-64. Epub 2019/04/13. [CrossRef]
- Dwivedi PD, Tripathi A, Ansari KM, Shanker R, Das M. Impact of nanoparticles on the immune system. J Biomed Nanotechnol. 2011;7(1):193-4. [CrossRef] [PubMed]
- Ray P, Haideri, N, Haque,I, Mohsmmed,O, Chakraborty,S, Banerjee,S, Quadir,M, Brinker,A.E., Banerjee,S.K. The Impact of Nanoparticles on the Immune System: A Gray Zone of Nanomedicine. J Immunological Sci. 2021;5(5):19-33. [CrossRef]
Figure 1.
Chemical structure of Tunicamycin. The molecule consists of various structural fragments crucial in its anti-cancer properties. The first fragment of interest is the aminosugar moiety, which consists of a hexose sugar called N, N'-diacetyl chitobiose. This fragment is responsible for the initial recognition and binding of Tunicamycin to its target enzyme, UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase (GPT). The interaction between the aminosugar and the enzyme is crucial for inhibiting the biosynthesis of N-linked glycoproteins, a process essential for cancer cell growth and metastasis. Another vital fragment is the nucleoside moiety, which consists of a thymidine derivative known as 4-(2-amino-2-deoxy-α-D-glucopyranosyl) thymine. It stabilizes the interaction between Tunicamycin and GPT, thereby enhancing the drug's potency. The third fragment of interest is the fatty acid chain, which consists of a 17-carbon isoprenoid structure, sometimes called decaprenyl phosphate. It is involved in anchoring the drug to the endoplasmic reticulum, the site of glycoprotein synthesis, facilitating its localization to the target enzyme. The fatty acid chain for various derivatives of Tunicamycin, composed of 14-17 carbon chain lengths, contributes to the overall hydrophobicity of Tunicamycin, which affects its pharmacokinetic properties. Finally, the unsaturated uridine ring system, which binding to the target enzyme, and contributes to its inhibitory potency.
Figure 1.
Chemical structure of Tunicamycin. The molecule consists of various structural fragments crucial in its anti-cancer properties. The first fragment of interest is the aminosugar moiety, which consists of a hexose sugar called N, N'-diacetyl chitobiose. This fragment is responsible for the initial recognition and binding of Tunicamycin to its target enzyme, UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase (GPT). The interaction between the aminosugar and the enzyme is crucial for inhibiting the biosynthesis of N-linked glycoproteins, a process essential for cancer cell growth and metastasis. Another vital fragment is the nucleoside moiety, which consists of a thymidine derivative known as 4-(2-amino-2-deoxy-α-D-glucopyranosyl) thymine. It stabilizes the interaction between Tunicamycin and GPT, thereby enhancing the drug's potency. The third fragment of interest is the fatty acid chain, which consists of a 17-carbon isoprenoid structure, sometimes called decaprenyl phosphate. It is involved in anchoring the drug to the endoplasmic reticulum, the site of glycoprotein synthesis, facilitating its localization to the target enzyme. The fatty acid chain for various derivatives of Tunicamycin, composed of 14-17 carbon chain lengths, contributes to the overall hydrophobicity of Tunicamycin, which affects its pharmacokinetic properties. Finally, the unsaturated uridine ring system, which binding to the target enzyme, and contributes to its inhibitory potency.
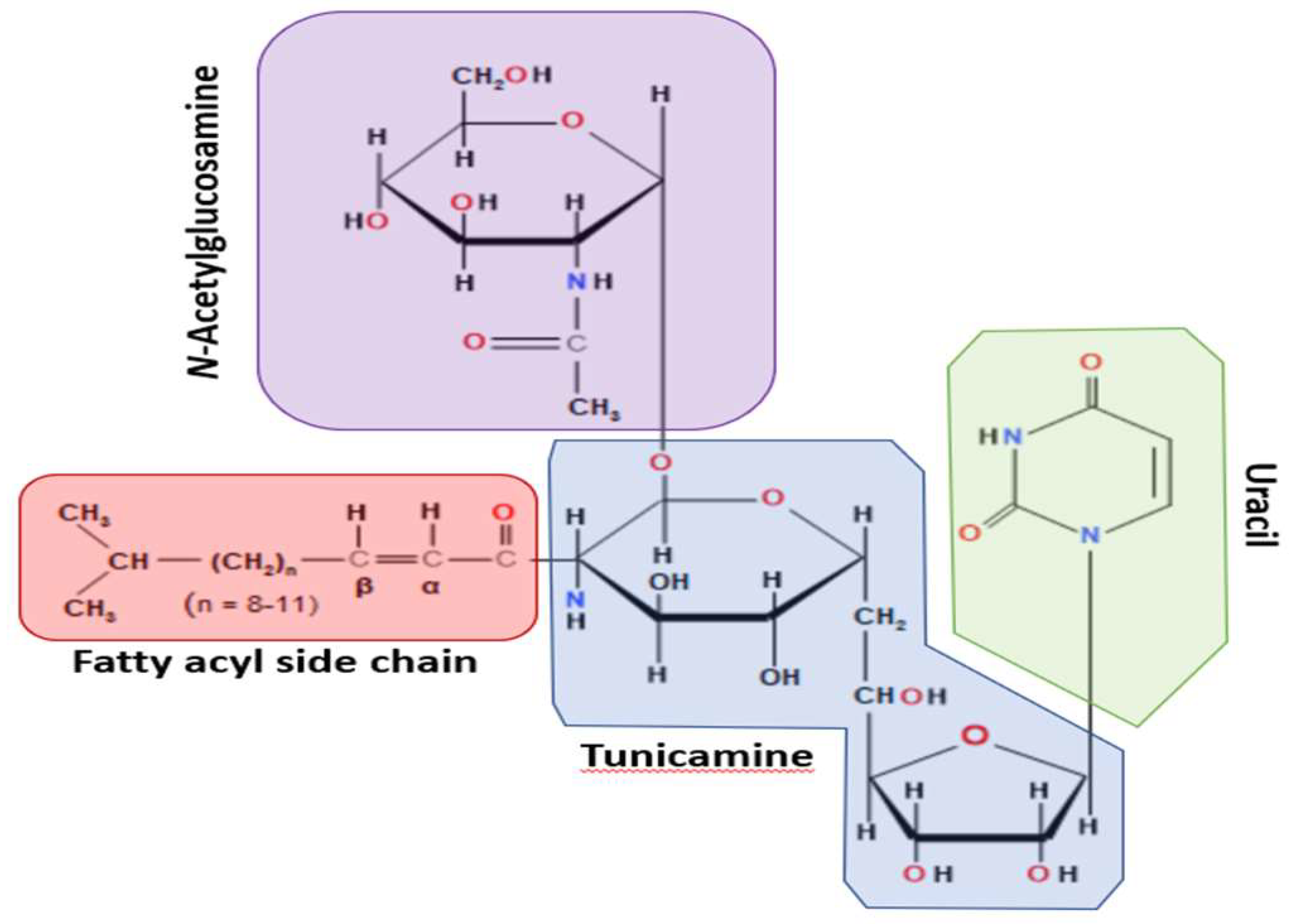
Figure 2.
The unfolded protein response (UPR) via TM.
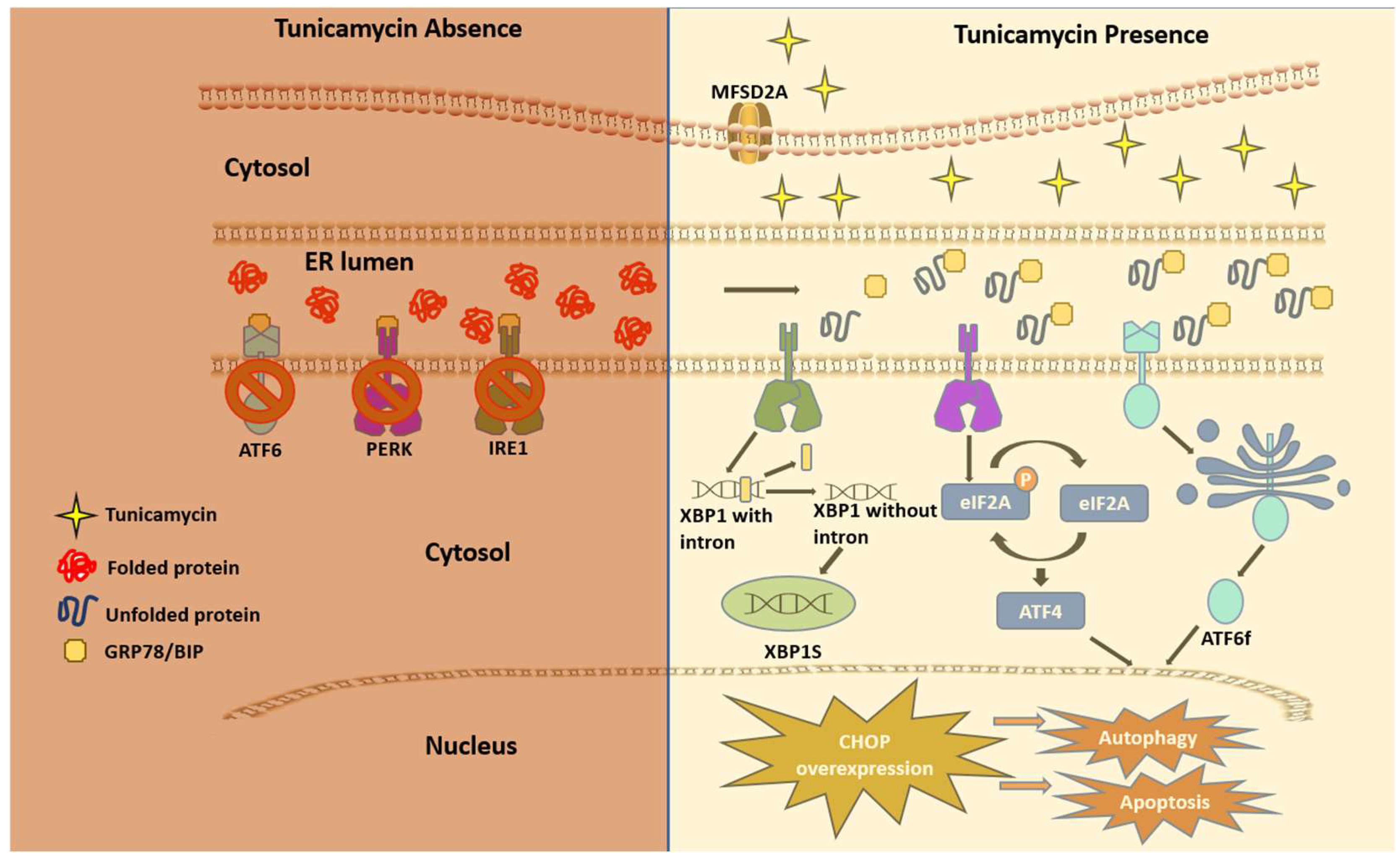
Figure 3.
The Therapeutic impact of Tunicamycin on various cancers. The Therapeutic impact of Tunicamycin on various cancers. Based on in vitro and preclinical animal studies, TM's antitumorigenic effects are found in the lungs, liver, breast, pancreas, and colons. Thus, TM can be an ideal drug to treat human cancers in combination with other antibodies or chemotherapies. .
Figure 3.
The Therapeutic impact of Tunicamycin on various cancers. The Therapeutic impact of Tunicamycin on various cancers. Based on in vitro and preclinical animal studies, TM's antitumorigenic effects are found in the lungs, liver, breast, pancreas, and colons. Thus, TM can be an ideal drug to treat human cancers in combination with other antibodies or chemotherapies. .
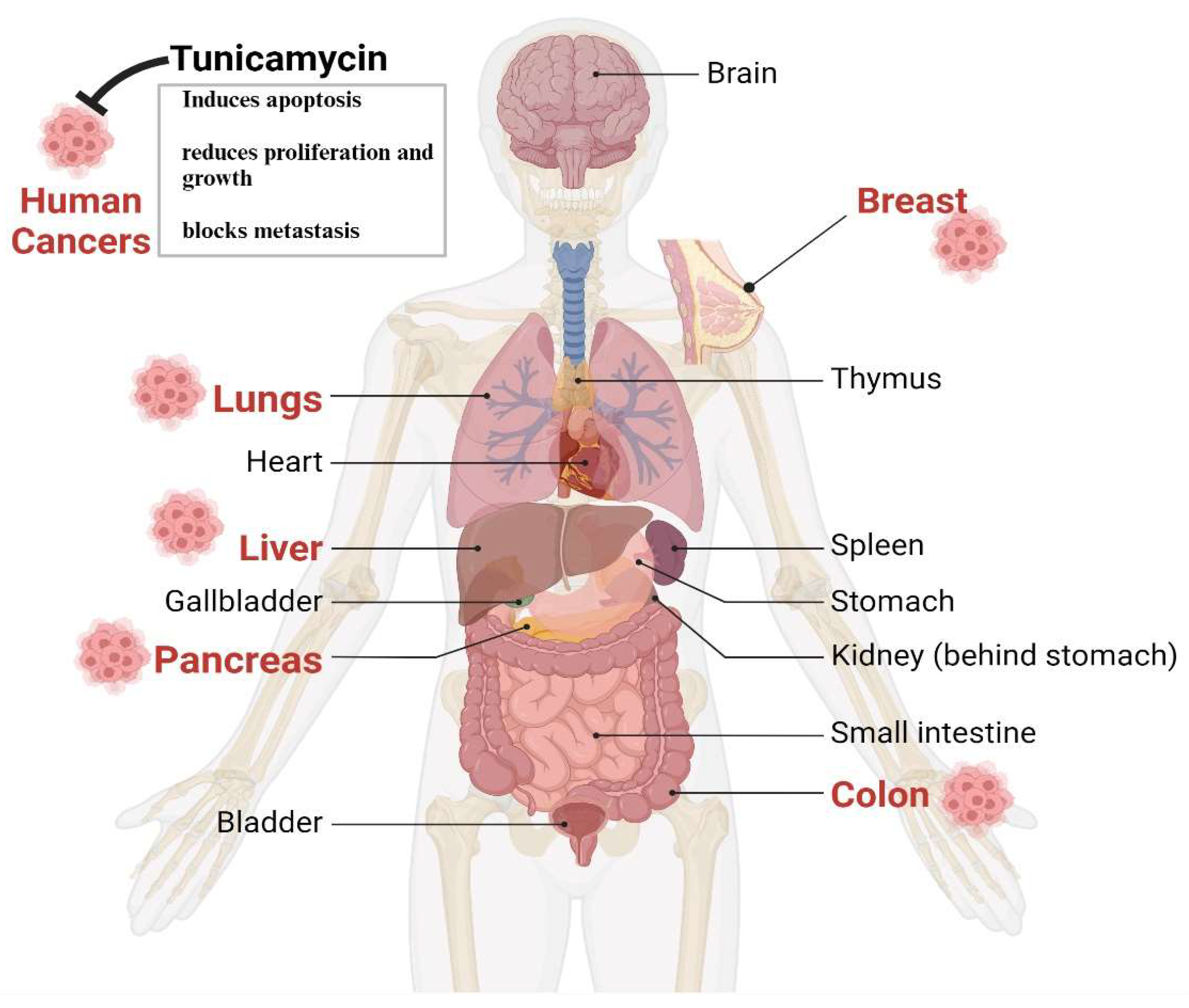
Figure 4.
Effect of Tunicamycin on tumor cell killing by blocking PD-1-PD-L1 axis inhibition. The antigen-producing cells (APC) promote the maturation/activation of naïve T cells. The active CD8+ T cells, via their receptors (TCR), bind with tumor cell-producing antigens with MHC I and activate tumor cell-killing mechanisms. However, aggressive tumor cells produced program cell death ligand 1 (PD-L1) that binds with its receptor PD-1 in CD8+ T cells and deactivates T cells, which decreases the antitumor activities of T cells. .
Figure 4.
Effect of Tunicamycin on tumor cell killing by blocking PD-1-PD-L1 axis inhibition. The antigen-producing cells (APC) promote the maturation/activation of naïve T cells. The active CD8+ T cells, via their receptors (TCR), bind with tumor cell-producing antigens with MHC I and activate tumor cell-killing mechanisms. However, aggressive tumor cells produced program cell death ligand 1 (PD-L1) that binds with its receptor PD-1 in CD8+ T cells and deactivates T cells, which decreases the antitumor activities of T cells. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



