
Submitted:
26 January 2024
Posted:
26 January 2024
You are already at the latest version
Abstract
Secondary acute myeloid leukemia (sAML) is a heterogeneous malignant hematopoietic disease that arises either from an antecedent hematologic disorder (AHD) including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), aplastic anemia (AA), or as a result of exposure to genotoxic chemotherapeutic agents or radiotherapy (therapy related AML, tAML). sAML is diagnosed when the number of blasts is ≥20% in the bone marrow or peripheral blood, and it is characterized by poor prognosis, resistance to therapy and low overall survival rate. With the recent advances in next generation sequencing technologies, our understanding of the molecular events associated with sAML evolution has significantly increased and opened new perspectives for the development of novel therapies. The genetic aberrations that are associated with sAML affect genes involved in processes such as splicing, chromatin modification and genome integrity. Moreover, non-coding RNAs’ emerged as an important contributing factor to leukemogenesis. For decades, the standard treatment for secondary AML has been the 7 + 3 regimen of cytarabine and daunorubicin which prolongs survival for several months, but modifications in either dosage or delivery has significantly extended that time. Apart from traditional chemotherapy, hematopoietic stem cell transplantation, CAR-T cell therapy and small molecule inhibitors have also emerged to treat sAML.
Keywords:
sAML
; AHD
; MPN
; MDS
; tAML
; 7+3 regimen
; AlloSCT
; CAR-T
; HSC
; LSC
1. Introduction
Acute myeloid leukemia (AML) is defined as a heterogeneous malignant clonal disorder of hematopoietic stem cells (HSC) and is the most common myeloid disorder among adults. This disease can be secondary (sAML) to either an AHD such as MPN and MDS or as a consequence of a prior treatment (tAML), or without an AHD history in the case of de novo AML [1,2,3]. MPNs lack cytopenia and are instead characterized by heightened differentiation of progenitor cells and are negative for the BCR-ABL fusion protein. They are divided into three main sub-categories: polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) [4]. MPN rates of progression to sAML vary by subtype: on average 15% of PMF patients, 8.35% of PV patients, and 1.85% of ET patients develop sAML over a ten-year period [5]. MDS are narrow clonal stem cell disorders characterized by heightened cytopenia in the blood and bone marrow due to apoptosis of hematopoietic progenitor cells, and one third of these syndromes progress to sAML [6,7]. AA is a rare, life-threatening bone marrow disorder characterized by deficiencies in hematopoietic cell production resulting from T-cell mediated autoimmunity. Like other AHDs, approximately 15-20% of AA patients over a ten-year period progress further to MDS/sAML. In addition to mutations and chromosomal abnormalities (Figure 1), other factors including telomere attrition, time to therapy, and the patient response to initial immunosuppressant treatment contribute to disease progression [8,9].
Unlike AHDs, sAML is a severe disease with a poor prognosis that has an overall survival time of 4.7 months and an event-free survival time of 2.9 months [27].The disease affects the elderly and the majority of diagnosed cases are over 65 years old: the median age of diagnosis is 67 years old with a third of patients over the age of 75. Common mutations that lead to the evolution of sAML are found in members of the spliceosome such as SRSF2, epigenetic modifiers including TET2, IDH1/2, ASXL1, and EZH2, or TP53 which maintains genomic integrity [28]. The aforementioned mutations are acquired on top of the mutations driving MDS or MPN development [29,30,31,32,33,34,35].
According to the 2016 WHO classification, patients are diagnosed with sAML when the percentage of myeloblasts in the bone marrow and/or peripheral blood is equal to or greater than 20% [36,37]. Although blast count has been set as the differentiator between the three phases, there are limitations to this method in that the blasts under examination, commonly through microscopy, are not easily distinguishable between normal samples and those of MDS and MPN patients. In addition to blast count, there are other indicators of progression to sAML, such as decreased apoptosis in the case of post-MDS sAML and increased cell proliferation for post-MPN sAML [38,39,40]. The vast majority of sAML patients progress from MDS (~85%) and upon exposure to therapy (~9%). MPN and AA contribution is lower with (~5%) and (~1%), respectively [5,7,41,42,43,44].
The outcome of sAML patients correlates with the mutational landscape. For example, patients with TP53 mutations, roughly 10-15%, have a worse outcome than those with wild type TP53. Not just due to the pernicious nature of these mutations, but also because of the co-occurring mutations including but not limited to IDH2 and NPM1. Due to the difference in mutation profiles between sAML and de novo AML, the treatment regimen has not been as successful when applied to sAML. For example, TP53 mutant patients had an overall survival of 8 months with induction therapy compared to 1.7 months for those without. The hazard ratio for having a TP53 mutation was 3.1-fold which is higher than either increasing age or performance status, although co-occurring mutations in FLT3 had a slightly smaller ratio of 3.01 [45]. The most recommended method for treating sAML is allogeneic stem cell transplantation (alloSCT) due to the highest probability of success [46], especially when compared to the more traditional method of 7 + 3, or 7 days of continuous dosing of cytarabine followed by 3 days of IV injection of daunorubicin, which has been the standard for decades [47]. However, direct comparison studies have shown that sAML patients are consistently less responsive to 7 + 3 treatment compared to de novo AML, and have a lower overall survival rate with this regimen, prompting the need for new therapies to be developed [48,49]. In this review, we discuss the recent advances in sAML progression, and we elaborate on the factors that drive the clonal evolution and discuss the different approaches used in the treatment of patients.
2. Pathophysiology of sAML
The WHO standard of a ≥20% blast count to differentiate between sAML and antecedent disorders is arbitrary like any threshold, but also has a potentially decisive impact on patients who may either display other characteristics of sAML with a blast count below the threshold or do not display characteristics of leukemogenesis despite having surpassed the threshold [50]. Therefore, this approach does not always assure diagnosis accuracy or reflect the complexity of leukemogenesis nor does it guarantee the optimal treatment for the patient. It is crucial to note that there are additional factors such as mutation type influencing diagnosis and treatment regimens. During the progression process, the immune system responds to the growth of the malignant cells. For example, Bauer et al. have shown a shift in immune cell populations between healthy donors’ bone marrow samples and those diagnosed with either MDS or sAML: there were neither CD3+CD8+ nor CD3+FOXP3+ T cells within a 25-micron radius in healthy bone marrow samples, but both of these populations were present in sAML patients at much higher levels. However, when comparing subsets of sAML, the outcomes were not uniform: in contrast to patients with TP53 mutations, patients with mutations in either signal transduction genes, chromatin modifiers, or splicing factors showed a significant increase in both populations [51].
One of the reasons why sAML is more common in older patients, especially over the age of 60, is the phenomenon of clonal hematopoiesis of indeterminate potential(CHIP), which is defined as having more than 2% mutant hematopoietic stem cells (HSC) without a history of either cytopenia or myeloid neoplasms. The risk for patients accumulates every year by 0.5-1%, thus most sAML patients skew older with some exceptions such as Fanconi anemia (FA) that progresses earlier. FA is a rare blood disorder characterized by a selective growth advantage to HSCs with an extra copy of 1q, leading to bone marrow depletion and an elevated risk of both MDS and later sAML [52,53]. However, in cases of cytopenia where patients do not exactly meet the criteria for MDS, they are diagnosed with clonal cytopenia of undetermined significance(CCUS) [54]. A 2023 study of UK Biobank patients found that patients diagnosed with either CCUS or CHIP had a significantly higher risk to develop MPNs and subsequently sAML: 1.74 for the former and 2.63 for the latter. For comparison, the risk for patients over 65 to develop MPN/sAML was 1.53 and the risk associated with the total number of mutations was 2.32. Non-genomic factors like red blood cell width distribution (RDW) over 15% or the mean corpuscular volume over 100 fL (MCV) had even higher risks: 3.63 and 4.03 [55]. RDW is also a biomarker for leukemic transformation: a higher RDW is not only used to distinguish MDS patients from healthy ones, but it is also a reliable predictor of leukemogenesis years after the initial MDS diagnosis. Higher RDW is associated with overall worse outcome in patients who have been treated with alloSCT and increased possibility to have passenger mutations in genes like NPM1 or ASXL1 [56].
In terms of cytogenetic risk for leukemic transformation, chromosomal mosaicism is also positively correlated with MPN formation: the 10-year cumulative incidence with mosaicism was 83% and 43% without it [55]. Moreover, chromosomal abnormalities impact diagnosis. For example, t(8;21) is associated with a favorable diagnosis, whereas, poor prognosis is associated with −7, inv(3)/t(3q)/del(3q), −7/del(7q), or complex karyotype (CK) with ≥3 abnormalities, which substantially increases the risk of leukemic progression [61,62]. A recent study has reported a case of a 44-year-old female with MDS/MPN where constitutional trisomy 21 was the only identified chromosomal abnormality [63]. Due to higher average age, the risk of an adverse karyotype is higher in sAML than in de novo. Compared to de novo AML, sAML patients have lower overall platelet and leukocyte counts, as well as a lower blast percentage in either bone marrow or peripheral blood [64]. Another study found that 81% of post-MDS sAML patients had a lower WBC count compared to 68% of MPN blast phase or 60% of de novo AML cases (Table 1) [65].
Another factor that contributes to sAML progression is inflammation through immune system dysregulation. Patients with autoimmune diseases (AID) are already at higher risk of developing sAML due to the elevated levels of inflammatory cytokines in the blood [66,67]. A recent study demonstrated that the increase in inflammation is particularly observed in MPN patients with TP53 mutations, either heterozygous or multi-hit, where the mutant myeloid cells gain a selective advantage over erythroid cells, especially those with WT TP53, which leads to a distortion in the ratio between erythroid and myeloid progenitor cells [68]. For example, TNF drives malignant clonal dominance by targeting healthy myeloid progenitor cells with both apoptotic and necroptotic signaling while malignant cells are left unaffected and able to proliferate through immuno-evasive mechanisms. Moreover, constitutive NFκB activity has been reported in both MPN/MDS and sAML patients [68,69,70]. In the case of MDS progressing to sAML, the chemokine receptor CCRL2, normally expressed in granulocytes, monocytes and NK cells is up-regulated in stem cells, which in turn stimulates IL-8 and the chemokine receptor CXCR2 [71,72]. On the other hand, IL-8 is a catalyst for several downstream pathways that promote proliferation, especially in tumors, including NFκB, MAPK, AKT, STAT3, and β-catenin. In a study conducted by Montes et al., compared to healthy donors, patients with MDS and sAML have significantly reduced counts of both CD4+ T lymphocytes and NK cells, with sAML having a higher count of CD4+ T lymphocytes than MDS, but still lower than healthy donors. This illustrates that at least the correlation between CD4+ T lymphocytes and MDS progression to sAML is not linear. In tandem with lower counts of proactive immune cells, programmed death ligand 1 (PD-L1) is upregulated, which suppresses the T-cell response to tumor growth and permits clonal expansion and metastasis of leukemic cells [73,74]. Patients with sAML have lower or similar expression levels of PD-L1 compared to MPN/MDS, with no difference between early and advanced stages of MDS; suggesting that the peak of PD-L1 expression results in a long-term suppression of the immune response that allows subsequent mutations to develop and trigger the progression to sAML [75]. In parallel, monocytic myeloid-derived suppressor cells (Mo-MDSC), another immuno-suppressive cell type, has been shown to have stronger positive correlation with the progression to sAML from MDS [74,76]. In addition, alteration in the extracellular matrix (ECM), and in particular the leucine-rich proteoglycan biglycan (BG), contributes to the heightened inflammatory environment observed in both MPN/MDS and sAML. BG is expressed in the bone marrow of both MDS and sAML patients but not in healthy individuals: it promotes cell signaling, bone mineralization, and differentiation. The presence of BG was positively correlated with activity of inflammasome components such as IL-1β, IL-18, and IFN-α. There was no significant difference in BG bone marrow expression between MDS and sAML patients. The hazard ratio of BG-high MDS patients versus BG-low patients for progression to sAML was 8.3 [77].
Another key feature of sAML is the increased self-renewal activity of pre- Leukemic Stem Cells (pre- LSC) through the WNT/β-catenin pathway activation during progression, which produces three main LSC phenotypes: multi-potent progenitor (MPP), lymphoid primed multi-potent progenitor (LMPP) and granulocyte-macrophage progenitor (GMP) [78]. Compared to de novo AML, sAML patients had higher amounts of MPP-like LSCs and LMPP-like LSCs, and this difference was more pronounced in post-MPN sAML. Post-MDS sAML patients had more GMP-like LSCs than post-MPN patients, but similar to de novo AML. The first two types of LSCs were strongly correlated with poor prognosis while GMP-like LSCs were more commonly seen in patients (either de novo or sAML) with either an intermediate or favorable prognosis. There was no difference in terms of LSC type distribution between patients younger than 65 and those older than 65 [79].
In contrast to increased pre-LSC activity in MPN patients progressing to sAML, there is a negative correlation between interferon (IFN) activity and risk of sAML. A study by de Castro et al. categorized MPN patients by both LSC and IFN activity and found that those with both the lowest IFN and highest LSC activity had the greatest risk of progression to sAML. Clonogenicity was significantly higher in this cohort compared to the rest of the study population, and the result was the same when comparing before and after transformation. The low IFN activity in these transforming cells also results in a more chaotic microenvironment where endothelial cells are dysregulated, and leukocytes are behaving abnormally while under increased oxidative stress [80].
The transition to sAML is accompanied with a shift in the clonal architecture inside the bone marrow. This shift is correlated with the number of acquired mutations during progression. Static shift occurs when mutations are acquired sequentially and the clones with the most mutations gradually dominate the bone marrow. Dynamic-S (for single nucleotide variant) shift occurs upon acquisition of multiple mutations that can be in multiple categories simultaneously (Table 2) and their rise to clonal dominance is expedited. Finally, the Dynamic-C (for chromosomal) shift is similar to Dynamic-S except that instead of gaining mutations, the clones acquire chromosomal abnormalities that confer a selective advantage (Table 3) [81]. Most genomic aberrations are either initiators in terms of clonal expansion and myeloid transformation or acquired after the process has begun [82].
3. Cytogenetics and mutational landscape of sAML
Like other cancers, AHD progression to sAML is a gradual process that is accompanied by chromosomal abnormalities and acquisition of mutations in genes involved in several processes such as signaling, splicing, cell cycle, and chromatin modification (Table 2 and Table 3). In MPNs, the most prevalent driver mutation is JAK2 V617F, which accounts for 98% of PV, 55-60% of ET, and MF cases. Frameshift mutations in calreticulin (CALR) represent 20%-25% of ET and MF patients. In MDS, the most common mutations affect the members of the spliceosome such as SF3B1, SRDF2, U2AF1, and ZRSR2; and DNA methylation and chromatin remodeling such as TET2, DNMT3A, IDH1/2, and ASXL1 [83]. Before progression to sAML, patients acquire mutations in other genes. Luque Paz et al. performed a molecular study by targeted sequencing on 49 PV and ET patients after leukemic transformation and they found that certain mutations, in particular spliceosome members such as SF3B1, were classified as “short-term” mutations that resulted in a rapid transformation, while other mutations, such as TP53 and ATM, were considered “long-term” as they took many years to occur but also had a poor prognosis upon transformation. TP53 requires both wild-type alleles to be mutated as opposed to other genes and thus it takes more time [84]. Makishima et al. analyzed the mutation landscape in 2250 MDS patients that evolved to sAML and they have shown that the number of mutations, their diversity and clone sizes significantly increased. Patients with FLT3, PTPN11, WT1, IDH1, NPM1, IDH2 and NRAS mutations were newly acquired and associated with faster sAML progression and a shorter overall survival. However, patients with mutations in genes such as TP53, GATA2, KRAS, RUNX1, STAG2, ASXL1, ZRSR2 and TET2 mutations had a weaker impact on sAML progression and overall survival [85].

Table 2.
Mutated genes implicated in leukemic transformation and clonal expansion.
| Category of Genes | Examples | Citations |
|---|---|---|
| Spliceosome | SRSF2, U2AF1, SF3B1 | [86,87] |
| DNA Methylation | DMNT3A, TET 1 / 2, IDH 1 / 2, | [88,89] |
| Activated Signaling | CALR, JAK2, PTPN11, TpoR, KRAS, FLT3, NRAS | [90,91] |
| Transcription Factors | RUNX1, NFE2, TP53 | [92,93,94,95] |
| Chromatin Modification | EZH2, ASXL1, NPM1 | [89,96] |
Despite the overlap in the genetic profile, the events accompanying leukemic transformation are not identical between the two disorders: post-MDS sAML is mainly initiated upon acquisition of mutations in: proteins involved in signaling (eg, K-Ras, N-Ras and FLT3), transcription factors (RUNX1, GATA2, CEBPA), or nucleophosmin 1 (NPM1) [97]. On the other hand, post-MPN sAML is mostly associated with the loss of TP53, RUNX1, IDH1/2, EZH2, and ASXL1 (Table 2) [84,98,99]. The rate of leukemic progression varies largely between the three subtypes of MPNs, with 20% being MF patients, 4% ET and 1% PV [100]. A higher proportion of MDS patients progress to sAML: roughly one third will undergo leukemic transformation over a ten-year period [5]. tAML patients usually acquire mutations in genes like TP53, TET2, DNMT3A, IDH2, NRAS, RUNX1, and SRSF2 before the initiation of therapy. After treatment, they gain mutations in FLT3, IDH1 and NPM1, which drive progression [101,102].
Alterations in the TP53 pathway are one of the main drivers of this process; however, the molecular mechanisms behind it are still obscure and require further investigations. TP53 loss by point mutations or chromosomal abnormalities such as gain of 1q that results in the amplification of MDM4, a p53 negative regulator, or deletion of 17p accounts for up to 50% of MPN cases evolving to sAML [19,25,83,92,103,104]. 1q gain is mostly found in PV patients; however, 17p deletions are common in MF patients [25]. TP53 mutations are also found in post-MDS sAML, but with a lower frequency (5%-10%). This frequency increases with age and in patients with complex karyotype or with a loss of chromosomes 5/5q, 7/7q, and 17/17p [105,106,107,108,109]. For tAML patients, TP53 bi-allelic mutations occur in 25%-50% of the cases making it the most frequently seen mutation in this disease [101,102]. sAML with TP53 mutations is highly aggressive and characterized by poor prognosis and short overall survival rate [110]. sAML patients usually lose both alleles of TP53 either by homozygous point mutations, or a point mutation with uniparental disomy (UPD) [19,92,104]. Monoallelic mutations are mostly found in the MDS/MPN-BP (blast phase) stages, and pretreatment stage in tAML; which forms a fertile ground for progression. Upon the loss of the second allele, transformation to sAML is accelerated, indicating a key role for TP53 in this process.
In a fraction of FA patients evolving to sAML, dysfunction in DNA repair proteins like BRCA2 as well as duplication 1q have been characterized [53]. Alternatively, other alterations trigger progression to MDS and sAML: either monosomy 7q or duplication of 3q which contains the secondary oncogene RUNX1. Both of these chromosomal abnormalities occur after bone marrow cells enter the blast phase [111]. Pezeshki et al. found that roughly 14% of FA patients progressed to sAML, significantly higher than other hematological disorders such as Shwachman-Diamond syndrome or Diamond-Blackfan anemia, and this was caused by the higher abundance of cytogenetic abnormalities [112]. The loss of 7q contributes to sAML because of the triune of LUC7-like proteins, especially LUC7L2, that interact with the spliceosome and regulate exonic splicing. Upon downregulation of LUC7-like genes, both intron retention and exon skipping increase. This is clinically relevant given that these genes are disproportionately expressed in the bone marrow and thymus, highlighting their importance [113].
Table 3.
Chromosomal abnormalities correlated with sAML transformation.
| Type of chromosomal abnormality |
Examples | Citations |
| Deletions | del(7q), del(5q), del(17p) | [107,114] |
| Duplications |
dup(1q), dup(3q), dup(11q), dup(17q) |
[115,116,117] |
| Translocations |
t(1;11)(q21;p15), t(10;11)(q22;q23), t(8;21) |
[60,118,119] |
|
Inversions |
inv(3)/t(3;3) |
[24] |
|
Monosomy |
-7 |
[87,120] |
|
Trisomy |
+8, +19, +21 |
[63,87,121] |
| Uniparental disomy |
UPD(9p), UPD(1p), UPD (17p) |
[75,92,122] |
Another important gene that is frequently mutated in sAML is NPM1, or nucleophosmin 1, which normally functions as a histone-binding, DNA-stabilizing factor as a response to UV-induced DNA damage, but when overexpressed, it contributes to uncontrolled cell proliferation. NPM1 is not enough to trigger the progression to sAML on its own, and any mutation in the NPM1 gene is generally preceded by a mutation in DNA methyltransferase 3A (DMNT3A), permitting clonal expansion of hematopoietic stem cells into myeloid progenitor cells. NPM1 mutant cells are characterized by down-regulation of TP53 activity and decreased apoptosis as well as Myc up-regulation. A third mutation in FLT3-ITD (fml-like tyrosine kinase 3) after NPM1 completes the path towards leukemogenesis, and patients with this combination of mutations, observed in 40% of those who have sAML derived from MDS, have a much poorer prognosis and shorter overall survival rate [2,64,123].
In addition, non-coding RNAs emerge as important players in leukemogenesis. For example, the miR-320 family of microRNAs (miRNAs), which are down-regulated in many types of cancer, are up-regulated in sAML. Compared to normal patients, both MDS patients with an intermediate-to-high risk for progression and sAML patients had significantly higher levels of these miRNAs expressed in the bone marrow. All miR-320 family members were negatively correlated with overall survival in MDS patients, but their exact contribution to leukemic progression remains elusive [124]. miR-196 is involved in MDS progression to sAML by contributing to both increased myeloid cell differentiation and proliferation as well as decreased apoptosis [125]. A miRNA microarray screening revealed a strong correlation between miRNAs associated with cytokine signaling activation, particularly the Toll-like receptor (TLR) family and interleukins, and progression of MDS [126]. Furthermore, the progression is also facilitated by mutations in certain miRNAs. A study performed on 326 patients undergoing alloSCT for sAML after a prior diagnosis of MDS revealed that mutations in miR-142 are recurrent in these patients [127]. Mutations are not the only source of miRNA alterations: deletions of entire chromosome sections also contribute to their loss of function: the deletion of the chromosome 7q32 coding for pro-apoptotic miRNAs in MDS patients has been strongly correlated with cell proliferation and progression [128]. Additionally, the lncRNA growth arrest-specific transcript 5 (GAS5) acts as both a negative regulator of the oncogenic miR-222 and a positive regulator of the tumor suppressor PTEN. Pavlovic et al. observed lower levels of GAS5 in sAML compared to healthy donors [129].
Extensive sequencing analysis revealed that RNA-binding proteins (RBPs) are essential in normal hematopoiesis, and their mutation is associated with 55% of sAML patients [86,87,130]. RBPs play an important role in RNA splicing, stability, translation, and localization; in addition to controlling the production of different isoforms, which has been reported to impact cancer development through regulation of different mechanisms such as proliferation and differentiation [130]. Moreover, they play key roles in miRNA biosynthesis and maturation. A recent study has reported an inverse correlation between the presence of mature miRNA and the progression to sAML. Bauer et al. observed down-regulation of the ribonucleases Dicer and Drosha in bone marrow samples from sAML patients and attributed heightened immature miRNA levels to the relative sparsity of these proteins [131]. RNA splicing factors SF3B1 and SRSF2, associated with exon skipping and nonsense-mediated decay of homeostatic proteins, contribute to the etiology of sAML and are associated with a poor prognosis in patients. Although elevated levels of mutations in these splicing proteins have been observed in sAML, as well as other myeloid malignancies, the reason as to why these mutations are elevated in these diseases has yet to be fully explained [86]. A major contributor to the pro-inflammatory switch between MPN/MDS and sAML is the change in isoform of adenosine deaminase acting on RNA 1 (ADAR1) from a constitutively active isoform to an isoform selective for inflammatory signaling, thus facilitating leukemogenesis. ADAR1 increases the risk of progression, but it does not act on its own. In tandem with another pro-inflammatory chemokine, apolipoprotein B mRNA editing enzyme catalytic polypeptide like type 3 (APOBEC3), ADAR1 promotes aberrant RNA editing, which enhances alternative splicing of STAT3 into its STAT3β proactive isoform that prevents β-catenin phosphorylation and degradation, allowing the Wnt pathway to continue driving proliferation and leukemogenesis [132].
4. Treatment of sAML
The standard of care for sAML patients is the 7 + 3 regimen, which has been improved through a liposomal delivery system CPX-351 [133]. However, its relative inadequacy towards sAML has propelled a drive towards developing more successful therapeutics. In addition to delivery, this regimen was also modified in an experimental study where cytarabine at a low dose (40 mg/m2 for 10 days compared to 100 mg/m2 for 7 days for 7 + 3) in combination with the cytotoxic purine analog cladribine. The objective of the low cytarabine study is to test the regimen for patients who were considered unfit for intensive chemotherapy. Overall survival for the entire cohort was seven months, which is promising considering that the average age of the cohort was 70 years old, and 40% of the patients had high risk genetic profiles. However, the survival time for those who had complete remission (CR) was 21 months, also adding to the optimistic prospects for this modified regimen [134]. Taking a more preemptive approach towards treatment, the tumor suppressor FBX011 was identified via CRISPR-Cas9 as a contributor to aberrant RNA splicing via EZH2 and cytokine-independent growth once it is inhibited. One candidate for preventing the down-regulation of FBX011 was bortezomib, a proteasome inhibitor already approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma, but it did not improve the overall survival of sAML patients in a randomized Phase 2 trial. Bortezomib was screened in conjunction with decitabine, a DNA-hypomethylating agent (HMA) currently approved for treatment of MDS. Decitabine has also been used for treating MDS patients that have crossed the 20% blast threshold and progressed towards sAML as an alternative to chemotherapy. HMAs have also been shown to significantly decrease the progression from MPNs to sAML by inducing a viral mimicry response to upregulate IFN activity and reduce LSC levels [80,135,136,137].
Another class of drugs used in combination with decitabine to treat sAML are inhibitors of BCL-2 like proteins, which help LSCs evade apoptotic mechanisms. Venetoclax (BCL-2 inhibitor) has been successfully tested for both de novo AML and tAML in terms of CR, with CR rates for these two AML variants above 70%. However, for post-MDS sAML, the hazard ratio of resistance to treatment is 2.01 compared to de novo AML, and this is likely influenced by two factors: the first is that some of the post-MDS sAML patients had previously received HMA treatment and the second is that some of these patients also had the RUNX1-RUNX1T1 fusion gene that confers resistance to HMA treatment [138]. Even with the combination of venetoclax and decitabine, it is possible for sAML patients to relapse because of the overexpression of the “don’t eat me” signal CD47. An anti-CD47 antibody, magrolimab, has been developed and is currently being tested on sAML patients in combination with venetoclax and decitabine in ongoing clinical trials [139]. Besides decitabine, another HMA, 5-azacitidine (AZA), has also been screened against post-MDS sAML, especially in patients with TP53 mutations. AZA and APR-246, a pro-apoptotic agent that restores the normal function of TP53, synergistically suppress AML cell growth by promoting cell cycle stagnation at the G0 phase and apoptosis. APR-246 also halted cell growth in the absence of AZA, but not to the same extent. Another pro-proliferation pathway, FLT3, was also inhibited by this combination. However, the effects of the AZA-APR-246 combination were reversed by the presence of the FLT3 ligand [140].
For post-MPN sAML in particular, there is the promising option of combining inhibitors for both the lysine demethylase LSD1 and the bromodomain and extra-terminal motifs (BET). Using CRISPR knockouts of LSD1 in post-MPN sAML, SET-2 cells demonstrated both increased apoptosis and differentiation compared to control cells. LSD inhibitors used in combination with either ruxolitinib or BET inhibitors do not develop any non-genetic resistance to either of those treatments in sAML xenografts in mice models. Moreover, either of these combinations are proven to be efficient on cells derived from sAML patients suffering from relapse post 3 + 7 treatment [141]. Another recently identified target, that also works synergistically with ruxolitinib and persists in ruxolitinib-resistant post-MPN sAML cells, is CDK9, a transcription-promoting enzyme that helps prolong the lifespan of otherwise short-lived mRNAs for oncogenes like c-Myc. When treated with a combination of a CDK9 inhibitor (NVP2) and ruxolitinib, these cells underwent higher levels of apoptosis accompanied with less chromatin accessibility, thus also leading to decreased Myc transcription [142,143].
Figure 3.
A comprehensive schematic representing several classes of drugs and their mode of action for sAML treatment.
Figure 3.
A comprehensive schematic representing several classes of drugs and their mode of action for sAML treatment.
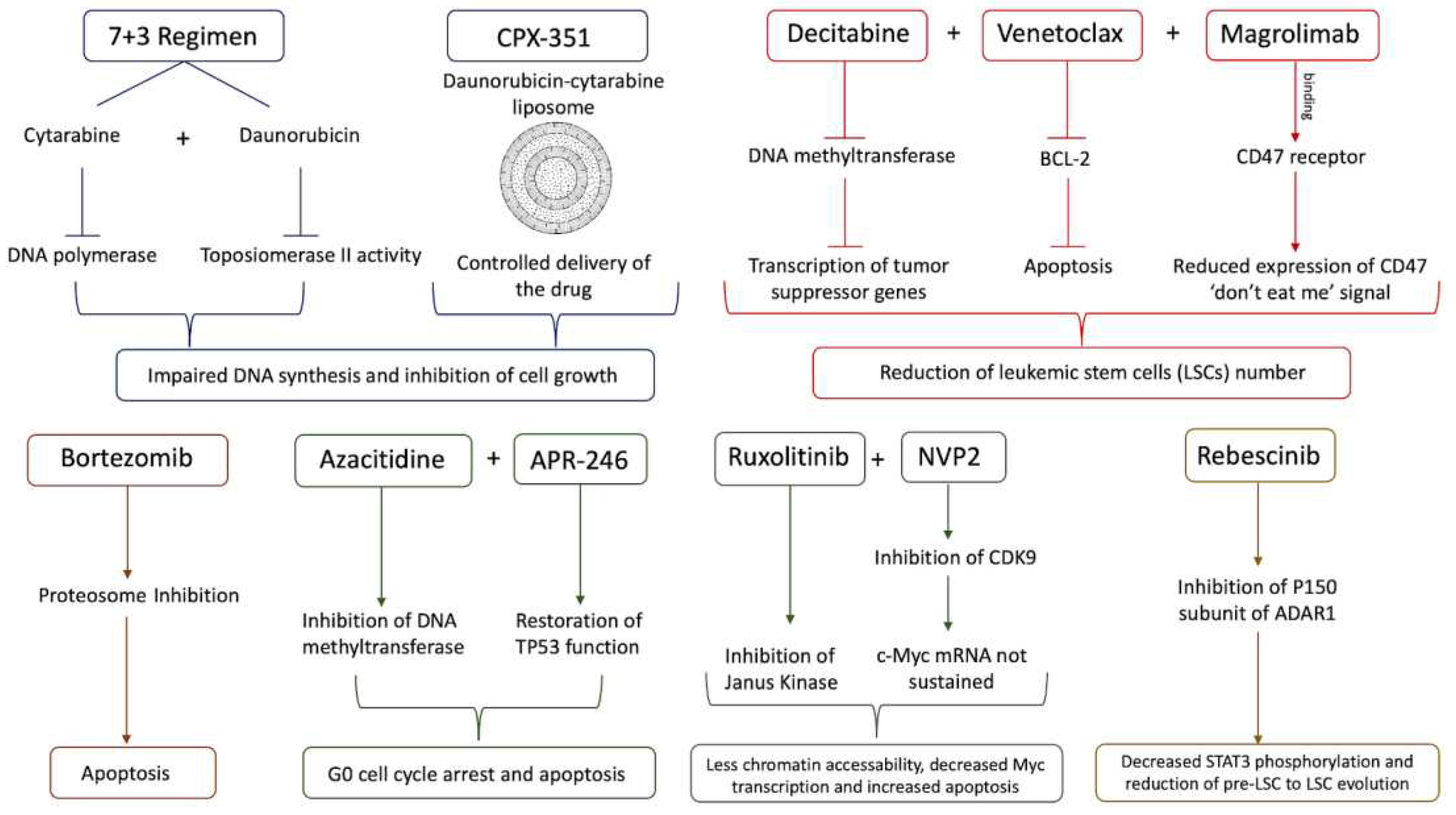
Recent studies present rebecsinib as a novel potential drug that targets LSCs. It specifically inhibits the p150 subunit of ADAR1, which is activated by the inflammatory cytokines and clonal expansion. Another recent comparative whole-genome and whole-transcriptome sequencing analysis of FACS purified pre-LSCs from MPN patients documented APOBEC3C upregulation, increased C to T mutational burden, and HSPC proliferation during evolution. Pre-LSC to LSC evolution is associated with STAT3 editing, STAT3β isoform switching, and increased ADAR1 p150 expression [132]. There was consistently decreased STAT3 phosphorylation and significantly improved survival of treated mice. These studies are notable because they did not just use samples from sAML patients but also from MDS and MPN as well, suggesting that rebecsinib could potentially work as a preventative measure for the progression to sAML and also as a treatment to prevent relapse for sAML patients. ADAR1 is a promising target because of both its strong stimulation of LSC proliferation in an immuno-evasive manner and its weak association with normal myelopoiesis [132,144,145,146].
Another relatively more efficient strategy for sAML treatment is alloSCT, but it has a generally lower response and overall survival rate compared to that of de novo AML. These worse indices are in spite of the higher incidence of graft versus host disease in de novo AML [147]. Nilsson et al. demonstrated the possibility of alloSCT as an alternative to the 7 + 3 regimen: the 5-year overall survival rate for sAML patients who had an AHD was 28% for those who underwent transplantation compared to 2% for those who underwent chemotherapy. Post-remission survival rates were significantly higher for sAML patients with alloSCT, but still lower than de novo AML (52% versus 65% respectively) [148]. tAML patients, however, since they have a higher risk of relapse because of the presence of comorbidities, show poor outcomes upon alloSCT treatment [21]. A recent study investigated the potential of a CPX-351 combination with alloSCT and showed that 70% of the patients had CR, and 35% of this cohort had TP53 mutations. Of those with TP53 mutations, 77% had CR. Cytogenetic risk also did not affect the overall remission rate, and the study group which did not receive alloSCT had a worse performance than those who did [149].
Figure 4.
A diagram illustrating alloSCT (left) and CAR-T cell therapy (right) treatments for sAML.
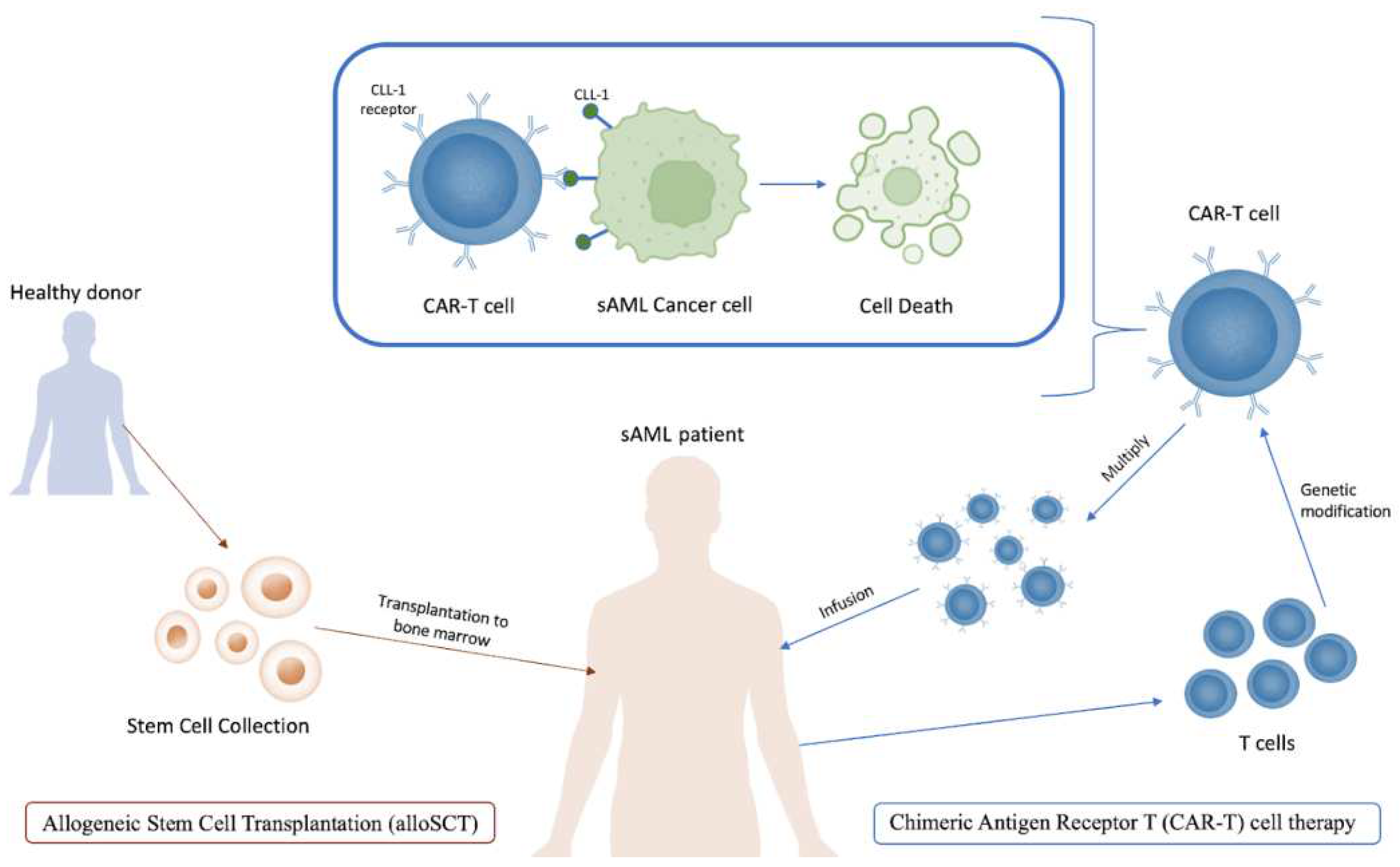
Chimeric Antigen Receptor T (CAR-T) cell therapy has recently emerged as a promising strategy for cancer treatment, since these cells are specifically engineered to bind to biomarkers more commonly expressed on cancer cells [150]. It has been successfully applied in the treatment of sAML in several studies. Zhang et al. targeted the biomarker CLL-1, or C-type lectin-like molecule 1, in one patient with sAML, and the outcome was a morphological, immunophenotypic and molecular CR for over 10 months [151]. It is important to note that the patient in this study was only 10 years old, and age may also be a factor in the future application of this therapy given that the average age of sAML patients is 70 years old. CLL-1, among other several biomarkers including lymphocyte activation molecule CD244 and IL-3 receptor CD123, is disproportionately expressed on LSCs and AML blast cells while simultaneously not detected on normal HSCs. The successes of CAR-T against acute lymphoblastic leukemia and non-Hodgkin lymphoma have also propelled interest in this therapy [148,149,151,152,153].
5. Perspectives
Despite the advances and discoveries laid out in this review that provide a greater understanding of the genetic and cytogenetic aberrations associated with sAML progression, more mechanistic studies are needed to uncover the molecular bases behind this process. This would accelerate the development of novel strategies to treat and prevent sAML progression. The successful efforts in the identification of the aberrations in genes involved in this disease should be extended to explore the role of lncRNAs, which have been shown to be involved in cancer etiology and resistance to therapy. Recently, a CRISPR-based study identified the most differentially expressed lncRNAs for AML patients treated with cytarabine through analysis of a corresponding MOLM14 cell line and revealed that lncRNAs associated with oxidative phosphorylation and fatty acid metabolism had the strongest correlation with resistance to treatment and renewed myeloid proliferation [154]. More studies are required to understand their mechanism of action and provide a complete picture of their implication in the leukemogenesis process and resistance to therapy.
CAR-T appears to be a very promising strategy especially after its success in AML. Recently, an improved version of CAR T, called modified or smartly reprogrammed CAR T cells has been developed to overcome toxicity issues in the original strategy [155,156,157,158]. Overall, joint efforts between the experts in the field would help in better understanding the disease, which would have a great impact on the development of novel customized therapeutic approaches.
Acknowledgments
Funding to IC is acknowledged from Scientific and Technological Research Council of Türkiye (TUBITAK 3501 grant).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Estey E, Döhner H (2006) Acute myeloid leukaemia. Lancet 368:1894–1907. [CrossRef] [PubMed]
- Higgins A, Shah MV (2020) Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes (Basel) 11:1–25. [CrossRef]
- Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH (2008) Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leuk 2008 222 22:240–248. [CrossRef]
- McMullin MF, Anderson LA (2020) Aetiology of Myeloproliferative Neoplasms. Cancers (Basel) 12:1–11. [CrossRef]
- Shahin OA, Chifotides HT, Bose P, Masarova L, Verstovsek S (2021) Accelerated Phase of Myeloproliferative Neoplasms. Acta Haematol 144:484–499. [CrossRef]
- Adès L, Itzykson R, Fenaux P (2014) Myelodysplastic syndromes. Lancet 383:2239–2252. [CrossRef]
- Dan C, Chi J, Wang L (2015) Molecular mechanisms of the progression of myelodysplastic syndrome to secondary acute myeloid leukaemia and implication for therapy. Ann Med 47:209–217. [CrossRef]
- Sun L, Babushok D V. (2020) Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood 136:36–49. [CrossRef]
- Wang L, Liu H (2019) Pathogenesis of aplastic anemia. Hematology 24:559–566. [CrossRef]
- Cook MR, Karp JE, Lai C (2022) The spectrum of genetic mutations in myelodysplastic syndrome: Should we update prognostication? eJHaem 3:301–313. [CrossRef]
- Heuser M, Gabdoulline R, Löffeld P, Dobbernack V, Kreimeyer H, Pankratz M, Flintrop M, Liebich A, Klesse S, Panagiota V, Stadler M, Wichmann M, Shahswar R, Platzbecker U, Thiede C, Schroeder T, Kobbe G, Geffers R, Schlegelberger B, Göhring G, Kreipe HH, Germing U, Ganser A, Kröger N, Koenecke C, Thol F (2017) Individual outcome prediction for myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia from MDS after allogeneic hematopoietic cell transplantation. Ann Hematol 96:1361–1372. [CrossRef]
- Hussein K, Van Dyke DL, Tefferi A (2009) Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol 82:329–338. [CrossRef]
- Rubin CM, Larson RA, Anastasi J, Winter JN, Thangavelu M, Vardiman JW, Rowley JD, Le Beau MM (1990) t(3;21)(q26;q22): a recurring chromosomal abnormality in therapy- related myelodysplastic syndrome and acute myeloid leukemia. Blood 76:2594–2598. [CrossRef]
- Andersen MK, Larson RA, Mauritzson N, Schnittger S, Jhanwar SC, Pedersen-Bjergaard J (2002) Balanced chromosome abnormalities inv(16) and t(15;17) in therapy-related myelodysplastic syndromes and acute leukemia: Report from an International Workshop†. Genes, Chromosom Cancer 33:395–400. [CrossRef]
- Kotsiafti A, Giannakas K, Christoforou P, Liapis K (2023) Progress toward Better Treatment of Therapy-Related AML. Cancers 2023, Vol 15, Page 1658 15:1658. [CrossRef]
- Hosono N (2019) Genetic abnormalities and pathophysiology of MDS. Int J Clin Oncol 24:885–892. [CrossRef]
- Cheng Y, Wang Y, Wang H, Chen Z, Lou J, Xu H, Wang H, Qian W, Meng H, Lin M, Jin J (2009) Cytogenetic profile of de novo acute myeloid leukemia: a study based on 1432 patients in a single institution of China. Leuk 2009 2310 23:1801–1806. [CrossRef]
- Heuser M, Schlarmann C, Dobbernack V, Panagiota V, Wiehlmann L, Walter C, Beier F, Ziegler P, Yun H, Kade S, Kirchner A, Huang L, Koenecke C, Eder M, Brümmendorf TH, Dugas M, Ganser A, Thol F (2014) Genetic characterization of acquired aplastic anemia by targeted sequencing. Haematologica 99:e165. [CrossRef]
- Milosevic JD, Puda A, Malcovati L, Berg T, Hofbauer M, Stukalov A, Klampfl T, Harutyunyan AS, Gisslinger H, Gisslinger B, Burjanivova T, Rumi E, Pietra D, Elena C, Vannucchi AM, Doubek M, Dvorakova D, Robesova B, Wieser R, Koller E, Suvajdzic N, Tomin D, Tosic N, Colinge J, Racil Z, Steurer M, Pavlovic S, Cazzola M, Kralovics R (2012) Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am J Hematol 87:1010–1016. [CrossRef]
- Maciejewski JP, Risitano A, Sloand EM, Nunez O, Young NS (2002) Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood 99:3129–3135. [CrossRef]
- Strickland SA, Vey N (2022) Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit Rev Oncol Hematol 171:103607. [CrossRef]
- Keung Y-K, Pettenati MJ, Cruz JM, Powell BL, Woodruff RD, Buss DH (2001) Bone Marrow Cytogenetic Abnormalities of Aplastic Anemia. J Hematol 66:167–171. [CrossRef]
- Van Gelder M, Schetelig J, Volin L, Maertens J, Socié G, Petersen E, Thomssen H, Biezen A, Brand R, Witte TM de, Kröger N (2009) Monosomal Karyotype Predicts Poor Outcome for MDS/sAML Patients with Chromosome 7 Abnormalities After Allogeneic Stem Cell Transplantation for MDS/sAML. A Study of the MDS Subcommittee of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Blood 114:293. [CrossRef]
- Birdwell C, Fiskus W, Kadia TM, DiNardo CD, Mill CP, Bhalla KN (2021) EVI1 dysregulation: impact on biology and therapy of myeloid malignancies. Blood Cancer J 2021 113 11:1–14. [CrossRef]
- Marcellino BK, Hoffman R, Tripodi J, Lu M, Kosiorek H, Mascarenhas J, Rampal RK, Dueck A, Najfeld V (2018) Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv 2:3581. [CrossRef]
- Thoennissen NH, Kawamata N, Lasho TL, Weiss T, Nowak D, Kato M, Takita J, Sanada M, Haferlach T, Mesa RA, Tefferi A, Müller-Tidow C, Ogawa S, Koeffler PH (2008) Genomic Changes Associated with Leukemic Transformation of Myeloproliferative Disorders. Blood 112:3371. [CrossRef]
- Venton G, Courtier F, Charbonnier A, D’incan E, Saillard C, Mohty B, Mozziconacci MJ, Birnbaum D, Murati A, Vey N, Rey J (2018) Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am J Hematol 93:330–338. [CrossRef]
- Dunbar AJ, Rampal RK, Levine R (2020) Leukemia secondary to myeloproliferative neoplasms. Blood 136:61–70. [CrossRef]
- Schwind S, Jentzsch M, Kubasch AS, Metzeler KH, Platzbecker U (2021) Myelodysplastic syndromes: Biological and therapeutic consequences of the evolving molecular aberrations landscape. Neoplasia 23:1101–1109. [CrossRef]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365:1054–1061. [CrossRef]
- James C, Ugo V, Casadevall N, Constantinescu SN, Vainchenker W (2005) A JAK2 mutation in myeloproliferative disorders: pathogenesis and therapeutic and scientific prospects. Trends Mol Med 11:546–554. [CrossRef]
- Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A (2006) MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108:3472–3476. [CrossRef]
- Pikman Y, Lee BH, Mercher T, Mcdowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, Deangelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL (2006) MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. [CrossRef]
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M, Silber Y, Dimitropoulou D, Bloxham D, Mudie L, Maddison M, Robinson B, Keohane C, Maclean C, Hill K, Orchard K, Tauro S, Du M-Q, Greaves M, Bowen D, Huntly BJP, Harrison CN, Cross NCP, Ron D, Vannucchi AM, Papaemmanuil E, Campbell PJ, Green AR (2013) Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med 369:2391–2405. [CrossRef]
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NCC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schönegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R (2013) Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N Engl J Med 369:2379–2390. [CrossRef]
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405. [CrossRef]
- Mesa RA, Verstovsek S, Cervantes F, Barosi G, Reilly JT, Dupriez B, Levine R, Le Bousse-Kerdiles MC, Wadleigh M, Campbell PJ, Silver RT, Vannucchi AM, Deeg HJ, Gisslinger H, Thomas D, Odenike O, Solberg LA, Gotlib J, Hexner E, Nimer SD, Kantarjian H, Orazi A, Vardiman JW, Thiele J, Tefferi A (2007) Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and. Leuk Res 31:737–740. [CrossRef]
- Jilg S, Reidel V, Müller-Thomas C, König J, Schauwecker J, Höckendorf U, Huberle C, Gorka O, Schmidt B, Burgkart R, Ruland J, Kolb HJ, Peschel C, Oostendorp RAJ, Götze KS, Jost PJ (2015) Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leuk 2016 301 30:112–123. [CrossRef]
- Saenz DT, Fiskus W, Manshouri T, Mill CP, Qian Y, Raina K, Rajapakshe K, Coarfa C, Soldi R, Bose P, Borthakur G, Kadia TM, Khoury JD, Masarova L, Nowak AJ, Sun B, Saenz DN, Kornblau SM, Horrigan S, Sharma S, Qiu P, Crews CM, Verstovsek S, Bhalla KN (2018) Targeting nuclear β-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leuk 2018 336 33:1373–1386. [CrossRef]
- Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A (2000) The role of apoptosis, proliferation, and the Bcl-2–related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 96:3932–3938. [CrossRef]
- Cogle CR (2015) Incidence and Burden of the Myelodysplastic Syndromes. Curr Hematol Malig Rep 10:272. [CrossRef]
- Vaht K, Göransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, Uggla B, Winiarski J, Ljungman P, Brune M, Andersson PO (2017) Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica 102:1683. [CrossRef]
- Prébet T, Gore SD, Thépot S, Esterni B, Quesnel B, Beyne Rauzy O, Dreyfus F, Gardin C, Fenaux P, Vey N (2012) Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br J Haematol 157:764. [CrossRef]
- Nilsson C, Linde F, Hulegårdh E, Garelius H, Lazarevic V, Antunovic P, Cammenga J, Deneberg S, Eriksson A, Jädersten M, Björkvall CK, Möllgård L, Wennström L, Ölander E, Höglund M, Juliusson G, Lehmann S (2023) Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications. Haematologica 108:1015. [CrossRef]
- Yanada M, Yamamoto Y, Iba S, Okamoto A, Inaguma Y, Tokuda M, Morishima S, Kanie T, Mizuta S, Akatsuka Y, Okamoto M, Emi N (2016) TP53 mutations in older adults with acute myeloid leukemia. Int J Hematol 103:429–435. [CrossRef]
- Michelis F V., Atenafu EG, Gupta V, Kim DD, Kuruvilla J, Lipton JH, Loach D, Seftel MD, Uhm J, Alam N, Lambie A, McGillis L, Messner HA (2015) Comparable outcomes post allogeneic hematopoietic cell transplant for patients with de novo or secondary acute myeloid leukemia in first remission. Bone Marrow Transplant 2015 507 50:907–913. [CrossRef]
- Rowe JM (2022) The “7+3” regimen in acute myeloid leukemia. Haematologica 107:3. [CrossRef]
- Hulegårdh E, Nilsson C, Lazarevic V, Garelius H, Antunovic P, Rangert Derolf Å, Möllgård L, Uggla B, Wennström L, Wahlin A, Höglund M, Juliusson G, Stockelberg D, Lehmann S (2015) Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: A report from the Swedish Acute Leukemia Registry. Am J Hematol 90:208–214. [CrossRef]
- Østgård LSG, Medeiros BC, Sengeløv H, Nørgaard M, Andersen MK, Dufva I, Friis LS, Kjeldsen E, Marcher CW, Preiss B, Severinsen M, Nørgaard JM (2015) Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J Clin Oncol 33:3641–3649. [CrossRef]
- Estey E, Hasserjian RP, Döhner H (2022) Distinguishing AML from MDS: a fixed blast percentage may no longer be optimal. Blood 139:323–332. [CrossRef]
- Bauer M, Vaxevanis C, Al-Ali K, Jaekel N, Le C, Naumann H, Schaffrath J, Rau A, Seliger B, Wickenhauser C (2021) Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34 + Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia. Cancers (Basel) 13:186. [CrossRef]
- Hasserjian RP, Steensma DP, Graubert TA, Ebert BL (2020) Clonal hematopoiesis and measurable residual disease assessment in acute myeloid leukemia. Blood 135:1729–1738. [CrossRef]
- Sebert M, Phanie Gachet S, Leblanc T, Bluteau D, Gis Peffault De Latour R, Correspondence JS (2023) Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in Fanconi anemia. [CrossRef]
- Malcovati L, Gallì A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, Elena C, Ferretti VV, Catricalà S, Bono E, Todisco G, Bianchessi A, Rumi E, Zibellini S, Pietra D, Boveri E, Camaschella C, Toniolo D, Papaemmanuil E, Ogawa S, Cazzola M (2017) Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 129:3371–3378. [CrossRef]
- Weeks LD, Niroula A, Neuberg D, Wong W, Lindsley RC, Luskin MR, Berliner N, Stone RM, DeAngelo DJ, Soiffer RJ, Uddin MM, Griffin G, Vlasschaert C, Gibson CJ, Jaiswal S, Bick AG, Malcovati L, Natarajan P, Ebert BL (2023) Prediction of Risk for Myeloid Malignancy in Clonal Hematopoiesis. NEJM Evid 2:. [CrossRef]
- Vucinic V, Ruhnke L, Sockel K, Rohnert MA, Backhaus D, Brauer D, Franke GN, Niederwieser D, Bornhauser M, Rollig C, Platzbecker U, Schwind S, Jentzsch M (2021) The diagnostic red blood cell distribution width as a prognostic factor in acute myeloid leukemia. Blood Adv 5:5584–5587. [CrossRef]
- Ye XP, Bao S, Gao HM, Guo Y, Wei YP (2016) A case of myeloproliferative neoplasm with a normal complete blood cell count: A novel problem of the JAK2 era. Oncol Lett 11:2134. [CrossRef]
- Kim SY, Park Y, Kim H, Kim J, Kwon GC, Koo SH (2018) Discriminating myelodysplastic syndrome and other myeloid malignancies from non-clonal disorders by multiparametric analysis of automated cell data. Clin Chim Acta 480:56–64. [CrossRef]
- Wang SY, Cheng WY, Mao YF, Zhu YM, Liu FJ, Ma TT, Shen Y (2019) Genetic alteration patterns and clinical outcomes of elderly and secondary acute myeloid leukemia. Hematol Oncol 37:456–463. [CrossRef]
- Rogers HJ, Wang X, Xie Y, Davis AR, Thakral B, Wang SA, Borthakur G, Cantu MD, Margolskee EM, Philip JKS, Sukhanova M, Bagg A, Bueso-Ramos CE, Orazi A, Arber DA, Hsi ED, Hasserjian RP (2020) Comparison of therapy-related and de novo core binding factor acute myeloid leukemia: A bone marrow pathology group study. Am J Hematol 95:799–808. [CrossRef]
- Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, Malcovati L, Cazzola M, Cermak J, Fonatsch C, Le Beau MM, Slovak ML, Krieger O, Luebbert M, Maciejewski J, Magalhaes SMM, Miyazaki Y, Pfeilstöcker M, Sekeres M, Sperr WR, Stauder R, Tauro S, Valent P, Vallespi T, Van De Loosdrecht AA, Germing U, Haase D (2012) Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120:2454–2465. [CrossRef]
- Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, Granada I, Hildebrandt B, Slovak ML, Ohyashiki K, Steidl C, Fonatsch C, Pfeilstöcker M, Nösslinger T, Valent P, Giagounidis A, Aul C, Lübbert M, Stauder R, Krieger O, Garcia-Manero G, Faderl S, Pierce S, Le Beau MM, Bennett JM, Greenberg P, Germing U, Haase D (2012) New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol 30:820–829. [CrossRef]
- O’Hagan Henderson S, Glaser A, Frietsch JJ, Hochhaus A, Hilgendorf I (2022) The incidental discovery of a constitutional trisomy 21 mosaicism in an adult female with myelodysplastic/myeloproliferative neoplasm. Ann Hematol 101:919. [CrossRef]
- Begna KH, Ali W, Naseema Gangat, Elliott MA, Al-Kali A, Litzow MR, Christopher Hook C, Wolanskyj-Spinner AP, Hogan WJ, Patnaik MM, Pardanani A, Zblewski DL, Chen D, He R, Viswanatha D, Hanson CA, Ketterling RP, Tefferi A (2021) Mayo Clinic experience with 1123 adults with acute myeloid leukemia. Blood Cancer J 2021 113 11:1–8. [CrossRef]
- Gupta V, Kim S, Hu ZH, Liu Y, Aljurf M, Bacher U, Beitinjaneh A, Cahn JY, Cerny J, Copelan E, Gadalla SM, Gale RP, Ganguly S, George B, Gerds AT, Gergis U, Hamilton BK, Hashmi S, Hildebrandt GC, Kamble RT, Kindwall-Keller T, Lazarus HM, Liesveld JL, Litzow M, Maziarz RT, Nishihori T, Olsson RF, Rizzieri D, Savani BN, Seo S, Solh M, Szer J, Verdonck LF, Wirk B, Woolfrey A, Yared JA, Alyea EP, Popat UR, Sobecks RM, Scott BL, Nakamura R, Saber W (2020) Comparison of outcomes of HCT in blast phase of BCR-ABL1− MPN with de novo AML and with AML following MDS. Blood Adv 4:4748–4757. [CrossRef]
- Elessa D, Zhao LP, de Oliveira RD, Maslah N, Soret-Dulphy J, Verger E, Marcault C, Parquet N, Fenaux P, Adès L, Raffoux E, Giraudier S, Fain O, Cassinat B, Kiladjian JJ, Mekinian A, Benajiba L (2023) Clinical features and genomic landscape of myeloproliferative neoplasm (MPN) patients with autoimmune and inflammatory diseases (AID). Leuk 2023 378 37:1741–1744. [CrossRef]
- Boddu PC, Zeidan AM (2019) MYELOID DISORDERS AFTER AUTOIMMUNE DISEASE. Best Pract Res Clin Haematol 32:74. [CrossRef]
- Rodriguez-Meira A, Norfo R, Wen S, Chédeville AL, Rahman H, O’Sullivan J, Wang G, Louka E, Kretzschmar WW, Paterson A, Brierley C, Martin JE, Demeule C, Bashton M, Sousos N, Moralli D, Subha Meem L, Carrelha J, Wu B, Hamblin A, Guermouche H, Pasquier F, Marzac C, Girodon F, Vainchenker W, Drummond M, Harrison C, Chapman JR, Plo I, Jacobsen SEW, Psaila B, Thongjuea S, Antony-Debré I, Mead AJ (2023) Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet 2023 559 55:1531–1541. [CrossRef]
- Mendez Luque LF, Blackmon AL, Ramanathan G, Fleischman AG (2019) Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr Hematol Malig Rep 14:145–153. [CrossRef]
- Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G (2006) Targeting NF-κB in hematologic malignancies. Cell Death Differ 2006 135 13:748–758. [CrossRef]
- Karantanos T, Teodorescu P, Perkins B, Christodoulou I, Esteb C, Varadhan R, Helmenstine E, Rajkhowa T, Paun BC, Bonifant C, Dalton WB, Gondek LP, Moliterno AR, Levis MJ, Ghiaur G, Jones RJ (2022) The role of the atypical chemokine receptor CCRL2 in myelodysplastic syndrome and secondary acute myeloid leukemia. Sci Adv 8:. [CrossRef]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, Pellagatti A, Boultwood J, Wickrema A, Yu Y, Will B, Wei S, Steidl U, Verma A (2015) IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 125:3144–3152. [CrossRef]
- Wang X, Teng F, Kong L, Yu J (2016) PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 9:5023. [CrossRef]
- Montes P, Bernal M, Campo LN, González-Ramírez AR, Jiménez P, Garrido P, Jurado M, Garrido F, Ruiz-Cabello F, Hernández F (2019) Tumor genetic alterations and features of the immune microenvironment drive myelodysplastic syndrome escape and progression. Cancer Immunol Immunother 68:2015–2027. [CrossRef]
- Milosevic Feenstra JD, Jäger R, Schischlik F, Ivanov D, Eisenwort G, Rumi E, Schuster M, Gisslinger B, Machherndl-Spandl S, Bettelheim P, Krauth MT, Keil F, Bock C, Cazzola M, Gisslinger H, Kralovics R, Valent P (2022) PD-L1 overexpression correlates with JAK2-V617F mutational burden and is associated with 9p uniparental disomy in myeloproliferative neoplasms. Am J Hematol 97:390–400. [CrossRef]
- Palumbo GA, Parrinello NL, Giallongo C, D’amico E, Zanghì A, Puglisi F, Conticello C, Chiarenza A, Tibullo D, Di Raimondo F, Romano A (2019) Monocytic Myeloid Derived Suppressor Cells in Hematological Malignancies. Int J Mol Sci 20:. [CrossRef]
- Vaxevanis CK, Bauer M, Subbarayan K, Friedrich M, Massa C, Biehl K, Al-Ali HK, Wickenhauser C, Seliger B (2023) Biglycan as a mediator of proinflammatory response and target for MDS and sAML therapy. Oncoimmunology 12:. [CrossRef]
- Rontauroli S, Carretta C, Parenti S, Bertesi M, Manfredini R (2022) Novel Molecular Insights into Leukemic Evolution of Myeloproliferative Neoplasms: A Single Cell Perspective. Int. J. Mol. Sci. 23. [CrossRef]
- Han H, Byun JM, Shin DY, Yoon SS, Koh Y, Hong J, Kim I, Lee C, Yoo H, Yun H, Kim MJ, Cho SI, Seong MW, Park SS (2021) Leukemic stem cell phenotype is associated with mutational profile in acute myeloid leukemia. Korean J Intern Med 36:401. [CrossRef]
- Castro FA de, Mehdipour P, Chakravarthy A, Ettayebi I, Loo Yau H, Medina TS, Marhon SA, Almeida FC de, Bianco TM, Arruda AGF, Devlin R, Figueiredo-Pontes LL de, Chahud F, Costa Cacemiro M da, Minden MD, Gupta V, De Carvalho DD (2023) Ratio of stemness to interferon signalling as a biomarker and therapeutic target of myeloproliferative neoplasm progression to acute myeloid leukaemia. Br J Haematol. [CrossRef]
- Guess T, Potts CR, Bhat P, Cartailler JA, Brooks A, Holt C, Yenamandra A, Wheeler FC, Savona MR, Cartailler J-P, Ferrell PB (2022) Distinct Patterns of Clonal Evolution Drive Myelodysplastic Syndrome Progression to Secondary Acute Myeloid Leukemia. Blood Cancer Discov 3:316–329. [CrossRef]
- Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, Bowman M, Famulare C, Patel MA, Mendez P, Ainali C, Demaree B, Delley CL, Abate AR, Manivannan M, Sahu S, Goldberg AD, Bolton KL, Zehir A, Rampal R, Carroll MP, Meyer SE, Viny AD, Levine RL (2020) Single cell mutation analysis of clonal evolution in myeloid malignancies. Nature 587:477. [CrossRef]
- Luque Paz D, Kralovics R, Skoda RC (2023) Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 141:1909–1921. [CrossRef]
- Paz DL, Jouanneau-Courville R, Riou J, Ianotto JC, Boyer F, Chauveau A, Renard M, Chomel JC, Cayssials E, Gallego-Hernanz MP, Pastoret C, Murati A, Courtier F, Rousselet MC, Quintin-Roue I, Cottin L, Orvain C, Thepot S, Chretien JM, Delneste Y, Ifrah N, Blanchet O, Hunault-Berger M, Lippert E, Ugo V (2020) Leukemic evolution of polycythemia vera and essential thrombocythemia: genomic profiles predict time to transformation. Blood Adv 4:4887–4897. [CrossRef]
- Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, Przychodzen BJ, Nagata Y, Meggendorfer M, Sanada M, Okuno Y, Hirsch C, Kuzmanovic T, Sato Y, Sato-Otsubo A, Laframboise T, Hosono N, Shiraishi Y, Chiba K, Haferlach C, Kern W, Tanaka H, Shiozawa Y, Gómez-Seguí I, Husseinzadeh HD, Thota S, Guinta KM, Dienes B, Nakamaki T, Miyawaki S, Saunthararajah Y, Chiba S, Miyano S, Shih LY, Haferlach T, Ogawa S, MacIejewski JP (2016) Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet 2016 492 49:204–212. [CrossRef]
- Taylor J, Lee SC, Stanley Lee CC, Sloan Kettering M (2019) Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes, Chromosom Cancer 58:889–902. [CrossRef]
- Kar SA, Jankowska A, Makishima H, Visconte V, Jerez A, Sugimoto Y, Muramatsu H, Traina F, Afable M, Guinta K, Tiu R V., Przychodzen B, Sakaguchi H, Kojima S, Sekeres MA, List AF, McDevitt MA, Maciejewski JP (2013) Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica 98:107. [CrossRef]
- Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, Schauer S, Döhner H, Döhner K (2011) DNMT3A mutations in myeloproliferative neoplasms. Leuk 2011 257 25:1217–1219. [CrossRef]
- Brune MM, Rau A, Overkamp M, Flaadt T, Bonzheim I, Schürch CM, Federmann B, Dirnhofer S, Fend F, Tzankov A (2021) Molecular Progression of Myeloproliferative and Myelodysplastic/Myeloproliferative Neoplasms: A Study on Sequential Bone Marrow Biopsies. Cancers 2021, Vol 13, Page 5605 13:5605. [CrossRef]
- Benton CB, Boddu PC, DiNardo CD, Bose P, Wang F, Assi R, Pemmaraju N, Devendra KC, Pierce S, Patel K, Konopleva M, Ravandi F, Garcia-Manero G, Kadia TM, Cortes J, Kantarjian HM, Andreeff M, Verstovsek S (2019) Janus kinase 2 variants associated with the transformation of myeloproliferative neoplasms into acute myeloid leukemia. Cancer 125:1855–1866. [CrossRef]
- Stengel A, Baer C, Walter W, Meggendorfer M, Kern W, Haferlach T, Haferlach C (2021) Mutational patterns and their correlation to CHIP-related mutations and age in hematological malignancies. Blood Adv 5:4426–4434. [CrossRef]
- Harutyunyan A, Klampfl T, Cazzola M, Kralovics R (2011) p53 Lesions in Leukemic Transformation. N Engl J Med 364:488–490. [CrossRef]
- Rampal R, Ahn J, Abdel-Wahaba O, Nahas M, Wang K, Lipson D, Otto GA, Yelensky R, Hricik T, McKenney AS, Chiosis G, Chung YR, Pandey S, Van Den Brink MRM, Armstrong SA, Dogan A, Intlekofer A, Manshouri T, Park CY, Verstovsek S, Rapaport F, Stephens PJ, Miller VA, Levine RL (2014) Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A 111:E5401–E5410. [CrossRef]
- Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, Wallrabenstein T, Kolbinger B, Köhne CH, Horst HA, Brossart P, Held G, Kündgen A, Ringhoffer M, Götze K, Rummel M, Gerstung M, Campbell P, Kraus JM, Kestler HA, Thol F, Heuser M, Schlegelberger B, Ganser A, Bullinger L, Schlenk RF, Döhner K, Döhner H (2016) RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leuk 2016 3011 30:2160–2168. [CrossRef]
- Jutzi JS, Bogeska R, Nikoloski G, Schmid CA, Seeger TS, Stegelmann F, Schwemmers S, Gründer A, Peeken JC, Gothwal M, Wehrle J, Aumann K, Hamdi K, Dierks C, Wang W, Döhner K, Jansen JH, Pahl HL (2013) MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J Exp Med 210:1003–1019. [CrossRef]
- Zarka J, Short NJ, Kanagal-Shamanna R, Issa GC (2020) Nucleophosmin 1 Mutations in Acute Myeloid Leukemia. Genes (Basel) 11:1–16. [CrossRef]
- Menssen AJ, Walter MJ (2020) Genetics of progression from MDS to secondary leukemia. Blood 136:50–60. [CrossRef]
- Delhommeau F, Dupont S, Valle V Della, James C, Trannoy S, Massé A, Kosmider O, Le Couedic J-P, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Bernard OA (2009) Mutation in TET2 in Myeloid Cancers. N Engl J Med 360:2289–2301. [CrossRef]
- Abdel-Wahab O, Pardanani A, Rampal R, Lasho TL, Levine RL, Tefferi A (2011) DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leuk 2011 257 25:1219–1220. [CrossRef]
- Goyal H, Chachoua I, Pecquet C, Vainchenker W, Constantinescu SN (2020) A p53-JAK-STAT connection involved in myeloproliferative neoplasm pathogenesis and progression to secondary acute myeloid leukemia. Blood Rev 42:100712. [CrossRef]
- Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, Nimer SD, Levine RL, Klimek VM (2013) Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 98:908–912. [CrossRef]
- Guerra VA, Yuanqing Y, Hsu J, Wang F, Alfayez M, Morita K, Song X, DiNardo CD, Konopleva MY, Borthakur GM, Montalban Bravo G, Zhang J, Little L, Gumbs C, Kantarjian HM, Garcia-Manero G, Futreal A, Takahashi K (2019) Comprehensive Analysis of Genotype and Prior Exposures in Therapy-Related Myeloid Neoplasms (t-MNs). Blood 134:458. [CrossRef]
- Rampal R, Ahn J, Abdel-Wahaba O, Nahas M, Wang K, Lipson D, Otto GA, Yelensky R, Hricik T, McKenney AS, Chiosis G, Chung YR, Pandey S, Van Den Brink MRM, Armstrong SA, Dogan A, Intlekofer A, Manshouri T, Park CY, Verstovsek S, Rapaport F, Stephens PJ, Miller VA, Levine RL (2014) Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A 111:E5401–E5410. [CrossRef]
- Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, Girsberger S, Lehmann T, Passweg J, Stern M, Beisel C, Kralovics R, Skoda RC (2014) Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 123:2220–2228. [CrossRef]
- Soenen V, Preudhomme C, Roumier C, Daudignon A, Luc Laı̈ J, Fenaux P (1998) 17p Deletion in Acute Myeloid Leukemia and Myelodysplastic Syndrome. Analysis of Breakpoints and Deleted Segments by Fluorescence In Situ. Blood 91:1008–1015. [CrossRef]
- Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, Von Lilienfeld-Toal M, Lübbert M, Fröhling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Döhner K, Döhner H (2012) TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 119:2114–2121. [CrossRef]
- Bahaj W, Kewan T, Gurnari C, Durmaz A, Ponvilawan B, Pandit I, Kubota Y, Ogbue OD, Zawit M, Madanat Y, Bat T, Balasubramanian SK, Awada H, Ahmed R, Mori M, Meggendorfer M, Haferlach T, Visconte V, Maciejewski JP (2023) Novel scheme for defining the clinical implications of TP53 mutations in myeloid neoplasia. J Hematol Oncol 16:1–12. [CrossRef]
- Rahmé R, Braun T, Manfredi JJ, Fenaux P (2023) TP53 Alterations in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Biomedicines 11:. [CrossRef]
- Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH, Garcia-Manero G, Craddock C, Sallman DA, Kantarjian HM (2022) TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov 12:2516. [CrossRef]
- Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, Wang SA, Bagg A, Barbui T, Branford S, Bueso-Ramos CE, Cortes JE, Dal Cin P, DiNardo CD, Dombret H, Duncavage EJ, Ebert BL, Estey EH, Facchetti F, Foucar K, Gangat N, Gianelli U, Godley LA, Gökbuget N, Gotlib J, Hellström-Lindberg E, Hobbs GS, Hoffman R, Jabbour EJ, Kiladjian JJ, Larson RA, Le Beau MM, Loh MLC, Löwenberg B, Macintyre E, Malcovati L, Mullighan CG, Niemeyer C, Odenike OM, Ogawa S, Orfao A, Papaemmanuil E, Passamonti F, Porkka K, Pui CH, Radich JP, Reiter A, Rozman M, Rudelius M, Savona MR, Schiffer CA, Schmitt-Graeff A, Shimamura A, Sierra J, Stock WA, Stone RM, Tallman MS, Thiele J, Tien HF, Tzankov A, Vannucchi AM, Vyas P, Wei AH, Weinberg OK, Wierzbowska A, Cazzola M, Döhner H, Tefferi A (2022) International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood 140:1200–1228. [CrossRef]
- Sebert M, Gachet S, Leblanc T, Rousseau A, Bluteau O, KIM R, Benabelali R, Sicre de Fontbrune F, Maillard L, Fedronie C, Vasquez N, Bertrand Y, Dalle J-H, Sigaux F, Chevret S, Socie G, de Thé H, Antoniewski C, Bluteau D, Peffault De Latour R, Soulier J (2021) Clonal Hematopoiesis Driven By MDM4 Amplification Defines a Canonical Route Towards Secondary MDS/AML in Fanconi Anemia Patients. Blood 138:860. [CrossRef]
- Pezeshki A, Podder S, Kamel R, Corey SJ (2017) Monosomy 7/del (7q) in inherited bone marrow failure syndromes: A systematic review. Pediatr Blood Cancer 64:e26714. [CrossRef]
- Daniels NJ, Hershberger CE, Gu X, Schueger C, DiPasquale WM, Brick J, Saunthararajah Y, Maciejewski JP, Padgett RA (2021) Functional analyses of human LUC7-like proteins involved in splicing regulation and myeloid neoplasms. Cell Rep 35:. [CrossRef]
- Awada H, Durmaz A, Gurnari C, Kishtagari A, Meggendorfer M, Kerr CM, Kuzmanovic T, Durrani J, Shreve J, Nagata Y, Radivoyevitch T, Advani AS, Ravandi F, Carraway HE, Nazha A, Haferlach C, Saunthararajah Y, Scott J, Visconte V, Kantarjian H, Kadia T, Sekeres MA, Haferlach T, Maciejewski JP (2021) Machine learning integrates genomic signatures for subclassification beyond primary and secondary acute myeloid leukemia. Blood 138:1885–1895. [CrossRef]
- Guglielmelli P, Calabresi L (2021) The MPL mutation. Int Rev Cell Mol Biol 365:163–178. [CrossRef]
- Chang L, Cui Z, Shi D, Chu Y, Wang B, Wan Y, Ma Q, Zhang R, Li H, Cheng X, Cheng T, Zhu X, Li C, Yuan W (2022) Polyclonal evolution of Fanconi anemia to MDS and AML revealed at single cell resolution. Exp Hematol Oncol 11:1–14. [CrossRef]
- Lee C, Kim HN, Kwon JA, Hwang J, Park JY, Shin OS, Yoon SY, Yoon J (2023) Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling. Cancers (Basel) 15:5289. [CrossRef]
- Tembrink M, Gerding WM, Wieczorek S, Mika T, Schroers R, Nguyen HP, Vangala D Ben, Nilius-Eliliwi V (2023) Novel NUP98::ASH1L Gene Fusion in Acute Myeloid Leukemia Detected by Optical Genome Mapping. Cancers (Basel) 15:2942. [CrossRef]
- Zhang X, Zhang Y, Wang C, Wang X (2023) TET (Ten-eleven translocation) family proteins: structure, biological functions and applications. Signal Transduct Target Ther 2023 81 8:1–20. [CrossRef]
- Ganster C, Müller-Thomas C, Haferlach C, Strupp C, Ogata K, Germing U, Hildebrandt B, Mallo M, Lübbert M, Müller C, Solé F, Götze KS, Vandenberghe P, Göhring G, Steinmetz T, Kröger N, Platzbecker U, Söling U, Raynaud S, Shirneshan K, Schanz J, Haase D (2019) Comprehensive analysis of isolated der(1;7)(q10;p10) in a large international homogenous cohort of patients with myelodysplastic syndromes. Genes, Chromosom Cancer 58:689–697. [CrossRef]
- Hebeda K, Boudova L, Beham-Schmid C, Orazi A, Kvasnicka HM, Gianelli U, Tzankov A (2021) Progression, transformation, and unusual manifestations of myelodysplastic syndromes and myelodysplastic-myeloproliferative neoplasms: lessons learned from the XIV European Bone Marrow Working Group Course 2019. Ann Hematol 100:117–133. [CrossRef]
- Gurban P, Mambet C, Botezatu A, Necula LG, Neagu AI, Matei L, Pitica IM, Nedeianu S, Chivu-Economescu M, Bleotu C, Ataman M, Mocanu G, Saguna C, Pavel AG, Stambouli D, Sepulchre E, Anton G, Diaconu CC, Constantinescu SN (2023) Leukemic conversion involving RAS mutations of type 1 CALR-mutated primary myelofibrosis in a patient treated for HCV cirrhosis: a case report. Front Oncol 13:1266996. [CrossRef]
- Zarka J, Short NJ, Kanagal-Shamanna R, Issa GC (2020) Nucleophosmin 1 Mutations in Acute Myeloid Leukemia. Genes (Basel) 11:1–16. [CrossRef]
- Wan C, Wen J, Liang X, Xie Q, Wu W, Wu M, Liu Z (2021) Identification of miR-320 family members as potential diagnostic and prognostic biomarkers in myelodysplastic syndromes. Sci Reports 2021 111 11:1–11. [CrossRef]
- Symeonidis A, Chatzilygeroudi T, Chondrou V, Sgourou A (2022) Contingent Synergistic Interactions between Non-Coding RNAs and DNA-Modifying Enzymes in Myelodysplastic Syndromes. Int J Mol Sci 2022, Vol 23, Page 16069 23:16069. [CrossRef]
- Lyu C, Liu K, Jiang Y, Wang T, Wang Y, Xu R (2021) Integrated analysis on mRNA microarray and microRNA microarray to screen immune-related biomarkers and pathways in myelodysplastic syndrome. 26:417–431. [CrossRef]
- Thol F, Scherr M, Kirchner A, Shahswar R, Battmer K, Kade S, Chaturvedi A, Koenecke C, Stadler M, Platzbecker U, Thiede C, Schroeder T, Kobbe G, Bug G, Ottmann O, Hofmann WK, Kröger N, Fiedler W, Schlenk R, Döhner K, Döhner H, Krauter J, Eder M, Ganser A, Heuser M (2015) Clinical and functional implications of microRNA mutations in a cohort of 935 patients with myelodysplastic syndromes and acute myeloid leukemia. Haematologica 100:e122. [CrossRef]
- Kirimura S, Kurata M, Nakagawa Y, Onishi I, Abe-Suzuki S, Abe S, Yamamoto K, Kitagawa M (2016) Role of microRNA-29b in myelodysplastic syndromes during transformation to overt leukaemia. Pathology 48:233–241. [CrossRef]
- Pavlovic D, Tosic N, Zukic B, Pravdic Z, Vukovic NS, Pavlovic S, Gasic V (2021) Expression Profiles of Long Non-Coding RNA GAS5 and MicroRNA-222 in Younger AML Patients. Diagnostics 2022, Vol 12, Page 86 12:86. [CrossRef]
- Qin T, Cheng Y, Wang X (2022) RNA-binding proteins as drivers of AML and novel therapeutic targets. 63:1045–1057. [CrossRef]
- Bauer M, Vaxevanis C, Heimer N, Al-Ali HK, Jaekel N, Bachmann M, Wickenhauser C, Seliger B (2020) Expression, Regulation and Function of microRNA as Important Players in the Transition of MDS to Secondary AML and Their Cross Talk to RNA-Binding Proteins. Int J Mol Sci 2020, Vol 21, Page 7140 21:7140. [CrossRef]
- Jiang Q, Isquith J, Ladel L, Alexandrov LB, Fisch KM, Jamieson Correspondence C, Mark A, Holm F, Mason C, He Y, Mondala P, Oliver I, Pham J, Ma W, Reynoso E, Ali S, Morris IJ, Diep R, Nasamran C, Xu G, Sasik R, Rosenthal SB, Birmingham A, Coso S, Pineda G, Crews L, Donohoe ME, Craig Venter J, Whisenant T, Mesa RA, Jamieson C (2021) Inflammation-driven deaminase deregulation fuels human pre-leukemia stem cell evolution. [CrossRef]
- Maakaron JE, Mims AS (2019) Daunorubicin-cytarabine liposome (CPX-351) in the management of newly diagnosed secondary AML: A new twist on an old cocktail. Best Pract Res Clin Haematol 32:127–133. [CrossRef]
- Budziszewska BK, Salomon-Perzyński A, Pruszczyk K, Barankiewicz J, Pluta A, Helbig G, Janowska A, Kuydowicz M, Bołkun Ł, Piszcz J, Patkowska E, Wątek M, Małecki P, Kościołek-Zgódka S, Cichocka E, Charliński G, Irga-Staniukiewicz A, Zaucha JM, Piekarska A, Gromek T, Hus M, Wójcik K, Raźny M, Sędzimirska M, Puła B, Giebel S, Grosicki S, Wierzbowska A, Lech-Marańda E (2021) Cladribine Combined with Low-Dose Cytarabine as Frontline Treatment for Unfit Elderly Acute Myeloid Leukemia Patients: Results from a Prospective Multicenter Study of Polish Adult Leukemia Group (PALG). Cancers 2021, Vol 13, Page 4189 13:4189. [CrossRef]
- Schieber M, Marinaccio C, Bolanos LC, Haffey WD, Greis KD, Starczynowski DT, Crispino JD (2020) FBXO11 is a candidate tumor suppressor in the leukemic transformation of myelodysplastic syndrome. Blood Cancer J 2020 1010 10:1–12. [CrossRef]
- Roboz GJ, Mandrekar SJ, Desai P, Laumann K, Walker AR, Wang ES, Kolitz JE, Powell BL, Attar EC, Stock W, Bloomfield CD, Kohlschmidt J, Mrózek K, Hassane DC, Garraway L, Jané-Valbuena J, Baltay M, Tracy A, Marcucci G, Stone RM, Larson RA (2018) Randomized trial of 10 days of decitabine ± bortezomib in untreated older patients with AML: CALGB 11002 (Alliance). Blood Adv 2:3608–3617. [CrossRef]
- Malik P, Cashen AF (2014) Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res 6:53–61. [CrossRef]
- Zong L, Yin M, Kong J, Zhang J, Song B, Zhu J, Xue S, Wu X, Wu D, Bao X, Qiu H (2023) Development of a scoring system for predicting primary resistance to venetoclax plus hypomethylating agents (HMAs) in acute myeloid leukemia patients. Mol Carcinog 62:1572–1584. [CrossRef]
- Haddad F, Daver N (2021) Targeting CD47/SIRPα in Acute Myeloid Leukemia and Myelodysplastic Syndrome: Preclinical and Clinical Developments of Magrolimab. J Immunother Precis Oncol 4:67. [CrossRef]
- Maslah N, Salomao N, Drevon L, Verger E, Partouche N, Ly P, Aubin P, Naoui N, Schlageter MH, Bally C, Miekoutima E, Rahmé R, Lehmann-Che J, Ades L, Fenaux P, Cassinat B, Giraudier S (2020) Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 105:1539. [CrossRef]
- Fiskus W, Mill CP, Nabet B, Perera D, Birdwell C, Manshouri T, Lara B, Kadia TM, DiNardo C, Takahashi K, Daver N, Bose P, Masarova L, Pemmaraju N, Kornblau S, Borthakur G, Montalban-Bravo G, Manero GG, Sharma S, Stubbs M, Su X, Green MR, Coarfa C, Verstovsek S, Khoury JD, Vakoc CR, Bhalla KN (2021) Superior efficacy of co-targeting GFI1/KDM1A and BRD4 against AML and post-MPN secondary AML cells. Blood Cancer J 2021 115 11:1–16. [CrossRef]
- Tibes R, Bogenberger JM (2019) Transcriptional Silencing of MCL-1 Through Cyclin-Dependent Kinase Inhibition in Acute Myeloid Leukemia. Front Oncol 9:1205. [CrossRef]
- Fiskus W, Manshouri T, Birdwell C, Mill CP, Masarova L, Bose P, Kadia TM, Daver N, DiNardo CD, Borthakur G, Khoury JD, Verstovsek S, Bhalla KN (2022) Efficacy of CDK9 inhibition in therapy of post-myeloproliferative neoplasm (MPN) secondary (s) AML cells. Blood Cancer J 2022 121 12:1–5. [CrossRef]
- Crews LA, Ma W, Ladel L, Pham J, Balaian L, Steel SK, Mondala PK, Diep RH, Wu CN, Mason CN, van der Werf I, Oliver I, Reynoso E, Pineda G, Whisenant TC, Wentworth P, La Clair JJ, Jiang Q, Burkart MD, Jamieson CHM (2023) Reversal of malignant ADAR1 splice isoform switching with Rebecsinib. Cell Stem Cell 30:250-263.e6. [CrossRef]
- Qing Y, Su R, Chen J (2021) RNA modifications in hematopoietic malignancies: a new research frontier. Blood 138:637–648. [CrossRef]
- Eisenberg E, Levanon EY (2018) A-to-I RNA editing — immune protector and transcriptome diversifier. Nat Rev Genet 2018 198 19:473–490. [CrossRef]
- Schmaelter AK, Labopin M, Socié G, Itälä-Remes M, Blaise D, Yakoub-Agha I, Forcade E, Cornelissen J, Ganser A, Beelen D, Labussière-Wallet H, Passweg J, Savani BN, Schmid C, Nagler A, Mohty M (2020) Inferior outcome of allogeneic stem cell transplantation for secondary acute myeloid leukemia in first complete remission as compared to de novo acute myeloid leukemia. Blood Cancer J 2020 103 10:1–9. [CrossRef]
- Nilsson C, Hulegårdh E, Garelius H, Möllgård L, Brune M, Wahlin A, Lenhoff S, Frödin U, Remberger M, Höglund M, Juliusson G, Stockelberg D, Lehmann S (2019) Secondary Acute Myeloid Leukemia and the Role of Allogeneic Stem Cell Transplantation in a Population-Based Setting. Biol Blood Marrow Transplant 25:1770–1778. [CrossRef]
- Guolo F, Fianchi L, Minetto P, Clavio M, Gottardi M, Galimberti S, Rizzuto G, Rondoni M, Bertani G, Dargenio M, Bilio A, Scappini B, Zappasodi P, Scattolin AM, Grimaldi F, Pietrantuono G, Musto P, Cerrano M, D’Ardia S, Audisio E, Cignetti A, Pasciolla C, Pavesi F, Candoni A, Gurreri C, Morselli M, Alati C, Fracchiolla N, Rossi G, Caizzi M, Carnevale-Schianca F, Tafuri A, Rossi G, Ferrara F, Pagano L, Lemoli RM (2020) CPX-351 treatment in secondary acute myeloblastic leukemia is effective and improves the feasibility of allogeneic stem cell transplantation: results of the Italian compassionate use program. Blood Cancer J 2020 1010 10:1–11. [CrossRef]
- Limongello R, Marra A, Mancusi A, Bonato S, Hoxha E, Ruggeri L, Hui S, Velardi A, Pierini A (2021) Novel Immune Cell-Based Therapies to Eradicate High-Risk Acute Myeloid Leukemia. Front Immunol 12:695051. [CrossRef]
- Zhang H, Gan WT, Hao WG, Wang PF, Li ZY, Chang LJ (2020) Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front Oncol 10:685. [CrossRef]
- Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, Schnorfeil FM, Krupka C, Lichtenegger FS, Liu X, Kerbs P, Schneider S, Metzeler KH, Spiekermann K, Hiddemann W, Greif PA, Herold T, Sadelain M, Subklewe M (2018) Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leuk 2018 331 33:64–74. [CrossRef]
- Sugita M, Galetto R, Zong H, Ewing-Crystal N, Trujillo-Alonso V, Mencia-Trinchant N, Yip W, Filipe S, Lebuhotel C, Gouble A, Hassane DC, Smith J, Roboz GJ, Guzman ML (2022) Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat Commun 2022 131 13:1–11. [CrossRef]
- Bester AC, Lee JD, Chavez A, Lee YR, Nachmani D, Vora S, Victor J, Sauvageau M, Monteleone E, Rinn JL, Provero P, Church GM, Clohessy JG, Pandolfi PP (2018) An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 173:649-664.e20. [CrossRef]
- Labanieh L, Majzner RG, Mackall CL (2018) Programming CAR-T cells to kill cancer. Nat Biomed Eng. [CrossRef]
- Marofi F, Rahman HS, Al-Obaidi ZMJ, Jalil AT, Abdelbasset WK, Suksatan W, Dorofeev AE, Shomali N, Chartrand MS, Pathak Y, Hassanzadeh A, Baradaran B, Ahmadi M, Saeedi H, Tahmasebi S, Jarahian M (2021) Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res Ther 2021 121 12:1–23. [CrossRef]
- Oliai C, Schiller G (2020) How to address second and therapy-related acute myelogenous leukaemia. Br J Haematol 188:116–128. [CrossRef]
- Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, Schmucker A, Reder J, Sentman CL, Gilham DE, Lehmann FF, Galinsky I, DiPietro H, Cummings K, Munshi NC, Stone RM, Neuberg DS, Soiffer R, Dranoff G, Ritz J, Nikiforow S (2019) Phase i trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. CancerImmunol Res 7:100–112. [CrossRef]
Figure 1.
Schematic representation of mutations, abnormalities and factors driving different hematological disorders and their progression to AML/sAML. Red: chromosomal abnormalities, blue: gene mutations, purple: therapeutic agents [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26].
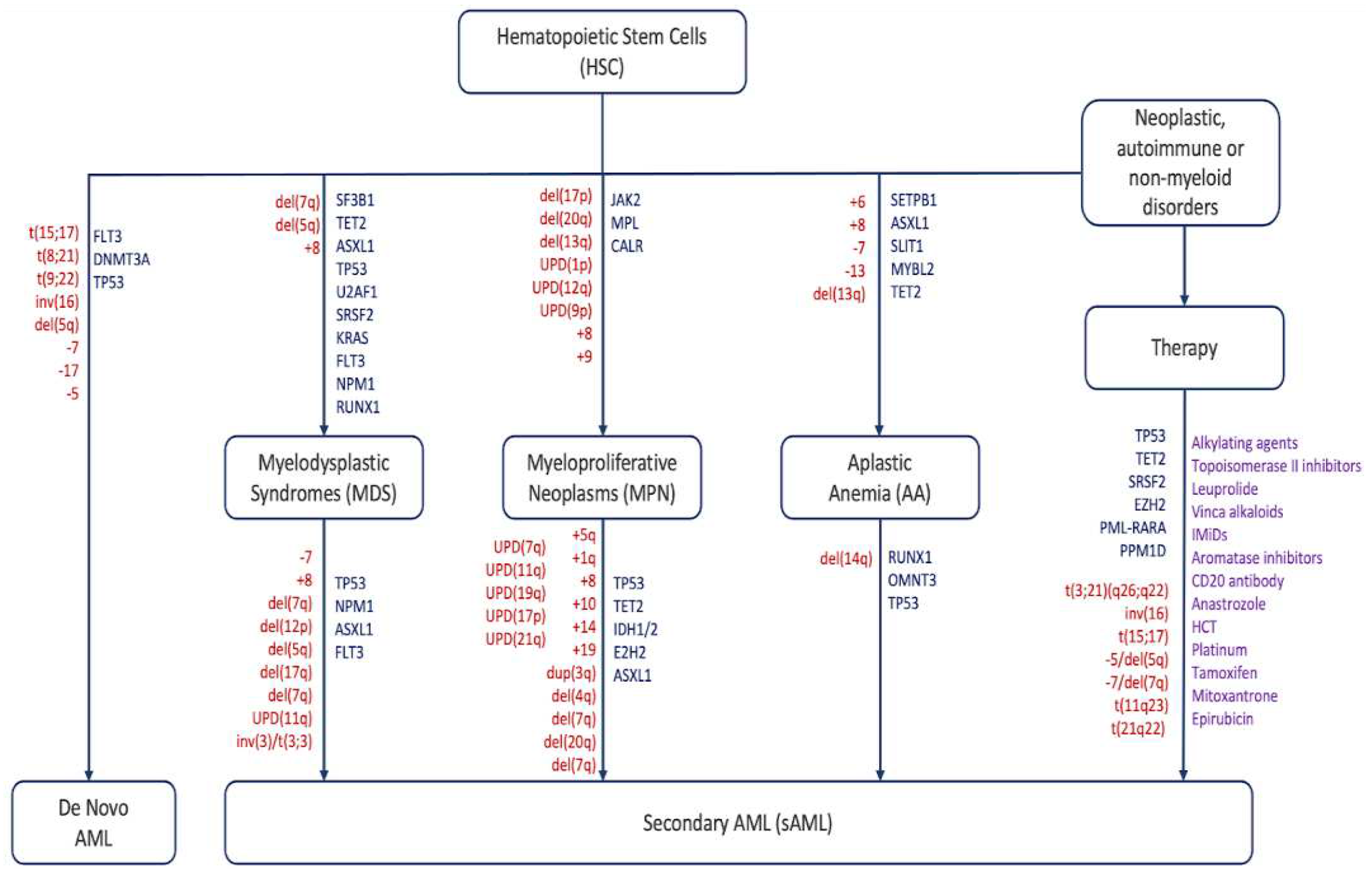
Figure 2.
Pie chart displaying estimated proportions of sAML patients by history based on studies monitoring leukemic transformation. Each color represents the proportion of sAML patients by class. Percentages based on average incidence per 100,000 people.
Figure 2.
Pie chart displaying estimated proportions of sAML patients by history based on studies monitoring leukemic transformation. Each color represents the proportion of sAML patients by class. Percentages based on average incidence per 100,000 people.
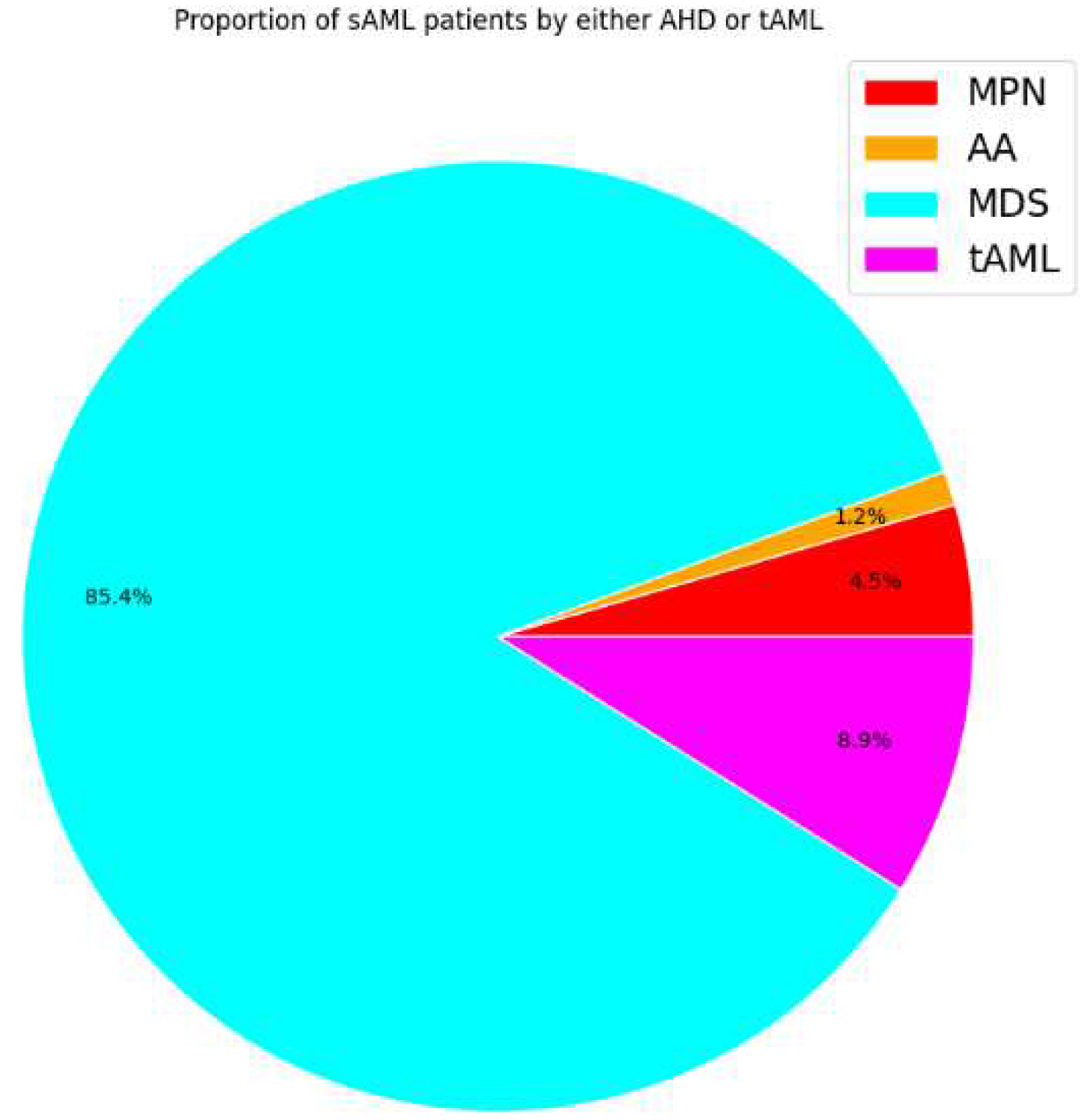
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



