Submitted:
28 January 2024
Posted:
29 January 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Viruses continue to provide vital insights into the field of control of gene expression. Viral transactivators, in concert with other viral and cellular proteins, regulate expression of self, other viruses, and host genes with profound effects on infected cells, underlying inflammation, control of immune responses, and pathogenesis. The multifunctional Tat proteins of lentiviruses (HIV-1, HIV-2, and SIV) transactivate gene expression by recruiting host proteins and binding to trans-acting responsive regions (TARs) in viral and host RNAs. SARS-CoV-2 nucleocapsid participates in early viral transcription, recruits similar cellular proteins, and shares intracellular, surface, and extracellular distribution with Tat. SARS-CoV-2 nucleocapsid interacting with the Replication-Transcription complex might, therefore, transactivate viral and cellular RNAs in transcription and reactivation of self and other viruses, acute and chronic pathogenesis, immune evasion, and viral evolution, constituting a potential pan-coronaviral therapeutic target. Here, we show by using primary and secondary structural comparisons that the leaders of SARS-CoV-2 and other coronaviruses contain TAR-like sequences in stem-loops 2 and 3, the coronaviral nucleocapsid C-terminal domains (N-CTDs) harbor a region of similarity to TAR-binding regions of lentiviral Tat proteins, and the coronaviral nonstructural protein 12 has a cysteine-rich metal binding, dimerization domain similar to that in lentiviral Tat proteins.
Keywords:
transactivation
; HIV
; SARS-CoV-2
; TAR
; Tat
; nucleocapsid
; RNA-dependent RNA polymerase
; HEMIX
; pan-coronaviral target
; long COVID
1. Introduction
Transcription of viruses requires a variety of viral and cellular proteins and is essential for viral genome amplification and reactivation. Viruses have a narrow window for effective replication and transmission, and have developed strategies to boost their transcription, gene expression and immune evasion by affecting various cellular pathways, protein and nucleic acid interactions, and gene expression. Transactivation of gene expression often encompassing intra- and transcellular effects is one such strategy, also contributing to transmission, acute and long-term pathogenesis, and reactivation from latency. The human immunodeficiency virus (HIV)-1, for instance, encodes Tat, a transactivator of gene expression discovered four decades ago in the Haseltine laboratory in cells infected with HIV-1, in which it stimulated by at least two orders of magnitude the synthesis of reporter genes placed under the control of the HIV-1 long-terminal repeat [1–3]. This activity was corroborated in an in vitro cell-free system [4,5]. Tat thus became the first non-prokaryotic transcription factor known to interact with RNA and emulate prokaryotic anti-termination factors, driving a positive feedback loop of gene expression.
HIV-1 Tat increases the processivity of the host’s RNA polymerase II [6,7] by recruiting the host’s super elongation complex (SEC) [8]. The SEC includes the heterodimer positive transcription elongation factor-b (P-TEFb) complex comprising Cyclin T1 and CDK9 [9–14], and the polymerase-associated factor 1 (PAF-1) complex [15,16] to the trans-acting responsive region (TAR). Located downstream of the initiation site for HIV-1 transcription (nucleotides +1 to +59), TAR adopts an A-form stem-loop structure with a bulge [4,17–25].
In the absence of Tat, short non-polyadenylated RNAs terminating at TAR accumulate [26], are translocated to the cytoplasm, and translated into Tat [27,28]. Tat then enters the nucleus and nucleolus and binds P-TEFb, increasing its components’ efficiency, including triggering CDK9 autophosphorylation [9,10,29]. Levels of viral full-length polyadenylated long messenger (m)RNAs thus increase significantly [26,30–32] via several mechanisms [33].
The Tat-P-TEFb complex stabilizes RNA polymerase II, allowing it to overcome premature termination of transcription resulting from the assembly of two multi-subunit complexes, the negative elongation factor (NELF) and the DRB sensitivity-inducing factor (DSIF) [34,35], and RNA polymerase II meeting TAR [36,37]. The transition from abortive to productive elongation involves CDK9-mediated phosphorylation of the carboxyl-terminal domain of the largest RNA polymerase II subunit RPB1 after Cyclin-T1’s binding to it facilitated by liquid-liquid phase formation [7,10,38–45] and by phosphorylation of NELF and DSIF components [16,34,46–48].
Tat antagonizes other negative elongation factors. For instance, the positively charged arginine-rich TAR-recognition motif of HIV Tat shows strong similarity to the N-terminal part of the 7SK-binding motif of the hexamethylene bisacetamide (HMBA)-inducible (HEXIM) proteins from evolutionarily distant species [49,50]. Without involving Cyclin T1, Tat binds to a TAR-like sequence in the 7SK small nuclear RNA [51] and replaces HEXIM1 [50] in the 7SK ribonucleoprotein (RNP). This interaction disassembles the kinase-inactive 7SK/HEXIM/P-TEFb negative transcriptional regulatory small nuclear RNP, which includes up to half of the cellular P-TEFb [52]. The result is increased nuclear levels of active P-TEFb and viral transcription and replication. As another way to favor gene expression, when nucleosomes obstruct the path of the elongating RNA polymerase II, Tat-P-TEFb can modify histones by recruiting histone acetyltransferases like the CBP/p300 complex to the viral promoter to activate nucleosome acetylation [37,47,53–55].
Beyond viral gene expression, HIV-1 Tat transactivates the expression of host cytokine genes [33], such as tumor necrosis factor (TNF)-β [56–58],interleukin ( IL)-6 [59,60], IL-8 [61], the C-X-C receptor (CXCR)4 chemokine [62], and the β-chemokine monocyte chemoattractant protein (MCP)-1 [63]. Tat can also inhibit cytokine expression, as in the case of IL-2 [64–66]. These effects are accomplished through TAR-like or non-TAR promoter regions that bind transcription regulators such as NF-κB and SP1 [14,67–69]. By binding to SP1, Tat can decrease the recruitment of SP1 to the promoter of the heat shock protein (HSP)70 binding protein (HSPBP)1 gene [70], and decrease its expression, thereby attenuating HSPBP1-mediated inhibition of viral replication [71]. Moreover, the cleavage and polyadenylation specificity factor (CPSF) interacts with the HIV-1 promoter to repress HIV-1gene expression, and HV-1 Tat interacts with CPSF, attenuating said repression [72].
The effects of Tat on viral and host cytokine gene expression and their regulators underlie acute and chronic clinical manifestations. Tat can also be found as a surface-bound or an extracellular protein with an Arginine-Glycine-Aspartic acid (RGD) integrin-binding motif encoded by a second exon, which does not contribute to the transcriptional activities of Tat but can contribute to neurological and vascular manifestations of HIV-1 infection [73–76]. For instance, HIV-1 Tat stimulates in a TAR-independent manner, the expression of some endogenous retrovirus type K (HML-2) proviruses [77,78] or endogenous viruses linked to inflammation, neurodegeneration, and oncogenesis [79,80]. HIV-1 can also transactivate viruses such as the human neurotropic John Cunningham (JC) virus, a polyomavirus, via TAR-like sequences [81] reactivating JCV and increasing the risk of developing progressive multifocal leukoencephalopathy, a neurodegenerative disorder.
Other than HIV-1, several viruses, including other lentiviruses, such as HIV-2 and simian immunodeficiency virus (SIV) [82,83], human T-lymphotropic virus types I and II [84,85], adenovirus [86], simian virus 40 [87], and human herpesviruses, such as human simplex virus [88] and Epstein-Barr virus (EBV) [89,90], among others, use transactivation of gene expression. Viruses such as EBV can transactivate HIV-1 gene expression [89]. We, therefore, assessed using a bioinformatics approach if the severe acute respiratory syndrome-coronavirus (SARS-CoV)-2 and other coronaviruses might contain TAR-like sequences and, if so, if they also encode a Tat-like transactivator and we found both to be in theory the case. We discuss the implications of these findings and other consistent evidence on replication, reactivation of self and other viruses, immune evasion, inflammation and cytokine storm, acute and chronic pathogenesis, and variant evolution of coronaviruses, which cause respiratory and systemic infections in humans and animals and are responsible for epidemics and pandemics. If these implications prove valid, targeting the coronaviral transactivation might be a promising pan-coronaviral therapeutic target.
2. Results
2.1. TAR-like sequences are present in the SARS-CoV-2 RNA leader
To determine if SARS-CoV-2 might also use gene expression transactivation, we first searched for a Tat-binding-like site in the SARS-CoV-2 leader and found the sequence AGAUCUG, which is identical to the HIV-1 and JCV TAR sequences to which HIV-1 Tat binds (AGAUCU, binding sequence underlined, with an additional guanosine residue), and spans the entire right stem half of stem-loop (SL) 2 up to the first nucleotide of SL3 (Figure 1). The secondary structure for the segment including the AGAUCUG sequence of the SARS-CoV-2 leader sequence is similar to that by which Tat recognizes TAR, i.e., with an indispensable first uridine in the bulge (AGAUCU) and G-C (AGAUCU) and A-U (AGAUCU) base pairs in the upper stem of the HIV-1 TAR stem-bulge-stem [21–23,25,91–96]. In JCV TAR, the Tat binding sequence is at its loop, not its bulge.
Adjacent to the identical Tat-binding sequence in the SARS-CoV-2 leader, there are two other regions of similarity to the TARs of HIV-1 and JCV; the proximal one (light blue, Figure 1) spans most of the rest of SL2, and the distal one (fuchsia, Figure1) spans most of the rest of SL3 including the transcription regulatory sequence (TRS). Although the SARS-CoV-2 leader lacks a Cyclin T1 binding sequence at the same position as HIV-1 TAR (loop), the complement of the Sarbecovirus SL1 loop and part of its second stem-half match the Cyclin T1 binding site sequence (7 of 10 nucleotides; Figure 1), which might provide a means for these coronaviral leaders to recruit Cyclin T1 if it can also bind the complement of its binding sequence. The JCV TAR also lacks a Cyclin T1 binding site at the same position as that of HIV-1 TAR but has a Cyclin T1 binding-like site in its bulge, which is also similar to the Sarbecovirus SL1 complement and is present in the HIV-1 TAR (Figure 1).
The similarities between the SARS-CoV-2 leader and the JCV and HIV-1 TARs are apparent not only at the primary structure but also at the secondary structure level. This is underscored in Figure 2, which shows the visualized secondary structures highlighting similarities among the stems of the hairpin (stem-loop) structures and the Tat-binding sites in the SARS-CoV-2 leader and the JCV and HIV-1 TARs.
2.2. The leaders of all genera of coronaviruses share the same regions of similarities as SARS-CoV-2 with the TARs of HIV-1, HIV-2, SIV, and the human 7SK small nuclear RNA
The regions of similarity between the HIV-1 and JCV TARs and the SARS-CoV-2 leader shown in Figure 1 are also present in the leaders of the other six β- and α-coronaviruses known to infect humans, in λ- and δ-coronaviruses, HIV-2 and SIV TARs and the human 7SK snRNA TAR (Figure 3).
Similarities shown in Figure 3 extend to β-coronaviruses infecting other species including, for example, bat (Accession numbers: MK211377, MK211378, KY770858, KY770859, KY417143, KY417146, KY417150) and civet (AY304486) for Sarbecoviruses; porcine (KY419113, DQ011855), rabbit (JN874562), bovine (NC_003405), rat (NC_026011, KM349742, NC_012936), and mouse (JQ173883, AF201929, AF208067, NC_001846) for Embecoviruses; and bat (KJ473821, NC_039207, NC_009019, DQ648794, EF065505, NC_009020, EF65510, EF065509) for Merbecoviruses. The regions of similarity are also well conserved among SARS-CoV-2 strains (for instance, NC-045512, MT324062, MT123290.1, MT019530.1, MT020781.2, MT184908.1, MT152824.1, MT123291.2, MT163718.1, LC528233.1).
The HIV-2 leader has a 123-nucleotide-long TAR sequence relative to the 59-nucleotide-long TAR of the more pathogenic HIV-1 [102]. Depending on the model, the HIV-2 TAR can fold into a three-stem-loop structure or a long stem-loop. The Tat binding site is located in a bulge region in both cases. There is heterogeneity among SIV TARs, as reflected in Figure 3. The TAR of the SIV CPZ strain is almost identical to that of HIV-1, while the TARs of the SIV MND and CPZ strains are closer in length and secondary structure to that of HIV-2. The Tat binding sites also differ among the TARs of HIV-1 (AGAUCU; binding site underlined), HIV-2 (AGAUUG), and SIV (AGAUCU or GGGUUG depending on the strain). A similar heterogeneity in potential Tat binding sites is seen among the leaders of the four genera of coronaviruses (Figure 3).
Leaders of coronaviruses other than those of SARS-CoV-2 and -1 do not have a region matching the complement of the Cyclin T1 binding site (with 7 or 6 nucleotides, respectively, out of 10 nucleotides; Figure 1). As shown in Figure 4, and in contrast to HIV-1 and JCV TARs (Figure 1), the similarity between the complement (antisense) of the HIV-2 TAR in the region corresponding to the Cyclin T1 binding site-like sequence (8 out of 10 nucleotides) extends to the sequence corresponding to the HIV-2 Tat binding site (AGAUUG) within the SL1 region in the SARS-CoV-2 leader.
2.3. The C-terminal domain of the SARS-CoV-2 nucleocapsid protein contains a nucleotide-binding arginine-rich and adjacent regions similar to the TAR binding site and adjacent regions in HIV-1, HIV-2, and SIV Tat and the human HEXIM proteins that bind to the 7SK snRNA TAR
The presence of TAR-like regions in coronaviral leaders led us to conduct an in silico search for a possible Tat protein homolog encoded by coronaviruses. As is the case with many RNA-binding proteins, the HIV-1 Tat protein contains an arginine-rich segment spanning amino acid residues 48-57 that binds TAR RNA through an arginine fork [103–108]. In general, arginine can interact with RNA nucleotide bases via highly specific hydrogen bonding and π-stacking, recognizing specific RNA stem-loops, internal loops, bulges, and purine quartets [22,93,107,109,110]. The basic region of Tat becomes partially or fully structured upon binding RNA and induces a conformational change in the RNA [106,107].
Using the BLAST algorithm, we compared the HIV-1 Tat protein and SARS-CoV-2 proteins, as well as amino acid sequences encoded by all six open reading frames in the sense and antisense SARS-CoV-2 genome. These analyses showed that the C-terminal domain of the nucleocapsid protein (N-CTD) of SARS-CoV-2 has a nucleic acid-binding, basic amino acid-rich region (amino acid residues 255 to 264) similar to the HIV-1 TAR binding region (e=0.02 for comparison between SARS-CoV-2 N and HIV-1 Tat; and 0.72 for comparisons between HIV-1 Tat and the translated sequence of sense open reading frame 2 of the SARS-CoV-2 genome). The similarity extends to adjacent regions of the SARS-CoV-2 N-CTD (Figure 5). Amino acids at both adjacent sides of the basic domain are important in increasing Tat-TAR binding affinity [93,111].
In Tat, the substitution of the basic amino acids Lysine and Arginine within the basic residue-rich binding region reduces binding affinity to TAR [111]. In the N-CTD of SARS-CoV-2, a 3D structural study revealed that some of the arginine and lysine residues in the basic amino acid-rich and adjacent region form a nucleic acid binding groove which constitutes one of the most positively charged regions of the N protein [112]. The basic amino acids that form the groove can interact with the negatively charged single- and double-stranded RNA or DNA in vitro without nucleotide sequence specificity, with ssRNA molecules as short as 7 base pairs expected to be well cradled by the groove [112–115]. For instance, a study showed that SARS-CoV-2 N bound a 17-mer single-stranded oligonucleotide (nucleotides 6-76: UGUCUCUAAACGAACU) spanning from the last base of the Tat-like binding site (underlined) to the end of the leader sequence [112].
Figure 5 shows that the five groove-forming basic amino acid positions within the basic amino acid-rich region are shared with the HIV-1 TAR (highlighted with red triangles), HEXIM proteins, and with some exceptions, with the other β-, and α- λ-, and δ-coronaviruses. Some substitutions in the basic amino acid-rich region among coronaviruses are also seen in the HIV-1 Tat proteins of pandemic HIV-1 group M subtypes and other lentiviral Tat proteins [111]. The three basic amino acid positions outside the basic amino acid-rich region that were characterized to contribute to the nucleic acid binding groove of SARS-CoV-2 are also present, albeit not as well preserved among coronaviruses, lentiviral Tat proteins, and HEXIM proteins.
Other β- and α-coronaviruses infecting humans and λ- and δ-coronaviruses have similar N-CTD regions. The similarity extends to the Tat proteins of HIV-2 and SIV and to the host HEXIM proteins, which bind to the 7SK snRNA TAR-like structure (Figure 5).
No or a negligible percentage (<0.0004%) of amino acid substitutions in the TAR binding-like region of SARS-CoV-2 N-CTD were detected in a GISAID database [GISAID - Initiative; 116-118] search of all SARS-CoV-2 lineages, including Alpha (B.1.1.7+ Q.*), Beta (B.1.351 + B.1.351.2 + B.1.351.3). Gamma (P.1 + P.1.*), Delta (B.1.617.2), Lambda (C.37 + C.37.1), Mu (B.1.621 + B.1.621.1), Omicron (B.1.529 + BA.*), BA.286 + BA.286.*, XBB.1.5 + XBB.1.5.*, XBB.1.16 + XBB.1.16.*, EG.5 + EG.5.*, BA.2.75 + BA.275.*, CH.1.1 + CH.1.1.*, XBB + XBB.*, XBB.1.9.1 + XBB.1.9.1.*, XBB.1.9.2 + XBB.1.9.2.*, XBB.2.3 + XBB.2.3.*, GH/490R (B.1.640 + B.1.640.*). Individual mutations were found in only poor-quality sequences.
The nucleocapsid protein is a substrate of cyclin-dependent kinase (CDK), glycogen synthase kinase, mitogen-activated protein kinase, and casein kinase II [119]. The binding of Cyclin T1 and CPK9 to the SARS-CoV-2 and -1 N proteins (amino acids 186 to 191) may recruit the P-TEFb heterodimer for transactivation of gene expression.
Among known RNA-binding proteins, RGG/RG motif-containing proteins [110,120] are the second most common in the human genome [110,120,121]. However, lentiviral Tat proteins and the N-CTDs of SARS-CoV-2 and most other coronaviruses contain only one copy of an RG within or in the region adjacent to the TAR or TAR-like binding sites.
RNA binding of the SARS-CoV-2 genome is also required for viral capsid formation [122]. The N-CTD is also referred to as the dimerization domain, which is needed because the instability of monomeric N-CTD [123]. The dimerization core required to form the viral capsid is distal to the nucleic acid-binding region shown in Figure 5.
The arginine-rich domain in HIV-1 Tat not only functions as an RNA binding domain (RBD) but also as a protein transduction domain (PTD) and a nuclear localization signal (NLS) [124]. SARS-CoV-2 and -1 have a nuclear localization signal at the end of N, and localization may be regulated by phosphorylation of the serine-arginine-rich region of N [125,126]. The N coronaviral protein colocalizes with replicase components early during infection [127], suggesting its early intervention in RNA transcription [128–130].
2.4. The interface region of the NSP12 protein of SARS-CoV-2 contains a cysteine-rich region that is similar to that present in HIV-1, HIV-2, and SIV Tat proteins
Another HIV-1 Tat-like feature revealed by the bioinformatics approach involves the highly conserved zinc-coordinating cysteine-rich region (7 cysteines out of 16 residues) that is essential for Tat function [131–135] via metal-linked dimers [136,137].
As shown in Figure 6, the interface region between the Nidovirus RNA-dependent RNA polymerase-associated nucleotidyl transferase (NiRAN) domain and the RNA-dependent RNA polymerase (RdRp) of the SARS-CoV-2 nonstructural protein (NSP)12 protein has the conserved cysteine-rich motif of HIV-1 Tat (e=0.002 when comparing HIV-1 and SARS-CoV-2 NSP12). This domain is present in the Tat proteins of HIV-1, HIV-2, and SIV within a larger region of similarity. The positions of dissimilarity between the HIV-1/SIV and HIV-2 Tat proteins tend to coincide with those with dissimilarity between HIV-1/SIV Tat and the interface regions between NiRAN domains and RdRps of coronaviruses. Although lentiviral Tat proteins have regions of similarities in two different coronaviral proteins, namely, the nucleocapsid protein and RNA-dependent polymerase protein, these two proteins, together with other nonstructural proteins, interact in the Replication-Transcription complex [138], where they may work in synchrony in transactivation of gene expression.
2.5. The β-arterivirus porcine reproductive and respiratory syndrome virus also contains a Tat-binding site identical to that in human 7SK snRNA and adjacent similarities, and its nucleocapsid has a basic amino acid-rich region similar to that in Tat
Arteriviruses have been grouped with coronaviruses under the order Nidovirales. The crystal structure of the SARS-CoV-1 nucleocapsid protein dimerization domain that partially overlaps with the nucleic acid binding domain revealed an evolutionary linkage between corona- and arteriviruses [123]. We, therefore, analyzed the leader of the β-arterivirus porcine reproductive and respiratory syndrome virus (PRRSV) and found it to contain a potential Tat-binding site (CCAUUG) identical to that of human 7SK snRNA and closest to HIV-2 and SIV TARs with UU dinucleotides as binding sites (AGAUUG and GGGUUG, respectively) (Figure 7A). Regions of similarity, including the Tat binding site, correspond to those shown in Figure 3. The coronaviruses with the closest similarities to HIV-1 Tat are the Sarbecoviruses SARS-CoV-2 and -1, with an identical AGAUCU Tat binding site extending the similarity to the rest of SL2 and having in SL1 a complement of the Cyclin T1-like binding site.
Figure 7B depicts the alignment of the PRRSV N with the Tat proteins of HIV-1, HIV-2, SIV, the human HEXIM1 and 2 proteins, and the coronaviral N-CTDs. The lentiviral Tat core region (highlighted in green before the red basic amino acid region in Figure 5) contains a Lysine (K) residue at position 41 in HIV-1 and SIV-1 Tat, and 70 in HIV-2 Tat that is essential for transactivation while the other residues in the core region are partially essential [133]. A lysine residue is similarly located in the N-CTD region of similarity of α-, β- and γ-coronaviruses analyzed and substituted for a negatively charged aspartic, acid, glutamic acid, or glutamine in human HEXIM1, δ-coronavirus, and the arterivirus PRRSV analyzed. The effects of such substitutions on binding affinities remain to be determined.
The zinc-coordinating cysteine-rich motif in lentiviral Tat proteins and the interface region of the coronaviral RNA-dependent RNA polymerase is absent in any of the known encoded proteins of PRRSV. These observations suggest that transactivation of gene expression might extend to arteriviruses, and possibly to the entire Nidovirales order, illustrating how dissimilar RNA and DNA viruses come up with similar strategies.
3. Discussion
The present study revealed the presence of TAR-like and Tat-like sequences in SARS-CoV-2, other coronaviruses, and an arterivirus infecting humans and animals. The TAR-like sequences are located in the leader regions and encompass stem-loop (SL) 2 and 3. The Tat-like sequences span the nucleic acid-binding and adjacent region of the CTDs of coronaviral nucleocapsid proteins and the N protein of an arterivirus, and a Zn-coordinating motif in the interface region between the NiRAN domain and the polymerase domain of coronaviral RNA-dependent polymerase (NSP12). The nucleocapsid and NSP12 proteins interact in the coronaviral Replication-Transcription complex and recruit similar transactivation-related host proteins as the lentiviral Tat proteins. These findings open up the possibility for coronaviruses and arteriviruses, both of the Nidovirales order, to join the ranks of other viruses that use transactivation of gene expression as a means to favor their transcription and replication and that of other viruses while participating in immune evasion and viral-associated acute and chronic pathogenicity.
Coronaviruses include pathogens of significant veterinary and medical importance. The emergence of a highly pathogenic human coronavirus in China in 2019 confirmed the long-held opinion that coronaviruses are important emerging and re-emerging pathogens [139]. The possibility of coronaviruses using transactivation of gene expression as a tactic to hijack their host’s cell machinery to their advantage would be particularly pertinent to transcribing and replicating their complex single-stranded, positive-sense, non-segmented, polycistronic genomes up to 37 kilobases, the largest among RNA viruses [140].
Transactivation of gene expression may have an ancient origin, as do coronaviruses [141,142]. Coronaviruses currently infecting humans are believed to have emerged repeatedly from zoonotic sources for the past one thousand years [141–143]. Alpha coronaviruses are the oldest known coronaviruses infecting humans, and alpha and beta coronaviruses are estimated to have separated from the gamma and delta ones, for which wild birds are the main reservoir [144,145], 300 million years ago, when the evolutionary line for mammals was estimated to have diverged from that for birds [141,142].
The extended evolutionary periods have allowed coronaviruses infecting humans the opportunity to evolve and adapt to their host, rendering it particularly challenging to design preventive and therapeutic interventions that will be effective and durable. Transactivation of gene expression would, therefore, be part of an elaborate replication strategy, enabling coronaviruses to efficiently synthesize various proteins essential for the multiple stages of their life cycle and interactions with host cells. This would also render the transactivation of gene expression a therapeutic target.
3.1. Implications of potential transactivation to transcription of SARS-CoV-2 and other coronaviruses
Transcription of SARS-CoV-2 and other coronaviruses takes place as a continuous process for open reading frame (ORF)1a/b in the proximal two-thirds of the genome and a discontinuous process unique among RNA viruses for the structural and accessory genes in the distal third of the genome [146]. The 5’-untranslated region (UTR) begins with a short 5’ leader sequence (nucleotides 1-69), added via discontinuous transcription to the 5'-end of all subgenomic RNAs encoding viral structural and accessory proteins. Although the 5’-UTR nucleotide sequences are somewhat divergent amongst coronaviruses, the secondary structure is highly conserved. SL2 is the most conserved structure in coronavirus 5′-UTRs [147]. Notably, SL2s from different coronaviruses can functionally replace each other, even across different genera [148]. We show that SL2 includes a Tat-like binding site and a proximal region of similarity with lentiviral and JCV TARs and human 7SK snRNA.
We also show that the nucleic acid binding domain of N-CTD of SARS-CoV-2 and other coronaviruses is similar to that of lentiviral Tat and host HEXIM1 and 2, which are known to bind to the TAR-like structure of 7SK snRNA. The N proteins of human coronaviruses bind viral RNA and play pivotal roles in packaging and transcribing viral RNA [149,150]. The N gene is highly conserved and stable, with 90% amino acid homology and minimal mutations over time. Coronaviral N, a 45 kDa protein in SARS-CoV-2, is composed of two separate domains, an N- and a C-terminal domains, both capable of binding RNA in vitro in SARS-CoVs [113,114,151]. However, each domain uses different mechanisms to bind RNA, with N-NTD likely preferring unpaired single-stranded regions and N-CTD preferring double-stranded RNA. It has been suggested that optimal RNA binding requires contributions from both domains [152,153], creating a local high density of N [154–157].
The more distal region of similarity shown here between coronavirus leaders and lentiviral and JCV TARs comprises the coronaviral SL3, which contains the transcription regulatory sequence (TRS) essential for the synthesis of subgenomic messenger RNAs by discontinuous transcription [148]. Unwinding of SL3 by the N-NTD might be necessary for TRS function, as in mouse hepatitis virus, most closely related to SARS-CoV-1 and -2 [155,158]. Unwinding of SL3 by N-NTD might also contribute to N-mediated transactivation, while N-NTD binding to SL4a, SL7, and SL8 is involved in viral packaging [159].
The highly structured SARS-CoV-2 SL3 binds with high affinity to the human T-cell intracellular antigen (TIA)1 protein [160], a ubiquitous RNA-binding protein (RBP)20 that plays multifunctional regulatory roles in gene expression, including transcription, alternative splicing, and translation of messenger RNAs, as well as in cell stress and viral infections [161]. TIA1 protein could interact with conserved SL3 RNA elements within other beta-coronavirus lineages [160]. How and if the NTD and CTD of the N protein interact with adjacent sequences in the coronaviral leader sequence and with other RNA-binding proteins, as well as the roles of these interactions on potential transactivation of gene expression and other functions, remains to be determined.
Our analysis also highlighted the presence of a shared zinc-coordinating cysteine-rich motif between lentiviral Tat and the interface region of the coronaviral RNA-dependent RNA polymerase protein. In lentiviral Tat, the cysteine-rich motif is required for dimerization, protein structure stabilization, and metal binding [108]. Beyond the RNA-dependent RNA polymerase (NSP12), the coronaviral replication-transcription complex includes processivity factors (NSP7-8), a helicase (NSP13), single-strand binding protein (NSP9), a proofreading exonuclease (NSP14), other cofactors (e.g., NSP10), and capping enzymes (e.g., NSP16). In addition to many viral nonstructural proteins, the presence of cell nuclear proteins and the coronaviral nucleocapsid protein in the coronaviral replication-transcription complex increases virus amplification efficacy [162], to which transactivation of gene expression might contribute based on the findings presented here. The complexity of the RTC, including N, is reminiscent of replisomes from DNA-based organisms and is potentially a consequence of the unusually large coronaviral genomes [162]. Extending the prokaryotic analogy, our comparisons of SARS-CoV-2 N-CTD against the GenBank database revealed its similarity (85.4%) to the amino-terminal portions of two bacterial site-specific DNA methyltransferases likely involved in epigenetic regulation of gene expression [163] (Figure 8).
Coronaviral N is the most copiously expressed protein during infection [164]. It is responsible for releasing nascent negative-strand RNA that promotes a template switch enabling the transcription of subgenomic RNAs [165]. Circularization or cyclization of the coronavirus genome, which places the 5’ with 3’ termini and their regulatory sequences [147,166] in proximity, also has been postulated as a necessary early requirement for synthesis of subgenomic negative-sense strands [129] (Figure 8). In SARS-CoV-1 and -2, two complementary segments in the genomic termini have been identified [167]. The 5’ sequence is 36 nucleotides long. It begins at position 60 from the 5’ terminal m7G cap structure of SARS-CoV-2, which corresponds to the last uridine of the sequence in the SARS-CoV-2 leader identical to the HIV-1 Tat binding sequence (AGAUCU). This 36-nucleotide segment includes the stem-loop structure SL3, which encompasses the TRS (ACGAAC) in the 5’ leader sequence and extends to the beginning of SL4. Another 36-nucleotide sequence complementary to the 5’sequence is located at the 3’ terminus immediately 5’ to the polyadenylation site (Figure 9).
In SARS-CoV-2, genome circularization would require complete opening of SL3 and disruption of the triple helix junction in the 3′-UTR. In agreement with this observation, SL3 of related β-coronaviruses was suggested to be weakly folded or unfolded [168,169]. Genome circularization plays an essential role in the replication of several RNA viruses, including flaviviruses [167,170]. Competition within cells between intact and defective viral genomes may underlie the evolutionary selection of genome circularization by ensuring that only genomes bearing intact 5’ and 3’ UTRs engage with the replication machinery. The SARS-CoV-2 genome cyclization results in a complete opening of the 5′ SL3 where the TRS-L resides, raising the possibility that genome cyclization regulates SARS-CoV-2 discontinuous transcription, as was previously suggested for mouse hepatitis virus [168]. In the setting of the linearized SL3, the N protein, by potentially binding through its CTD to the Tat-like binding sequence, may mediate the transactivation of subgenomic RNA expression.
The finding of potential circularization sequences only in the termini of SARS-CoV-2 and -1 RNA genomes raises the question of how other coronaviruses replicate without said sequences. In bovine coronavirus, the N protein can act as a bridge to facilitate interaction between the 5'- and 3'-ends of its genome, leading to circularization of the genome [171]. A protein bridge composed of cap-binding protein, eIF4E, eIF4G, and poly (A) binding protein may also mediate circularization and subsequent initiation of replicase gene translation [172–181]. The presence of the circularization sequences in SARS-CoV-2 and -1 might help stabilize such protein bridges, potentially translate into increased replication competence, and contribute to the transactivation of gene expression of SARS-CoV-1 and SARS-CoV-2. Determining which viral and cellular factors contribute to the replication complexes and how the putative RNA-RNA interaction between genomic termini contributes to facilitating or stabilizing RNA-protein and protein-protein interactions in subgenomic RNA synthesis.
The discontinuous transcription process encounters a decision problem when reaching the TRS: transcription stops and switches to the leader TRS to produce shorter subgenomic mRNAs, or transcription continues through the TRS to generate longer subgenomic mRNAs and genomic RNAs. The greater abundance of shorter than longer subgenomic mRNAs suggests that transcription might switch upon reaching the 3′ end of the body TRS to produce shorter subgenomic RNAs [181]. However, the virus has to pass the body TRS in an appropriate proportion to produce enough longer subgenomic mRNAs and genomic RNA essential to the life cycle. Studies have investigated several cis-regulating elements and trans-regulating factors involved in this process [182].
The N protein might also participate in the discontinuous transcription of subgenomic mRNAs because the depletion of N from the replicon reduces the synthesis of subgenomic mRNA but not genomic RNA [129]. In the mouse hepatitis virus JHM strain, nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enable the transition from discontinuous to continuous transcription [183]. Phosphorylation of viral N by host glycogen synthase kinase-3 (GSK-3) is required for template switching. GSK-3 inhibition selectively reduces the generation of genomic RNA and longer subgenomic mRNAs but not shorter subgenomic mRNAs.
N phosphorylation allows the recruitment of the RNA helicase DDX1 to the phosphorylated-N-containing complex, facilitating template readthrough and enabling longer subgenomic mRNA synthesis. DDX1 is a member of the DEAD-box protein family, the largest family of the superfamily 2 (SF2) helicases [184]. DDX1 knockdown or loss of helicase activity markedly reduces the levels of longer subgenomic mRNAs. DDX1 has been identified as a member of the cellular interactomes for the IBV N protein [185], suggesting that the interaction between DDX1 and N for regulating viral RNA synthesis could be a general phenomenon in coronaviruses.
The interaction between pS197-N and DDX1 could also function as a regulator to prevent the assembly of higher-order ribonucleoprotein complexes. Using a phospho-specific pS197-N Ab, a study [183] identified that the N protein is phosphorylated immediately after synthesis but is dephosphorylated in virions. Therefore, binding with pS197-N/DDX1 might impede packaging genomic RNA into mature virions. Similar dephosphorylation patterns for N proteins in assembled virions were observed in several other viruses [186–188]. Coronaviruses employ a unique strategy for the transition from discontinuous to continuous transcription to ensure balanced subgenomic mRNAs and full-length genomic RNA synthesis [150]. This transition would also influence N-CTD-mediated transactivation of gene expression.
As mentioned, coronavirus RNA synthesis is connected with the formation of double-membrane vesicles and convoluted membranes [127], thanks partly to the intervention of the N protein via liquid-liquid phase separation (LLPS). One of the RNA-binding proteins that host cells use to control viral infections is the RGG-containing motif cellular nucleic acid-binding protein (CNBP). CRBP binds SARS-CoV-2 RNA and competes with N to prevent the formation of liquid-liquid phase separation condensates for viral replication [189]. Phosphorylated CNBP also translocates to the nucleus and binds the interferon-β (IFN-β) enhancer together with IFN-regulatory factor 3 (IRF-3) to turn on the transcription of type I IFNs and antiviral responses. Conversely, the RNA-binding region in N-CTD inhibits IFN-β production probably by shielding viral RNAs from recognition by cellular pattern recognition receptors (PPRs) [190].
The liquid-liquid phase separation that the SARS-CoV-2 N protein undergoes after binding to viral RNA recruits the key kinases of NF-κB signaling TAK1 and IKK complex enhancing NF-κB activation [191]. The hCoV-OC43 N protein potentiates NF-κB activation by binding to one of its negative regulators, the microRNA (miR)-9 [192]. Infectious bronchitis virus and porcine epidemic diarrhea virus upregulate cFOS expression, and other AP-1 transcription factors, regulating coronaviral-induced apoptosis and favoring viral replication [193]. These effects can contribute to pathogenesis, such as inflammation and cytokine storm, by favoring the expression of cytokines, which could also be the potential transactivation activity of N.
The TAR-like features of the SARS-CoV-2 leader may mediate a competition with host 7SR snRNA, which would generate a higher proportion of the free form of the P-TEFb complex comprising Cyclin T1 and CDK available to bind to N and phosphorylate it, favoring its transport to the cytoplasm to participate in transactivation of gene expression in compartmentalized settings.
Discontinuous transcription in coronaviruses involves transcription-regulating sequence (TRS)-dependent template switching, in which the leader TRS-L (ACGAAC core in β-coronaviruses) [191] interacts with homologous TRS-body (B) elements upstream of viral genes in the last third of the genome. An in vitro study using a SARS-CoV-1 replicon expressing a luciferase reporter under the control of a TRS-B derived from the region preceding the M gene showed that N, X and SUD domains of NSP3, and NSP12 provided in trans stimulated the replicon reporter activity, indicating that these proteins may regulate coronavirus replication and transcription [194].
3.2. Implications of potential transactivation to variant evolution of SARS-CoV-2 and other coronaviruses
A systematic analysis [195] described yet another aspect of genome variation by β- and α-coronaviruses by documenting the presence of intragenomic rearrangements involving segments of the 5’-leader sequence in geographically and temporally diverse isolates of SARS-CoV-2 (Figure 10). The intragenomic rearrangements could modify the carboxyl-termini of the ORF8 (also in Rhinolophus bat Sarbecovirus β-coronaviruses) and ORF7b proteins; the serine-arginine-rich region of the nucleocapsid protein, generating the well-characterized R203K/G204R paired mutation; and two sites of the NiRAN domain of the RNA-dependent RNA polymerase (NSP12). Interestingly, the latter two rearrangements bring the Tat-like binding site and, therefore, the possibility of transactivation of gene expression to a region of N preceding that encoding the TAR binding-like segment or of NSP12 preceding the Zn-coordinating domain shared with HIV-1 Tat.
Beyond SARS-CoV-2, similar rearrangements of 5’-UTR leader sequence segments, including the TRS-L, were found in all subgenera of β-coronaviruses except for Hibecovirus (possibly secondary to the availability of only three sequences in GenBank). These rearrangements were in the intergenic region between ORFs 3 and 4a and at the distal end of ORF4b of the Merbecovirus MERS-CoV; intergenic regions in the Embecoviruses hCoV-OC43 (between S and Ns5) and hCoV-HKU-1 (between S and NS4); and in the distal end that encodes the Y1 cytoplasmic tail domain of NSP3 of Nobecoviruses of African Rousettus and Eidolon bats. Intragenomic rearrangements were also found in α-coronaviruses in NSP2 (Luchacovirus subgenus), nucleocapsid (Nyctacovirus subgenus), and ORF5b or ORF4b (Decacovirus subgenus). No rearrangements involving 5’-UTR sequences were detected for the β-coronavirus SARS-CoV-1; the other 12 subgenera of α-coronaviruses including hCoV-229E and hCoV-NL63 infecting humans; or δ (Andecovirus, Buldecovirus, and Herdecovirus subgenera) and γ coronaviruses (Brangacovirus, Cegacovirus, and Igacovirus subgenera).
The study thus highlighted an intragenomic source of variation involving duplication, inversion (in two α-coronavirus subgenera), and translocation of 5’-UTR sequences to the body of the genome with potential implications on gene expression and immune escape of α- and β-coronaviruses in humans and bats causing mild-to-moderate or severe disease in endemic, epidemic, and pandemic settings. The intragenomic rearrangements involving 5’-UTR sequences, which in several cases affect highly conserved genes with a low propensity for recombination, may underlie the generation of variants homotypic with those of concern or interest and with potentially differing pathogenic profiles. Intragenomic rearrangements are yet another example of the tremendous genomic flexibility of coronaviruses, which underlies changes in transmissibility, immune escape, and virulence documented during the SARS-CoV-2 pandemic [195]. The possibility that they may facilitate the transactivation of expression of specific subgenomic RNAs adds to their relevance. Increased levels of the nucleocapsid protein, as well as those of the overlapping reading frame OR9b and ORF6, have been associated with increased immune evasion of Alpha, Delta, and Omicron SARS-CoV-2 variants of concern [196,197].
3.3. Implications of potential intra- and trans-cellular transactivation to acute SARS-CoV-2 pathology and long COVID and their treatment
The N-CTD nucleic acid binding domain may also transactivate other viruses containing TAR-like sequences, similar to the lentiviral Tat binding domains to TAR and as exemplified by the transactivation of JCV by HIV-1 Tat leading to the development of progressive multifocal leukoencephalopathy. However, there would be subtleties in the affinity level of Tat- or Tat-like-binding to TAT- or TAR-like sequences. For instance, HIV-2 Tat binds TAR from HIV-1 with low affinity and only partially activates the HIV-1 LTR, while HIV-1 Tat binds TAR-1 and TAR-2 RNAs with similar affinity, fully trans-activating HIV-1 and HIV-2 LTR [27,101,198–200].
HIV-1 Tat residues 30 to 55 interact with the SP1 transcription factor, mediate its phosphorylation, and increase HIV-1 LTR-driven gene expression [201,202]. N-CTD of coronaviruses may also bind SP1 based on the primary sequence similarities shown in Figure 5 between the HIV-1 Tat core and the basic amino acid-rich region with the nucleic acid-binding and adjacent regions of coronaviral N-CTD. The SARS-CoV-1 N-CTD is the main component of N to bind to the host translation elongation factor (EF)1α and inhibit host cell proliferation, which would also favor viral replication [190,203].
The effects of binding of coronaviral N-CTD to noncoding RNAs, as is the case with binding of HIV-1 Tat to 7SK snRNA, may increase the levels of specific transcriptional factors, in the case of HIV-1, the P-TEFb complex composed of Cyclin T1 and CDK, which in turn activate gene expression of viral and cellular genes. Coronaviral N can bind to the Cyclin/CDK complex, which could render the need for a Cyclin T1 binding site in the coronaviral leader unnecessary. However, it is possible that the N-CTD binds to the Tat-binding-like site in coronaviral RNA and activates transcription by displacing host or viral proteins that may bind to SL2, which is highly conserved among coronaviruses, similar to the effects of HIV-1 Tat binding to 7SK snRNA.
Another mechanism that has been documented in SARS-CoV-2 and -1 to free active P-TEFb from the 7Sk snRNP complex involves the NSP7 protein, which together with NSP8 assists the RNA-dependent RNA polymerase (NSP12) in the Replication-Transcription-Complex, with which N interacts, to catalyze the synthesis of viral RNA [204,205]. SARS-CoV-2 and -1 NSP7 interacts with the 7SK small nuclear ribonucleoprotein (7SK snRNP) complex comprising La-related protein (LARP7), methyl-phosphate capping enzyme (MEPCE), and HEXIM1, which sequesters P-TEFb. The interaction between NSP7 and the 7SK snRNP releases active P-TEFb, which is critical for the replication of several viruses [206]. Based on the findings of the present study, coronaviral N-CTD could also bind to 7SK snRNA and free active P-TEFb, which could then bind to N. This possibility is further underscored by the finding of an HIV Tat-binding sequence identical to that in 7SK snRNA in the leader of the coronavirus-related arterivirus PRRSV and of a region in the PRRSV N similar to coronaviral N-CTD including the TAR-like binding region (Figure 7). SARS-CoV-2 NSP7 shows no similarities to PRRSV proteins, lentiviral Tat, or human HEXIM1.
Beyond intracellular functions, the N proteins of SARS-CoV-2, hCoV-OC43, and mouse hepatitis virus are abundantly expressed on the surface of infected and neighboring cells, where the first two have been shown to inhibit leukocyte chemotaxis by binding to chemokines [207–209]. N can also sequester cytokines, possibly favoring viral transmission [207]. Like HIV-1 Tat, coronaviral N is exported from cells by a noncanonical process [210] and binds to the surfaces of other cells through electrostatic interactions with heparan sulfate proteoglycans.
Once in the extracellular compartment, HIV-1 Tat, through the same basic domain that in the intracellular environment binds to nucleic acids and is involved in translocation to the nucleus, binds to cell surface heparan sulfate proteoglycans, and is internalized by various cell types. This process requires the integrity of cell membrane lipid rafts and mainly occurs through caveolar endocytosis. This property is being used for the Tat-mediated delivery of fused heterologous proteins, nanoparticles, liposomes, phage and viral vectors, and plasmid DNA [211–213].
Extracellular Tat has been linked to chronic complications. Secreted Tat protein can be detected in cerebrospinal fluid, sera, and tissues of HIV-infected people, even in those without detectable viral load [214]. In addition to the HIV-1 surface glycoprotein gp160, extracellular Tat protein plays an important role in the development and progression of HIV-1-associated neurocognitive disorder (HAND), whose spectrum ranges from asymptomatic neurocognitive impairment, symptomatic mild neurocognitive disorder, to HIV-associated dementia [215].
HIV-1 Tat protein leads to the death of neuronal cells due to immune activation and rapid as well as sustained production of cytokines, mainly TNFα, in macrophages and microglia [216]. There is a synergistic effect of HIV-1 Tat and TNF-α to promote neuronal death [217]. In our comparisons combining primary and secondary structures, we detected a similarity between the SARS-CoV-2 leader and the reverse sequence of the TNF-α leader, which may also result in a synergistic interaction between the SARS-CoV-2 and TNF-α leaders (Figure 11). In particular, the sequences of the loops in the SL1 and SL2 of the SARS-CV-2 leader are identical to those in the SL1-like and SL2-like loops and beyond (as highlighted in lighter colors in Figure 11) in the reverse TFα leader [218]. It might be possible that the SARS-CoV-2 leader also shares strategies to favor its gene expression with the TNF-α promoter if NSP1, which, by binding to SL1 protects SARS-CoV-2 RNA from degradation before its translation, also protects TNF-α RNA via its leader sequence.
Neurotoxicity of Tat protein is also linked with the presence of R57 residue in the basic domain of Tat, and polymorphisms at this juncture can show prominent effect on increasing the neurotoxic potential along with transactivation of the protein [214]. Tat secretion, therefore, represents an attractive target to attenuate HAND. The R57 residue is also present in the region of similarity with coronaviral N-CTD except for the porcine deltacoronavirus where it is substituted for a glutamine (Q) residue.
SARS-CoV-2 replication might occur for several months after the initial infection as evinced by the detection of subgenomic RNA, a marker of recent virus replication; the isolation of replication-competent SARS-CoV-2 from respiratory and non-respiratory tissues [219–221]; and the existence of viral reservoirs for SARS-CoV-2 [222,223]. More specifically, these studies have demonstrated the persistence of viral antigens, viral RNA, and whole virus in the brain, sinus, adrenal glands, kidneys, gut, lymph nodes, spleen, lungs, heart, and fungiform papillae in taste buds, which can underlie symptoms through direct viral cytopathic effects; regional inflammation; triggering an immune response causing an elevated and prolonged state of generalized inflammation; and prompting autoimmunity in pediatric or adult populations, independently from severity of acute disease [224,225]. Extracellular coronaviral N could have similar effects as HIV-1 Tat in terms of toxicity to the nervous system or other organs, even though neither virus can infect neurons. Thus far, attention has focused on the extracellular SARS-CoV-2 protein. Experimentally defining if SARS-CoV-2 includes transactivation of gene expression will shed light on pathophysiological processes driven by its protein products in various organ systems.
In terms of treatment prospects, potential small molecule inhibitors of the Tat-TAR interaction have been identified over almost three decades; however, none has shown sufficient potency and selectivity [226–229]. HIV-1 nucleocapsid-fused chimeric proteins, namely nucleocapsid-HEXIM1-Tat and HEXIM1-Tat-nucleocapsid, have anti-HIV effects by inhibiting both HIV-1 transcription and packaging [230]. A similar approach could be used based on the SARS-CoV-2 nucleocapsid protein. However, the regulatory sequences shared among viruses and the multiple functions of host proteins involved pose a selectivity limitation as therapeutic targets.
The active form of P-TEFb, which drives transcription, elongation, and transactivation of gene expression in HIV-1 and possibly also coronaviruses and arteriviruses, binds bromodomain-containing protein 4 (BRD4). Bromodomain and extra-terminal motif (BET) inhibitors induce P-TEFb release and are latency-reversal agents in HIV infection [205,231]. Latency-promoting agents could help manage the post-acute infectious syndromes seen with HIV-1, SARS-CoV-2 (long COVID), and other viruses and microbes.
Further insights into the workings of the nucleocapsid and the Replication-Transcription-Complex of coronaviruses and other nidoviruses are warranted to develop novel treatments for acute infection and to design latency-regulating strategies to deal with viral persistence and long-term consequences of infection. As discussed, three coronaviral proteins, namely nucleocapsid through its nucleic acid binding-C-terminal domain interacting with Tar-lie sequences in coronaviral leaders, NSP12 via the metal-binding, dimerization domain in the interface region preceding the RNA-dependent polymerase region, and NSP7 by allowing to free P-TEFb complex from the 7SK ribonucleoprotein would contribute to the potential transactivation activity of SARS-CoV-2, other coronaviruses, and the arterivirus PRRSV. All proteins are part of a larger coronaviral Replication-Transcription Complex.
4. Materials and Methods
4.1. Detection of HIV-1 TAR-like sequences in the SARS-CoV-2 leader, and expansion to other lentiviral, JCV, and human 7SK snRNA TARs
We used manual inspection and the BLASTN program [232] to determine the presence of a Tat-binding-like site and adjacent regions of similarity between HIV-1 TAR, the closely associated JCV TAR, and the leader of SARS-CoV-2 (β-coronavirus, genus Sarbecovirus). Upon finding regions of similarity, we expanded the search to include HIV-2 and SIV TARs, the human 7SK snRNA TAR, and the leaders of the six other β-coronaviruses (Sarbecovirus SARS-CoV-1, Embecoviruses hCoV-OC43 and -HKU1; and Merbecovirus MERS-CoV) and α-coronaviruses (Setracovirus hCoV-NL63, and Duvinacovirus hCoV-229E) known to infect humans, and of representatives of γ- and δ- coronaviruses, all associated with epidemics or pandemics.
We used the Rfam database (http://rfam.xfam.org/covid-19) with the curated Stockholm files containing untranslated region (UTR) sequences, alignments, and consensus RNA secondary structures of major genera of Coronoviridae; the representative RefSeq sequences for each genus obtained from the International Committee on Taxonomy of Viruses (ICTV) taxonomy Coronaviridae Study Group [233]; the reference sequences in the GenBank and NCBI Virus databases; and listings in publications involving phylogenetic analyses of alpha-, delta-, and gamma-coronaviruses from NCBI Taxonomy [234,235] to derive the leaders of coronaviruses of all genera for analysis. Variations in lentiviral TAR and Tat sequences were obtained from the literature.
Multiple alignments and sequence similarities derived from the manual and BLASTN assessment were also evaluated using CLUSTAL Omega (https://www.ebi.ac.uk/services) [236]. Primary structure alignments shown were selected based on shared secondary structural features that were determined based on alignment with lentiviral Tat and as described next.
4.2. Secondary structural mapping of the regions of similarity between HIV TAR-like and coronaviral leader sequences
Because lentiviral Tat binding involves both primary and secondary structural features of the TAR sequences to which they bind, RNA secondary structures of the HIV-1, HIV-2, SIV, and JCV TARs, the leaders of SARS-CoV-2, other coronaviruses, and the arterivirus PRRSV, and the human 7SK snRNA TAR, were derived from the literature and also visualized using forna, a force directed graph layout (ViennaRNA Web services) [237–239]. Similar nucleotides were highlighted by designated color to the region of similarity to assess distribution in stem, loop, and bulge regions. Complementary (antisense) sequences were also analyzed, particularly those involving SARS-CoV-2/-1 SL1, because of the presence of the complement of a Cyclin T1 binding-like site in Sarbecoviruses.
4.3. Detection of HIV-1 Tat-like sequences among SARS-CoV-2 proteins
The HIV-1 Tat protein sequence was compared against SARS-CoV-2 proteins deposited in the GenBank® database using the Basic Alignment Search Tool (BLAST)P® (Protein BLAST: search protein databases using a protein query (nih.gov) [240] for SARS-CoV-2 and SARS-CoV-related viral proteins encoding similar stretches. All nonredundant translated CDS + PDB + SwissProt + PRF, excluding environmental samples from WGS projects, were searched, specifying severe acute respiratory syndrome coronavirus 2 as the organism. We also compared HIV-1 Tat against sequences in the six reading frames translated from the SARS-CoV-2 genome in the sense and antisense directions. The results of comparisons against known and uncharacterized open reading frames were compatible in terms of the significance of the similarity between HIV-1 Tat and the SARS-CoV-2 N-CTD domain or the SARS-CoV-2 interface region of the RNA-dependent RNA polymerase.
The assess the degree of conservation of the HIV-1 Tat-like N-CTD nucleic binding basic amino acid-rich region of SARS-CoV-2, we used the Global Initiative on Sharing All Influenza Data (GISAID) EpiFlu™ database of SARS-CoV-2 sequences (GISAID - Initiative) [116–118] querying each amino acid position.
We extended the analyses to the HIV-2 and SIV Tat proteins, the human HEMIX1 and 2 proteins, the coronaviral N-CTDs, interface regions of RNA-dependent RNA polymerase of coronaviruses of the four genera, and the N protein of β-arterivirus porcine reproductive and respiratory syndrome virus. We used the same verification methods for multiple alignments for protein sequences as described above for nucleotide sequences.
5. Conclusions
The presence of TAR- and Tat-like sequences in SARS-CoV-2, other coronaviruses, and the arterivirus PRRSV, suggests that nidoviruses may use transactivation of gene expression as another strategy to favor their transcription and replication using the host’s cell machinery. As summarized in Figure 12, three proteins would be involved in transactivation, all part of the Replication-Transcription-Complex, namely the nucleocapsid protein via its C-terminal domain with a nucleic acid binding region similar to that in lentiviral Tats, NSP12 with a metal-binding, dimerization region in the interface region preceding the RNA-dependent RNA polymerase domain similar to that in lentiviral Tats, and NSP7 through its proven ability to bind to the 7SK ribonucleoprotein and liberate active P-TEFb.
Similarities to HIV-1 TAR and Tat were higher among Sarbecoviruses, Embecoviruses and Merbecoviruses and included the presence of a region of similarity with the Cyclin T1 binding site in stem-loop 1 of the Sarbecoviruses SARS-CoV-2 and -1. Transactivation in nidoviruses could affect not only replication but also immune evasion, pathogenesis, transmission, and viral evolution, constituting a potential broad-spectrum therapeutic target.
Author Contributions
Conceptualization, methodology, formal analysis, data curation, writing—original draft preparation, writing—review and editing, R.P. and W.A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data generated and its sources are included in the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sodroski, J.; Patarca, R.; Rosen, C.; Wong-Staal, F.; Haseltine, W. (1985). Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science (New York, N.Y.) 1985, 229, 74–77. [Google Scholar] [CrossRef]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.Z.; Popovic, M.; Arya, S.; Gallo, R.C.; Haseltine, W. A. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science (New York, N.Y.) 1985, 227, 171–173. [Google Scholar] [CrossRef]
- Dayton, A.I.; Sodroski, J.G.; Rosen, C.A.; Goh, W.C.; Haseltine, W.A. The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell 1986, 44, 941–947. [Google Scholar] [CrossRef]
- Okamoto, T.; Wong-Staal, F. Demonstration of virus-specific transcriptional activator(s) in cells infected with HTLV-III by an in vitro cell-free system. Cell 1986, 47, 29–35. [Google Scholar] [CrossRef]
- Okamoto, T.; Benter, T.; Josephs, S.F.; Sadaie, M.R.; Wong-Staal, F. Transcriptional activation from the long-terminal repeat of human immunodeficiency virus in vitro. Virology 1990, 177, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.; Kwak, Y.T.; Nee, E.; Guo, J.; García-Martínez, L.F.; Gaynor, R.B. Cyclin T1 domains involved in complex formation with Tat and TAR RNA are critical for tat-activation. J Mol Biol 1999, 288, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef] [PubMed]
- He, N., Zhou, Q. New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC). Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology 2011, 6, 260-268. [CrossRef]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B,R. Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc Natl Acad Sci U S A 1999, 96, 7791-7796. [CrossRef]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol Cell Biol 2000, 20, 6958–69. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Cao, H.; Rana, T.M. Specific HIV-1 TAR RNA loop sequence and functional groups are required for human cyclin T1-Tat-TAR ternary complex formation. Biochemistry 2002, 41, 6391–7. [Google Scholar] [CrossRef]
- Karn, J.; Stoltzfus, C. M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2012, 2, a006916. [Google Scholar] [CrossRef]
- Asamitsu, K.; Okamoto, T. The Tat/P-TEFb protein-protein interaction determining transcriptional activation of HIV. Curr Pharm Des 2017, 23, 4091–4097. [Google Scholar] [CrossRef]
- Asamitsu, K.; Fujinaga, K.; Okamoto, T. HIV tat/P-TEFb interaction: a potential target for novel anti-HIV therapies. Molecules 2018, 23, 933. [Google Scholar] [CrossRef]
- Chameettachal, A.; Mustafa, F.; Rizvi, T.A. Understanding Retroviral Life Cycle and its Genomic RNA Packaging. Journal of molecular biology 2023, 435, 167924. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, A.; Krasnopolsky, S.; Taube, R. Super elongation complex promotes early HIV transcription and its function is modulated by P-TEFb. Transcription 2017, 8, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.K.; Gallo, R.C.; Hahn, B.H.; Shaw, G.M.; Popovic, M.; Salahuddin, S.Z.; Wong-Staal, F. Homology of genome of AIDS-associated virus with genomes of human T-cell leukemia viruses. Science 1984, 225, 927–930. [Google Scholar] [CrossRef]
- Rosen, C.A.; Sodroski, J.G.; Haseltine, WA. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 1985, 41, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Jeang, K.T. Trans activation of human immunodeficiency virus type 1 is sequence specific for both the single-stranded bulge and loop of the trans-acting-responsive hairpin: a quantitative analysis. J Virol 1989, 63, 5501–5504. [Google Scholar] [CrossRef]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A.; Valerio, R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci U S A 1989, 86, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M.; Ampe, C.; Schultz, S.C.; Steitz, T.A.; Crothers, D.M. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science (New York, N.Y.) 1990, 249, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M.; Crothers, D.M. RNA recognition by Tat-derived peptides: interaction in the major groove? Cell 1991, 66, 577–588. [Google Scholar] [CrossRef]
- Sumner-Smith, M.; Roy, S.; Barnett, R.; Reid, L.S.; Kuperman, R.; Delling, U.; Sonenberg, N. Critical chemical features in trans-acting-responsive RNA are required for interaction with human immunodeficiency virus type 1 Tat protein. Journal of virology 1991, 65, 5196–5202. [Google Scholar] [CrossRef]
- Karn, J. Tackling Tat. J Mol Biol 1999, 293, 235-254. [CrossRef]
- Levintov, L.; Vashisth, H. Structural and computational studies of HIV-1 RNA. RNA biology 2024, 21, 1–32. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Rhim, H.; Rice, A.P. TAR RNA binding properties and relative transactivation activities of human immunodeficiency virus type 1 and 2 Tat proteins. J Virol 1993, 67, 1110–1121. [Google Scholar] [CrossRef]
- Cullen, B.R. RNA-sequence-mediated gene regulation in HIV-1. Infect Agents Dis 1994, 3, 68–76. [Google Scholar]
- Roebuck, K.A.; Saifuddin, M. Regulation of HIV-1 transcription. Gene Expr 1999, 8, 67–84. [Google Scholar]
- Kessler, M.; Mathews, M.B. Premature termination and processing of human immunodeficiency virus type 1-promoted transcripts. J Virol 1992, 66, 4488–4496. [Google Scholar] [CrossRef]
- Ratnasabapathy, R.; Sheldon, M.; Johal, L.; Hernandez, N. The HIV-1 long terminal repeat contains an unusual element that induces the synthesis of short RNAs from various mRNA and snRNA promoters. Genes Dev 1990, 4, 2061–2074. [Google Scholar] [CrossRef]
- Toohey, M.G.; Jones, KA. In vitro formation of short RNA polymerase II transcripts that terminate within the HIV-1 and HIV-2 promoter-proximal downstream regions. Genes Dev 1989, 3, 265–282. [Google Scholar] [CrossRef]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; Buratowski, S.; Handa, H. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes & development, 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Wada, T.; Orphanides, G.; Hasegawa, J.; Kim, D.K.; Shima, D.; Yamaguchi, Y.; Fukuda, A.; Hisatake, K.; Oh, S.; Reinberg, D.; Handa, H. FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Molecular cell 2000, 5, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Kasten, M.; De Falco, G.; Micheli, P.; Khalili, K.; Giordano, A. Regulatory functions of Cdk9 and of cyclin T1 in HIV tat transactivation pathway gene expression. J Cell Biochem 1999, 75, 357–368. [Google Scholar] [CrossRef]
- Marcello, A.; Zoppé, M.; Giacca, M. Multiple modes of transcriptional regulation by the HIV-1 Tat transactivator. IUBMB Life 2001, 51, 175–181. [Google Scholar] [CrossRef]
- Isel, C.; Karn, J. Direct evidence that HIV-1 Tat stimulates RNA polymerase II carboxyl-terminal domain hyperphosphorylation during transcriptional elongation. J Mol Biol 1999, 290, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; Xiao, H.; Rich, E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem 1999, 274, 28837–28840. [Google Scholar] [CrossRef]
- Zhou, M.; Halanski, M.A.; Radonovich, M.F.; Kashanchi, F.; Peng, J.; Price, D.H.; Brady, J.N. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol Cell Biol 2000, 20, 5077–5086. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol Cell Biol 2002, 22, 4622–4637. [Google Scholar] [CrossRef]
- Hamasaki, T.; Okamoto, M.; Baba, M. Identification of novel inhibitors of human immunodeficiency virus type 1 replication by in silico screening targeting cyclin T1/Tat interaction. Antimicrob Agents Chemother 2013, 57, 1323–1331. [Google Scholar] [CrossRef]
- Lu, H.; Yu, D.; Hansen, A.S.; Ganguly, S.; Liu, R.; Heckert, A.; Darzacq, X.; Zhou, Q. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 2018, 558, 318–323. [Google Scholar] [CrossRef]
- Yan, Y.; Tang, Y.D.; Zheng, C. When cyclin-dependent kinases meet viral infections, including SARS-CoV-2. J Med Virol 2022, 94, 2962–2968. [Google Scholar] [CrossRef]
- Fujinaga, K., Huang, F., Peterlin, B.M. P-TEFb: The master regulator of transcription elongation. Mol Cell 2023, 83, 393-403. [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Molecular and cellular biology 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Cho, S.; Schroeder, S.; Ott, M. CYCLINg through transcription: Post-translational modifications of P-TEFb regulate transcription elongation. Cell Cycle 2010, 9, 1697–1705. [Google Scholar] [CrossRef]
- Egloff, S. CDK9 keeps RNA polymerase II on track. Cellular and molecular life sciences : CMLS, 2021, 78, 5543–5567. [CrossRef]
- Yik, J.H.; Chen, R.; Pezda, A.C.; Samford, C.S.; Zhou, Q. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Molecular and cellular biology 2004, 24, 5094–5105. [Google Scholar] [CrossRef]
- Muniz, L.; Egloff, S.; Ughy, B.; Jády, B.E.; Kiss, T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog 2010, 6, e1001152. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Molecular cell, 2010, 38, 439–451. [Google Scholar] [CrossRef]
- Yang, Z.; Yik J.H.; Chen, R.; He, N; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005, 19, 535-545. [CrossRef]
- Nekhai, S.; Jeang, K.T. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: role of cellular factors for Tat and Rev. Fut. Microbiol 2006, 1, 417–426. [Google Scholar] [CrossRef]
- Vardabasso, C.; Manganaro, L.; Lusic, M.; Marcello, A.; Giacca, M. The histone chaperone protein nucleosome assembly protein-1 (hNAP-1) binds HIV-1 Tat and promotes viral transcription. Retrovirology 2008, 5, 8. [Google Scholar] [CrossRef]
- Easley, R.; Van Duyne, R.; Coley, W.; Guendel, I.; Dadgar, S.; Kehn-Hall, K.; Kashanchi, F. Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim Biophys Acta 2010, 1799, 275–85. [Google Scholar] [CrossRef]
- Sastry, K.J.; Reddy, H.R.; Pandita, R.; Totpal, K.; Aggarwal, B.B. HIV-1 tat gene induces tumor necrosis factor-beta (lymphotoxin) in a human B-lymphoblastoid cell line. The Journal of biological chemistry 1990, 265, 20091–20093. [Google Scholar] [CrossRef]
- Buonaguro, L.; Buonaguro, F.M.; Giraldo, G.; Ensoli, B. The human immunodeficiency virus type 1 Tat protein transactivates tumor necrosis factor beta gene expression through a TAR-like structure. Journal of virology, 1994, 68, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Brother, M.B.; Chang, H.K.; Lisziewicz, J.; Su, D.; Murty, L.C.; Ensoli, B. Block of Tat-mediated transactivation of tumor necrosis factor beta gene expression by polymeric-TAR decoys. Virology 1996, 222, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Scala, G.; Ruocco, M.R.; Ambrosino, C.; Mallardo, M; Giordano, V.; Baldassarre, F.; Dragonetti, E.; Quinto, I.; Venuta, S. The expression of the interleukin 6 gene is induced by the human immunodeficiency virus 1 TAT protein. J Exp Med 1994, 179, 961-971. [CrossRef]
- Ambrosino, C.; Ruocco, M.R.; Chen, X.; Mallardo, M.; Baudi, F.; Trematerra, S.; Quinto, I.; Venuta, S.; Scala, G. HIV-1 Tat induces the expression of the interleukin-6 (IL6) gene by binding to the IL6 leader RNA and by interacting with CAAT enhancer-binding protein beta (NF-IL6) transcription factors. The Journal of biological chemistry, 1997, 272, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Lovett, J.L.; Mueller, L.; Verdin, E. Superinduction of IL-8 in T cells by HIV-1 Tat protein is mediated through NF-kappaB factors. J Immunol 1998, 160, 2872–2880. [Google Scholar] [CrossRef]
- Bohn-Wippert, K., Tevonian, E.N., Megaridis, M.R., Dar, R.D. Similarity in viral and host promoters couples viral reactivation with host cell migration. Nature communications 2017, 8, 15006. [CrossRef]
- Lim, S.P.; Garzino-Demo, A. The human immunodeficiency virus type 1 Tat protein up-regulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of SP-1, AP-1, and NF-kappaB consensus sites. Journal of virology 2000, 74, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Farina, M.; Maroder, M.; Alesse, E.; Screpanti, I.; Frati, L.; Gulino, A. Human immunodeficiency virus type-1 tat enhances interleukin-2 promoter activity through synergism with phorbol ester and calcium-mediated activation of the NF-AT cis-regulatory motif. Biochem Biophys Res Commun 1994, 205, 467-474.
- González, E.; Punzón, C.; González, M.; Fresno, M. HIV-1 Tat inhibits IL-2 gene transcription through qualitative and quantitative alterations of the cooperative Rel/AP1 complex bound to the CD28RE/AP1 composite element of the IL-2 promoter. Journal of immunology (Baltimore, Md. : 1950), 2001, 166, 4560–4569. [Google Scholar] [CrossRef] [PubMed]
- Anastasopoulou, S.; Georgakopoulos, T.; Mouzaki, A. HIV-1 Transcriptional Activator Tat Inhibits IL2 Expression by Preventing the Presence of Pol II on the IL2 Promoter. Biomolecules 2023, 13, 881. [Google Scholar] [CrossRef] [PubMed]
- Patarca, R.; Heath, C.; Goldenberg, G.J.; Rosen, C.A., Sodroski, J.G., Haseltine, W.A., Hansen, U.M. Transcription directed by the HIV long terminal repeat in vitro. AIDS research and human retroviruses 1987, 3, 41–55. [CrossRef]
- Taylor, J.P.; Pomerantz, R.J.; Raj, G.V.; Kashanchi, F.; Brady, J.N.; Amini, S.; Khalili, K. Central nervous system-derived cells express a kappa B-binding activity that enhances human immunodeficiency virus type 1 transcription in vitro and facilitates TAR-independent transactivation by Tat. Journal of virology 1994, 68, 3971–3981. [Google Scholar] [CrossRef]
- Yang, L.; Morris, G.F.; Lockyer, J.M.; Lu, M.; Wang, Z.; Morris, C.B. Distinct transcriptional pathways of TAR-dependent and TAR-independent human immunodeficiency virus type-1 transactivation by Tat. Virology 1997, 235, 48–64. [Google Scholar] [CrossRef]
- Iyer, K.; Mitra, A.; Mitra, D. Identification of 5' upstream sequence involved in HSPBP1 gene transcription and its downregulation during HIV-1 infection. Virus research 2023, 324, 199034. [Google Scholar] [CrossRef]
- Chaudhary, P.; Khan, S.Z.; Rawat, P.; Augustine, T.; Raynes, D.A.; Guerriero, V.; Mitra, D. HSP70 binding protein 1 (HspBP1) suppresses HIV-1 replication by inhibiting NF-κB mediated activation of viral gene expression. Nucleic Acids Res 2016, 44(4):1613-1629. [CrossRef]
- de la Vega, L.; Sánchez-Duffhues, G.; Fresno, M.; Schmitz, M.L.; Muñoz, E.; Calzado, M.A. The 73 kDa subunit of the CPSF complex binds to the HIV-1 LTR promoter and functions as a negative regulatory factor that is inhibited by the HIV-1 Tat protein. Journal of molecular biology 2007, 372, 317–330. [Google Scholar] [CrossRef]
- Verhoef, K.; Bauer, M.; Meyerhans, A.; Berkhout, B. On the role of the second coding exon of the HIV-1 Tat protein in virus replication and MHC class I downregulation. AIDS research and human retroviruses 1998, 14, 1553–1559. [Google Scholar] [CrossRef]
- Cafaro, A.; Barillari, G.; Moretti, S.; Palladino, C.; Tripiciano, A.; Falchi, M.; Picconi, O.; Pavone Cossut, M.R.; Campagna, M.; Arancio, A.; Sgadari, C.; Andreini, C.; Banci, L.; Monini, P.; Ensoli, B. HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication. International journal of molecular sciences, 2020, 22, 317. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Niu, F.; Hu, G.; Guo, M.L.; Sil, S.; Buch, S. HIV Tat-mediated induction of autophagy regulates the disruption of ZO-1 in brain endothelial cells. Tissue barriers 2020, 8, 1748983. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.V.; Joseph, S.B.; Dittmer, D.P.; Mackman, N. Cardiovascular Disease and Thrombosis in HIV Infection. Arteriosclerosis, thrombosis, and vascular biology 2023, 43, 175–191. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.J.; Swanson, M.D.; Contreras-Galindo, R.; Cookinham, S.; King, S.R.; Noel, R.J.Jr; Kaplan, M.H.; Markovitz, D.M. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J Virol 2012, 86, 7790–7805. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.J.; Cavalcoli, J.D.; Sartor, M.A.; Contreras-Galindo, R.; Meng, F.; Dai, M.; Dube, D.; Saha, A.K.; Gitlin, S.D.; Omenn, G.S.; Kaplan, M.H.; Markovitz, D.M. Regulation of the human endogenous retrovirus K (HML-2) transcriptome by the HIV-1 Tat protein. J Virol 2014, 88, 8924–8935. [Google Scholar] [CrossRef]
- Römer, C. Viruses and Endogenous Retroviruses as Roots for Neuroinflammation and Neurodegenerative Diseases. Front Neurosci 2021, 15, 648629. [Google Scholar] [CrossRef]
- Dopkins, N.; Nixon, D.F. Activation of human endogenous retroviruses and its physiological consequences. Nat Rev Mol Cell Biol 2023, Oct 23. [CrossRef]
- Chowdhury, M.; Taylor, J.P.; Chang, C.F.; Rappaport, J.; Khalili, K. Evidence that a sequence similar to TAR is important for induction of the JC virus late promoter by human immunodeficiency virus type 1 Tat. J Virol 1992, 66, 7355–7361. [Google Scholar] [CrossRef]
- Arya, S.K.; Gallo, R.C. Human immunodeficiency virus type 2 long terminal repeat: analysis of regulatory elements. Proc Natl Acad Sci U S A 1988, 85, 9753–9757. [Google Scholar] [CrossRef]
- Viglianti, G.A.; Mullins, J.I. Functional comparison of transactivation by simian immunodeficiency virus from rhesus macaques and human immunodeficiency virus type 1. J Virol 1988, 62, 4523–4532. [Google Scholar] [CrossRef] [PubMed]
- Haseltine, W.A.; Sodroski, J.; Patarca, R.; Briggs, D.; Perkins, D.; Wong-Staal, F. Structure of 3' terminal region of type II human T lymphotropic virus: evidence for new coding region. Science (New York, N.Y.) 1984, 225, 419–421. [Google Scholar] [CrossRef]
- Sodroski, J.; Trus, M.; Perkins, D.; Patarca, R.; Wong-Staal, F.; Gelmann, E.; Gallo, R.; Haseltine, W.A. Repetitive structure in the long-terminal-repeat element of a type II human T-cell leukemia virus. Proceedings of the National Academy of Sciences of the United States of America 1984, 81, 4617–4621. [Google Scholar] [CrossRef]
- Nevins, J.R.; Raychaudhuri, P.; Yee, A.S.; Rooney, R.J.; Kovesdi, I.; Reichel, R. Transactivation by the adenovirus E1A gene. Biochemistry and cell biology = Biochimie et biologie cellulaire 1988, 66, 578–583. [Google Scholar] [CrossRef]
- Brady, J.; Bolen, J.B.; Radonovich, M.; Salzman, N.; Khoury, G. Stimulation of simian virus 40 late gene expression by simian virus 40 tumor antigen. Proceedings of the National Academy of Sciences of the United States of America 1984, 81, 2040–2044. [Google Scholar] [CrossRef]
- Diaz, J.J.; Dodon, M.D.; Schaerer-Uthurralt, N.; Simonin, D.; Kindbeiter, K.; Gazzolo, L.; Madjar, J.J. Post-transcriptional transactivation of human retroviral envelope glycoprotein expression by herpes simplex virus Us11 protein. Nature 1996, 379, 273–277. [Google Scholar] [CrossRef]
- Scala, G.; Quinto, I.; Ruocco, M.R.; Mallardo, M.; Ambrosino, C.; Squitieri, B.; Tassone, P.; Venuta, S. Epstein-Barr virus nuclear antigen 2 transactivates the long terminal repeat of human immunodeficiency virus type 1. Journal of virology 1993, 67, 2853–2861. [Google Scholar] [CrossRef]
- Hung, C.C.; Kuo, CW; Wang, WH; Chang, T.H.; Chang, P.J.; Chang L.K.; Liu, S.T. Transcriptional activation of Epstein-Barr virus BRLF1 by USF1 and Rta. The Journal of general virology 2015, 96, 2855–2866. [CrossRef]
- Roy, S.; Delling, U.; Chen, C.H.; Rosen, C.A.; Sonenberg, N. A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated trans-activation. Genes & development 1990, 4, 1365–1373. [Google Scholar] [CrossRef]
- Delling, U.; Reid, L.S.; Barnett, R.W.; Ma, M.Y.; Climie, S.; Sumner-Smith, M.; Sonenberg, N. Conserved nucleotides in the TAR RNA stem of human immunodeficiency virus type 1 are critical for Tat binding and trans activation: model for TAR RNA tertiary structure. Journal of virology 1992, 66, 3018–3025. [Google Scholar] [CrossRef]
- Churcher, M.J.; Lamont, C.; Hamy, F.; Dingwall, C.; Green, S.M.; Lowe, A.D.; Butler, J.G.; Gait, M.J.; Karn, J. High affinity binding of TAR RNA by the human immunodeficiency virus type-1 tat protein requires base-pairs in the RNA stem and amino acid residues flanking the basic region. J Mol Biol 1993, 230, 90–110. [Google Scholar] [CrossRef]
- Baker, B.; Muckenthaler, M.; Vives, E.; Blanchard, A.; Braddock, M.; Nacken, W.; Kingsman, A.J.; Kingsman, S.M. Identification of a novel HIV-1 TAR RNA bulge binding protein. Nucleic acids research 1994, 22, 3365–3372. [Google Scholar] [CrossRef]
- Aboul-ela, F.; Karn, J.; Varani, G. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. Journal of molecular biology 1995, 253, 313–332. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Gait, M.J.; Ivanovskaya, M.G. RNA recognition and regulation of HIV-1 gene expression by viral factor Tat. Biochemistry. Biokhimiia 1998, 63, 489–503. [Google Scholar] [PubMed]
- Lalonde, M.S.; Lobritz, M.A.; Ratcliff, A.; Chamanian, M.; Athanassiou, Z.; Tyagi, M.; Wong, J.; Robinson, J.A.; Karn, J.; Varani, G.; Arts, E.J. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLoS Pathog 2011, 7, e1002038. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.S.; Bonn-Breach, R.; Wedekind, J.E. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J Biol Chem 2019, 294, 9326–9341. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y., Yan, W., Hall, A.B., Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol Biol Evol 2021, 38, 1241-1248. [CrossRef]
- Berkhout, B. Structural features in TAR RNA of human and simian immunodeficiency viruses: a phylogenetic analysis. Nucleic acids research 1992, 20, 27–31. [Google Scholar] [CrossRef]
- Berkhout, B.; Gatignol, A.; Silver, J.; Jeang, K.T. Efficient trans-activation by the HIV-2 Tat protein requires a duplicated TAR RNA structure. Nucleic Acids Res 1990, 18, 1839–1846. [Google Scholar] [CrossRef]
- Pachulska-Wieczorek, K.; Purzycka, K.J.; Adamiak, R.W. New, extended hairpin form of the TAR-2 RNA domain points to the structural polymorphism at the 5' end of the HIV-2 leader RNA. Nucleic Acids Res 2006, 34, 2984–2997. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.K.; Guo, C.; Josephs, S.F.; Wong-Staal, F. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 1985, 229, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Laspia, M.F.; Rice, A.P.; Mathews, M.B. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 1989, 59, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Shida, H.I.; Maki, M.A.; Hatanaka, M.A. Effects of a highly basic region of human immunodeficiency virus Tat protein on nucleolar localization. J Virol 1990, 64, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Calnan, B.J.; Biancalana, S.; Hudson, D.; Frankel, A.D. Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes & development 1991, 5, 201-210. [CrossRef]
- Calnan, B.J.; Tidor, B.; Biancalana, S.; Hudson, D.; Frankel, A.D. Arginine-mediated RNA recognition: the arginine fork. Science 1991, 252, 1167–1171. [Google Scholar] [CrossRef]
- Chiozzini, C.; Toschi, E. HIV-1 TAT and immune dysregulation in aids pathogenesis: a therapeutic target. Curr. Drug Targets 2016, 17, 33–45. [Google Scholar] [CrossRef]
- Tan, R.; Chen, L.; Buettner, J.A.; Hudson, D.; Frankel, A.D. RNA recognition by an isolated alpha helix. Cell 1993, 73, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.N.; Jin, H. The RGG motif proteins: Interactions, functions, and regulations. Wiley Interdiscip Rev RNA 2023, 14, e1748. [Google Scholar] [CrossRef]
- Gotora, P.T.; van der Sluis, R.; Williams, M.E. HIV-1 Tat amino acid residues that influence Tat-TAR binding affinity: a scoping review. BMC Infect Dis 2023, 23, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zeng, R.; von Brunn, A.; Lei, J. Structural characterization of the C-terminal domain of SARS-CoV-2 nucleocapsid protein. Mol Biomed 2020, 1, 2. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. Journal of molecular biology 2007, 368, 1075–1086. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, L.; Petros, A.M.; Gunasekera, A.; Liu, Z.; Xu, N.; Hajduk, P.; Mack, J.; Fesik, S.W.; Olejniczak, E.T. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 2004, 43, 6059–6063. [Google Scholar] [CrossRef]
- Zinzula, L.; Basquin, J.; Bohn, S.; Beck, F.; Klumpe, S.; Pfeifer, G.; Nagy, I.; Bracher, A.; Hartl, F.U.; Baumeister, W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid-19 severe acute respiratory syndrome coronavirus 2. Biochem Biophys Res Commun 2021, 538, 54–62. [Google Scholar] [CrossRef]
- Khare, S., Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N; Ho, J.; Lee, R.T.; Yeo, W.; Curation Team GC; Maurer-Stroh, S. GISAID’s Role in Pandemic Response. China CDC Weekly 2021, 3, 1049-1051. [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: from vision to reality. EuroSurveillance 2017, 22. [Google Scholar] [CrossRef]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.; Lal, S.K. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. Journal of virology 2005, 79, 11476–11486. [Google Scholar] [CrossRef] [PubMed]
- Thandapani, P.; O'Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG motif. Mol Cell 2013, 50, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ozdilek, B.A.; Thompson, V.F.; Ahmed, N.S.; White, C.I.; Batey, R.T.; Schwartz, J.C. Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res 2017, 45, 7984–7996. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zandi, R. Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding. Viruses 2022, 14, 2089. [Google Scholar] [CrossRef]
- Yu, I.M.; Oldham, M.L.; Zhang, J.; Chen, J. Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between corona- and arteriviridae. The Journal of biological chemistry 2006, 281, 17134–17139. [Google Scholar] [CrossRef]
- Hauber, J.; Malim, M.H.; Cullen, B.R. Mutational analysis of the conserved basic domain of human immunodeficiency virus tat protein. J Virol 1989, 63, 1181–1187. [Google Scholar] [CrossRef]
- Peng, T.Y.; Lee, K.R.; Tarn, W.Y. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. The FEBS journal 2008, 275, 4152–4163. [Google Scholar] [CrossRef]
- Adly, A.N.; Bi, M.; Carlson, C.R.; Syed, A.M.; Ciling, A.; Doudna, J.A.; Cheng, Y.; Morgan, D.O. Assembly of SARS-CoV-2 ribonucleosomes by truncated N∗ variant of the nucleocapsid protein. J Biol Chem 2023, 299, 105362. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S.; Koetzner, C.A.; Kerr, C.A.; Heo, Y. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. Journal of virology 1994, 68, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, S.; Cruz, J.L.; Sola, I.; Mateos-Gómez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. Journal of virology 2010, 84, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; Galán, C.; Enjuanes, L. The nucleoprotein is required for efficient coronavirus genome replication. Journal of virology 2004, 78, 12683–12688. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.A.; Harrich, D.; Pearson, L.; Mitsuyasu, R.; Gaynor, R.B. Functional domains required for tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J 1988, 7, 3143–3147. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.R.; Mukhopadhyaya, R.; Benaissa, Z.N.; Pavlakis, G.N.; Wong-Staal, F. Conservative mutations in the putative metal-binding region of human immunodeficiency virus tat disrupt virus replication. AIDS Res Hum Retroviruses 1990, 6, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Kuppuswamy, M.; Subramanian, T.; Srinivasan, A.; Chinnadurai, G. Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res 1989, 17, 3551–3561. [Google Scholar] [CrossRef]
- Ruben, S.; Perkins, A.; Purcell, R.; Joung, K.; Sia, R.; Burghoff, R.; Haseltine, W.A.; Rosen, C.A. Structural and functional characterization of human immunodeficiency virus tat protein. J Virol 1989, 63, 1–8. [Google Scholar] [CrossRef]
- Rice, A.P.; Carlotti, F. Structural analysis of wild-type and mutant human immunodeficiency virus type 1 Tat proteins. J Virol 1990, 64, 6018–6026. [Google Scholar] [CrossRef]
- Frankel, A.D.; Bredt, D.S.; Pabo, C.O. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science 1988, 240, 70–73. [Google Scholar] [CrossRef]
- Frankel, A.D.; Chen, L.; Cotter, R.J.; Pabo, C.O. Dimerization of the tat protein from human immunodeficiency virus: a cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc Natl Acad Sci U S A 1988, 85, 6297–6300. [Google Scholar] [CrossRef]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V'kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. Journal of virology 2020, 94, e01925–19. [Google Scholar] [CrossRef]
- Mulabbi, E.N.; Tweyongyere, R.; Byarugaba, DK. The history of the emergence and transmission of human coronaviruses. Onderstepoort J Vet Res 2021, 88, e1–e8. [Google Scholar] [CrossRef]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Advances in virus research 1997, 48, 1–100. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Chu, D.K.; Peiris, J.S.; Kosakovsky Pond, S.L.; Poon, L.L. A case for the ancient origin of coronaviruses. J Virol 2013, 87, 7039–7045. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Forero-Muñoz, N.R.; Muylaert, R.L.; Seifert, S.N.; Albery, G.F.; Becker, D.J.; Carlson, C.J.; Poisot T. The coevolutionary mosaic of bat betacoronavirus emergence risk, Virus Evolution, 2023, vead079. [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Experimental Biology and medicine 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Holmes, E.C. Wild birds as reservoirs for diverse and abundant gamma- and deltacoronaviruses. FEMS microbiology reviews 2020, 44, 631–644. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annual review of virology 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Olsthoorn, R.C. Group-specific structural features of the 5'-proximal sequences of coronavirus genomic RNAs. Virology 2010, 401, 29–41. [Google Scholar] [CrossRef]
- Madhugiri, R.; Karl, N.; Petersen, D.; Lamkiewicz, K.; Fricke, M.; Wend, U.; Scheuer, R.; Marz, M.; Ziebuhr, J. Structural and functional conservation of cis-acting RNA elements in coronavirus 5'-terminal genome regions. Virology 2018, 517, 44–55. [Google Scholar] [CrossRef]
- Ma, Y.; Tong, X.; Xu, X.; Li, X.; Lou, Z.; Rao, Z. Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell 2010, 1, 688–697. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Takeda, M.; Chang, C.K.; Ikeya, T.; Güntert, P.; Chang, Y.H.; Hsu, Y.L.; Huang, T.H.; Kainosho, M. Solution structure of the c-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method. Journal of molecular biology, 2008, 380, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Sue, S.C.; Yu, T.H.; Hsieh, C.M.; Tsai, C.K.; Chiang, Y.C.; Lee, S.J.; Hsiao, H.H.; Wu, W.J.; Chang, W.L.; Lin, C.H.; Huang, T.H. Modular organization of SARS coronavirus nucleocapsid protein. Journal of biomedical science 2006, 13, 59–72. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Identification of in vivo-interacting domains of the murine coronavirus nucleocapsid protein. Journal of virology 2009, 83, 7221–7234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Syed, A.M.; Khalid, M.M.; Nguyen, A.; Ciling, A.; Wu, D.; Yau, W.M.; Srinivasan, S.; Esposito, D.; Doudna, J.A.; Piszczek, G.; Ott, M.; Schuck, P. Assembly reactions of SARS-CoV-2 nucleocapsid protein with nucleic acid. bioRxiv [Preprint]. 2023 Nov 23:2023.11.22.568361. [CrossRef]
- Korn, M.; Dhamotharan, K.; Jeffries, C.M.; Schlundt, A. The preference signature of the SARS-CoV-2 Nucleocapsid NTD for its 5’-genomic RNA elements. Nat Commun 2023, 14, 3331. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Molecular cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef]
- Estelle, A.B.; Forsythe, H.M.; Yu, Z.; Hughes, K.; Lasher, B.; Allen, P.; Reardon, P.N.; Hendrix, D.A.; Barbar, E.J. RNA structure and multiple weak interactions balance the interplay between RNA binding and phase separation of SARS-CoV-2 nucleocapsid. PNAS nexus 2023, 2, pgad333. [Google Scholar] [CrossRef]
- Grossoehme, N.E.; Li, L.; Keane, S.C.; Liu, P.; Dann, C.E. 3rd.; Leibowitz, J.L.; Giedroc, D.P. Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J Mol Biol 2009, 394, 544–557. [Google Scholar] [CrossRef]
- Carlson, C.R.; Adly, A.N.; Bi, M.; Howard, C.J.; Frost, A.; Cheng, Y.; Morgan, D.O. Reconstitution of the SARS-CoV-2 ribonucleosome provides insights into genomic RNA packaging and regulation by phosphorylation. The Journal of biological chemistry 2022, 298, 102560. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, C.; Izquierdo, J.M. T-cell intracellular antigens in health and disease. Cell cycle (Georgetown, Tex.) 2015, 14, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3' and 5' ends. Virus Res 2015, 206, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Sexton, N.R.; Denison, M.R. Thinking Outside the Triangle: Replication Fidelity of the Largest RNA Viruses. Annu Rev Virol 2014, 1, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Lu, S.; Wang, Y.; He, L.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; Yang, Q.; Wu, Y.; Zhang, S.; Huang, J.; Mao, S.; Ou, X.; Sun, D.; Tian, B.; Cheng, A. Bacterial DNA methyltransferase: A key to the epigenetic world with lessons learned from proteobacteria. Frontiers in microbiology 2023, 14, 1129437. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.A.; Hiscox, J.A. Characterisation of the RNA binding properties of the coronavirus infectious bronchitis virus nucleocapsid protein amino-terminal region. FEBS letters 2006, 580, 5993–5998. [Google Scholar] [CrossRef] [PubMed]
- Caruso, Í.P.; Sanches, K.; Da Poian, A.T.; Pinheiro, A.S.; Almeida, F.C.L. Dynamics of the SARS-CoV-2 nucleoprotein N-terminal domain triggers RNA duplex destabilization. Biophys J 2021, 120, 2814–2827. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; Liu, F.; Yu, X.; Li, S.; Liu, Q.; Yang, X.L.; Guo, M.; Li, X.; Chen, M.; Shi, Z.L.; Lan, K.; Chen, Y.; Zhou, Y. The SARS-CoV-2 subgenome landscape and its novel regulatory features. Molecular cell 2021, 81, 2135–2147.e5. [Google Scholar] [CrossRef]
- Ziv, O.; Gabryelska, M.M.; Lun, A.T.L.; Gebert, L.F.R.; Sheu-Gruttadauria, J.; Meredith, L.W.; Liu, Z.Y.; Kwok, C.K.; Qin, C.F.; MacRae, I.J.; Goodfellow, I.; Marioni, J.C.; Kudla, G.; Miska, E.A. COMRADES determines in vivo RNA structures and interactions. Nat Methods 2018, 15, 785–788. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Liu, P.; Makkinje, N.; Williamson, S.T.; Leibowitz, J.L.; Giedroc, D.P. Structural lability in stem-loop 1 drives a 5' UTR-3' UTR interaction in coronavirus replication. J Mol Biol 2008, 377, 790–803. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary structure of the SARS-CoV-2 5'-UTR. RNA biology 2021, 18, 447–456. [Google Scholar] [CrossRef]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3' untranslated region of flavivirus RNAs and potential cyclization sequences. J Mol Biol 1987, 198, 33–41. [Google Scholar] [CrossRef]
- Lo, C.Y.; Tsai, T.L.; Lin, C.N.; Lin, C.H.; Wu, H.Y. Interaction of coronavirus nucleocapsid protein with the 5'- and 3'-ends of the coronavirus genome is involved in genome circularization and negative-strand RNA synthesis. The FEBS journal 2019, 286, 3222–3239. [Google Scholar] [CrossRef]
- Goebel, S.J.; Hsue, B.; Dombrowski, T.F.; Masters, P.S. Characterization of the RNA components of a putative molecular switch in the 3' untranslated region of the murine coronavirus genome. J Virol 2004, 78, 669–682. [Google Scholar] [CrossRef]
- Xue, X.; Yang, H.; Shen, W.; Zhao, Q.; Li, J.; Yang, K.; Chen, C.; Jin, Y.; Bartlam, M.; Rao, Z. Production of authentic SARS-CoV M(pro) with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. Journal of molecular biology 2007, 366, 965–975. [Google Scholar] [CrossRef]
- Jonassen, C.M.; Jonassen, T.O.; Grinde, B. A common RNA motif in the 3' end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. The Journal of general virology 1998, 79, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M.Jr.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS biology 2005, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, J.F.; Hogue, B.G. Host protein interactions with the 3' end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. Journal of virology 2000, 74, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Tarun, S.Z.Jr.; Wells, S.E.; Deardorff, J.A.; Sachs, A.B. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proceedings of the National Academy of Sciences of the United States of America 1997, 94, 9046–9051. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, T.; Arya, S.; Chan, S.H.; Qi, S.; Dai, N.; Misra, A.; Park, J.G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; Martinez-Sobrido, L.; Gupta, Y.K. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun 2020, 11, 3718. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Ivanov, K.A.; Putics, Á.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; Gorbalenya, A.E.; Ziebuhr, J. Mechanisms and enzymes involved in SARS coronavirus genome expression. The Journal of general virology 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome research 2019, 29, 1545–1554. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Regulation of relative abundance of arterivirus subgenomic mRNAs. Journal of virology 2004, 78, 8102–8113. [Google Scholar] [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuñiga, S.; Enjuanes, L. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA biology 2011, 8, 237–248. [Google Scholar] [CrossRef]
- Wu, H.Y.; Guan, B.J.; Su, Y.P.; Fan, Y.H.; Brian, D.A. Reselection of a genomic upstream open reading frame in mouse hepatitis coronavirus 5'-untranslated-region mutants. Journal of virology 2014, 88, 846–858. [Google Scholar] [CrossRef]
- Byrd, A.K.; Raney, K.D. Superfamily 2 helicases. Frontiers in bioscience (Landmark edition) 2012, 17, 2070–2088. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Munday, D.; Bickerton, E.; Britton, P.; Rodgers, M.A.; Whitehouse, A.; Zhou, E.M.; Hiscox, J.A. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J Virol 2013, 87, 9486–9500. [Google Scholar] [CrossRef] [PubMed]
- Cartier, C.; Sivard, P.; Tranchat, C.; Decimo, D.; Desgranges, C.; Boyer, V. Identification of three major phosphorylation sites within HIV-1 capsid. Role of phosphorylation during the early steps of infection. The Journal of biological chemistry 1999, 274, 19434–19440. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.I.; Puustinen, P.; Gabrenaite, R.; Vihinen, H.; Rönnstrand, L.; Valmu, L.; Kalkkinen, N.; Mäkinen, K. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. The Plant cell 2003, 15, 2124–2139. [Google Scholar] [CrossRef] [PubMed]
- Law, L.M.; Everitt, J.C.; Beatch, M.D.; Holmes, C.F.; Hobman, T.C. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J Virol 2003, 77, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lei, X.; Jiang, Z.; Humphries, F.; Parsi, K.M.; Mustone, N.J.; Ramos, I.; Mutetwa, T.; Fernandez-Sesma, A.; Maehr, R.; Caffrey, D.R.; Fitzgerald, K.A. Cellular nucleic acid-binding protein restricts SARS-CoV-2 by regulating interferon and disrupting RNA-protein condensates. Proc Natl Acad Sci U S A 2023, 120, e2308355120. [Google Scholar] [CrossRef]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus genes 2011, 42, 37–45. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Cai, S.; Zhuang, Z.; Zhao, Z.; Jin, S.; Xie, W.; Zhou, L.; Zhang, L.; Zhao, J.; Cui, J. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-κB hyper-activation and inflammation. Signal transduction and targeted therapy 2021, 6, 167. [Google Scholar] [CrossRef]
- Lai, F.W.; Stephenson, K.B.; Mahony, J.; Lichty, B.D. Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-κB activation. Journal of virology, 2014, 88, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.X.; Liang, J.Q.; Zhu, Q.C.; Dai, G.; Li, S.; Fung, T.S. Liu, D.X. Gammacoronavirus Avian Infectious Bronchitis Virus and Alphacoronavirus Porcine Epidemic Diarrhea Virus Exploit a Cell-Survival Strategy via Upregulation of cFOS to Promote Viral Replication. Journal of virology 2021, 95, e02107-20. [CrossRef]
- Pan, J.; Peng, X.; Gao, Y.; Li, Z.; Lu, X.; Chen, Y.; Ishaq, M.; Liu, D.; Dediego, M.L.; Enjuanes, L.; Guo, D. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PloS one 2008, 3, e3299. [Google Scholar] [CrossRef] [PubMed]
- Patarca, R.; Haseltine, W.A. Intragenomic rearrangements involving 5'-untranslated region segments in SARS-CoV-2, other betacoronaviruses, and alphacoronaviruses. Virology journal 2023, 20, 36. [Google Scholar] [CrossRef] [PubMed]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; Ragazzini, R.; Jungreis, I.; Ummadi, M.; Rojc, A.; Turner, J.; Bischof, M.L.; Obernier, K.; Braberg, H.; Soucheray, M.; Richards, A.; … Krogan, N.J. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Reuschl, A.K.; Thorne, L.G.; Whelan, M.V.X.; Ragazzini, R.; Furnon, W.; Cowton, V.M.; De Lorenzo, G.; Mesner, D.; Turner, J.L.E.; Dowgier, G.; Bogoda, N.; Bonfanti, P.; Palmarini, M.; Patel, A.H.; Jolly, C.; Towers, G.J. Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants. Nature microbiology 2024, Advance online publication. [CrossRef]
- Emerman, M.; Guyader, M.; Montagnier, L.; Baltimore, D.; Muesing, MA. The specificity of the human immunodeficiency virus type 2 transactivator is different from that of human immunodeficiency virus type 1. EMBO J 1987, 6, 3755–60. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Bain, E.S.; Luciw, P.A.; Peterlin, B.M. Structure, sequence, and position of the stem-loop in tar determine transcriptional elongation by tat through the HIV-1 long terminal repeat. Genes Dev 1989, 3, 547–558. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, L.F.; Mavankal, G.; Peters, P.; Wu-Baer, F.; Gaynor, R.B. Tat functions to stimulate the elongation properties of transcription complexes paused by the duplicated TAR RNA element of human immunodeficiency virus 2. J Mol Biol 1995, 254, 350–63. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; Chun, R.; Lin, N.H.; Gatignol, A.; Glabe, C.G.; Fan, H. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol 1993, 67, 6224–6233. [Google Scholar] [CrossRef]
- Chun, R.F.; Semmes, O.J.; Neuveut, C.; Jeang, K.T. Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J Virol 1998, 72, 2615–2629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Wang, Q.; Liu, X.; Li, X.; Li, P.; Ma, Q.; Cao, C. The nucleocapsid protein of severe acute respiratory syndrome coronavirus inhibits cell cytokinesis and proliferation by interacting with translation elongation factor 1alpha. J Virol 2008, 82, 6962–6971. [Google Scholar] [CrossRef] [PubMed]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc Natl Acad Sci U S A 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed]
- El Baba, R.; Herbein, G. Management of epigenomic networks entailed in coronavirus infections and COVID-19. Clin Epigenetics 2020, 12, 118. [Google Scholar] [CrossRef]
- Zaborowska, J.; Isa, N.F.; Murphy, S. P-TEFb goes viral. BioEssays : news and reviews in molecular, cellular and developmental biology, 2016, 38 Suppl 1, S75–S85. [CrossRef]
- López-Muñoz, A.D.; Santos, J.J.S.; Yewdell, J.W. Cell surface nucleocapsid protein expression: A betacoronavirus immunomodulatory strategy. Proc Natl Acad Sci U S A 2023, 120, e2304087120. [Google Scholar] [CrossRef]
- Nakanaga, K.; Yamanouchi, K.; Fujiwara, K. Protective effect of monoclonal antibodies on lethal mouse hepatitis virus infection in mice. Journal of virology, 1986, 59, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, J.; Cainelli-Gebara, V.; Mercier, G.; Mansour, S.; Talbot, P.J.; Lussier, G.; Oth, D. Protection from mouse hepatitis virus type 3-induced acute disease by an anti-nucleoprotein monoclonal antibody. Brief report. Archives of virology 1987, 97, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Sparn, C.; Meyer, A.; Saleppico, R.; Nickel, W. Unconventional secretion mediated by direct protein self-translocation across the plasma membranes of mammalian cells. Trends in biochemical sciences 2022, 47, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Giacca, M. Transcellular protein transduction using the Tat protein of HIV-1. Advanced drug delivery reviews, 2005, 57, 597–608. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 2001, 276, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.P.; Ajasin, D.O.; Ramasamy, S.; DesMarais, V.; Eugenin, E.A.; Prasad, V.R. A naturally occurring polymorphism in the HIV-1 tat basic domain inhibits uptake by bystander cells and leads to reduced neuroinflammation. Sci. Rep 2019, 9, 3308. [Google Scholar] [CrossRef] [PubMed]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: role in bystander toxicity. Frontiers in Cellular and Infection Microbiology 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Nema, V. HIV-Associated Neurotoxicity: The Interplay of Host and Viral Proteins. Mediators Inflamm 2021, 2021, 1267041. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Geiger, J.D. Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor- α-mediated neurotoxicity. Neurobiology of Disease 2007, 26, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, HE; Lipton, S.A. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death and Differentiation 2005, 12, 878–892. [CrossRef]
- Nedwin, G.E.; Naylor, S.L.; Sakaguchi, A.Y.; Smith, D.; Jarrett-Nedwin, J.; Pennica, D.; Goeddel, D.V.; Gray, P.W. Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization. Nucleic acids research 1985, 13, 6361–6373. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.R.; Ramelli, S.C.; Grazioli. A; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; Ko, S.H.; Platt, A.P.; Burbelo, P.D.; Quezado, M.; Pittaluga, S.; Purcell, M.; Munster, V.J.; Belinky, F.; Ramos-Benitez, M.J.; Boritz, E.A.; Lach, I.A.; Herr, D.L.; Rabin, J.; Saharia, K.K.; Madathil, R.J.; Tabatabai, A.; Soherwardi, S.; McCurdy, M.T.; NIH COVID-19 Autopsy Consortium; Peterson, K.E.; Cohen, J.I.; de Wit, E.; Vannella, K.M.; Hewitt, S.M.; Kleiner, D.E.; Chertow, D.S. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758-763. [CrossRef]
- Patel, K.P.; Vunnam, S.R.; Patel, P.A.; Krill, K.L.; Korbitz, P.M.; Gallagher, J.P.; Suh, J.E.; Vunnam, R.R. Transmission of SARS-CoV-2: An update of current literature. Eur. J. Clin. Microbiol. Infect. Dis 2020, 39, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.; Kaushal, S.; Yeo, D. Enteric involvement of coronaviruses: Is faecal-oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol. Hepatol 2020, 5, 335–337. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; Kornilov, S.A.; Scherler, K.; Pavlovitch-Bedzyk, A.J.; Dong, S.; Lausted, C.; Lee, I.; Fallen, S.; Dai, C.L.; Baloni, P.; Smith, B.; Duvvuri, V.R.; Anderson, K.G.; Li, J.; Yang, F.; Duncombe, C.J.; McCulloch, D.J.; Rostomily, C.; Troisch, P.; Zhou, J.; Mackay, S.; DeGottardi, Q.; May, D.H.; Taniguchi, R.; Gittelman, R.M.; Klinger, M.; Snyder, T.M.; Roper, R.; Wojciechowska, G.; Murray, K.; Edmark, R.; Evans, S.; Jones, L.; Zhou, Y.; Rowen, L.; Liu, R.; Chour, W.; Algren, H.A.; Berrington, W.R.; Wallick, J.A.; Cochran, R.A.; Micikas, M.E.; ISB-Swedish COVID-19 Biobanking Unit; Wrin, T.; Petropoulos, C.J.; Cole, H.R.; Fischer, T.D.; Wei, W.; Hoon, D.S.B.; Price, N.D.; Subramanian, N.; Hill, J.A.; Hadlock, J.; Magis, A.T.; Ribas, A.; Lanier, L.L.; Boyd, S.D.; Bluestone, J.A.; Chu, H.; Hood, L.; Gottardo, R,; Greenberg, P.D.; Davis, M.M.; Goldman, J.D.; Heath, J.R. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881-895.e20. [CrossRef]
- Castanares-Zapatero, D.; Chalonm, P.; Kohn, L.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; Maertens de Noordhout, C.; Primus-de Jong, C.; Cleemput, I.; Van den Heede, K. Pathophysiology and mechanism of long COVID: a comprehensive review. Ann Med 2022, 54, 1473–1487. [Google Scholar] [CrossRef]
- Perumal, R.; Shunmugam, L.; Naidoo, K.; Abdool Karim, S.S.; Wilkins, D.; Garzino-Demo, A.; Brechot, C.; Parthasarathy, S.; Vahlne, A.; Nikolich, J.Ž. Long COVID: a review and proposed visualization of the complexity of long COVID. Frontiers in immunology 2023, 14, 1117464. [Google Scholar] [CrossRef]
- Buonsenso, D.; Martino, L.; Morello, R.; Mariani, F.; Fearnley, K.; Valentini, P. Viral persistence in children infected with SARS-CoV-2: current evidence and future research strategies. The Lancet. Microbe 2023, S2666-5247(23)00115-5. [CrossRef]
- Cupelli, L.A.; Hsu, M.C. The human immunodeficiency virus type 1 Tat antagonist, Ro 5-3335, predominantly inhibits transcription initiation from the viral promoter. J Virol 1995, 69, 2640–2643. [Google Scholar] [CrossRef]
- Hwang, S.; Tamilarasu, N.; Kibler, K.; Cao, H.; Ali, A.; Ping, Y.H.; Jeang, K.T.; Rana, T.M. Discovery of a small molecule Tat-trans-activation-responsive RNA antagonist that potently inhibits human immunodeficiency virus-1 replication. J Biol Chem 2003, 278, 39092–39103. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Afshar, M.; Varani, G.; Murchie, A.I.; Karn, J.; Lentzen, G.; Drysdale, M.; Bower, J.; Potter, A.J.; Starkey, I.D.; Swarbrick, T.; Aboul-ela, F. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic "hot spots". J Mol Biol 2004, 336, 343–56. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.: Schutt, A.D.; Holly, M.; Slice, L.W.; Sherman, M.I.; Richman, D.D.; Potash, M.J.; Volsky, D.J. Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science 1991, 254, 1799-802. [CrossRef]
- Kim, H.I.; Kim, G.N.; Yu, K.L.; Park, S.H.; You, J.C. Identification of Novel Nucleocapsid Chimeric Proteins Inhibiting HIV-1 Replication. Int J Mol Sci 2022, 23, 12340. [Google Scholar] [CrossRef] [PubMed]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Frontiers in microbiology 2020, 10, 3060. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: a better web interface. Nucleic Acids Res 2008, 36(Web Server issue): W5-W9. [CrossRef]
- ICTV Coronaviridae Study Group. International Committee on Taxonomy of Viruses (ICTV). 2021. Available from. https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/223/coronaviridae-figures.
- Nikolaidis, M.; Markoulatos, P.; van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The neighborhood of the spike gene is a hotspot for modular intertypic homologous and non-homologous recombination in coronavirus genomes. Mol Biol Evol 2022, msab 292. [CrossRef]
- Tsoleridis, T.; Chappell, J.G.; Onianwa, O.; Marston, D.A.; Fooks, A.R.; Monchatre-Leroy, E.; Umhang, G.; Müller, M.A.; Drexler, J.F.; Drosten, C.; Tarlinton, R.E.; McClure, C.P.; Holmes, E.C.; Ball, J.K. Shared Common Ancestry of Rodent Alphacoronaviruses Sampled Globally. Viruses 2019, 11, 125. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Research 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Research 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. "ViennaRNA Package 2.0", Algorithms for Molecular Biology 2011, 6, 26. [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Similarities among HIV-1 TAR, JCV TAR, and the SARS-CoV-2 leader. The three regions of similarity are shown in light blue (SARS-CoV-2; NC_045512), dark blue (identical to Tat binding site in HIV-1 and JCV TARs), and fuchsia in the primary and secondary structures. Differing nucleotides among HIV subtypes [97,98] are shown above the HIV-1 sequence. Cyclin T1 binding sites in HIV-1 and JCV TARs are shown in red. The complement of the HIV-1 Cyclin T1 binding site matching SL1 of SARS-CoV-2 is shown in green. SARS-CoV-2 TRS is underlined and in italics [99].
Figure 1.
Similarities among HIV-1 TAR, JCV TAR, and the SARS-CoV-2 leader. The three regions of similarity are shown in light blue (SARS-CoV-2; NC_045512), dark blue (identical to Tat binding site in HIV-1 and JCV TARs), and fuchsia in the primary and secondary structures. Differing nucleotides among HIV subtypes [97,98] are shown above the HIV-1 sequence. Cyclin T1 binding sites in HIV-1 and JCV TARs are shown in red. The complement of the HIV-1 Cyclin T1 binding site matching SL1 of SARS-CoV-2 is shown in green. SARS-CoV-2 TRS is underlined and in italics [99].
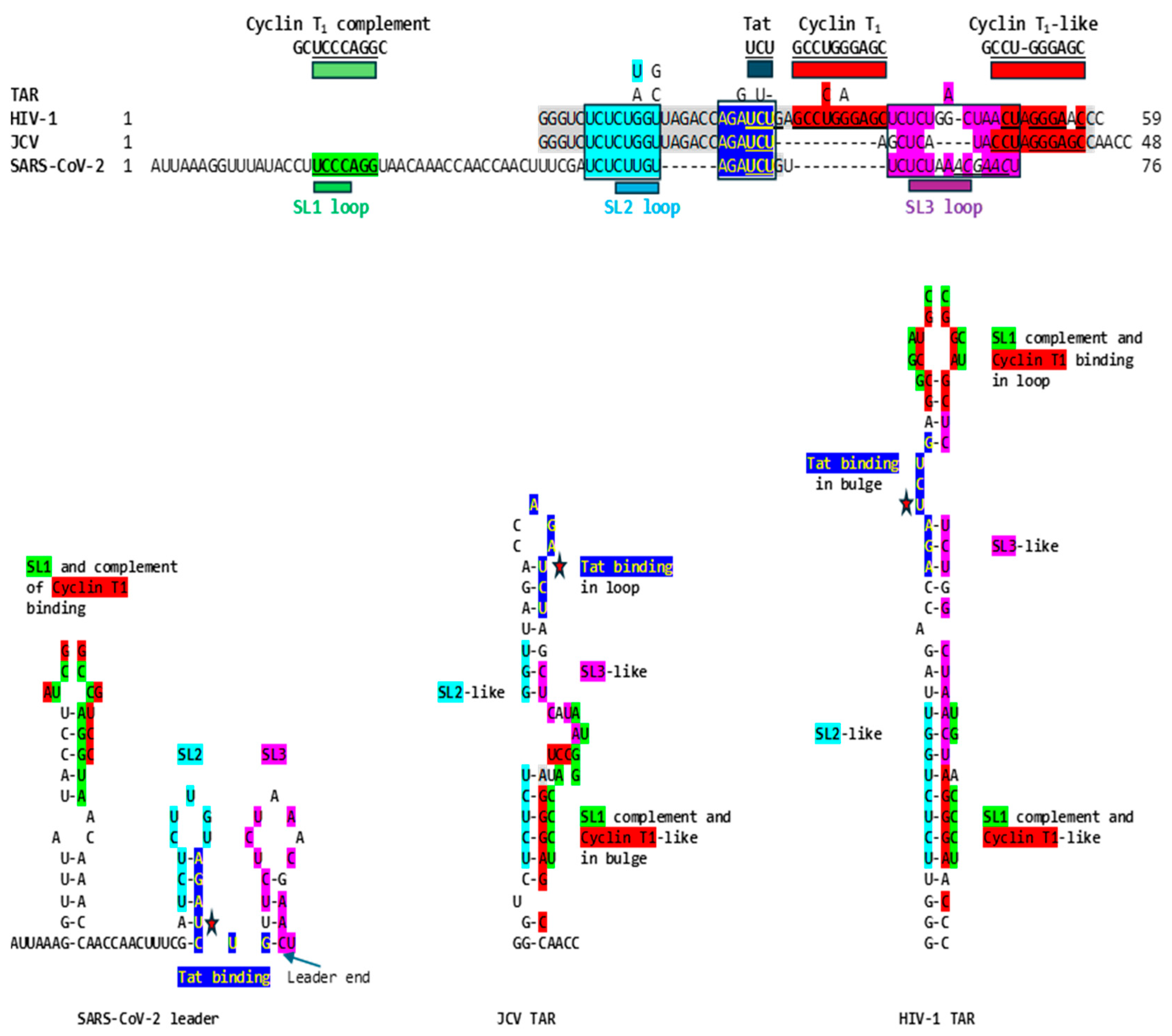
Figure 2.
Similarities among the stems in the hairpin structures of SARS-CoV-2 leader, JCV TAR, and HIV-1 TAR. Regions similar to the stems in SL1 and SL2 are shown using dark and light green and blue lines, respectively. Their primary sequences are aligned on the right. The light blue arrows depict the Tat binding site in the three sequences, remaining putative for the SARS-CoV-2 leader.
Figure 2.
Similarities among the stems in the hairpin structures of SARS-CoV-2 leader, JCV TAR, and HIV-1 TAR. Regions similar to the stems in SL1 and SL2 are shown using dark and light green and blue lines, respectively. Their primary sequences are aligned on the right. The light blue arrows depict the Tat binding site in the three sequences, remaining putative for the SARS-CoV-2 leader.
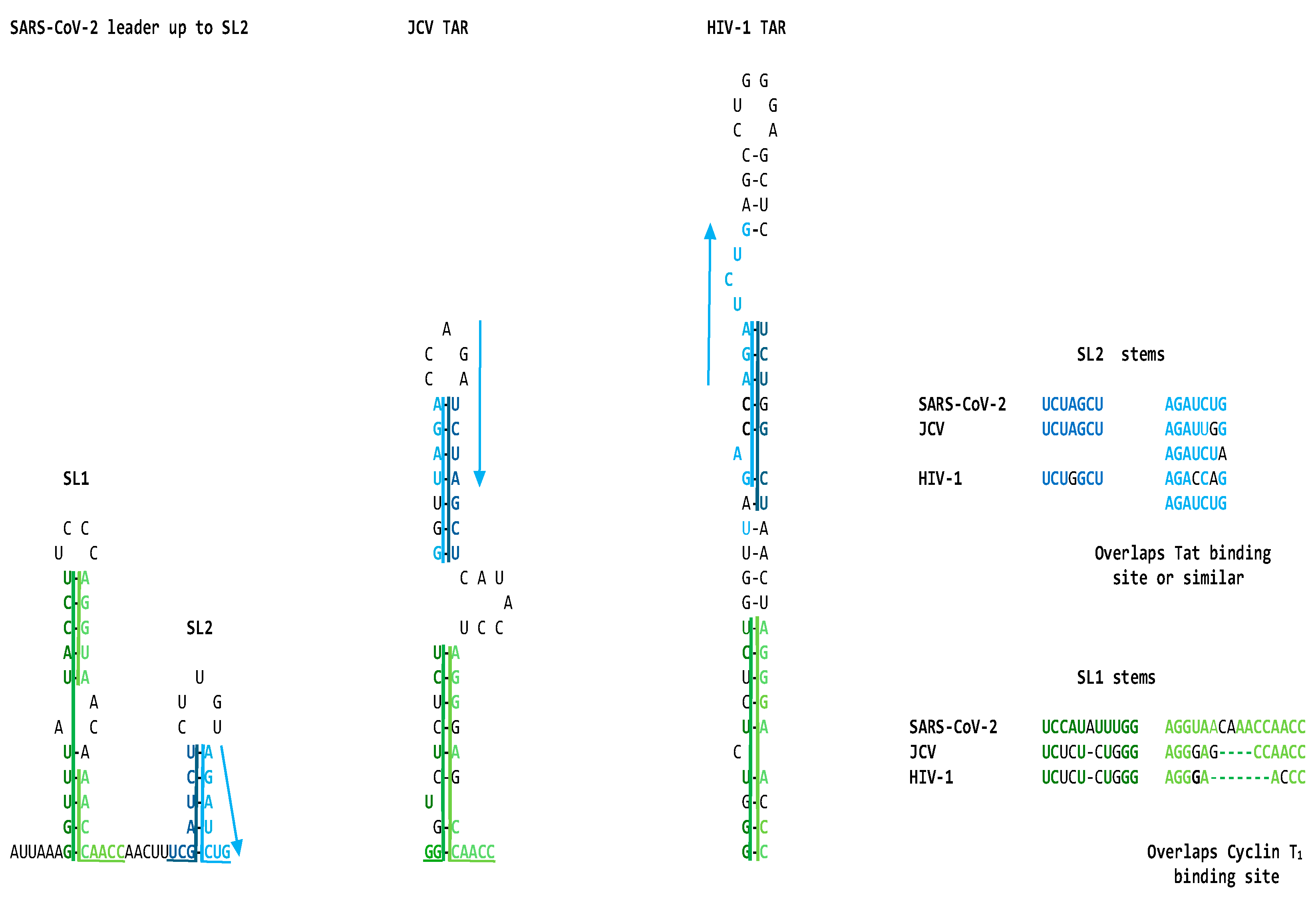
Figure 3.
Conservation among the seven coronaviruses infecting humans and gamma- and delta coronaviruses of regions of similarity with lentiviral TARs and human 7SK snRNA. Nucleotide variation among HIV-1 TARs or SIV TARs are indicated at the top and bottom, respectively. Coronaviral TRSs [99] are in italics and underlined. Accession numbers: SARS-CoV-2: NC_045512; SARS-CoV-1: NC_004718; hCoV-OC43: KJ958218; hCoV-HKU-1: MH940245; MERS-CoV:NC_019843; hCoV-NL63: MW202337.1; hCoV-229E: KU291448; IBV: NC_001451; Porcine deltacoronavirus: KR265862; human 7SK snRNA [50]; HIV-1, HIV-2 and SIV TARs [97,100,101].
Figure 3.
Conservation among the seven coronaviruses infecting humans and gamma- and delta coronaviruses of regions of similarity with lentiviral TARs and human 7SK snRNA. Nucleotide variation among HIV-1 TARs or SIV TARs are indicated at the top and bottom, respectively. Coronaviral TRSs [99] are in italics and underlined. Accession numbers: SARS-CoV-2: NC_045512; SARS-CoV-1: NC_004718; hCoV-OC43: KJ958218; hCoV-HKU-1: MH940245; MERS-CoV:NC_019843; hCoV-NL63: MW202337.1; hCoV-229E: KU291448; IBV: NC_001451; Porcine deltacoronavirus: KR265862; human 7SK snRNA [50]; HIV-1, HIV-2 and SIV TARs [97,100,101].
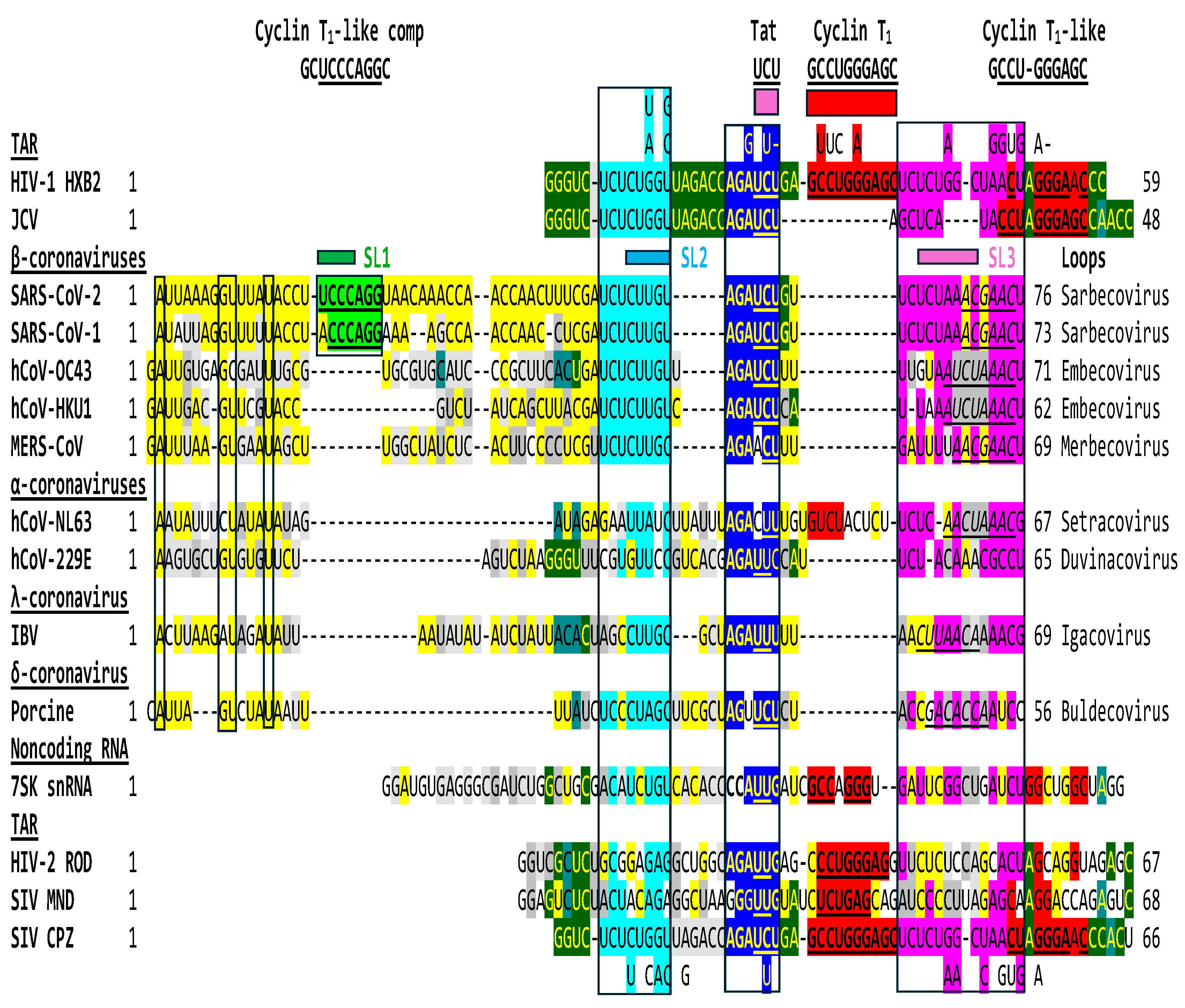
Figure 4.
HIV-2 TAR complement (3’ half of antisense corresponding to 5’ half of sense) shows regions of similarity (Cyclin T1-binding-like site and Tat-binding site) with SL1 of SARS-CoV-2 and -1 (5’ half of sense), in contrast to HIV-1 and JCV TARs that show regions of similarity to SL2 and SL3 (3’ half of sense). HIV-2 accession number: NC_001722.
Figure 4.
HIV-2 TAR complement (3’ half of antisense corresponding to 5’ half of sense) shows regions of similarity (Cyclin T1-binding-like site and Tat-binding site) with SL1 of SARS-CoV-2 and -1 (5’ half of sense), in contrast to HIV-1 and JCV TARs that show regions of similarity to SL2 and SL3 (3’ half of sense). HIV-2 accession number: NC_001722.
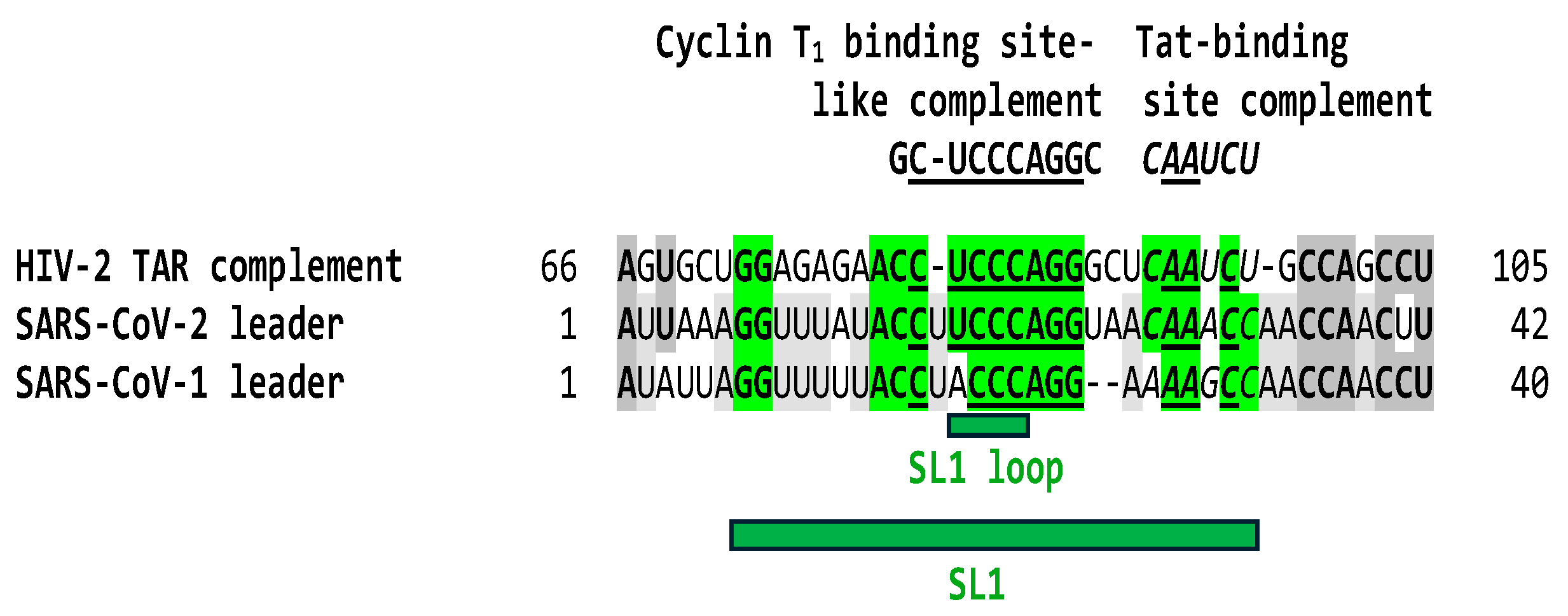
Figure 5.
TAR binding-like region in the C-terminal domains of the nucleocapsid proteins (N-CTDs) of SARS-CoV-2 and other coronaviruses. The N-CTD region of similarity to lentiviral Tat and human HEXIM1 & 2 proteins is shown in red. Differing amino acids among epidemic group M HIV-1 subtypes are shown above the HIV-1 Tat sequence [97]. Triangles indicate basic amino acids that form a positive charge groove for nucleic acid binding in SARS-CoV-2 N-CTD [112]. Accession: HIV-2: AAA76845.1; SIV: AEK79597.1; HEXIM1: NP_006451.1; HEXIM2: NP_653209.1; SARS-CoV-2 & -1: YP_009724397.2, YP_009825061.1; hCoV-OC43: AIX09803.1; hCoV-HKU1: AYN64565.1; MERS-CoV: YP_009047211.1; hCoV-NL63: UDL16983.1; hCoV-229E: AOG74787.1; Avian infectious bronchitis virus: NP_040838.1; Porcine deltacoronavirus: AML40885.1.
Figure 5.
TAR binding-like region in the C-terminal domains of the nucleocapsid proteins (N-CTDs) of SARS-CoV-2 and other coronaviruses. The N-CTD region of similarity to lentiviral Tat and human HEXIM1 & 2 proteins is shown in red. Differing amino acids among epidemic group M HIV-1 subtypes are shown above the HIV-1 Tat sequence [97]. Triangles indicate basic amino acids that form a positive charge groove for nucleic acid binding in SARS-CoV-2 N-CTD [112]. Accession: HIV-2: AAA76845.1; SIV: AEK79597.1; HEXIM1: NP_006451.1; HEXIM2: NP_653209.1; SARS-CoV-2 & -1: YP_009724397.2, YP_009825061.1; hCoV-OC43: AIX09803.1; hCoV-HKU1: AYN64565.1; MERS-CoV: YP_009047211.1; hCoV-NL63: UDL16983.1; hCoV-229E: AOG74787.1; Avian infectious bronchitis virus: NP_040838.1; Porcine deltacoronavirus: AML40885.1.
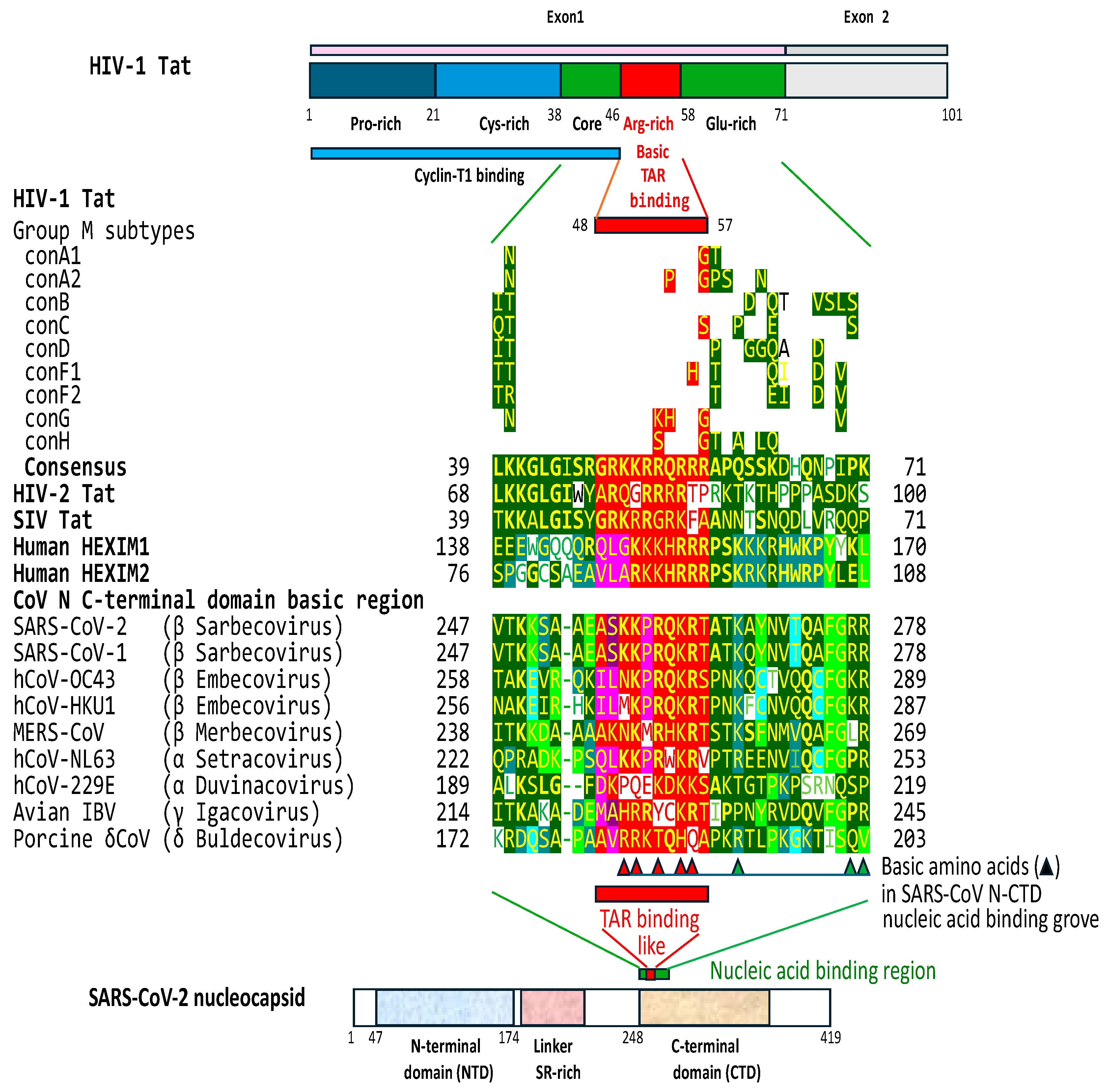
Figure 6.
Similarity between Zinc-coordinating domains of HIV-1, HIV-2, and SIV Tat (cysteine-rich region) proteins and the interface region of SARS-CoV-2 NSP12 and equivalent regions in other coronaviruses. The metal binding motif consisting of cysteine and histidine residues in yellow with a dark blue background is conserved among coronaviruses. Similarities between lentiviral Tat proteins with interface regions of coronaviruses, and among the latter are shown in yellow letters with a dark and light green background, respectively. Differing amino acids among epidemic group M HIV-1 subtypes are shown above the HIV-1 Tat sequence [97].
Figure 6.
Similarity between Zinc-coordinating domains of HIV-1, HIV-2, and SIV Tat (cysteine-rich region) proteins and the interface region of SARS-CoV-2 NSP12 and equivalent regions in other coronaviruses. The metal binding motif consisting of cysteine and histidine residues in yellow with a dark blue background is conserved among coronaviruses. Similarities between lentiviral Tat proteins with interface regions of coronaviruses, and among the latter are shown in yellow letters with a dark and light green background, respectively. Differing amino acids among epidemic group M HIV-1 subtypes are shown above the HIV-1 Tat sequence [97].
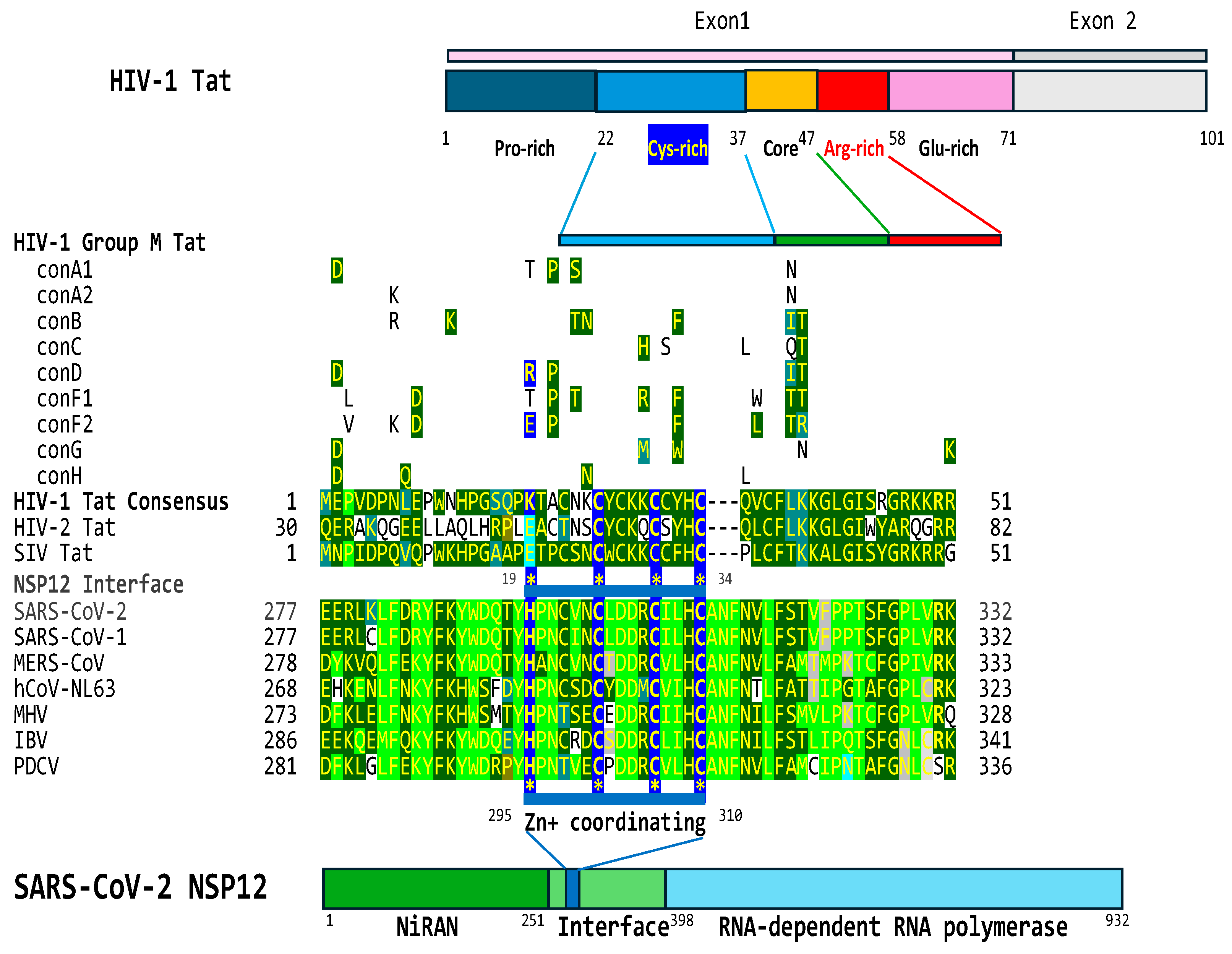
Figure 7.
A. Alignment of the PRRSV leader sequence (NC_001961) with human 7SK snRNA, and TARs of HIV-2 ROD strain and SIV MND strain. B. Alignment of the PRRSV N protein (NP_047413.1) with lentiviral Tat proteins, human HEXIM1 and 2, and coronaviral N-CTDs.
Figure 7.
A. Alignment of the PRRSV leader sequence (NC_001961) with human 7SK snRNA, and TARs of HIV-2 ROD strain and SIV MND strain. B. Alignment of the PRRSV N protein (NP_047413.1) with lentiviral Tat proteins, human HEXIM1 and 2, and coronaviral N-CTDs.
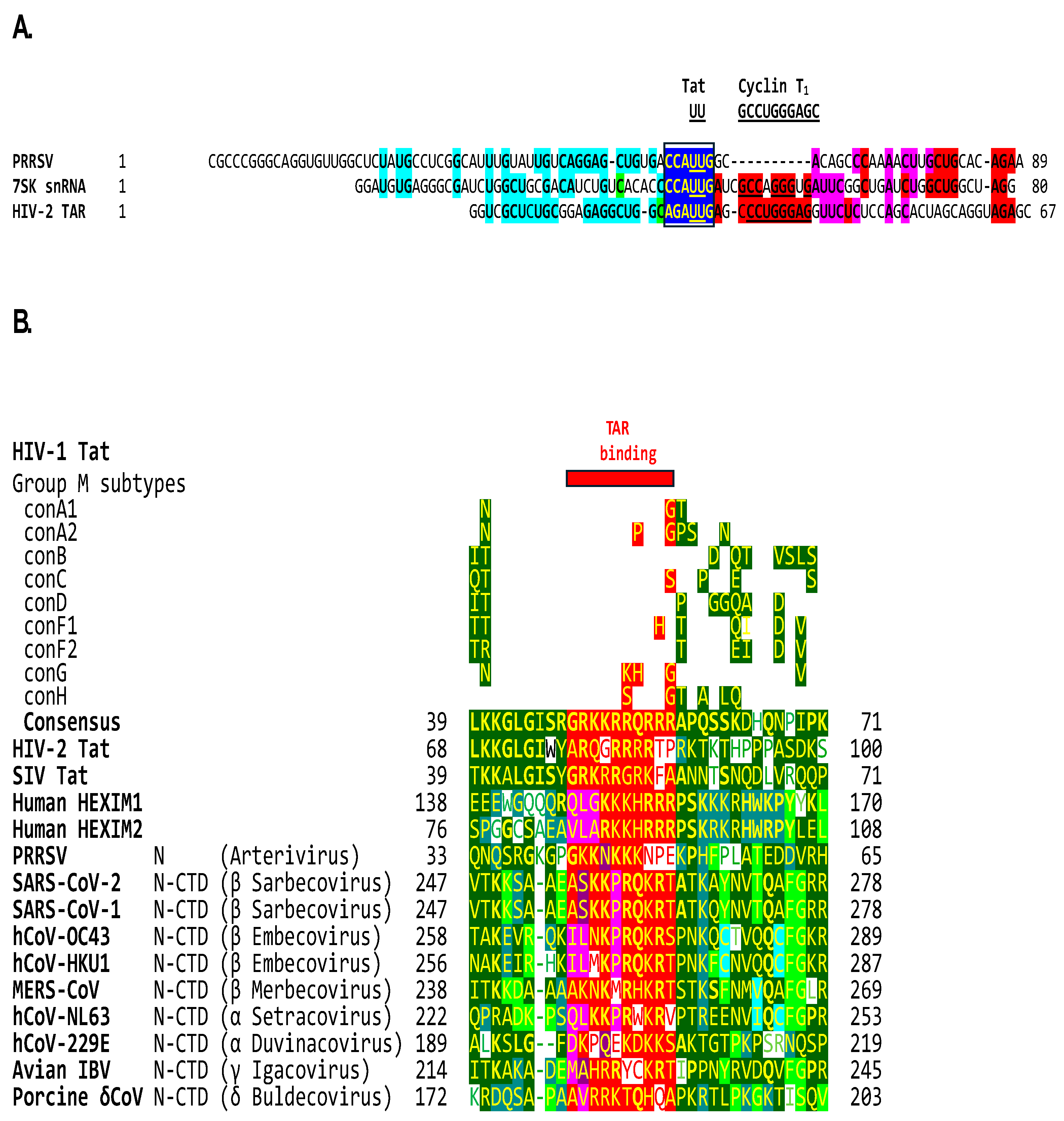
Figure 8.
Alignment among a prokaryotic site-specific DNA-methyltransferase (Candidatus Kaiserbacterium) and the N-CTD nucleic acid-binding regions of SARS-CoV-2 and -1 and MERS-CoV. Identical (bold letters) and conservative substitutions are shown as yellow letters with a dark green background. Accession numbers for the bacterial sequence shown and for that of Candidatus Nomurabacteria are MCA9362859.1 and MCB9810153.1.
Figure 8.
Alignment among a prokaryotic site-specific DNA-methyltransferase (Candidatus Kaiserbacterium) and the N-CTD nucleic acid-binding regions of SARS-CoV-2 and -1 and MERS-CoV. Identical (bold letters) and conservative substitutions are shown as yellow letters with a dark green background. Accession numbers for the bacterial sequence shown and for that of Candidatus Nomurabacteria are MCA9362859.1 and MCB9810153.1.
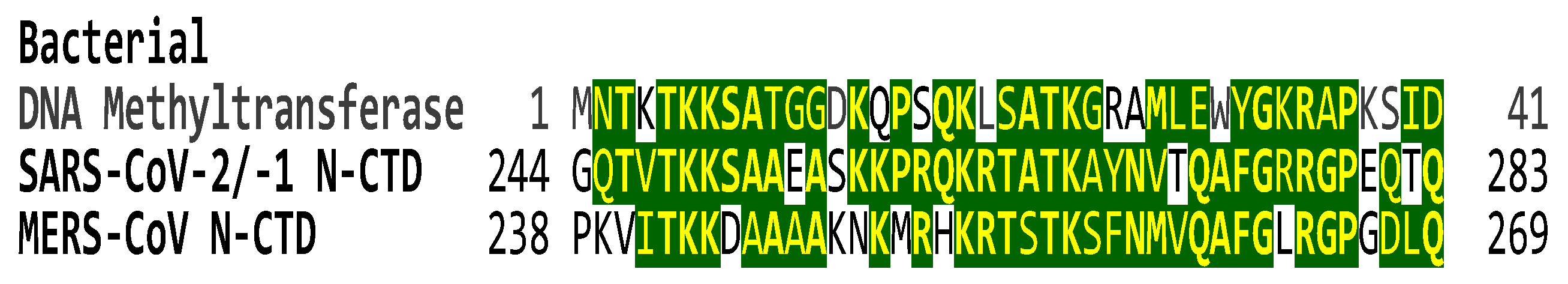
Figure 9.
Conserved circularization sequences (CCSs) in 5’- and 3’ genomic termini of SARS-CoV-2 (Wuhan reference, NC_045512). The segment spanning between the last nucleotide of the potential Tat-binding site and the end of the SARS-CoV-2 leader encompasses half of the CCS in the SARS-CoV-2 leader. The underlined guanosine (G) is replaced by an adenosine (A) in SARS-CoV-1. The blue lines represent the beginning and end of the circularized viral genome.
Figure 9.
Conserved circularization sequences (CCSs) in 5’- and 3’ genomic termini of SARS-CoV-2 (Wuhan reference, NC_045512). The segment spanning between the last nucleotide of the potential Tat-binding site and the end of the SARS-CoV-2 leader encompasses half of the CCS in the SARS-CoV-2 leader. The underlined guanosine (G) is replaced by an adenosine (A) in SARS-CoV-1. The blue lines represent the beginning and end of the circularized viral genome.
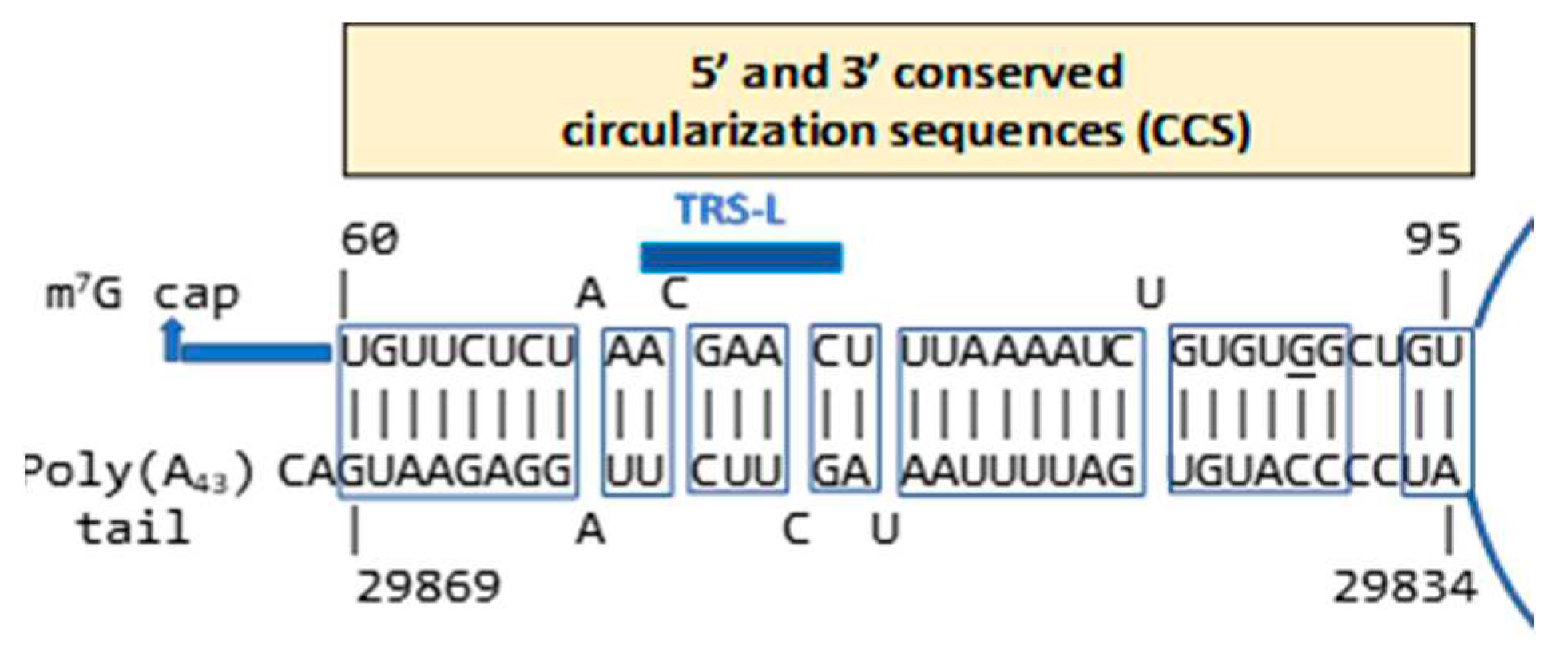
Figure 10.
Intragenomic rearrangements in SARS-CoV-2 bring TAR-like sequences to regions preceding or within genes such as N and NSP12 preceding the lentiviral Tat-like sequences. Segments of the SARS-CoV-2 leader sequence encompassing the Tat-binding site and SL3, and in some cases the rest of SL2, are involved in intragenomic rearrangements in SARS-CoV-2, which would bring the Tat binding site to regions usually preceding or within several viral genes.
Figure 10.
Intragenomic rearrangements in SARS-CoV-2 bring TAR-like sequences to regions preceding or within genes such as N and NSP12 preceding the lentiviral Tat-like sequences. Segments of the SARS-CoV-2 leader sequence encompassing the Tat-binding site and SL3, and in some cases the rest of SL2, are involved in intragenomic rearrangements in SARS-CoV-2, which would bring the Tat binding site to regions usually preceding or within several viral genes.

Figure 11.
Comparison between SARS-CoV-2 SL1 and SL2 and the reverse of the TNF-α promoter. TNF-α sequence starts 3 nucleotides from the start codon to the transcription initiation site [218].
Figure 11.
Comparison between SARS-CoV-2 SL1 and SL2 and the reverse of the TNF-α promoter. TNF-α sequence starts 3 nucleotides from the start codon to the transcription initiation site [218].
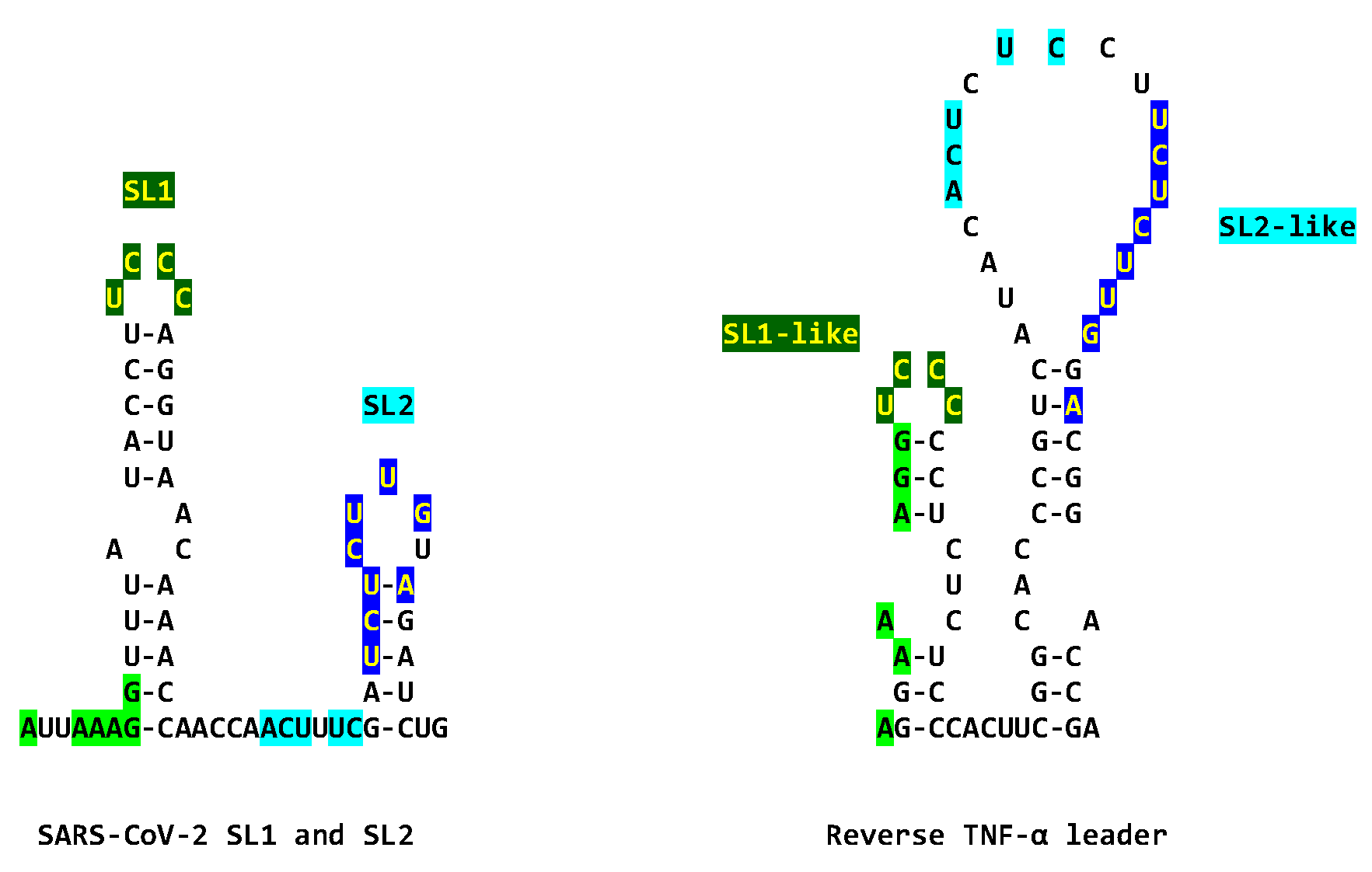
Figure 12.
SARS-CoV-2 proteins with Tat-like structure and functions, and TAR-like sequences. Replication-Transcription-Complex (orange) includes NSP12 (RNA-dependent RNA polymerase, RdRp), NSP7, and NSP8. The nucleocapsid protein interacts with NSP12 and NSP3. Beyond the nucleocapsid, NSP3 interacts with NSP12 and NSP13. The TAR-like sequences in the SARS-CoV-2 leader are indicated in light and dark blue in stem-loop (SL)2 and in fuchsia in SL3. The complement of SL1 that matches the Cyclin T1 binding site in HIV-1 TAR is indicated in green.
Figure 12.
SARS-CoV-2 proteins with Tat-like structure and functions, and TAR-like sequences. Replication-Transcription-Complex (orange) includes NSP12 (RNA-dependent RNA polymerase, RdRp), NSP7, and NSP8. The nucleocapsid protein interacts with NSP12 and NSP3. Beyond the nucleocapsid, NSP3 interacts with NSP12 and NSP13. The TAR-like sequences in the SARS-CoV-2 leader are indicated in light and dark blue in stem-loop (SL)2 and in fuchsia in SL3. The complement of SL1 that matches the Cyclin T1 binding site in HIV-1 TAR is indicated in green.
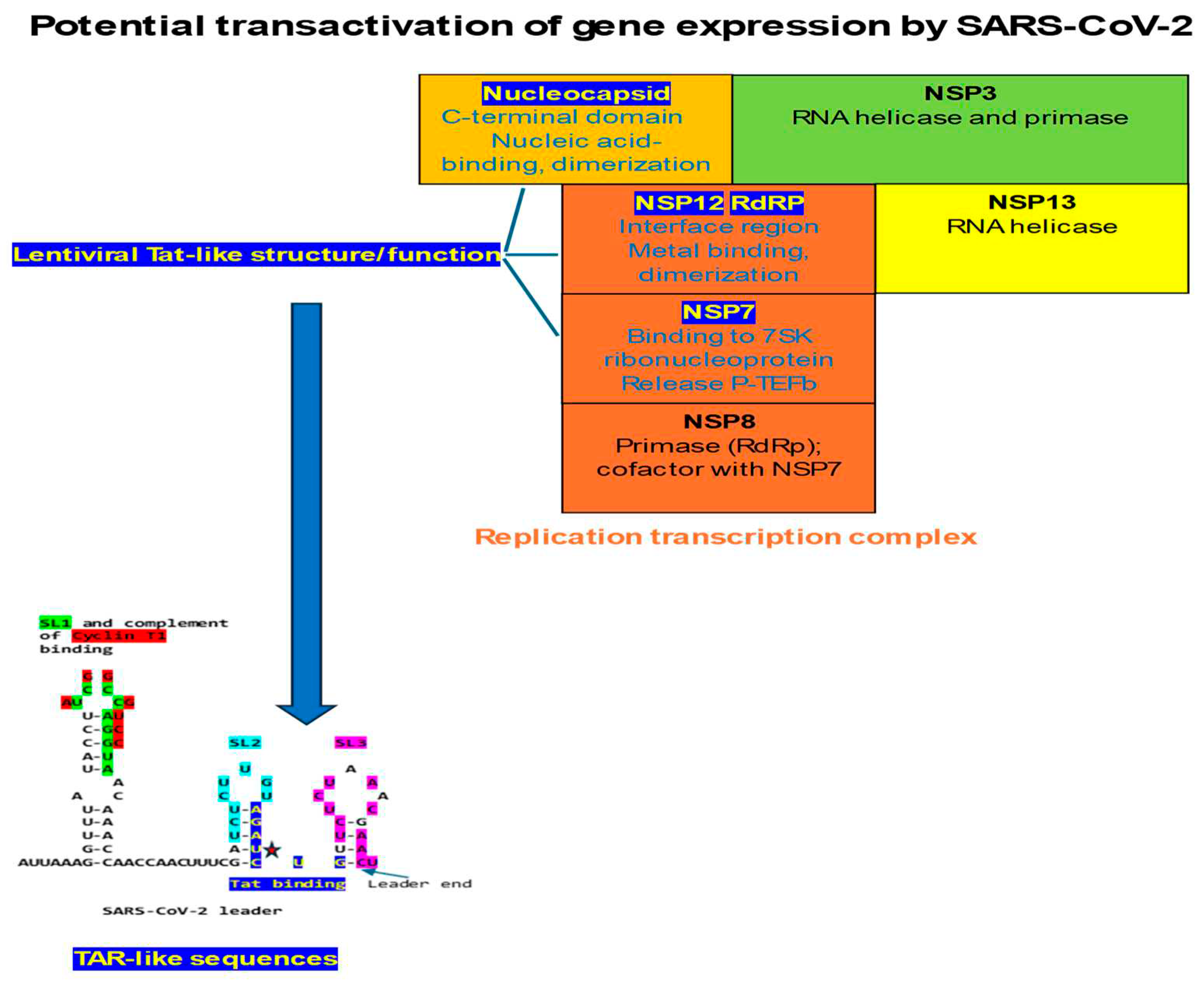
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



