
Submitted:
26 January 2024
Posted:
29 January 2024
You are already at the latest version
Abstract
In this paper the thermodynamic and reactivity study of light alcohol fuels was prepared, based on experimental and numerical results. We also tested the influence of water addition on fundamental properties of the combustion reactivity dynamics in closed vessels, like the maximum explosion pressure, maximum rate of pressure rise and the explosion delay time of alcohol-air mixtures. Substances that we investigated were: methanol, ethanol, n-propanol and iso-propanol. All experiments were conducted at initial conditions of 323.15 K and 1 bar in a 20 dm3 closed testing vessel. We investigated the reactivity and thermodynamic properties during the combustion of liquid fuel-air mixtures with equivalence ratios between 0.3-0.7 as well as some admixtures with water, to observe water mitigation effects. All light alcohols samples were prepared at the same initial conditions on a volumetric basis by mixing the pure components. The volumetric water content of the admixtures was varied from 10 to 60 vol%. The aim of water addition was to investigate the influence of thermodynamic properties of light alcohols and to find out to which extent a water addition may accomplish mitigation of combustion dynamics and thermodynamic reactivity.
Keywords:
thermodynamic reactivity
; combustion process
; light alcohols
; methanol
; ethanol
; n-propanol
; iso-propanol
1. Introduction
At present, the global energy industry is strongly dependent on the fossil fuels [1-5]. Over 80% of the global energy production comes from the petroleum, coal and the natural gas combustion. As the resources of fossil fuels are definitely exhaustible, there is a need to find the applicable solution to substitute the fossil fuels in conventional combustion systems. Moreover, from the ecological point of view, the conventional fuels that are used in both automotive and power industries have the poor impact on the environment and generates the environmental pollution. The current condition of the atmosphere requires the limitation in the emission of the factors that are ecologically harmful. Lower alcohols appear to have a great potential to be a supplement in a mixtures with the conventional fuels or even as independent fuels in combustion systems that are currently used [6]. All of that caused the raising interests in combustion properties of alcohols, but despite of that, still there are relatively small number of published papers concerning the experimental data. Most of published results present the overview of general phenomena of alcohol combustion process or it is mainly focused on the ability of alcohol use in conventional combustion systems. For example, Sarathy et al. [7] prepared a comprehensive review of fundamental combustion chemistry of alcohol fuels. They considered the studies of alcohol fuels in shock tubes, rapid compression machines, jet-stirred or flow reactors and also laminar flames. Three to eight carbon alcohols appear to have the boiling points near the mid-point of gasoline or diesel fuel range, so they can be easily blended with petroleum fuels with relatively high concentrations and with little impact on the fuel distillation curve. Due to high octane rating and thus lower propensity for ignition, alcohols allows the higher pressure operating conditions for SI engines without knocking phenomenon. Some alcohols can also improve the fuel economy: the four carbon alcohols tend to have the LHVs close to that of gasoline. The higher alcohols have less difficulty with the cold-start of the engine because of the lower specific latent heat of vaporization. Alcohols can also be successfully applied in modern LTC and DI SI engines in which the high sensitivity fuels can be of great importance. Authors focused on the chemical kinetics of alcohol fuels during combustion. There are many processes during the combustion, such as the reactions in the flame region, resulting in the heat release, reactions that control the ignition process, combustion pollution formation, which can take place when the temperature and the pressure rapidly changes. These processes can strongly depend on the chemical kinetics, ruled by the temperature, pressure, concentrations of reactants and products. Therefore the complex kinetic chemical reactivity models are also needed to understand the thermodynamic properties of alcohols as good energy fuels. By thermodynamic and reactivity properties, we mean the ability to describe the combustion phenomenon is a wide range, what implies great number of experiments due to the changes of the elementary reactions with the temperature, pressure and composition. For example, the reactions between the hydrogen atoms and the fuel molecules are dominant when there is the fuel reactivity. Many reactions are important only in certain ranges of the temperature and negligible in others during the thermodynamic process. The development of the alcohol combustion models requires the understanding of the basis of the hydrocarbon oxidation process in closed combustion systems like vessels, chambers, engines, turbines, etc. [7]. Li and others investigated the explosion characteristics of alcohol-air mixtures [8]. Explosion characteristics of five alcohol-air mixtures were investigated, including some of light alcohol fuels: ethanol, 1-butanol, 1-pentanol, 2-pentanol and 3-pentanol. They performed the combustion experiments under several different initial conditions: three temperature values, three pressure values and equivalence ratios (ϕ) between 0.8 and 1.8. The experimental data was composed of the constant volume cylinder vessel with the centrally located electrodes, data acquisition system and the inlet and exhaust system. Experiments showed that during the thermodynamic process (combustion dynamics), the fundamental combustion reactivity parameters, like peak explosion pressure (Pmax) is decreased with the temperature increase, while the other parameter, like the maximum rate of pressure rise (dP/dt)max varies pretty much and the time intervals between the ignition and peak explosion pressure decreased. The adiabatic flame temperature and the flame speed have the maximum values at the alcohol mixture equivalence ratio of 1.1, which can correspond to the peak values of the explosion pressures at the equivalence ratios of 1.0-1.2. The maximum rate of explosion pressure rise and the deflagration index KG are sensitive to the temperature changes as the key important parameters of combustion reactivity. The whole time of the combustion phenomenon increases with the decrease of temperature and the pressure increasing, while the flame speed decreases. Among all pentanol-air mixtures, 1-pentanol gives the largest flame speed and the highest adiabatic temperature an yields the higher Pmax and also (dP/dt)max and KG. Among the ethanol, butanol and pentanol mixtures with air, the ethanol gives the highest (dP/dt)max while 1-pentanol gives the lowest, but the difference is relatively small. Then the value of Pmax decreases monotonically in rich fuel mixtures of pentanol, butanol and ethanol. For lean mixtures the ethanol gives the lowest adiabatic temperature and the highest flame speed [8]. Weber and others focused on the auto-ignition of n-butanol [9]. The goal of this study was to provide the auto-ignition data, as another fundamental thermodynamic reactivity property, of n-butanol at elevated pressures and low temperatures. Auto-ignition delay measurements were performed in a rapid compression machine (RCM), which compresses mixed fuel and oxidizer to a given temperature and pressure (over 25-35 ms). The uncertainty in the compressed temperature is dependent of the initial conditions of the experiment. It was found that the ignition delay decreases monotonically with the increase of compressed temperature. Two-stage ignition was not noted. The total uncertainty in the compressed temperature was about 0.7-1.7%. The reactivity increase due to increasing equivalence ratio (due to increasing fuel mole fraction) was noted. The uncertainty is estimated at about 5 ms in the ignition delay time. Results demonstrate that the higher pressure of the experiment the shorter ignition delays are. Th fuel and oxygen concentrations can decrease the ignition delay time. Zhu and others investigated the ignition delay times of 1-butanol using two methods: conventional one and the constrained-reaction-volume strategy [11]. The 1-butanol-O2 -N2 ignition delay times was measured, at various temperatures, pressures and equivalence ratios, behind the shock waves. The well-known Stanford University high-purity and high-pressure shock tube (HPST) was used. Pressure during ignition was recorded by five transducers and time-interval counters measured the incident shock speed. They observed that while using the conventional filling method, at the highest temperature (1014 K), the pressure trace was flat, and then rapidly and smoothly rose exponentially to ignition (sharp or strong ignition). At the lowest temperature (792 K), the pressure trace was flat only for 2 ms and then slowly rose to ignition (mild ignition). There were multiple rumps or humps at the pre-ignition pressure rise, that can be regarded as a mild-to-strong transition ignition mode. Such pre-ignition effects are pressure dependent. Experiments performed with use of the CRV showed, that the energy release during chemical induction dissipate instead of creating pressure ramp - absence of any detonation like pressure ringing. The ignition delay time was longer than in conventional-filling experiment. Remote ignition did not occur. CRV approach enables the unambiguous, quantitative modelling of ignition delay time, using P, H gas-dynamic constraints. Conventional-filling experiments with pre-ignition perturbations are difficult to interpret and it is uncertain which gas-dynamic model to use in simulations. CRV experiment concept resolved this problem. There are more confident comparisons of data with simulations from existing 1-butanol detailed reaction mechanism possible. This literature review indicates clearly that there is a huge need for experimental and numerical data on thermodynamic and reactivity properties of light alcohols as the potential for energy carrier and also mitigation strategies and technologies in case of accidental release of alcohol vapours, when mixing with air, can provide serious hazards to people, environment and the infrastructure in form of fires, explosions. Knowledge of thermodynamic and reactivity data of light alcohol fuels can support the energy market in building proper strategies how to use such fuels in good and safe technologies in the process industries. Furthermore, based on the literature review it is now clear that there is also a huge gap of knowledge of the thermodynamic reactivity data for the combustion process of very low equivalence ratios of alcohol-air mixtures (e.g. ϕ < 0,8). These kind of mixtures can play an important role as the addition to traditional fuels to improve the combustion process and make the positive influence on the environmental pollution.
2. Experimental study
All experiments were conducted in the 20-liter combustion testing vessel shown in Figure 1. The vessel enables us to investigate the deflagration mode of combustion, often call the explosion phenomenon as well as associated explosion parameters of alcohol-air mixtures, including explosion pressure (Pex), maximum rate of explosion pressure rise (dp/dt)max or explosion delay time (tdel). It was equipped with ignition system including exploding wire, pressure measurement system comprising the pressure sensor and the pressure signal recording system as well as temperature measurement system with two thermocouples: one at the bottom plate and one at the vessel wall. Also it was equipped with the vacuum pump, a device for magnetic mixing mounted near the top of the vessel and safety measures to prevent premature ignition during the preparation of combustible mixtures.
Using our combustion vessel, we were ready to perform deflagration (explosion) experiments even at initial temperatures of up to 393.15 K. At this temperature condition the combustion vessel is capable of withstanding the maximum explosion pressure of 16 bar (the design pressure is 20 bar). We recorded experimental data at a sample rate of 150 kHz. The measurement range of the dynamic pressure sensor was 13.8 bar. Pressure and temperature data are recorded and processed by an ANKO dedicated software, involving data acquisition. The combustion vessel was equipped with an injection device to permit liquid sample into the testing vessel. Measurements were carried out with initially quiescent combustible mixtures at initial conditions of 323.15 K and 1 bar. The air was used as the oxidizer and the light alcohol samples tested were following:
- methanol, ethanol, n-propanol and iso-propanol at ϕ = 0.3 – 0.7;
- mixtures of alcohols and water, such that the volumetric content of the latter ranged from 10 and 60 vol%.
Liquid light alcohol samples were admitted into the combustion vessel mixed with air, including ϕ = 0.3 – 0.7 as determined at experimental conditions. Specific details on the samples, their composition and volumetric water content are given in Table A.1. Each sample was tested at least three times. The procedure to create initially quiescent combustible mixtures in the combustion vessel was strongly followed. First, liquid sample volumes of 4 cm3 (ϕ = 0.3), 6 cm3 (ϕ = 0.5) and 8 cm3 (ϕ = 0.7), including alcohol-air mixtures as well as alcohol-water-air mixtures were rendered at initial experimental conditions. Next, the combustion vessel was heated up to a temperature of 323.15 K and evacuated to such an extent that the pressure became less than 1 millibar. Also, the dedicated injection device was applied to provide the liquid samples of light alcohols going at the bottom of the combustion vessel. The heating plate was mounted and kept at temperature of 333.15 K. The magnetic mixing device was also deployed for at least 3 minutes to allow the liquid sample of light alcohols to evaporate. The heating plate’s temperature was decreased to 323.15 K and the air was slowly administered to bring the pressure in the testing vessel to a pressure of 1 bar. During this step, the magnetic stirring device was continuously used to ensure thermal homogeneity throughout the combustion vessel and assist mixing within the combustible mixtures. After disengaging the magnetic stirrer, a time span of at least 5 minutes was permitted to elapse prior to ignition. A spark was deployed to ignite the combustible mixtures to deflagration mode of combustion process (explosion phenomenon). The pressure development during the combustion reaction dynamics was measured by means of a piezoelectric pressure transducer while the pressure signal was recorded at a sample rate of 150 kHz by the data acquisition system. The combustion vessel was properly cleaned and thoroughly flushed with air after each experiment. All experimental combustion dynamics curves thus experimentally obtained are presented in Figures 2. Nevertheless, there are also some exemplary plots of thermodynamic reactivity parameters during the experiments of tested alcohols, including the explosion pressure (Pex), the maximum rate of pressure rate (dP/dt)max and the explosion delay time (tdel) and also the influence of water addition on thermodynamic reactivity of alcohol-air mixtures. Below figures were created based on the arithmetic mean of the values measured during the tests of each volume sample (3 times). Our plots show the dynamics of the deflagration mode of combustion phenomenon in closed vessel, indicating some factors that can strongly influence the thermodynamic reactivity of alcohol-air mixtures as well as with water addition.
Figure 2.
Pressure increase of 6 cm3 of methanol and 20% water addition (ϕ = 0.5).
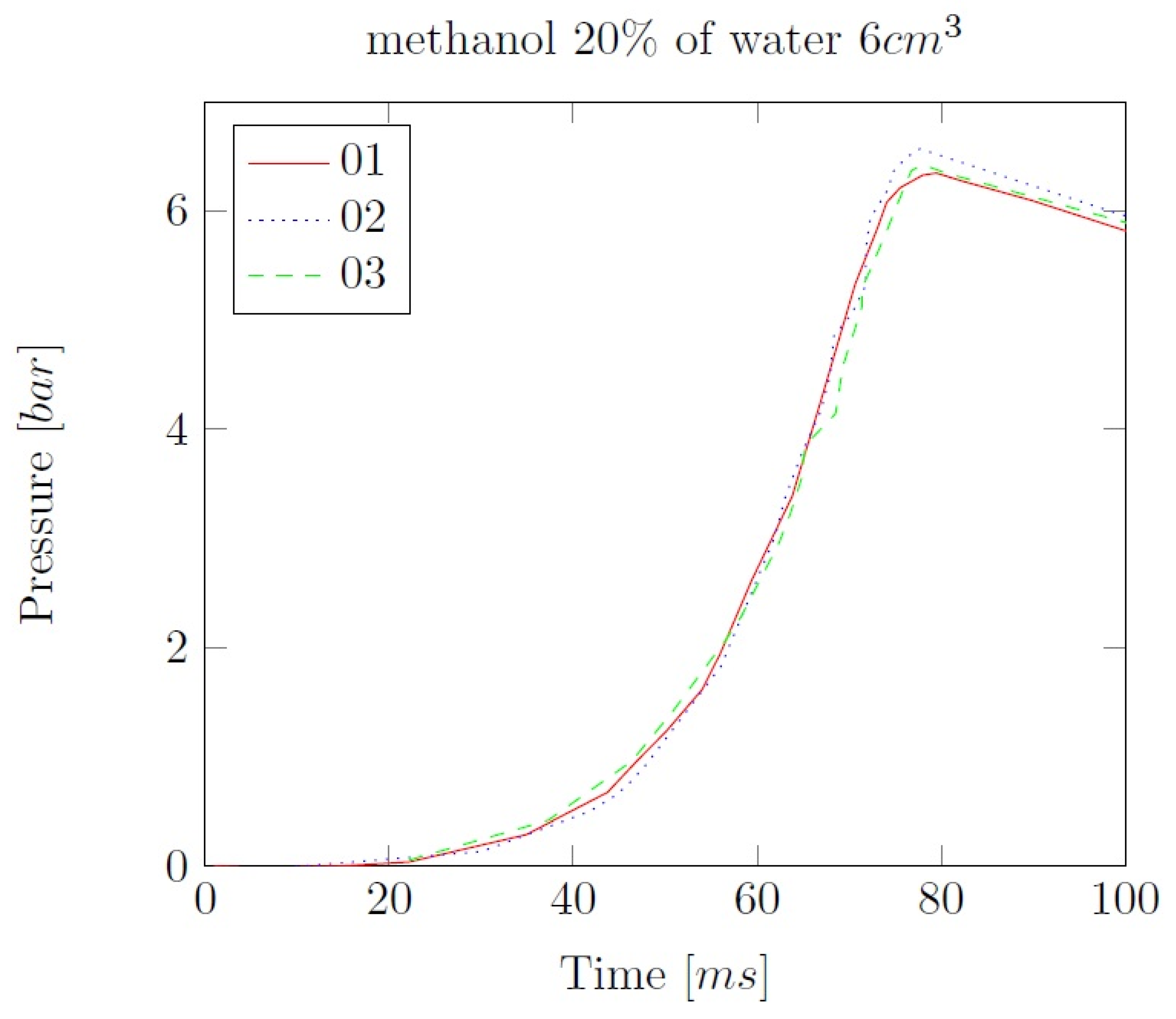
Methanol-water mixtures appeared to be combustible in the range between 0% to 60% of water addition, except for the 4 cm3 samples. The exemplary profile of the pressure during the experiment is showed in the Figure 2. Usually, the pressure runs during the test of certain sample were similar, with several exceptions during the test of extreme amounts of water addition and volumes of alcohol samples. The explosion pressure of 6.88 bar was reached during the test of 4 cm3 (ϕ = 0.3) of pure methanol-air mixture. The maximum rate of pressure rise of 365.29 bar/sec was observed during the same reaction, what indicates that this is the most reactive one between all methanol-water mixtures. The reaction of 4 cm3 of pure alcohol-air mixture appeared to be the most reactive, where the delay time was about 69 ms. The dependence of the reaction delay times on the water addition in methanol-air mixtures is illustrated in the Figure 3.
The explosion delay time values of methanol-air mixtures (ϕ = 0.3) increased with the water concentration increase. For the 6 cm3 samples (ϕ = 0.5), tdel reached minimum in 30% of water addition in the mixture and for the 8 cm3 samples (ϕ = 0.7), tdel is minimum, when there is 20% of water concentration in the mixture. Ethanol-water mixtures appeared to be combustible in the range between 0% to 60% of water addition. The exemplary profiles of the combustion pressure during the experiment is showed in the Figure 4.
Usually, the pressure profiles during the test of certain ethanol sample were similar, except several exceptions during the test of extreme amounts of water addition and volumes of samples. The results obtained during the tests of 8 cm3 of pure alcohol (ϕ = 0.7) was random and only one test qualified as the explosion phenomenon (with the overpressure more than 0.5 bar). Thus, the estimation of explosion delay time or Pex and (dP/dt)max is meaningless. The explosion pressure of 6.70 bar was reached during the test of 4 cm3 of pure ethanol (ϕ = 0.3). The maximum rate of pressure rise of about 260.05 bar/sec was for the same mixture but with 10% water content, which indicates that it was the most reactive one between all ethanol-water mixtures. The reaction of 4 cm3 of 10%-water mixture appeared to be the most reactive one, with tdel about 79 ms.
Figure 5.
Influence of the water addition on the explosion delay time of ethanol-air mixtures.
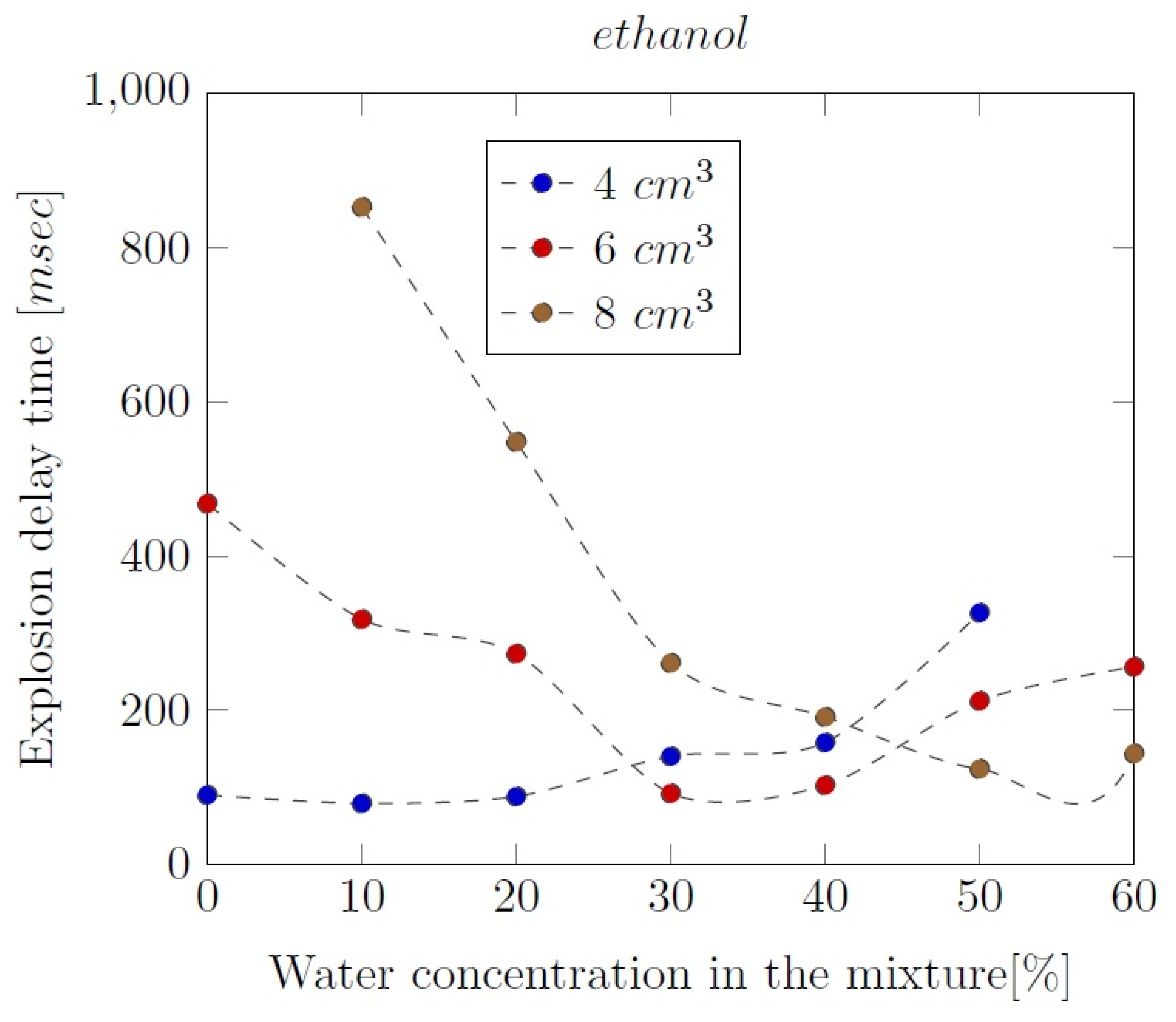
The dependence of the reaction delay times on the water addition of ethanol-air mixtures is shown in Figure 5. In case of the 4 cm3 samples of n-propanol (ϕ = 0.3), the reaction delay time is increasing with the increase of the water concentration in sample. For the 6 cm3 of n-propanol samples (ϕ = 0.5), the tdel reached minimum at 30-40% of water concentration. Explosion delay time decreases with the increase of water concentration in 8 cm3 of n-propanol samples (ϕ = 0.7). The exemplary profiles of the pressure is showed in the Figure 6.
Usually, the pressure profiles during the test of certain sample were similar, except several tests with the extreme amounts of water addition and volumes of samples. The explosion pressure of n-propanol-air mixtures was 6.57 bar, including the equivalence ratio ϕ = 0.3. The maximum rate of pressure rise of 260 bar/sec was observed for ϕ = 0,3. It indicates that this mixture was the most reactive one of all n-propanol-water mixtures tested during this study.
Figure 7.
Influence of the water addition on the explosion delay time of n-propanol-air mixtures.
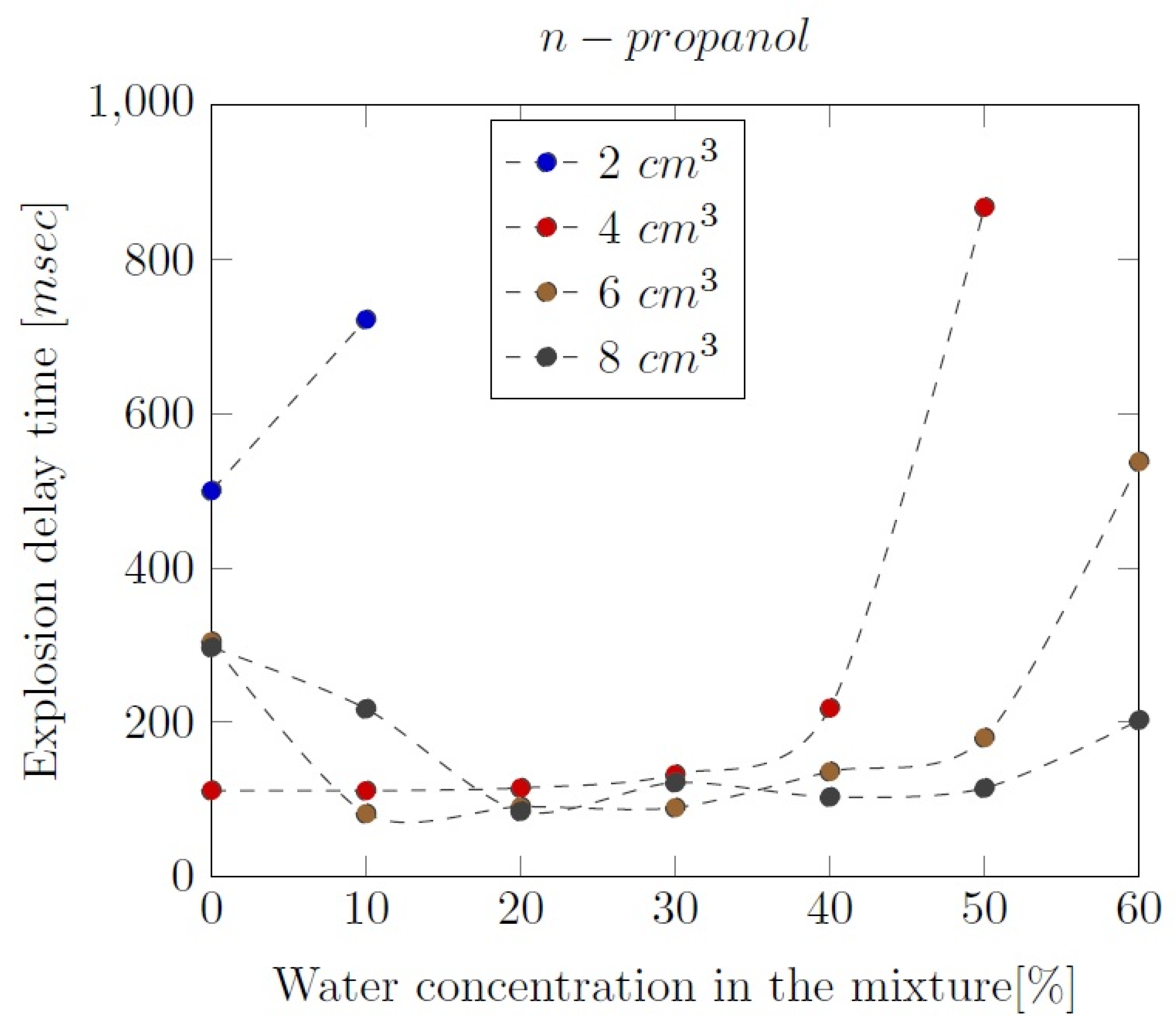
The reaction of 6 cm3 of 10%-water mixture appeared as the most reactive one, the delay time was about 81 ms. The dependence of the reaction delay times on the water addition in the samples is shown in Figure 7. In case of lower volumes of iso-propanol-air mixtures, the tdel increased with the water concentration increasing. In case of the higher volumes of iso-propanol-air mixtures, tdel initially decreased, then remained constant and finally decreasing again, with 40% of water addition. The exemplary profiles of the pressure for iso-propanol-air mixtures is showed in the Figure 8.
The maximum explosion pressure was recorded as 5.87 bar for 4 cm3 of 10%-water mixture. The maximum rate of pressure rise was observed as 248 bar/sec for ϕ = 0.3. It indicates that this iso-propanol-air mixture was the most reactive one of all n-propanol mixtures. The reaction of 4 cm3 and 10%-water mixture appeared to be the most reactive one, with the tdel about 88 ms. The dependence of the explosion delay time on the water addition is shown in Figure 9. In case of lower volumes of iso-propanol-mixtures, the tdel increased with the water concentration increasing. For higher volumes, the tdel initially decreased, then reached minimum at 30% of water addition and then finally increased.
Every profiles of explosion pressure and the maximum rate of pressure rise were prepared as the arithmetic average of the experimental results for each alcohol-air mixtures. From Figure 10 it can be observed that for methanol-air mixtures with ϕ = 0.3, the highest explosion pressure was recorded.
Experimental results of the Pex of all mixtures with ϕ = 0.3 are similar, except the explosion pressure of iso-propanol, which is slightly lower than the other results. In general, the values of the Pex of ethanol-water mixtures are the highest for all variants of water concentration. The Pex of the methanol-air mixtures decrease the fastest way with the water addition increasing. In case of all propanol-water mixtures, the results of the explosion pressure of n-propanol are slightly higher than those of the iso-propanol. This can be found as the fundamental reason of thermodynamic reactivity difference between propanol-air mixtures. The results of maximum rate of pressure rise for the mixtures with ϕ = 0.3 are shown on Figure 11. The highest (dP/dt)max values were reached during the thermodynamic reaction of methanol-air mixture with no water. However, the (dP/dt)max of the methanol-water mixtures is very sensitive to the water addition and sharply decreases with the water concentration increase. For the results of ethanol-air mixtures, the (dP/dt)max values appear to be the highest among the maximum rates of pressure rise of all alcohol-air mixtures, with ϕ = 0.3.
On Figure 12 there is a comparison of tdel results. Between 0-20% of water content, the tdel values are similar for all alcohol-air mixtures. Then the tdel starts to increase. The smallest values are obtained for the ethanol-water mixtures.
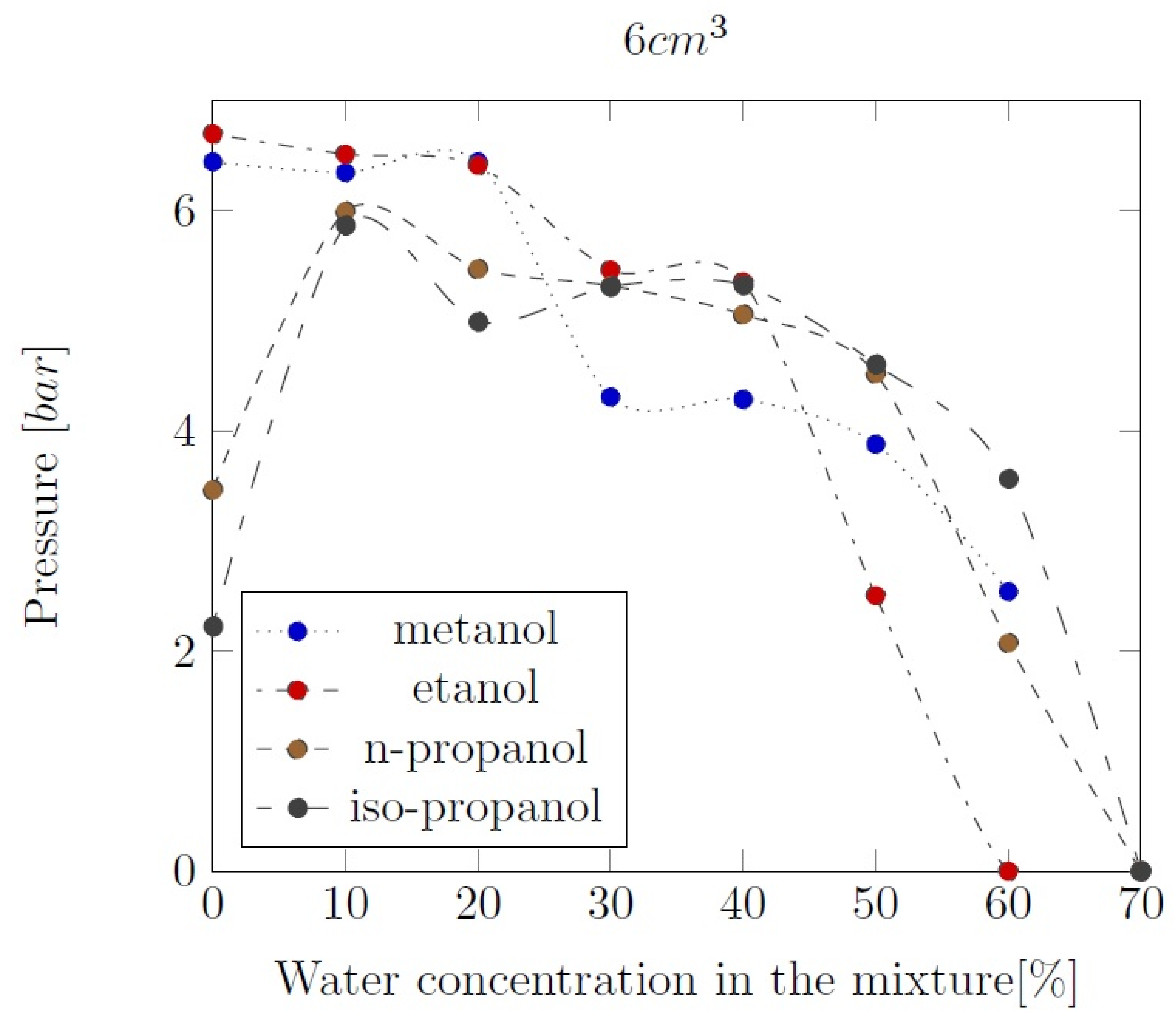
In Figure 13 we present the results of the explosion pressure of all alcohol-air mixtures, with ϕ = 0.5. It shows that the explosion pressure peak values of methanol and ethanol decrease with the water addition increase. For n-propanol and iso-propanol with no water, the peak values of Pex are the lowest ones. Between 0-30% of water addition, the ethanol-air mixtures reach the highest Pex for all tested alcohol-air mixtures. Experimental results also clearly show that for alcohol-air mixtures with the 50-60% of water addition, the highest Pex values appear for the iso-propanol-air mixtures and the lowest for the ethanol-air mixtures.
Figure 13.
Influence of water addition on the explosion pressure of all alcohol-air mixtures (ϕ = 0.5).
Figure 13.
Influence of water addition on the explosion pressure of all alcohol-air mixtures (ϕ = 0.5).
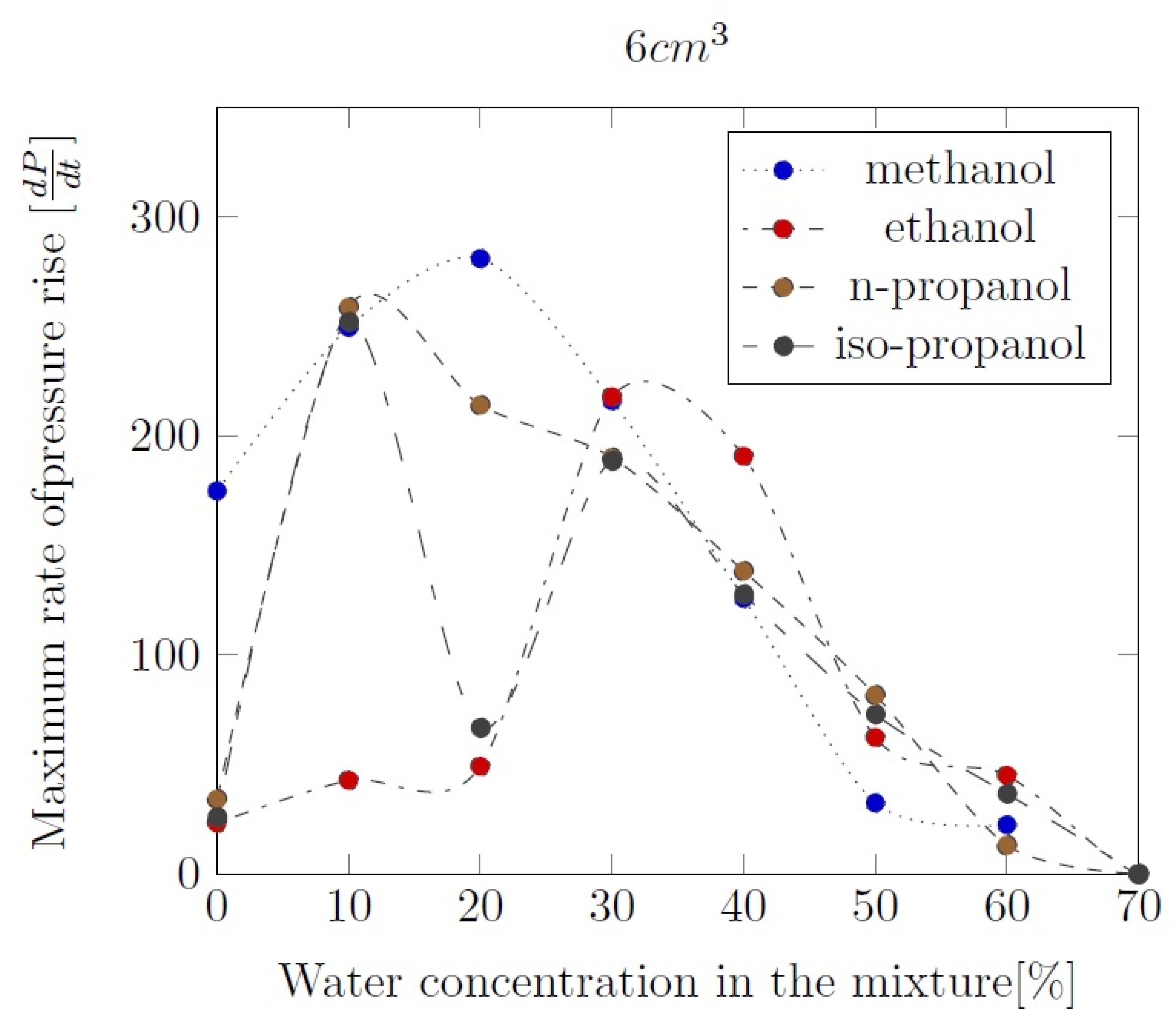
In Figure 14 we present the results of the maximum rate of explosion pressure rise for all alcohol-air mixtures with ϕ = 0.5. Between 0-30% of the water content in the mixtures, the (dP/dt)max of methanol-air mixtures are the highest and the lowest values are for the ethanol-air mixtures. But between 30-70% of the water content the above relations are opposite. This is also very important observation for the thermodynamic reactivity of these alcohols, where the number of C and H in the chemical structure is of great importance. The unexpected low value of (dP/dt)max of the 20% of the water content in iso-propanol-air mixtures can be probably assumed as a random error during the experiments, because these results do not fit to any thermo-physical or chemical reactivity theories. For all alcohol-air mixtures, the water addition also increases the maximum rate of explosion pressure rise.
Figure 14.
Influence of the water addition on (dP/dt)max for all alcohol-air mixtures (ϕ = 0.5).
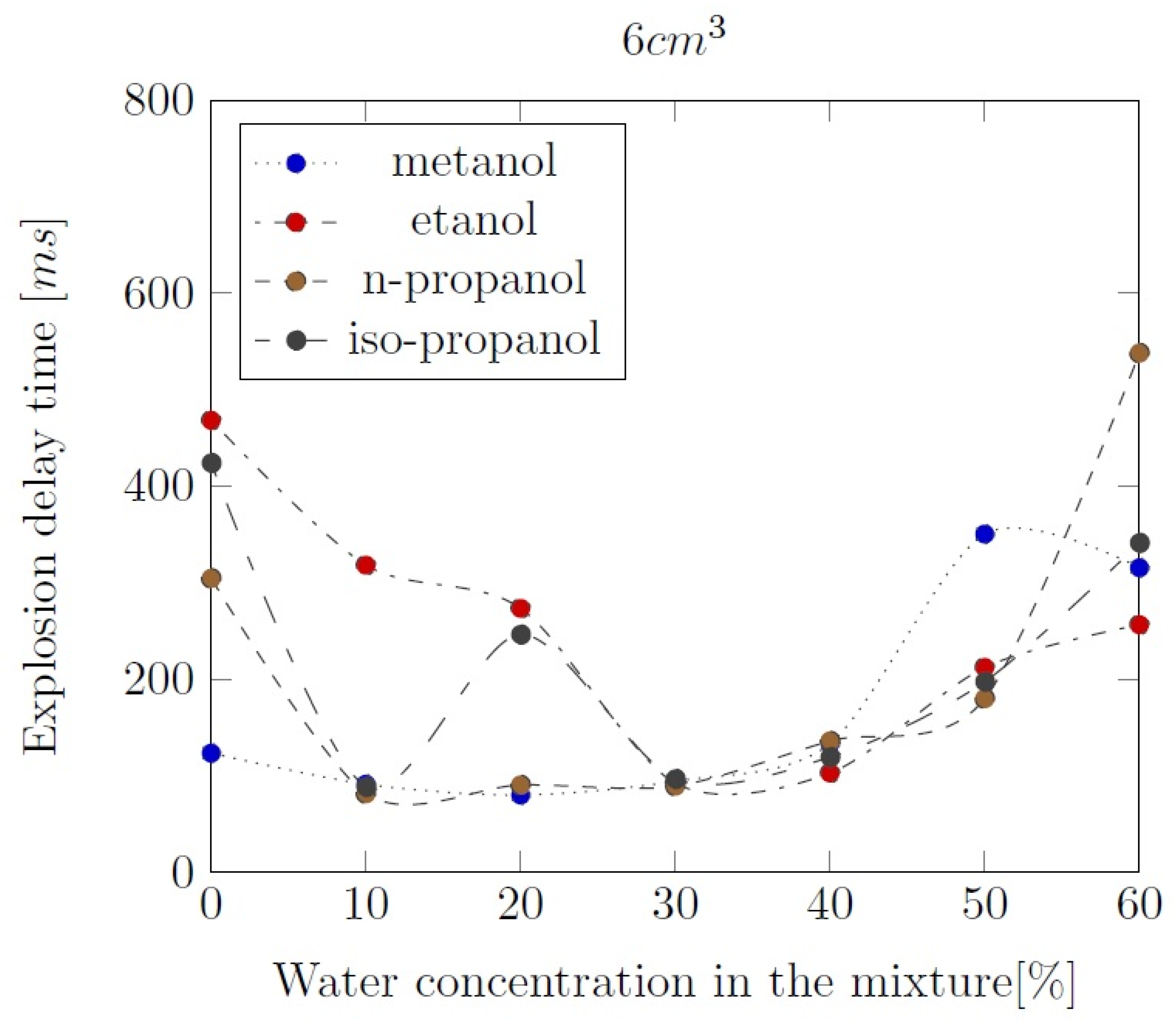
In Figure 15 we present the results of explosion delay time for all tested mixtures. The lowest values appear at methanol-water mixtures, especially during the tests of alcohol-rich samples. When it is 30% of water addition in mixtures of equivalence ratio of 0.5, values of explosion delay time for all alcohols are extremely close to each other.
Figure 15.
Influence of the water addition on explosion delay time for all alcohol-air mixtures (ϕ = 0.5).
Figure 15.
Influence of the water addition on explosion delay time for all alcohol-air mixtures (ϕ = 0.5).
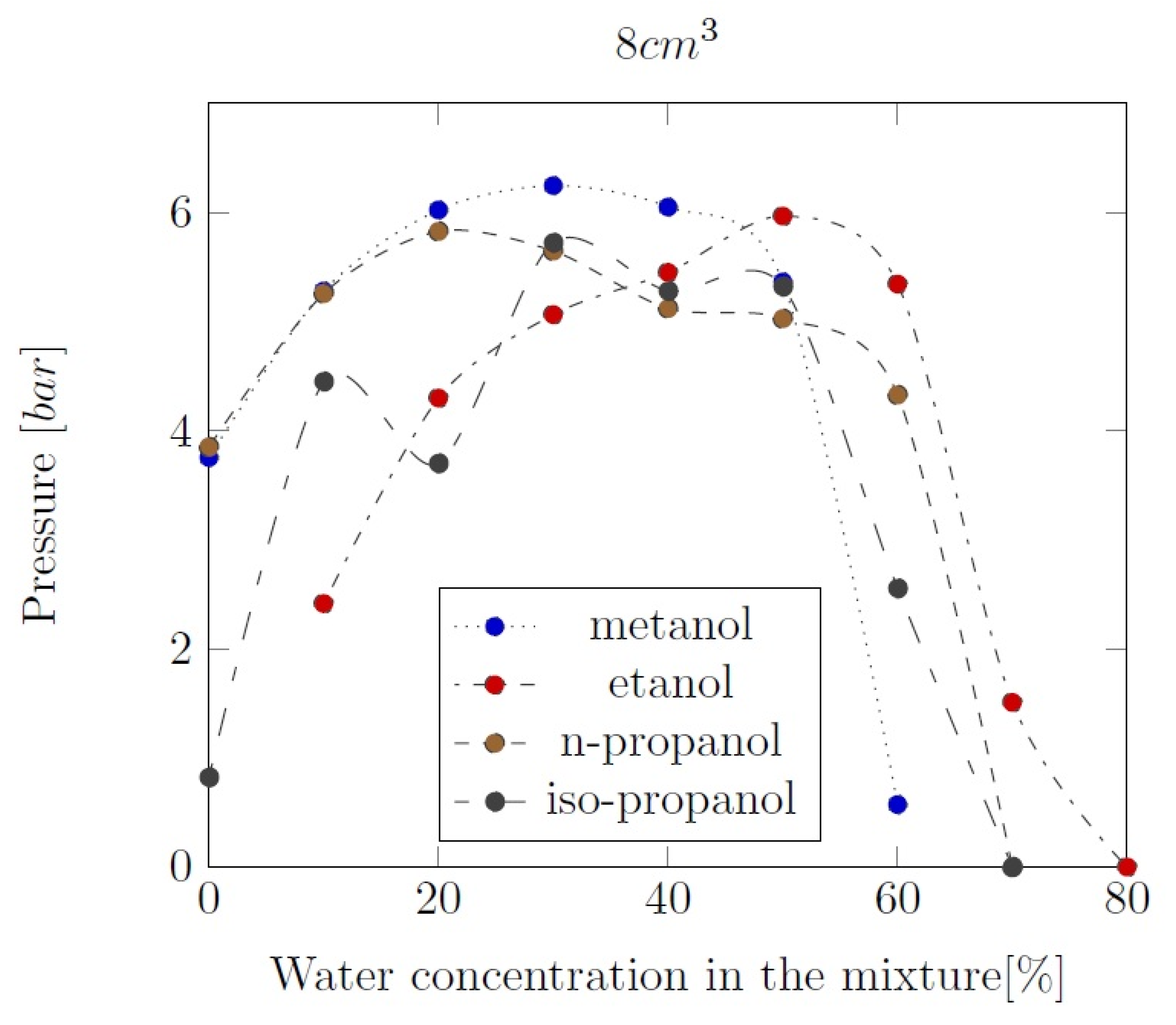
The values of explosion pressure for 8 cm3 samples (ϕ = 0.7) are shown in Figure 16. Ethanol has the widest range of flammability, as even the 70% of water addition samples have thermodynamically reacted as the deflagration mode of combustion. Up to 40% of water addition, the methanol-air mixtures reach the highest explosion pressure, but then it started to decrease. For ethanol-air mixtures, the situation is quite opposite. For all alcohol-air mixtures with ϕ = 0.7, the water addition causes the explosion pressure increase relatively to the same as Pex of the pure substance.
Figure 16.
Influence of the water addition on explosion pressure for all alcohol-air mixtures (ϕ = 0.7).
Figure 16.
Influence of the water addition on explosion pressure for all alcohol-air mixtures (ϕ = 0.7).
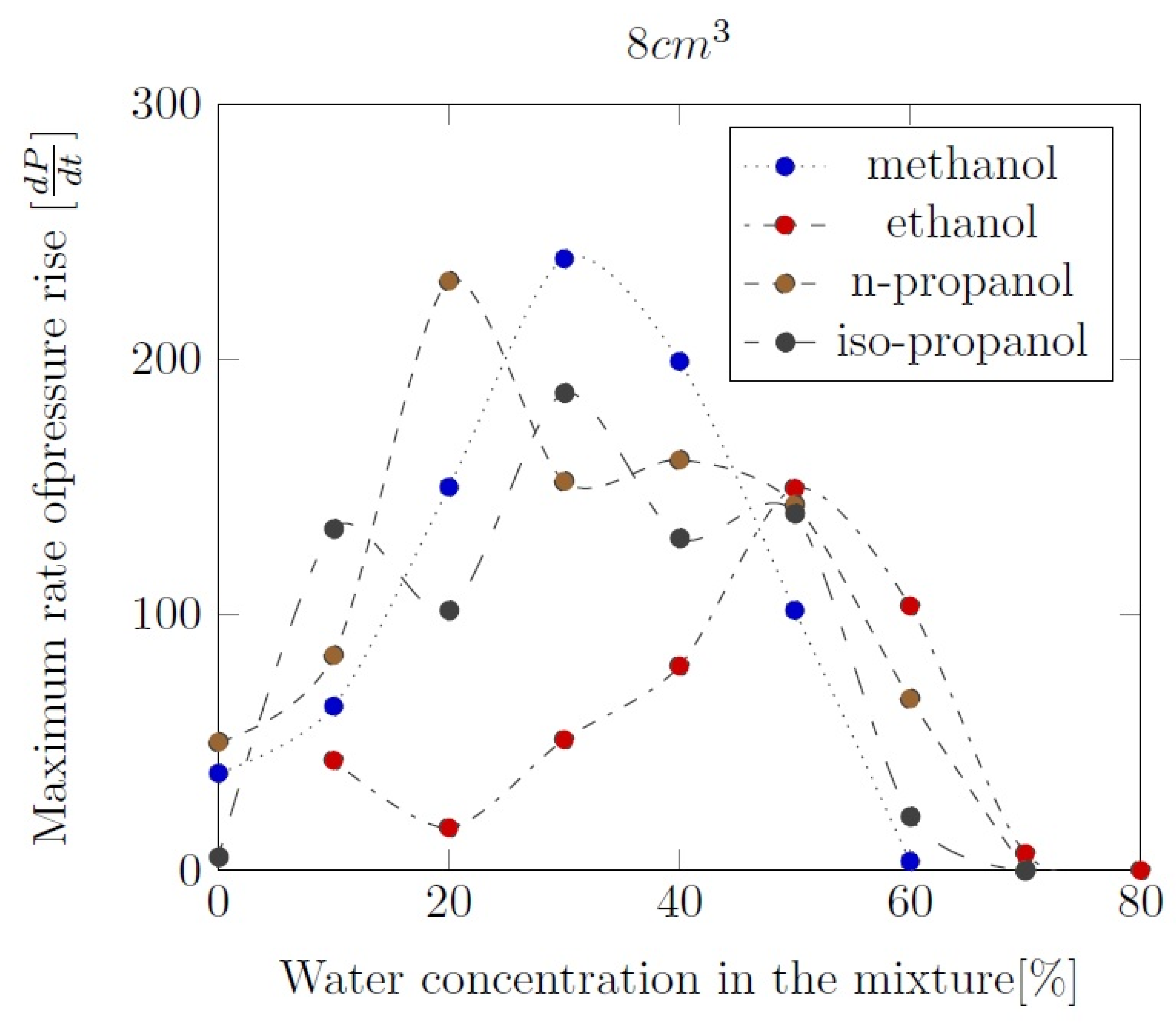
In Figure 17 we observe the certain dispersion of the values of the maximum rate of pressure rise during the combustion of the 8 cm3 samples (ϕ = 0.7). The (dP/dt)max values are mostly the lowest for the ethanol-water mixtures. The (dP/dt)max of the methanol-water mixtures monotonically increases with the water addition increase until the 30% of water concentration in the mixture and then monotonically decreases. It is much more complicated in case of the n-propanol and iso-propanol samples. In general for all alcohol-air mixtures, we observed the increase of the (dP/dt)max values, together with the water addition increase.
For the tdel results for all alcohol-mixtures at ϕ = 0.7, all of these values are quite similar to each other when the water addition reaches up to 50%. In case of alcohol-rich mixtures tdel is the shortest for the n-propanol mixtures. The idea of our experiments was to investigate the thermodynamic reactivity of alcohol-air mixtures, including water addition. In order to take a deeper look on the chemical composition of alcohol-water-air mixtures, it is needed to perform some kind of reactivity calculations based on the same initial conditions. That is why based on our experimental results we also prepared the thermodynamic analysis of the liquid phase composition of alcohol water mixtures as well as the density analysis of the liquid samples, the excess volume of liquid mixtures and also the calculations of vapor pressures of the pure liquids, to observe more details in the thermodynamic reactivity of light alcohol mixtures with air and water.
Figure 17.
Influence of water addition on maximum rate of pressure rise (dP/dt)max for all alcohol-air mixtures (ϕ = 0.7).
Figure 17.
Influence of water addition on maximum rate of pressure rise (dP/dt)max for all alcohol-air mixtures (ϕ = 0.7).
3. Liquid phase composition of the alcohol-water mixtures
As stated before, liquid samples were created by mixing the alcohols with water at thermodynamic conditions of 298.15 K and 1 bar. Relevant physical properties of the pure substances at these conditions are given in Table 1. These properties were used to compute the sample features in Table A.1: the volume of the alcohol –H2O admixtures , the volume ratio of H2O to alcohol prior to mixing q, the alcohol mass fraction in the liquid and gaseous phases (ȳAlc and ỹAlc), the H2O mass fraction in the liquid and gaseous phases ( and ), the alcohol mole fraction in the liquid and gaseous phases (Alc and Alc), and, the H2O mole fraction in the liquid and gaseous phases (and ).

Table 1.
Molecular mass (M) density (ρ), critical temperature (Tc), critical pressure (Pc), critical volume (Vc), acentric factor (ω),boiling point (Tb) and vapor pressure (PSat) of CH3OH, C2H5OH, n-C3H7OH, i-C3H7OH, H2O and air [1, 2].
Table 1.
Molecular mass (M) density (ρ), critical temperature (Tc), critical pressure (Pc), critical volume (Vc), acentric factor (ω),boiling point (Tb) and vapor pressure (PSat) of CH3OH, C2H5OH, n-C3H7OH, i-C3H7OH, H2O and air [1, 2].
| Substance | M (kg mol−1) | ρ(†) (kg m−3) |
Tc(⊕) (K) |
Pc(⊕) (bar) |
Vc(⊕) (cm3 mol−1) |
ω (-) |
Tb(‡) (K) |
PSat (∗) (bar) |
Fl. lim.(y) (vol%) |
|---|---|---|---|---|---|---|---|---|---|
| CH3OH | 32.04·10−3 | 787.2 | 512.16 | 80.92 | 117.88 | 0.565 | 338.15 | 0.17 | 6 – 37 |
| C2H5OH | 46.07·10−3 | 787.3 | 513.9 | 61.37 | 167.10 | 0.649 | 351.15 | 0.079 | 3 – 19 |
| n-C3H7OH | 60.10·10−3 | 802.0 | 536.8 | 51.70 | 218.41 | 0.629 | 370.15 | 0.028 | 2 – 14 |
| i-C3H7OH | 60.10·10−3 | 782.7 | 508.3 | 47.62 | 220.10 | 0.665 | 355.15 | 0.061 | 2 – 12 |
| H2O | 18.015·10−3 | (a)997.05 | 647.10 | 220.64 | 56.02 | 0.344 | (a)373.13 | (a)0.0317 | |
| Air | (b)28.964·10−3 | 1.161 | (c)132.63 | (c)37.858 | 92.35 |
(†)At 298.15 K and 1 bar. (‡)At 1 bar. (*) At 298.15 K. (⊕)These quantities permit the calculation of the critical density ρc = MPc/RTc and the critical compressibility Zc = PcVc/RTc. (y)Flammability limits in air at 298.15 K and 1 bar as tabulated in Ref. [1]. Notice that the upper flammability limit in air supersedes the vapor pressure of the pure substances at these conditions. (a)From Ref. [3]. (b)From Ref. [4]. (c)From Ref. [5].
- ➢
- Equations (1) to (3) become invalid for calculating mixture properties such as the liquid sample density and specific volume. Instead, equation (5) should be applied, provided that VE is known on an a priori basis from experiments or theoretical predictions.
- ➢
- The partial molar volumes of the species in the mixture are no longer equal to the molar volumes of the pure species: ≠ VAlc and ≠ . Furthermore, for real mixtures and become nonlinear functions of and . Equation (12) further on, establishes a thermodynamic relationship between the partial molar volume of a species, , and the total volume V of a mixture as a function of varying composition. Hence, although equation (4) remains valid for both ideal and real mixtures, its application to the latter requires precise a priori knowledge of the nonlinear dependence of ), i.e. ), in equation (12), on the mixture composition.
For a mixture comprising m components, thermodynamics [18, 20] provides a formal mathematical connection between an extensive total property Mt (= ntM) and the corresponding partial molar properties i . That is:
where M and i are intensive properties. The molar property M and the partial molar property i are functions of the intensive properties P, T, and m mole fractions = ni/nt. A practical relationship between i, M and may be obtained by expanding equation (6) into
and using = 1 so that
The fact that there are only m-1 independent mole fractions (because the sum up to unity) permits the total differential of M to be expressed as
where the summation over k excludes i and the index l indicates that all mole fractions other than xi and xk are held constant. Combined with the constant nj≠i restriction, division by dni yields:
for k ≠ i. Given that xk = nk/nt and hence
combination of equations (8), (10) and (11) finally results into
As stated earlier, application equation (4) to the sample volume requires accurate a priori knowledge of the )-term.
Since the samples were created by mixing the pure liquids on a volumetric basis, it would be convenient to characterize them as the volumetric water to alcohol ratio, q, for further thermodynamic and reactivity studies. But the nonlinear dependence of VE on the binary mixture composition (Figure 18) renders q arbitrary. Nonetheless the volumetric ratio q is kept in the second column of Table A.1 to characterize the samples for the sake of bookkeeping. Unequivocal characterization of the samples requires aforementioned quantities to be known.
Using the physical properties in Table 1 it is straightforward to compute the alcohol and water mass fractions {ȳAlc, } and mole fractions {Alc, } in the liquid phase when q, VAlc and/or prior to mixing are known. These liquid phase mass and mole fractions can be obtained via
When q is known beforehand, equations (16) to (18) permit the calculation of ȳAlc, , Alc and from the physical properties listed in Table 1 only. The values thus obtained are presented in the third, fourth, seventh and eighth column of Table A.1. The liquid phase mass fractions {ȳAlc, } and mole fractions {Alc, } are interchangeable via
The same interchangeability applies equally well to the vapor phase mass and mole fractions through the substitutions {ȳAlc ← ỹAlc ← ; Alc ← Alc; ← } into equations (19) to (22).
The binary alcohol-water mixtures were administered into the combustion vessel on a volumetric basis at initial conditions of 298.15 K and 1 bar. While the sample volumes are known, the density of these liquid mixtures, ρS is affected by the excess volume VE. To determine the species mass and mole fractions in the liquid and vapor phase there are two possibilities:
- ➢
- The application of models that predict ρS directly.
- ➢
- The deployment of equation (5) in conjunction with models that predict VE.
4. Density of the liquid samples and the excess volume of liquid mixtures
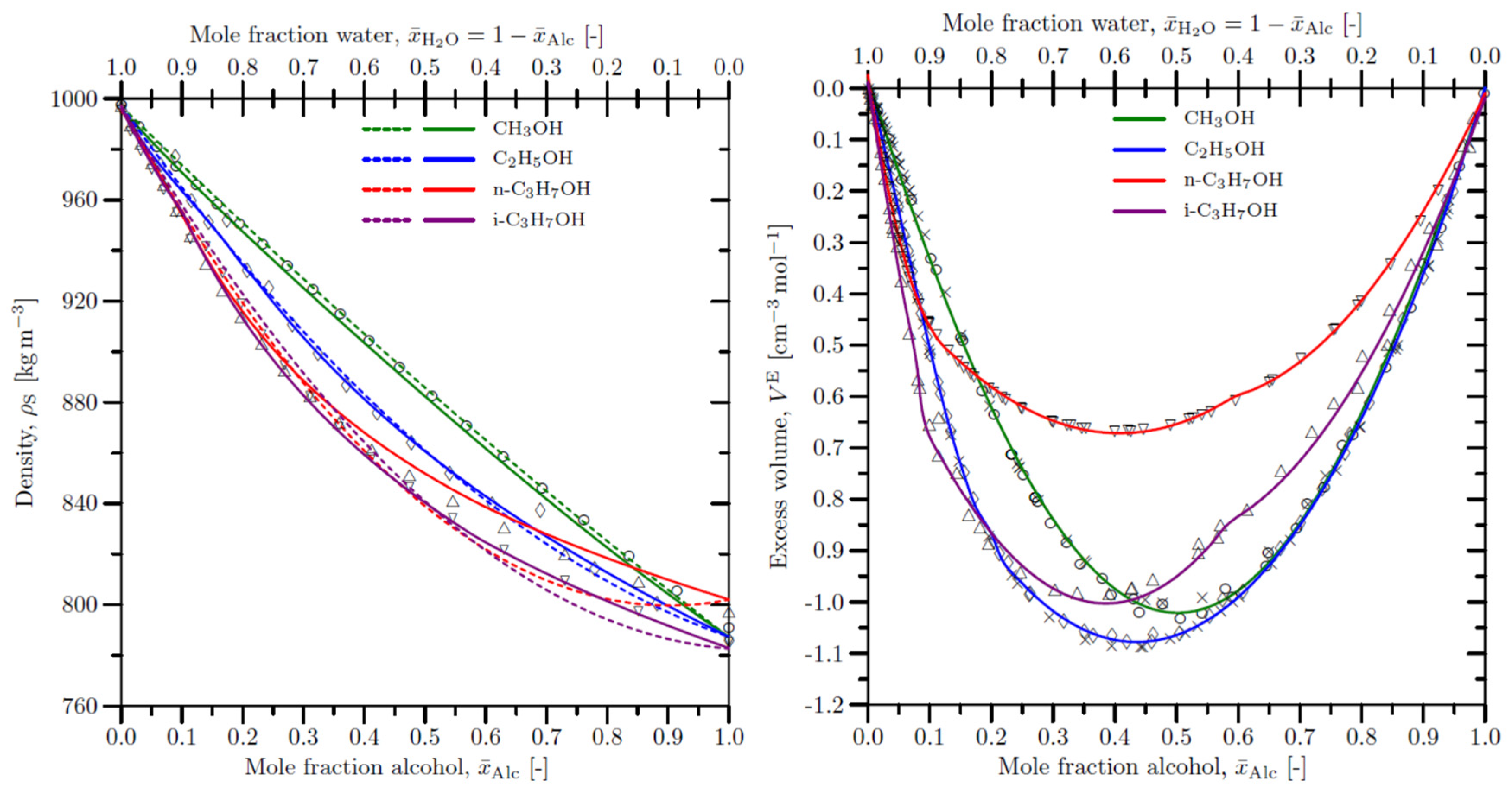
It is fortuitous that tabulations of experimental liquid density and excess volume data exist for the alcohol-water mixtures studied in this work (see Figure 18).
Table 2.
Polynomials for interpolation between the experimental ρS and VE data in Figure 2. The polynomial coefficients were determined by the Levenberg-Marquardt method [21,22,23].
| Polynomial: VE = ρAlc Alc + ρH2O(1 − Alc) + a0 Alc(1 − Alc) (13) | |||||
| Mixture | Range | a0 | |||
| CH3OH – H2O | 0.0 ≤ Alc ≤ 1.0 | -25.2·100 | |||
| C2H5OH – H2O | 0.0 ≤ Alc ≤ 1.0 | -124.7·100 | |||
| n-C3H7OH – H2O | 0.0 ≤ Alc ≤ 1.0 | -242.1·100 | |||
| i-C3H7OH – H2O | 0.0 ≤ Alc ≤ 1.0 | -19.6·101 | |||
| Polynomial: VE = a0 Alc + a1(1 − Alc) + a2 Alc(1 − Alc) + a3 2 (1 − Alc)2 (14)Alc | |||||
| Mixture | Range | a0 | a1 | a2 | a3 |
| CH3OH – H2O | 0.0 ≤ Alc ≤ 1.0 | 0.3·10−3 | 17.0·10−3 | -370.1·10−2 | -16.7·10−1 |
| C2H5OH – H2O | 0.0 ≤ Alc < 0.2 0.2 ≤ Alc ≤ 1.0 |
25.2·100 1.3·10−3 |
8.0·10−3 -41.6·10−2 |
-29.5·100 -36.3·10−1 |
-46.6·100 8.1·10−1 |
| n-C3H7OH – H2O | 0.0 ≤ Alc < 0.1 0.1 ≤ Alc < 0.6 0.6 ≤ Alc ≤ 1.0 |
20.7·100 5.9·10−2 -10.1·10−3 |
25.6·10−3 -324.8·10−3 16.0·10−2 |
-28.4·100 -208.3·10−2 -27.3·10−1 |
0.0 0.0 0.0 |
| i-C3H7OH – H2O | 0.0 ≤ Alc < 0.1 0.1 ≤ Alc < 0.6 0.6 ≤ Alc ≤ 1.0 |
2.4·100 5.0·10−1 -1.9·10−2 |
12.3·10−3 -41.1·10−2 -1.4·10−1 |
-10.2·100 -39.8·10−1 -31.9·10−1 |
0.0 0.0 0.0 |
Since these experimental data are available for discrete values of the mixture composition, it would be helpful to have a mathematical relationship to reconstruct the liquid densities of the samples studied in this paper. Although various models have been proposed to predict the density of pure liquids and liquid mixtures directly [18, 24–28], their deployment as a correlation for interpolating experimental data turns out to be laborious, prone to curve fitting inaccuracies and susceptible to polynomial oscillations. For this reason it was attempted to deploy a polynomial of the form ρS = a0Alc + a1(1 – Alc) + a2Alc(1 – Alc) +… (1 – Alc)n–1 whereby its order was kept to a minimum.
When the excess volume VE is known, the density of the liquid samples S can be obtained from equation (5) by noting that VS = [Alc ΜAlc + (1 – Alc)]/ ρS and Vi = Μi/ρi. Henceforth:
in conjunction with equation (17), Table 1 and equation (14).
5. Vapor pressures of the pure liquids
To quantify the composition of the gaseous binary alcohol-air and ternary alcohol-water-air mixtures, it is necessary to know the vapor pressure of the pure substances. The Clapeyron equation [18, 52, 53], which is an exact thermodynamic relation, provides a fundamental connection between the vapor pressure of a pure substance and varying temperature:
However, despite its derivation from first principles and usefulness for many purposes, this expression does not represent PSat versus T data sufficiently well. For it predicts a linear dependence of PSat on 1/T whereas all experimental data in Figure, when plotted against 1/T, exhibit a deviation from linearity. For the accurate representation of vapor-pressure data, and to overcome accuracy limitations of equation (25) over a wide range of temperatures for a large number of species, various equations of greater complexity have been proposed. Examples are the Antoine equation [2, 18, 20]:
and the Riedel equation [18, 20]:
where A, B, C, D and E in aforementioned expressions are substance specific model constants whose values are readily available in tabulations for many species. There is an even more accurate formula for interpolation between PSat values that are reasonably spaced. That is the so called extended Antoine equation [2]:
Where:
Figure 19.
Experimental vapor pressures from Refs. [1, 8, 29–51]: (○) CH3OH, (◊) C2H5OH, (▽) n-C3H7OH, (△) i-C3H7OH, (●) H2O. Solid black curve: extended Antoine equation (28). Solid red curve: Wagner equation (30). Solid blue curve: Lee-Kesler equation (32). The coefficients of the extended Antoine and Wagner equations are given in Table 3.
Figure 19.
Experimental vapor pressures from Refs. [1, 8, 29–51]: (○) CH3OH, (◊) C2H5OH, (▽) n-C3H7OH, (△) i-C3H7OH, (●) H2O. Solid black curve: extended Antoine equation (28). Solid red curve: Wagner equation (30). Solid blue curve: Lee-Kesler equation (32). The coefficients of the extended Antoine and Wagner equations are given in Table 3.
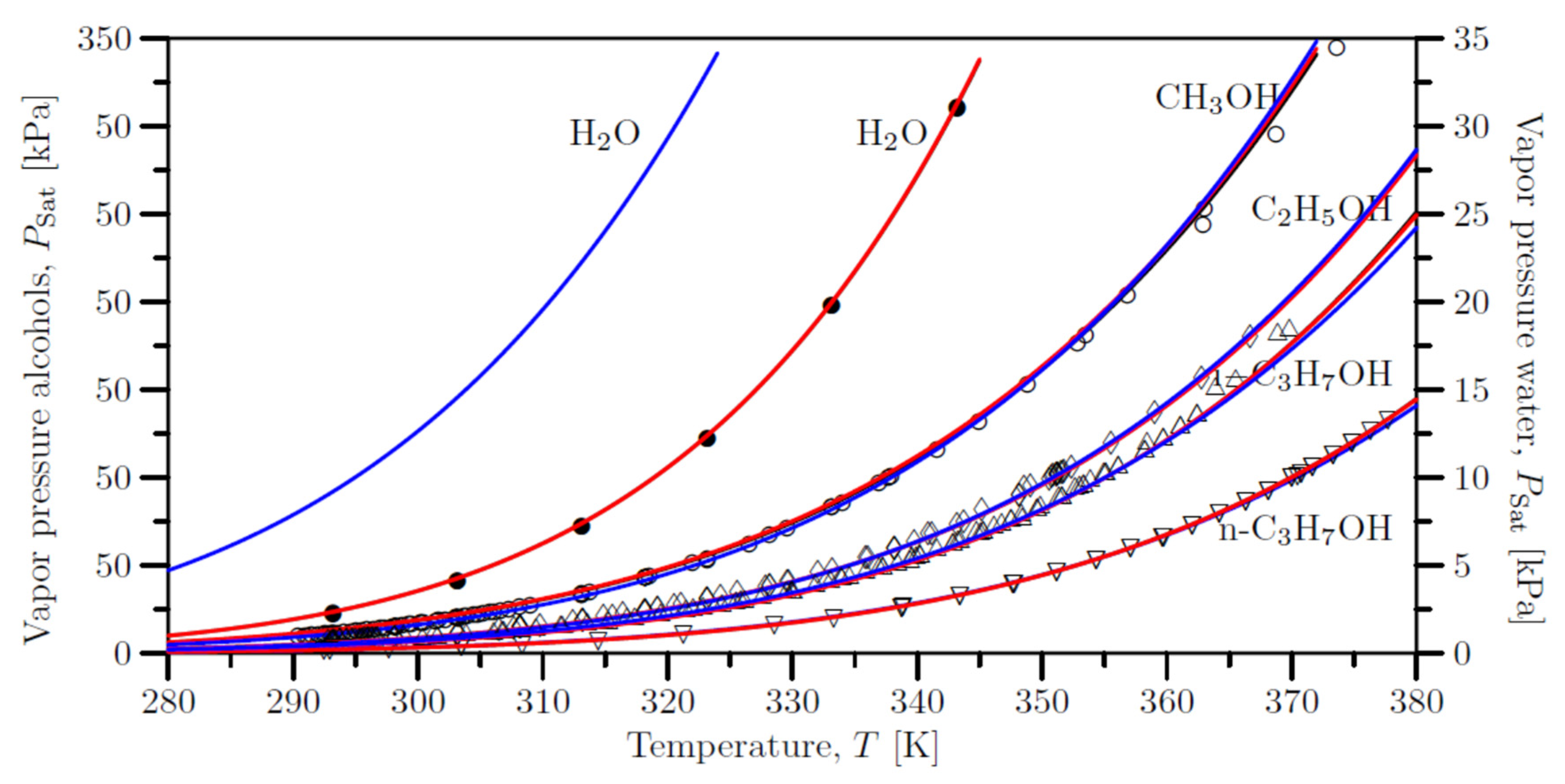
Table 3.
Coefficients of the extended Antoine equation (28) and the Wagner equation (30) for the pure sample constituents [2].
Table 3.
Coefficients of the extended Antoine equation (28) and the Wagner equation (30) for the pure sample constituents [2].
| Species | Extended Antoine equation | Wagner equation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | E | F | t0 | n | p | q | a | b | c | d | |
| CH3OH | 5.20277 | 1580.080 | 239.500 | – | – | – | – | 8 | 12 | -8.63571 | 1.17982 | -2.4790 | -1.0240 |
| C2H5OH | 5.33675 | 1648.220 | 230.918 | – | – | – | – | 8 | 12 | -8.68587 | 1.17831 | -4.8762 | 1.5880 |
| n-C3H7OH | 4.99991 | 512.940 | 205.807 | – | – | – | – | 8 | 12 | -8.53706 | 1.96214 | -7.6918 | 2.9450 |
| i-C3H7OH | 5.24268 | 1580.920 | 219.610 | – | – | – | – | 8 | 12 | -8.73656 | 2.16240 | -8.70785 | 4.77927 |
| H2O | 5.11564 | 1687.537 | 230.17 | – | – | – | – | 3 | 6 | -7.77224 | 1.45684 | -2.71942 | -1.41336 |
In the aforesaid expression PSat is in bar, T in K, D= 0.43429, and, the exponents p and q assume distinct values for alcohols and water. For alcohols {p = 8, q =12} whereas {p = 3, q =6} for water. The substance specific values of A, B, C, E, F, t0 and n are given in Table 3. Notice that the extended Antoine equation is a superset of the classical Antoine equation and the Riedel equation. The set of coefficients for the species involved in this work causes it to become identical to the latter two equations. Nonetheless the extended Antoine equation is resorted to because the required coefficients are provided in its 10log-form by Ref. [2]. The reason for resorting to Antoine-type equations is their ability to handle polar species. Another model for correlating experimental vapor pressures is the Wagner equation [2, 54–57]:
where
where PSat is in the same units as Pc and T in K. For water only the last two terms in equation (30) are cτ3 and dτ6. This expression too is capable of handling polar species. Figure 3 shows an inter-comparison between the values predicted by the extendend Antoine equation, the Wagner equation and experimental PSat data retrieved from the literature. Evidently, despite their very distinct mathematical forms, there is very close agreement amongst the models. The black and red solid curves in Figure 19 are almost indistinguishable. Both models are also able to correlate the experimental data accurately. In the present work the extended Antoine equation was deployed for the calculation and interpolation of the species vapor pressures. With non-polar substances the Lee-Kesler [58] equation:
where:
would also suffice. Here ω denotes the acentric factor and {a0=5.92714, a1= –6.09648, a2= – 1.28862, a3=0169347; b0=15.2518, b1= –15.6875, b2= –13.4721, b3=0.43577}. The solid blue curves in Figure 3 show a comparison between saturation pressures predicted by the Lee-Kesler equation and experimental data.
6. Conclusions
In our paper, we presented the results of experimental studies and numerical calculations on the thermodynamic reactivity of selected mixtures of alcohol fuels with air, as well as the addition of water to these mixtures. A review of the literature on the subject of this knowledge, clearly indicates that the experimental data in question are lacking, especially for lower equivalence ratios of alcohol-air mixtures, e.g. ϕ < 0.8. Therefore, the conducted research supplements the lack of knowledge in this area. Substances that we investigated were: methanol, ethanol, n-propanol and iso-propanol. All experiments were conducted at initial conditions of 323.15 K and 1 bar in a 20 dm3 closed testing combustion vessel. We investigated the reactivity and thermodynamic properties during the combustion of liquid fuel-air mixtures at ϕ = 0.3-0.7 as well as some admixtures with water, to observe water mitigation effects. Our experiments were conducted by igniting gaseous binary alcohol-air mixtures and also alcohol-water-air mixtures to the deflagration mode of combustion process at initial conditions of 323.15 K and 1 bar, whereby the liquid-vapor equilibrium existed in the explosion chamber prior to ignition. The analysis of the experimental results let us draw some conclusions about the influence of water addition on three basic thermodynamic reactivity parameters. In case of propanol-water mixtures, the values of the explosion pressure of n-propanol are slightly higher than those of the iso-propanol. This phenomenon clearly indicates the theory of the branching process of alcohol molecules for the thermodynamic reactivity. The n-propanol has the linear structure and thus it is more thermodynamically reactive than the branched iso-propanol. It can be also observed that the greater the volume of the sample the less orderly composed results are recorded. In case of higher volume samples, it was observed that thermodynamic reactivity of methanol mixtures was very dynamic between of 10-30% of water addition to the mixtures, while the dynamics of the ethanol mixtures reactions was rather less intensive. The smallest explosion delay time varies with the volume of alcohol-air mixtures. For 4 cm3 samples (ϕ = 0.3), the explosion delay time is the shortest during the tests of alcohol-air mixtures with no water content. For 6 cm3 (ϕ = 0.5) and 8 cm3 (ϕ = 0.7) samples the profiles of explosion delay time can be found as more complicated and the extreme points are different for each alcohol. Furthermore, in the chemical structure of investigated mixtures, the functional group is the hydroxyl group (OH), which is also responsible for thermodynamic reactivity of all alcohol-air mixtures. This group has two reactive covalent bonds, the C–O bond and the O–H bond. The electronegativity of oxygen is substantially greater than that of carbon and hydrogen. Regarding thermodynamic reactivity analysis based on experimental results of investigated light alcohol-air mixtures and water addition, we may conclude that quantification of the binary and ternary vapor compositions (the vapor phase mass fractions, ỹAlc and , and the vapor phase mole fractions, Alc and ) is more challenging as well as it requires the deployment of phase equilibrium theory and models. Based on our calculations of light alcohols, we may state that there was a modest deviation of less than 4% in saturation pressures based on our calculations. But for water there was a very large difference between prediction and experimental observations. The reason for this discrepancy is that the acentric factor, meant to correlate interactions amongst molecular force fields that deviate from spherical symmetry to improve the accuracy of corresponding-states correlations, becomes progressively inaccurate with increasing polarity of the molecules involved. For water there were specific equations whose predictions deviate less than 0.4% from experimental observations in the temperature range from 293.15 K to 323.15 K: the August-Roche-Magnus equation [59], the Tetens equation [60], the Buck equation [61] and the Goff-Gratch equation [62]. Also, like many hydrocarbon derivatives, methanol, ethanol and propanol undergoes combustion, when combined with heat and oxygen. This reaction releases energy, carbon dioxide, and water. Addition of water into alcohol-air reactive system can make a strong influence on thermodynamic properties of light alcohols and dramatically decrease the combustion reactivity of such fuels. Knowledge of thermodynamic and reactivity data of light alcohol fuels can support the energy market in building proper strategies how to use such fuels in good and safe technologies in the process industries.
Acknowledgments
Authors want to spread the acknowledgement to the ANKO company in Poland and Mr. Andrzej Kołaczkowski who produced and delivered our combustion testing vessel and supporting devices. This work was funded by the research project supported by program „Excellence initiative – Research University” for the AGH University of Krakow.
Conflicts of Interest
authors declare no conflicts of interest.
Appendix A. Thermodynamic quantities and properties
Table 1.
Sample compositions and properties of methanol, ethanol, n-propanol and i-propanol admixtures with water.
Table 1.
Sample compositions and properties of methanol, ethanol, n-propanol and i-propanol admixtures with water.
| Methanol – water | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (cm3) | q (vol%) |
(-) |
(-) |
(-) | (-) |
(-) |
(-) |
(-) |
(-) |
(cm3 mol-1) | ρs (kg m-3) | Ignition | ||||||||||
| 4.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 10 | 0.888 | 0.112 | - | - | 0.817 | 0.183 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 20 | 0.798 | 0.202 | - | - | 0.691 | 0.309 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 30 | 0.725 | 0.275 | - | - | 0.599 | 0.401 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 40 | 0.664 | 0.336 | - | - | 0.528 | 0.472 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 50 | 0.612 | 0.388 | - | - | 0.472 | 0.528 | - | - | - | - | 2×n | ||||||||||
| 4.0 | 60 | 0.568 | 0.432 | - | - | 0.427 | 0.573 | - | - | - | - | 3×n | ||||||||||
| 6.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 10 | 0.888 | 0.112 | - | - | 0.817 | 0.183 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 20 | 0.798 | 0.202 | - | - | 0.691 | 0.309 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 30 | 0.725 | 0.275 | - | - | 0.599 | 0.401 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 40 | 0.664 | 0.336 | - | - | 0.528 | 0.472 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 50 | 0.612 | 0.388 | - | - | 0.472 | 0.528 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 60 | 0.568 | 0.432 | - | - | 0.427 | 0.573 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 10 | 0.888 | 0.112 | - | - | 0.817 | 0.183 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 20 | 0.798 | 0.202 | - | - | 0.691 | 0.309 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 30 | 0.725 | 0.275 | - | - | 0.599 | 0.401 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 40 | 0.664 | 0.336 | - | - | 0.528 | 0.472 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 50 | 0.612 | 0.388 | - | - | 0.472 | 0.528 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 60 | 0.568 | 0.432 | - | - | 0.427 | 0.573 | - | - | - | - | 3×y | ||||||||||
| Ethanol – water | ||||||||||||||||||||||
| (cm3) | q (vol%) |
(-) |
(-) |
(-) |
(-) |
Alc(-) | (-) |
(-) |
(-) | (cm3 mol-1) | ρs (kg m-3) | Ignition | ||||||||||
| 4.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 10 | 0.888 | 0.112 | - | - | 0.757 | 0.243 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 20 | 0.798 | 0.202 | - | - | 0.609 | 0.391 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 30 | 0.725 | 0.275 | - | - | 0.509 | 0.491 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 40 | 0.664 | 0.336 | - | - | 0.438 | 0.562 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 50 | 0.612 | 0.388 | - | - | 0.384 | 0.616 | - | - | - | - | 2×y | ||||||||||
| 4.0 | 60 | 0.568 | 0.432 | - | - | 0.341 | 0.659 | - | - | - | - | 3×n | ||||||||||
| 6.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 10 | 0.888 | 0.112 | - | - | 0.757 | 0.243 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 20 | 0.798 | 0.202 | - | - | 0.609 | 0.391 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 30 | 0.725 | 0.275 | - | - | 0.509 | 0.491 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 40 | 0.664 | 0.336 | - | - | 0.438 | 0.562 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 50 | 0.612 | 0.388 | - | - | 0.384 | 0.616 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 60 | 0.568 | 0.432 | - | - | 0.341 | 0.659 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 10 | 0.888 | 0.112 | - | - | 0.757 | 0.243 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 20 | 0.798 | 0.202 | - | - | 0.609 | 0.391 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 30 | 0.725 | 0.275 | - | - | 0.509 | 0.491 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 40 | 0.664 | 0.336 | - | - | 0.438 | 0.562 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 50 | 0.612 | 0.388 | - | - | 0.384 | 0.616 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 60 | 0.568 | 0.432 | - | - | 0.341 | 0.659 | - | - | - | - | 3×y | ||||||||||
| n-Propanol – water | ||||||||||||||||||||||
| (cm3) | q (vol%) |
(-) |
(-) |
(-) | (-) |
Alc (-) |
(-) |
(-) | (-) | (cm3 mol-1) | ρs (kg m-3) | Ignition | ||||||||||
| 4.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 10 | 0.888 | 0.112 | - | - | 0.708 | 0.292 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 20 | 0.798 | 0.202 | - | - | 0.548 | 0.452 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 30 | 0.725 | 0.275 | - | - | 0.447 | 0.553 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 40 | 0.664 | 0.336 | - | - | 0.378 | 0.622 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 50 | 0.612 | 0.388 | - | - | 0.327 | 0.673 | - | - | - | - | 2×n | ||||||||||
| 4.0 | 60 | 0.568 | 0.432 | - | - | 0.288 | 0.712 | - | - | - | - | 3×n | ||||||||||
| 6.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 10 | 0.888 | 0.112 | - | - | 0.708 | 0.292 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 20 | 0.798 | 0.202 | - | - | 0.548 | 0.452 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 30 | 0.725 | 0.275 | - | - | 0.447 | 0.553 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 40 | 0.664 | 0.336 | - | - | 0.378 | 0.622 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 50 | 0.612 | 0.388 | - | - | 0.327 | 0.673 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 60 | 0.568 | 0.432 | - | - | 0.288 | 0.712 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 10 | 0.888 | 0.112 | - | - | 0.708 | 0.292 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 20 | 0.798 | 0.202 | - | - | 0.548 | 0.452 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 30 | 0.725 | 0.275 | - | - | 0.447 | 0.553 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 40 | 0.664 | 0.336 | - | - | 0.378 | 0.622 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 50 | 0.612 | 0.388 | - | - | 0.327 | 0.673 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 60 | 0.568 | 0.432 | - | - | 0.288 | 0.712 | - | - | - | - | 3×y | ||||||||||
| i-Propanol – water | ||||||||||||||||||||||
| (cm3) | q (vol%) |
(-) |
(-) |
(-) | (-) |
Alc (-) |
(-) |
(-) |
(-) | (cm3 mol-1) | ρs (kg m-3) | Ignition | ||||||||||
| 4.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 10 | 0.888 | 0.112 | - | - | 0.703 | 0.297 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 20 | 0.798 | 0.202 | - | - | 0.542 | 0.458 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 30 | 0.725 | 0.275 | - | - | 0.441 | 0.559 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 40 | 0.664 | 0.336 | - | - | 0.372 | 0.628 | - | - | - | - | 3×y | ||||||||||
| 4.0 | 50 | 0.612 | 0.388 | - | - | 0.322 | 0.678 | - | - | - | - | 2×n | ||||||||||
| 4.0 | 60 | 0.568 | 0.432 | - | - | 0.283 | 0.717 | - | - | - | - | 3×n | ||||||||||
| 6.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 10 | 0.888 | 0.112 | - | - | 0.703 | 0.297 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 20 | 0.798 | 0.202 | - | - | 0.542 | 0.458 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 30 | 0.725 | 0.275 | - | - | 0.441 | 0.559 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 40 | 0.664 | 0.336 | - | - | 0.372 | 0.628 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 50 | 0.612 | 0.388 | - | - | 0.322 | 0.678 | - | - | - | - | 3×y | ||||||||||
| 6.0 | 60 | 0.568 | 0.432 | - | - | 0.283 | 0.717 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 0 | 1.000 | 0.000 | - | - | 1.000 | 0.000 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 10 | 0.888 | 0.112 | - | - | 0.703 | 0.297 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 20 | 0.798 | 0.202 | - | - | 0.542 | 0.458 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 30 | 0.725 | 0.275 | - | - | 0.441 | 0.559 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 40 | 0.664 | 0.336 | - | - | 0.372 | 0.628 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 50 | 0.612 | 0.388 | - | - | 0.322 | 0.678 | - | - | - | - | 3×y | ||||||||||
| 8.0 | 60 | 0.568 | 0.432 | - | - | 0.283 | 0.717 | - | - | - | - | 3×y | ||||||||||
Appendix B. Experimental explosion pressure curves of alcohol-air and alcohol-water-air mixtures.
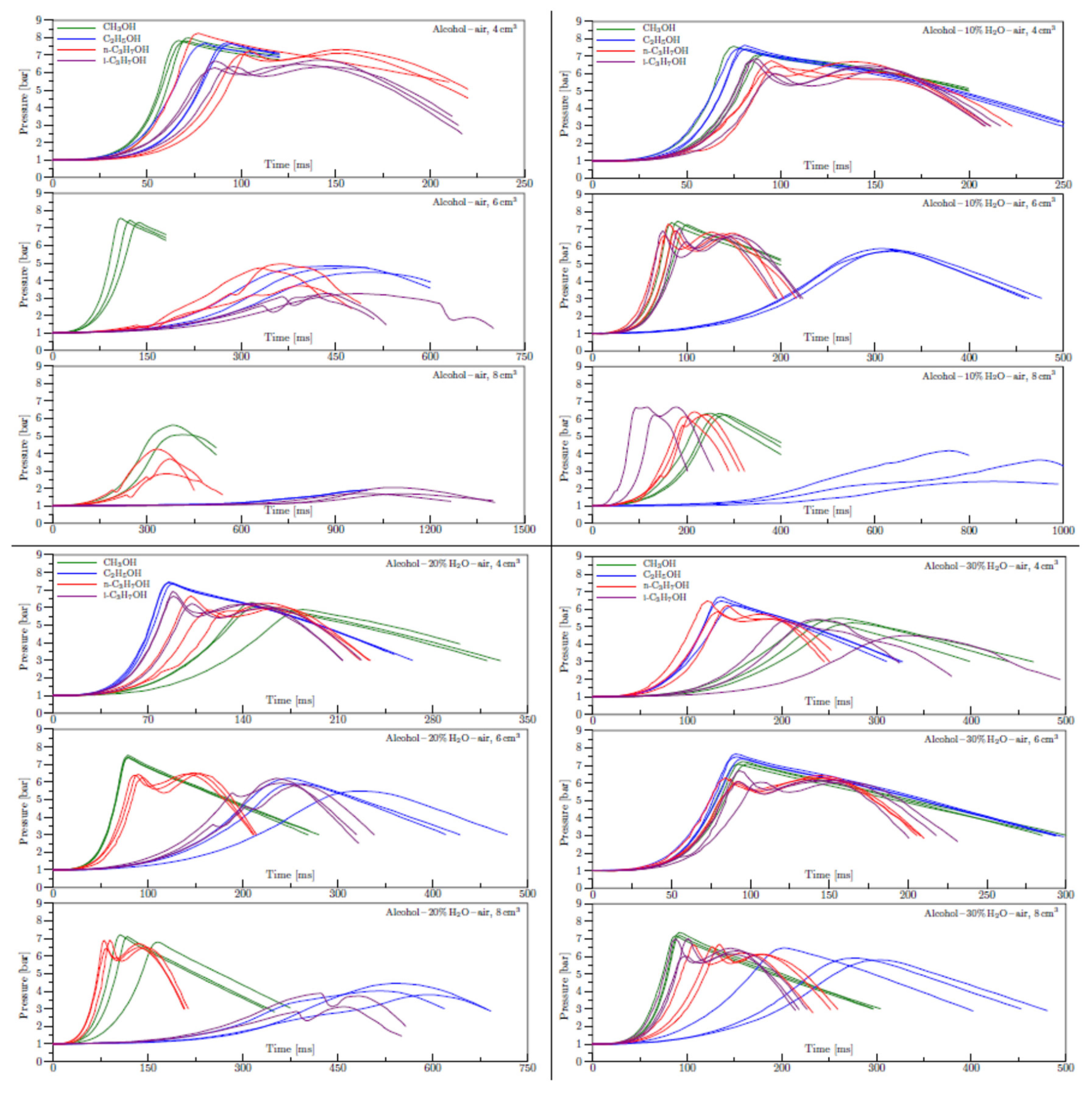
Figure B1. Explosion pressure curves of alcohol – air (upper left), alcohol – 10%H2O– air (upper right), alcohol – 20%H2O– air (lower left) and alcohol – 30%H2O– air (lower right) mixtures at initial conditions of 1 bar and 323.15 K.
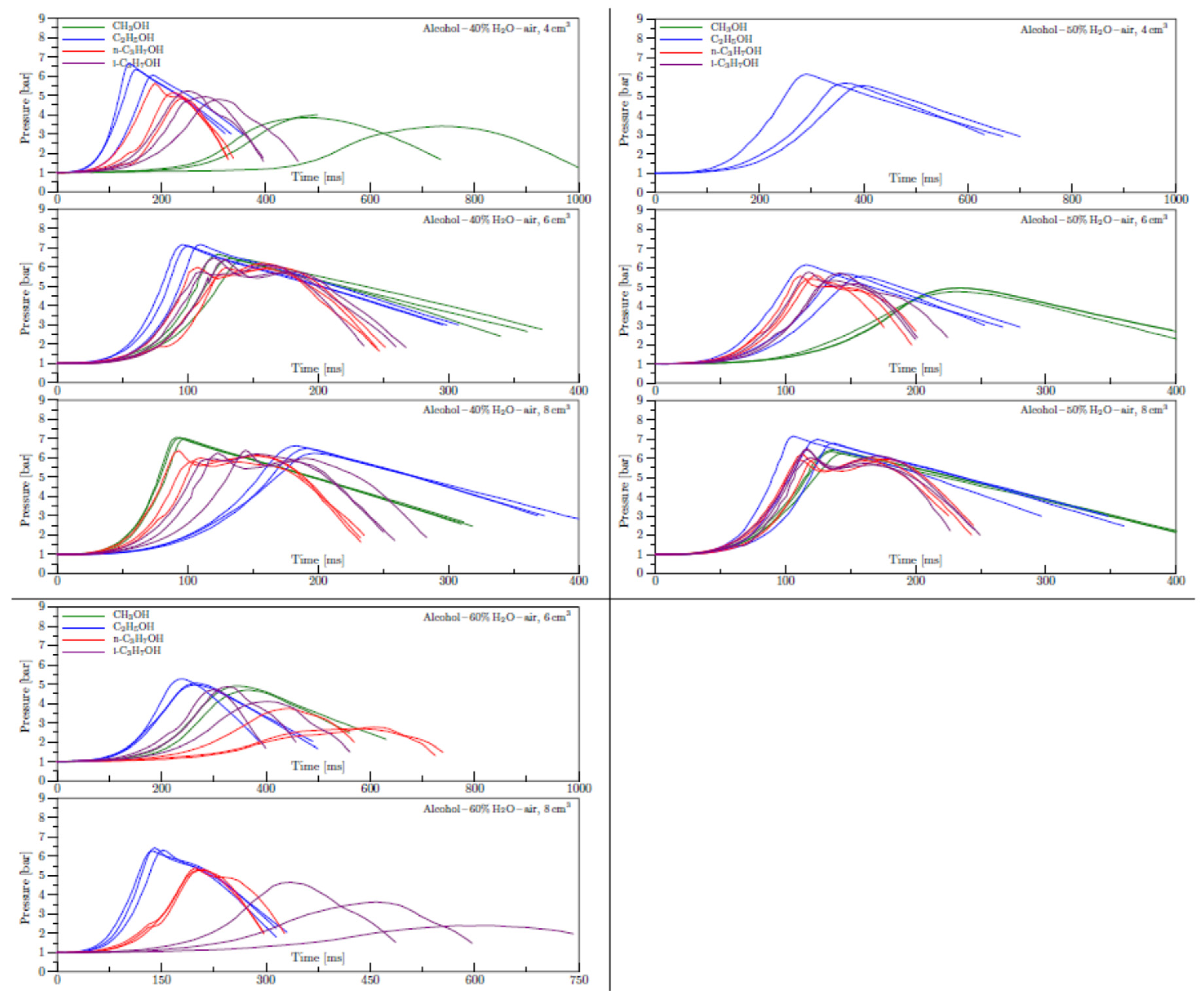
Figure B2. Explosion pressure curves of alcohol – 40%H2O– air (upper left), alcohol – 50%H2O– air (upper right), and alcohol – 60%H2O– air (lower left) mixtures at initial conditions of 1 bar and 323.15 K.
References
- Lide D.R., editor. CRC Handbook of Chemistry and Physics. CRC Press, 84th edition, 2004.
- Poling, B.E., Prausnitz J.M., and O’Connell J.P. The Properties of Gases and Liquids. McGraw-Hill, fifth edition, 2001.
- Perry, R.H., Green D.W., and Maloney J.O. Perry’s Chemical Engineers’ Handbook. McGraw-Hill, New York, seventh edition, 1999.
- The Dortmund Data Bank. Thermophysical properties of pure components and their mixtures. http://www.ddbst.com/, 2018. Publicly available online resource.
- Arce, A., Blanco A., Soto A., and Vidal I. Densities, refractive indices, and excess molar volumes of the ternary systems water + methanol + 1-octanol and water + ethanol + 1-octanol and their binary mixtures at 298.15 K. Journal of Chemical Engineering Data 1993, 38, 336–340. [CrossRef]
- Vilcu, R. and Simion A. Grandeurs d’exces par des Mesures Acoustiques, dans les Systemes Binaires de Liquides. Revue Roumaine de Chimie 1976, 21, 177–186.
- S. Mani Sarathy, Patrick Oßwald, Nils Hansen, and Katharina Kohse-Hoinghaus. Alcohol combustion chemistry. Progress in Energy and Combustion Science, 2014.
- Qianqian Li, Yu Cheng, and Zuohua Huang. Comparative assessment of the explosion characteristics of alcohol-air mixtures. Journal of Loss Prevention in the Process Industries, 2015.
- Bryan, W. Weber, Kamal Kumar, Yu Zhang, and Chih-Jen Sung. Autoignition of n-butanol at elevated pressure and low to intermediate temperature. Combustion and Flame, 2011.
- Zhu Yangye, David Frank Davidson, and Ronald K. Hanson. 1-butanol ignition delay times at low temperatures: An application of the constrained-reaction-volume strategy. Combustion and Flames, 2013.
- Douheret, G., Khadir A, and Pal A. Thermodynamic characterization of the water + methanol system, at 298.15 K. Thermochimica Acta 1989, 142, 219–243. [CrossRef]
- Grolier, J.-P.E. and Wilhelm E. Excess volumes and excess heat capacities of water + ethanol at 298.15 K. Fluid Phase Equilibria 1981, 6, 283–287. [CrossRef]
- Ott, J.B., Sipowska J.T., Gruszkiewicz M.S., and Woolley A.T. Excess volumes for (ethanol + water) at the temperatures (298.15 and 348.15) K and pressures (0.4, 5, and 15) MPa and at the temperature 323.15 K and pressures (5 and 15) MPa. The Journal of Chemical Thermodynamics 1993, 25, 307–318. [CrossRef]
- Davis, M.I. and Ham E.S. Part 2. Comparisons of the propanol isomers in their aqueous mixtures. Thermochimica Acta 1991, 190, 251–258.
- Langdon, W.M. and Keyes D.B. Isopropyl alcohol-water system. Industrial & Engineering Chemistry 1943, 35, 459–464.
- Yamamoto, H., Ichikawa K., and Tokunaga J. Solubility of helium in methanol + water, ethanol + water, 1-propanol + water, and 2-propanol + water solutions at 25°C. Journal of Chemical Engineering Data 1994, 39, 155–157. [CrossRef]
- Benson, G.C. and Kiyohara O. Thermodynamics of aqueous mixtures of nonelectrolytes. I. Excess volumes of water – n-alcohol mixtures at several temperatures. Journal of Solution Chemistry 1980, 9, 791–804. [CrossRef]
- Smith, J.M., Van Ness H.C., and Abbott M.M. Introduction to Chemical Engineering Thermodynamics. McGraw-Hill, New York, seventh edition, 2007.
- Battino, R. Volume changes on mixing for binary mixtures of liquids. Chemical Reviews 1971, 71, 5–45. [CrossRef]
- Abbott, M.M. and Van Ness H.C. Theory and problems of thermodynamics. Schaum’s outline series. McGraw-Hill, New York, 1972.
- Press, W.H., Teukolsky S.A., Vetterling W.T., and Flannery B.P. Numerical Recipes, The Art of Scientific Computing. Cambridge University Press, Cambridge, third edition, 2007.
- Marquardt, D.W. An algorithm for least-squares estimation of nonlinear parameters. SIAM Journal on Applied Mathematics 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Dahoe, A.E., Skjold T, Roekaerts D.J.E.M., Pasman H.J., Eckho R.K., Hanjalic K., and Donze M. On the application of the Levenberg-Marquardt method in conjunction with an explicit Runge-Kutta and an implicit Rosenbrock method to assess burning velocities from confined deflagrations. Flow, Turbulence and Combustion 2013, 91, 281–317.
- Rackett, H.G. Equation of state for saturated liquids. Journal of Chemical & Engineering Data 1970, 15, 514–517. [Google Scholar]
- Lydersen, A.L., Greenkorn R.A., and Hougen O.A. Generalized thermodynamic properties of pure fluids. Engineering Experiment Station Report 4, 1955. College of Engineering, University of Wisconsin, Madison, Wisconsin.
- Gunn, R.D. and Yamada T. A corresponding states correlation of saturated liquid volumes. AIChE Journal 1971, 17, 1341–1345. [CrossRef]
- Yen, L.C. and Woods S.S. A generalized equation for computer calculation of liquid densities. AIChE Journal 1966, 12, 95–99. [CrossRef]
- Yamada T. and Gunn R. Saturated liquid molar volumes. The Rackett equation. Journal of Chemical & Engineering Data 1973, 18, 234–236.
- The NIST ChemistryWebBook. Data compiled under the Standard Reference Data Program. https://webbook.nist.gov/, 2018. The National Institute of Standards and Technology (NIST). Publicly available online resource.
- Kotsarenko, A.A. and Yarym-Agaev N.L. Sättigungsdampfdruck von Methanol im Temperaturbereich von 251.9 bis 298.15 k. Izvestiya Vysshikh Uchebnykh Zavedenii: Neft i gaz, pages 59–61, 1990.
- Gibbard H.F. and Creek J.L. Vapor pressure of methanol from 288.15 to 337.65 k. Journal of Chemical & Engineering Data 1974, 19, 308–310.
- Dever, D.F. Dever D.F., Finch A., and Grunwald E. Vapor pressure of methanol from 288.15 to 337.65 k. The Journal of Physical Chemistry 1974, 59, 668–669. [Google Scholar] [CrossRef]
- Chun K.W. and Davison R.R. Thermodynamic properties of binary mixtures of triethylamine with methyl and ethyl alcohol. Journal of Chemical & Engineering Data 1972, 17, 307–310.
- Ambrose, D. and Sprake C.H.S. Thermodynamic properties of organic oxygen compounds. XXV. Vapour pressures and normal boiling temperatures of aliphatic alcohols. The Journal of Chemical Thermodynamics 1970, 2, 631–645. [CrossRef]
- Ambrose, D., Sprake C.H.S., and Townsend R. Thermodynamic properties of organic oxygen compounds. XXXVII. Vapour pressures of methanol, ethanol, pentan-1-ol, and octan-1-ol from the normal boiling temperature to the critical temperature. The Journal of Chemical Thermodynamics 1975, 7, 185–190. [CrossRef]
- Hirata M. and Suda S. Vapor pressure on methanol in high pressure regions. The Journal of Chemical Thermodynamics 1967, 31, 339–342.
- Mishchenko K.P. and Subbotina V.V. Dampfdruck von Ethanol bei Temperaturen von 4 bis 46°c. Zhurnal Pikladnoi Khimii 1967, 40, 1156–1159.
- Kahlbaum, G.W.A. and von Wirkner C.G. Studien uber Dampfspannkraftsmessungen. Monograph, 1897. Reprint in 2012 under ISBN-number 9785883729552.
- Scatchard G. and Raymond C.L. II. Chloroform-ethanol mixtures at 35, 45 and 55°C. Journal of the American Chemical Society 1938, 60, 1278–1287. [CrossRef]
- Scatchard G. and Satkiewicz F.G. XII. The system ethanol-cyclohexane from 5 to 65°C. Journal of the American Chemical Society 1964, 86, 130–133. [CrossRef]
- Kretschmer C.B. and Wiebe R. Liquid-vapor equilibrium of ethanol-toluene solutions. Journal of the American Chemical Society 1949, 71, 1793–1797. [CrossRef]
- Dejoz A., Gonzalez-Alfaro V., Miguel P.J., and Vazquez M.I. Isobaric vapor-liquid equilibria of tetrachloroethylene + 1-propanol and + 2-propanol at 20 and 100 kpa. Journal of Chemical & Engineering Data 1996, 41, 1361–1365.
- Shulgin I.L., Belousov V.P., and Baglai A.K. Eine tensimetrische Untersuchungsmethode des Dampf-flussig-Gleichgewichtes in binaren Systemen. Termodinamika Organicheskoi Soedinenii 1989, 32, 32–35.
- Barr-David F. and Dodge B.F. The systems ethanol–water and 2-propanol–water. Journal of Chemical & Engineering Data 1959, 4, 107–121.
- Daubert T.E., Jalowka J.W., and Goren V. Vapor pressure of 22 pure industrial chemicals. AIChE Symposium Series 1987, 32, 128–156.
- Ambrose D. and Townsend R. Thermodynamic properties of organic oxygen compounds. the critical properties and vapour pressures, above five atmospheres, of six aliphatic alcohols. Part IX. Journal of the Chemical Society 1963, 54, 3614–3625.
- Biddiscombe D.P., Collerson R.R., Handley R., Herington E.F.G., Martin J.F., and Sprake C.H.S. Thermodynamic properties of organic oxygen compounds. Part XIII. Journal of the Chemical Society 1963, 54, 1954–1957.
- Kemme H.R. and Kreps S.I. Vapor pressure of primary n-alkyl chlorides and alcohols. Journal of Chemical & Engineering Data 1969, 14, 98–102.
- Gudkov A.N., Fermor N.A., and Smirnov N.I. Communication I. Zurnal Prikladnoj Chimii 1964, 37, 2204–2210.
- Bridgeman O.C. and Aldrich E.W. Vapor pressure tables for water. Journal of Heat Transfer 1964, 86, 279–286. [CrossRef]
- Stull, D.R. Vapor pressure of pure substances. Organic and inorganic compounds. Industrial & Engineering Chemistry 1947, 39, 517–540. [Google Scholar]
- Clapeyron, E. M´emoire sur la Puissance Motrice de la Chaleur. Journal de l’Ecole Royale Polytechnique 1834, 14, 153–191. [Google Scholar]
- Wisniak, J. Historical development of the vapor pressure equation from Dalton to Antoine. Journal of Phase Equilibria 2001, 22, 622–630. [Google Scholar] [CrossRef]
- Wagner, W. New vapour pressure measurements for argon and nitrogen and a new method for establishing rational vapour pressure equations. Cyrogenics 1973, 13, 470–482. [Google Scholar] [CrossRef]
- Wagner, W. A new correlation method for thermodynamic data applied to the vapor-pressure curve of argon, nitrogen, and water. In: Watson J.T.R. (editor). IUPACThermodynamic Tables Project Centre, London, 1977.
- Ambrose, D. The corelation and estimation of vapour pressures IV. Observations on Wagner’s method of fitting equations to vapour pressures. The Journal of Chemical Thermodynamics 1986, 18, 45–51. [Google Scholar] [CrossRef]
- Ambrose, D. Vapour pressures and critical temperatures and critical pressures of some alkanoic acids: C1 to C10. The Journal of Chemical Thermodynamics 1987, 19, 505–519. [Google Scholar] [CrossRef]
- Lee, B.I. Lee B.I. and Kesler M.G. A generalized thermodynamic correlation based on three-parameter corresponding states. AIChE Journal 1975, 21, 510–527. [Google Scholar] [CrossRef]
- Alduchov O.A. and Eskridge R.E. Improved Magnus form approximation of saturation vapor pressure. Journal of Applied Meteorology 1996, 35, 601–619. [CrossRef]
- Alduchov O.A. and Eskridge R.E. U¨ ber einige meteorologische Begriffe. Zeitschrift fur Geophysik 1930, 6, 207–309.
- Buck, A.L. New equations for computing vapor pressure and enhancement factor. Journal of Applied Meteorology 1981, 20, 1527–1532. [Google Scholar] [CrossRef]
- Murphy D.M. and Koop T. Review of the vapour pressures of ice and supercooled water for atmospheric applications. Quarterly Journal of the Royal Meteorological Society 2005, 131, 1539–1565. [CrossRef]
- Dethlefsen C., Sørensen P.G., and Hvidt A. Excess volumes of propanol-water mixtures at 5, 15, and 25°C. Journal of Solution Chemistry 1984, 13, 191–202. [CrossRef]
- Soetens J.C. and Bopp P.A. Water-methanol mixtures: simulations of mixing properties over the entire range of mole fractions. The Journal of Physical Chemistry B 2015, 119, 8593–8599. [CrossRef]
- McGlashan M.L. and Williamson A. Isothermal liquidvapor equilibria for system methanol-water. Journal of Chemical Engineering Data 1976, 21, 196–199. [CrossRef]
- Van Ness, H.C. Thermodynamics in the treatment of vapor/liquid equilibrium (VLE) data. Pure and Applied Chemistry 1995, 67, 859–872. [Google Scholar] [CrossRef]
Figure 1.
Photograph (left) and schematic (right) of the 20-liter combustion vessel provided by ANKO.
Figure 1.
Photograph (left) and schematic (right) of the 20-liter combustion vessel provided by ANKO.

Figure 3.
Influence of the water addition on the explosion delay time of methanol-air mixtures.
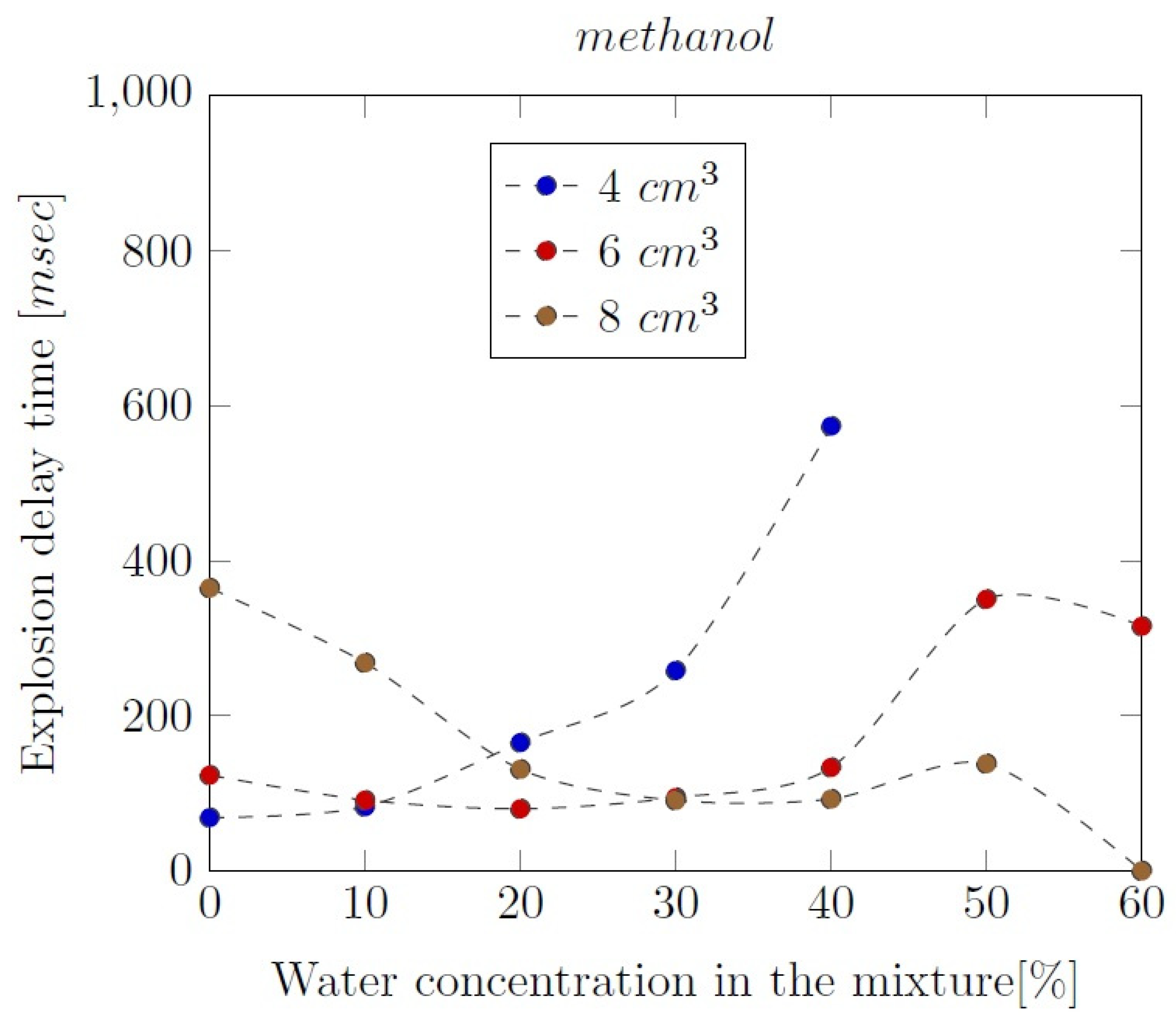
Figure 4.
Pressure increase of 6 cm3 of ethanol and 20% water addition (ϕ = 0.5).
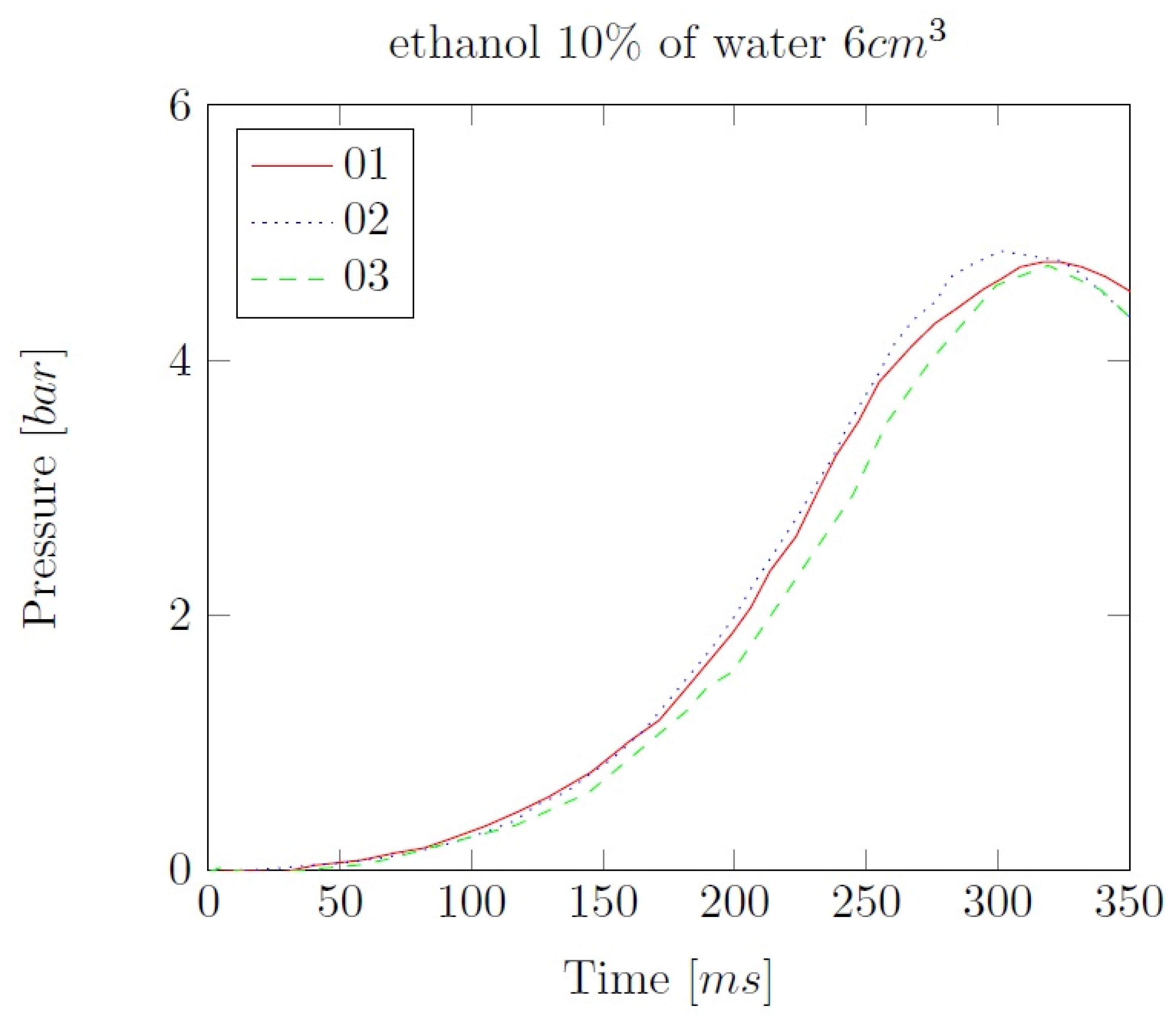
Figure 6.
Pressure increase of 4 cm3 of n-propanol and 20% water addition (ϕ = 0.3).
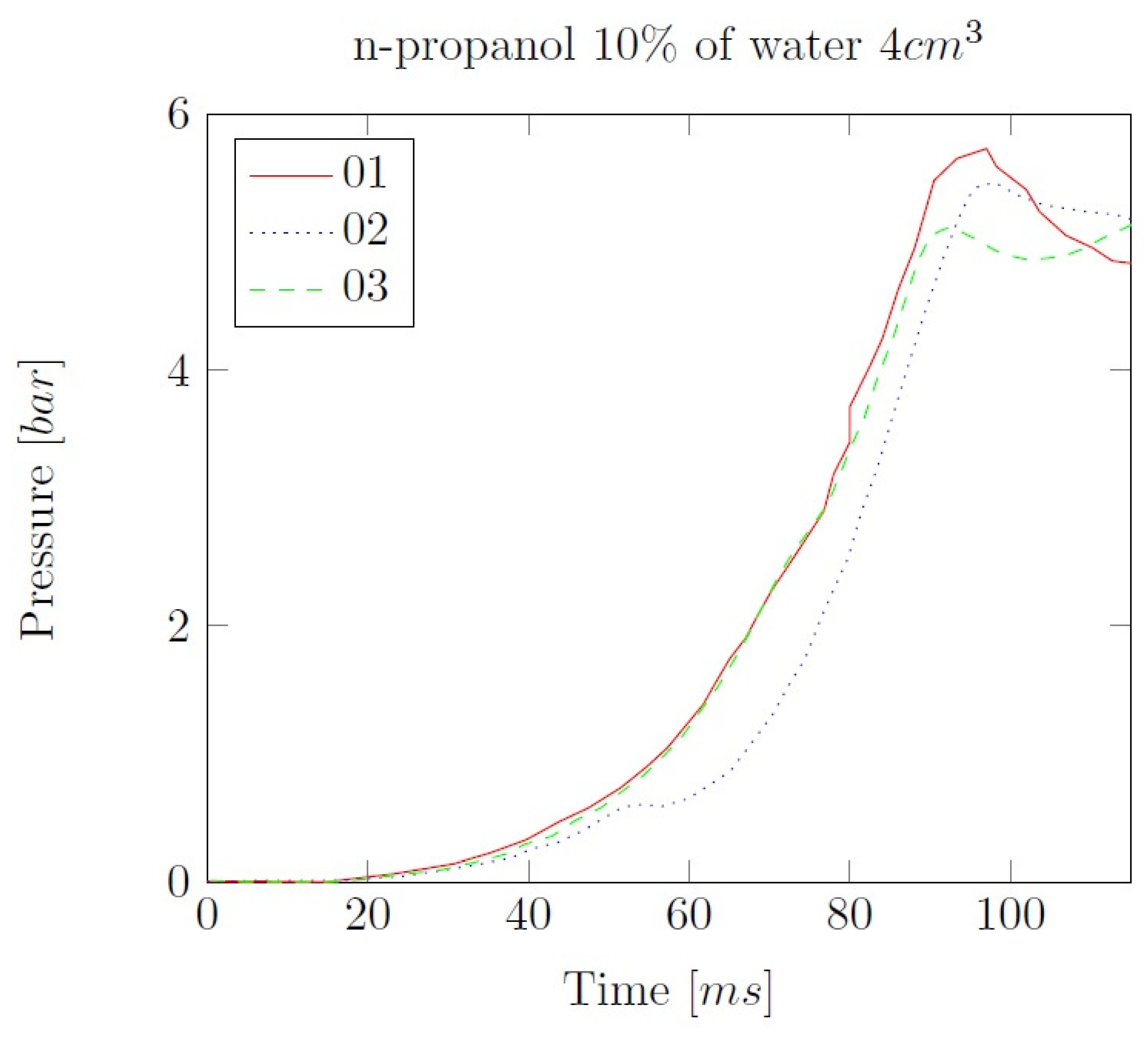
Figure 8.
Pressure increase of 4 cm3 of iso-propanol and 20% water addition (ϕ = 0.3).
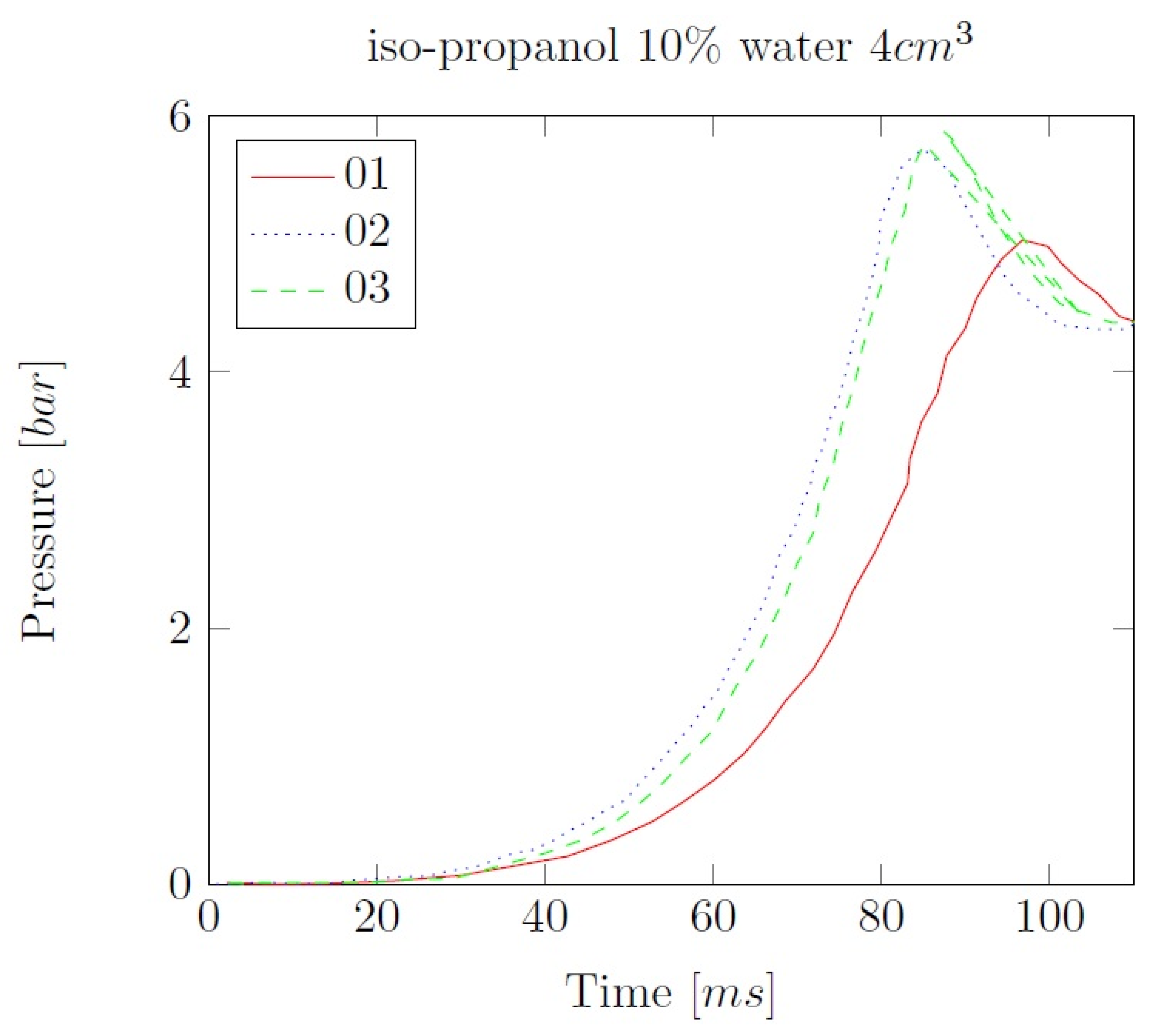
Figure 9.
Influence of the water addition on the explosion delay time of iso-propanol mixtures.
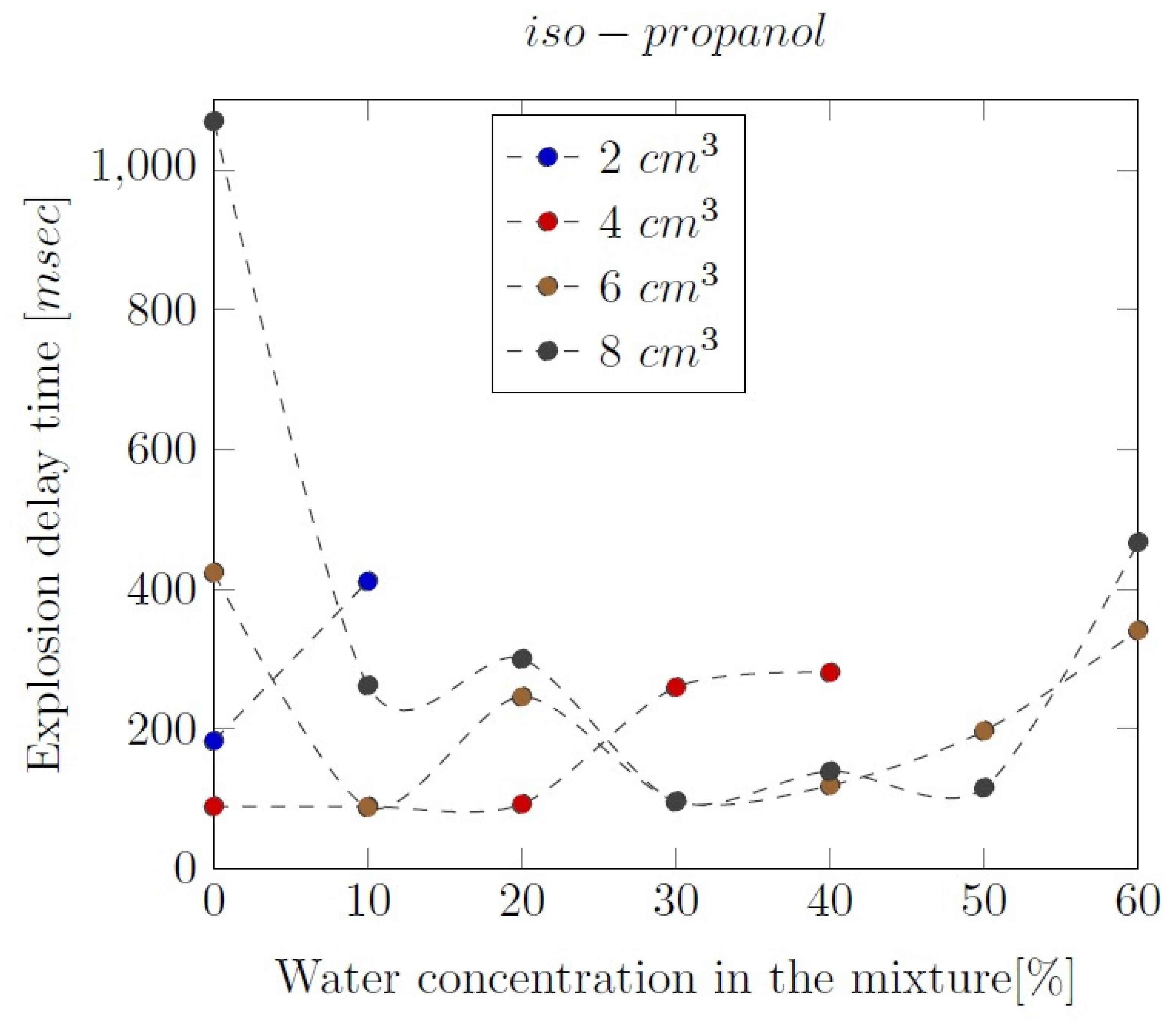
Figure 10.
Influence of water addition on the explosion pressure for investigated alcohol-air mixtures (ϕ = 0.3).
Figure 10.
Influence of water addition on the explosion pressure for investigated alcohol-air mixtures (ϕ = 0.3).
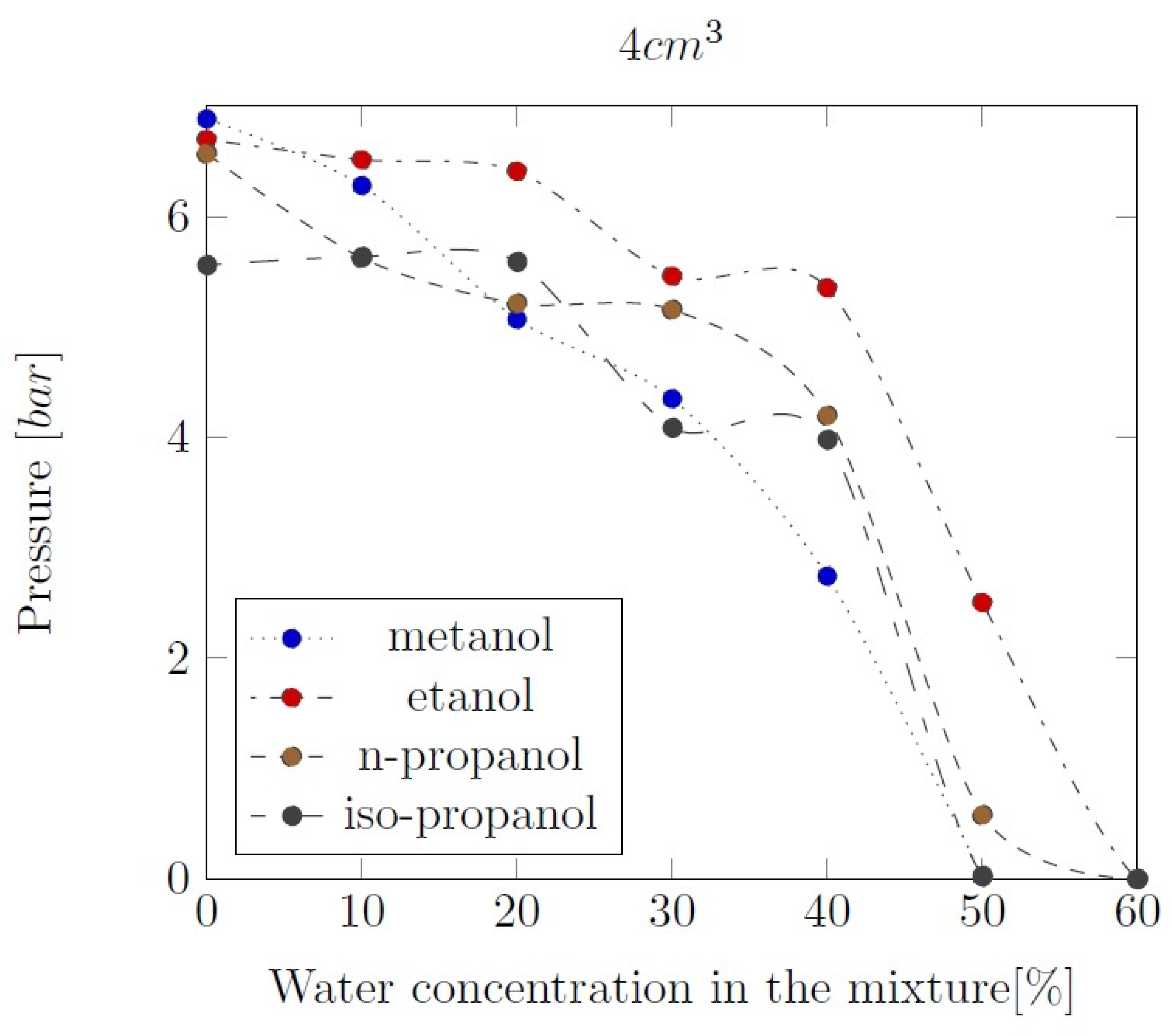
Figure 11.
Influence of water addition on the (dP/dt)max for all alcohol-air mixtures (ϕ = 0.3).
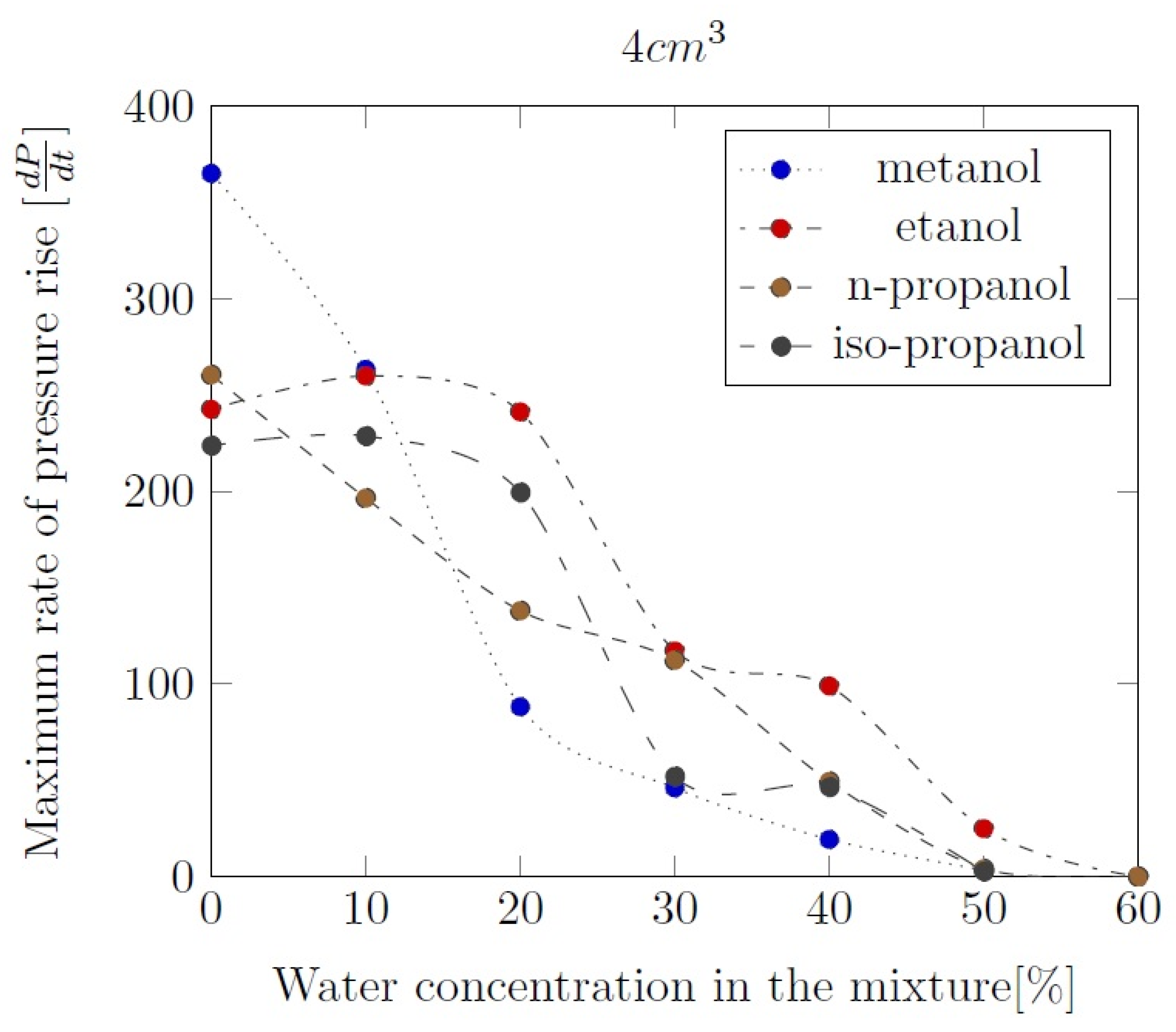
Figure 12.
Influence of water addition on the explosion delay time for all alcohol-air mixtures (ϕ = 0.3). .
Figure 12.
Influence of water addition on the explosion delay time for all alcohol-air mixtures (ϕ = 0.3). .
Figure 18.
Left. Density ρS of binary alcohol –water liquid samples. Experimental data from Ref. [7]: (○) CH3OH – H2O at T = 293.15 K and 1 bar, (◊) C2H5OH – H2O at T = 298.15 K and 1 bar, (▽) n-C3H7OH – H2O at T = 293.15 K and 1 bar, (△) i-C3H7OH – H2O at T = 303.15 K and 1 bar. The dashed colored curves are polynomial interpolations based on equation (13). The solid colored curves are interpolations based on equations (24) and (14). Right. Excess volumes VE of binary alcohol – water liquid mixtures at T = 298.15 K and 1 bar. Experimental data from Refs. [8,9,10,11,12,13,14,15,16,17]: (○) CH3OH – H2O, (◊) C2H5OH – H2O, (▽) n-C3H7OH – H2O, (△) i-C3H7OH – H2O. The solid colored curves are polynomial interpolations based on equation (14). Additional data collected from the literature [17] but not included in the determination of the polynomial coefficients: (×).
Figure 18.
Left. Density ρS of binary alcohol –water liquid samples. Experimental data from Ref. [7]: (○) CH3OH – H2O at T = 293.15 K and 1 bar, (◊) C2H5OH – H2O at T = 298.15 K and 1 bar, (▽) n-C3H7OH – H2O at T = 293.15 K and 1 bar, (△) i-C3H7OH – H2O at T = 303.15 K and 1 bar. The dashed colored curves are polynomial interpolations based on equation (13). The solid colored curves are interpolations based on equations (24) and (14). Right. Excess volumes VE of binary alcohol – water liquid mixtures at T = 298.15 K and 1 bar. Experimental data from Refs. [8,9,10,11,12,13,14,15,16,17]: (○) CH3OH – H2O, (◊) C2H5OH – H2O, (▽) n-C3H7OH – H2O, (△) i-C3H7OH – H2O. The solid colored curves are polynomial interpolations based on equation (14). Additional data collected from the literature [17] but not included in the determination of the polynomial coefficients: (×).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



