
Submitted:
17 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
Cancer is a disease caused by the abnormal growth of cells in different parts of body is one of the top causes of death globally. Microarray gene expression data plays a critical role in the identification and classification of cancer tissues. Due to recent advancements in Machine Learning (ML) techniques, researchers are analyzing gene expression data using a variety of such techniques to model the progression rate & treatment of cancer patients with great effect. But high dimensionality alongside the presence of highly correlated columns in gene expression datasets leads to computational difficulties. This paper aims to propose the use of ML classification techniques- Linear Discriminant Analysis (LDA) & Random Forest (RF) for classifying five types of cancer (breast cancer, kidney cancer, colon cancer, lung cancer and prostate cancer) based on high dimensional microarray gene expression data. Principal component analysis (PCA) was used for dimensionality reduction, and principal component scores of the raw data for classification. Six distinct categorization performance measures were used to evaluate these approaches; RF method provided us with higher accuracy than LDA method. The method and results of this article should be helpful to researchers who are dealing with many genes in microarray data.
Keywords:
principal components analysis
; Linear Discriminant Analysis
; Random Forest
; precision
; recall
; F1
; AUC
; macro-averaged AUC
; micro-averaged AUC
1. Introduction
Cancer is a disease which can start almost anywhere in the human body, in which some of the body’s trillion cells grow uncontrollably and spread to other parts of the body. There are over 200 types of cancer such as colon, liver, ovarian and breast etc. [1,2]. In 2023, 1,958,310 new cancer cases and 609,820 cancer deaths were projected in the United States. [3]. This prompts a clear understanding of the underlying mechanism and characteristics of this potentially fatal disease alongside identifying the most significant genes responsible for it.
Cancer can alter the gene expression profile of the body cells. Therefore, microarray data is utilized in clinical diagnosis to recognize down or up the regulated gene expression, which is the reason for generating new biomarkers, and leading to cancer disease [4]. Microarray data analysis has been a popular approach for diagnosing cancer, and DNA microarray is a technology used to collect data on large numbers of various gene expressions at the same time [5,6]. The classification and identification of gene expression using DNA microarray data is an effective tool for cancer diagnosis and prognosis for specific cancer subtypes. Gene expression analysis can assure medical experts whether a patient suffers from cancer within a relatively shorter time than traditional methods. Recently, its analysis has emerged as an important means for addressing the fundamental challenges associated with cancer diagnosis and drug discovery [7,8]. Analysis of gene expression data involves the identification of informative genes, [9] and [10] demonstrates that cancer classification can be improved by identifying informative genes which in turn can be used to accurately predict new sample classes.
Machine learning (ML) is a branch of artificial intelligence (AI) that enables computers to “self-learn” or obtain information from training data; recognize patterns in data and develop their own predictions, improving over time without being explicitly programmed [11]. Medical researchers and clinicians are utilizing several ML techniques on medical data sets to construct an intelligent diagnosis system [12]. Massive volume of data is being generated in the medical industry thanks to the digital revolution in information technology. ML techniques are highly suited for analyzing these massive data sets, and multiple algorithms have been used to diagnose various diseases [13,14,15]. Numerous research has been done to classify cancer using microarray gene expression data. Golub et al. [16] suggested a strategy based on expression profiles generated by microarrays. According to ML theory, classification outcomes are dependent on the features of the input set, the training algorithm, and the system's capacity to adapt to the original data. It is necessary to evaluate the behavior of various classifiers on provided data.
Recently, several classification approaches were created in the ML domain, and many of them were utilized in cancer classification [17]. However, there are several difficulties possible to face in the microarray classification process like (a) The microarray genes expression data constitutes many highly correlated genes for just a small sample size. The small number of cancer samples compared with the number of features can degrade the performance of the classifier and increase the risk of over-fitting. (b) Various uncertainties associated with the process of acquiring microarray data, for example, fabrication, image processing etc., resulting in unexplained fluctuation in the data. (c) The majority of genes in the microarray date are redundant for classifying diverse tissue types [18,19].
The earliest detection of cancer is among the most efficient approaches to reduce cancer-related death [20,21,22,23]. The microarray's primary characteristic is its greater number of genes (p) in comparison to the number of tissues (n) [24]. In most gene expression studies selection of relevant genes to differentiate between patients with and without cancer is a common task [25,26,27,28,29,30]. Due to overestimation & various linearity issues it is difficult to categorize high-dimensional microarray data (p > n) using statistical approaches [31,32]. There is no single optimal method to examine microarray data, with its continually evolving analysis methods [33]. Various supervised and unsupervised ML techniques have also been adopted to identify the most significant genes [34,35,36].In microarray gene expression analysis, gene selection or feature selection (FS) is utilized to improve cancer classification performance while using fewer samples, eliminate undesired & repetitive attributes from data and ultimately counter the curse of dimensionality by identifying the most informative genes to enhance disease prediction accuracy [37,38]. ML and dimensionality reduction techniques also perform exceptionally well at classifying biologic data [39,40,41]. Hence it may be beneficial to use feature selection methods which can address the challenges arising from high data dimensionality and small sample size.
2. Related Work
ML can assist in automating intelligent processes, increasing development efficiency and accuracy, and lowering costs [42]. Over the years ML-based classifiers have been widely used in classification of cancer sub-types. Several studies tried to assess whether ML can help in oncology care, by investigating the applications of ML in cancer risk stratification, diagnoses, and medication development [17,43,44,45]. According to those studies, ML can help in cancer prediction and diagnosis by analyzing pathology profiles and imaging studies.
BRCA (Breast Cancer gene) genes produce proteins that help repair damaged DNA and are referred to as tumor suppressor genes since certain changes in these genes can cause cancer [46]. People born with a certain variant of BRCA tend to develop cancer at early ages. Chang, Dalpatadu, Phanord and Singh [47] fitted a Bayesian Logistic Regression model for prediction of breast cancer using the Wisconsin Diagnosis Breast Cancer (WDBC) data set [48] which was downloaded from the UCI Machine Learning Repository; precision, recall and F1-measures of 0.93, 0.89, and 0.91 were reported for the training data, and 0.87, 0.91, 0.89 for the test data, respectively.HER2 protein accelerates breast cancer cell growth and HER2 positive patients when treated with medicines which attack the HER2 protein. Gene expression patterns of HER2 are quite complex and pose a challenge to pathologists. Cordova et al. (2023) developed a new interpretable ML method in immunohistochemistry for accurate HER2 classification and obtained high precision (0.97) and high accuracy (0.89) using immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) data [49].
Kidney renal cell carcinoma (KIRC) is the most prevalent type of kidney cancer, with a survival rate of less than 5 years and 338,000 estimated number of new cases each year [50]. ICD profile of KIRC. Wang et al. (2023) correlated the immunogenic cell death (ICD) of KIRK with the heterogeneity and therapeutic complexity which is useful for developing optimal immunotherapy strategy for KIRC patients [51].
A common cancerous tumor in the digestive track is colon adenocarcinoma (COAD) and is commonly associated with fatty acids [52]; diagnosis of COAD is difficult as there are hardly any early symptoms. Li et al. (2017) used a genetic algorithm and the k-nearest neighbors clustering method to determine genes which can accurately classify samples as well as class subtypes for a TCGA RNA-seq dataset of 9066 cancer patients and 602 normal samples [53].
Lung adenocarcinoma (LUAD) is a common form of lung cancer which also gets detected in the middle/late stages and therefore is hard to treat [54]. Yang et al. (2022) used a dataset of gene expression profiles from 515 tumor samples and 59 normal tissues and split the dataset into two significantly different clusters; they further showed that using age, gender, pathological stages, and risk score as predictors of LUAD increased the prediction accuracy measures [55]. Liu, Lei, Zhang, and Wang (2022) used cluster analysis on enrichment scores of 12 stemness signatures to identify three LUAD subtypes, St-H, St-M and St-L for six different datasets [56].
Prostate adenocarcinoma (PRAD) is common in elderly men, and patients suffering from PRAD typically have good prognosis [57]. Khosravi et al. (2021) used Deep Learning ML models on an MRI dataset from 400 subjects with suspected prostate cancer combined with histological data and reported high accuracies [58].
PCA is an exploratory multivariate statistical technique for simplifying complex data sets [59,60,61]. It has been used in a wide range of biomedical problems, including the analysis of microarray data in search of outlier genes [62], analysis of other types of expression data [63,64] as well as cancer classification [65]. AK Oladejo, TO Oladele, YK Saheed (2018) presented two methods of dimension reduction: feature extraction (FE) and FS; one-way Anova for FE and PCA was utilized for FS [66]. The Support vector machine (SVM) and k-nearest neighbor (K-NN) were used for the classification of leukemia genome data. The obtained results gave an accuracy of 90% for SVM and 81.67% for K-NN.
MO Adebiyi, MO Arowolo, MD Mshelia, OO Olugbara (2022) applied the machine learning algorithms of RF and the SVM with the feature extraction method of LDA to the Wisconsin Breast Cancer Dataset [67]. The SVM with LDA and RF with LDA yielded accuracy results of 96.4% and 95.6% respectively. Evidence from this study shows that better prediction is crucial and can benefit from machine learning methods. This research has validated the use of feature extraction in predicting a diagnostic system for breast cancer when compared to the existing literature.
Ak, Muhammet Fatih (2020) utilized the Wisconsin Breast Cancer Dataset [48] for the comparison of most of the major machine-learning procedures for detection and diagnosis [69]. Supervised learning-decision tree, RF, multilayer perception, SVM, and linear regression (LR) were compared in both the classification and regression categories. The results revealed that under the classification algorithm, the SVM provides high accuracy; however, under the regression methodology, multilayer perception regression delivers reduced errors. Díaz-Uriarte, Ramón (2006) investigated the implication of RF for classification of microarray data (including multi-class problems) and propose a new method of gene selection in classification problems based on RF [70]. The study used simulated and nine microarray data sets and demonstrated that random forest has comparable performance to other classification methods, including diagonal discriminant analysis (DLDA), KNN, and SVM, and that the new gene selection procedure yields very small sets of genes without compromising predictive accuracy.
AC Tan, D Gilbert (2003) classified cancer using gene expression data using three distinct tree-based supervised ML techniques [71]. Seven different categories of cancer data were classified using bagged and boosted decision trees (DT) alongside C4.5 DT. The bagging DT outperforms the other two. A Sharma, S Imoto, S Miyano, V Sharma (2012) proposed a Null space-based feature selection method for gene expression data in terms of supervised classification. [72]. Scatter matrices-generated null space information were utilized as a feature selection method in removing the duplicate gene expressions. After effectively lowering the dimension of the features, classification was performed using three different types of classifiers: SVM, naïve Bayes (NB), and LDA.
Degroeve, De Baets, Van de Peer and Rouz´e (2002) created a balanced train and set by randomly selecting 1000 positive instances and 1000 negative and created a test data with 281 positive and 7505 negative instances and another test data set with 281 positive and 7643 negative instances; they used SVM classifier, a NB classifier, and a traditional method for feature selection for predicting splice site and obtained improved performance. Precision obtained for these datasets ranged in 93-98% range, but the recall and F1-measures were in 25-49% range [73]. Peng, Li and Liu (2006) compared various methods of gene selection over four microarray gene expression datasets and showed that the hybrid method works well on the four datasets [74].
Sharma and Paliwal (2008) used Gradient LDA method for three small microarray gene expression datasets: acute leukemia, small round blue-cell tumor (SRBCT) and lung adenocarcinoma and have obtained higher accuracies than some competing methods [75]. Bar-Joseph, Gitter and Simon (2012) provided a discussion of how time-series gene expression data is used for identification of activated genes in biological processes and describe how basic patterns lead to gene expression programs [76]. Cho et al. (2004) proposed a modified kernel Fisher discriminant analysis (KFDA) for the analysis of the hereditary breast cancer dataset [77]. The KFDA classifier employed the mean-squared-error as the gene selection criterion. D Huang (2009) evaluated the classification performance of LDA, prediction analysis for microarrays (PAM), shrinkage centroid regularized discriminant analysis (SCRDA), shrinkage linear discriminant analysis (SLDA) and shrinkage diagonal discriminant analysis (SDDA) by applying these methods to six public cancer gene expression datasets [78].
Dwivedi (2018) used the method of Artificial Neural Network (ANN) for classification of acute cases of lymphoblastic leukemia and myeloid leukemia and reported over 98% overall classification accuracy [79]. Sun et al. (2019) used the genome deep learning method to analyze 6,083 samples from the Whole Exon Sequencing mutations with 12 types of cancer and 1991 non-cancerous samples from the 1000 Genome Project and obtained overall classification accuracies ranging in 70% - 97% [80]. A survey of feature selection literature for gene expression microarray data analysis based on a total of 132 research articles [81] was conducted by Alhenawi, Al-Sayyed, Hudaib and Mirjalili (2022). Khatun et al. (2023) developed an ensemble rank-based feature selection method (EFSM) and a weighted average voting scheme to overcome the problems posed by high dimensionality of microarray gene expression data [82]. They obtained overall classification accuracies of 100% (leukemia), 95% (colon cancer), and 94.3% for the 11-tumor dataset. Osama, Shaban and Ali (2023) have provided a review of ML methods for cancer classification of microarray gene expression data; data pre-processing and feature selection methods including filter, wrapper, embedded, ensemble, and hybrid algorithms [83].
Kabir et al. (2023) compared two different dimension reduction techniques—PCA, and autoencoders for the selection of features in a prostate cancer classification analysis. Two machine learning methods—neural networks and SVM—were further used for classification. The study showed that the classifiers performed better on the reduced dataset [84]. Another study Adiwijaya et al. (2018) utilized PCA dimension reduction method that includes the calculation of variance proportion for eigenvector selection followed by the classification methods, SVM and Levenberg-Marquardt Backpropagation (LMBP) algorithm. Based on the tests performed, the classification method using LMBP was more stable than SVM [10].
Kharya, S., D. Dubey, and S. Soni (2013) compared the accuracy of the SVM, ANN, Naive Bayes classifier, and AdaBoost tree to identify a potent model for breast cancer prediction as observational research [85]. PCA was used to reduce dimensionality. The study found that, when compared to techniques like decision trees, regression trees, and so on, ANN came out to be the one with the most reliable approach in making real-time predictions and prognoses. Rana et al (2015) used machine learning classification algorithms, which use stored historical data to learn from and forecast new input categories, benign and malignant tumors [86]. According to this study, the random forest model demonstrated the highest accuracy of 96% to detect different cancers.
Based on previous research, the general scheme in the process of classification of microarray data for the detection of cancer can be conducted via preprocessing the data and dimensionality reduction followed by cancer classification.
3. Materials and Methods
In this article, we have used the Linear Discriminant Analysis (LDA) classifier [87] and the random forest (RF) classifier [88] on an 801 rows x 20531 columns (genes) dataset of patients with five cancer types: BRCA, KIRC, COAD, LUAD and PRAD; the dataset has no missing values. Variables in this dataset are RNA-Seq gene expression levels measured by illumina HiSeq platform. The variables are dummy named gene XX. This dataset (gene expression cancer RNA-Seq) was downloaded from the UCI Machine Learning Repository [89]. The statistical software package R (2023) was used for all data analyses and visualizations [90]. We computed Principal Component (PC) scores [91] of the data and performed the 5-level classification on an increasing number of PC's and obtained excellent classification results using just the first two components PC1 and PC2. Five cancer types are described below.
We will next provide brief descriptions of the methods of data analysis and the common measures of accuracy used in multi-level classification.
3.1. Principal Components Analysis (PCA)
PCA is a dimension-reduction technique which creates new and uncorrelated linear combinations of original variables (principal components); the values of the principal components are called PC-Scores and can be used in place of the original variables for further analyses such as Multiple Linear Regression (MLR) or Discrimination and Classification. Using PC-Scores instead of original variables as predictors eliminates the problem of multicollinearity. PCA was performed using the correlation matrix which normalizes the input variables.
3.2. Linear Discriminant Analysis (LDA)
LDA is itself a dimension-reduction technique which is used for separating a dataset into 2 or more subgroups, and for classification of new data into these subgroups. LDA is typically one of the methods used for multi-level classification problems. The LDA method involves computing separating hyperplanes for classification purposes [92] (pp. 587–590). We used the function prcompfast of the R-package Morpho to first perform PCA of the gene expression microarray dataset at hand and the PC-Scores were used as input variables for LDA. All computations were performed on a Windows 10 PC with AMD Ryzen Threadpiper1950X 16-Core Processor and 128 GB usable RAM.
3.3. Random Forest (RF)
The RF method is a decision-tree based method that can be used for classification (categorical response) or regression (continuous response) problems. It randomly selects a subset of rows (samples) and a subset of columns (features) at a time and fits decision trees a very large number of times to predict Y and then uses a voting mechanism to predict Y values. Random forest is known to be highly accurate [93].
3.4. Training and Test Datasets
In ML literature, it is common practice to randomly split the available dataset into Training and Test datasets and report the accuracy measures of prediction for both datasets. Typically, higher accuracy measures are obtained for the training set than the test set. The entire raw dataset was used to compute PC-Scores by using the fast-PCA method of the R-package Morpho. A dataset of 801 rows and 25 PC-scores was created, and then this dataset of PC-scores was randomly split into an 80% training set and 20% test set. The LDA and RF methods were used on the training set of PC-Scores and the accuracy measures given below were computed for both training and test sets.
3.5. Accuracy Measures for Multi-Level Classification
All accuracy measures are computed from the confusion matrix which is a cross-tabulation of observed Y and predicted Y values.
- Overall Accuracy (OA) = sum of diagonal elements of CM/sum of all elements of CM
- Precision_j = j-th diagonal element of CM/sum of j-th column of CM
- Recall_j = j-th diagonal element of CM/sum of j-th row of CM
- F1_j = harmonic mean of Precision_j and Recall_j
The following accuracy measures are computed for each level by calculating the one vs all binary confusion matrices:
- Area Under the Curve (AUC)
- Macro- and micro-averages of AUC
- Explanations of the accuracy measures and computational details are provided in [52].
4. Results
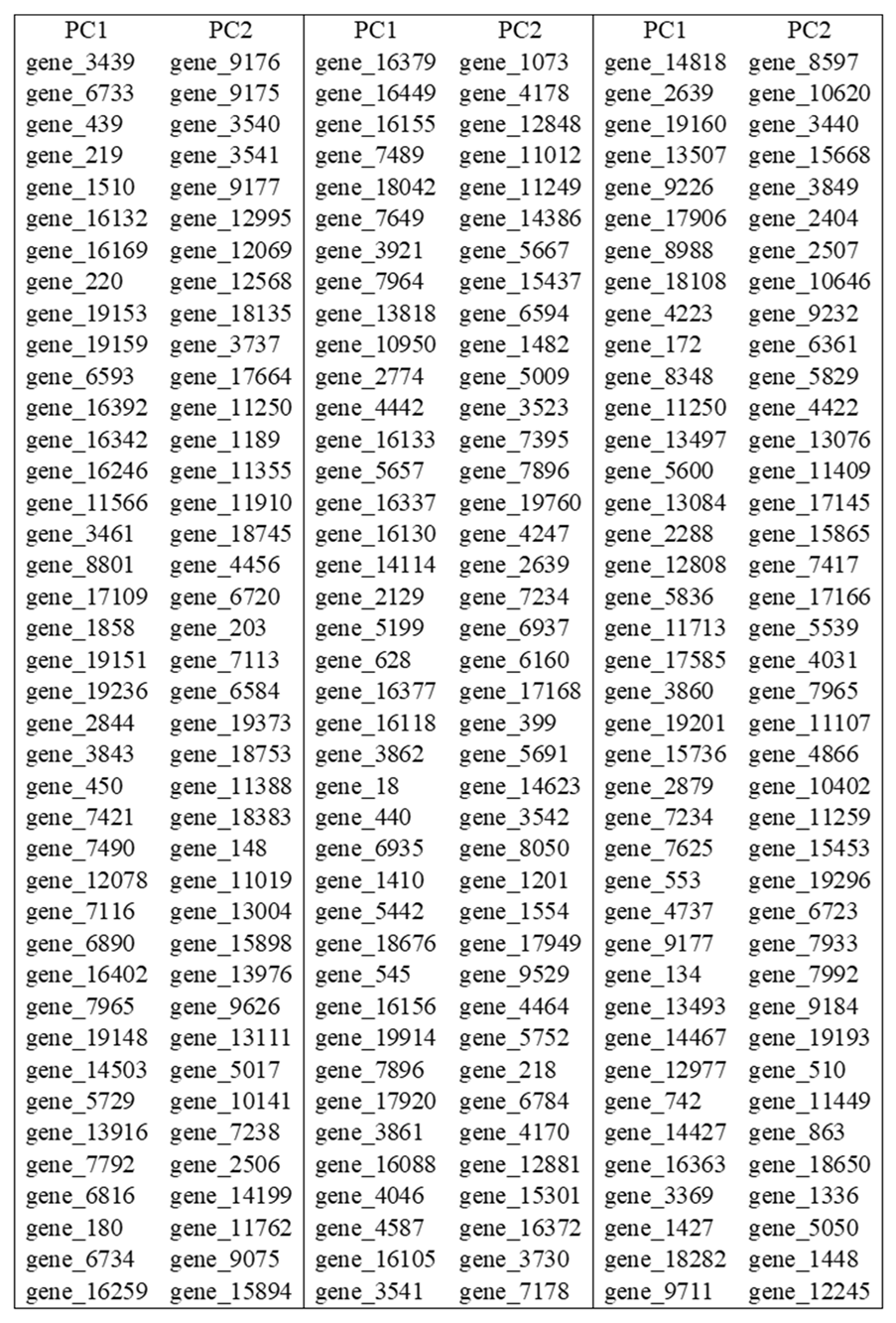
PCA was run on the entire 801 rows x 20531 genes data set, and trial-and-error showed that just the first two principal components were sufficient for classification purposes. The genes with highest absolute loadings are shown in Table 9.
A scatterplot of the first two PC-scores for the entire dataset is shown in Figure 1. A clear separation between BRCA and KIRC cancer sub-types with some overlap between COAD, LUAD and PRAD is seen in Figure 1.
Figure 1.
Scatterplot of PC2 vs PC1 for the Entire Data.
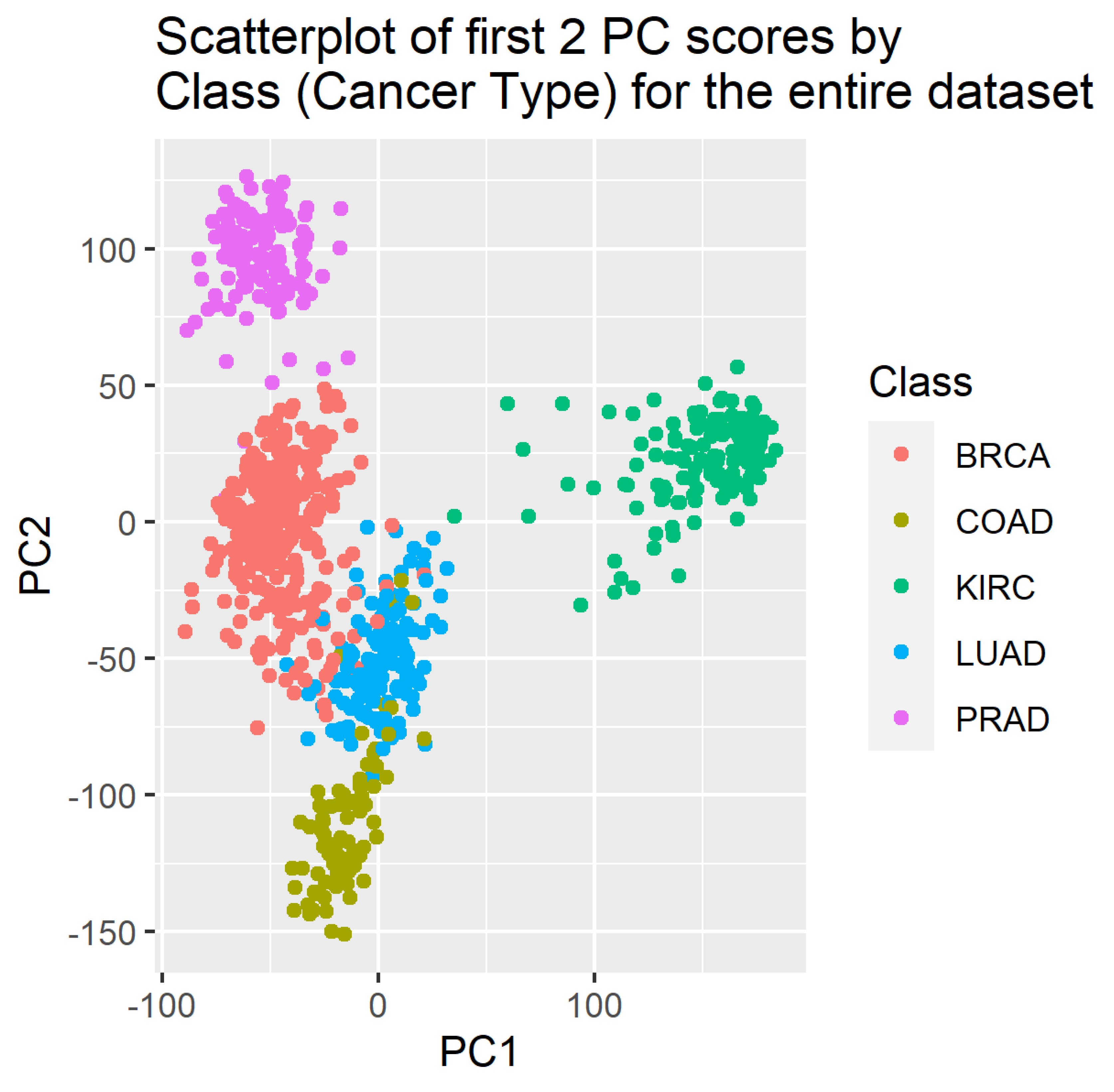
Figure 2, Figure 3, Figure 4 and Figure 5 show plots of the confusion matrices for the LDA and RF classifiers for training and test sets, respectively. Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7 and Table 8 show that all measures of multi-level accuracy are high for both training and test datasets and both LDA and RF methods.
Accuracy Measures for the LDA Classifier for Training Data
Figure 2.
Confusion Matrix Plot for the LDA Classifier – Training Data.


Table 1.
Precision, Recall, F1 and AUC Measures for the LDA Classifier – Training Data.
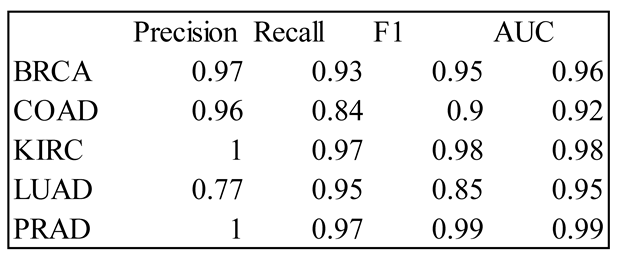
Table 2.
Macro- and Micro-Averaged AUC Measures for the LDA Classifier – Training Data.

Accuracy Measures for the LDA Classifier for Test Data
Figure 3.
Confusion Matrix Plot for the LDA Classifier – Test Data.
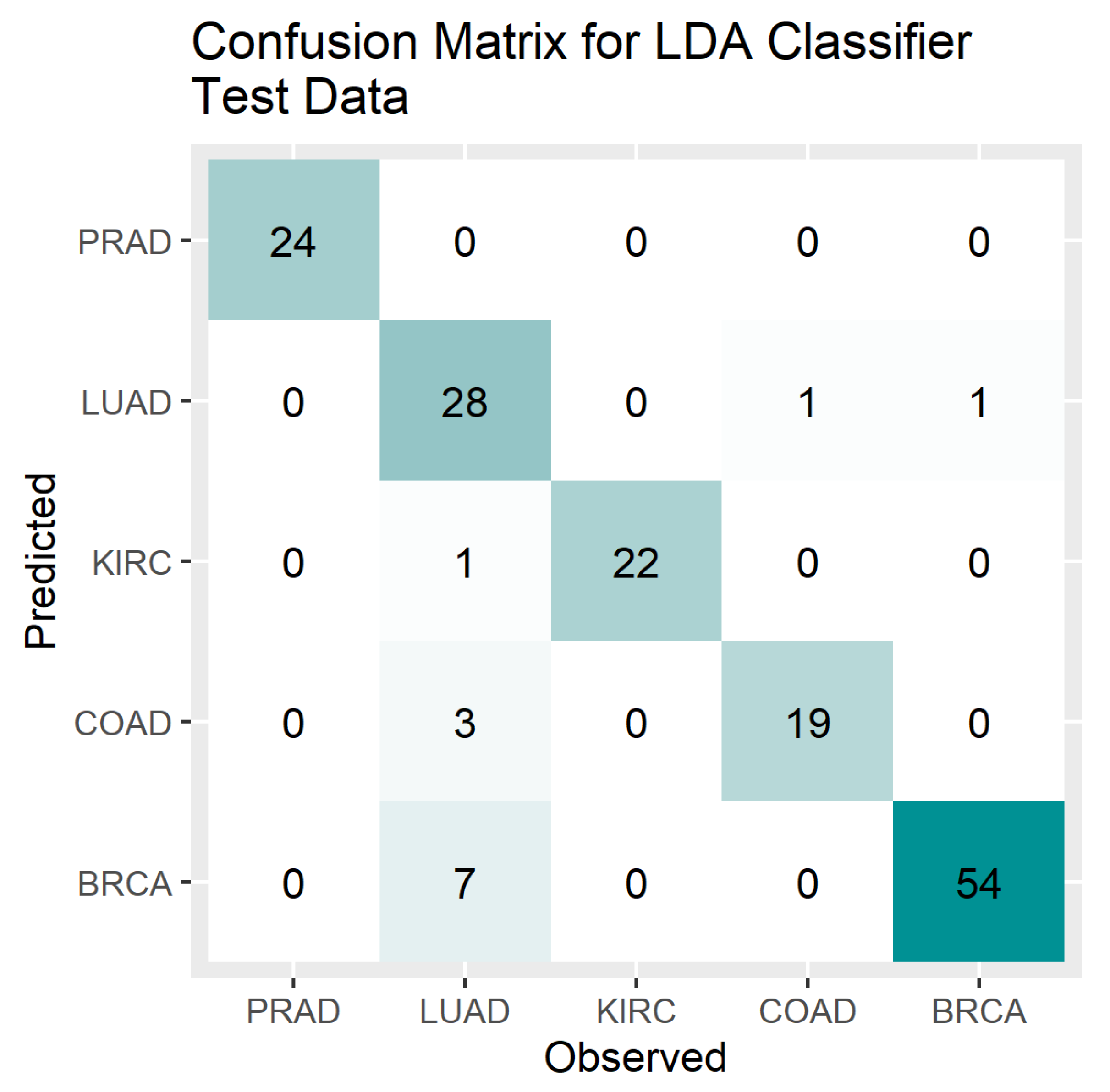
Table 3.
Confusion Matrix Plot and Accuracy Measures for the LDA Classifier – Test Data.
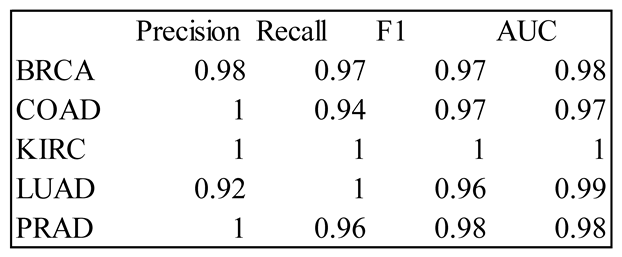
Table 4.
Macro and Micro averaged AUC for the LDA Classifier – Test Data.

Accuracy Measures for the RF Classifier for Training Data
Figure 4.
Confusion Matrix Plot for the RF Classifier – Training Data.
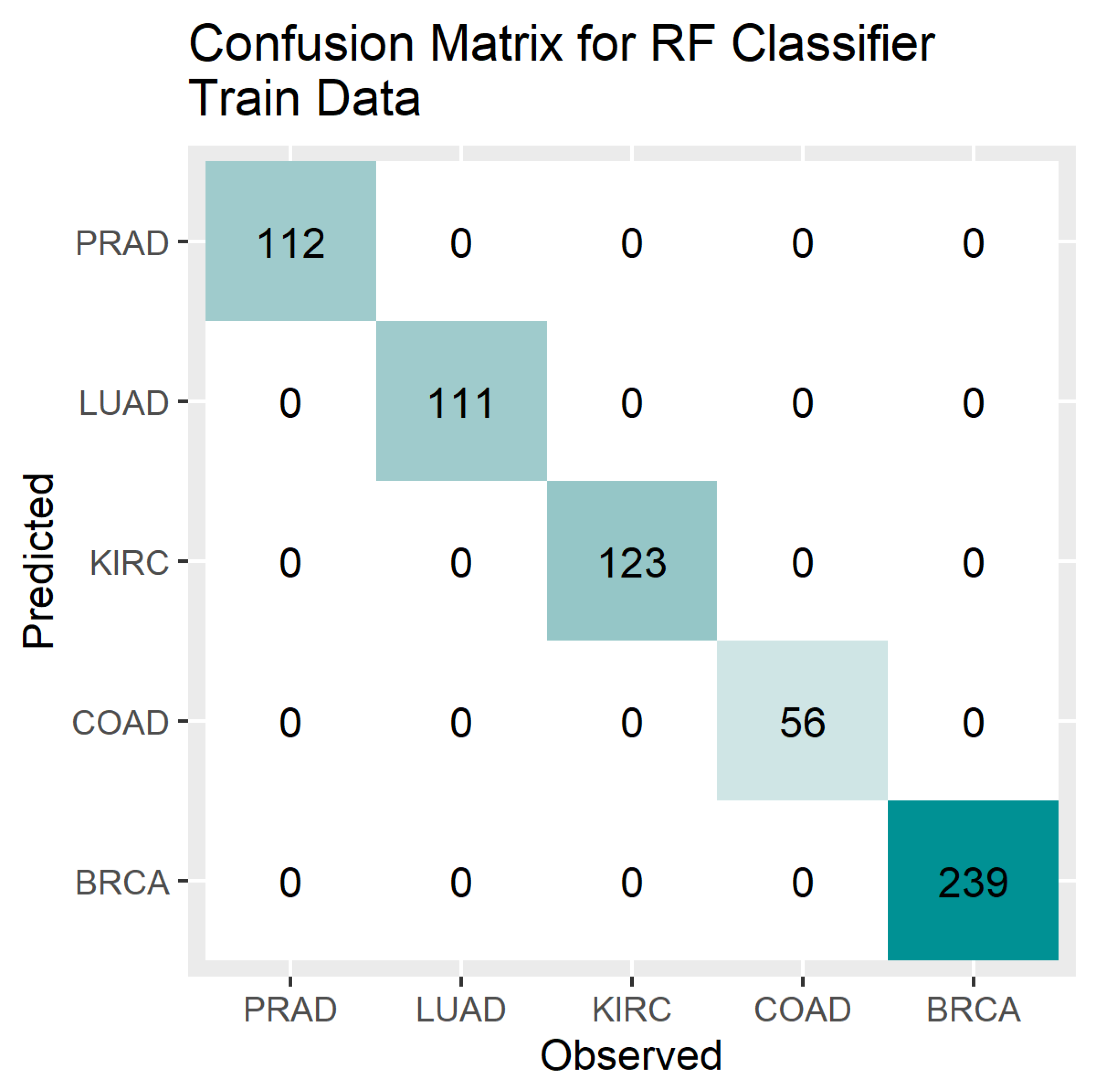
Table 5.
Precision, Recall, F1 and AUC Measures for the RF Classifier – Training Data.
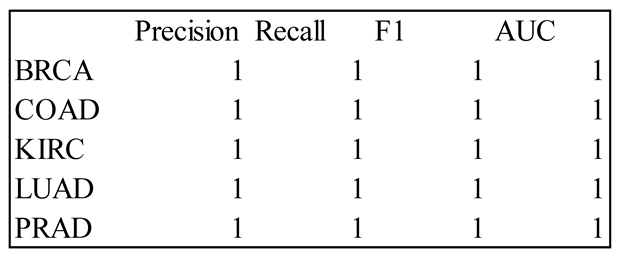
Table 6.
Macro and Micro averaged AUC for the RF Classifier – Training Data.

Accuracy Measures for the RF Classifier for Test Data
Figure 5.
Confusion Matrix Plot for the RF Classifier – Test Data.

Table 7.
Precision, Recall, F1 and AUC Measures for the RF Classifier – Test Data.
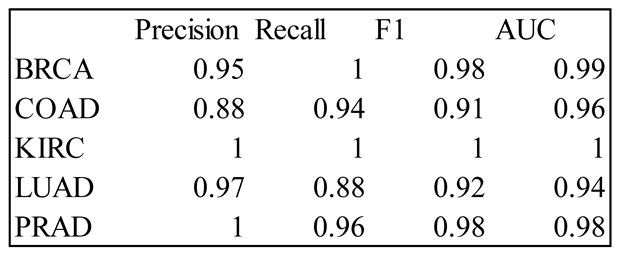
Table 8.
Macro and Micro averaged AUC for the RF Classifier – Test Data.

In Table 9 we provide the variables (genes) with high absolute loadings on the first two PC-scores; such a table can be very useful for selection of features (genes).
Table 9.
Significant genes with highest absolute loadings on the first two PC-scores.
5. Discussion
We have demonstrated successful application of PCA for dimensionality reduction on a dataset with a very large number of genes collected from a much smaller number of subjects. PCA results showed that the first 50 components (PC) cumulatively explained 71% of all variability present in the 801 × 20532 gene expression data, with the first two PC’s explaining only 26% of total variability. The first two PC’s, however, were sufficient for classification of cancer sub-types with high accuracy. This can be seen from the plot of the first two components by of cancer sub-type. LDA was able to classify each of the five cancer-subtypes with high accuracies except for LUAD which had a precision of 77% for the training set. The RF method was able to classify each sub-type with very high accuracy. The PCA loadings on 20532 genes were sorted in order of magnitude and genes (features) important for classification were identified. Our results are not generalizable, but the proposed classification method should be very helpful to researchers and clinicians working with gene expression microarray data of very high dimensionality. It should be noted that high accuracy is achieved by the LDA and the RM classifiers using just the first two PC-Scores even though only 26% of variability was explained by the first two components.
Author Contributions
Conceptualization, DM. and AKS; methodology, DM. and AKS.; validation, DM, DP, RD, LG, AS.; formal analysis, DM, AK, RD.; investigation, DM.; DM, AK, DP, LG; .; data curation, DM.; writing—original draft preparation, DM.; writing—review and editing, DM.; DM, AK, DP, LG.; visualization, DM.; supervision, AKS and DP.; project administration, AKS and DP. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
“Not applicable” since secondary data is used.
Informed Consent Statement
“Not applicable” for studies not involving humans.
Data Availability Statement
The microarray gene expression data is available at https://archive.ics.uci.edu/dataset/401/gene+expression+cancer+rna+seqmicroarray-202.
Conflicts of Interest
All authors declare no conflicts of interest.
References
- Alladi, Subha Mahadevi, Vadlamani Ravi, and Upadhyayula Suryanarayana Murthy. "Colon cancer prediction with genetic profiles using intelligent techniques." Bioinformation 3, no. 3 (2008): 130. [CrossRef]
- Alon, Uri, Naama Barkai, Daniel A. Notterman, Kurt Gish, Suzanne Ybarra, Daniel Mack, and Arnold J. Levine. "Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays." Proceedings of the National Academy of Sciences 96, no. 12 (1999): 6745-6750. [CrossRef]
- Siegel, Rebecca L., Kimberly D. Miller, Nikita Sandeep Wagle, and Ahmedin Jemal. "Cancer statistics, 2023." Ca Cancer J Clin 73, no. 1 (2023): 17-48. [CrossRef]
- Slonim, Donna K. "From patterns to pathways: Gene expression data analysis comes of age." Nature genetics 32, no. 4 (2002): 502-508. [CrossRef]
- Harrington, Christina A., Carsten Rosenow, and Jacques Retief. "Monitoring gene expression using DNA microarrays." Current opinion in Microbiology 3, no. 3 (2000): 285-291. [CrossRef]
- Schena, Mark, Dari Shalon, Ronald W. Davis, and Patrick O. Brown. "Quantitative monitoring of gene expression patterns with a complementary DNA microarray." Science 270, no. 5235 (1995): 467-470. [CrossRef]
- Brewczyński, Adam, Beata Jabłońska, Agnieszka Maria Mazurek, Jolanta Mrochem-Kwarciak, Sławomir Mrowiec, Mirosław Śnietura, Marek Kentnowski, Zofia Kołosza, Krzysztof Składowski, and Tomasz Rutkowski. "Comparison of selected immune and hematological parameters and their impact on survival in patients with HPV-related and HPV-unrelated oropharyngeal Cancer." Cancers 13, no. 13 (2021): 3256. [CrossRef]
- Munkácsy, Gyöngyi, Libero Santarpia, and Balázs Győrffy. "Gene expression profiling in early breast cancer—Patient stratification based on molecular and tumor microenvironment features." Biomedicines 10, no. 2 (2022): 248. [CrossRef]
- Siang, Tan Ching, Ting Wai Soon, Shahreen Kasim, Mohd Saberi Mohamad, Chan Weng Howe, Safaai Deris, Zalmiyah Zakaria, Zuraini Ali Shah, and Zuwairie Ibrahim. "A review of cancer classification software for gene expression data." International Journal of Bio-Science and Bio-Technology 7, no. 4 (2015): 89-108. [CrossRef]
- Adiwijaya, Wisesty Untari, E. Lisnawati, Annisa Aditsania, and Dana Sulistiyo Kusumo. "Dimensionality reduction using principal component analysis for cancer detection based on microarray data classification." Journal of Computer Science 14, no. 11 (2018): 1521-1530. [CrossRef]
- Alpaydin, Ethem. Introduction to machine learning. MIT press, 2020. [CrossRef]
- Sidey-Gibbons, Jenni AM, and Chris J. Sidey-Gibbons. "Machine learning in medicine: A practical introduction." BMC medical research methodology 19 (2019): 1-18. [CrossRef]
- Erickson, Bradley J., Panagiotis Korfiatis, Zeynettin Akkus, and Timothy L. Kline. "Machine learning for medical imaging." radiographics 37, no. 2 (2017): 505-515. [CrossRef]
- Mahmood, Nasir, Saman Shahid, Taimur Bakhshi, Sehar Riaz, Hafiz Ghufran, and Muhammad Yaqoob. "Identification of significant risks in pediatric acute lymphoblastic leukemia (ALL) through machine learning (ML) approach." Medical & Biological Engineering & Computing 58 (2020): 2631-2640. [CrossRef]
- Kononenko, Igor. "Machine learning for medical diagnosis: History, state of the art and perspective." Artificial Intelligence in medicine 23, no. 1 (2001): 89-109. [CrossRef]
- Golub, Todd R., Donna K. Slonim, Pablo Tamayo, Christine Huard, Michelle Gaasenbeek, Jill P. Mesirov, Hilary Coller et al. "Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring." science 286, no. 5439 (1999): 531-537. [CrossRef]
- Kourou, Konstantina, Themis P. Exarchos, Konstantinos P. Exarchos, Michalis V. Karamouzis, and Dimitrios I. Fotiadis. "Machine learning applications in cancer prognosis and prediction." Computational and structural biotechnology journal 13 (2015): 8-17. [CrossRef]
- Wang, Xujing, Martin J. Hessner, Yan Wu, Nirupma Pati, and Soumitra Ghosh. "Quantitative quality control in microarray experiments and the application in data filtering, normalization and false positive rate prediction." Bioinformatics 19, no. 11 (2003): 1341-1347. [CrossRef]
- Mohamad, Mohd Saberi, Sigeru Omatu, Michifumi Yoshioka, and Safaai Deris. "An approach using hybrid methods to select informative genes from microarray data for cancer classification." In 2008 Second Asia International Conference on Modelling & Simulation (AMS), pp. 603-608. IEEE, 2008. [CrossRef]
- Sung, Hyuna, Jacques Ferlay, Rebecca L. Siegel, Mathieu Laversanne, Isabelle Soerjomataram, Ahmedin Jemal, and Freddie Bray. "Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries." CA: A cancer journal for clinicians 71, no. 3 (2021): 209-249. [CrossRef]
- Reid, Alison, Nick de Klerk, and Arthur W. Musk. "Does exposure to asbestos cause ovarian cancer? A systematic literature review and meta-analysis." Cancer epidemiology, biomarkers & prevention 20, no. 7 (2011): 1287-1295. [CrossRef]
- Ünver, Halil Murat, and Enes Ayan. "Skin lesion segmentation in dermoscopic images with combination of YOLO and grabcut algorithm." Diagnostics 9, no. 3 (2019): 72. [CrossRef]
- Maniruzzaman, Md, Md Jahanur Rahman, Benojir Ahammed, Md Menhazul Abedin, Harman S. Suri, Mainak Biswas, Ayman El-Baz, Petros Bangeas, Georgios Tsoulfas, and Jasjit S. Suri. "Statistical characterization and classification of colon microarray gene expression data using multiple machine learning paradigms." Computer methods and programs in biomedicine 176 (2019): 173-193. [CrossRef]
- Kalina, Jan. "Classification methods for high-dimensional genetic data." Biocybernetics and Biomedical Engineering 34, no. 1 (2014): 10-18. [CrossRef]
- Lee, Jae Won, Jung Bok Lee, Mira Park, and Seuck Heun Song. "An extensive comparison of recent classification tools applied to microarray data." Computational Statistics & Data Analysis 48, no. 4 (2005): 869-885. [CrossRef]
- Yeung, Ka Yee, Roger E. Bumgarner, and Adrian E. Raftery. "Bayesian model averaging: Development of an improved multi-class, gene selection and classification tool for microarray data." Bioinformatics 21, no. 10 (2005): 2394-2402. [CrossRef]
- Jirapech-Umpai, Thanyaluk, and Stuart Aitken. "Feature selection and classification for microarray data analysis: Evolutionary methods for identifying predictive genes." BMC bioinformatics 6 (2005): 1-11. [CrossRef]
- Hua, Jianping, Zixiang Xiong, James Lowey, Edward Suh, and Edward R. Dougherty. "Optimal number of features as a function of sample size for various classification rules." Bioinformatics 21, no. 8 (2005): 1509-1515. [CrossRef]
- Li, Yi, Colin Campbell, and Michael Tipping. "Bayesian automatic relevance determination algorithms for classifying gene expression data." Bioinformatics 18, no. 10 (2002): 1332-1339. [CrossRef]
- Díaz-Uriarte, Ramón. "Supervised methods with genomic data: A review and cautionary view." Data analysis and visualization in genomics and proteomics (2005): 193-214. [CrossRef]
- Piao, Yongjun, Minghao Piao, Kiejung Park, and Keun Ho Ryu. "An ensemble correlation-based gene selection algorithm for cancer classification with gene expression data." Bioinformatics 28, no. 24 (2012): 3306-3315. [CrossRef]
- Chen, Kun-Huang, Kung-Jeng Wang, Kung-Min Wang, and Melani-Adrian Angelia. "Applying particle swarm optimization-based decision tree classifier for cancer classification on gene expression data." Applied Soft Computing 24 (2014): 773-780. [CrossRef]
- Akay, Mehmet Fatih. "Support vector machines combined with feature selection for breast cancer diagnosis." Expert systems with applications 36, no. 2 (2009): 3240-3247. [CrossRef]
- Brahim-Belhouari, Sofiane, and Amine Bermak. "Gaussian process for nonstationary time series prediction." Computational Statistics & Data Analysis 47, no. 4 (2004): 705-712. [CrossRef]
- Bray, Freddie, Jacques Ferlay, Isabelle Soerjomataram, Rebecca L. Siegel, Lindsey A. Torre, and Ahmedin Jemal. "Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries." CA: A cancer journal for clinicians 68, no. 6 (2018): 394-424. [CrossRef]
- Cai, Jie, Jiawei Luo, Shulin Wang, and Sheng Yang. "Feature selection in machine learning: A new perspective." Neurocomputing 300 (2018): 70-79. [CrossRef]
- Jain, Anil, and Douglas Zongker. "Feature selection: Evaluation, application, and small sample performance." IEEE transactions on pattern analysis and machine intelligence 19, no. 2 (1997): 153-158. [CrossRef]
- Wang, Yu, Igor V. Tetko, Mark A. Hall, Eibe Frank, Axel Facius, Klaus FX Mayer, and Hans W. Mewes. "Gene selection from microarray data for cancer classification—A machine learning approach." Computational biology and chemistry 29, no. 1 (2005): 37-46. [CrossRef]
- Liu, Kun-Hong, Muchenxuan Tong, Shu-Tong Xie, and Vincent To Yee Ng. "Genetic programming based ensemble system for microarray data classification." Computational and mathematical methods in medicine 2015 (2015). [CrossRef]
- Bhonde, Swati B., and Jayashree R. Prasad. "Performance analysis of dimensionality reduction techniques in cancer detection using microarray data." Asian Journal For Convergence In Technology (AJCT) ISSN-2350-1146 7, no. 1 (2021): 53-57. [CrossRef]
- Sun, Xiaoxiao, Yiwen Liu, and Lingling An. "Ensemble dimensionality reduction and feature gene extraction for single-cell RNA-seq data." Nature communications 11, no. 1 (2020): 5853. [CrossRef]
- Rowe, Raymond C., and Ronald J. Roberts. "Artificial intelligence in pharmaceutical product formulation: Knowledge-based and expert systems." Pharmaceutical Science & Technology Today 1, no. 4 (1998): 153-159. [CrossRef]
- Yu, Chaoran, and Ernest Johann Helwig. "The role of AI technology in prediction, diagnosis and treatment of colorectal cancer." Artificial Intelligence Review 55, no. 1 (2022): 323-343. [CrossRef]
- Kumar, Yogesh, Surbhi Gupta, Ruchi Singla, and Yu-Chen Hu. "A systematic review of artificial intelligence techniques in cancer prediction and diagnosis." Archives of Computational Methods in Engineering 29, no. 4 (2022): 2043-2070. [CrossRef]
- McKinney, Scott Mayer, Marcin Sieniek, Varun Godbole, Jonathan Godwin, Natasha Antropova, Hutan Ashrafian, Trevor Back et al. "International evaluation of an AI system for breast cancer screening." Nature 577, no. 7788 (2020): 89-94. [CrossRef]
- Mersch, Jacqueline, Michelle A. Jackson, Minjeong Park, Denise Nebgen, Susan K. Peterson, Claire Singletary, Banu K. Arun, and Jennifer K. Litton. "Cancers associated with BRCA 1 and BRCA 2 mutations other than breast and ovarian." Cancer 121, no. 2 (2015): 269-275. [CrossRef]
- Chang, Michael, Rohan J. Dalpatadu, Dieudonne Phanord, and Ashok K. Singh. "Breast cancer prediction using bayesian logistic regression." Biostatistics and Bioinformatics 2, no. 3 (2018): 1-5. [CrossRef]
- Wolberg, William, W. Street, and Olvi Mangasarian. "Breast cancer wisconsin (diagnostic)." UCI Machine Learning Repository 414 (1995): 415. [CrossRef]
- Cordova, Claudio, Roberto Muñoz, Rodrigo Olivares, Jean-Gabriel Minonzio, Carlo Lozano, Paulina Gonzalez, Ivanny Marchant, Wilfredo González-Arriagada, and Pablo Olivero. "HER2 classification in breast cancer cells: A new explainable machine learning application for immunohistochemistry." Oncology Letters 25, no. 2 (2023): 1-9. [CrossRef]
- Hu, Fuyan, Wenying Zeng, and Xiaoping Liu. "A gene signature of survival prediction for kidney renal cell carcinoma by multi-omic data analysis." International journal of molecular sciences 20, no. 22 (2019): 5720. [CrossRef]
- Wang, Licheng, Yaru Zhu, Zhen Ren, Wenhuizi Sun, Zhijing Wang, Tong Zi, Haopeng Li et al. "An immunogenic cell death-related classification predicts prognosis and response to immunotherapy in kidney renal clear cell carcinoma." Frontiers in Oncology 13 (2023): 1147805. [CrossRef]
- Yue, Fu-Ren, Zhi-Bin Wei, Rui-Zhen Yan, Qiu-Hong Guo, Bing Liu, Jing-Hui Zhang, and Zheng Li. "SMYD3 promotes colon adenocarcinoma (COAD) progression by mediating cell proliferation and apoptosis." Experimental and Therapeutic Medicine 20, no. 5 (2020): 1-1. [CrossRef]
- Li, Yuanyuan, Kai Kang, Juno M. Krahn, Nicole Croutwater, Kevin Lee, David M. Umbach, and Leping Li. "A comprehensive genomic pan-cancer classification using The Cancer Genome Atlas gene expression data." BMC genomics 18 (2017): 1-13. [CrossRef]
- Liu, Yangyang, Lu Liang, Liang Ji, Fuquan Zhang, Donglai Chen, Shanzhou Duan, Hao Shen, Yao Liang, and Yongbing Chen. "Potentiated lung adenocarcinoma (LUAD) cell growth, migration and invasion by lncRNA DARS-AS1 via miR-188-5p/KLF12 axis." Aging (Albany NY) 13, no. 19 (2021): 23376. [CrossRef]
- Yang, Jian, Zhike Chen, Zetian Gong, Qifan Li, Hao Ding, Yuan Cui, Lijuan Tang et al. "Immune landscape and classification in lung adenocarcinoma based on a novel cell cycle checkpoints related signature for predicting prognosis and therapeutic response." Frontiers in Genetics 13 (2022): 908104. [CrossRef]
- Liu, Qian, Jiali Lei, Xiaobo Zhang, and Xiaosheng Wang. "Classification of lung adenocarcinoma based on stemness scores in bulk and single cell transcriptomes." Computational and Structural Biotechnology Journal 20 (2022): 1691-1701. [CrossRef]
- Zhao, Xin, Daixing Hu, Jia Li, Guozhi Zhao, Wei Tang, and Honglin Cheng. "Database mining of genes of prognostic value for the prostate adenocarcinoma microenvironment using the cancer gene atlas." BioMed research international 2020 (2020). [CrossRef]
- Khosravi, Pegah, Maria Lysandrou, Mahmoud Eljalby, Qianzi Li, Ehsan Kazemi, Pantelis Zisimopoulos, Alexandros Sigaras et al. "A deep learning approach to diagnostic classification of prostate cancer using pathology–radiology fusion." Journal of Magnetic Resonance Imaging 54, no. 2 (2021): 462-471. [CrossRef]
- Basilevsky, Alexander T. Statistical factor analysis and related methods: Theory and applications. John Wiley & Sons, 2009. [CrossRef]
- Everitt, Brian, and Graham Dunn. Applied multivariate data analysis. Vol. 2. London: Arnold, 2001. [CrossRef]
- Pearson, Karl. "LIII. On lines and planes of closest fit to systems of points in space." The London, Edinburgh, and Dublin philosophical magazine and journal of science 2, no. 11 (1901): 559-572. [CrossRef]
- Hilsenbeck, Susan G., William E. Friedrichs, Rachel Schiff, Peter O'Connell, Rhonda K. Hansen, C. Kent Osborne, and Suzanne AW Fuqua. "Statistical analysis of array expression data as applied to the problem of tamoxifen resistance." Journal of the National Cancer Institute 91, no. 5 (1999): 453-459. [CrossRef]
- Vohradsky, Jiří, Xin-Ming Li, and Charles J. Thompson. "Identification of procaryotic developmental stages by statistical analyses of two-dimensional gel patterns." Electrophoresis 18, no. 8 (1997): 1418-1428. [CrossRef]
- Craig, J. C., J. H. Eberwine, J. A. Calvin, B. Wlodarczyk, G. D. Bennett, and R. H. Finnell. "Developmental expression of morphoregulatory genes in the mouse embryo: An analytical approach using a novel technology." Biochemical and molecular medicine 60, no. 2 (1997): 81-91. [CrossRef]
- Liu, JingJing, WenSheng Cai, and XueGuang Shao. "Cancer classification based on microarray gene expression data using a principal component accumulation method." Science China Chemistry 54 (2011): 802-811. [CrossRef]
- Oladejo, Ayomikun Kubrat, Tinuke Omolewa Oladele, and Yakub Kayode Saheed. "Comparative evaluation of linear support vector machine and K-nearest neighbour algorithm using microarray data on leukemia cancer dataset." Afr. J. Comput. ICT 11, no. 2 (2018): 1-10. https://afrjcict.net/2017/08/29/african-journal-of-computing-ict/.
- Adebiyi, Marion Olubunmi, Micheal Olaolu Arowolo, Moses Damilola Mshelia, and Oludayo O. Olugbara. "A linear discriminant analysis and classification model for breast cancer diagnosis." Applied Sciences 12, no. 22 (2022): 11455. [CrossRef]
- Ak, Muhammet Fatih. "A comparative analysis of breast cancer detection and diagnosis using data visualization and machine learning applications." In Healthcare, vol. 8, no. 2, p. 111. MDPI, 2020. [CrossRef]
- Díaz-Uriarte, Ramón, and Sara Alvarez de Andrés. "Gene selection and classification of microarray data using random forest." BMC bioinformatics 7 (2006): 1-13. [CrossRef]
- Tan, Aik Choon, and David Gilbert. "Ensemble machine learning on gene expression data for cancer classification." (2003). URL: http://bura.brunel.ac.uk/handle/2438/3013.
- Sharma, Alok, Seiya Imoto, Satoru Miyano, and Vandana Sharma. "Null space based feature selection method for gene expression data." International Journal of Machine Learning and Cybernetics 3 (2012): 269-276. [CrossRef]
- Degroeve, Sven, Bernard De Baets, Yves Van de Peer, and Pierre Rouzé. "Feature subset selection for splice site prediction." Bioinformatics 18, no. suppl_2 (2002): S75-S83.7. [CrossRef]
- Peng, Yanxiong, Wenyuan Li, and Ying Liu. "A hybrid approach for biomarker discovery from microarray gene expression data for cancer classification." Cancer informatics 2 (2006): 117693510600200024. [CrossRef]
- Sharma, Alok, and Kuldip K. Paliwal. "Cancer classification by gradient LDA technique using microarray gene expression data." Data & Knowledge Engineering 66, no. 2 (2008): 338-347. [CrossRef]
- Bar-Joseph, Ziv, Anthony Gitter, and Itamar Simon. "Studying and modelling dynamic biological processes using time-series gene expression data." Nature Reviews Genetics 13, no. 8 (2012): 552-564. [CrossRef]
- Cho, Ji-Hoon, Dongkwon Lee, Jin Hyun Park, and In-Beum Lee. "Gene selection and classification from microarray data using kernel machine." FEBS letters 571, no. 1-3 (2004): 93-98. [CrossRef]
- Huang, Desheng, Yu Quan, Miao He, and Baosen Zhou. "Comparison of linear discriminant analysis methods for the classification of cancer based on gene expression data." Journal of experimental & clinical cancer research 28 (2009): 1-8. [CrossRef]
- Dwivedi, Ashok Kumar. "Artificial neural network model for effective cancer classification using microarray gene expression data." Neural Computing and Applications 29 (2018): 1545-1554. [CrossRef]
- Sun, Yingshuai, Sitao Zhu, Kailong Ma, Weiqing Liu, Yao Yue, Gang Hu, Huifang Lu, and Wenbin Chen. "Identification of 12 cancer types through genome deep learning." Scientific reports 9, no. 1 (2019): 17256. [CrossRef]
- Alhenawi, Esra'A., Rizik Al-Sayyed, Amjad Hudaib, and Seyedali Mirjalili. "Feature selection methods on gene expression microarray data for cancer classification: A systematic review." Computers in Biology and Medicine 140 (2022): 105051. [CrossRef]
- Khatun, R., Akter, M., Islam, M.M., Uddin, M.A., Talukder, M.A., Kamruzzaman, J., Azad, A.K.M., Paul, B.K., Almoyad, M.A.A., Aryal, S. and Moni, M.A., 2023. Cancer classification utilizing voting classifier with ensemble feature selection method and transcriptomic data. Genes, 14(9), p.1802. [CrossRef]
- Osama, Sarah, Hassan Shaban, and Abdelmgeid A. Ali. "Gene reduction and machine learning algorithms for cancer classification based on microarray gene expression data: A comprehensive review." Expert Systems with Applications 213 (2023): 118946. [CrossRef]
- Kabir, Md Faisal, Tianjie Chen, and Simone A. Ludwig. "A performance analysis of dimensionality reduction algorithms in machine learning models for cancer prediction." Healthcare Analytics 3 (2023): 100125. [CrossRef]
- Kharya, S., D. Dubey, and S. Soni. "Predictive machine learning techniques for breast cancer detection." International journal of computer science and information Technologies 4, no. 6 (2013): 1023-8.
- Rana, Mandeep, Pooja Chandorkar, Alishiba Dsouza, and Nikahat Kazi. "Breast cancer diagnosis and recurrence prediction using machine learning techniques." International journal of research in Engineering and Technology 4, no. 4 (2015): 372-376. [CrossRef]
- Johnson, Richard Arnold, and Dean W. Wichern. "Applied multivariate statistical analysis." (2002). https://books.google.com/books?id=gFWcQgAACAAJ.
- Genuer, Robin, Jean-Michel Poggi, Robin Genuer, and Jean-Michel Poggi. Random forests. Springer International Publishing, 2020. [CrossRef]
- Frank, Andrew. "UCI machine learning repository." http://archive. ics. uci. edu/ml (2010). [CrossRef]
- Team, R. Core. "R: A language and environment for statistical computing. R Foundation for Statistical Computing." (No Title) (2013). https://www.R-project.org/.
- Jolliffe, Ian T., and Jorge Cadima. "Principal component analysis: A review and recent developments." Philosophical transactions of the royal society A: Mathematical, Physical and Engineering Sciences 374, no. 2065 (2016): 20150202. [CrossRef]
- Hastie, Trevor, Robert Tibshirani, Jerome H. Friedman, and Jerome H. Friedman. The elements of statistical learning: Data mining, inference, and prediction. Vol. 2. New York: Springer, 2009. [CrossRef]
- Molin, Nicole L., Clifford Molin, Rohan J. Dalpatadu, and Ashok K. Singh. "Prediction of obstructive sleep apnea using Fast Fourier Transform of overnight breath recordings." Machine Learning with Applications 4 (2021): 100022. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



