
Submitted:
29 January 2024
Posted:
30 January 2024
You are already at the latest version
Abstract
The homeostasis of cellular calcium is fundamental for many physiological activities, while calcium levels maintain inhomogeneous within cells. During the onset of asthma, epithelial and inflammatory cells secrete platelet derived growth factor (PDGF), inducing proliferation and migration of airway smooth muscle (ASM) to the epidermal layer, narrowing the airway. The regulation of ASM cells by PDGF is closely related to the conduction of calcium signals. In this work, we generated subcellular-targeted FRET biosensors to investigate calcium regulations at different compartments of ASM cells. PDGF-induced cytoplasmic calcium [Ca2+]C increase was attributed from both extracellular calcium influx and endoplasmic reticulum (ER) calcium [Ca2+]ER release, which was partially regulated by PLC-IP3R pathway. Interestingly, removal of extracellular calcium influx led to inhibited ER calcium release, likely through inhibitory effect on calcium-dependent activation of ER ryanodine receptor. Inhibition of L-type calcium channel on plasma membrane, or SERCA pump on ER resulted in both reduced [Ca2+]C and [Ca2+]ER from PDGF stimulation, while IP3R channel inhibition led to reduced [Ca2+]C only. Inhibited SERCA pump caused immediate [Ca2+]C increase and [Ca2+]ER decrease, indicating an active calcium exchange between cytosol and ER storage in resting cells. PDGF-induced calcium at outer mitochondrial membrane sub-region showed similar regulatory response to cytosolic calcium, not influenced by inhibition of mitochondrial calcium uniporter channel. Therefore, our work identified calcium flow pathways among extracellular medium, cell cytosol, and ER by regulatory calcium channels. Particularly, extracellular calcium flow has an essential function to fully activate ER calcium release.
Keywords:
calcium signal
; calcium channels
; FRET biosensor
; subcellular calcium regulation
; platelet derived growth factor (PDGF)
; airway smooth muscle cells
Running title: FRET study of subcellular calcium regulations
1. Introduction
Calcium ions (Ca2+), as one ubiquitous messenger in cells, are one of the most important biochemical signals involved in many cellular physiological processes. Changes in Ca2+ concentration often lead to changes in downstream related kinase levels, thereby regulating physiological activities, such as cell differentiation[1,2], apoptosis[3], gene expression[4], etc. The calcium signal transduction in cells is precise and diverse, and similar trends may lead to different outcomes. The interaction between organelles and the action of calcium channels jointly maintains the dynamic balance of intracellular calcium levels. The intracellular Ca2+ homeostasis is an important foundation for maintaining normal physiological activities of cells. Abnormal calcium signaling can lead to the occurrence of diseases. Asthma, a chronic airway disease, often accompanied by shortness of breath and airway remodeling during onset[5,6]. At the same time, airway epithelial cells and inflammatory cells secrete PDGF to promote the contraction and proliferation of airway smooth muscle (ASM) cells[7,8]. The regulation of ASM cells largely relies on the conduction of calcium signals[9].
In fact, the distribution of Ca2+ within cells is not uniform, and there is a certain concentration gradient. Among them, the endoplasmic reticulum (ER) is the largest intracellular organelle related to Ca2+ storage[10]. Under normal physiological conditions, the level of Ca2+ in the ER can reach over a thousand times of the Ca2+ concentration in cytoplasm[11]. Excessive or insufficient calcium levels in ER can promote unfolded protein reaction response and cause cell apoptosis[12]. Mitochondria also have the function of storing and releasing calcium, and play an important role in the conduction of calcium signals[13]. The enrichment of calcium into the mitochondrial matrix can promote mitochondrial ATP production, but excessive calcium concentration can also induce cell apoptosis[14]. In cytoplasm, the increase in calcium levels is mainly due to the action of calcium channels on the plasma membrane and the release of calcium stored in organelles. The uptake of extracellular calcium by cells mainly depends on several calcium channels for controlling calcium influx on the plasma membrane, such as L-type calcium channels[15], and store-operated Ca2+(SOC) channels[16].
The regulation of calcium signaling by cells receiving extracellular cytokine stimulation is inseparable from the G protein coupled receptor (GPCR) on the plasma membrane. GPCR can capture the stimulation of cytokines and convert them into the activation of intracellular phospholipase C (PLC)[17]. Phosphorylated PLC can further promote the production of inositol-1,4,5 tris-phosphonate (IP3)[18]. IP3 receptor (IP3R) is predominantly located on the ER, and induction by IP3 can promote the release of calcium from the ER, leading to an upregulation of cytoplasmic calcium levels[19,20]. In addition, there is another calcium release channel on the ER, the ryanodine receptor [21], and the ER organelles maintain high calcium level through SERCA pump uptake of calcium from cytoplasm[22]. The uptake of calcium by mitochondria depends on related calcium transporters, such as mitochondrial calcium uniporter (MCU)[23]. In addition, studies have shown that mitochondrial associated membranes (MAMs), the contact part between the outer and ER membranes of mitochondria, are involved in calcium signaling between the ER and mitochondria[24,25].
The changes in intracellular calcium signals are a rapidly changing process, sometimes recovering from calcium oscillations to a resting state in just a few tens of seconds. Traditional chemical fluorescent dyes for calcium verification often have inaccuracies, significant errors, and cannot reflect changes in subcellular calcium levels in a timely manner. Fluorescence resonance energy transfer (FRET) is an emerging technique in recent decades, with timeliness and the ability to observe changes in kinase levels at the cellular or subcellular level in a targeted manner[26,27]. FRET-based calcium biosensors enable real-time visualization of changes in intracellular and subcellular calcium levels. Calmodulin (CaM) is the core part of the construction of calcium biosensors[28]. CaM has affinity for calcium ions, which can cause certain conformational changes after binding to calcium ions, resulting in spatial positional changes in the fluorescence fragments at both ends of the sensor[29], and generating FRET changes. By adding localization peptides[30], FRET biosensors can achieve calcium detection at subcellular levels.
In this paper, we used FRET technique for real-time monitoring calcium levels in cytoplasm, ER, mitochondria outer membrane (Out-Mito) and mitochondrial matrix (mito-matrix) by four versions of FRET biosensors. They are cytoplasmic calcium (Cyto-Ca2+), ER calcium (ER-Ca2+), mitochondrial outer membrane calcium (Out-Mito-Ca2+), and mito-matrix calcium (Mito-Ca2+) biosensors. These FRET calcium biosensors were proteins consisting of four core parts including ECFP, CaM, M13 peptide and YPet[31], while specific signals peptides can help target the biosensor proteins to subcellular locations (diagram shown in Figure 1A). When the calmodulin (CaM) in FRET biosensors bind to calcium in cells, CaM can interact with M13 to change the spatial position of the two fluorescence fragments, hence the FRET changes were generated[32]. In this work, by combining FRET biosensors with several common calcium channels’ inhibitors, the PDGF-induced flow mechanism of calcium signaling at different compartments of ASM cells was explored. Our work shows that PDGF stimulation promotes calcium influx through the plasma membrane and calcium release from the ER, leading to increase of cytosolic calcium level and mitochondrial uptake of calcium. Moreover, the extracellular calcium influx through plasma membrane affected ER calcium release, which is not entirely dependent on the PLC-IP3R pathway.
2. Materials and Methods
2.1. Cell culture
The airway smooth muscle (ASM) cells used in our experiments were purchased from Beina Biotech Co., which are derived from the ASM of female Sprague Dawley rats aged 6-8 weeks. Primary rat ASM cells were cultured in a low sugar medium (DMEM, Sigma Aldrich) containing 10% fetal bovine serum (FBS, Thermo), and maintained in a humid incubator at 37℃ with 5% CO2.
2.2. Chemical reagents
Dimethyl sulfoxide (DMSO) and platelet derived growth factor (PDGF) were purchased from Beyotime Biotechnology; 2-Amino-ethoxydiphenyl borate (2-APB), and nifedipine were from Sigma-Aldrich; Thapsigargin and U73122 from MedChemExpress; Ruthenium red (RuR) from Macklin; Fibronectin and calcium free culture medium from Thermo Fisher Scientific.
2.3. Constructions of calcium FRET plasmids
The four versions of calcium FRET biosensor plasmids include cytoplasmic calcium (Cyto-Ca2+), ER calcium (ER-Ca2+), mitochondria outer membrane calcium (Out-Mito-Ca2+) and mito-matrix calcium (Mito-Ca2+) biosensors (listed in Figure 1A). For Cyto-Ca2+ biosensor, the FRET expression plasmid was using pcDNA3.1-Ca2+-YPet version reported in our previous work[31].
For ER-Ca2+ biosensor, the expression plasmid was generated using the pcDNA3.1-Ca2+-YPet and pRSETb-Ca2+-YPet plasmids as templates. By the PCR primer design, the DNA sequence of ER-targeting signal peptide (MLLPVLLLGLLGAAAD) [33] was added to the 5' end of the forward primer behind Hind III restriction enzyme site while the ER retention peptide (KDEL) sequence was added to the 5' end of the reverse primer before EcoR I site. The DNA fragment, which contains Ca2+-YPet portion along with the two signal peptides, was amplified by PCR using pRSETb-Ca2+-YPet plasmid as the template. After the PCR product was purified using the gel extraction kit (Vazyme, DC301-01), the fragment “ER-ECFP-CaM-M13-YPet-KDEL” and pcDNA3.1-Ca2+-YPet plasmid were both double digested using Hind III and EcoR I. The digested PCR fragment and pcDNA3.1 vector were ligated together to generate ER-Ca2+ biosensor.
Similarly, for Out-Mito-Ca2+ biosensor, the signal peptide DAKAP1 encoding sequence[34] was added to the 5' end of the forward primer behind Hind III. The DAKAP1-ECFP-CaM-M13-YPet fragment was amplified by PCR, and ligated into pcDNA3.1 vector after digestion with Hind III and EcoR I to generate the Out-Mito-Ca2+ biosensor.
For Mito-Ca2+ biosensor, we obtained the mitochondrial matrix-targeting signal DNA fragment by gene synthesis service, which contains the signal peptide sequence as four folds of COX8 (4xMSVLTPLLLRGLTGSARRLPVPRAKIHSLGDP) and a linker peptide (RSGSAKDPT)[35]. The 4xCOX8-linker fragment and pcDNA3.1-ECFP-CaM-M13-YPet were double digested with Nhe I and Hind III, and ligated together to generate the Mito-Ca2+ biosensor.
2.4. FRET plasmid transfection
According to the instruction of Lipofectamine 3000 (Invitrogen), the appropriate number of ASM cells were incubated into a 12-well plate overnight in advance, and then FRET plasmids (1.5 μg per well) were transfected into cells by using Lipofectamine 3000 regents. After 8-10 hours, culture medium was replaced with a new low sugar DMEM medium containing 10% FBS. After 36 hours of transfection, ASM cells were digested with Accutase Cell Dissociation Reagent (Thermo) and inoculated into a confocal dish (NEST) precoated with fibronectin. Then the cells were subjected to starvation treatment in medium containing 1% FBS for 16-20 hours. Subsequently, FRET imaging experiments were conducted.
2.5. Intracellular FRET imaging of the calcium biosensors
The Zeiss microscope imaging system has a multi-position function and is equipped with a cell culture chamber (Zeiss). In the FRET microscope system, the filter parameters of the ECFP and FRET channels are excitation (436 ± 10 nm), dichroic mirror (455 nm), and emission (480 ± 20 nm) for ECFP, and emission (535 ± 15 nm) for YPet. In our imaging experiments, an x100 Oil objective was chosen to acquire the FRET images, and the ASM cell samples were placed in the cell culture chamber. By controlling the fast switch between ECFP and FRET channels through Zeiss software system, the image data of ASM cells from both channels were collected almost simultaneously. During the experimental process, fluorescence images were collected at interval of 1 min for 20 minutes duration.
Inhibitor pretreatments include adding appropriate concentrations of reagents such as DMSO (<0.1% v/v), 2-APB (100 μM), nifedipine (10 μM), and thapsigargin (10 μM), or RuR (10 μM) in advance, followed by incubation for 1 hour before cell imaging. To add PDGF stimulation (50 ng/ mL) in the middle, 1 mL medium containing PDGF was injected into the dish through a guiding microtube without disruption of the imaging process. In the experimental group with calcium free culture, L-glutamine (0.4 mL) and sodium pyruvate (0.2 mL) were added to a calcium free medium (19.4 mL) in advance. Before performing live cell imaging, removed the culture medium, washed three times with PBS (phosphate buffered saline), and then added pre-prepared calcium-free culture medium immediately before placed on the microscope.
2.6. Data processing
Quantitative analysis of image data was conducted using the FRET image analysis software FluoCell 6.0.0[36]. Principally, after background subtraction, the ratio of the fluorescence intensity was calibrated between ECFP and FRET channels by pixel to pixel, and ratiometric images were acquired along with FRET/ECFP ratio data. The data statistical analysis was conducted in GraphPad Prism 6.0 software. The time-course curves of FRET ratio (Mean ± S.E.M.), and the graphs with scattering dots (Mean ± S.D.) along with the differences between each two data groups were analyzed.
Student’s t-tests were conducted between the control group and an experimental group, and multiple rounds of t-tests were conducted on variable experimental conditions. *, **, ***, and **** indicate p value < 0.05, 0.01, 0.001, and 0.0001 for significant difference, while 'ns' indicates no significant difference.
3. Results
3.1. Extracellular calcium entry affects both calcium upregulation in cytoplasm and calcium release from ER storage
When asthma attacks, it is accompanied by narrowing of the airway and shortness of breath. PDGF is an important growth factor that induces ASM cells proliferation and migration into the epidermal layer[9]. Studies have shown that PDGF can significantly cause an increase of calcium concentration in the cytoplasm[2]. Here, we used PDGF as inducer and FRET Cyto-Ca2+ biosensor for real-time monitoring calcium level in the cytosol of ASM cells. As shown in Figure 1B&D (the time-course shown in Movie S1), after stimulation with PDGF, calcium ions in the cell cytosol [Ca2+]C was detected with a rapid increase by Cyto-Ca2+ biosensor. PLC plays an important role in promoting calcium release from the ER via IP3-IP3R pathway, and U73122 is a common PLC inhibitor. By pretreating ASM cells with U73122, PDGF-induced upregulation of cytoplasmic calcium signaling in cells was inhibited to a certain extent (Figure 1B&D). This indicates that the PLC pathway plays a role in calcium signaling induced by PDGF.
To check whether cytoplasmic calcium increase depends on calcium flow through plasma membrane channels from the medium, we studied the effect with calcium-free culture medium. [Ca2+]C increase induced by PDGF was severely inhibited when without calcium in the medium (Figure 1C&D). After pretreating ASM cells with U73122, PDGF-induced [Ca2+]C was further reduced in calcium-free medium (Figure 1C&D). Statistical quantifications confirmed that PDGF-induced cytoplasmic calcium levels and the percentage changes were regulated by both PLC signal and calcium influx from the medium (Figure 1E&F).
Endoplasmic reticulum (ER) is the largest calcium storage site in cells, which contributes mostly to the rapid change of cytoplasmic calcium levels[10]. By targeting the calcium biosensor into ER, it allows to monitor calcium level at the subcellular organelles. Here, we attempted to investigate the relationships of calcium exchanges among the cell cytosol, the ER and the extracellular medium by using ER-Ca2+ biosensor. As shown in Figure 1G&I (the time-course in Movie S2), PDGF stimulation can lead to a dramatic decrease in ER calcium concentration [Ca2+]ER, in corresponding to the [Ca2+]C increase (Figure 1B&C). In comparison, the pretreatment of cells with U73122 resulted in a greater decrease in [Ca2+]ER (Figure 1G&I), indicating that the pre-inhibition of PLC pathway reduced the concentration of free calcium in ER. In calcium-free culture medium, the decreasing rate of [Ca2+]ER was apparently reduced in comparison to normal culture medium, in corresponding to the reduced increase in cytoplasmic calcium (Figure 1H&I). Statistical quantifications demonstrated that under calcium free medium, the calcium level in ER storage was higher and the change rate was lower in comparison to that under the normal culture (Figure 1J&K). Surprisingly, inhibition of PLC signaling reduced both [Ca2+]C and [Ca2+]ER (Figure 1D&E, I&G), which may be resulted from inhibited extracellular calcium flow through the plasma membrane. These data demonstrated that PDGF-induced [Ca2+]C increase require both extracellular calcium influx and calcium release from ER storage, while ER calcium release was kindly dependent on extracellular calcium influx.
3.2. PDGF-induced increase of cytoplasmic calcium is regulated by ER calcium release and extracellular calcium flow through their calcium channels
Regarding the crucial roles of calcium exchanges between different compartments in mediating cytoplasmic calcium level, we checked the involvements of several common calcium channels, such as L-type calcium channels on plasma membrane (nifedipine as inhibitor), IP3R calcium channel (2-APB) and SERCA calcium pump (thapsigargin) on ER membrane.
In comparison with the control group, the pretreated cells with nifedipine for one hour resulted in largely inhibited increase of [Ca2+]C induced by PDGF stimulation (Figure 2A-C), similar to the observation in calcium-free medium (Figure 1C&D). This result confirmed that calcium flow through the plasma membrane had essential role in cytoplasmic calcium increase. Pretreated inhibition of ER IP3R channel with 2-APB or ER SERCA pump with thapsigargin also substantially inhibited [Ca2+]C increase (Figure 2A-C). Particularly with SERCA inhibition, the cytosolic calcium response to PDGF stimulation almost disappeared (Figure 2A-C). These data indicates that calcium release from ER storage is critical for PDGF-induced cytoplasmic calcium.
Correspondingly, we also checked the effects of these channel inhibitors on PDGF-induced ER calcium release. Compared to the control group, ASM cells pretreated with nifedipine showed extremely low [Ca2+]ER after PDGF stimulation (Figure 2E&F), indicating that ER calcium was nearly emptied to supply cytoplasmic calcium when extracellular calcium flow was inhibited. [Ca2+]ER was similar to control group when IP3R channel on ER was inhibited by 2-APB (Figure 2E&F). However, [Ca2+]ER became extremely low when inhibiting SERCA pump on ER (Figure 2E&F), indicating that active uptake of calcium was essential to maintain the ER calcium storage and supply cytoplasmic calcium. These observations were further confirmed by statistical comparisons of ER calcium FRET levels at 14 min, and the percentages of FRET changes after PDGF stimulations (Figure 2G&H). Therefore, the active calcium exchanges among the extracellular medium, cell cytosol and ER storage are crucial for appropriate calcium signal mediations in cells.
Regarding that one-hour pre-incubation with the inhibitors had remarkedly changed the basal [Ca2+]ER in ER storage (Figure 2E&F), we examined the effect of these inhibitors upon immediate additions before FRET imaging. In comparison to the long pre-incubation ones, PDGF-induced [Ca2+]C showed similar changes under immediate additions (Figure 3A-D). Interestingly, immediate inhibition of SERCA pump with thapsigargin caused a basal increase of [Ca2+]C, implying an inhibitory flux of cytosolic calcium back into ER storage (Figures 3A&B, S1). Reasonably, long incubation with thapsigargin would lead calcium release into the extracellular medium, as constant high [Ca2+]C isn’t normal for cells. PDGF-induced ER calcium changes also showed similar trends with immediate additions of the inhibitors in comparison to one-hour pre-incubation ones (Figure 3E-H). The basal [Ca2+]ER in ER storage with immediate addition of thapsigargin was less different from the control group (Figure 3E&F), in comparison to the remarkedly changed basal [Ca2+]ER under one-hour pre-incubation conditions (Figure 3A&B).
3.4. The regulation of mitochondrial calcium by calcium channels
Mitochondrial organelles are also important in regulating calcium signals and thus maintaining calcium homeostasis for physiological function including ATP production[37]. The calcium uptake is mostly through mitochondrial calcium uniporter complex (MCU) consisting of multiple subunits[38]. We further generated mitochondria-targeting biosensors to investigate calcium exchanges between mitochondria and the calcium pools. By adding signal peptides to Cyto-Ca2+ biosensor, we developed two versions by localizing the biosensor in mitochondrial matrix (Mito-Ca2+) and on the outer mitochondrial membrane (Out-Mito-Ca2+)[34,35] (Figure 1A).
The outer mitochondrial membrane is facing to the cytoplasm, and the Out-Mito-Ca2+ biosensor measured largely similar changes in calcium level to the Cyto-Ca2+ one. As shown in Figure 4A&C, PDGF induced calcium increase at outer mitochondrial membrane sub-region ([Ca2+]OM) displaying a sustaining Ca2+ signal, and inhibition of PLC pathway with U73122 had little effect. When ASM cells were in calcium-free medium, the [Ca2+]OM increase was markedly reduced, and almost completely inhibited by further addition of U73122 (Figure 4B&C), similar to the responses of cytosolic [Ca2+]C (Figure 1B-D). The observations were confirmed by statistical quantifications and comparisons among these few experimental conditions (Figure 4D). When inhibited the membrane L-type or ER IP3R calcium channels with nifedipine or 2-APB, PDGF-induced [Ca2+]OM increase measured by Out-Mito-Ca2+ biosensor was markedly reduced in contrast to the control group (Figure 4E-G, Movie S3). Inhibition of MCU channel on mitochondria with Ruthenium red (RuR) didn’t change PDGF-induced [Ca2+]OM increase (Figure 4E-G). Hence, PDGF-induced calcium level at the outer mitochondrial membrane sub-region showed similar regulatory response to the cytosolic one.
We further tried to measure the calcium level within the mitochondrial matrix (mito-matrix) by Mito-Ca2+ biosensor. In our experiments, the mito-matrix-localized biosensor protein didn’t achieve bright fluorescence as the other three versions. We didn’t present the time courses of FRET data in considering that the images didn’t reach the same quality as the others in this work.
3.5. Characterization of calcium exchanges at cellular compartments
Calcium concentrations are inhomogeneous in cells [40,41]. The subcellular-located calcium biosensors allow to measure calcium levels and study the regulatory mechanisms at the different cellular compartments. Our measurements by the FRET biosensors showed that ER had the highest calcium level, which FRET ratio reached 3.9±0.046, followed by cytosolic calcium (1.6±0.022) and mitochondrial outer membrane calcium (1.3±0.021), and mitochondrial matrix calcium was the lowest (0.79±0.018) among them (Figure 5A). This contrast trend at the cellular compartments was still kept after PDGF stimulation for 5 min (Figure 5B). As a note, the FRET ratio and calcium concentration are not in a linear relationship. The data indicated that mitochondrial calcium had own regulatory mechanism, and showed a lower calcium level compared to cytosolic one.
We further applied inhibitors of calcium channels to check the calcium flow between the subcellular compartments without PDGF stimulation. It was found that inhibition of ER calcium uptake with thapsigargin (ER calcium pump inhibitor) caused a rapid increase in [Ca2+]C (Figure 5B&C), which was in corresponding to a sharp decrease in [Ca2+]ER (Figure 5D&E). Inhibition of L-type calcium channel on the plasma (by nifedipine), or IP3R channel on the ER (by 2-APB) didn’t cause significant difference in changes of calcium levels, compared to the control group (Figure 5B-E). Hence, the calcium exchanges between the cytosol and ER storage are very dynamic and active in cells.
We also tried inhibition of mitochondrial calcium uniporter (MCU) channel on mitochondria with Ruthenium Red (RuR) to check the calcium exchanges between mitochondria and the cytoplasm. The inhibition of MCU channel didn’t cause significant difference in the change of cytosolic calcium level [Ca2+]C compared to the control group (Figure 5F-H), whereas RuR treatment reduced calcium release from ER storage with PDGF stimulation (Figure 5I-K). It was reported that RuR also inhibits ryanodine receptor (RyR) located on ER for calcium release[42], of which is consistent in our observation (Figure 5I-K). Therefore, mitochondria uptake calcium through own calcium channels, and maintains a relatively low calcium level. We didn’t find evidence showing a Ca2+-pool within mitochondria to regulate Ca2+ level in the cytoplasm.
4. Discussion
The intracellular calcium homeostasis is extremely important and closely related to many cellular physiological activities [43,44,45]. PDGF can also regulate physiological activities such as cell proliferation, migration, and differentiation by altering intracellular calcium levels[9]. We investigated PDGF-induced shuttles of intracellular calcium among different cellular compartments by generating calcium FRET biosensors tagged with subcellular-targeted peptides.
Previous studies have shown that PDGF stimulation increases calcium influx[46]. We confirmed that PDGF-induced cytosolic calcium increase is also related to the regulation of the ER. Our experiments demonstrated that extracellular calcium flow through the plasma membrane can affect the calcium regulations of cytoplasm, ER, and mitochondria. When the calcium in the extracellular environment is emptied, PDGF-induced [Ca2+]C increase in cytoplasm, and the release of calcium from the ER are significantly inhibited (Figure 1). After decreased ER calcium release, the cytoplasm lose important sources of calcium signaling (Figure 2 and Figure 3). Therefore, the calcium signaling changes caused by PDGF are mainly provided by the extracellular environment and ER, flowing towards the cytoplasm including mitochondria.
Cells have a certain self-protection mechanism, and when calcium overload occurs in the ER, certain pathways are activated to maintain intracellular calcium homeostasis[47,48]. In addition, we also observed changes in intracellular calcium signal flow after inhibiting IP3R, L-type calcium pathway, and SERCA calcium pump. The inhibition of IP3R can reduce the upregulation of cytoplasmic and mitochondrial calcium caused by PDGF. However, there is not much inhibitory effect on the calcium release from the ER. This may be regulated by another calcium release channel ryanodine receptor on ER membrane. Additionally, our recent work reported that L-type calcium channel is not relevant with mechanical stretch-activated ERK via calcium signals[49]. Hence, chemical and biomechanical activations of intracellular calcium may be through different sets of calcium channels on the plasma membrane.
After a certain inhibition of calcium influx in cells, the average calcium changes in cytoplasm, and mitochondria decreased significantly, which is consistent with the experimental results in calcium free culture medium. Surprisingly, PDGF-induced ER calcium release was attenuated with reserved higher [Ca2+]ER after immediate switch to calcium free culture medium (Figure 1I&J). This indicates that extracellular calcium influx may enhance ER release of calcium. This observation can be attributed to calcium-dependent activation of ryanodine receptor (RyR) resulting in ER calcium release[50]. Our data proved that extracellular calcium influx effects ER calcium release and is essential for PDGF-induced full activation of cytosolic calcium signals in cells.
The inhibition of SERCA calcium pump initially leads to an increase in cytoplasmic calcium levels, but gradually decreases over time and becomes unresponsive to PDGF stimulation (Figure 2 and Figure 3) [51]. Our experimental data confirmed the process for calcium release from ER into the cytosol and further exported into the medium when addition of SERCA inhibitor (Figure 5C-F). Hence, the dynamic and active calcium shuttling between ER storage and cytoplasm is essential to maintain calcium homeostasis in cells.
The calcium level near the outer mitochondrial membrane shows similar responses to PDGF-induced cytosolic calcium, which was not influenced by the mitochondrial MCU channel inhibition with RuR (Figure 4). In addition, treatment with RuR inhibitor didn’t interfere with PDGF-induced cytosolic calcium increase, but significantly reduce ER's release of calcium (Figure 5G-L), likely through RuR inhibition of ryanodine receptors on ER[42]. Hypothetically, the extracellular calcium might have a compensative role in the cytosolic calcium from the less ER release with RuR treatment.
5. Conclusions
Based on our study with subcellular-targeted FRET biosensors along with precious reports, the calcium exchanges and regulations among different compartments of the cells are summarized in Figure 6. As shown in the schematics, PDGF-activated PDGFR signaling mediates extracellular calcium flow through plasma membrane (which further activates calcium release from endoplasmic reticulum (ER) storage as an amplifying effect) and ER calcium release through PLC-IP3-IP3R and RyR pathways, which resulted in cytosolic [Ca2+]C increase; ER uptakes calcium through SERCA pump to keep the calcium homeostasis in cells, and mitochondria uptake calcium through MCU channels. When without calcium in the medium, upon PDGF stimulation, no extracellular calcium entry leads to inhibited ER calcium release and reduced cytosolic [Ca2+]C increases.
Author Contributions
M.O. and L.D. designed the research; B.Z. performed the majority of experiments; B.Z., C.L. and M.O. carried the major data organization and analysis; C.L., J.F., Q.Z. and S.Z. helped with project discussion, experiments, or reagent preparations; L.D. provided the setups of equipment; B.Z., C.L., M.O. and L.D. prepared the paper.
Acknowledgments
The cytosolic calcium FRET biosensor is from Professor Yingxiao Wang Lab (University of Southern California); the work was assisted with technical help from Yan Pan, Jingjing Li, and Lei Liu (Changzhou University); the illustration of Figure 7 was produced by Yang Jin (Chongqing University). This project was supported financially by National Natural Science Foundation of China (NSFC12372312, 11872129), and Project of “Jiangsu Specially-appointed Professor” (M.O.); National Natural Science Foundation of China (NSFC12272063) (L.D.).
Statement for No Conflict of Interest
The authors declare no conflicts of interest with the contents of this article.
References
- Bidaux, G., et al., Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J Clin Invest, 2007. 117(6): p. 1647-57. [CrossRef]
- Egan, C.G., et al., PDGF-induced signaling in proliferating and differentiated vascular smooth muscle: effects of altered intracellular Ca2+ regulation. Cardiovasc Res, 2005. 67(2): p. 308-16. [CrossRef]
- Patergnani, S., et al., Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int J Mol Sci, 2020. 21(21). [CrossRef]
- Pingel, J., et al., Altered gene expression levels of genes related to muscle function in adults with cerebral palsy. Tissue Cell, 2022. 76: p. 101744. [CrossRef]
- Du, X., et al., Research progress in the mechanism of calcium ion on contraction and relaxation of airway smooth muscle cells. J Recept Signal Transduct Res, 2021. 41(2): p. 117-122. [CrossRef]
- Wang, I.Y., et al., A mathematical analysis of agonist- and KCl-induced Ca(2+) oscillations in mouse airway smooth muscle cells. Biophys J, 2010. 98(7): p. 1170-81. [CrossRef]
- Pepe, C., et al., Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol, 2005. 116(3): p. 544-9. [CrossRef]
- Kardas, G., et al., Role of Platelet-Derived Growth Factor (PDGF) in Asthma as an Immunoregulatory Factor Mediating Airway Remodeling and Possible Pharmacological Target. Front Pharmacol, 2020. 11: p. 47. [CrossRef]
- Spinelli, A.M., et al., Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch, 2012. 464(5): p. 481-92. [CrossRef]
- Wang, W.A., L.B. Agellon, and M. Michalak, Organellar Calcium Handling in the Cellular Reticular Network. Cold Spring Harb Perspect Biol, 2019. 11(12). [CrossRef]
- Case, R.M., et al., Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signalling system. Cell Calcium, 2007. 42(4-5): p. 345-50. [CrossRef]
- Xu, C., B. Bailly-Maitre, and J.C. Reed, Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest, 2005. 115(10): p. 2656-64. [CrossRef]
- Giacomello, M., et al., Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ, 2007. 14(7): p. 1267-74. [CrossRef]
- Bonora, M., et al., Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene, 2015. 34(12): p. 1475-86. [CrossRef]
- Catterall, W.A., Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol, 2000. 16: p. 521-55. [CrossRef]
- Prakriya, M. and R.S. Lewis, Store-Operated Calcium Channels. Physiol Rev, 2015. 95(4): p. 1383-436.
- Mykytyn, K. and C. Askwith, G-Protein-Coupled Receptor Signaling in Cilia. Cold Spring Harb Perspect Biol, 2017. 9(9). [CrossRef]
- Berridge, M.J., Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta, 2009. 1793(6): p. 933-40. [CrossRef]
- Berridge, M.J., The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev, 2016. 96(4): p. 1261-96. [CrossRef]
- Schmitz, E.A., H. Takahashi, and E. Karakas, Structural basis for activation and gating of IP3 receptors. Nature Communications, 2022. 13(1). [CrossRef]
- Ozawa, T., Ryanodine-sensitive Ca2+ release mechanism in non-excitable cells (Review). Int J Mol Med, 2001. 7(1): p. 21-5. [CrossRef]
- Mekahli, D., et al., Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol, 2011. 3(6). [CrossRef]
- Kamer, K.J. and V.K. Mootha, The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol, 2015. 16(9): p. 545-53. [CrossRef]
- Marchi, S., S. Patergnani, and P. Pinton, The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta, 2014. 1837(4): p. 461-9. [CrossRef]
- Madec, A.M., et al., Role of mitochondria-associated endoplasmic reticulum membrane (MAMs) interactions and calcium exchange in the development of type 2 diabetes. Int Rev Cell Mol Biol, 2021. 363: p. 169-202. [CrossRef]
- Wang, Y., J.Y. Shyy, and S. Chien, Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng, 2008. 10: p. 1-38. [CrossRef]
- Ouyang, M., et al., Sensitive FRET Biosensor Reveals Fyn Kinase Regulation by Submembrane Localization. ACS Sens, 2019. 4(1): p. 76-86. [CrossRef]
- Chin, D. and A.R. Means, Calmodulin: a prototypical calcium sensor. Trends Cell Biol, 2000. 10(8): p. 322-8. [CrossRef]
- Tay, L.H., O. Griesbeck, and D.T. Yue, Live-cell transforms between Ca2+ transients and FRET responses for a troponin-C-based Ca2+ sensor. Biophys J, 2007. 93(11): p. 4031-40. [CrossRef]
- Martin, M.E. and K.G. Rice, Peptide-guided gene delivery. Aaps j, 2007. 9(1): p. E18-29. [CrossRef]
- Ouyang, M.X., et al., Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proceedings of the National Academy of Sciences of the United States of America, 2008. 105(38): p. 14353-14358. [CrossRef]
- Miyawaki, A., et al., Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature, 1997. 388(6645): p. 882-887. [CrossRef]
- Kim, T.J., et al., Distinct mechanisms regulating mechanical force-induced Ca²⁺ signals at the plasma membrane and the ER in human MSCs. Elife, 2015. 4: p. e04876. [CrossRef]
- Mehta, S., et al., Calmodulin-controlled spatial decoding of oscillatory Ca2+ signals by calcineurin. Elife, 2014. 3: p. e03765. [CrossRef]
- Ashrafi, G., et al., Molecular Tuning of the Axonal Mitochondrial Ca(2+) Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron, 2020. 105(4): p. 678-687.e5. [CrossRef]
- Qin, Q., et al., for Ratiometric and High-Throughput Live-Cell Image Visualization and Quantitation. Frontiers in Physics, 2019. 7. [CrossRef]
- Giorgi, C., S. Marchi, and P. Pinton, The machineries, regulation and cellular functions of mitochondrial calcium. Nature Reviews Molecular Cell Biology, 2018. 19(11): p. 713-730. [CrossRef]
- Fan, M.R., et al., Structure and mechanism of the mitochondrial Ca uniporter holocomplex. Nature, 2020. 582(7810): p. 129-+. [CrossRef]
- Hamblin, M.R., Mechanisms and Mitochondrial Redox Signaling in Photobiomodulation. Photochemistry and Photobiology, 2018. 94(2): p. 199-212. [CrossRef]
- Bagur, R. and G. Hajnóczky, Intracellular Ca Sensing: Its Role in Calcium Homeostasis and Signaling. Molecular Cell, 2017. 66(6): p. 780-788. [CrossRef]
- Schulte, A. and R. Blum, Shaped by leaky ER: Homeostatic Ca fluxes. Frontiers in Physiology, 2022. 13. [CrossRef]
- Van Petegem, F., Ryanodine Receptors: Structure and Function. Journal of Biological Chemistry, 2012. 287(38): p. 31624-31632. [CrossRef]
- Cui, C., et al., Targeting calcium signaling in cancer therapy. Acta Pharm Sin B, 2017. 7(1): p. 3-17. [CrossRef]
- Berridge, M.J., M.D. Bootman, and H.L. Roderick, Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol, 2003. 4(7): p. 517-29. [CrossRef]
- Mahn, K., et al., Ca(2+) homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax, 2010. 65(6): p. 547-52. [CrossRef]
- Emter, C.A. and D.K. Bowles, Store-operated Ca(2+) entry is not essential for PDGF-BB induced phenotype modulation in rat aortic smooth muscle. Cell Calcium, 2010. 48(1): p. 10-8. [CrossRef]
- Zhao, S., et al., ER Ca(2+) overload activates the IRE1α signaling and promotes cell survival. Cell Biosci, 2023. 13(1): p. 123. [CrossRef]
- Schulte, A. and R. Blum, Shaped by leaky ER: Homeostatic Ca(2+) fluxes. Front Physiol, 2022. 13: p. 972104. [CrossRef]
- Fang, X., et al., FRET Visualization of Cyclic Stretch-Activated ERK via Calcium Channels Mechanosensation While Not Integrin β1 in Airway Smooth Muscle Cells. Frontiers in Cell and Developmental Biology, 2022. 10. [CrossRef]
- Kamishima, T. and J.M. Quayle, Ca-induced Ca release in cardiac and smooth muscle cells. Biochemical Society Transactions, 2003. 31: p. 943-946. [CrossRef]
- Iida-Tanaka, N., et al., Involvement of intracellular Ca(2+) in the regulatory volume decrease after hyposmotic swelling in MDCK cells. J Pharmacol Sci, 2007. 104(4): p. 397-401. [CrossRef]
Figure 1.
PDGF-induced calcium changes in cell cytoplasm and ER storage. The calcium concentrations were measured by calcium FRET biosensors. (A) The depictions of the four versions of calcium FRET biosensors, as described in the Methods. (B, C) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF (50 ng/mL) in ASM cells pretreated with DMSO (as control) or U73122 (10 μM) in normal culture medium (B), or in calcium-free culture medium (C). (D) Quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) in the ASM cells under the (B, C) conditions in normal or Ca2+-free medium. (E, F) Statistical comparisons of peak values of FRET/ECFP ratio (E), and the FRET change rates (F) from the quantified curves in (D). The sample sizes of Cyto-Ca2+ FRET measurements for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 55, 40, 72, 66, respectively. (G, H) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells pretreated with DMSO or U73122 in normal medium (G) or Ca2+-free medium (H). (I, J, K) Quantified time-course curves of ER calcium FRET ratio (I), and the statistical comparisons of peak values of FRET/ECFP ratio (J) and the FRET change rates (K) under the various conditions of (G, H). The sample sizes of ER-Ca2+ FRET measurements for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 58, 47, 45, 47, respectively.
Figure 1.
PDGF-induced calcium changes in cell cytoplasm and ER storage. The calcium concentrations were measured by calcium FRET biosensors. (A) The depictions of the four versions of calcium FRET biosensors, as described in the Methods. (B, C) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF (50 ng/mL) in ASM cells pretreated with DMSO (as control) or U73122 (10 μM) in normal culture medium (B), or in calcium-free culture medium (C). (D) Quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) in the ASM cells under the (B, C) conditions in normal or Ca2+-free medium. (E, F) Statistical comparisons of peak values of FRET/ECFP ratio (E), and the FRET change rates (F) from the quantified curves in (D). The sample sizes of Cyto-Ca2+ FRET measurements for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 55, 40, 72, 66, respectively. (G, H) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells pretreated with DMSO or U73122 in normal medium (G) or Ca2+-free medium (H). (I, J, K) Quantified time-course curves of ER calcium FRET ratio (I), and the statistical comparisons of peak values of FRET/ECFP ratio (J) and the FRET change rates (K) under the various conditions of (G, H). The sample sizes of ER-Ca2+ FRET measurements for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 58, 47, 45, 47, respectively.
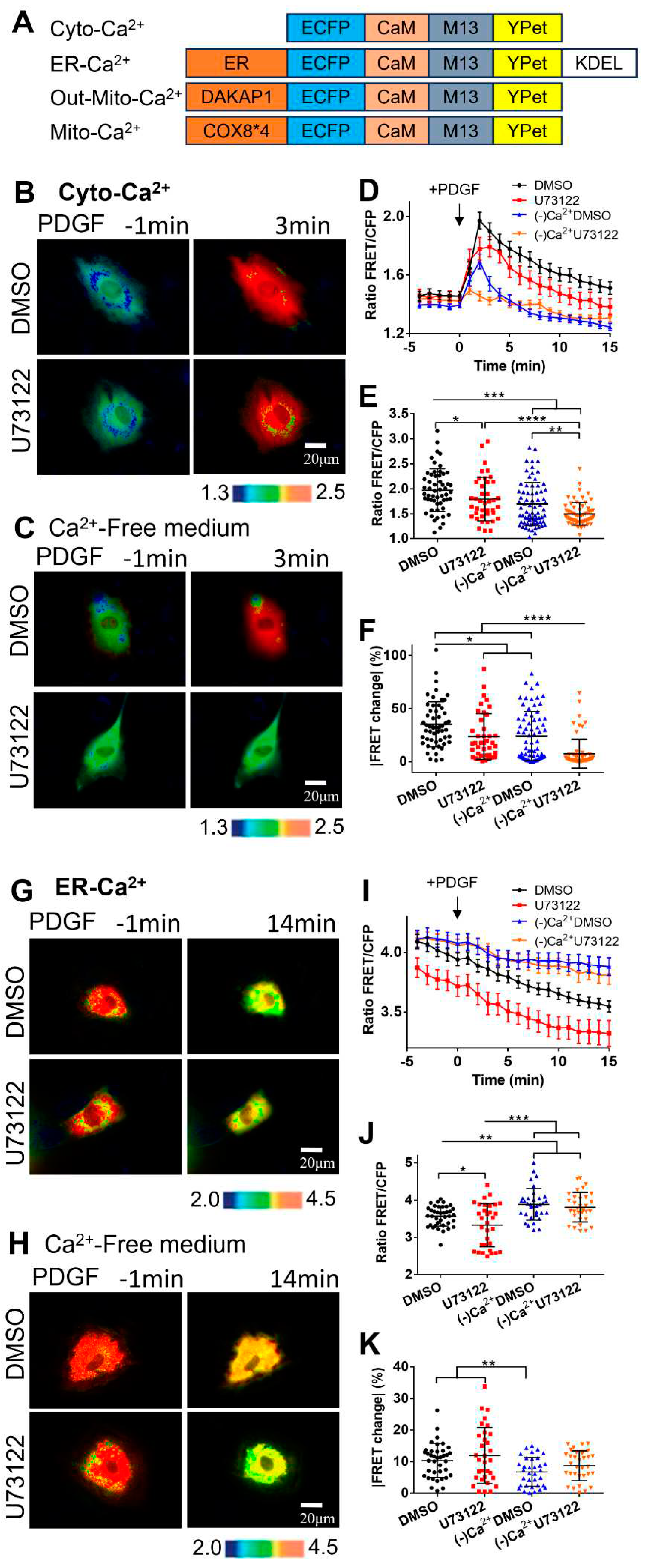
Figure 2.
PDGF-induced calcium changes in cell cytoplasm and ER storage with one-hour pre-incubation of calcium channel inhibitors. (A) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF in ASM cells pretreated for one hour with DMSO (as control), 2-APB (100 μM), nifedipine (10 μM), and thapsigargin (10 μM). (B) Quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) in the ASM cells under the (A) conditions. (C, D) Statistical comparisons of peak values of FRET/ECFP ratio (C), and the FRET change rates (D) from the quantified curves in (B). The sample sizes of Cyto-Ca2+ FRET measurements for DMSO, 2-APB, nifedipine, and thapsigargin are 66, 53, 49, 51, respectively. (E) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells pretreated with DMSO, 2-APB, nifedipine, and thapsigargin. (F, G, H) Quantified time-course curves of ER calcium FRET ratio (F), and the statistical comparisons of peak values of FRET/ECFP ratio (G) and the FRET change rates (H) under the various conditions of (E). The sample sizes of ER-Ca2+ FRET measurements for DMSO, 2-APB, nifedipine, and thapsigargin are 53, 52, 50, 71, respectively.
Figure 2.
PDGF-induced calcium changes in cell cytoplasm and ER storage with one-hour pre-incubation of calcium channel inhibitors. (A) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF in ASM cells pretreated for one hour with DMSO (as control), 2-APB (100 μM), nifedipine (10 μM), and thapsigargin (10 μM). (B) Quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) in the ASM cells under the (A) conditions. (C, D) Statistical comparisons of peak values of FRET/ECFP ratio (C), and the FRET change rates (D) from the quantified curves in (B). The sample sizes of Cyto-Ca2+ FRET measurements for DMSO, 2-APB, nifedipine, and thapsigargin are 66, 53, 49, 51, respectively. (E) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells pretreated with DMSO, 2-APB, nifedipine, and thapsigargin. (F, G, H) Quantified time-course curves of ER calcium FRET ratio (F), and the statistical comparisons of peak values of FRET/ECFP ratio (G) and the FRET change rates (H) under the various conditions of (E). The sample sizes of ER-Ca2+ FRET measurements for DMSO, 2-APB, nifedipine, and thapsigargin are 53, 52, 50, 71, respectively.
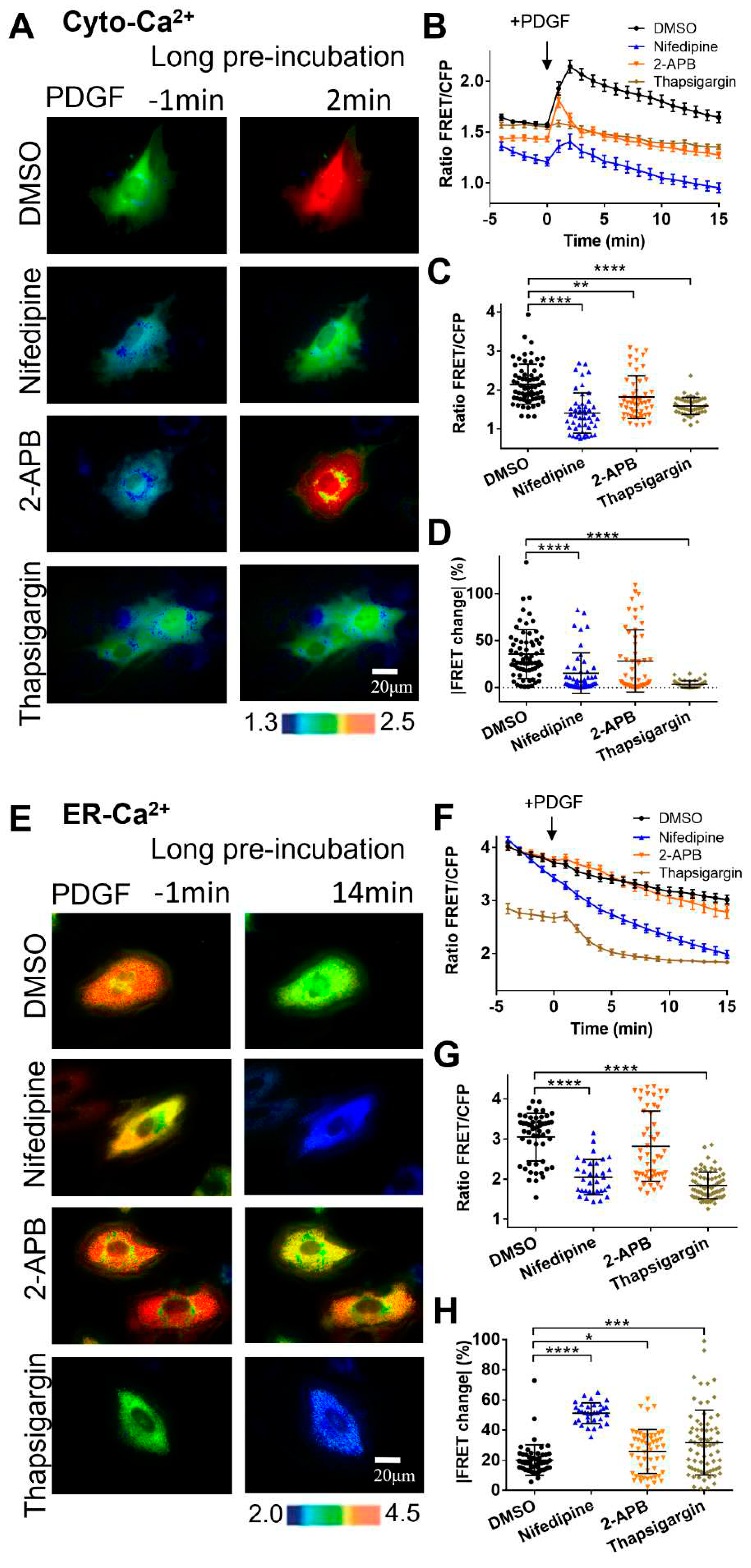
Figure 3.
PDGF-induced calcium changes in cell cytosol and ER storage under immediate addition of calcium channel inhibitors before microscopic imaging. (A-D) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF in ASM cells with immediate addition of DMSO (as control), 2-APB (100 μM), nifedipine (10 μM), and thapsigargin (10 μM) (A), the corresponding quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) (B), and statistical comparisons of peak values of FRET ratio (C), and the FRET change rates (D). The sample sizes of Cyto-Ca2+ FRET for DMSO, 2-APB, nifedipine, and thapsigargin are 37, 36, 32, 32, respectively. (E-H) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells with immediate addition of DMSO, 2-APB, nifedipine, and thapsigargin (E), quantified time-course curves of ER calcium FRET ratio (F), and the statistical comparisons of peak values of FRET/ECFP ratio (G) and the FRET change rates (H). The sample sizes of ER-Ca2+ FRET for DMSO, 2-APB, nifedipine, and thapsigargin are 37, 42, 38, 38, respectively.
Figure 3.
PDGF-induced calcium changes in cell cytosol and ER storage under immediate addition of calcium channel inhibitors before microscopic imaging. (A-D) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF in ASM cells with immediate addition of DMSO (as control), 2-APB (100 μM), nifedipine (10 μM), and thapsigargin (10 μM) (A), the corresponding quantified time-course curves of cytoplasmic calcium FRET ratio (FRET/ECFP) (B), and statistical comparisons of peak values of FRET ratio (C), and the FRET change rates (D). The sample sizes of Cyto-Ca2+ FRET for DMSO, 2-APB, nifedipine, and thapsigargin are 37, 36, 32, 32, respectively. (E-H) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF in ASM cells with immediate addition of DMSO, 2-APB, nifedipine, and thapsigargin (E), quantified time-course curves of ER calcium FRET ratio (F), and the statistical comparisons of peak values of FRET/ECFP ratio (G) and the FRET change rates (H). The sample sizes of ER-Ca2+ FRET for DMSO, 2-APB, nifedipine, and thapsigargin are 37, 42, 38, 38, respectively.
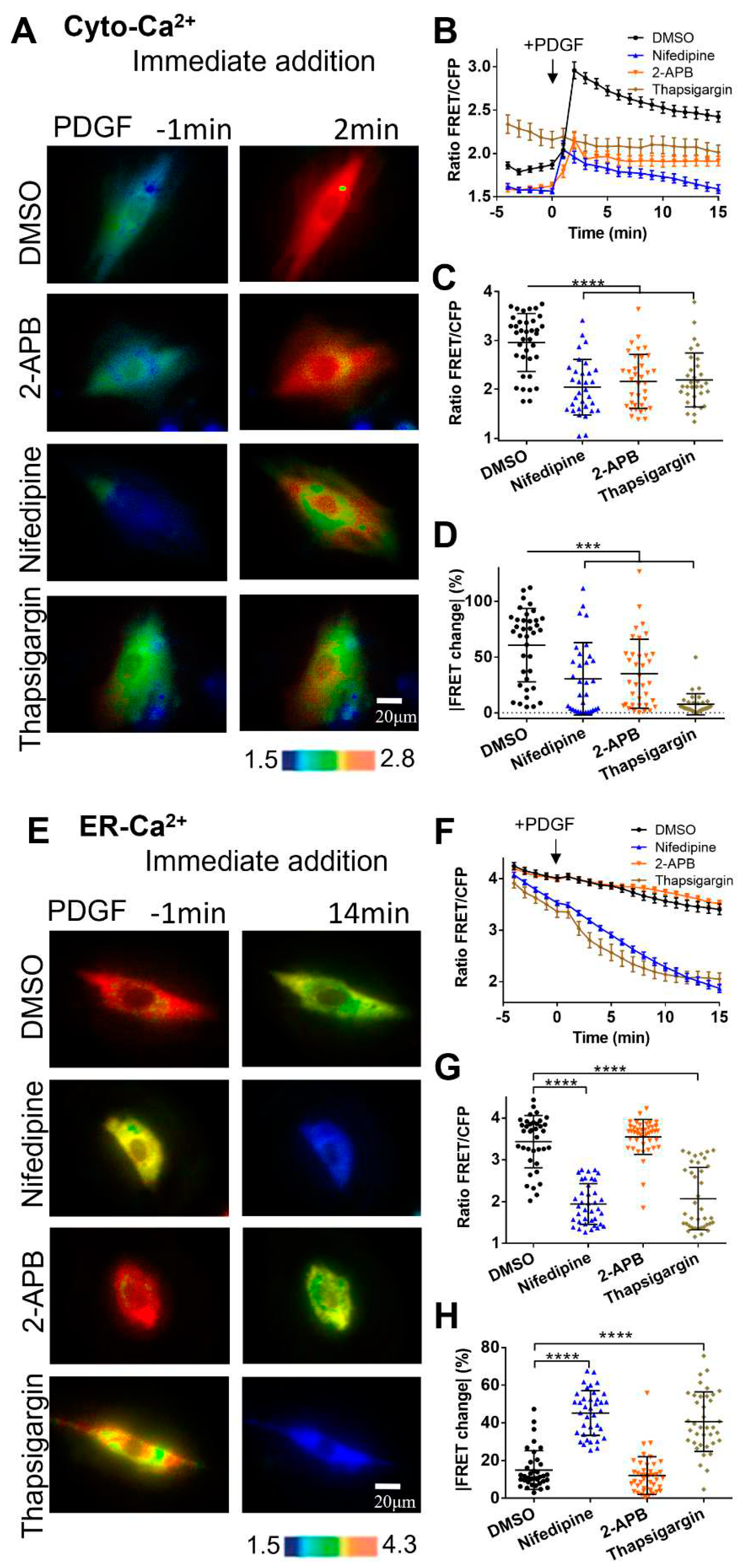
Figure 4.
PDGF-induced calcium changes at the outer mitochondrial membrane measured by Out-Mito-Ca2+ FRET biosensor. (A, B) PDGF-induced ratiometric FRET images of ASM cells pretreated with DMSO or U73122 (10 μM) in normal culture medium (A), or calcium-free medium (B). (C-D) Quantified time-course curves of Out-Mito-Ca2+ FRET ratio (C), and statistical comparisons of peak values of FRET/ECFP ratio (D) under the (A, B) conditions. The sample sizes for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 33, 33, 40, 33, respectively. (E) Ratiometric FRET images of Out-Mito-Ca2+ biosensor induced by PDGF in ASM cells with one-hour pre-incubation of DMSO, 2-APB (100 μM), nifedipine (10 μM), and RuR (10 μM). (F, G) Quantified time-course curves of Out-Mito-Ca2+ FRET ratio (F), and statistical comparisons of peak values of FRET/ECFP ratio (G). The sample sizes of Out-Mito-Ca2+ FRET for DMSO, 2-APB, nifedipine, thapsigargin, and RuR are 48, 38, 36, 36, respectively.
Figure 4.
PDGF-induced calcium changes at the outer mitochondrial membrane measured by Out-Mito-Ca2+ FRET biosensor. (A, B) PDGF-induced ratiometric FRET images of ASM cells pretreated with DMSO or U73122 (10 μM) in normal culture medium (A), or calcium-free medium (B). (C-D) Quantified time-course curves of Out-Mito-Ca2+ FRET ratio (C), and statistical comparisons of peak values of FRET/ECFP ratio (D) under the (A, B) conditions. The sample sizes for DMSO, U73122, (-)Ca2+/DMSO, and (-)Ca2+/U73122 are 33, 33, 40, 33, respectively. (E) Ratiometric FRET images of Out-Mito-Ca2+ biosensor induced by PDGF in ASM cells with one-hour pre-incubation of DMSO, 2-APB (100 μM), nifedipine (10 μM), and RuR (10 μM). (F, G) Quantified time-course curves of Out-Mito-Ca2+ FRET ratio (F), and statistical comparisons of peak values of FRET/ECFP ratio (G). The sample sizes of Out-Mito-Ca2+ FRET for DMSO, 2-APB, nifedipine, thapsigargin, and RuR are 48, 38, 36, 36, respectively.
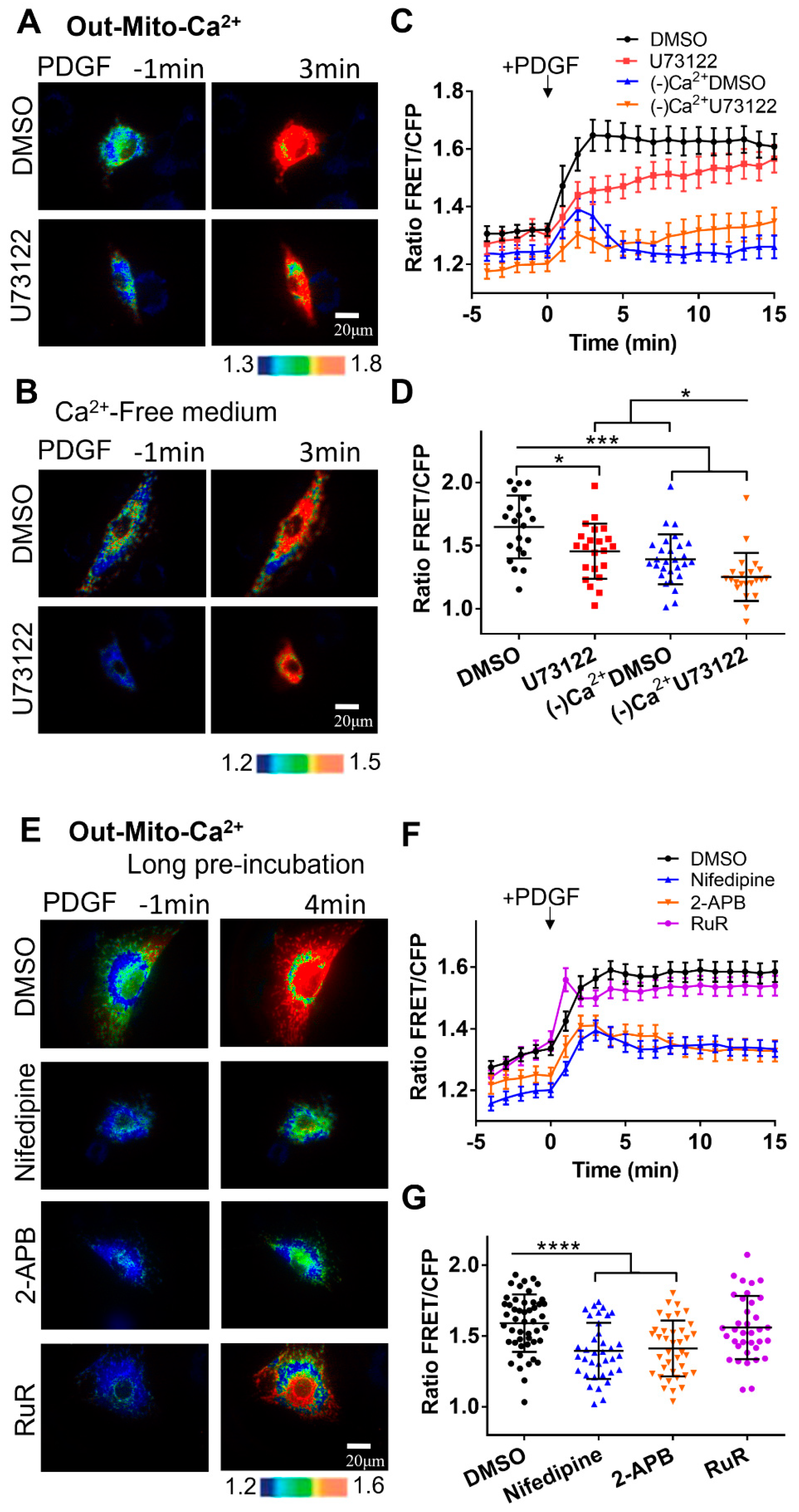
Figure 5.
Characterization of calcium levels at cellular compartments. (A, B) Statistical comparisons of calcium FRET levels at cellular cytoplasm (Cyto-Ca2+), ER (ER-Ca2+), and outer mitochondrial membrane (Out-Mito Ca2+in resting ASM cells (A) or with PDGF stimulation (at 5 min) (B). The ratio values and sample sizes for without/with PDGF stimulations: Cyto-Ca2+ (1.6 ± 0.022, N=87)/(2.05 ± 0.05, N=87), ER-Ca2+ (3.9 ± 0.046, N=78)/(3.7 ± 0.043, N=78), and Out-Mito-Ca2+ (1.3 ± 0.021, N=48)/(1.58 ± 0.032, N=48), respectively. (C-F) The time courses (average values) and FRET ratio comparisons of cytosolic calcium (peak values) (C, D) and ER (at 25 min) (E, F) calcium levels in ASM cells treated with DMSO, nifedipine, 2-APB, and thapsigargin. (G-I) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF (G), the time courses of cytosolic calcium FRET ratio (H), and statistical comparison of peak value of FRET/ECFP ratio (I) in ASM cells pretreated with DMSO, or RuR (10 μM). (J-L) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF (J), the time courses (K) and statistical comparison (at 14 min) of FRET/ECFP ratio (L) in ASM cells pretreated with DMSO, or RuR (10 μM).
Figure 5.
Characterization of calcium levels at cellular compartments. (A, B) Statistical comparisons of calcium FRET levels at cellular cytoplasm (Cyto-Ca2+), ER (ER-Ca2+), and outer mitochondrial membrane (Out-Mito Ca2+in resting ASM cells (A) or with PDGF stimulation (at 5 min) (B). The ratio values and sample sizes for without/with PDGF stimulations: Cyto-Ca2+ (1.6 ± 0.022, N=87)/(2.05 ± 0.05, N=87), ER-Ca2+ (3.9 ± 0.046, N=78)/(3.7 ± 0.043, N=78), and Out-Mito-Ca2+ (1.3 ± 0.021, N=48)/(1.58 ± 0.032, N=48), respectively. (C-F) The time courses (average values) and FRET ratio comparisons of cytosolic calcium (peak values) (C, D) and ER (at 25 min) (E, F) calcium levels in ASM cells treated with DMSO, nifedipine, 2-APB, and thapsigargin. (G-I) Ratiometric FRET images of Cyto-Ca2+ biosensor induced with PDGF (G), the time courses of cytosolic calcium FRET ratio (H), and statistical comparison of peak value of FRET/ECFP ratio (I) in ASM cells pretreated with DMSO, or RuR (10 μM). (J-L) Ratiometric FRET images of ER-Ca2+ biosensor induced with PDGF (J), the time courses (K) and statistical comparison (at 14 min) of FRET/ECFP ratio (L) in ASM cells pretreated with DMSO, or RuR (10 μM).
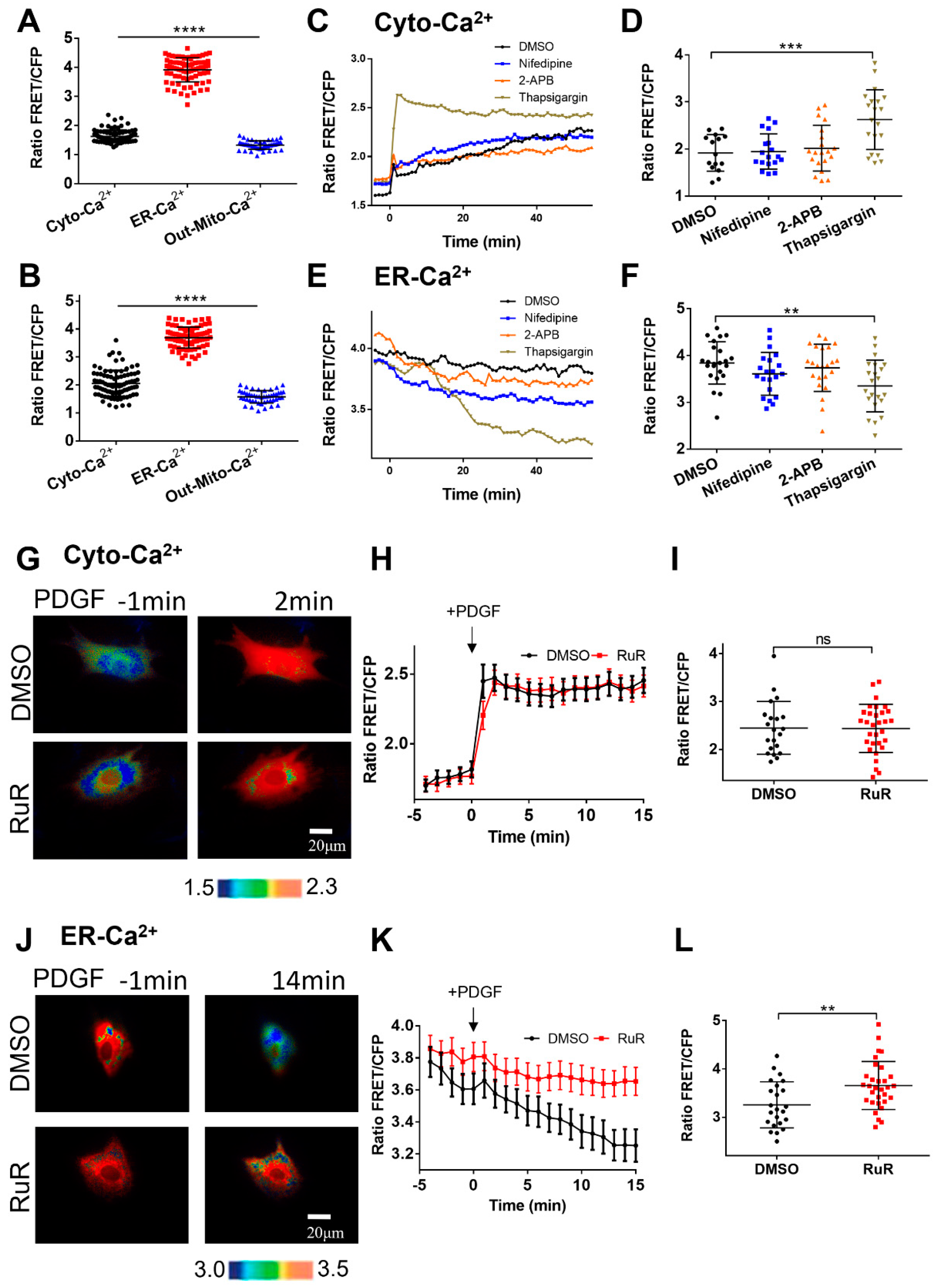
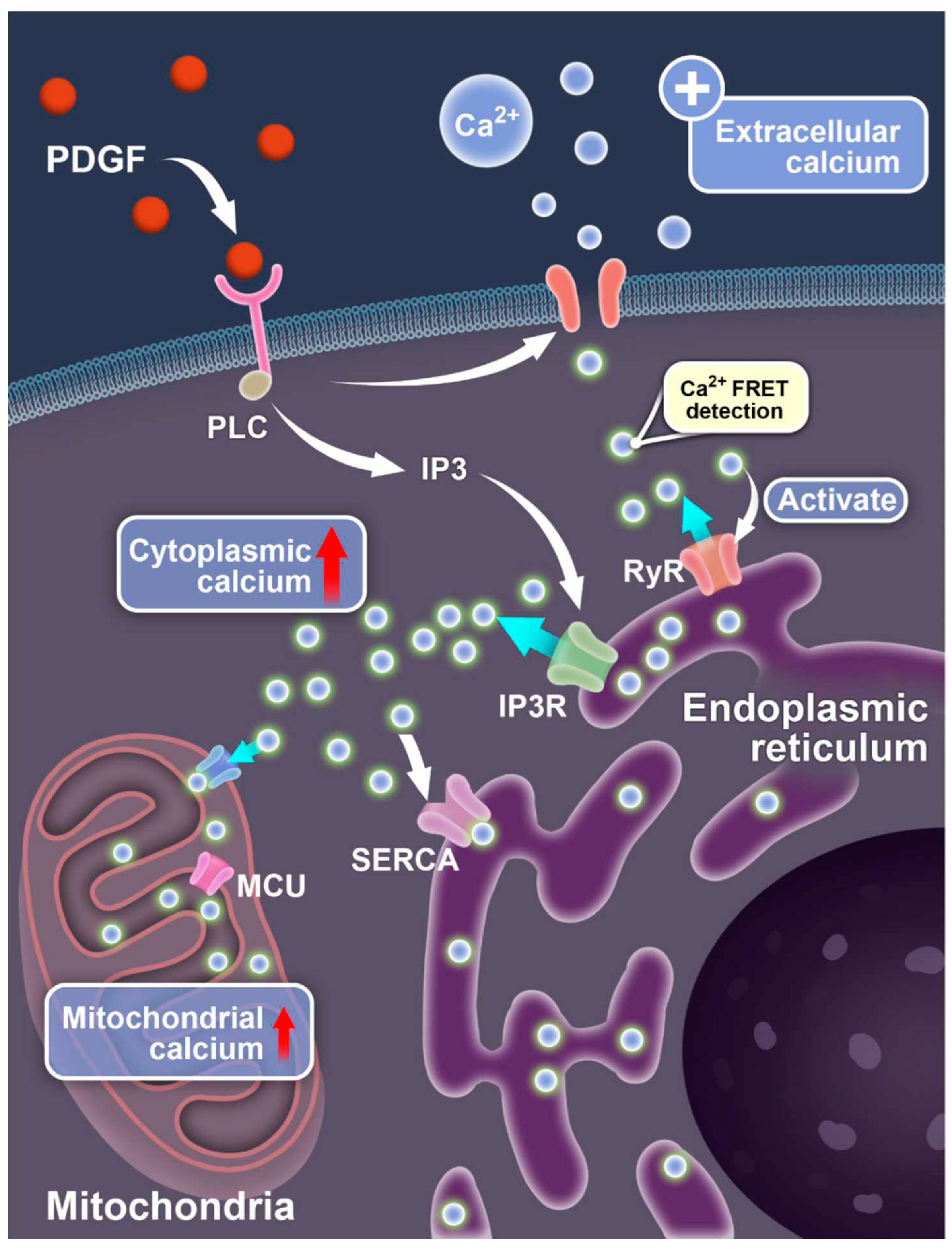
Figure 6.
Illustration for PDGF-induced calcium exchanges between different compartments of the cells. The diagrams show the signaling-mediated Ca2+ ion flow pathways between the extracellular medium, cell cytosol, endoplasmic reticulum, and mitochondria.
Figure 6.
Illustration for PDGF-induced calcium exchanges between different compartments of the cells. The diagrams show the signaling-mediated Ca2+ ion flow pathways between the extracellular medium, cell cytosol, endoplasmic reticulum, and mitochondria.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



